

Summary
Novel strategies are needed to identify drug targets and treatments for the COVID-19 pandemic. The altered gene expression of virus-infected host cells provides an opportunity to specifically inhibit viral propagation via targeting the synthetic lethal and synthetic dosage lethal (SL/SDL) partners of such altered host genes. Pursuing this disparate antiviral strategy, here we comprehensively analyzed multiple in vitro and in vivo bulk and single-cell RNA-sequencing datasets of SARS-CoV-2 infection to predict clinically relevant candidate antiviral targets that are SL/SDL with altered host genes. The predicted SL/SDL-based targets are highly enriched for infected cell inhibiting genes reported in four SARS-CoV-2 CRISPR-Cas9 genome-wide genetic screens. We further selected a focused subset of 26 genes that we experimentally tested in a targeted siRNA screen using human Caco-2 cells. Notably, as predicted, knocking down these targets reduced viral replication and cell viability only under the infected condition without harming noninfected healthy cells.
Subject areas: Drugs, Virology, Synthetic biology
Graphical abstract
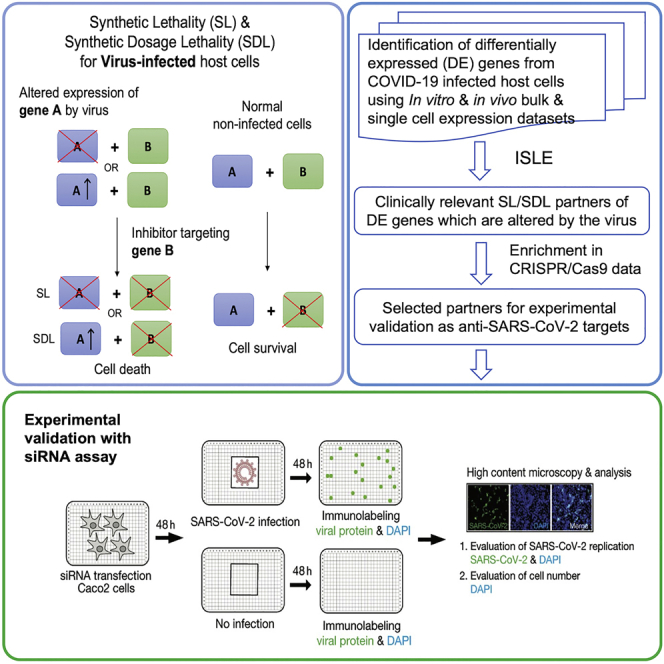
Highlights
-
•
Identified anti-SARS-CoV-2 targets using synthetic lethality from infected datasets
-
•
Predicted targets are enriched by infected cell inhibiting genes from CRISPR/Cas9 data
-
•
Experimental validation of selected SL targets in siRNA assay from human Caco-2 cells
-
•
Predicted targets are made publicly available for in vivo testing and validation
Drugs; Virology; Synthetic biology
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by the novel coronavirus SARS-CoV-2 together with its emerging new variants in 2022 has resulted in hundreds of millions of infected people with millions of deaths worldwide (WHO Coronavirus Disease (COVID-19) Dashboard, 2022). Up to this date, there are several thousand registered clinical trials of anti-COVID-19 therapies worldwide (International Clinical Trials Registry Platform, 2022), with remdesivir being the only drug approved by the United States Food and Drug Administration (FDA) and the drug regulatory agencies of several other countries (Beigel et al., 2020). However, the clinical benefit of remdesivir in most COVID-19 patients is still modest (Pan et al., 2021). A total of 14 therapies, including virus-neutralizing monoclonal antibodies bamlanivimab and etesevimab (Mahase, 2020), sotrovimab, the combination of casirivimab and imdevimab, interleukin-6 receptor inhibitor Tocilizumab and Janus kinase (JAK) inhibitor baricitinib (in combination with remdesivir), molnupiravir, the combination of nirmatrelvir and ritonavir have obtained Emergency Use Authorization (EUA) from the U.S. FDA (U. S. Food and Drug Administration, 2021). Dexamethasone and other corticosteroids have been recommended for the control of COVID-19-related symptoms (Ledford, 2020). Numerous preclinical studies have conducted anti-SARS-CoV-2 drug repurposing as well as genetic screening of varied scales, for example (Daniloski et al., 2021; Riva et al., 2020; Wei et al., 2021), supplemented by other studies providing computational target predictions, for example (Cheng et al., 2021a, 2021b; Tilocca et al., 2020; Zhou et al., 2020a, 2020b). Two mRNA vaccines from Pfizer-BioNTech and Moderna have been approved by the U.S. FDA (U. S. Food and Drug Administration, 2021). Despite all these efforts, there is an urgent unmet need to identify additional anti-SARS-CoV-2 targets and drugs to resolve the COVID-19 crisis (Jomah et al., 2020).
Existing COVID-19 antiviral strategies are either directly targeting the virus or indirectly targeting the virus via host modulation. Although the direct targeting of the viral life cycle to inhibit its proliferation remains challenging (Pan et al., 2021), numerous recent studies have contributed to the understanding of the biology of SARS-CoV-2 infection, for example (Bojkova et al., 2020; Forster et al., 2020; Paules and Fauci, 2021; V'Kovski et al., 2021), to pursue host-directed therapies. A lot of interest has been focused on targeting ACE2 and TMPRSS2, the virus entry receptors (Muralidar et al., 2021; Ragia and Manolopoulos, 2020) and the search for drugs targeting these genes is underway (Hoffmann et al., 2021; Lin et al., 2021). Majority of these host-directed antiviral strategies aim to protect host cells from virus-induced death, including our recently published strategy of rescuing the infected host cells via targeting the host metabolism (Cheng et al., 2021a). In contrast to these “host cell protection” approaches, a conceptually different “host cell inhibition” strategy has been proposed (Mast et al., 2020) and demonstrated with poliovirus (Navare et al., 2020). In this latter strategy, the aim is to selectively inhibit the viability or even kill the virus-infected host cells (preferentially in the early stages of viral infection), thereby diminishing the capacity of these cells to sustain viral proliferation and propagation. This can be achieved utilizing synthetic lethality (SL), a type of genetic interaction where reduced viability or cell death occurs upon the inactivation of a pair of genes but not by either gene alone. More specifically, viruses are known to alter the expression and activity of many host genes and by identifying and targeting the “SL partner genes” of genes downregulated by the virus, the virus-infected cells can be specifically targeted, sparing noninfected healthy cells. A variant of SL, called synthetic dosage lethality (SDL) where decreased cell viability occurs only upon the overexpression of one gene and the inactivation of a second gene (Kroll et al., 1996), can similarly be utilized to specifically target the infected cells based on the host genes that are upregulated by the virus. Although we infer the SL and SDL partners of genes differentially expressed in infected vs control cells, we group them together (annotated as SL/SDL genes) in the following downstream analyses as we are searching for antiviral targets that are directly altered by the virus, whose inhibition (irrespective if they are SL or SDL) could selectively kill infected cells. Adopting this distinct SL/SDL-based approach (see Figure 1A), we systematically identify candidate targets for selective killing of host infected cells and further test the predicted candidate targets with in vitro assays.
Figure 1.

Synthetic lethality, differentially expressed genes from SARS-CoV-2 infected host cells and their identified clinically relevant SL/SDL partners
(A) An illustration of the concept of synthetic lethality (SL and SDL, left-hand side) and its application to the context of antiviral infection (right-hand side).
(B) An illustration of the overall workflow we used to identify SL/SDL-based candidate targets for anti-SARS-Cov-2 by integrating different in vitro (vt) and in vivo (vv), including single-cell (sc) datasets. Highlighted modules are: (1) Intersection of ISLE-predicted SL/SDL partners of the SARS-CoV-2-induced differentially expressed (DE) genes in all the denoted vt, vv, and sc datasets yielded 454 genes (vt-vv-sc). (2) These 454 vt-vv-sc genes are shown to be enriched for strong hits from two different CRISPR-Cas9 screens (cr) in the SARS-CoV-2 infection setting (see main text for details), and the intersection with the hits from the CRISPR screens yielded 140 final candidate SL/SDL-based anti-SARS-CoV-2 targets (vt-vv-sc-cr). (3) These 140 candidates are further filtered based on their enrichment for genes whose knockdowns decrease cell viability in a genome-wide siRNA screen we performed earlier. (4) We further selected a subset of 26 targets from genome-wide siRNA screen and validated experimentally via a small-scale targeted siRNA screen.
(C) A heatmap showing the extent of overlaps between the differentially expressed (DE) genes in SARS-CoV-2 infected samples vs noninfected controls identified from different datasets, as measured by the odds ratio of enrichment between each pair of datasets, which is encoded by the color and also labeled in the cells within the heatmap. The dataset labels are as follows: vt.Vero (Vero E6 cell samples from Riva et al., 2020), vt.A549 (A549ACE2 cell samples from Blanco-Melo et al., 2020), vv.Swab (COVID-19 patient nasopharyngeal swab samples from Butler et al., 2021; Lieberman et al., 2020), sc.Chua (single-cell data of nasopharyngeal swab and bronchial samples from Chua et al., 2020), and sc.Liao (single-cell data of bronchoalveolar lavage fluid samples from Liao et al., 2020).
(D) A heatmap showing the extent of overlaps between the SL/SDL partner genes identified with ISLE based on the different datasets, with the same dataset labels as in (C).
(E) The negative log10-transformed Benjamini-Hochberg-adjusted p values (Padj) from Fisher’s exact tests (X axis) for the enrichment between the 454 consensus ISLE-identified SL/SDL-based candidate targets and different validation gene sets, including: genes with strong negative log fold-changes identified in the CRISPR-Cas9 screen in SARS-CoV-2-infected Vero E6 cells from (Wei et al., 2021) (vero CRISPR), such genes in another CRISPR-Cas9 screen in SARS-CoV-2-infected A549ACE2 cells (Daniloski et al., 2021) at two different multiplicities of infection (Human A549 MOI1 CRISPR and Human A549 MOI3 CRISPR, respectively), and human genes interacting with SARS-CoV-2 proteins identified in (Gordon et al., 2020; Stukalov et al., 2021) (SARS-CoV-2 PPI). The black-dotted line corresponding to the cutoff of adjusted p < 0.05.
(F) The negative log10-transformed Benjamini-Hochberg-adjusted p values (Padj) (X axis) resulting from a GSEA enrichment analysis between the 140 candidates and genes with strong negative log fold-changes identified in the CRISPR-Cas9 screen in SARS-CoV-2-infected human Huh-7.5.1 hepatoma cells from (Wang et al., 2021) (Huh7.5.1-Wang) and genes in another CRISPR-Cas9 screens in SARS-CoV-2-infected human Huh-7.5 hepatoma cells (Schneider et al., 2021), at 37°C and 33°C (Huh-7.5-Schneider.37C and Huh-7.5-Schneider.33C, corresponding to two physiologically relevant temperatures of the lower and upper airways, respectively). The black-dotted line corresponding to the cutoff of adjusted p < 0.05. These two CRISPR-Cas9 screens were not used to generate the list of 140 targets.
Previously, we have developed a computational pipeline named ISLE (identification of clinically relevant synthetic lethality) to identify clinically relevant SL/SDL genetic interactions by mining large-scale genomic and transcriptomic data across human cancers (Lee et al., 2018). Applying ISLE, we have previously successfully identified a Gαq-driver mutation as marker for FAK inhibitor in uveal melanoma (Feng et al., 2019) and a synergistic combination of asparaginase and MAPK inhibitors for treating melanoma and pancreatic cancer (Pathria et al., 2019). Although the ISLE algorithm was originally designed to predict SL in cancers, we have recently found that some SL gene pairs predicted by ISLE are also functional in noncancerous tissues (Cheng et al., 2021b), reflecting the conservation of the wiring of molecular networks of human cells more generally. Therefore, we reasoned that ISLE can also be valuable in predicting SL gene pairs for noncancer contexts. Adopting this conjecture, here we applied ISLE to a large cohort of both in vitro and in vivo SARS-CoV-2 RNA-sequencing datasets to identify SL/SDL-based anti-SARS-CoV-2 targets. We then set to combine these predictions with the results of large-scale SARS-CoV-2 in vitro genome-wide CRISPR and siRNA screens. Focusing on a selected list of 26 predicted targets, we tested them experimentally in a small-scale targeted siRNA screen and demonstrated their efficacy in inhibiting SARS-CoV-2 replication. As these targets are supported by analyzing in vivo data, these are more likely to be of translational relevance.
Results
Overview of our strategy for SL/SDL-based anti-SARS-CoV-2 target prediction
Our analysis proceeds in four steps: (A) First, we analyzed multiple published host cell gene expression datasets (Figure 1B) involving in vitro and in vivo human samples of SARS-CoV-2 infection to identify the strongest significantly differentially expressed (DE) genes in response to viral infection. (B) Second, applying the ISLE pipeline, we identified the most robust clinically relevant SL/SDL partner of these DE genes in human cells, as potential SL/SDL-based anti-SARS-CoV-2 targets. We show that these 454 predicted targets (Figure 1B, module 1) are enriched with genes whose knockout strongly reduced cell viability specifically in several SARS-CoV-2 infected CRISPR-Cas9 genetic screens, and thus could be potentially targeted to selectively kill infected cells and reduce viral spread. (C) We further filtered and prioritized a smaller list of 140 predicted targets based on support by several CRISPR screens (Figure 1B, module 2) and a high throughput genome-wide siRNA screen conducted and analyzed in our lab (Figure 1B, module 3). (D) Finally, we employed a network-based simulated annealing algorithm to further prioritize 26 highly ranked targets (Figure 1B, module 4) and conducted a small-scale targeted siRNA screen to experimentally test these targets in greater depth. A stepwise detailed description of the analysis performed, and the results obtained in each step are as follows.
Identification of host genes differentially expressed after SARS-CoV-2 infection
To identify the host cell genes whose expression is altered upon SARS-CoV-2 infection, we collected and analyzed multiple transcriptomic datasets of SARS-CoV-2 infection (Figure 1B). These include bulk RNA-seq data of in vitro samples from Vero E6 cells (Riva et al., 2020) and human A549 cells with exogenous ACE2 expression (A549ACE2) (Blanco-Melo et al., 2020) as well as in vivo nasopharyngeal swab samples from COVID-19 patients (Butler et al., 2021; Lieberman et al., 2020). For in vivo analysis, we additionally used single-cell RNA-seq data of nasopharyngeal and bronchial samples (Chua et al., 2020) and bronchoalveolar lavage fluid (BALF) samples (Liao et al., 2020). For each of these datasets, we identified the significantly differentially expressed (DE) genes in the SARS-CoV-2-infected samples vs matched virus-negative controls (FDR<0.05 with additional dataset-specific criteria, see STAR Methods). For the single-cell datasets, we performed the DE analysis only in the epithelial cells, as these represent a primary target for SARS-CoV-2 infection, thus being our focus for SL/SDL-based antiviral strategy. By comparing the identified DE genes across datasets with Fisher’s exact tests (STAR Methods), we find varied but overall higher-than-random pairwise similarities (i.e., odds ratio >1, Figure 1C). The lowest odds ratio (2.7, adjusted p = 2.66E-03) is found between the in vitro A549ACE2 cell dataset and the in vivo single-cell RNA-seq samples (the Chua et al. dataset), and the highest odds ratio (12.4, adjusted p = 1.33E-38) is seen between the in vivo single-cell (the Liao et al. dataset) and in vivo bulk swab datasets. Reassuringly, the DE genes identified from the in vivo bulk and single-cell RNA-seq datasets have reasonably good overlaps (Figure 1C). We performed pathway enrichment analysis of the DE genes from each dataset (STAR Methods; Table S1) and noticed that although these genes are different across datasets, they are enriched for functionally similar pathways (e.g., platelet aggregation plaque formation pathway in human A549ACE2 dataset and platelet activation signaling and aggregation in in vivo single-cell Chua et al. dataset; Table S1).
Identification of the SL/SDL partners of SARS-CoV-2-induced DE genes
Next, we predicted the SL/SDL partners of the identified DE genes, as they constitute potential antiviral targets that can selectively impair the function of infected host cells and thus limit their capacity to support viral proliferation (list of SL/SDL pairs of each dataset are given in Table S2). Specifically, we applied the ISLE algorithm (Lee et al., 2018) to predict the SL partners of the genes significantly downregulated by SARS-CoV-2 infection in the aforementioned datasets. For the upregulated genes, we analogously predicted their SDL partners with a modified version of ISLE (STAR Methods). Interestingly, the SL/SDL partners identified across datasets with FDR cutoff 0.1 exhibit overall higher similarities than those similarities observed among the DE genes, with a minimum pairwise odds ratio of 4.3 (adjusted p = 1.48E-118; Figure 1D). In particular, the overlap in the SL/SDL partner genes between the in vitro A549ACE2 cell dataset and the in vivo single-cell RNA-seq samples (in Chua et al. dataset) is higher than the overlap observed between their corresponding DE genes (SL odds ratio of 4.3 compared to corresponding DE odds ratio of 2.7). This suggests that the DE genes identified across the datasets may be functionally similar, resulting in higher chances of forming SL/SDL interactions with the same genes, which is indeed supported by the pathway enrichment results of the DE genes as described previously (Table S1). Reassured by the overall similarity of the SL/SDL partner genes across datasets, we then selected a subset of 454 SL/SDL partner genes that is common to all in vitro and in vivo datasets (Table S2F, see details in STAR Methods; illustrated in Figure 1B, module 1), which we focus on further analysis.
Initial screening and filtering of predicted SL/SDL-based targets with published CRISPR-Cas9 genetic screens
To further filter the 454 predicted SL/SDL-based targets, we collected data from two published CRISPR-Cas9 genetic screens that aim to identify host genes modulating SARS-CoV-2 replication. These include a screen in the Vero E6 cell line (Wei et al., 2021) and another in the human A549ACE2 cell line at two different multiplicities of infections (MOIs; Daniloski et al., 2021). We note that these studies seek to identify targets whose knockout promotes cell survival by reducing the virus-induced cytopathic effects; however, our strategy aims to identify genes whose knockout specifically reduces the viability of infected cells. Thus, we expect that our predictions would be enriched in genes whose knockout results in strong cell dropouts in these genetic screens. Indeed, we find that our 454 predicted SL/SDL-based target genes are significantly enriched for cell viability-reducing genes (adjusted p values from Fisher’s exact tests range from 2.38E-02 to 4.00E-04, Figure 1E; STAR Methods). The 454 SL/SDL-based targets are also enriched for host proteins that interact with SARS-CoV-2 viral proteins (Fisher’s exact test adjusted p = 1.10E-02, Figure 1E; data of virus-interacting proteins is from (Gordon et al., 2020; Stukalov et al., 2021), indicating that the predicted targets are closely relevant to SARS-CoV-2 biology and infection. Among the 454 predicted targets, 140 genes are significantly strong cell viability-reducing genes (Wei et al., 2021, P = 1E-05; Daniloski et al., 2021, p = 0.002 for MOI1 and p = 0.02 for MOI3) when depleted in both CRISPR-Cas9 screen datasets (illustrated in Figure 1B, module 2; Figure 1E). These genes are also significantly enriched among the genes whose knockout reduced cell viability in two other recently published genome-wide CRISPR-Cas9 knockout screens in human hepatoma cells (Wang et al., 2021) p = 1.09E-04; and (Schneider et al., 2021) p = 3.73E-03 for the screen at 33°C and p = 4.95E-04 for the screen at 37°C; Figure 1F). We thus focused our further analysis on these 140 candidate targets as listed in Table S3, ranked by a weighted DE score (WDS) that considers both the number of DE genes genetically interacting with each SL/SDL partner and the DE fold change (STAR Methods).
Among those 140 targets, those that are inhibited by existing drugs are listed in Figure 2A (Table S3B), together with these drugs’ mechanisms of action as potential candidates for drug repurposing. Among these inhibitory drugs, warfarin and dicoumarol, targeting VKORC1 are FDA approved. There is one clinical trial for Covid-19 patients on chronic treatment with anticoagulant warfarin vs who are not receiving (ClinicalTrials.gov: NCT04518735). Other experimental drugs are undergoing clinical trials either for cancer or other diseases. Notably, quercetin, targeting CEBPB is specifically undergoing 12 clinical trials (such as ClinicalTrials.gov: NCT04844658, NCT04578158, and NCT04377789) for Covid-19 patients. A pathway enrichment analysis of these 140 SL/SDL-based targets (STAR Methods, Table S3E) shows enrichment in pathways including cell cycle processes, stress responses, DNA break repair processes, RNA metabolic processes, and RNA polymerase II transcriptions (Figure 2B). The DE genes are enriched in pathways including complement cascade, ion channel transport, innate immune processes, cytokine signaling, interferon signaling, and neutrophil degranulation (Figure 2C, Table S3F). A pairwise pathway enrichment analysis (STAR Methods; Figure 2D) shows that many SL/SDL interactions include cell-cell communications, innate immune system, posttranslational protein modification (i.e., columns with high numbers of significant p values in Figure 2D, Table S3G). The pathway combination involving the largest number of gene pairs is between the innate immune system (DE genes) and the cell cycle (SL/SDL partners) (involving 9 pairs, adjusted p = 5.63E-04). These results suggest that the cellular immune responses induced by SARS-CoV-2 infection can potentially control the cells’ fate when targeted in conjunction with the central pathways regulating cell proliferation and nucleic acid/protein metabolism.
Figure 2.

Candidate SL/SDL partner genes
(A) A panel of candidate SL/SDL-based known drug targets with an inhibitory mechanism of action. These drugs can be potentially repurposed to treat SARS-CoV-2 infection.
(B) Pathway enrichment results for the 140 candidate SL/SDL-based targets. A bar plot showing the negative log10-transformed Benjamini-Hochberg-adjusted p values (Padj) from Fisher’s exact tests (X axis) for the top pathways from the Reactome database (Fabregat et al., 2018) enriched by these SL/SDL-based targets. The color of the bars encodes the number of overlapping genes within each pathway. The black-dotted line corresponds to the cutoff of adjusted p < 0.05.
(C) A similar plot as in (b) showing the top pathways enriched by the SARS-CoV-2-induced differentially expressed (DE) genes that form SL/SDL interactions with the 140 candidate target genes.
(D) A heatmap illustrating the enrichment of DE gene – SL/SDL partner gene pairs formed by the 140 candidate targets in various combinations of pathways. Significant pathway combinations with adjusted p < 0.1 are shown here. Pathways of the DE genes are given on the horizontal axis (columns) and pathways of the SL/SDL partner genes are given on the vertical axis (rows) of the heatmap. Sizes of the circles correspond to their odds ratio of enrichment and color of the circles correspond to the negative log10 adjusted P values of a pathway combination.
(E) Boxplot of one-sided Wilcoxon rank-sum test results for average cell number between 135 candidate genes and the rest of the genes in the genome-wide siRNA screen. P value is shown in between the boxes.
In the screens analyzed previously, the cell viability-decreasing effects observed are potentially confounded by virus-induced cytopathic effects. Therefore, we additionally analyzed a very recent imaging-based genome-wide siRNA screen that we performed previously in human Caco-2 cells (data deposited at https://figshare.com/s/4117ac39b1d21b56f5e6), where SARS-CoV-2 replication and cell number after each individual target gene knockdown were measured independently via anti-SARS-CoV-2 antibody and DAPI staining, respectively. 135 of our 140 predicted candidate genes were covered within the siRNA library of this screen (Table S3H). First, across these genes, we found that knockdowns resulting in lower cell numbers tend to also have lower percentage of SARS-CoV-2-positive cells (Spearman’s ρ=0.16, p < 2.2E-16), supporting the notion that reducing the viability of the virus-infected cells can indeed inhibit virus replication. Second, we find that the knockdown of these 135 predicted genes indeed results in significantly lower cell numbers compared to the knockdown of the rest of the genes in the screen (one-sided Wilcoxon rank-sum test p = 0.002, Figure 2E). Reassuringly, within the 135 SL/SDL candidates, the candidate genes with lower cell viability (relative cell number <1.0) have higher weighted DE score (one-sided Wilcoxon rank-sum test p = 0.032), and similarly, the candidates with lower percentage of SARS-CoV-2-positive cells (Z score<0) also have higher weighted DE score (one-sided Wilcoxon rank-sum test p = 0.057; STAR Methods). These results suggest that the SL/SDL-based predictions and the associated weighted DE score measuring the strength of SL/SDL effects can facilitate the identification of anti-SARS-CoV-2 targets that function via specifically reducing the viability of the infected host cells.
Network identification and experimental testing of 26 top-ranked SL/SDL targets
Next, we aimed to further narrow down the list of targets that we should further test in an additional, dedicated genomic screen. To this end, we set to first identify the subset of SL/SDL interactions with higher relevance to SARS-CoV-2 infection. Using simulated annealing, we identified an optimal SARS-CoV-2 specific SL subnetwork that maximizes the correlation between viral inhibition scores measured in the genome-wide siRNA screen conducted in house and the SL-based weighted DE scores of the target genes it includes (see STAR Methods). To experimentally validate the effect of the resulting 26 candidates on SARS-CoV-2 replication and cell viability, we performed an additional imaging-based siRNA assay in human intestinal epithelial Caco-2 cells. These cells were selected for these assays as they harbor endogenous expression on ACE2 and TMPRSS2 and are highly permissible to SARS-CoV-2 infection, they are amenable to high-throughput siRNA transfection, and the gastrointestinal epithelium is a target for SARS-CoV-2 infection in vivo (Xiao et al., 2020). The individual knockdown of each of the 26 genes was performed in a 384-well plate format using four replicates per target (Figure 3A). The percentage of virus-infected cells after target knockdown (reflecting viral load) was quantified via immunofluorescence staining with anti-SARS-CoV-2 antibody. Nontargeting (scrambled) siRNAs were used as negative control, and siRNAs targeting ACE2 and TMPRSS2 (two genes essential for viral entry) were used as positive controls (Figure 3B; STAR Methods). The total cell number after gene knockdown (reflecting cell viability) was measured based on DAPI staining, using siRNAs targeting essential genes for the survival of the host cells (Toxic siRNA) as positive controls for cell viability (Figure 3C).
Figure 3.

Experimental validation of selected synthetic lethal partners as anti-SARS-CoV-2 targets
(A) Workflow of targeted siRNA screens in human Caco-2 cells with 4 replicates for each targeted gene. Cells were transfected with siRNAs and then either infected with SARS-CoV-2 (MOI = 0.1) or left noninfected. The number of cells (viability) after each gene knockdown was measured via DAPI staining (number of DAPI+ objects) and viral replication was evaluated via anti-SARS-CoV-2 antibody (percentage of SARS-CoV-2+ cells).
(B) Scatterplot of viral infection in terms of average (n = 4) log fold change of SARS-CoV-2 infected cells (log2FC infection values) of 26 SL/SDL targets (gray dots) relative to the scrambled siRNA (green dots). Positive controls ACE2 and TMPRSS2 are marked by red dots and Toxic siRNA with blue dots. P value from one-sided Wilcoxon rank-sum test between the scrambled siRNA vs. the rest of the genes is shown in the plot.
(C) Scatterplot of cell viability in terms of average (n = 4) normalized count of cell numbers from the DAPI-stained images of 26 SL/SDL targets relative to the scrambled siRNA. P value from one-sided Wilcoxon rank-sum test between the scrambled siRNA vs. the rest of the genes is shown in the plot.
(D) Representative images showing viral (SARS-CoV-2, green) and DAPI (DNA, blue) staining. Shown are cells treated with scrambled, siRNAs targeting positive controls (ACE2, TMPRSS2, and toxic siRNA), or 3 top target genes (VKORC1, MED8, and EIF4G1), and then either infected with SARS-CoV-2 for 48 h (top panel) or left noninfected (bottom panel). Scale bar = 10 μm. Reduction in cell numbers in infected versus noninfected cells following the knockdown of predicted SL/SDL targets (when the same target gene is knocked down for both conditions).
(E) Barplot of cell viabilities (average normalized cell numbers out of 4 replicates for each gene relative to scrambled) of siRNAs targeting each of the 26 SL/SDL targets and controls are plotted for both infected (blue) and noninfected (pink) conditions. One-sided Wilcoxon signed-rank test p value of the average normalized cell numbers of each knocked-down target between two conditions is shown.
We first established the antiviral efficacy of the predicted targets; the individual knockdown of the 26 predicted SL/SDL targets resulted in an overall reduction in viral replication (p = 6.57E-07 ; Figure 3B) and reduced cell viability (p = 0.047; Figure 3C) compared to scrambled siRNAs. Of note, the knockdown of ACE2 and TMPRSS2 also markedly reduced viral replication, consistent with their previously recognized potential as effective drug targets (Hoffmann et al., 2021; Lin et al., 2021). Second, we tested if knockdown of the predicted targets indeed selectively reduces cell viability in infected cells but not in noninfected cells. To this end, we quantified the number of DAPI-positive cells after knockdown of the 26 predicted SL/SDL targets in both infected and noninfected conditions (Figure 3A). Reassuringly, we found that the cell numbers after knockdown of the SL/SDL targets, including VKORC1, MED8, and EIF4G1 but not of the non-SL target ACE2, were significantly reduced in the infected compared to noninfected conditions (p = 3.302E-11, Figures 3D and 3E), testifying to the selective killing induced by the predicted SL/SDL targets. Overall, 24 out of 26 targets showed reduced viral load compared to the control and reduced cell viability in the infected condition compared to the noninfected state (Figure 3E; Table S4A).
Among our 26 predicted targets, the top three emerging in our screen are VKORC1, an anticoagulant that binds to ORF7a of SARS-CoV-2 (Holcomb et al., 2020; Janssen and Walk, 2020) and targeted by warfarin (Irwin et al., 2021); MED8, a gene required for the activation of transcription (Boube et al., 2002) and EIF4G1, known for its role in cell growth, proliferation, and differentiation (Ramírez-Valle et al., 2008) (Figure 3D). Some of the validated targets are known to be involved in the context of other viral infections (Table S4B) – such as, CDC25B promotes influenza A virus replication (Cui et al., 2018).
Discussion
Given the current lack of effective treatment options for COVID-19, identifying new drug targets for the virus causing this rapidly spreading disease with emerging new variants is highly critical. In this work, we predicted potential clinically relevant SL/SDL-based anti-SARS-CoV-2 targets. We further tested our initial list of 454 emerging predictions using two published CRISPR-Cas9 genetic screens in two different cell lines (Vero E6 and human A549 with exogenous ACE2 expression) infected with SARS-CoV-2. By intersecting our predicted targets with the hits from the CRISPR screens, we further narrowed down this initial list to that of 140 clinically relevant candidate targets. A network-based analysis of these 140 targets using the results of a genome-wide siRNA screen that we have previously performed led to the selection of a smaller subset of 26 genes, which we further experimentally tested and validated via a small-scale targeted siRNA screen in human Caco-2 cells, thereby identifying targets that selectively reduce cell viability only in infected cells and demonstrating the utility of our synthetic lethality approach. Our analysis involves the integration of multiple published gene expression datasets of SARS-CoV-2 infection, both in vitro and in vivo, finding an overall moderate but robust level of concordance of virus-induced DE genes, especially and importantly across the in vivo datasets. This analysis encompasses some of the largest available transcriptomic datasets of COVID-19 patients (Butler et al., 2021; Lieberman et al., 2020) as well as single-cell datasets, which adds to the clinical relevance of our predicted targets. Many additional in vitro transcriptomics datasets on SARS-CoV-2 infection are available (like in human hepatoma cells or in non-small cell lung cancer cells), but we have chosen to build up our 140 targets list based on the two particular datasets of Vero E6 and A549ACE2 cells, as the same cell lines were used in the two CRISPR-Cas9 genetic screens that we used for validation. Reassuringly, we identified the significantly differentially expressed (DE) genes in the SARS-CoV-2 infected samples vs matched controls in Calu3 cell lines (available from (Blanco-Melo et al., 2020), as we did for A549ACE2 and Vero E6 cells. We then did an enrichment test for the overlapping DE genes between Calu3 and A549ACE2 and between Calu3 and Vero E6 cell lines (odds ratio and adjusted p values computed using the Fisher’s exact test; similar to the analysis done in Figure 1C) and found higher-than-random pairwise enrichment. Therefore, the relevant transcriptomic analyses are comparable between these cell lines.
Our study is motivated by the SL-based antiviral strategy proposed by (Mast et al., 2020) with preliminary proof-of-concept application for poliovirus demonstrated in (Navare et al., 2020). This strategy aims to specifically reduce the viability or impair the normal functioning of the infected host cells, thereby suppressing their capacity to support viral proliferation (Mast et al., 2020). To tease apart these two factors, we specifically employed an imaging-based siRNA assay where both viral replication and cell counts after gene knockdown were measured. In our previously reported genome-wide screen using this method, a positive correlation between these two variables was observed, supporting the inhibition of host cell viability as a potential antiviral strategy. In the targeted siRNA screen conducted in this study, a similar correlation was observed, where the knockdown of our 26 candidate targets significantly reduced viral replication accompanied by a reduction in cell number. However, importantly, knocking down these genes did not harm the noninfected cells. This reduction in cell viability in infected cells is likely not driven by virus-induced cytopathic effects (CPE), as cells treated with the negative control scrambled siRNA showed significantly higher rates of infection and higher cell number counts than cells treated with targeting siRNAs (Figures 3B–3D). The Pearson correlation value between log2FC infection and cell number in infected cells was in the positive range (p value = 0.1235). Further in vivo validations are required to translate these in vitro validated predictions to the clinic.
In summary, our study represents a first of its kind proof-of-concept application of a synthetic lethality inference approach to predict anti-SARS-CoV-2 targets in a systematic genome-wide manner. We provide a list of potential targets that are supported by multiple lines of evidence, importantly, emerging from the integrative analysis of both in vitro and in vivo data, giving rise to the hope that they then are more likely to be translationally relevant and help in the development of new drugs for treating COVID-19.
Limitations of the study
To predict the SL/SDL partner genes, we applied the ISLE algorithm which was originally designed to identify synthetic lethal partners in the cancer setting (Lee et al., 2018). Therefore, an important assumption underlying our approach is that many of the SL/SDL interactions identified by ISLE for pan-cancer analysis (i.e., SL/SDLs which are functional across many cancer types) are also functionally relevant in normal noncancerous cells because of conservation in the cellular network. We have demonstrated evidence supporting this assumption in a recent study (Cheng et al., 2021b), although not all partners identified by ISLE are functional outside the context of cancer. At least partially mitigating this limitation, the final 140 candidates target list we reported represents a high-confidence subset filtered based on COVID-19 patient data supported by the CRISPR-Cas9/siRNA genetic screens in the SARS-CoV-2 setting, and these candidates also have considerable literature support for their relevance to SARS-CoV-2. Of note, gene knockdowns (via siRNA or CRISPR-Cas9) and small molecule inhibition aimed at the same targets may yet lead to different phenotypic effects. Distinct SL/SDL partners may act via different biological mechanisms and their elucidation is obviously a huge endeavor that lies beyond the scope of this study. Moreover, targeting SL/SDL partners may potentially lead to both cell death and cell-cycle arrest/reduced growth. Obviously, further in vivo validations are required for the potential future therapy development based on these targets, specifically testing the potential toxicity issues that may arise with killing of infected cells. Even if such toxicity arises on a serious scale, SL-based treatments may possibly still prove to be useful at early stages of infection.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-SARS-CoV-2 N protein rabbit polyclonal antibody | kind gift from Dr. Adolfo Garcia-Sastre (Icahn School of Medicine at Mount Sinai) | N/A |
| Alexa Fluor 488-conjugated anti-rabbit secondary antibody | ThermoFisher | RRID: AB_2910224 |
| Bacterial and virus strains | ||
| SARS-CoV-2 USA-WA1/2020 | BEI resources | NR-52281 |
| Biological samples | ||
| Heat-inactivated fetal bovine serum | Gibco | |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine RNAiMAX transfection reagent | ThermoFisher | 13778150 |
| OptiMEM media | ThermoFisher | |
| PFA | Boston BioProducts | |
| Triton X-100 | Sigma | |
| DAPI (4,6-diamidine-2-phenylindole) | KPL | 5930-0006 |
| DMEM media | Gibco | |
| Penicillin | Fisher Scientific | |
| streptomycin | Fisher Scientific | |
| BSA | Sigma | |
| Deposited data | ||
| SARS-CoV-2 infected RNAseq data, Vero E6 cells | Riva et al., 2020 | GSE153940 |
| SARS-CoV-2 infected RNAseq data, A549 cells with exogenous ACE2 | Blanco-Melo et al., 2020 | GSE147507 |
| SARS-CoV-2 infected RNAseq data, Calu3 cell line | Blanco-Melo et al., 2020 | GSE147507 |
| SARS-CoV-2 infected RNAseq data, nasopharyngeal swab samples from human COVID-19 patients | Butler et al., 2021 | https://www.nature.com/articles/s41467-021-21361-7#Sec46 |
| SARS-CoV-2 infected RNAseq data, nasopharyngeal swab samples from human COVID-19 patients | Lieberman et al., 2020 | https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000849 |
| SARS-CoV-2 infected RNAseq data, single-cell data of nasopharyngeal and bronchial samples from COVID-19 patients | Chua et al., 2020 | https://doi.org/10.6084/m9.figshare.12436517 |
| SARS-CoV-2 infected RNAseq data, single-cell data of bronchoalveolar lavage fluid from COVID-19 patients | Liao et al., 2020 | https://github.com/zhangzlab/covid_balf |
| CRISPR-Cas9 genetic screening data, Vero E6 cell line | Wei et al., 2021 | https://doi.org/10.1016/j.cell.2020.10.028 |
| CRISPR-Cas9 genetic screening data, human alveolar basal epithelial carcinoma A549 cell line with exogenous ACE2 expression | Daniloski et al., 2021 | https://doi.org/10.1016/j.cell.2020.10.030 |
| Genome-wide siRNA screen in Caco-2 cells infected with SARS-CoV-2 | Figshare | https://figshare.com/s/4117ac39b1d21b56f5e6 |
| Human proteins reported to interact with SARS-CoV-2 viral proteins | Gordon et al., 2020 | https://www.nature.com/articles/s41586-020-2286-9 |
| Human proteins reported to interact with SARS-CoV-2 viral proteins | Stukalov et al., 2021 | https://www.nature.com/articles/s41586-021-03493-4 |
| Experimental models: Cell lines | ||
| Human Caco-2 cells | ATCC | ATCC HTB-37 |
| Vero E6 cell line | ATCC | ATCC CRL-1586 |
| Software and algorithms | ||
| ISLE | Lee et al., 2018 | https://github.com/jooslee/ISLE/ |
| Drugbank | Wishart et al., 2018 | https://go.drugbank.com/ |
| Pathway annotations from MSigDB v7.2 | Liberzon et al., 2011 | http://www.gsea-msigdb.org/gsea/msigdb/collections.jsp |
| Columbus v2.5 | Perkin Elmer | https://www.perkinelmer.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Eytan Ruppin (eytan.ruppin@nih.gov).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
Caco-2 (ATCC HTB-37) are epithelial cells isolated from colon tissue from a 72-year-old, white, male with colorectal adenocarcinoma. Cells were cultured in Eagle’s Minimum Essential Medium (EMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 1 mM Sodium Pyruvate (Life Technologies), 2 mM L-Glutamine (Fisher Scientific), 10 mM HEPES (Fisher Scientific), and 100 U/ml penicillin–100 μg/ml streptomycin (Fisher Scientific). Cells were grown at 37°C in 5% CO2. Cells are authenticated by the ATCC and were tested regularly and confirmed to be free of mycoplasma contamination.
Viral strains
SARS-CoV-2 USA-WA1/2020 strain, isolated from an oropharyngeal swab from a patient with a respiratory illness who developed clinical disease (COVID-19) in January 2020 in Washington, USA, was obtained from BEI Resources (NR-52281). The virus was propagated in Vero E6 (ATCC CRL-1586) cells and stored at −80°C in aliquots. Plaque forming unit (PFU) assays were performed to titrate the cultured virus. All experiments involving live SARS-CoV-2 followed the approved standard operating procedures of the Biosafety Level 3 facility at the Sanford Burnham Prebys Medical Discovery Institute.
Method details
Differential expression analysis of transcriptomic data on SARS-CoV-2 infection
Several RNA-sequencing datasets of SARS-CoV-2-infected and matched control samples were obtained as follows: Vero E6 cells (Riva et al., 2020) from the GEO database, dataset ID GSE153940; A549 cells with exogenous ACE2 (A549ACE2 (Blanco-Melo et al., 2020) from GEO, GSE147507), nasopharyngeal swab samples from human COVID-19 patients (Butler et al., 2021; Lieberman et al., 2020), from the Supplementary Materials of the respective publications), single-cell data of nasopharyngeal and bronchial samples from COVID-19 patients (Chua et al., 2020) from FigShare https://doi.org/10.6084/m9.figshare.12436517), and single-cell data of bronchoalveolar lavage fluid from COVID-19 patients (Liao et al., 2020) obtained following instructions in https://github.com/zhangzlab/covid_balf.
For the Vero E6 and A549ACE2 data, we performed differential expression (DE) analysis of virus-infected vs control samples with the DESeq2 R package (Love et al., 2014). Significant DE genes with FDR<0.05 were identified and further filtered by dataset-specific log fold-change cutoffs so as to get a reasonable number of such DE genes for further analysis. Specifically, 123 down (log fold-change < −1.0) and 171 up-regulated (log fold-change > 2.0) genes were identified from the Vero E6 dataset, 98 down (log fold-change < −1.5) and 172 up-regulated (log fold-change > 2.0) genes were identified from the A549ACE2 dataset. For the dataset from (Butler et al., 2021), we used the virus-positive vs negative DE results provided by the authors that correspond to the analysis of 10 million human mapped reads with correction of potential confounders (batch effect and deconvolution of cell proportions). Similarly, we also used the DE results provided by the authors for the (Lieberman et al., 2020) dataset. We combined the significant DE genes (FDR<0.05) with absolute log fold-change values greater than 1.0 from both datasets for further downstream analysis; this set includes 121 down and 166 up-regulated genes. For single-cell dataset from (Chua et al., 2020; Liao et al., 2020), we only focused on epithelial cells (based on the cell type annotation provided by the authors) as the virus-infected cells relevant to our target prediction approach and performed DE analysis for virus-positive vs negative samples using the Seurat R package (Stuart et al., 2019), specifically, for nasopharyngeal swab and bronchial basal cells from (Chua et al., 2020) and bronchoalveolar lavage fluid epithelial cells from (Liao et al., 2020). Significant DE genes (FDR<0.05) with absolute log fold-change values greater than 0.5 were selected, resulting in 166 down and 181 up-regulated genes from the (Chua et al., 2020) dataset and 196 down and 320 up-regulated genes from the (Liao et al., 2020) dataset.
Identification of SL/SDL partners of the SARS-CoV-2-altered genes
For the significant DE genes identified from each of the above datasets, we identified their SL partners (for down-regulated genes) using the ISLE algorithm (Lee et al., 2018) (Table S2). ISLE was originally designed for SL prediction in the cancer setting, but as discussed in the main text here we apply it to identify potential SLs for antivirus. ISLE involves four steps analyzing gene essentiality data in cancer cell lines, tumor molecular profiles, cancer patient survival data, and gene phylogeny data to identify clinically relevant SL interactions (described in detail in Lee et al., 2018). For the up-regulated DE genes, we instead identified their SDL partner genes also using ISLE but with minor modifications to account for the directional difference between SL and SDL. For example, in the third step of ISLE analyzing cancer patient survival data, while the original ISLE algorithm tests for the association of concomitant down-regulation of a pair of genes with better patient survival as a feature of SL, in the modification for SDL we instead tested for the association of down-regulation of one gene and the up-regulation of another gene in a gene pair with better patient survival, which corresponds to the SDL definition. We used FDR cutoff 0.1 to select significant SL/SDL partners. We ranked the ISLE-predicted partner genes by their P values from the patient survival analysis. Significant partner genes (a total of 454 genes, Table S2F) that are common to all analyzed datasets were taken for further downstream analysis.
Prioritization of targets using genome-wide CRISPR-Cas9 genetic screening data in SARS-CoV-2-infected cells
We obtained two currently available genome-wide CRISPR-Cas9 genetic screening data aiming to identify host genes regulating SARS-CoV-2 infection, one from Vero E6 cell line (Wei et al., 2021) and the other from human alveolar basal epithelial carcinoma A549 cell line with exogenous ACE2 expression (A549ACE2; Daniloski et al., 2021). The complete results of sgRNA differential expression were obtained from the Supplementary Materials of the two publications. To validate that our predicted targets are indeed SL with SARS-CoV-2 infection, i.e., the inhibition of these genes can lead to more cell dropout in the virus-infected cells than in control cells, we tested for enrichment of genes showing negative log fold-changes in the CRISPR-Cas9 screens with the GSEA method (Subramanian et al., 2005) as implemented in the R package fgsea (Korotkevich et al., 2021). Specifically, the log fold-change values across genes from each CRISPR-Cas9 screen were ranked, and GSEA was applied to the ranked list with the “gene set” being the 454 predicted SL/SDL-based targets common to all datasets. To obtain our final list of 140 candidate targets, we intersect the 454 consensus predictions with the genes showing the strongest negative log fold-changes in the CRISPR-Cas9 screens. These strongest CRISPR hits were selected as follows: In the Vero E6 dataset, we selected 680 genes with negative mean log2 fold-changes with FDR<0.1. The A549ACE2 dataset contains results from two screens with different multiplicities of infections (MOIs), and we selected 2811 genes from the lower MOI screen and 1653 genes from the higher MOI screen whose absolute log fold-changes are lesser than −0.5. The intersection of all selected CRISPR genes with the 454 consensus SL/SDL-based targets gives the final list of 140 candidate target genes (Table S3).
Calculation of weighted DE score
Each of 140 candidates have the same or different significant DE genes from six different transcriptomic datasets - in vitro Vero E6 (Riva et al., 2020), in vitro A549ACE2 cells (Blanco-Melo et al., 2020), two in vivo Bulk Swab datasets (Butler et al., 2021; Lieberman et al., 2020) and two in vivo single-cell RNA-seq datasets (Chua et al., 2020; Liao et al., 2020). We hypothesized that the effect of knockdown of these SL/SDL-based targets is dependent upon the synthetic lethality strength, as reflected by the extent of perturbation of DE genes by the virus (log fold change) in their respective transcriptomic datasets. We considered the median absolute log fold change of a DE gene in all six datasets (if it is available). If a DE gene is down-regulated in the dataset where it is considered to be significant, then all down-regulated log fold changes in other datasets will contribute to the final median value, and similar will be the treatment for up-regulated cases. For a candidate gene g, its weighted DE score (Sg) was calculated as in Equation 1,
| (Equation 1) |
Where, ‘g’ is synthetic lethal partner of ‘n’ number of DE genes (D) and median log fold changes (in absolute value) of Di from six different transcriptomic datasets were considered according to the status (either up-regulated or down-regulated) of the significant DE gene corresponding to the SL/SDL pair.
Virus-interacting genes and pathway enrichment analysis on the predicted SL/SDL-based target genes
We also tested for the enrichment between the 454 predicted targets with human proteins reported to interact with SARS-CoV-2 viral proteins (Gordon et al., 2020; Stukalov et al., 2021) (data obtained from the Supplementary Materials of these publications) with Fisher’s exact test using the fisher.test function from the R software. Similarly, Fisher’s exact tests were used to test for pathway enrichment of the final SL/SDL-based candidate targets (Table S3E) and their corresponding DE genes (Table S3F) using pathway annotations from the Reactome database (Fabregat et al., 2018) collected from MSigDB v7.2 (Liberzon et al., 2011). Pathways with more than three genes overlapping with the candidate target set were considered. P values across pathways were corrected for multiple testing with the Benjamini-Hochberg method.
Enrichment of pathway combinations for DE-S(D)L partner gene pairs
Fisher’s exact tests were used to test the enrichment of the SL/SDL gene pairs in each possible pathway combination (i.e. “pathway pair”) (Table S3G). Specifically, pathway annotation of single genes from the Reactome database (Fabregat et al., 2018) collected from MSigDB v7.2 (Liberzon et al., 2011) was used to create all possible pathway pairs and their member gene pairs. The “background” set for the enrichment was the gene pairs formed between the virus-induced DE genes and all genome-wide genes. P values were corrected with the Benjamini-Hochberg method.
Annotation of the final SL/SDL-based candidate target gene list
The predicted targets (140 candidates) were mapped to known drugs (clinically approved and experimental/investigational) that target (i.e. inhibit) them using drug target annotation from DrugBank (Wishart et al., 2018). We manually searched the literature on each of the 26 candidate genes to assess their relevance to SARS-CoV-2 or other viral infection based on prior knowledge. Detailed annotation with regard to literature support for the candidate genes are provided in Table S4B.
Genome-wide siRNA screen
We used our previously performed genome-wide siRNA screen, carried out in human Caco-2 cells to identify host cell factors that affect the replication of SARS-CoV-2 (data deposited at https://figshare.com/s/4117ac39b1d21b56f5e6).
Selection of 26 genes for experimental validation
Out of 140 candidate genes, we have cell viability and viral replication information for 135 SL/SDL-based targets. The size of our synthetic lethal network for 135 candidates including DE genes from all six different transcriptomic datasets was 854. We used the simulated annealing method to maximize the correlation between viral inhibition from our genome-wide siRNA screen and the SL/SDL-based weighted DE scores, which resulted in an optimal SARS-CoV-2 specific SL subnetwork. We finally selected 26 target genes from this subnetwork where percentage of SARS-CoV-2-positive cells was reduced (z-score < 0) in the previously performed genome-wide siRNA screen.
‘Optim’ function in R was used to optimize the whole SL network, using variable network sizes and seed and using 10,000 iterations for each run. All other parameters were used as default in the function. Different sizes of networks were optimized for minimization of correlation (Spearman’s ρ in R) between weighted DE score and cell viability from siRNA screen for candidate genes present in that network. The resulting SL network was used to calculate the correlation between weighted DE score and percentage of SARS-CoV-2-positive cells of those candidate genes in the selected network. The aim of this in silico experiment was to extract a SARS-CoV-2 specific SL network where cell viability and viral replication will be lower upon knocking down the candidate genes in that network.
Targeted siRNA screen
Screen
To evaluate the effect of the 26 predicted targets on viral replication and cell viability, a targeted siRNA screen was conducted using human Caco-2 cells (ATCC). siRNAs targeting these genes were obtained from the Dharmacon ON-TARGETplus SMARTpool library and individually arrayed in 384-well plates at a concentration of 12.5 nM per well. First, siRNAs were incubated with 0.1 μL Lipofectamine RNAiMAX transfection reagent (ThermoFisher) diluted in 9.90 μl Opti-MEM media (ThermoFisher) for 20 min at room temperature. Then, 3,000 Caco-2 cells diluted in cell growth media (see below) were seeded on top and incubated for 48 h at 37°C with 5% CO2 conditions. Cells were then either infected with SARS-CoV-2 (isolate USA-WA1/2020) at a multiplicity of infection (MOI) 0.1 or left non-infected. After an incubation period of 48 h at 37°C with 5% CO2 conditions, cells were fixed with 4% PFA (Boston BioProducts) for 4 h at room temperature. Cells were then washed twice with PBS prior to 20 min permeabilization with 0.5% Triton X-100 and 1 h blocking with 3% BSA (Sigma) at room temperature. Cells were then incubated for 2 h at room temperature with primary anti-SARS-CoV-2 N protein rabbit polyclonal antibody (kind gift from Dr. Adolfo Garcia-Sastre), followed by three PBS washes and 1 h incubation with Alexa Fluor 488-conjugated anti-rabbit secondary antibody (A-11008, ThermoFisher) diluted in 3% BSA. Cells were washed three times with PBS and then stained with DAPI (4,6-diamidine-2-phenylindole, KPL). Plates were sealed and stored at 4°C until imaging. Caco-2 cells growth media: 40 μL DMEM media (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), and 50 U/mL penicillin - 50 μg/mL streptomycin (Fisher Scientific).
Analysis
The resulting number of cells and SARS-CoV-2 replication after each individual target knockdown was measured using high-content imaging. Images were acquired using the IC200 imaging system (Vala Sciences), and then analyzed using the analysis software Columbus v2.5 (Perkin Elmer). The number of DAPI+ objects was used to calculate the number of cells in the well, and the number of SARS-CoV-2+ to calculate the percentage of infected cells. The average percentage of infected cells for each target knockdown (n=4) was normalized to that of the negative control scrambled and used to calculate the log2FC infection.
Quantification and statistical analysis
Differential expression analyses were performed using R library DEseq2 (Love et al., 2014). Gene enrichment analysis was done using R package fgsea (Korotkevich et al., 2021). Fisher’s exact test was performed using the fisher.test function from the R software. P values were corrected with the Benjamini-Hochberg method. ‘Optim’ function in R was used to optimization of the network. Correlation between two data were calculated using Spearman’s ρ in R software.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, NCI, CCR; and used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). We acknowledge and thank the National Cancer Institute for providing financial and infrastructural support. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS- Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281. This work was also supported by the following grants to the Sanford Burnham Prebys Medical Discovery Institute: DoD: W81XWH-20-1-0270; DHIPC: U19 AI118610; Fluomics/NOSI: U19 AI135972. K.C. and S.S. are supported by the NCI-UMD Partnership for Integrative Cancer Research Program.
Author contributions
L.R.P., K.C., N.U.N., S.S., and E.R. conceived the study, collected the data, designed the computational analyses, performed the analyses, and wrote the paper. L.M.S. and S.C. designed the experimental analyses and wrote the paper. Y.P., L.R., X.Y. performed experimental analyses. F.S. and J.S.L. helped in computational analyses. All authors approved the publication of this manuscript.
Declaration of interests
E.R. is a co-founder of Medaware, Metabomed, and Pangea Therapeutics (divested from the latter). E.R. serves as a nonpaid scientific consultant to Pangea Therapeutics, a company developing a precision oncology SL-based multi-omics approach. The other authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104311.
Contributor Information
Sumit K. Chanda, Email: schanda@sbpdiscovery.org.
Eytan Ruppin, Email: eytan.ruppin@nih.gov.
Supporting citations
The following reference appears in the Supplemental Information: Beura et al., 2011; Cencic et al., 2011; Chapagain., 2020; Chen et al., 2020; Dutta et al., 2020; Nehme et al., 2020; Salvatori et al., 2020; Turjya et al., 2020; Weinstein et al., 2013; Wu et al., 2020; Younan et al., 2018; Zhang et al., 2018.
Supplemental information
Pathway enrichment analysis (FDR <0.1) for SL/SDL partner genes, DE genes and combination pathway enrichment analysis for DE gene-SL/SDL partner gene pairs for 140 candidates. List of 135 candidates overlapping with genome-wide siRNA screen.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
References
- Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., et al. Remdesivir for the treatment of covid-19 - final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura L.K., Dinh P.X., Osorio F.A., Pattnaik A.K. Cellular poly(c) binding proteins 1 and 2 interact with porcine reproductive and respiratory syndrome virus nonstructural protein 1β and support viral replication. J. Virol. 2011;85:12939–12949. doi: 10.1128/jvi.05177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Münch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583:469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boube M., Joulia L., Cribbs D.L., Bourbon H.M. Evidence for a mediator of RNA polymerase II transcriptional regulation conserved from yeast to man. Cell. 2002;110:143–151. doi: 10.1016/s0092-8674(02)00830-9. [DOI] [PubMed] [Google Scholar]
- Butler D., Mozsary C., Meydan C., Foox J., Rosiene J., Shaiber A., Danko D., Afshinnekoo E., MacKay M., Sedlazeck F.J., et al. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 2021;12:1660. doi: 10.1038/s41467-021-21361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencic R., Desforges M., Hall D.R., Kozakov D., Du Y., Min J., Dingledine R., Fu H., Vajda S., Talbot P.J., Pelletier J. Blocking eIF4E-eIF4G interaction as a strategy to impair coronavirus replication. J. Virol. 2011;85:6381–6389. doi: 10.1128/jvi.00078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapagain P. Potential role of cellular senescence on coronavirus infections. Preprints. 2020 doi: 10.20944/preprints202004.0532.v1. [DOI] [Google Scholar]
- Chen D., Sun W., Li J., Wei B., Liu W., Wang X., Song F., Chen L., Yang J., Yu L. Serum cystatin C and coronavirus disease 2019: a potential inflammatory biomarker in predicting critical illness and mortality for adult patients. Mediators Inflamm. 2020;2020:1–10. doi: 10.1155/2020/3764515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K., Martin-Sancho L., Pal L.R., Pu Y., Riva L., Yin X., Sinha S., Nair N.U., Chanda S.K., Ruppin E. Genome-scale metabolic modeling reveals SARS-CoV-2-induced metabolic changes and antiviral targets. Mol. Syst. Biol. 2021;17 doi: 10.15252/msb.202110260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K., Nair N.U., Lee J.S., Ruppin E. Synthetic lethality across normal tissues is strongly associated with cancer risk, onset, and tumor suppressor specificity. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abc2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- Cui L., Mahesutihan M., Zheng W., Meng L., Fan W., Li J., Ye X., Liu W., Sun L. CDC25B promotes influenza a virus replication by regulating the phosphorylation of nucleoprotein. Virology. 2018;525:40–47. doi: 10.1016/j.virol.2018.09.005. [DOI] [PubMed] [Google Scholar]
- Daniloski Z., Jordan T.X., Wessels H.H., Hoagland D.A., Kasela S., Legut M., Maniatis S., Mimitou E.P., Lu L., Geller E., et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell. 2021;184:92–105.e116. doi: 10.1016/j.cell.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta N.K., Mazumdar K., Gordy J.T. The nucleocapsid protein of SARS-CoV-2: a target for vaccine development. J. Virol. 2020;94 doi: 10.1128/jvi.00647-20. e00647-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gkx1132. D649-d655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X., Arang N., Rigiracciolo D.C., Lee J.S., Yeerna H., Wang Z., Lubrano S., Kishore A., Pachter J.A., König G.M., et al. A platform of synthetic lethal gene interaction networks reveals that the GNAQ uveal melanoma oncogene controls the hippo pathway through FAK. Cancer Cell. 2019;35:457–472.e5. doi: 10.1016/j.ccell.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster P., Forster L., Renfrew C., Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. U S A. 2020;117:9241–9243. doi: 10.1073/pnas.2004999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Hofmann-Winkler H., Smith J.C., Krüger N., Arora P., Sørensen L.K., Søgaard O.S., Hasselstrøm J.B., Winkler M., Hempel T., et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine. 2021;65:103255. doi: 10.1016/j.ebiom.2021.103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb D., Alexaki A., Hernandez N., Laurie K., Kames J., Hamasaki-Katagiri N., Komar A.A., DiCuccio M., Kimchi-Sarfaty C. Potential impact on coagulopathy of gene variants of coagulation related proteins that interact with SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.09.08.272328. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Clinical Trials Registry Platform (ICTRP) COVID-19 trials. 2022. https://www.who.int/clinical-trials-registry-platformhttps://www.who.int/ictrp/COVID19-web.csv
- Irwin M.N., Adie S., Sandison K., Alsomairy S.A., Brancaccio A. Warfarin dose requirements in adults hospitalized with COVID-19 infection: a retrospective case series. J. Pharm. Pract. 2021 doi: 10.1177/08971900211000705. 8971900211000705. [DOI] [PubMed] [Google Scholar]
- Janssen R., Walk J. Vitamin K epoxide reductase complex subunit 1 (VKORC1) gene polymorphism as determinant of differences in Covid-19-related disease severity. Med. Hypotheses. 2020;144:110218. doi: 10.1016/j.mehy.2020.110218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomah S., Asdaq S.M.B., Al-Yamani M.J. Clinical efficacy of antivirals against novel coronavirus (COVID-19): a review. J. Infect. Public Health. 2020;13:1187–1195. doi: 10.1016/j.jiph.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- Kroll E.S., Hyland K.M., Hieter P., Li J.J. Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics. 1996;143:95–102. doi: 10.1093/genetics/143.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H. Coronavirus breakthrough: dexamethasone is first drug shown to save lives. Nature. 2020;582:469. doi: 10.1038/d41586-020-01824-5. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Das A., Jerby-Arnon L., Arafeh R., Auslander N., Davidson M., McGarry L., James D., Amzallag A., Park S.G., et al. Harnessing synthetic lethality to predict the response to cancer treatment. Nat. Commun. 2018;9:2546. doi: 10.1038/s41467-018-04647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman N.A.P., Peddu V., Xie H., Shrestha L., Huang M.L., Mears M.C., Cajimat M.N., Bente D.A., Shi P.Y., Bovier F., et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Li Y., Zhang Y., Liu Z., Mu X., Gu C., Liu J., Li Y., Li G., Chen J. Ceftazidime is a potential drug to inhibit SARS-CoV-2 infection in vitro by blocking spike protein-ACE2 interaction. Signal Transduct. Target. Ther. 2021;6:198. doi: 10.1038/s41392-021-00619-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahase E. Covid-19: FDA authorises neutralising antibody bamlanivimab for non-admitted patients. BMJ. 2020;371:m4362. doi: 10.1136/bmj.m4362. [DOI] [PubMed] [Google Scholar]
- Mast F.D., Navare A.T., van der Sloot A.M., Coulombe-Huntington J., Rout M.P., Baliga N.S., Kaushansky A., Chait B.T., Aderem A., Rice C.M., et al. Crippling life support for SARS-CoV-2 and other viruses through synthetic lethality. J. Cell Biol. 2020;219 doi: 10.1083/jcb.202006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidar S., Gopal G., Visaga Ambi S. Targeting the viral-entry facilitators of SARS-CoV-2 as a therapeutic strategy in COVID-19. J. Med. Virol. 2021;93:5260–5276. doi: 10.1002/jmv.27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navare A.T., Mast F.D., Olivier J.P., Bertomeu T., Neal M., Carpp L.N., Kaushansky A., Coulombe-Huntington J., Tyers M., Aitchison J.D. Viral protein engagement of GBF1 induces host cell vulnerability through synthetic lethality. bioRxiv. 2020 doi: 10.1101/2020.10.12.336487. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme J., Borghesan M., Mackedenski S., Bird T.G., Demaria M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell. 2020;19 doi: 10.1111/acel.13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H., Peto R., Henao-Restrepo A.M., Preziosi M.P., Sathiyamoorthy V., Abdool Karim Q., Alejandria M.M., Hernández García C., Kieny M.P., Malekzadeh R., et al. Repurposed antiviral drugs for Covid-19 - interim WHO solidarity trial results. N. Engl. J. Med. 2021;384:497–511. doi: 10.1056/NEJMoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G., Lee J.S., Hasnis E., Tandoc K., Scott D.A., Verma S., Feng Y., Larue L., Sahu A.D., Topisirovic I., et al. Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat. Cell Biol. 2019;21:1590–1603. doi: 10.1038/s41556-019-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules C.I., Fauci A.S. COVID-19: the therapeutic landscape. Med (N Y) 2021;2:493–497. doi: 10.1016/j.medj.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragia G., Manolopoulos V.G. Inhibition of SARS-CoV-2 entry through the ACE2/TMPRSS2 pathway: a promising approach for uncovering early COVID-19 drug therapies. Eur. J. Clin. Pharmacol. 2020;76:1623–1630. doi: 10.1007/s00228-020-02963-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Valle F., Braunstein S., Zavadil J., Formenti S.C., Schneider R.J. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008;181:293–307. doi: 10.1083/jcb.200710215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L., Yuan S., Yin X., Martin-Sancho L., Matsunaga N., Pache L., Burgstaller-Muehlbacher S., De Jesus P.D., Teriete P., Hull M.V., et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020;586:113–119. doi: 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori G., Luberto L., Maffei M., Aurisicchio L., Roscilli G., Palombo F., Marra E. SARS-CoV-2 SPIKE PROTEIN: an optimal immunological target for vaccines. J. Transl. Med. 2020;18:222. doi: 10.1186/s12967-020-02392-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider W.M., Luna J.M., Hoffmann H.H., Sánchez-Rivera F.J., Leal A.A., Ashbrook A.W., Le Pen J., Ricardo-Lax I., Michailidis E., Peace A., et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell. 2021;184:120–132.e14. doi: 10.1016/j.cell.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukalov A., Girault V., Grass V., Karayel O., Bergant V., Urban C., Haas D.A., Huang Y., Oubraham L., Wang A., et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature. 2021;594:246–252. doi: 10.1038/s41586-021-03493-4. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilocca B., Britti D., Urbani A., Roncada P. Computational immune proteomics approach to target COVID-19. J. Proteome Res. 2020;19:4233–4241. doi: 10.1021/acs.jproteome.0c00553. [DOI] [PubMed] [Google Scholar]
- Turjya R.R., Khan M.A.A.K., Mir Md Khademul Islam A.B. Perversely expressed long noncoding RNAs can alter host response and viral proliferation in SARS-CoV-2 infection. Future Virol. 2020;15:577–593. doi: 10.2217/fvl-2020-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U. S. Food and Drug Administration Coronavirus (COVID-19) update: FDA authorizes monoclonal antibodies for treatment of COVID-19. 2021. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19
- V'Kovski P., Kratzel A., Steiner S., Stalder H., Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Simoneau C.R., Kulsuptrakul J., Bouhaddou M., Travisano K.A., Hayashi J.M., Carlson-Stevermer J., Zengel J.R., Richards C.M., Fozouni P., et al. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell. 2021;184:106–119.e14. doi: 10.1016/j.cell.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Alfajaro M.M., DeWeirdt P.C., Hanna R.E., Lu-Culligan W.J., Cai W.L., Strine M.S., Zhang S.M., Graziano V.R., Schmitz C.O., et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell. 2021;184:76–91.e13. doi: 10.1016/j.cell.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Coronavirus Disease (COVID-19) Dashboard . World Health Organization; 2022. https://covid19.who.int/ [Google Scholar]
- Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gkx1037. D1074-d1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Chen Y., Xia H., Wang C., Tan C.Y., Cai X., Liu Y., Ji F., Xiong P., Liu R., et al. Transcriptional and proteomic insights into the host response in fatal COVID-19 cases. Proc. Natl. Acad. Sci. U S A. 2020;117:28336–28343. doi: 10.1073/pnas.2018030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F., Tang M., Zheng X., Liu Y., Li X., Shan H. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology. 2020;158:1831–1833.e1833. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younan P., Iampietro M., Santos R.I., Ramanathan P., Popov V.L., Bukreyev A. Disruption of phosphatidylserine synthesis or trafficking reduces infectivity of ebola virus. J. Infect. Dis. 2018;218 doi: 10.1093/infdis/jiy489. S475-s485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Lan Y., Li M.Y., Lamers M.M., Fusade-Boyer M., Klemm E., Thiele C., Ashour J., Sanyal S. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. 2018;23:819–831.e815. doi: 10.1016/j.chom.2018.05.005. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Wang F., Tang J., Nussinov R., Cheng F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit Health. 2020;2:e667–e676. doi: 10.1016/s2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pathway enrichment analysis (FDR <0.1) for SL/SDL partner genes, DE genes and combination pathway enrichment analysis for DE gene-SL/SDL partner gene pairs for 140 candidates. List of 135 candidates overlapping with genome-wide siRNA screen.
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.