

Abstract
Synthetic opioids have been implicated as the single greatest contributor to rising drug-related fatalities in recent years. This study evaluated mu-opioid receptor (MOR) mediated effects of seven fentanyl-related substances that have emerged in the recreational drug marketplace, and for which there are no existing or only limited in vivo data. Adult male Swiss Webster mice were administered fentanyl-related substances and their effects on nociception and locomotion as compared to MOR agonist standards were observed. In locomotor activity tests, morphine (100, 180 mg/kg), fentanyl (1, 10 mg/kg), beta-methylfentanyl (10 mg/kg), para-methoxyfentanyl (10 mg/kg), fentanyl carbamate (100 mg/kg), and 3-furanylfentanyl (10 mg/kg), elicited significant (p ≤ 0.05) dose-dependent increases in locomotion. However, para-methylfentanyl and beta′-phenylfentanyl did not produce significant effects on locomotion at doses up to 100 mg/kg and phenylfentanyl (100 mg/kg) significantly decreased locomotion. In warm-water tail-withdrawal tests, all substances produced significant dose-dependent increases in antinociception with increasing ED50 values (95% CI) of fentanyl [0.08 mg/kg (0.04–0.16)] > para-methoxyfentanyl [0.43 mg/kg (0.23–0.77)] > 3-furanylfentanyl [0.51 mg/kg (0.36–0.74)] > beta-methylfentanyl [0.74 mg/kg (0.64–0.85)] > para-methylfentanyl [1.92 mg/kg (1.48–2.45)] > fentanyl carbamate [5.59 mg/kg (4.11–7.54)] > morphine [7.82 mg/kg (5.42–11.0)] > beta′-phenylfentanyl [19.4 mg/kg (11.0–34.4)] > phenylfentanyl [55.2 mg/kg (33.5–93.0)]. Naltrexone (1 mg/kg) increased ED50 values several fold with decreasing magnitudes of para-methylfentanyl (63.1×) > para-methoxyfentanyl (22.5×) > beta′-phenylfentanyl (21.0×) > 3-furanylfentanyl (20.6×) > beta-methylfentanyl (19.2×) > phenylfentanyl (5.23×) > fentanyl (3.95×) > fentanyl carbamate (2.21×) > morphine (1.48×). These findings expand upon in vivo results from previous studies and establish that the effects of these fentanyl related-related substances are at least in part mediated by the MOR.
Keywords: Analgesia, Analog, Fentanyl, Locomotion, Mice, Opioid
1. Introduction
Fentanyl is a synthetic drug that produces its effects primarily via activation of the mu-opioid receptor (MOR) and has become increasingly problematic for its role in accidental overdose in the United States (Al-Hasani and Bruchas, 2011; Drug Enforcement Administration, 2017a, 2017b, 2020; Hedegaard et al., 2018; Hedegaard et al., 2019; Pathan and Williams, 2012; Spencer et al., 2019). Clinically, fentanyl is administered to patients as an analgesic for acute and chronic pain and as an anesthetic in combination with other complementary drugs (Bailey et al., 1985; Mather, 1983; Peng and Sandler, 1999; Scholz et al., 1996). Recreationally, fentanyl and related substances are consumed by persons with opioid use disorder (OUD) for their euphoric effects and alleviation of opioid withdrawal symptoms resulting from physical dependence (Comer and Cahill, 2019; Kuczynska et al., 2018). Non-fatal intoxications in persons with OUD involving fentanyl and its analogs have steadily increased in prevalence (Arfken et al., 2017; Chhabra et al., 2021; Kenney et al., 2018; Martinez et al., 2020; Ochalek et al., 2020; Ochalek et al., 2019). Fatalities involving fentanyl and related substances have also surged in recent years (Kuhlman Jr. et al., 2003; Martin et al., 2006; Thompson et al., 2007). In 2013, there were 3105 deaths in the United States that involved synthetic opioids that increased over 10-fold to 36,359 in 2019 (Centers for Disease Control and Prevention, 2020). Moreover, synthetic opioids, including fentanyl-related substances, were involved in 72.9% of the 49,860 drug-related deaths in the United States in 2019 (Centers for Disease Control and Prevention, 2020). Given the increasing prevalence of both fatalities and non-fatal intoxications involving synthetic opioids, this study sought to evaluate the effects of fentanyl-related substances that are emerging in the recreational drug marketplace for which are no existing or only limited in vivo data.
We previously reported the effects of seven emerging fentanyl-related substances on nociception and locomotion in mice for isobutyrylfentanyl, crotonylfentanyl, valerylfentanyl, para-fluorobutyrylfentanyl, para-methoxybutyrylfentanyl, thiophenefentanyl, and benzodioxolefentanyl (Varshneya et al., 2019). We demonstrated that these fentanyl-related substances elicit significant dose-dependent antinociception and that most, but not all substances tested, also elicit significant dose-dependent hyperlocomotion. We found that these effects were at least in part mediated by the MOR as indicated by significant rightward shifts in antinociceptive dose-effect curves following pretreatment with naltrexone, a relatively selective MOR antagonist. Here we report that other structurally-related fentanyls will elicit MOR-mediated effects in mice. Given the relevance of these emerging substances to public health, their characterization will be useful for regulatory scientists and policy makers (e.g., drug scheduling), forensic toxicologists (e.g., post-mortem toxicological analysis), and clinicians (e.g., patient care following non-fatal intoxications).
This study was designed to test two related hypotheses to further understand the various structural determinants of potency and efficacy for MOR-mediated effects by fentanyl-related substances. First, that fentanyl-related substances will elicit antinociception in mice in warm-water tail-withdrawal tests similar to prototypical MOR agonists. Second, that fentanyl-related substances will elicit hyperlocomotion in mice in locomotor activity tests similar to prototypical MOR agonists. To test these hypotheses, seven fentanyl-related substances and two MOR agonist standards were evaluated to determine if they would elicit prototypical opioid-like effects including antinociception, as measured by increases in tail-withdrawal latency in warm-water tail-withdrawal tests, and hyperlocomotion, as measured by increases in distance traveled in locomotor activity tests. To further investigate the opioid-like mechanism responsible for antinociception, naltrexone was administered as a pretreatment in separate experiments to test for antagonism. The drugs evaluated were morphine and fentanyl as MOR agonist standards, and beta-methylfentanyl, para-methylfentanyl, para-methoxyfentanyl, fentanyl carbamate, 3-furanylfentanyl, phenylfentanyl, and beta′-phenylfentanyl as representative fentanyl-related substances. These drugs were chosen in part because of their appearance in the recreational drug marketplace and partly because several of them and their isomers or metabolites had been identified in fatal and non-fatal intoxications (Drug Enforcement Administration, 2021a, 2021b; European Monitoring Centre for Drugs and Drug Addiction, 2017; Krotulski and Logan, 2018; Misailidi et al., 2017; O’Donnell et al., 2017; Strayer et al., 2018; World Health Organization Expert Committee on Drug Dependence, 2017; Zawilska, 2017). This study clarifies whether these fentanyl-related substances have MOR agonist-like antinociceptive and hyperlocomotor effects similar to known abused opioids.
2. Methods
2.1. Drugs
(1) Morphine, (4R,4aR,7S,7aR,12bS)-3-methyl-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinoline-7,9-diol sulfate pentahydrate, was provided by the National Institute on Drug Abuse (Bethesda, MD, USA) Drug Supply Program. (2) Fentanyl, N-(1-phenethylpiperidin-4-yl)-N-phenylpropionamide citrate, was obtained from Sigma-Aldrich (St. Louis, MO, USA). Fentanyl-related substances: (3) beta-methylfentanyl; N-phenyl-N-(1-(2-phenylpropyl)piperidin-4-yl) propionamide hydrochloride, (4) para-methylfentanyl; N-(1-phenethylpiperidin-4-yl)-N-(p-tolyl)propionamide hydrochloride, (5) para-methoxyfentanyl; N-(4-methoxyphenyl)-N-(1-phenethylpiperidin-4-yl) propionamide hydrochloride, (6) fentanyl carbamate; ethyl (1-phenethylpiperidin-4-yl)(phenyl)carbamate, (7) 3-furanylfentanyl; N-(1-phenethylpiperidin-4-yl)-N-phenylfuran-3-carboxamide hydrochloride, (8) phenylfentanyl; N-(1-phenethylpiperidin-4-yl)-N-phenylbenzamide hydrochloride, and (9) beta′-phenylfentanyl; N-(1-phenethylpiperidin-4-yl)-N,3-diphenylpropanamide were synthesized by the Cayman Chemical Company (Ann Arbor, MI, USA) and provided by the United States Drug Enforcement Administration. (10) Naltrexone, (4R,4aS,7aR,12bS)-3-(cyclopropylmethyl)-4a,9-dihydroxy-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one hydrochloride, was obtained from Sigma-Aldrich (St. Louis, MO, USA). Drugs were obtained as dry powders and either dissolved in sterile saline (Fisher Scientific, Hampton, NH, USA) or suspended in 0.5% methylcellulose (Sigma-Aldrich, St. Louis, MO, USA) in deionized water and were administered subcutaneously (SC) in a volume equivalent to 10 ml/kg body weight. Chemical structures for drugs 1–9 are shown in Fig. 1.
Fig. 1.
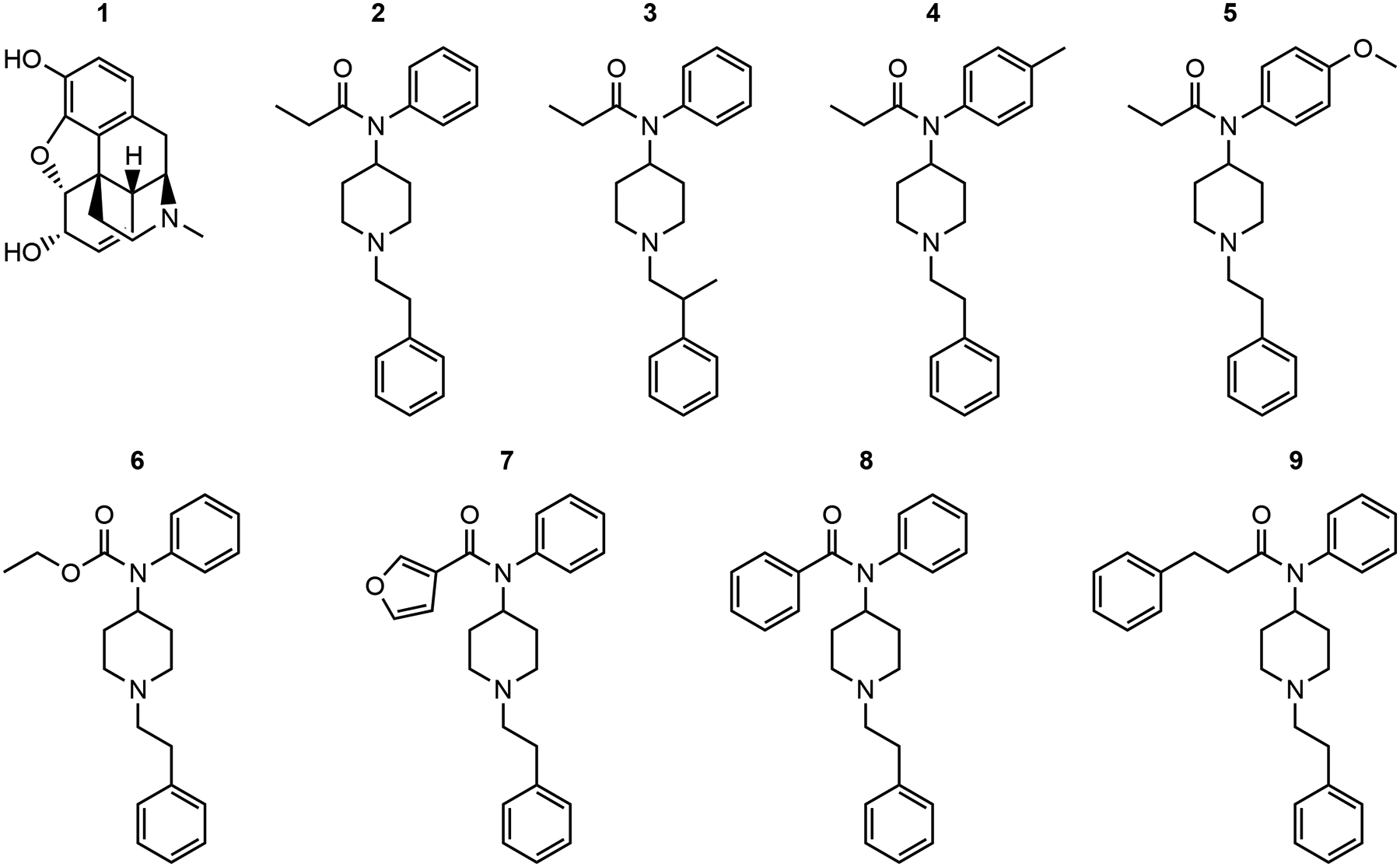
Chemical structures of (1) morphine, (2) fentanyl, (3) beta-methylfentanyl, (4) para-methylfentanyl, (5) para-methoxyfentanyl, (6) fentanyl carbamate, (7) 3-furanylfentanyl, (8) phenylfentanyl, and (9) beta′-phenylfentanyl.
2.2. Subjects
Adult male Swiss Webster mice (N = 512; Crl:CFW(SW), Charles River Laboratories, Raleigh, NC, USA) weighing ~25–50 g at the time of testing were housed four subjects per cage in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities. Subjects had ad libitum access to food (Teklad 7012 Rodent Diet; Envigo, Madison, WI, USA) and tap water. The vivarium was maintained at 22 °C ± 2 °C and 45%–50% humidity, with lights set to a 12-h light/dark cycle (lights on at 0600) and testing occurred during the light phase. Subjects were typically tested on weekdays between the hours of 1000 and 1600. Subjects were acclimated to the vivarium for at least one week before commencing experiments and were drug-naive before testing. All procedures were carried out in accordance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011). The experimental protocol was approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
2.3. Measurement of locomotion
Locomotor activity tests were conducted in eight commercially obtained, automated activity monitoring devices each enclosed in sound-and light-attenuating chambers that recorded movement via computer-controlled circuitry (AccuScan Instruments, Columbus, OH, USA). The interior of each device was divided into separate 20 × 20 × 30 cm fields permitting the independent and simultaneous measurement of two subjects. Sixteen photobeam sensors per axis were spaced 2.5 cm apart along the walls of the chamber and detected movement. A fan mounted in each test chamber provided ventilation and masking noise. On a test day, subjects were transported to the laboratory where they acclimated for ~30 min. Subjects were injected (SC) with either vehicle or drug and immediately placed in the test chambers where their movement was recorded for 120 min. Doses of fentanyl-related substances were selected to include a sufficient number of doses such that: (1) maximum mean effects between at least two doses significantly differed from one another (as defined by non-overlapping SEMs); and (2) at least at one time interval, the effects of at least one dose was significantly different (p ≤ 0.05) from vehicle; or (3) until 100 mg/kg, SC was tested. Based on these criteria, fentanyl-related substances were tested at doses of 0.1, 1, and 10 (and 100 if necessary) mg/kg, SC. Fentanyl (0.1, 1, and 10 mg/kg, SC) and morphine (1, 10, 100, and 180 mg/kg, SC) were tested as comparators (Janssen, 1975). The total distance traveled (cm) within each 10-min bin during the experimental session was recorded for each mouse. All locomotor activity tests used a between-subjects, acute dose design.
2.4. Measurement of nociception
A commercial water bath (Model # JBN5 US; Grant Instruments Ltd., Cambridge, UK) was used to assess nociception. At least 24 h before testing, subjects were handled and habituated to a restraint tube crafted from a cotton-lined surgical drape during two 5-min habituation periods. Subjects were placed in the restraint tube and tested for tail-withdrawal latencies under ambient temperature conditions to ensure they did not withdraw their tail in response to non-noxious stimuli, reducing false positive risk. The distal 3 cm of the tail was immersed into a bath containing 21 ± 0.5 °C water for three trials separated by a 2-min inter-trial interval. Subjects that withdrew their tail before 10 s had elapsed in more than one trial were not tested further. The tail-withdrawal latencies were measured with a digital stopwatch (Model # 14-649-7; Fisher Scientific, Pittsburgh, PA, USA).
Subjects that met inclusion criteria for testing (i.e., not excluded for failing tests under ambient temperature conditions) were evaluated using a cumulative dose procedure. Subjects were first tested for their baseline tail-withdrawal latencies using 50 °C water, followed immediately by injections (SC) of the drug’s vehicle and saline (naltrexone’s vehicle). After the designated pretreatment interval (10 min for fentanyl and all fentanyl-related substances; 20 min for morphine due to pharmacokinetic differences), the tail-withdrawal latency was re-determined for each subject and followed immediately by an injection of the lowest dose of drug (Varshneya et al., 2019). After the pretreatment interval had elapsed, subjects were tested again for tail-withdrawal latencies and injected with the next dose of drug. The process of assessing tail-withdrawal latencies and administering the next cumulative dose proceeded until the highest cumulative dose was tested. For example, acute doses tested were 0.01, 0.09, and 0.9 mg/kg, SC fentanyl resulting in cumulative doses of 0.01, 0.1, and 1.0 mg/kg, SC fentanyl (Fig. 2). After the highest cumulative dose was administered, time course testing commenced with tail-withdrawal latencies assessed at 10-min intervals for 60 min and then at 30-min intervals up to 120 min. A 10-s cutoff time was imposed across all assessments to minimize potential tissue damage. All warm-water tail-withdrawal tests used a within-subjects, cumulative dose design.
Fig. 2.
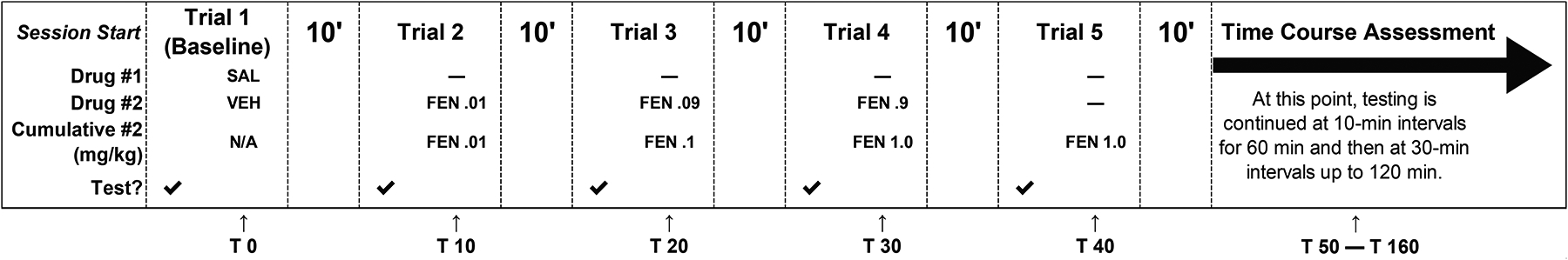
Schematic of the cumulative dose procedure for antinociceptive assessments of fentanyl-related substances in warm-water tail-withdrawal tests (FEN; fentanyl used as example). SAL and VEH refer to pretreatment of saline (i.e., naltrexone’s vehicle; control for antagonism experiments) and fentanyl’s vehicle (saline), respectively. Acute doses tested were 0.01, 0.09, and 0.9 mg/kg, SC fentanyl resulting in cumulative doses of 0.01, 0.1, and 1.0 mg/kg, SC fentanyl. Time course testing began 10 min after behavioral measurement of the highest cumulative dose. Time course assessments were performed at 10-min intervals for 60 min and then at 30-min intervals up to 120 min. “T X” indicates time in minutes following an injection of saline.
In separate experiments, drug-naive subjects were tested to evaluate naltrexone’s ability to attenuate the antinociceptive effects of each drug. Naltrexone (1 mg/kg, SC) was co-administered with the respective agonist vehicle, then testing proceeded as previously described except that higher agonist doses (not exceeding 100 mg/kg, SC) were also tested for their ability to surmount antagonist effects, and time course evaluations were not conducted. For example, acute doses tested were 0.01, 0.09, 0.9, 2.2, and 6.8 mg/kg, SC fentanyl resulting in cumulative doses of 0.01, 0.1, 1.0, 3.2, and 10 mg/kg, SC fentanyl (Fig. 3).
Fig. 3.
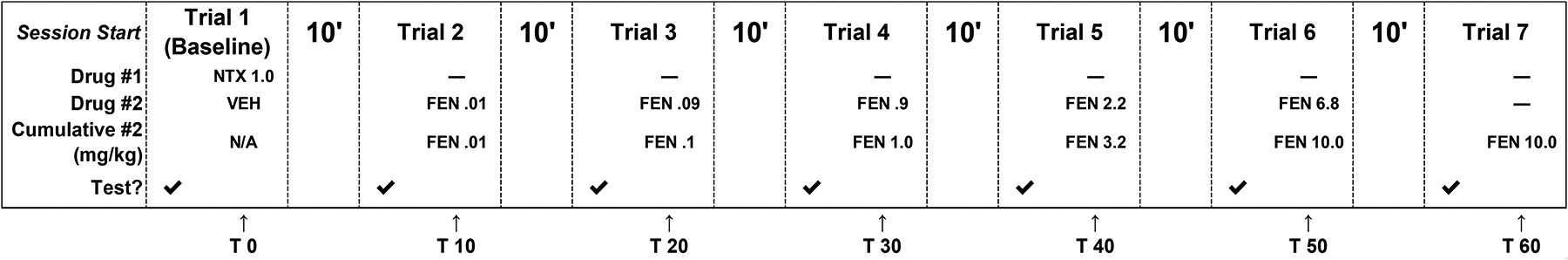
Schematic of the cumulative dose procedure for antinociceptive assessments of fentanyl-related substances following a 1 mg/kg, SC naltrexone (NTX) pretreatment (FEN; fentanyl used as example) in warm-water tail-withdrawal tests. Acute doses tested were 0.01, 0.09, 0.9, 2.2, and 6.8 mg/kg, SC fentanyl resulting in cumulative doses of 0.01, 0.1, 1.0, 3.2, and 10 mg/kg, SC fentanyl. “T X” indicates time in minutes following an injection of naltrexone.
2.5. Data analysis
In locomotor activity tests, distance traveled (cm) was the primary dependent variable and is shown as mean values (± SEM) for groups of subjects at each drug dose. Statistical significance was assessed by appropriate one-way or two-way analyses of variance (ANOVA). Fisher’s LSD post-hoc analyses were used for all pairwise comparisons. Dose-effect curves for locomotion were further analyzed to determine the estimated dose required for eliciting 100 m of travel using nonlinear regression ([Agonist] vs. response – Variable slope, Y = Bottom + (XĤillslope) * (Top-Bottom) / (XĤillSlope + EC50HillSlope)) with the exception of fentanyl and para-methoxyfentanyl that generated an inverted U-shaped curve relating distance traveled to dose and were modeled with a lognormal distribution (Y = (A / X) * exp(−0.5 * (ln(X / GeoMean) / ln(GeoSD))^2) where parameters ‘A’ and ‘GeoMean’ were constrained to values > 0, and parameter ‘GeoSD’ was constrained to values > 1). Para-methylfentanyl, phenylfentanyl, and beta′-phenylfentanyl did not produce marked effects on locomotion and therefore were not appropriate for modeling.
In warm-water tail-withdrawal tests, tail-withdrawal latencies were transformed into the percentage of maximum possible effect (%MPE) [100 × (post-treatment latency baseline latency) / (maximum possible latency – baseline latency)] where the maximum possible latency was 10 s. %MPEs are shown as mean values (± SEM) for groups of subjects at each drug dose. Statistical significance for dose-effect analyses was assessed by one-way analysis of variance (ANOVA). Fisher’s LSD post-hoc tests were used for all pairwise comparisons. Dose-effect curves for antinociception were further analyzed using nonlinear regression (Compounds 1–6: [Agonist] vs. normalized response — Variable slope, Y = 100 * (XĤillSlope)/(EC50ĤillSlope + (XĤill-Slope)); Compounds 7–9:[Agonist] vs. normalized response, Y = 100 * X / (EC50 + X)) from which ED50 values (interpolated 50% effective dose) with 95% asymmetrical (likelihood) confidence interval (95% CI) values were calculated for each agonist in the presence or absence of a naltrexone pretreatment. Time course effects for antinociception were analyzed using nonlinear regression (Compounds 1–5, 7: [Inhibitor] vs. normalized response — Variable slope, Y = 100 / (1 + (IC50 / X)ĤillSlope); Compound 6: [Inhibitor] vs. response — Variable slope, Y = Bottom + (Top Bottom) / (1 + (IC50 / X)ĤillSlope); Compounds 8 and 9: [Inhibitor] vs. response — Variable slope, Y = Bottom + (Top – Bottom) / (1 + (IC50 / X)ĤillSlope), where parameter ‘Bottom’ was constrained to values greater than zero). Constraints were applied to all models such that EC50 and IC50 values were presumed to be greater than zero. For each drug, the lowest dose that produced complete (operationally defined as ≥80% MPE) or maximum antinociception in saline-pretreated subjects was compared to the %MPE value obtained with the identical agonist dose in subjects pretreated with 1 mg/kg, SC naltrexone using unpaired t-tests. Comparisons were considered statistically significant if p ≤ 0.05. Analyses were performed with software (GraphPad Prism 9.1.1 (225) for Microsoft Windows 10 ×64; GraphPad Software, San Diego, CA, USA).
3. Results
3.1. Results from locomotor activity tests
Fig. 4 (filled circles, right ordinates) shows distance traveled (cm) as a function of dose for each drug in locomotor activity tests. Morphine [100, 180 mg/kg; F(4, 59) = 17.06, p < 0.0001], fentanyl [1, 10 mg/kg; F(3, 60) = 28.76, p < 0.0001], beta-methylfentanyl [10 mg/kg; F(3, 28) = 10.47, p < 0.0001], para-methoxyfentanyl [10 mg/kg; F(4, 34) = 6.986, p = 0.0003], fentanyl carbamate [100 mg/kg; F(4, 43) = 6.264, p = 0.0005], and 3-furanylfentanyl [10 mg/kg; F(3, 28) = 24.58, p < 0.0001] produced significant increases (p ≤ 0.05) in distance traveled relative to vehicle at least at one dose tested. Generally, as dose increased, distance traveled increased, except for both fentanyl and para-methoxyfentanyl, for which an inverted U-shaped dose-response function related distance traveled to increasing dose. Unlike the other drugs, para-methylfentanyl [F(4, 43) = 0.7226, p = 0.5813], beta′-phenylfentanyl [F(4, 43) = 1.412, p = 0.2461], and phenylfentanyl did not significantly increase locomotor activity; phenylfentanyl [100 mg/kg; F(4, 43) = 4.136, p = 0.0064] produced a significant decrease in locomotion relative to vehicle.
Fig. 4.
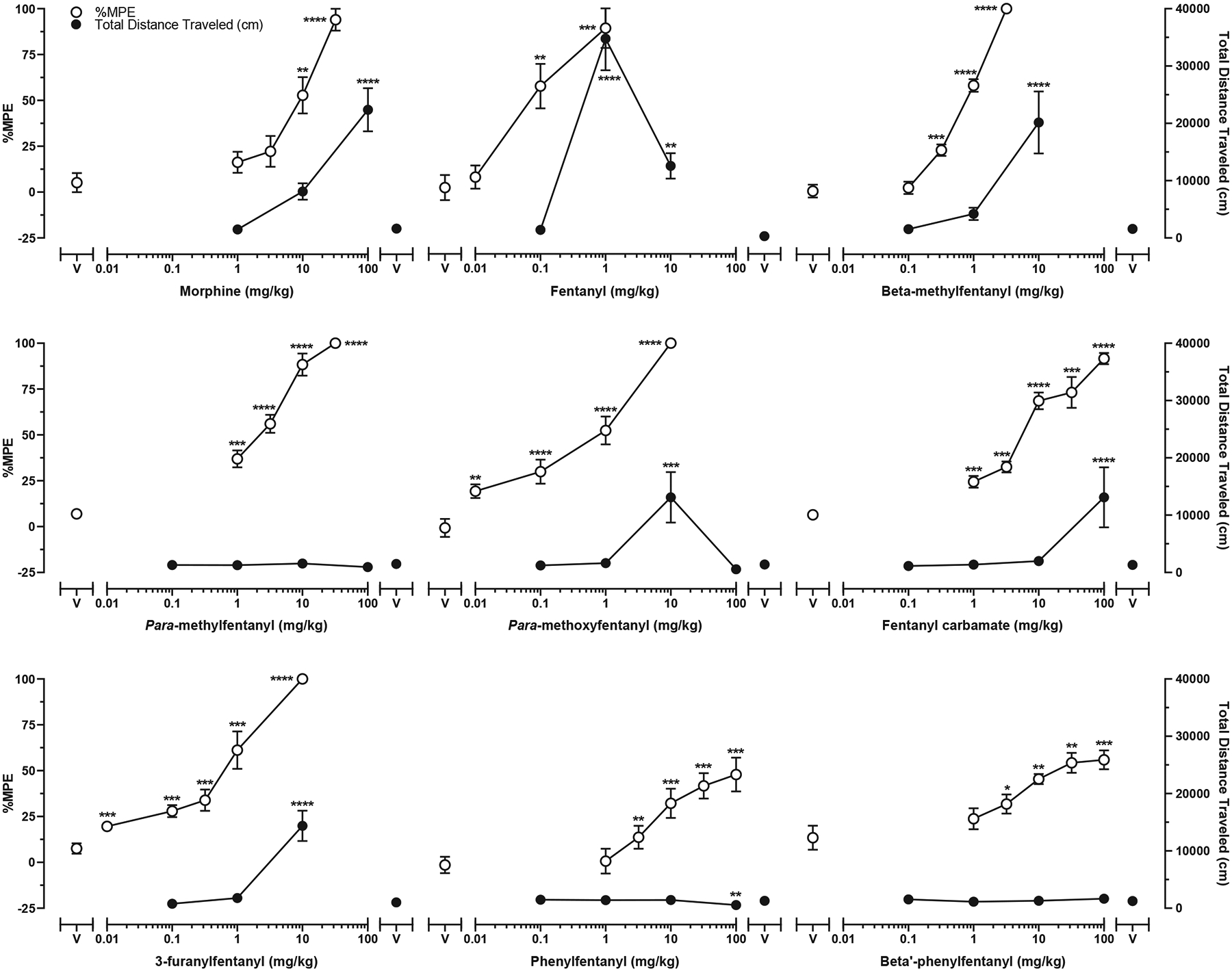
Cumulative dose effects in warm-water tail-withdrawal tests (open circles, left ordinate) and acute dose effects in locomotor activity tests (filled symbols, right ordinate). Symbols above “V” represent data from saline + vehicle conditions in warm-water tail-withdrawal tests and data from vehicle conditions in locomotor activity tests. Symbols representing data from warm-water tail-withdrawal tests are the mean (± SEM) %MPE for n = 8 mice per dose. Symbols representing data from locomotor activity tests are the mean (± SEM) distance traveled (cm) for n = 8–16 mice per dose. Significant differences between drug and vehicle are indicated by asterisks: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Table 1 shows the maximum total distance traveled, maximum total distance traveled as a percentage of fentanyl’s maximum effect, and estimated dose (mg/kg) required to elicit a level of effect equal to 100 m of travel. The highest level of effect was observed for subjects administered fentanyl (1 mg/kg, SC) which elicited 34,905 ± 5355 cm of travel in the 120-min test session. The rank order potency based upon the estimated dose required to induce 100 m of distanced traveled was: fentanyl > beta-methylfentanyl > para-methoxyfentanyl > 3-furanylfentanyl > morphine > fentanyl carbamate ⋙ para-methylfentanyl = phenylfentanyl = beta′-phenylfentanyl. Fig. 5 shows distance traveled as a function of dose and time for each drug.
Table 1.
Results from locomotor activity tests.
| # | Drug | Maximum effect (distance traveled in cm ± SEM) | Maximum effect as a % of fentanyl’s maximum effect | Estimated dose (mg/kg) required to elicit 100 m of travel |
|---|---|---|---|---|
| 1 | Morphine | 22,381 ± 3785 | 64.1 ± 10.8 | 15.3 |
| 2 | Fentanyl | 34,905 ± 5355 | 100 ± 15.3 | 0.23 |
| 3 | Beta-methylfentanyl | 20,165 ± 5413 | 57.8 ± 15.5 | 1.74 |
| 4 | Para-methylfentanyl | 1555 ± 632 | 4.46 ± 1.81 | N/A |
| 5 | Para-methoxyfentanyl | 13,109 ± 4412 | 37.6 ± 12.6 | 3.63 |
| 6 | Fentanyl carbamate | 13,107 ± 5250 | 37.6 ± 15.0 | 72.3 |
| 7 | 3-Furanylfentanyl | 14,375 ± 2638 | 41.2 ± 7.56 | 4.09 |
| 8 | Phenylfentanyl | 1498 ± 273 | 4.29 ± 0.78 | N/A |
| 9 | Beta′-phenylfentanyl | 1660 ± 158 | 4.76 ± 0.45 | N/A |
Efficacy estimates are expressed as maximum effect (total distance traveled in cm) and maximum effect as a % of fentanyl’s maximum effect.
Potency estimates are expressed as the dose (mg/kg) required to produce a level of effect equal to 100 m of travel.
The mean maximum effect (distance traveled in cm ± SEM) of all vehicle controls was 1399 ± 62.
Data are mean ± SEM for n = 8–16 mice per dose.
N/A: impossible to estimate based upon the slope of the dose-effect curve.
Fig. 5.
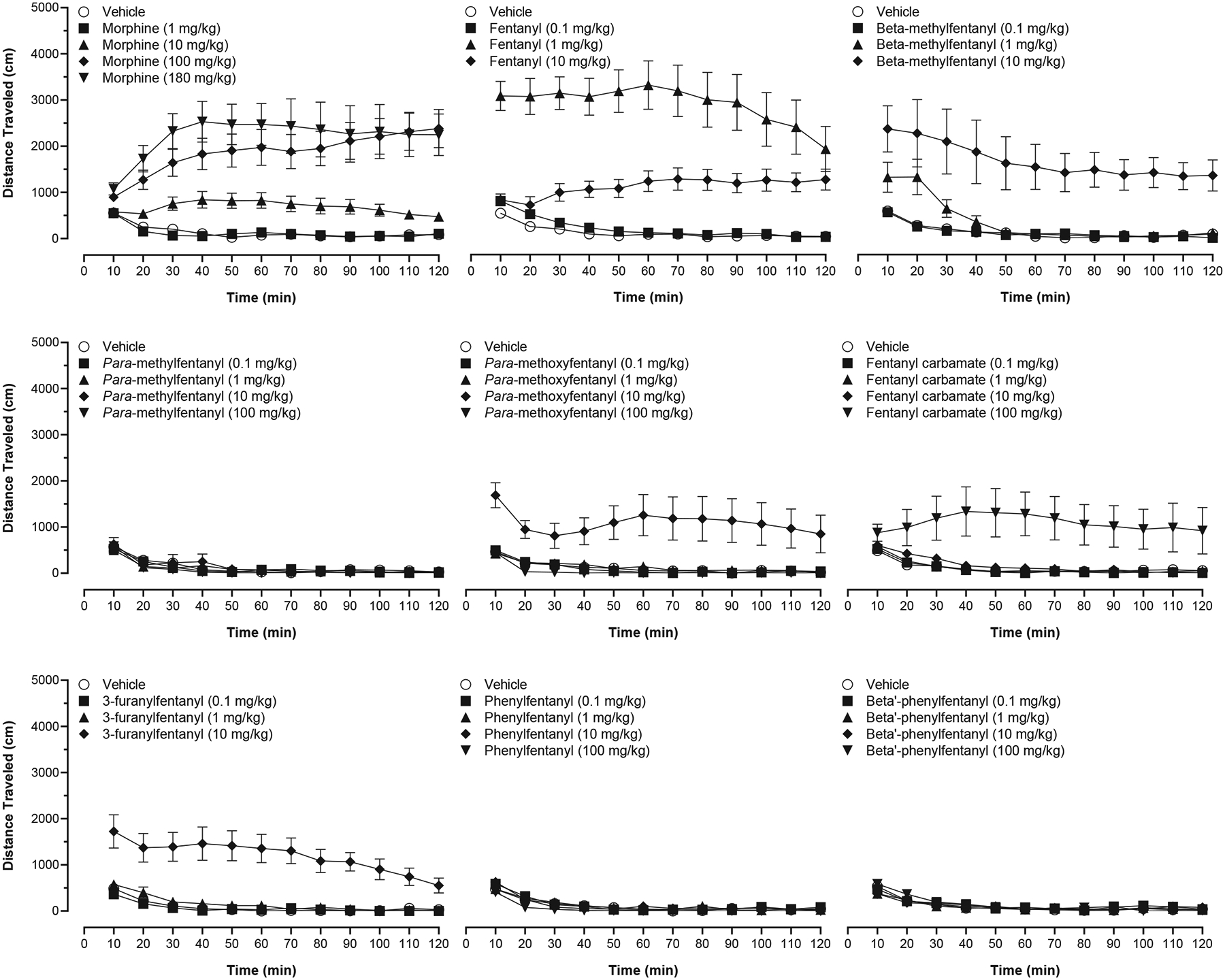
Time course effects in locomotor activity tests for (1) morphine, (2) fentanyl, (3) beta-methylfentanyl, (4) para-methylfentanyl, (5) para-methoxyfentanyl, (6) fentanyl carbamate, (7) 3-furanylfentanyl, (8) phenylfentanyl, and (9) beta′-phenylfentanyl. Symbols represent the mean (± SEM) distance traveled (cm) in 10-min bins for n = 8–16 mice per dose.
3.2. Results from warm-water tail-withdrawal tests
Fig. 4 (unfilled circles, left ordinate) shows %MPE as a function of dose for each drug in warm-water tail-withdrawal tests. As dose increased, %MPE increased for all drugs. Table 2 shows efficacy (%Emax values) and potency (ED50 values) estimates for antinociception in warm-water tail-withdrawal tests and potency ratios to both morphine and fentanyl for each drug. The ED50 values for antinociception showed a rank order of potency of fentanyl (ED50 = 0.08 mg/kg) > para-methoxyfentanyl (ED50 = 0.43 mg/kg) > 3-furanylfentanyl (ED50 = 0.51 mg/kg) > beta-methylfentanyl (ED50 = 0.74 mg/kg) > para-methylfentanyl (ED50 = 1.92 mg/kg) > fentanyl carbamate (ED50 = 5.59 mg/kg) > morphine (ED50 = 7.82 mg/kg) > beta′-phenylfentanyl (ED50 = 19.4 mg/kg) > phenylfentanyl (ED50 = 55.2 mg/kg).
Table 2.
Results from warm-water tail-withdrawal tests.
| # | Drug | %Emax ± SEM | Agonist ED50 mg/kg (95% CI) | Agonist + Antagonist ED50 mg/kg (95% CI) | Fold change (agonist + antagonist / agonist) | Potency ratio to morphine | Potency ratio to fentanyl |
|---|---|---|---|---|---|---|---|
| 1 | Morphine | 94.0 ± 5.98 | 7.82 (5.42–11.0) | 11.6 (5.98–23.8) | 1.48 | 1.00 | 0.01 |
| 2 | Fentanyl | 89.2 ± 10.8 | 0.08 (0.04–0.16) | 0.32 (0.15–0.62) | 3.95 | 97.7 | 1.00 |
| 3 | Beta-methylfentanyl | 100 | 0.74 (0.64)-0.85) | 14.2 (8.27–31.0) | 19.2 | 10.6 | 0.11 |
| 4 | Para-methylfentanyl | 100 | 1.92 (1.48–2.45) | 121 (62.7–345) | 63.1 | 4.07 | 0.04 |
| 5 | Para-methoxyfentanyl | 100 | 0.43 (0.23–0.77) | 9.62 (3.81–38.2) | 22.5 | 18.3 | 0.19 |
| 6 | Fentanyl carbamate | 91.6 ± 3.06 | 5.59 (4.11–7.54) | 12.4 (7.72–20.9) | 2.21 | 1.40 | 0.01 |
| 7 | 3-Furanylfentanyl | 100 | 0.51 (0.36–0.74) | 10.6 (5.29–21.0) | 20.6 | 15.3 | 0.16 |
| 8 | Phenylfentanyl | 48.0 ± 9.24 | 55.2 (33.5–93.0) | 289 (187–515) | 5.23 | 0.14 | 0.0015 |
| 9 | Beta′-phenylfentanyl | 55.9 ± 5.14 | 19.4 (11.0–34.4) | 407 (187–2294) | 21.0 | 0.40 | 0.0041 |
Efficacy estimates are expressed as %Emax (maximum %MPE).
Potency estimates are expressed as ED50 (mg/kg) values for drug alone, ED50 values for drug following a 1 mg/kg, SC naltrexone pretreatment, fold change between (agonist + antagonist)/(agonist) potency, drug potency ratio to morphine, and drug potency ratio to fentanyl.
Data are mean ± SEM or 95% confidence intervals for n = 8 mice per dose.
Fig. 6 shows %MPE for the highest cumulative dose of each drug as a function of time in warm-water tail-withdrawal tests. The time to peak effect for all drugs occurred within 10 min. Antinociceptive effects deteriorated most rapidly for phenylfentanyl (100 mg/kg), beta′-phenylfentanyl (100 mg/kg), and beta-methylfentanyl (3.2 mg/kg) in that order (i.e., effects for drugs listed first, dissipated first). Antinociceptive effects by 3-furanylfentanyl (10 mg/kg), para-methoxyfentanyl (10 mg/kg), para-methylfentanyl (32 mg/kg), morphine (32 mg/kg), fentanyl (1 mg/kg), and fentanyl carbamate (100 mg/kg) persisted longer than the other drugs and were still above ~50% levels at 120 min following drug administration. Both phenylfentanyl and beta′-phenylfentanyl were the least efficacious fentanyl-related substances in this series each with a MPE of about 50% at a dose of 100 mg/kg, SC.
Fig. 6.
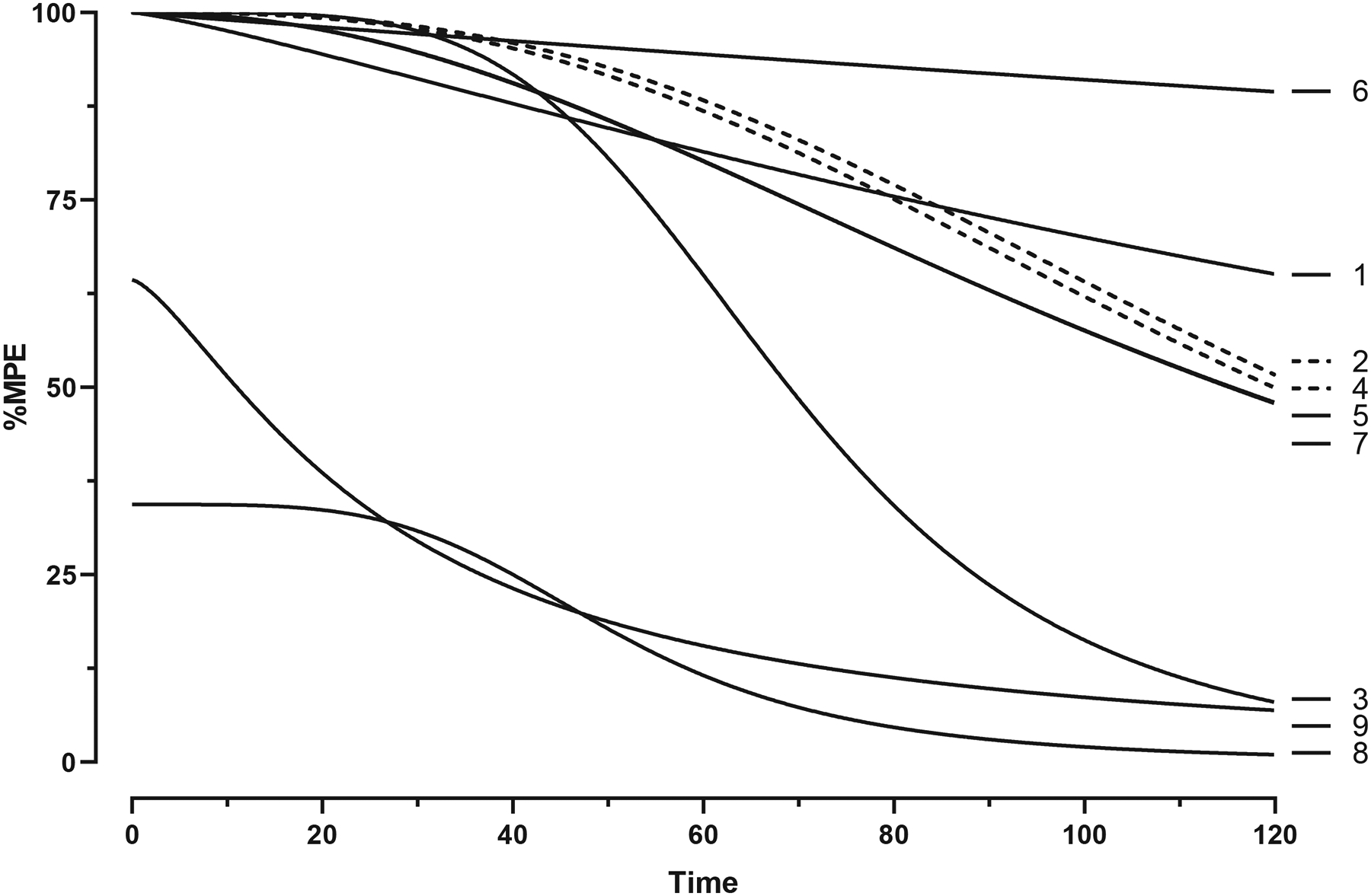
Time course effects of the highest cumulative dose of each drug in warm-water tail-withdrawal tests, i.e., (1) 32 mg/kg morphine, (2) 1 mg/kg fentanyl, (3) 3.2 mg/kg beta-methylfentanyl, (4) 32 mg/kg para-methylfentanyl, (5) 10 mg/kg para-methoxyfentanyl, (6) 100 mg/kg fentanyl carbamate, (7) 10 mg/kg 3-furanylfentanyl, (8) 100 mg/kg phenylfentanyl, and (9) 100 mg/kg beta′-phenylfentanyl. Data are represented as curves determined by nonlinear regression with n = 8 mice per dose. Data for drugs (5) and (7) were modeled by identical curves and therefore are represented by a single line.
The effects of a saline or 1 mg/kg, SC naltrexone pretreatment on antinociception produced by the highest cumulative dose of each drug are shown in Fig. 7 and Table 2. Naltrexone attenuated the antinociceptive effects of each drug, but relative levels of antagonism varied across drugs. Naltrexone increased antinociceptive ED50 values several fold in decreasing magnitudes of para-methylfentanyl (63.1×) > para-methoxyfentanyl (22.5×) > beta′-phenylfentanyl (21.0×) > 3-furanylfentanyl (20.6×) > beta-methylfentanyl (19.2×) > phenylfentanyl (5.23×) > fentanyl (3.95×) > fentanyl carbamate (2.21×) > morphine (1.48×).
Fig. 7.
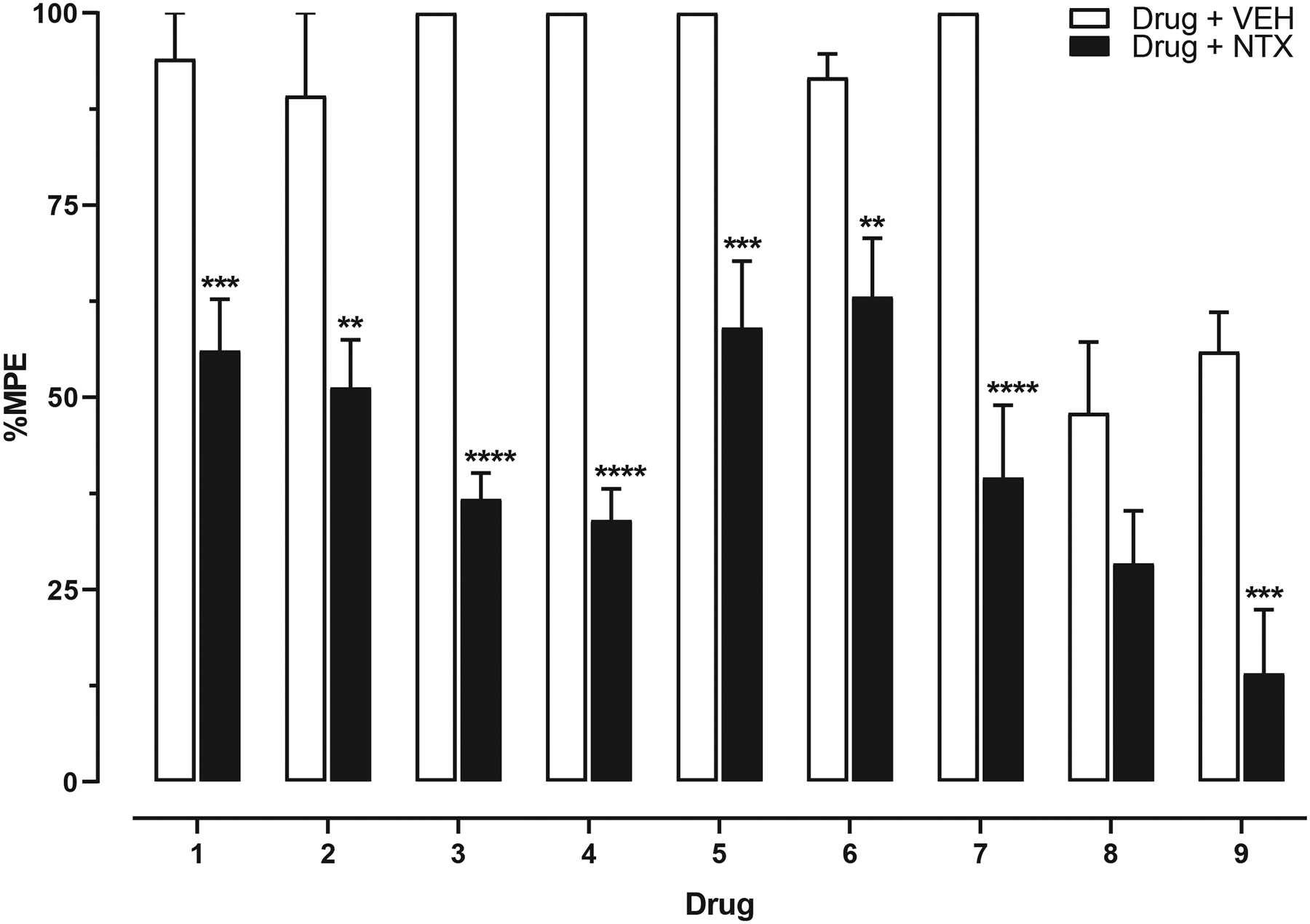
Effects of a saline or 1 mg/kg, SC naltrexone pretreatment on antinociception produced by the highest cumulative dose of each drug in warm-water tail-withdrawal tests, i.e., (1) 32 mg/kg morphine, (2) 1 mg/kg fentanyl, (3) 3.2 mg/kg beta-methylfentanyl, (4) 32 mg/kg para-methylfentanyl, (5) 10 mg/kg para-methoxyfentanyl, (6) 100 mg/kg fentanyl carbamate, (7) 10 mg/kg 3-furanylfentanyl, (8) 100 mg/kg phenylfentanyl, and (9) 100 mg/kg beta′-phenylfentanyl. Bars represent the mean (± SEM) %MPE of n = 8 mice per dose. Significant differences between drug + saline (SAL; open bars) and drug + naltrexone (NTX; filled bars) are indicated by asterisks: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
4. Discussion
This study adds to our knowledge of fentanyl-related substances and represents the first reported assessments of 3-furanylfentanyl and phenylfentanyl on nociception and locomotion in mice. There are only limited existing data for the remaining drugs beta-methylfentanyl, para-methylfentanyl, para-methoxyfentanyl, fentanyl carbamate, and beta′-phenylfentanyl. In locomotor activity tests, all drugs, except for para-methylfentanyl, phenylfentanyl, and beta′-phenylfentanyl, elicited significant dose-dependent hyperlocomotion in mice. In warm-water tail-withdrawal tests, all drugs produced significant dose-dependent antinociception with the apparent potency to do so of fentanyl > para-methoxyfentanyl > 3-furanylfentanyl > beta-methylfentanyl > para-methylfentanyl > fentanyl carbamate > morphine > beta′-phenylfentanyl > phenylfentanyl. To further explore the role of the MOR in mediating the in vivo effects of these fentanyl-related substances, we measured the antinociceptive effects of these drugs in warm-water tail-withdrawal tests with a naltrexone pretreatment. These tests demonstrated a significant attenuation of antinociception indicating that their effects are at least in part MOR-mediated. Overall, this research demonstrates that fentanyl-related substances, like morphine and fentanyl, can elicit antinociception and hyperlocomotion in mice, and supports the need for additional testing of other structurally-related drugs to elucidate their effects and mechanisms of action.
Several groups have reported the effects of a subset of the fentanyl-related substances tested in this study. Beta-methylfentanyl (R4471) was first reported to have an ED50 of 0.5 mg/kg, SC in mouse hot plate tests (22× more potent than morphine) and a LD50 of 120 mg/kg, SC (TI = 240) in mice (Janssen, 1964, 1975). We reported that beta-methylfentanyl has an ED50 of 0.74 mg/kg, SC (95% CI: 0.64–0.85) in mouse warm-water tail-withdrawal tests (10× more potent than morphine) and that a 1 mg/kg, SC naltrexone pretreatment antagonized the antinociceptive effects of beta-methylfentanyl, increasing the ED50 to 14.2 mg/kg, SC (95% CI: 8.27–31.0). In locomotor activity tests, beta-methylfentanyl (10 mg/kg, SC) elicited a maximum effect equal to 57.8% of fentanyl’s maximum. Consistent with these findings, beta-methylfentanyl exhibited a Ki of 14 ± 1 nM and an Emax of 86 ± 3% in CHO cells expressing the human MOR, indicating that beta-methylfentanyl is a full agonist although with a lower affinity for the MOR than fentanyl (Ki = 1.6 ± 0.4 nM) (Hassanien et al., 2020). Collectively, these results suggest that beta-methylfentanyl elicits antinociception and hyperlocomotion in mice through a MOR mechanism.
Para-methylfentanyl (R4408) was first reported to have an ED50 of 1.3 mg/kg, SC in mouse hot plate tests (8.5× more potent than morphine) and a LD50 of 95 mg/kg, SC (TI = 73) in mice (Janssen, 1964, 1975). Casy and Huckstep (1988) reported an ED50 of 0.31 mg/kg, IV for para-methylfentanyl in rat tail-withdrawal tests while Higashikawa and Suzuki (2008) found that para-methylfentanyl had an ED50 of 0.220 mg/kg, PO and elsewhere in the same report an ED50 of 0.261 mg/kg, PO. Woods et al. (1987) observed that para-methylfentanyl (NIH 10489) was similar to morphine’s potency in the displacement of etorphine binding, and in the mouse vas deferens preparation para-methylfentanyl appeared to be a MOR agonist. However, beta-funaltrexamine, a selective, irreversible MOR antagonist did not diminish the maximum response by para-methylfentanyl suggesting that its effects may be partially mediated by a non-opioid receptor mechanism (Woods et al., 1987). We reported that para-methylfentanyl has an ED50 of 1.92 mg/kg, SC (95% CI: 1.48–2.45) in mouse warm-water tail-withdrawal tests (4× more potent than morphine) and that a 1 mg/kg, SC naltrexone pretreatment antagonized the antinociceptive effects of para-methylfentanyl, increasing the ED50 to 121 mg/kg, SC (95% CI: 62.7–345). In locomotor activity tests, para-methylfentanyl did not elicit increases in locomotion even up to 100 mg/kg, SC. Interestingly, however, para-methylfentanyl substituted for morphine in rat drug discrimination tests with an ED50 of 0.27 mg/kg, SC (Gatch, 2020). Moreover, para-methylfentanyl exhibited a Ki of 4.2 ± 0.7 nM and an Emax of 31 ± 3% in CHO cells expressing the human MOR (Hassanien et al., 2020) as well as a Ki of 0.205 ± 0.031 nM and an Emax of 65.8 ± 1.8% in CHO cells expressing the rat MOR, indicating that para-methylfentanyl is a high affinity, moderate efficacy MOR partial agonist (Drug Enforcement Administration–Veterans Affairs (DEA-VA) Interagency Agreement, 2019). The doses required for eliciting 100 m of distance traveled in locomotor activity tests for para-methylfentanyl, and other fentanyl-related substances tested, were typically 10- to 30-fold greater than the ED50 values for producing antinociception in warm-water tail-withdrawal tests. This could be explained by differences in efficacy threshold requirements for each test, locomotor activity tests having a higher efficacy threshold than warm-water tail-withdrawal tests. Taken together, these results suggest that para-methylfentanyl elicits antinociception at least in part through a MOR mechanism, but may lack the necessary efficacy for eliciting hyperlocomotion in mice.
Para-methoxyfentanyl (R4480) was first reported to have an ED50 of 2.0 mg/kg, SC in mouse hot plate tests (5.5× more potent than morphine) and a LD50 of 140 mg/kg, SC (TI = 70) in mice (Janssen, 1964, 1975). Woods et al. (1987) reported that para-methoxyfentanyl (NIH 10490) was similar to morphine in potency for displacing etorphine binding. In the mouse vas deferens preparation, para-methoxyfentanyl was reported to be a relatively selective MOR agonist with a potency less than that of morphine (Woods et al., 1987). Aceto et al. (1987) reported an ED50 of 0.5 mg/kg, SC (95% CI: 0.2–1.5) in mouse tail-flick tests and an ED50 of 0.1 mg/kg, SC (95% CI: 0.05–0.17) in mouse phenylquinone tests. In single dose-suppression tests in morphine dependent monkeys deprived of morphine for 15 h, a dose of 0.5 mg/kg, IM para-methoxyfentanyl completely substituted for morphine by suppressing withdrawal signs for ~150 min following its administration (Aceto et al., 1987). Consistent with these findings, para-methoxyfentanyl substituted for morphine in rat drug discrimination tests with an ED50 of 0.15 mg/kg, SC (Gatch, 2020). Moreover, para-methoxyfentanyl exhibited a Ki of 0.79 ± 0.25 nM and an Emax of 79.6 ± 6.0% indicating in CHO cells expressing the MOR, indicating that para-methoxyfentanyl is a full MOR agonist (Drug Enforcement Administration–Veterans Affairs (DEA-VA) Interagency Agreement, 2019). We reported that para-methoxyfentanyl has an ED50 of 0.43 mg/kg, SC (95% CI: 0.23–0.77) in mouse warm-water tail-withdrawal tests (18× more potent than morphine) and that a 1 mg/kg, SC naltrexone pretreatment antagonized the antinociceptive effects of para-methoxyfentanyl, increasing the ED50 to 9.62 mg/kg, SC (95% CI: 3.81–38.2). In locomotor activity tests, para-methoxyfentanyl (10 mg/kg, SC) elicited a maximum effect equal to 37.6% of fentanyl’s maximum. Collectively, these results indicate that para-methoxyfentanyl elicits antinociception and hyperlocomotion in mice through a MOR mechanism.
Fentanyl carbamate (R4416) was first reported to have an ED50 of 1.0 mg/kg, SC in mouse hot plate tests (11× more potent than morphine) and a LD50 of >80 mg/kg, SC (TI > 80) in mice (Janssen, 1964, 1975). We reported that fentanyl carbamate has an ED50 of 5.59 mg/kg, SC (95% CI: 4.11–7.54) in mouse warm-water tail-withdrawal tests (1.4× more potent than morphine), and that a 1 mg/kg, SC naltrexone pretreatment antagonized the antinociceptive effects of fentanyl carbamate, increasing the ED50 to 12.4 mg/kg, SC (95% CI: 7.72–20.9). In locomotor activity tests, fentanyl carbamate (100 mg/kg, SC) elicited a maximum effect equal to 37.6% of fentanyl’s maximum. Taken together, these results suggest that fentanyl carbamate elicits antinociception and hyperlocomotion in mice through a MOR mechanism.
Our findings represent the first assessments of 3-furanylfentanyl on nociception and locomotion in mice. We reported that 3-furanylfentanyl has an ED50 of 0.51 mg/kg, SC (95% CI: 0.36–0.74) in mouse warm-water tail-withdrawal tests (15.3× more potent than morphine) and that a 1 mg/kg, SC naltrexone pretreatment antagonized the antinociceptive effects of 3-furanylfentanyl, increasing the ED50 to 10.6 mg/kg, SC (95% CI: 5.29–21.0). In locomotor activity tests, 3-furanylfentanyl (10 mg/kg, SC) elicited a maximum effect equal to 41.2% of fentanyl’s maximum. Consistent with these findings, 3-furanylfentanyl substituted for oxycodone in mouse drug discrimination tests with an ED50 of 0.093 mg/kg, SC (95% CI: 0.047–0.138) (Walentiny et al., 2019). Further, 3-furanylfentanyl substituted for morphine in rat drug discrimination tests with an ED50 of 0.042 mg/kg, SC (95% CI: 0.017–0.063) (Walker, 2019). Moreover, 3-furanylfentanyl exhibited a Ki of 0.442 ± 0.096 nM and an Emax of 58.5 ± 9.3% in CHO cells expressing the rat MOR, indicating that 3-furanylfentanyl is a high affinity MOR agonist with moderate efficacy (Eshleman et al., 2020). Collectively, these results suggest that 3-furanylfentanyl elicits antinociception and hyperlocomotion in mice via a MOR mechanism.
Our findings also represent the first assessments phenylfentanyl on nociception and locomotion in mice. We reported that phenylfentanyl has an ED50 of 55.2 mg/kg, SC (95% CI: 33.5–93.0) in mouse warm-water tail-withdrawal tests, however, phenylfentanyl’s maximum effect was only 48% MPE at a dose of 100 mg/kg, SC. A naltrexone pretreatment antagonized the antinociceptive effects of phenylfentanyl, increasing the ED50 to 289 mg/kg, SC (95% CI: 187–515). In locomotor activity tests, phenylfentanyl did not elicit increases in locomotion even up to 100 mg/kg, SC. Beta′-phenylfentanyl produced a similar pattern of effects to those of phenylfentanyl in assessments of nociception and locomotion, which is not surprising given their structural similarities. We reported that beta′-phenylfentanyl has an ED50 of 19.4 mg/kg, SC (95% CI: 11.0–34.4) in mouse warm-water tail-withdrawal tests, however, beta′-phenylfentanyl’s maximum effect was only 49.1% MPE at a dose of 100 mg/kg, SC. A naltrexone pretreatment antagonized the antinociceptive effects of beta′-phenylfentanyl, increasing the ED50 to 407 mg/kg, SC (95% CI: 187–2294). Beta′-phenylfentanyl exhibited only transient effects in warm-water tail-withdrawal tests and therefore its results obtained using the cumulative dose regimen should be interpreted with caution. However, our findings that beta′-phenylfentanyl exhibits relatively weak effects in warm-water tail-withdrawal tests are generally consistent with those of Zhu et al. (1981) who reported that beta′-phenylfentanyl produced no analgesic effects up to 10 mg/kg, IP in mouse hot plate tests (Zhu et al., 1981). Similar to phenylfentanyl, beta′-phenylfentanyl did not elicit increases in locomotion even up to 100 mg/kg, SC, but did substitute for morphine in rat drug discrimination tests with an ED50 of 0.46 mg/kg, SC suggesting that it has sufficient efficacy to elicit subjective-like effects, but not increases in locomotor activity (Gatch, 2020). Phenylfentanyl exhibited a Ki of 3.55 ± 0.99 nM and an Emax of 8.8 ± 2.2% in CHO cells expressing the rat MOR, indicating that phenylfentanyl, as a very low efficacy partial agonist, could antagonize the effects of a full agonist (Eshleman et al., 2020). Taken together, these results suggest that both beta′-phenylfentanyl and phenylfentanyl are low efficacy MOR agonists that have sufficient efficacy to elicit weak effects in drug discrimination and warm-water tail-withdrawal tests, but do not meet the efficacy threshold requirements for eliciting hyperlocomotion in locomotor activity tests.
In summary, fentanyl, morphine, and most of the fentanyl-related substances tested were efficacious MOR agonists as determined by their effects on nociception and locomotion in mice. Phenylfentanyl and beta′-phenylfentanyl were lowest efficacy agonists tested, producing little to no effects relative to the other fentanyl-related substances. To our knowledge, the effects of 3-furanylfentanyl and phenylfentanyl on nociception and locomotion had never before been published. Consistent with findings by others, the results of this study indicate that these fentanyl-related substances elicit their effects via the MOR, however, differences in the ability of naltrexone to reverse the effects of specific drugs suggests that receptors other than MOR may contribute, although additional experiments are needed to test this hypothesis. This study adds to the existing preclinical literature describing the pharmacology of these drugs and prompts new questions about their other effects. Overall, these findings confirm the role of the MOR in mediating the effects of these drugs and support the need for future tests concerning their respiratory depressant effects.
Acknowledgements
Research reported in this publication was supported by the National Institute on Drug Abuse of the National Institutes of Health (T32DA007027) and by the Drug Enforcement Administration of the United States Department of Justice (DJD-17-HQ-P-0641). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, United States Department of Justice, Johns Hopkins University, or Virginia Commonwealth University.
Footnotes
Declaration of competing interest
TDW and LRA were employees of the Drug Enforcement Administration at the time of writing. The remaining authors have no relevant conflicts of interest to declare.
References
- Aceto M, Bowman E, Harris L, May E, 1987. Dependence studies of new compounds in the rhesus monkey, rat and mouse. In: National Institute on Drug Abuse Research Monograph Series Problems of Drug Dependence 1987 (Proceedings of the 49th Annual Scientific Meeting. The Committee on Problems of Drug Dependence, Inc.). [Google Scholar]
- Al-Hasani R, Bruchas MR, 2011. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115 (6), 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arfken CL, Suchanek J, Greenwald MK, 2017. Characterizing fentanyl use in methadone-maintained clients. J. Subst. Abus. Treat 75, 17–21. [DOI] [PubMed] [Google Scholar]
- Bailey PL, Wilbrink J, Zwanikken P, Pace NL, Stanley TH, 1985. Anesthetic induction with fentanyl. Anesth. Analg 64 (1), 48–53. [PubMed] [Google Scholar]
- Casy AF, Huckstep MR, 1988. Structure-activity studies of fentanyl. J. Pharm. Pharmacol 40 (9), 605–608. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, 2020. Multiple Cause of Death 1999–2019 on CDC WONDER Online Database. released in 2020. Data are from the Multiple Cause of Death Files, 1999–2019, as compiled from data provided by the 57 vital statistics jurisdictions through the Vital Statistics Cooperative Program. National Center for Health Statistics. [Google Scholar]
- Chhabra N, Rizvanolli L, Rasin A, Marsden G, Hinami K, Aks SE, 2021. A cross-sectional analysis of fentanyl analog exposures among living patients. Am. J. Drug Alcohol Abuse 1–6. [DOI] [PubMed] [Google Scholar]
- Comer SD, Cahill CM, 2019. Fentanyl: receptor pharmacology, abuse potential, and implications for treatment. Neurosci. Biobehav. Rev 106, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011. Guide for the Care and Use of Laboratory Animals. Institute for Laboratory Animal Research; Division on Earth and Life Studies; National Research Council of the National Academies. [Google Scholar]
- Drug Enforcement Administration, 2017a. NFLIS Brief: Fentanyl and Fentanyl-Related Substances Reported in NFLIS, 2015–2016. National Forensic Laboratory Information System. [Google Scholar]
- Drug Enforcement Administration, 2017b. NFLIS Brief: Fentanyl, 2001–2015. NFLIS Brief: Fentanyl, 2001–2015 (March 2017). [Google Scholar]
- Drug Enforcement Administration, 2020. NFLIS-DRUG 2019 Annual Report. United States Department of Justice, Springfield, Virginia, United States of America. [Google Scholar]
- Drug Enforcement Administration, 2021a. Schedules of Controlled Substances: Placement of 10 Specific Fentanyl-related Substances in Schedule I, Docket No. DEA-476
- Drug Enforcement Administration, 2021b. Schedules of Controlled Substances: Placement of Four Specific Fentanyl-related Substances in Schedule I, Docket No. DEA-806
- Drug Enforcement Administration–Veterans Affairs (DEA-VA) Interagency Agreement, 2019. Binding and Functional Activity at Delta, Kappa and Mu Opioid Receptors. In Vitro Receptor and Transporter Assays for Abuse Liability Testing for the DEA by the VA. [Google Scholar]
- Eshleman AJ, Nagarajan S, Wolfrum KM, Reed JF, Nilsen A, Torralva R, Janowsky A, 2020. Affinity, potency, efficacy, selectivity, and molecular modeling of substituted fentanyls at opioid receptors. Biochem. Pharmacol 182, 114293. [DOI] [PubMed] [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction, 2017. Furanylfentanyl.EMCDDA–Europol Joint Report on a new psychoactive substance: N-phenyl-N-[1-(2-phenylethyl) piperidin-4-yl]-furan-2-carboxamide (furanylfentanyl). EMCDDA–Europol Joint Reports. [Google Scholar]
- Gatch MB, 2020. Discriminative Stimulus Effects of Fentanyl Analogs, College on Problems of Drug Dependence. Virtual. [Google Scholar]
- Hassanien SH, Bassman JR, Perrien Naccarato CM, Twarozynski JJ, Traynor JR, Iula DM, Anand JP, 2020. In vitro pharmacology of fentanyl analogs at the human mu opioid receptor and their spectroscopic analysis. Drug Test Anal 12 (8), 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard H, Bastian BA, Trinidad JP, Spencer M, Warner M, 2018. Drugs most frequently involved in drug overdose deaths: United States, 2011–2016. In: National Vital Statistics Reports, Hyattsville, MD. [PubMed] [Google Scholar]
- Hedegaard H, Bastian BA, Trinidad JP, Spencer MR, Warner M, 2019. Regional differences in the drugs most frequently involved in drug overdose deaths: United States, 2017. In: National Vital Statistics Reports. Hyattsville, MD. [PubMed] [Google Scholar]
- Higashikawa Y, Suzuki S, 2008. Studies on 1-(2-phenethyl)-4-(N-propionylanilino) piperidine (fentanyl) and its related compounds. VI. Structure-analgesic activity relationship for fentanyl, methyl-substituted fentanyls and other analogues. Foren. Toxicol 26 (1), 1–5. [Google Scholar]
- Janssen PAJ, 1964. Compose a activite pharmacologique a base de 1-arylalcoyl-4-(Narylalcanamido)-piperidines. N.V. Research Laboratorium, France. [Google Scholar]
- Janssen PAJ, 1975. N-(1-Arylalkyl-4-piperidyl)-Narylamide, deren Saureadditionssalze und Verfahren zu ihrer Herstellung. Janssen Pharmaceutica, Germany. [Google Scholar]
- Kenney SR, Anderson BJ, Conti MT, Bailey GL, Stein MD, 2018. Expected and actual fentanyl exposure among persons seeking opioid withdrawal management. J. Subst. Abus. Treat 86, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotulski AJ, Logan BK, 2018. Phenylfentanyl, Toxicology Analytical Reports. The Center for Forensic Science Research & Education. [Google Scholar]
- Kuczynska K, Grzonkowski P, Kacprzak L, Zawilska JB, 2018. Abuse of fentanyl: an emerging problem to face. Forensic Sci. Int 289, 207–214. [DOI] [PubMed] [Google Scholar]
- Kuhlman JJ Jr., McCaulley R, Valouch TJ, Behonick GS, 2003. Fentanyl use, misuse, and abuse: a summary of 23 postmortem cases. J. Anal. Toxicol 27 (7), 499–504. [DOI] [PubMed] [Google Scholar]
- Martin TL, Woodall KL, McLellan BA, 2006. Fentanyl-related deaths in Ontario, Canada: toxicological findings and circumstances of death in 112 cases (2002–2004). J. Anal. Toxicol 30 (8), 603–610. [DOI] [PubMed] [Google Scholar]
- Martinez S, Jones JD, Brandt L, Campbell ANC, Abbott R, Comer SD, 2020. The increasing prevalence of fentanyl: a urinalysis-based study among individuals with opioid use disorder in New York City. Am. J. Addict 30 (1), 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather LE, 1983. Clinical pharmacokinetics of fentanyl and its newer derivatives. Clin. Pharmacokinet 8 (5), 422–446. [DOI] [PubMed] [Google Scholar]
- Misailidi N, Papoutsis I, Nikolaou P, Katselou M, Spiliopoulou C, Athanaselis S, 2017. Furanylfentanyl: another fentanyl analogue, another hazard for public health. Foren. Toxicol 36 (1), 1–11. [Google Scholar]
- Ochalek TA, Parker MA, Higgins ST, Sigmon SC, 2019. Fentanyl exposure among patients seeking opioid treatment. J. Subst. Abus. Treat 96, 23–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochalek TA, Cumpston KL, Wills BK, Gal TS, Moeller FG, 2020. Nonfatal opioid overdoses at an urban emergency department during the COVID-19 pandemic. JAMA 324 (16), 1673–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JK, Halpin J, Mattson CL, Goldberger BA, Gladden RM, 2017. Deaths involving fentanyl, fentanyl analogs, and U-47700 — 10 states, July–December 2016. MMWR Morb. Mortal. Wkly Rep 66 (43), 1197–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan H, Williams J, 2012. Basic opioid pharmacology: an update. Br. J. Pain 6 (1), 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng PW, Sandler AN, 1999. A review of the use of fentanyl analgesia in the management of acute pain in adults. Anesthesiology 90 (2), 576–599. [DOI] [PubMed] [Google Scholar]
- Scholz J, Steinfath M, Schulz M, 1996. Clinical pharmacokinetics of alfentanil, fentanyl and sufentanil. An update. Clin. Pharmacokinet 31 (4), 275–292. [DOI] [PubMed] [Google Scholar]
- Spencer MR, Warner M, Bastian BA, Trinidad JP, Hedegaard H, 2019. Drug overdose deaths involving fentanyl, 2011–2016. In: National Vital Statistics Reports. Hyattsville, MD. [PubMed] [Google Scholar]
- Strayer KE, Antonides HM, Juhascik MP, Daniulaityte R, Sizemore IE, 2018. LCMS/MS-based method for the multiplex detection of 24 fentanyl analogues and metabolites in whole blood at sub ng mL−1 concentrations. ACS Omega 3 (1), 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JG, Baker AM, Bracey AH, Seningen J, Kloss JS, Strobl AQ, Apple FS, 2007. Fentanyl concentrations in 23 postmortem cases from the hennepin county medical examiner’s office. J. Forensic Sci 52 (4), 978–981. [DOI] [PubMed] [Google Scholar]
- Varshneya NB, Walentiny DM, Moisa LT, Walker TD, Akinfiresoye LR, Beardsley PM, 2019. Opioid-like antinociceptive and locomotor effects of emerging fentanyl-related substances. Neuropharmacology 151, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walentiny DM, Moisa LT, Beardsley PM, 2019. Oxycodone-like discriminative stimulus effects of fentanyl-related emerging drugs of abuse in mice. Neuropharmacology 150, 210–216. [DOI] [PubMed] [Google Scholar]
- Walker EA, 2019. Preclinical assessment of novel psychoactive substances: opioids. In: International Narcotics Research Conference. New York City, New York, USA. [Google Scholar]
- Woods JH, Medzihradsky F, Smith C, Winger G, Gmerek D, 1987. Evaluation of new compounds for opioid activity: 1987 annual report. In: National Institute on Drug Abuse Research Monograph Series Problems of Drug Dependence 1987 (Proceedings of the 49th Annual Scientific Meeting The Committee on Problems of Drug Dependence, Inc.). [Google Scholar]
- World Health Organization Expert Committee on Drug Dependence, 2017. Furanyl fentanyl. In: World Health Organization Critical Review Reports. Geneva, Switzerland. [Google Scholar]
- Zawilska JB, 2017. An expanding world of novel psychoactive substances: opioids. Front. Psychiat 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YQ, Ge GL, Fang SN, Zhu YC, Dai QY, Tan ZY, Huang ZM, Chen XJ, 1981. Studies on potent analgesics. I. Synthesis and analgesic activity of derivatives of fentanyl (author’s transl). Yao Xue Xue Bao 16 (3), 199–210. [PubMed] [Google Scholar]