

Abstract
A number of previously undescribed (1–7) and structurally related known (8–17) isobenzofuran-type polyketides were obtained from the fermentation of Penicillium commune P-4-1, an endophytic fungus isolated from the fresh trunk bark of the critically endangered conifer Abies beshanzuensis. Beshanzoides A–D (1–4, resp.) feature a cycloheptanone-containing isobenzofuran ring system hitherto unknown, which might be biosynthesized via two steps of aldol reactions starting from a common co-occurring isobenzofuran-type polyketide as the precursor. The new structures were elucidated by spectroscopic methods, electronic circular dichroism data, and single crystal X-ray diffraction analyses. Beshanzoide E (5) showed antimicrobial activity (MIC: 16 μg mL−1) against Staphylococcus aureus, whereas (±)-strobide A (10) inhibited (MIC: 16 μg mL−1) Candida albicans. Cyclopaldic acid (12) and 3-O-methyl-cyclopaldic acid (13) exhibited inhibitory effects against acetyl-CoA carboxylase 1 (ACC1) with IC50 values of 0.96 and 11.77 μM, respectively. Compound 12 also inhibited (IC50: 7.56 μM) ATP-citrate lyase (ACL).
Four unprecedented cycloheptanone-containing and some related known bioactive polyketides were isolated from an endophytic fungus associated with the critically endangered conifer Abies beshanzuensis.
Introduction
The loss of plant diversity significantly exacerbates the complications in the discovery of new natural products (NPs)-derived drugs owing to rare and endangered plants (REPs) being better botanical sources.1 An important action for the conservation of REPs is to provide valuable resources for researchers for new chemical entities (NCEs).1,2 Pinaceae is one of the top privileged drug-prolific plant families.1b However, a considerable part of Pinaceae plants became ‘vulnerable (VN)’, ‘endangered (EN)’, ‘critically endangered (CR), or even ‘extinct (EX)’. There were 39 species listed in the China Plant Red Data Book published in 1992,3 and the situation is increasingly getting worse.4,5 To make the best of these valuable resources, we keep paying special attention to REPs in Pinaceae with the hope of stimulating active protection and utilization of these fragile plant species in a sustainable way, especially by investigating the renewable needles and twigs from those large trees distributed and managed in the wild or cultivated in botanic gardens.5
Abies beshanzuensis M. H. Wu, among the above mentioned 39 species endemic to China, has been regarded as one of the 12 CR-grade plant species in the world by IUCN (International Union for Conservation of Nature and Natural Resources) since 1987.3,6 Only three living A. beshanzuensis trees nowadays grow in the wild at the summit of Baishanzu Mountain located at Zhejiang Province.3,6 In a preceding work, several rare Abies sesquiterpenoids with interesting structures and bio-activities were obtained from the mass-limited (400 g only) shed barks,7 and the findings immediately attracted much attention among synthetic chemists and pharmacologists.8
Microbes are always important sources for drug discovery.1b,9 Microorganisms living in special environments have been found to yield a large number of bioactive NPs with novel skeletons.10 The significances of chemistry and biology of plant endophytic fungi have been well documented.11 REPs species generally possess extended lifespans and survive in unique habitats,3,6 which hints at the existence of special endophytic fungi that could yield unique bioactive secondary metabolites.11c,12 The plant sample of A. beshanzuensis is too precious but extremely limited to be collected on a large scale for further phytochemical investigation, we thereafter turned to its endophytic fungi.13 This conifer (growing up in a special environmental setting) is still very attractive and promising, blow people away. It encouraged us to carry out a further research of its endophytic fungus.
In this study, we investigate the secondary metabolites of Penicillium commune P-4-1, an endophytic fungus isolated from the fresh trunk bark of the title plant. As a result, seven previously undescribed (1–7) and 10 structurally related known (8–17) isobenzofuran-type polyketides (Fig. 1 and 2) were isolated and characterized. Reported herein are the isolation and structure elucidation along with the biological evaluation of their antimicrobial and inhibitory effects against acetyl-CoA carboxylase1 (ACC1) and ATP-citrate lyase (ACL).
Fig. 1. Chemical structures of compounds 1–4.
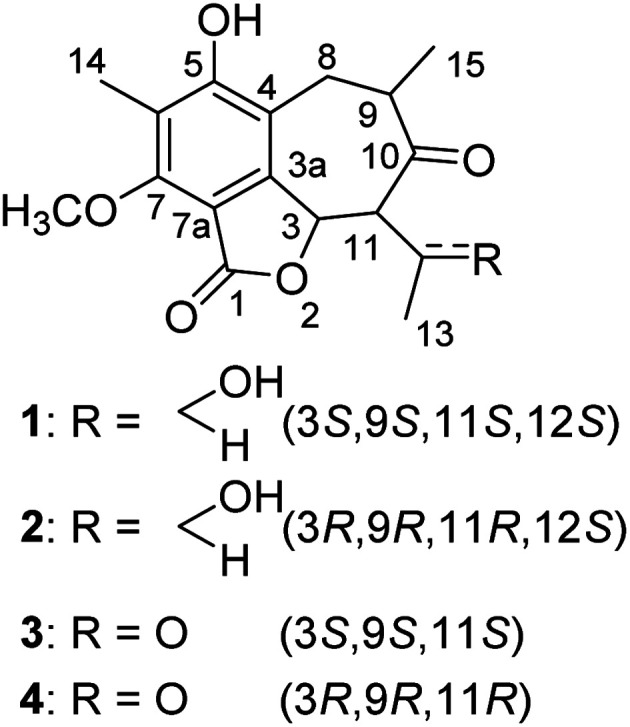
Fig. 2. Chemical structures of compounds 5–17.

Results and discussion
A fungal colony designated as P-4-1 was isolated from the fresh trunk bark of A. beshanzuensis and initially propagated on potato dextrose agar. DNA sequencing and comparison of the ITS rDNA region of P-4-1 to known fungi in the BLAST database suggested it belongs to Penicillium commune. This strain was cultivated on autoclaved solid rice medium at 28 °C for 30 days. The EtOAc extract of the culture was successively subjected to a series of column chromatography (CC) to afford 17 compounds (Fig. 1 and 2). By comparing their observed and reported spectroscopic data and physicochemical properties, the known ones were identified as 7-carboxy-4-hydroxy-6-methoxy-5-methylphthalide (8),14 3,5-dihydroxy-7-methoxy-4-(methoxymethyl)-6-methyl-1(3H)-isobenzofuranone (9), (±)-strobide A (10),15 5,7-dihydroxy-4,6-dimethyl-1(3H)-isobenzofuranone (11),16 cyclopaldic acid (12),15 3-O-methyl-cyclopaldic acid (13),17 5-hydroxy-4-(hydroxymethyl)-7-methoxy-6-methylphthalide (14),12b 4-hydroxy-6-methoxy-5-methyl-1(3H)-isobenzofuranone (15),18 nidulol (16),19 and 5-hydroxy-7-methoxy-4-methylphthalide (17),20 respectively.
Beshanzoide A (1) was obtained as a white powder. The molecular formula C17H20O6 of 1 was determined by its HRESIMS (m/z 321.1329 [M + H]+; calcd for C17H21O6, 321.1333) and 13C-NMR data (Table 1), indicating eight indices of hydrogen deficiency (IHDs). The IR spectrum revealed the presence of hydroxyl (3469 cm−1) and carbonyl groups (1719 and 1702 cm−1). The 1H-NMR data of 1 (Table 1) displayed two secondary methyl groups [δH 1.45 (3H, d, J = 6.5 Hz), 1.15 (3H, d, J = 6.1 Hz)], one tertiary methyl group [δH 2.18 (3H, s)], one methoxy group [δH 3.95 (3H, s)], and two oxymethine resonances [δH 6.31 (1H, d, J = 10.6 Hz), 4.26 (1H, qd, J = 6.5, 2.7 Hz)]. In addition, proton signals assigned to a methylene group [δH 3.03 (1H, br dd, J = 16.6, 5.0 Hz), 2.60 (1H, br dd, J = 16.6, 12.8 Hz)] and two methine protons [δH 3.61 (1H, m), 2.47 (1H, dd, J = 10.6, 2.7 Hz)] were also readily distinguished. The 13C-NMR spectrum of 1 with the aid of HSQC NMR experiment showed 30 carbon resonances (Table 1) classified as four methyls (δC 62.3, 23.1, 16.3, 9.0), one methylene (δC 35.5), four methines (δC 75.5, 70.5, 66.7, 42.5), six quaternary carbons (δC 160.2, 157.8, 148.9, 119.3, 116.1, 109.7), an ester carbonyl (δC 167.8), and a ketonic carbonyl (δC 213.5). The aforementioned 1D NMR data indicated that 1 is partly similar to the co-occurring (±)-strobide A (10),15 an isobenzofuranone derivative. This was further confirmed by the HMBC cross-peaks from H-3 to C-1/C-3a, from H3-14 to C-5/C-6/C-7, and from OCH3 to C-7. In addition, the 1H–1H COSY correlations of H2-8/H-9/H3-15 and H-3/H-11/H-12/H3-13 as well as the HMBC correlations from H2-8 to C-3a/C-4/C-5/C-10, from H3-15 to C-8/C-9/C-10, from H-11 to C-3/C-3a/C-10/C-12, and from H-3 to C-10 suggested the isobenzofuranone ring should be integrated into a bicyclo[5.4.0]undecane backbone (Fig. 3). Therefore, the planar structure of 1 was defined as the first representative of a 5/6/7 tricyclic isobenzofuranone-type skeleton.
1H and 13C NMRa (δ in ppm, J in Hz) of 1–4.
| No. | 1 | 2 | 3 (4) | |||
|---|---|---|---|---|---|---|
| δ H b | δ C c | δ H b | δ C c | δ H b | δ C c | |
| 1 | — | 167.8 | — | 167.8 | — | 167.4 |
| 3 | 6.31 d (10.6) | 75.5 | 6.29 d (10.6) | 76.8 | 6.41 d (10.7) | 75.9 |
| 3a | — | 148.9 | — | 148.6 | — | 147.5 |
| 4 | — | 116.1 | — | 115.8 | — | 115.4 |
| 5 | — | 160.2 | — | 160.1 | — | 160.2 |
| 6 | — | 119.3 | — | 119.2 | — | 119.5 |
| 7 | — | 157.8 | — | 157.7 | — | 157.8 |
| 7a | — | 109.7 | — | 109.7 | — | 109.6 |
| 8 | 2.60 br dd (16.6, 12.8) | 35.5 | 2.58 br dd (17.0, 12.8) | 35.8 | 2.71 br dd (17.1, 12.0) | 35.0 |
| 3.03 br dd (16.6, 5.0) | 3.00 br dd (17.0, 5.2) | 3.09 br dd (17.1, 4.7) | ||||
| 9 | 3.61 m | 42.6 | 3.64 m | 42.6 | 3.57 m | 42.4 |
| 10 | — | 213.5 | — | 212.0 | — | 205.8 |
| 11 | 2.47 dd (10.6, 2.7) | 66.7 | 2.30 dd (10.6, 1.9) | 66.7 | 4.02 d (10.7) | 73.4 |
| 12 | 4.26 qd (6.5, 2.7) | 70.5 | 4.44 qd (6.4, 1.9) | 67.7 | — | 201.0 |
| 13 | 1.45 d (6.5) | 23.1 | 1.27 d (6.4) | 22.2 | 2.34 s | 30.7 |
| 14 | 2.18 s | 9.0 | 2.17 s | 9.0 | 2.19 s | 9.1 |
| 15 | 1.15 d (6.1) | 16.3 | 1.13 d (6.3) | 16.0 | 1.14 d (6.3) | 16.6 |
| 7-OMe | 3.95 s | 62.3 | 3.95 s | 62.3 | 3.96 s | 62.3 |
Assignments were made by a combination of 1D and 2D NMR experiments.
Measured in acetone-d6 (600 MHz).
Measured in acetone-d6 (150 MHz).
Fig. 3. Observed key 2D NMR correlations of 1.

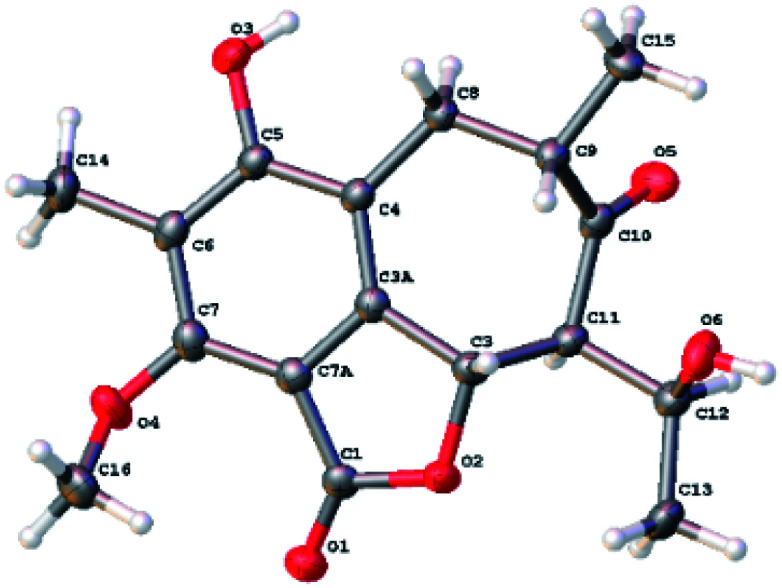
The relative configuration of 1 was determined by analysis of the proton–proton coupling constants (Table 1) and NOESY interactions (Fig. 3). The magnitude of JH-3/H-11 (10.6 Hz) indicated that H-3 and H-11 adopted trans-axial orientations. The clear NOESY correlation between H-3 /H-9 (Fig. 3) revealed their co-facial relationship and both to be β-oriented. Finally, a single-crystal X-ray diffraction experiment with Ga Kα radiation for 1 unambiguously defined its absolute configuration [absolute structure parameter: 0.01(15)] to be (3S,9S,11S,12S) (Fig. 4).
Fig. 4. The ORTEP drawing of 1.
Based on its HRESIMS (m/z 321.1326 [M + H]+; calcd for C17H21O6, 321.1333) and 13C-NMR data (Table 1), the molecular formula C17H20O6 of beshanzoide B (2) was found to be the same as compound 1. The 1H- and 13C-NMR data of compound 2 highly resembled those of 1 (Table 1), indicating that both 2 and 1 share the same planar structure (Fig. 1). Similarly, the large coupling constant (J = 10.6 Hz) between of H-3 and H-11 indicated both protons adopted trans-axial orientations. The NOESY correlation of H-3/H-9 indicated that 2 and 1 even have the same relative configurations at C-3, C-9, and C-11; however, the ECD spectra of 2 and 1 displayed totally reversed Cotton effects (CEs) around 225 and 290 nm (Fig. 5), exciting by the chromophores of isobenzofuranone ring21 and cycloheptanone.22 The absolute configurations at C-3, C-9, and C-11 in 2 were hence elucidated as 3R,9R,11R, respectively. Both 1 and 2 are not enantiomers, otherwise they should have the same set of NMR data. This means that the absolute configuration at C-12 in 2 should be consistent with 1. In fact, the chemical shifts of H-12 (δH 4.44 for 2; 4.26 for 1) and C-12 (δC 67.7 for 2; 70.5 for 1) were obviously differentiated with each other (Table 1), which was attributed to the contrary neighboring stereogenic center (C-11: R in 2, S in 1). Thus, compounds 2 and 1 are diastereoisomers, and the absolute configuration of 2 is determined as 3R,9R,11R,12S.
Fig. 5. Experimental ECD spectra of compounds 1–4.

Beshanzoides C (3) and D (4) were firstly isolated as a mixture of enantiomers possessing a molecular formula of C17H18O6 determined by an [M + H]+ ion at m/z 319.1184 in the HRESIMS and 13C NMR data (Table 1), indicating 3 (4) has one more IHD than compounds 1 and 2. Consistent with this, the 1H- and 13C-NMR spectroscopic data of 3 (4) revealed the presence of a ketocarbonyl group rather than the oxymethine at C-12 in 1 and 2. This was corroborated by the HMBC correlations from H-3/H-11/H3-13 to C-12. Like 1 and 2, the coupling constant of JH-3/H-11 (10.7 Hz) and the NOE correlation of H-3/H-9 in compounds 3 and 4 indicated that they share the same relative configurations at C-3, C-9, and C-11. Interestingly, as H-11 connects with two ketonic carbonyl groups (C-10 and C-12), the hydrogen atom becomes reactive and was partly replaced by the deuterium atom in the deuterated solvents, which weakened the intensity of the signal of H-11 in the 1H-NMR spectrum to some extent (the relative integral reads 0.28). As a result, H-3 (δH 6.41) appeared to be a doublet ridden by a singlet. This phenomenon of deuteration involving a keto–enol tautomerism has been also found for ivorenoid A, a limonoid previously isolated from Khaya ivorensis.23
The enantiomeric mixture 3 (4) were subsequently proven by the flat ECD curve and the near-zero specific rotation value. The following racemic resolution by chiral HPLC separation afforded the enantiomers with equal amount. Finally, the absolute configurations of purified 3 and 4 were assigned as 3S, 9S, 11S and 3R, 9R, 11R, respectively, by comparing their ECD spectra with those of 1 and 2 (Fig. 5). As expected, these were further supported by their opposite optical rotation values {[α]25D +25.0 (c 0.02, MeOH) for 3; −25.0 (c 0.02, MeOH) for 4}.
With a protonated ion peak at m/z 253.0703 [M + H]+ in the HRESIMS, beshanzoide E (5) was found to have the molecular formula of C12H12O6. Comparative analysis of its NMR spectroscopic data (Table 2) with those of to the co-occurring 7-carboxy-4-hydroxy-6-methoxy-5-methylphthalide (8), an isobenzofuranone derivative just recently isolated from marine-derived fungus Aspergillus sp. IMCASMF180035,14 revealed that the structure of 5 is almost the same as that of 8 except for an additional methoxy group [δH 3.91 (3H, s), δC 53.1] appearing in 5. The connection of this methoxy group with C-9 was confirmed by the HMBC correlation from δH 3.91 to δC 167.9 (C-9). Therefore, compound 5 were defined as a methylated derivative of 8, which might be an artifact formed during the isolation.
1H and 13C NMRa (δ in ppm, J in Hz) of 5, 6, 8, and 9.
| No. | 5 | 6 | 8 | 9 | |||||
|---|---|---|---|---|---|---|---|---|---|
| δ H b | δ C c | δ H b | δ H d | δ C c | δ H b | δ C c | δ H b | δ C c | |
| 1 | 171.2 | 171.7 | 171.2 | 169.1 | |||||
| 3 | 5.26 s | 69.3 | 5.18 s | 5.15 s | 68.5 | 5.26 s | 69.2 | 6.52 s | 97.6 |
| 3a | 122.5 | 135.8 | 121.8 | 148.0 | |||||
| 4 | 153.2 | 133.2 | 152.7 | 116.1 | |||||
| 5 | 126.5 | 152.7 | 126.6 | 163.1 | |||||
| 6 | 158.8 | 120.6 | 158.4 | 121.8 | |||||
| 7 | 118.2 | 152.8 | 119.5 | 158.8 | |||||
| 7a | 130.4 | 108.6 | 130.3 | 110.2 | |||||
| 8 | 2.25 s | 9.9 | 2.15 s | 2.06 s | 9.0 | 2.25 s | 9.9 | 4.71 s | 67.7 |
| 9 | 167.9 | 169.3 | 2.16 s | 8.9 | |||||
| 6-OMe | 3.80 s | 62.7 | 3.84 s | 62.6 | |||||
| 7-OMe | 3.88 s | 3.78 s | 62.5 | 3.97 s | 62.5 | ||||
| 8-OMe | 3.43 s | 58.7 | |||||||
| 9-OMe | 3.91 s | 53.1 | |||||||
| 4-OH | 9.39 s | ||||||||
| 5-OH | 9.56 s | ||||||||
Assignments were made by a combination of 1D and 2D NMR experiments.
Measured in CD3OD (400 MHz).
Measured in CD3OD (150 MHz).
Measured in DMSO-d6 (600 MHz).
The molecular formula C10H10O5 of beshanzoide F (6) was determined by its HRESIMS (m/z 233.0418 [M + Na]+, calcd for C10H10O5Na, 233.0420) and 13C NMR data (Table 2). The 1H-NMR spectrum (Table 2) showed signals for two equivalent oxymethylene protons (δH 5.18, 2H, s), one methoxyl group (δH 3.88, 3H, s) and one methyl group (δH 2.15, 3H, s). The 13C-NMR spectrum displayed one ester carbonyl (δC 171.7), six quaternary aromatic (δC 152.8, 152.7, 135.8, 133.2, 120.6 and 108.6), one methylene (δC 68.5), one methoxy (δC 62.5), and one methyl group (δC 9.0). The above data indicated that compound 6 shares the same carbon skeleton with 5, while four substituent groups on aromatic ring in 6 are two hydroxyls, one methyl, and one methoxy. The key HMBC correlations between H-3 (δH 5.18) and C-4 (δC 133.2) and from the methyl group to C-5 (δC 152.7), C-6 (δC 120.6) and C-7 (δC 152.8) suggested that the methyl group is attached to C-6 in 6. However, the location of the methoxy group remains unknown. Another set of 1H-NMR data were, thus, acquired in DMSO-d6 with two hydroxyl proton signals appearing at δH 9.39 and δH 9.56, which were not highly chelated by intramolecular hydrogen bond. The NOE correlations of 4-OH (δH 9.39)/H-3 (δH 5.15), 5-OH (δH 9.56)/H-8 (δH 2.06), and 7-OMe (δH 3.78)/H-8 (δH 2.06) revealed that the methoxy was connected to C-7, and the two hydroxyls were connected to C-4 and C-5, respectively.
Beshanzoide G (7) was isolated as a pair of HPLC inseparable mixture with one set of NMR data. The molecular formula C22H22O11 of 7 was deduced from its HRESIMS (m/z 485.1044 [M + Na]+, calcd for C22H22O11Na, 485.1054). The 1H-NMR spectrum provided one oxymethine (6.55, 1H, s), two equivalent oxymethylene protons (δH 4.83, 2H, s), one methoxyl group (δH 3.96, 3H, s) and one methyl group (δH 2.13, 3H, s). The 13C NMR spectrum showed one ester carbonyl (δC 169.0), six quaternary aromatic (δC 163.3, 158.9, 148.2, 121.7, 116.3, and 110.1), one methine (δC 97.6), one methylene (δC 65.6), one methoxy (δC 62.5), and one methyl group (δC 9.0).26 Based on the above data, compound 7 was proposed to have a completely symmetric framework. Compared with the reported compound (±)-strobide A (10),15 the signals are similar except for the appearance of two equivalent oxymethylene protons linked with the benzene ring in 7 instead of such a methyl group in 10. Therefore, compound 7 might be structured as a dimer by two (±)-strobide A fractions linked by an ether bond. The HMBC correlations from H-8 (δH 4.83) to C-3a (δC 148.2), C-4 (δC 116.3) and C-5 (δC 163.3), from H-9 (δH 2.13) to C-5 (δC 163.3), C-6 (δC 121.7), and C-7 (δC 158.9), as well as from 7-OCH3 (δH 3.96) to C-7 (δC 158.9) confirmed the substituents in 7. Additional HMBC correlations from H-8 (δH 4.83) to C-8′ (δC 65.6) ensure the ether linkage from C-8 to C-8′. The optical rotation value is zero, which suggested an equal proportion of C-3/C-3′ enantiomers. The phenomenon of related naturally occurring racemic phthalide mixtures have been well documented for (±)-strobide A15 and 3-O-methyl cyclopolic acid.17 A detailed explanation was given in 1992 by K. K. Ogilvie et al.24
The molecular formula C12H14O6 of compound 9 (ref. 26) was determined by its HRESIMS. The 1H-NMR and 13C-NMR spectrum data (Table 2) indicated that the methylene in 9 is connected with an methoxy rather than forming a dimer, which was confirmed by the HMBC correlation from 8-OCH3 (δH 3.43) to C-8 (δC 67.7). This structure can be found in SciFinder database (CAS # 1083201-20-0), but there is no reference available.
Beshanzoides A–D (1–4) are the first representatives of polyketides possessing a cycloheptanone-containing isobenzofuran ring system. A plausible biogenetic pathway for them is proposed as shown in Scheme 1. Briefly, the co-occurring cyclopaldic acid (12) could be regarded as the biogenetic precursor of 1–4. Similar to the proposed bio-synthesis pathway of stealthin F,25 a butanone was connected to 12 through an aldol reaction. After dehydration, an ACP (acyl carrier protein) joined to extend the chain and form a structure of 10, 12-dione, which is regarded as a common intermediate in the formation of fatty acid and polyketide.26 A following intramolecular aldol reaction would generate compounds 3 and 4 possessing a seven membered ring. 3 and 4 could be reduced by a ketoreductase to yield compounds 1 and 2, respectively.26
Scheme 1. A plausible biosynthetic pathway for compounds 1–4.

Naturally occurring compounds from fungi (mainly polyketides and alkaloids) have been often reported to have antibiotic and anti-tumor bioactivities.11,13,15 Therefore, all the isolates were evaluated for their anti-bacteria, anti-fungi and cytotoxicity activities. In the anti-bacteria screening, growth inhibition against Escherichia coli, Salmonella typhimurium, Shigella flexneri (Gram-negative), and Staphylococcus aureus (Gram-positive) were tested (Table S1†), while amoxicillin and chloromycetin were used as positive controls. Only compound 5 was found to inhibit S. aureus with an MIC value of 16 μg mL−1. Meanwhile, Candida albicans SC5314 was taken as the researching strain in the anti-fungus screening (Table S1,† fluconazole was used as positive control). Compound 10 exhibited an MIC value of 16 μg mL−1 against C. albicans. The cytotoxicity of each isolate against the human colon cancer RKO cell lines was evaluated using the CCK8 method,12a,27 but none of them displayed cytotoxicity against the RKO cell line at 10 μM.
In addition, as part of an on-going project towards the discovery of novel NPs-derived agents for treatments of hyperlipidemia and metabolic disorders associated with the abnormal glucose and lipid metabolism (e.g., T2DM),5,28 all the isolates were also tested for their inhibitory activity against acetyl-coenzyme A carboxylase alpha transcript variant 1 (ACC1) and adenosine triphosphate (ATP)-citrate lyase (ACL), two potential drug targets for both fatty acid and cholesterol biosynthesis.29 As a result, compounds 12 and 13 displayed inhibitory activity against ACC1 with IC50 values of 0.96 ± 0.11 and 11.77 ± 1.72 μM, respectively. ND-630 (CAS # 1434635-54-7)30 was used as the positive control (IC50: 1.56 ± 0.07 nM). Compound 12 also showed significant inhibition activity against ACL with an IC50 value of 7.56 ± 1.38 μM. BMS-303141 (CAS # 943962-47-8)31 was used as the positive control (IC50: 0.20 ± 0.01 μM).
Conclusions
All things considered, most studies on plant-derived secondary metabolites and/or their bioactivities are primarily focusing on the macroscopic organisms (e.g., leaves, twigs, and barks) of these plants.5,7 As a result, microbial communities, especially of conifers remain largely untouched and are underexplored resources for chemical and biological diversities. In addition, the populations of some medicinally valuable conifers are risking extinction due to the subsequent deforestation along with environmental changes. In an effort to protect these precious plants by gleaning valuable secondary metabolites from their endophytes, four unprecedented cycloheptanone-containing polyketides (beshanzoides A–D, 1–4) and a few structurally related new derivatives (5–7) were isolated and characterized from Penicillium commune P-4-1, an endophytic fungus isolated from the critically endangered plant Abies beshanzuensis. The discovery of structurally and biologically interesting polyketides extended the diversities of the secondary metabolites from endophytic fungi. It is the first time that isobenzofuranone derivatives were found to show remarkable inhibitory activity against ACC1 and ACL. We believe more glamorous discoveries will be unraveled from the endophytic fungi of this critically endangered and promising conifer. Additionally, investigations of the fungi associated with REPs would provide an alternative for NPs-derived drug discovery and an ideal strategy for utilization and protection of REPs themselves.
Experimental
General experimental procedure
Optical rotations were determined using a Rudolf Autopol IV polarimeter. UV absorptions were observed on a Hitachi U-2900E while IR spectra were recorded on an Avatar 360 ESP FTIR spectrometers. ECD spectra were recorded on a JASCO-810 spectropolarimeter. All NMR experiments were measured with Bruker Avance III 400 MHz or 600 MHz spectrometers. Chemical shifts are shown in δ (ppm) and the residual solvent peaks of CD3OD, acetone-d6, or DMSO-d6 were used as references. X-ray data were collected on a Bruker D8 Venture diffractometer (Ga Kα). HRESIMS were measured on an AB SCIEX Triple TOF 5600 spectrometer. Semi-preparative HPLC was performed on a Waters e2695 system with both 2998 prominence diode array (PDA) detector and 2424 evaporative light scattering detector (ELSD). A Waters Sunfire C18 column (5 μm, 250 × 10 mm) was equipped for purifications. The chiral HPLC separations were operated on a Shimadzu LC-20AT system with an SPD-M20A PDA detector and a Daicel Chiralpak IC column (5 μm, 250 × 4.6 mm). Column chromatography (CC) was carried out with silica gel (100–200 and 200–300 mesh, Qingdao Marine Chemical Co. Ltd., China), MCI gel CHP20P (75–150 μm, Mitsubishi Chemical Industries, Japan), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Sweden). Thin layer chromatography (TLC) was performed on pre-coated plates (HSGF254, Yantai Jiangyou Silica Gel Development Co., Ltd., Yantai, China). TLC spots were visualized under both 254 and 365 nm UV light and by heating after sprayed with 10% (v/v) H2SO4/EtOH reagent.
Fungal material
The fresh trunk bark of A. beshanzuensis was collected from the Baishanzu Mountain located at Lishui, Zhejiang Province, in April 2014. The plant sample was identified by Dr Ke-Jun Cheng (Lishui Institute of Agricultural Science, China). The bark was airlifted to the laboratory in ice packs within 24 h and stored at 4 °C until processed. The endophytic fungus P-4-1 was isolated from the bark and identified as Penicillium commune by comparing its ITS sequence (GenBank Accession No. MW721174) with corresponding sequences in public sequence databases (NCBI).
Fermentation, extraction, and isolation
The Penicillium commune P-4-1 was cultivated at 28 °C on autoclaved solid rice culture medium in 86 Erlenmeyer flasks (500 mL), each containing 60 g of rice and 80 mL of ddH2O. After 30 days' incubation, mediums were dried and soaked in EtOAc (60 min × 3 times) to afford a dark brown EtOAc extract (91.6 g). The EtOAc extract was subjected to a silica gel column (100–200 mesh) chromatography with a gradient-elution system, PE–EtOAc (20 : 1 → 10 : 1 → 5 : 1 → 2 : 1 → 1 : 1, neat EtOAc, v/v) and EtOAc–MeOH (1 : 1 → 0 : 1, v/v) to obtain seven fractions (Fr. 1 to Fr. 7), according to TLC analysis. Fr. 2 (528 mg) was separated over silica gel (PE–acetone, 5 : 1 → 2 : 1 → 1 : 1 → neat acetone) to give five subfractions (Fr. 2-A to Fr. 2-E). From Fr. 2-C (110 mg), compound 11 (13.6 mg, tR = 8.7 min) was fractioned by chromatograph column over Sephadex LH-20 (MeOH) followed by semi-preparative HPLC (MeCN–H2O 32 : 68). Fr. 2-D (21 mg) was purified by semi-preparative HPLC (MeCN–H2O 33 : 67), and compound 15 (3.5 mg, tR = 11.5 min) was isolated. Fr. 3 (3.1 g) was subjected to a silica gel column (PE–acetone, 10 : 1 → 5 : 1 → 2 : 1 → 1 : 1) to give Fr. 3-A to Fr. 3-D. Fr. 3-C (78 mg) was further fractionated by and semi-preparative HPLC (MeCN–H2O 32 : 68) and compound 16 (2.5 mg, tR = 12.0 min) was yielded. Fr. 4 (8.2 g) was fractioned into eight sub-fractions (Fr. 4-A to Fr. 4-H) by an MCI gel column with a gradient of MeOH–H2O (30% → 50% → 70% → 90% → 100% MeOH). By employing semi-preparative HPLC purification system (MeCN–H2O, 22 : 78), compounds 10 (1.2 mg, tR = 19.2 min) and 6 (1.5 mg, tR = 18.2 min) were obtained from Fr. 4-C (13 mg). Fr.4-F (340 mg) was further chromatographed by silica gel to afford seven subfractions (Fr. 4-F-1 to Fr. 4-F-7). Compounds 12 (3.0 mg, tR = 19.6 min) and 14 (0.5 mg, tR = 12.9 min) were purified from Fr. 4-F-5 (26 mg) while compounds 1 (5.0 mg, tR = 18.0 min) and 2 (3.0 mg, tR = 22.0 min) purified from Fr. 4-F-7 (33 mg) by semi-preparative HPLC (MeCN–H2O 36 : 64 /MeCN–H2O 32 : 68). In a similar way, compounds 13 (6.2 mg, tR = 10.0 min; MeCN–H2O, 55 : 45) and 3 (4) (2.1 mg, tR = 16.2 min; MeCN–H2O, 43 : 57) were isolated from Fr.4-G (400 mg). Fr. 5 (2.1 g) was separated by silica gel column (PE–acetone, 10 : 1 → 5 : 1 → 2 : 1 → 1 : 1), followed by semi-preparative HPLC (MeCN–H2O, 51 : 49) to get compound 17 (1.0 mg, tR = 5.0 min). Separation of Fr. 6 (5.2 g) on an MCI column with a stepwise gradient elution of MeOH–H2O (50 : 50 → 70 : 30 → 90 : 10 → 100 : 0) collected seven sub-fractions (Fr. 6-A to Fr. 6-G). Compound 8 (30 mg) was recrystallized from Fr. 6-A (200 mg). Fr. 6-E (130 mg) was further fractioned by silica gel with PE–acetone system (4 : 1 → 3 : 1 → 2 : 1 → 1 : 1) into twelve parts (Fr. 6-E-1 to Fr. 6-E-12). Semi-preparative HPLC afforded compounds 9 (1.9 mg, tR = 10.5 min), 5 (1.7 mg, tR = 10.5 min), and 7 (3.1 mg, tR = 13.2 min) from Fr. 6-E-4 (11 mg; MeCN–H2O, 29 : 71), Fr. 6-E-5 (24 mg; MeCN–H2O, 33 : 67), and Fr. 6-E-10 (7 mg; MeCN–H2O, 32 : 68), respectively. The mixture of enantiomers 3 and 4 (1.2 mg) was purified by chiral HPLC (hexane-isopropanol, 78 : 22). Compounds 3 (0.5 mg, tR = 22.6 min) and 4 (0.5 mg, tR = 24.9 min) were obtained.
Compound characterization
Beshanzoide A (1) white powder, mp 206.4–208.9 °C, [α]25D +80.9 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 216 (4.3), 262 (3.9) nm; ECD (c 1.00 × 10−4 M, MeOH) λmax (Δε) 225 (−5.6), 290 (+5.1) nm; IR (film) νmax: 3469 (br), 1719, 1702, 1589, 1275, 1116 and 1024 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 321 [M + H]+, 343 [M + Na]+; HRESIMS m/z 321.1329 [M + H]+(calcd for C17H21O6, 321.1333, Δ = −1.0 ppm).
Beshanzoide B (2) white powder, [α]25D −89.0 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 216 (4.1), 262 (3.7) nm; ECD (c 1.80 × 10−4 M, MeOH) λmax (Δε) 225 (+10.5), 291 (−8.4) nm; 1H and 13C NMR data, see Table 1; ESIMS m/z 321 [M + H]+, 343 [M + Na]+; HRESIMS m/z 321.1326 [M + H]+(calcd for C17H21O6, 321.1333, Δ = −2.1 ppm).
Beshanzoide C (3) white powder, [α]25D +25.0 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 216 (3.9), 262 (3.5) nm; ECD (c 1.30 × 10−4 M, MeOH) λmax (Δε) 222 (−1.6), 237 (−1.9), 307 (+7.1) nm; 1H and 13C NMR data, see Table 1; ESIMS m/z 319 [M + H]+; HRESIMS m/z 319.1184 [M + H]+ (calcd for C17H19O6, 319.1176, Δ = 2.4 ppm).
Beshanzoide D (4) white powder, [α]25D −25.0 (c 0.02, MeOH); UV (MeOH) see beshanzoide C (3); ECD (c 1.30 × 10−4 M, MeOH) λmax (Δε) 221 (+2.2), 237 (+1.6), 305 (−7.2) nm; 1H and 13C NMR data, see Table 1; ESIMS and HRESIMS see beshanzoide C (3).
Beshanzoide E (5) white powder; UV (MeOH) λmax (log ε) 214 (4.2), 250 (3.6), 296 (3.3) nm; 1H and 13C NMR data, see Table 2; ESIMS m/z 253 [M + H]+, 275 [M + Na]+; HRESIMS m/z 253.0703 [M + H]+ (calcd for C12H13O6, 253.0707, Δ = −1.6 ppm).
Beshanzoide F (6) white powder; UV (MeOH) λmax (log ε) 214 (4.2), 264 (3.8), 296 (3.5) nm; 1H and 13C NMR data, see Table 2; ESIMS m/z 211 [M + H]+, 233 [M + Na]+; HRESIMS m/z 233.0418 [M + Na]+ (calcd for C10H10O5Na, 233.0420, Δ = −1.0 ppm).
Beshanzoide G (7) white powder; UV (MeOH) λmax (log ε) 218 (4.6), 262 (4.2) nm; 1H NMR (CD3OD, 400 MHz): δ 2.13 (6H, s, H-9/9′), 3.96 (6H, s, 7/7′-OCH3), 4.83 (4H, s, H-8/8′), 6.55 (2H, s, H-3/3′); 13C NMR (150 MHz, CD3OD): δ 9.0 (C-9/9′), 62.5 (7/7′-OCH3), 65.6 (C-8/8′), 97.6 (C-3/3′), 110.1 (C-7a/7a′), 116.3 (C-4/4′), 121.7 (C-6/6′), 148.2 (C-3a/3a′), 158.9 (C-7/7′), 163.3 (C-5/5′), 169.0 (C-1/1′). ESIMS m/z 485 [M + Na]+; HRESIMS m/z 485.1044 [M + Na]+ (calcd for C22H22O11Na, 485.1054, Δ = −2.2 ppm).
7-Carboxy-4-hydroxy-6-methoxy-5-methylphthalide (8) white powder, UV (MeOH) λmax (log ε) 212 (4.4), 250 (3.8), 298 (3.5) nm; 1H and 13C NMR data (in DMSO-d6), see lit. 14; 1H and 13C NMR data (in CD3OD), see Table 2; ESIMS m/z 261 [M + Na]+; HRESIMS m/z 261.0368 [M + Na]+ (calcd for C11H10O6Na, 261.0370, Δ = −0.4 ppm).
3,5-Dihydroxy-7-methoxy-4-(methoxymethyl)-6-methyl-1(3H)-isobenzofuranone (9) white powder; UV (MeOH) λmax (log ε) 218 (4.2), 260 (3.8) nm; 1H and 13C NMR data, see Table 2 ESIMS m/z 277 [M + Na]+; HRESIMS m/z 277.0676 [M + Na]+ (calcd for C12H14O6Na, 277.0683, Δ = −2.4 ppm).
X-ray crystallographic analysis
Colorless needle crystal of 1 was obtained from acetone-d6. The structure was solved by the SheIXT structure solution program using Least Squares calculations on F2. Crystallographic data of compound 1 has been deposited in the Cambridge Crystallographic Data Centre as CCDC-2071749.† The data can be obtained free of charge viawww.ccdc.cam.ac.uk.
X-ray crystallographic data for 1
M = 320.33, orthorhombic, space group P212121, a = 5.5497(2) Å, b = 15.7041(5) Å, c = 17.1259(6) Å, α = β = γ = 90.00°, V = 1492.57(9) Å3, Z = 4, ρcalcd = 1.426 Mg m−3, μ (Ga Kα) = 1.34139 Å, crystal size 0.1 × 0.03 × 0.02 mm3, F(000) = 680, 7158 reflections collected, 2689 independent reflections (Rint = 0.0354), R1 = 0.0477 [I > 2σ(I)], wR2 = 0.1112 [I > 2σ(I)], R1 = 0.0555 (all data), wR2 = 0.1180 (all data), goodness of fit = 1.128, Flack parameter = 0.01(15).
Antimicrobial assays
The antibiotic properties of compounds 1–17 against Escherichia coli, Salmonella typhimurium, Shigella flexneri (Gram-negative), Staphylococcus aureus (Gram-positive) and Candida albicans (fungi) were evaluated by microdilution method. Amoxicillin and chloromycetin were the positive controls for antibacterial assays, and fluconazole was the antifungal positive control. Test bacteria or fungi were counted and seeded at the appropriate concentrations in sterile 96-well microplates. Isolated metabolites and positive controls were individually tested at 64, 32, 16, 8, 4, 2, 1, 0.5, 0.25 and 0.125 μg mL−1. Assays were incubated at 35 °C for 16 (bacteria) or 24 (fungi) hours. MIC (minimum inhibitory concentration) was the lowest concentration of the tested compounds that could completely inhibit the growth of bacteria (fungi). The MIC values were determined by a combination of naked eyes observations and optical density (OD) measurements.
ACC1 inhibitory activity assay
ADP-Glo™ Kinase Assay (Promega, Madison, WI) was used to assess the activity of ACC1. The kinase assay was carried out in a 384-well plate (ProxiPlateTM-384 Plus, PerkinElmer) with 2.0 μL of ACC1 and 1.0 μL of test compounds with different concentrations and pre-incubate for 0.5 hours at room temperature. Then the 2.0 μL of ATP was added to each well and kept for 60 min at room temperature. After the enzymatic reaction, 2.5 μL of ADP-Glo reagent was added to each well to terminate the kinase reaction and deplete the unconsumed ATP within 60 min at room temperature. In the end, 5.0 μL of kinase detection reagent (reagent 2) was added to each well and incubated for 1 h to simultaneously convert ADP to ATP. The luminescence signal was measured by a PerkinElmer En-Vision reader (PerkinElmer, MA, USA). The known inhibitor ND-630 (ref. 30) was used as the positive control.
ACL inhibitory activity assay
The same as the ACC1 inhibitory activity assay, the assay was performed using ADP-Glo™ Kinase Assay (Promega, Madison, WI). The ACL inhibition activity was quantified by the amount of ADP generated of the enzymatic reaction.31 The kinase assay was carried out in a 384-well plate (ProxiPlateTM-384 Plus, PerkinElmer) in a volume of 5 μL reaction mixture, containing 2.0 μL of ACL, 2.0 μL of ATP, and 1.0 μL of test compounds with different concentrations. Reactions in each well was kept going for 30 min under 37 °C. After that, 2.5 μL of ADP-Glo™ reagent was added to terminate the kinase reaction and deplete the unconsumed ATP within 60 min at room temperature. Finally, 5.0 μL of kinase detection reagent (reagent 2) was added to each well and incubated for 1 h to simultaneously convert ADP to ATP. The luminescent signal was measured using an En-Vision multilabel plate reader (PerkinElmer, MA, USA) and BMS-303141 (ref. 31) was taken as the positive control.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by NSFC grants (No. 21937002, 81773599, 21772025).
Electronic supplementary information (ESI) available: Observed key HMBC and NOESY correlations of compounds 5–9; 1D/2D NMR and HR-ESIMS spectra of compounds 1–9; 1D NMR of compounds 10–17; ITS sequence of Penicillium commune P-4-1 strain. CCDC 2071749. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08377e
Notes and references
- (a) Ibrahim M. A. Na M. Oh J. Schinazi R. F. McBrayer T. R. Whitaker T. Doerksen R. J. Newman D. J. Zachos L. G. Hamann M. T. Proc. Natl. Acad. Sci. U. S. A. 2013;110:16832–16837. doi: 10.1073/pnas.1311528110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu F. Qin C. Tao L. Liu X. Shi Z. Ma X. H. Jia J. Tan Y. Cui C. Lin J. S. Tan C. Y. Jiang Y. Y. Chen Y. Z. Proc. Natl. Acad. Sci. U. S. A. 2011;108:12943–12948. doi: 10.1073/pnas.1107336108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Butchart S. H. M. Walpole M. Collen B. van Strien A. Scharlemann J. P. W. Almond R. E. A. Baillie J. E. M. Bomhard B. Brown C. Bruno J. Carpenter K. E. Carr G. M. Chanson J. Chenery A. M. Csirke J. Davidson N. C. Dentener F. Foster M. Galli A. Galloway J. N. Genovesi P. Gregory R. D. Hockings M. Kapos V. Lamarque J.-F. Leverington F. Loh J. McGeoch M. A. McRae L. Minasyan A. Morcillo M. H. Oldfield T. E. E. Pauly D. Quader S. Revenga C. Sauer J. R. Skolnik B. Spear D. Stanwell-Smith D. Stuart S. N. Symes A. Tierney M. Tyrrell T. D. Vié J.-C. Watson R. Science. 2010;328:1164–1168. doi: 10.1126/science.1187512. [DOI] [PubMed] [Google Scholar]
- Fox J. L. Science. 1984;226:150. doi: 10.1126/science.226.4671.150.a. [DOI] [PubMed] [Google Scholar]
- Fu L.-K. and Jin J.-M., China Plant Red Data Book: Rare and Endangered Plants I, Science Press, Beijing and New York, 1992 [Google Scholar]
- López-Pujol J. Zhang F.-M. Ge S. Biodivers. Conserv. 2006;15:3983–4026. doi: 10.1007/s10531-005-3015-2. [DOI] [Google Scholar]
- (a) Xiong J. Zhou P.-J. Jiang H.-W. Huang T. He Y.-H. Zhao Z.-Y. Zang Y. Choo Y.-M. Wang X. J. Chittiboyina A. G. Pandey P. Hamann M. T. Li J. Hu J.-F. Angew. Chem., Int. Ed. 2021;60:22270–22275. doi: 10.1002/anie.202109082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang T. Ying S.-H. Li J.-Y. Chen H.-W. Zang Y. Wang W.-X. Li J. Xiong J. Hu J.-F. Phytochemistry. 2020;169:112184. doi: 10.1016/j.phytochem.2019.112184. [DOI] [PubMed] [Google Scholar]
- Yang Y., Zhang D., Luscombe D., Liao W., Farjon A., Katsuki T., Xiang Q. and Li N., Abies beshanzuensis, The IUCN Red List of Threatened Species, 2013, e.T32318A2814360; DOI: 10.2305/IUCN.UK.2013-1.RLTS.T32318A2814360.en [DOI] [Google Scholar]
- Hu C.-L. Xiong J. Li J.-Y. Gao L.-X. Wang W.-X. Cheng K.-J. Yang G.-X. Li J. Hu J.-F. Eur. J. Org. Chem. 2016;10:1832–1835. doi: 10.1002/ejoc.201600165. [DOI] [Google Scholar]
- Davis D. C. Hoch D. G. Wu L. Abegg D. Martin B. S. Zhang Z.-Y. Adibekian A. Dai M. J. J. Am. Chem. Soc. 2018;140:17465–17473. doi: 10.1021/jacs.8b07652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Keller N. P. Nat. Rev. Microbiol. 2019;17:167–180. doi: 10.1038/s41579-018-0121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bills G. F. Gloer J. B. Microbiol. Spectrum. 2016;4:1–32. doi: 10.1128/microbiolspec.FUNK-0009-2016. [DOI] [PubMed] [Google Scholar]
- (a) Newman D. J. Cragg G. M. J. Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]; (b) Yang G.-X. Ma G.-L. Li H. Huang T. Xiong J. Hu J.-F. Chin. J. Nat. Med. 2018;16:881–906. doi: 10.1016/S1875-5364(18)30131-6. [DOI] [PubMed] [Google Scholar]
- (a) Kharwar R. N. Mishra A. Gond S. K. Stierle A. Stierle D. Nat. Prod. Rep. 2011;28:1208–1228. doi: 10.1039/C1NP00008J. [DOI] [PubMed] [Google Scholar]; (b) Zhang H. W. Song Y. C. Tan R. X. Nat. Prod. Rep. 2006;23:753–771. doi: 10.1039/B609472B. [DOI] [PubMed] [Google Scholar]; (c) Strobel G. Daisy B. Microbiol. Mol. Biol. Rev. 2003;67:491–502. doi: 10.1128/MMBR.67.4.491-502.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ma G.-L. Guo N. Wang X.-L. Li J. M. Jin Z.-X. Han Y. Q. Dong S.-Z. Xiong J. Hu J.-F. Bioorg. Chem. 2020;105:104445. doi: 10.1016/j.bioorg.2020.104445. [DOI] [PubMed] [Google Scholar]; (b) McMullin D. R. Green B. D. Prince N. C. Tanney J. B. Miller J. D. J. Nat. Prod. 2017;80:1475–1483. doi: 10.1021/acs.jnatprod.6b01157. [DOI] [PubMed] [Google Scholar]
- Zhu Y.-X. Peng C. Ding W. X. Hu J.-F. Li J. Y. Nat. Prod. Res. 2020 doi: 10.1080/14786419.2020.1844700. [DOI] [PubMed] [Google Scholar]
- Song F. H. Lin R. Yang N. Jia J. Wei S. Z. Han J. H. Li J. P. Bi H. K. Xu X. L. Antibiotics. 2021;10:377. doi: 10.3390/antibiotics10040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullin D. R. Tanney J. B. McDonald K. P. Miller J. D. Phytochem. Lett. 2019;29:17–24. doi: 10.1016/j.phytol.2018.10.016. [DOI] [Google Scholar]
- Matsuda Y. Wakimoto T. Mori T. Awakawa T. Abe I. J. Am. Chem. Soc. 2014;136:15326–15336. doi: 10.1021/ja508127q. [DOI] [PubMed] [Google Scholar]
- Achenbach H. Muehlenfeld A. Weber B. Kohl W. Brillinger G.-U. Z. Naturforsch., B: Anorg. Chem., Org. Chem. 1982;37:1091–1097. doi: 10.1515/znb-1982-0823. [DOI] [Google Scholar]
- Achenbach H. Müehlenfeld A. Brillinger G.-U. Liebigs Ann. Chem. 1985;8:1596–1628. doi: 10.1002/jlac.198519850808. [DOI] [Google Scholar]
- Fujita M. Yamada M. Nakajima S. Kawai K.-I. Nagai M. Chem. Pharm. Bull. 1984;32:2622–2627. doi: 10.1248/cpb.32.2622. [DOI] [PubMed] [Google Scholar]
- Valente A. M. M. P. Ferreira A. G. Daolio C. Filho E. R. Boffo E. F. Souza A. Q. L. Sebastianes F. L. S. Melo I. S. An. Acad. Bras. Cienc. 2013;85:487–496. doi: 10.1590/S0001-37652013005000024. [DOI] [PubMed] [Google Scholar]
- Ge Y. Han Z. B. Wang Z. Feng C.-G. Zhao Q. Lin G.-Q. Ding K. L. Angew. Chem., Int. Ed. 2018;57:13140–13144. doi: 10.1002/anie.201807639. [DOI] [PubMed] [Google Scholar]
- Lightner D. A. Docks E. L. Tetrahedron. 1979;35:713–720. doi: 10.1016/0040-4020(79)87022-2. [DOI] [Google Scholar]
- Wu W.-B. Zhang H. Liu H.-C. Dong S.-H. Wu Y. Ding J. Yue J.-M. Tetrahedron. 2014;70:3570–3575. doi: 10.1016/j.tet.2014.04.007. [DOI] [Google Scholar]
- Tsantrizos Y. S. Ogilvie K. K. Watson A. K. Can. J. Chem. 1992;70:2276–2284. doi: 10.1139/v92-286. [DOI] [Google Scholar]
- Huang C. S. Yang C. F. Fang Z. J. Zhang L. P. Zhang W. J. Zhu Y. G. Zhang C. S. Mar. Drugs. 2019;17:150. doi: 10.3390/md17030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallimore A. R. Nat. Prod. Rep. 2009;26:266–280. doi: 10.1039/B807902C. [DOI] [PubMed] [Google Scholar]
- Hu L. Cai X. Dong S. Z. Zhen Y. J. Hu J. D. Wang S. J. Jiang J. W. Huang J. W. Han Y. Q. Qian Y. Yuan Y. Q. Hu W. H. J. Med. Chem. 2020;63:6959–6978. doi: 10.1021/acs.jmedchem.0c00079. [DOI] [PubMed] [Google Scholar]
- Wan J. Zang Y. Xiao D.-A. Li N. Li J. M. Jin Z.-X. Chen D.-L. Xiong J. Li J. Hu J.-F. RSC Adv. 2020;10:3343–3356. doi: 10.1039/C9RA09542J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Verschueren K. H. G. Blanchet C. Felix J. Dansercoer A. De Vos D. Bloch Y. Van Beeumen J. Svergun D. Gutsche I. Savvides S. N. Verstraete K. Nature. 2019;568:571–575. doi: 10.1038/s41586-019-1095-5. [DOI] [PubMed] [Google Scholar]; (b) Endo Y. Onodera A. Obata-Ninomiya K. Koyama-Nasu R. Asou H. K. Ito T. Yamamoto T. Kanno T. Nakajima T. Ishiwata K. Kanuka H. Tumes D. J. Nakayama T. Nat. Metab. 2019;1:261–275. doi: 10.1038/s42255-018-0025-4. [DOI] [PubMed] [Google Scholar]; (c) Burker A. C. Huff M. W. Curr. Opin. Lipidol. 2017;28:193–200. doi: 10.1097/MOL.0000000000000390. [DOI] [PubMed] [Google Scholar]
- Harriman G. Greenwood J. Bhat S. Huang X. Y. Wang R. Y. Paul D. Tong L. Saha A. K. Westlin W. F. Kapeller R. Harwood Jr H. J. Proc. Natl. Acad. Sci. U. S. A. 2016;113:E1796–E1805. doi: 10.1073/pnas.1520686113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner S. K. Hanai J. Bai S. Jernigan F. E. Oki M. Komaba C. Shuto E. Sukhatme V. P. Sun L. J. Eur. J. Med. Chem. 2017;126:920–928. doi: 10.1016/j.ejmech.2016.12.018. [DOI] [PubMed] [Google Scholar]