

ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to pose an enormous threat to economic activity and public health worldwide. Previous studies have shown that the nonstructural protein 5 (nsp5, also called 3C-like protease) of alpha- and deltacoronaviruses cleaves Q231 of the NF-κB essential modulator (NEMO), a key kinase in the RIG-I-like receptor pathway, to inhibit type I interferon (IFN) production. In this study, we found that both SARS-CoV-2 nsp5 and SARS-CoV nsp5 cleaved NEMO at multiple sites (E152, Q205, and Q231). Notably, SARS-CoV-2 nsp5 exhibited a stronger ability to cleave NEMO than SARS-CoV nsp5. Sequence and structural alignments suggested that an S/A polymorphism at position 46 of nsp5 in SARS-CoV versus SARS-CoV-2 may be responsible for this difference. Mutagenesis experiments showed that SARS-CoV-2 nsp5 (S46A) exhibited poorer cleavage of NEMO than SARS-CoV-2 nsp5 wild type (WT), while SARS-CoV nsp5 (A46S) showed enhanced NEMO cleavage compared with the WT protein. Purified recombinant SARS-CoV-2 nsp5 WT and SARS-CoV nsp5 (A46S) proteins exhibited higher hydrolysis efficiencies than SARS-CoV-2 nsp5 (S46A) and SARS-CoV nsp5 WT proteins in vitro. Furthermore, SARS-CoV-2 nsp5 exhibited stronger inhibition of Sendai virus (SEV)-induced interferon beta (IFN-β) production than SARS-CoV-2 nsp5 (S46A), while introduction of the A46S substitution in SARS-CoV nsp5 enhanced suppression of SEV-induced IFN-β production. Taken together, these data show that S46 is associated with the catalytic activity and IFN antagonism by SARS-CoV-2 nsp5.
IMPORTANCE The nsp5-encoded 3C-like protease is the main coronavirus protease, playing a vital role in viral replication and immune evasion by cleaving viral polyproteins and host immune-related molecules. We showed that both SARS-CoV-2 nsp5 and SARS-CoV nsp5 cleave the NEMO at multiple sites (E152, Q205, and Q231). This specificity differs from NEMO cleavage by alpha- and deltacoronaviruses, demonstrating the distinct substrate recognition of SARS-CoV-2 and SARS-CoV nsp5. Compared with SARS-CoV nsp5, SARS-CoV-2 nsp5 encodes S instead of A at position 46. This substitution is associated with stronger catalytic activity, enhanced cleavage of NEMO, and increased interferon antagonism of SARS-CoV-2 nsp5. These data provide new insights into the pathogenesis and transmission of SARS-CoV-2.
KEYWORDS: severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), nonstructural protein 5 (nsp5), 3C-like protease, catalytic activity, NF-κB essential modulator (NEMO), interferon
INTRODUCTION
A sudden outbreak of coronavirus disease 2019 (COVID-19) caused by an emerging virus, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), has disturbed normal public activities worldwide and poses an enormous threat to public health (1–4). The typical symptoms of COVID-19 include fever, cough, and pneumonia, while severe cases can lead to death (5, 6). As of 2 March 2022, 440,172,104 individuals have been infected with SARS-CoV-2, of whom 5,972,607 have died (database at https://coronavirus.jhu.edu/). Although the genome of SARS-CoV-2 shares 82% identity with that of the highly lethal SARS-CoV that caused an outbreak in 2003, SARS-CoV-2 has a lower fatality rate but faster spread (7–10). Understanding the features of SARS-CoV-2 that contribute to its lower virulence and stronger transmissibility may help in identifying targeted therapies against SARS-CoV-2 infection.
Both SARS-CoV-2 and SARS-CoV are enveloped, positive-sense, single-stranded RNA viruses belonging to the genus Betacoronavirus of the family Coronaviridae (11, 12). The genomes of both viruses contain at least seven open reading frames (ORFs). The two larger ORFs located downstream from the 5′ untranslated region, ORF1a and ORF1b, encode two viral replicase polyprotein precursors, 1a and 1b, that are proteolytically processed to yield 16 mature nonstructural proteins (nsps) (1, 13–16). The 3C-like protease (3CLpro, nsp5) and the papain-like cysteine protease (PLpro, nsp3) are responsible for cleavage of polyproteins (17–19). Eleven sites in polyproteins are cleaved by 3C-like protease to yield mature functional proteins (20, 21). Thus, nsp5 plays an indispensable role in viral replication and is an attractive target for anticoronavirus drugs (22–24). Previous studies have shown that coronavirus nsp5 possesses a nonclassical catalytic dyad (His41-Cys145) involved in hydrolysis and shows a preference for Q at position P1 of the substrate (25–27). The recognition characteristics of nsp5 have been investigated extensively, but the detailed catalytic mechanisms remain poorly understood. Understanding the catalytic mechanism of SARS-CoV-2 nsp5 will help us understand SARS-CoV-2 replication.
In addition to its essential role in viral replication, coronavirus nsp5 plays an important role in escaping the host immune response by targeting immune-related molecules. For instance, the nsp5 of porcine deltacoronavirus (PDCoV)—a member of genus Deltacoronavirus—targets signal transducer and activator of transcription 2 (STAT2) and mRNA-decapping enzyme 1a (DCP1A) to disrupt interferon (IFN)-induced JAK-STAT signaling and the antiviral activity of DCP1A (28). The nsp5 of porcine epidemic diarrhea virus (PEDV)—a member of the genus Alphacoronavirus—suppresses type I IFN production by cleaving NF-κB essential modulator (NEMO) at Q231, and a similar mechanism is used by PDCoV nsp5 (29, 30). Recent studies demonstrated that SARS-CoV-2 nsp1, nsp6, nsp13, nsp14, nsp15, and ORF6 can suppress IFN-β promoter activity (31–33). However, whether SARS-CoV-2 nsp5 contributes to escape from innate immune responses and plays roles in COVID-19 pathogenesis and transmission remains unclear.
In this study, we found that both SARS-CoV-2 and SARS-CoV nsp5 antagonize IFN-β production and target NEMO at multiple sites. Moreover, SARS-CoV-2 nsp5 shows enhanced cleavage efficiency of NEMO compared with SARS-CoV nsp5, resulting in stronger suppression of IFN production. Remarkably, residue S46 within SARS-CoV-2 nsp5 is responsible for its enhanced catalytic activity and IFN antagonism compared with SARS-CoV nsp5. Learning the catalysis and IFN antagonism mechanism of SARS-CoV-2 nsp5 may help in understanding the pathogenesis and transmission of SARS-CoV-2.
RESULTS
SARS-CoV-2 nsp5 and SARS-CoV nsp5 cleave NEMO at multiple sites.
Previous studies have shown that PEDV (Alphacoronavirus) nsp5 and PDCoV (Deltacoronavirus) nsp5 cleave NEMO to antagonize type I IFN production (29, 30). To investigate whether SARS-CoV-2 nsp5 and SARS-CoV nsp5 also target NEMO for cleavage, HEK-293T cells were cotransfected with a plasmid encoding FLAG-tagged NEMO and increasing amounts of expression vectors encoding hemagglutinin (HA)-tagged SARS-CoV-2/SARS-CoV nsp5. Western blotting showed that NEMO cleavage increased following transfection with increasing amounts of plasmids encoding SARS-CoV-2 nsp5 or SARS-CoV nsp5 (Fig. 1A and B). Surprisingly, three cleaved bands were apparent in cells transfected with expression vectors encoding SARS-CoV-2 nsp5 or SARS-CoV nsp5, indicating that both nsp5s cleaved NEMO at a minimum of three sites. This pattern differs from that of PEDV nsp5- or PDCoV nsp5-mediated NEMO cleavage (29, 30).
FIG 1.

Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) nsp5 and SARS-CoV nsp5 target NF-κB essential modulator (NEMO) at multiple sites. (A and B) HEK-293T cells were cotransfected with various amounts of expression vectors encoding SARS-CoV-2 nsp5 (0.5 μg, 0.25 μg, 0.125 μg, and 0.0625 μg) (A) or various amounts of expression vectors encoding SARS-CoV nsp5 (2 μg, 1 μg, 0.5 μg, and 0.25 μg) (B) along with a plasmid encoding FLAG-tagged NEMO. The transfected cells were lysed 28 h after transfection and then analyzed by Western blotting. (C) HEK-293T cells were cotransfected with equal amounts of expression vectors encoding SARS-CoV nsp5, SARS-CoV-2 nsp5, or empty vector together with a plasmid encoding FLAG-tagged NEMO. After 28 h, the cells were harvested and assessed by Western blotting. Band intensity was quantified by densitometry using ImageJ software. The relative abundance of NEMO cleavage products and nsp5 were normalized to levels of β-actin. A final ratio was calculated of the relative amount of cleaved NEMO product standardized to the amount of nsp5. All experiments were performed at least three times, and one representative experiment was presented.
To further compare SARS-CoV-2 nsp5- and SARS-CoV nsp5-mediated NEMO cleavage, HEK-293T cells were cotransfected with equal amounts of expression vectors encoding HA-tagged SARS-CoV-2 nsp5 or SARS-CoV nsp5 and FLAG-tagged NEMO. As shown in Fig. 1C, the sizes of the three NEMO cleavage products were similar in cells transfected with SARS-CoV-2 nsp5 and SARS-CoV nsp5, indicating that they recognized similar cleavage sites. Interestingly, quantitative analysis of the cleaved bands showed that the gray values of NEMO cleavage products in SARS-CoV-2 nsp5-mediated NEMO cleavage were higher than that in SARS-CoV nsp5-mediated NEMO cleavage, indicating that SARS-CoV-2 nsp5 exhibited stronger cleavage of NEMO than SARS-CoV nsp5 (Fig. 1C).
E152, Q205, and Q231 are the sites of SARS-CoV-2 and SARS-CoV nsp5-mediated NEMO cleavage.
Coronavirus nsp5 has been reported to prefer Q at the P1 position for proteolytic cleavage (34). To explore whether SARS-CoV-2 nsp5 also possesses this recognition characteristic, the polyprotein junctions cleaved by SARS-CoV-2 nsp5 were analyzed and presented as sequence logos. As shown in Fig. 2A, SARS-CoV-2 nsp5 showed a preference for Q at the P1 position. Considering the recognition preference for P1-Q and the size of the two larger cleavage bands (between 25 kDa and 35 kDa), NEMO mutants in which Q residues between 25 kDa and 35 kDa were replaced with A or K were generated (Fig. 2B). HEK-293T cells were cotransfected with expression vectors encoding NEMO mutants (Q218A, Q229K, Q231A, Q236A/Q239A, and Q259A) and SARS-CoV-2 nsp5. As shown in Fig. 2B, the largest cleavage product (∼30 kDa) disappeared in cells cotransfected with expression vectors encoding NEMO (Q231A) and SARS-CoV-2 nsp5, whereas three cleavage bands were still present in cells cotransfected with plasmids encoding the other NEMO mutants and SARS-CoV-2 nsp5 (Fig. 2C). This result indicated that NEMO Q231, which was identified in previous studies of PEDV nsp5 and PDCoV nsp5, is a site of SARS-CoV-2 nsp5-mediated NEMO cleavage. To further examine the second band resulting from SARS-CoV-2 nsp5-mediated NEMO cleavage, HEK-293T cells were cotransfected with expression vectors encoding each of four NEMO mutants (Q198A, Q201A, Q205A, and Q207A) and SARS-CoV-2 nsp5. As shown in Fig. 2D, the second cleavage band was absent in cells cotransfected with plasmids encoding SARS-CoV-2 nsp5 and NEMO Q205A, indicating that Q205 is the second site of SARS-CoV-2 nsp5-mediated NEMO cleavage.
FIG 2.

E152, Q205, and Q231 are three sites of SARS-CoV-2- and SARS-CoV nsp5-mediated NEMO cleavage. (A) Amino acid sequence logo of polyprotein junctions cleaved by SARS-CoV-2 nsp5. The sequence logo for the substrate was generated using WebLogo3 (http://weblogo.threeplusone.com/). (B) Schematic representation of NEMO and its mutants. (C to E) HEK-293T cells were cotransfected with expression vectors encoding SARS-CoV-2 nsp5 and FLAG-tagged wild-type (WT) NEMO or NEMO mutants Q218A, Q229K, Q231A, Q236A/Q239A, or Q259A (C), NEMO mutants Q198A, Q201A, Q205A, or Q207A (D), or NEMO mutants Q120A, Q132A, Q133A, Q134A, Q145A, or Q154A (E). At 28 h posttransfection, the cells were collected and assessed by Western blotting with corresponding antibodies. (F) HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged WT NEMO or NEMO mutants (E137A/E152A/E155A) and a plasmid encoding SARS-CoV-2 nsp5. The cells were treated as described in panel C. (G) HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged WT NEMO or a NEMO triple mutant (E152A/Q205A/Q231A) and a plasmid encoding SARS-CoV-2 nsp5. The cells were treated as described in panel C. (H) HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged NEMO or NEMO mutants (E152A, Q205, Q231) and a plasmid encoding SARS-CoV nsp5. At 28 h posttransfection, cells were treated as described in panel C. (I) HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged WT NEMO or a NEMO triple mutant (E152A/Q205A/Q231A) and a plasmid encoding SARS-CoV nsp5. Subsequently, the cells were treated as described in panel C. All experiments were performed at least three times, and one representative experiment is presented.
We next identified the smallest band resulting from SARS-CoV-2 nsp5-mediated NEMO cleavage. Considering the size of the smallest band is approximately 15 kDa, all Q residues spanning between amino acids 132 to 155 of NEMO were analyzed and mutated into A. Subsequently, HEK-293T cells were cotransfected with expression vectors encoding wild type (WT) or mutants NEMO (Q132A/Q133A/Q134A/Q145A/Q154A) and SARS-CoV-2 nsp5. Western blotting showed that no cleavage bands disappeared in cells expressing any of the NEMO mutants (Fig. 2E), suggesting that SARS-CoV-2 nsp5 may not recognize the conserved P1-Q to target this site. Interestingly, the expression level of the smallest band in cells cotransfected with plasmids encoding NEMO Q145A/Q154A and SARS-CoV-2 nsp5 was significantly reduced compared with that in cells expressing NEMO WT or other NEMO mutants (Fig. 2E). Thus, we speculated that the cleavage site might be near Q145 and Q154. Our previous work showed that equine arteritis virus (EAV) nsp4 (the ortholog of SARS-CoV-2 nsp5) also targets NEMO for proteolytic cleavage and that scission occurred at four sites, E166, E171, Q205, and E349 (35). To test whether SARS-CoV-2 nsp5 also cleaves NEMO by recognizing P1-E, HEK-293T cells were cotransfected with expression vectors encoding NEMO mutants (E137A/E152A/E155A) and SARS-CoV-2 nsp5. As excepted, the smallest cleavage product disappeared in cells cotransfected with expression vectors encoding NEMO (E152A) and SARS-CoV-2 nsp5 (Fig. 2F), suggesting that E152 was the third site of SARS-CoV-2 nsp5-mediated NEMO cleavage. To further validate whether the three sites identified are targeted by SARS-CoV-2 nsp5, HEK-293T cells were cotransfected with expression vectors encoding a triple NEMO mutant (E152A/Q205A/Q231A) and SARS-CoV-2 nsp5. As shown in Fig. 2G, SARS-CoV-2 nsp5 failed to cleave NEMO (E152A/Q205A/Q231A), indicating that E152, Q205, and Q231 are three sites of SARS-CoV-2 nsp5-mediated NEMO cleavage.
Because similar NEMO bands could be observed following cleavage by SARS-CoV-2 nsp5 and SARS-CoV nsp5 (Fig. 1C), we further investigated whether the three cleavage sites were targeted by SARS-CoV nsp5 to hydrolyze NEMO. To this end, HEK-293T cells were cotransfected with expression vectors encoding NEMO mutants (E152A, Q205A, or Q231A) and SARS-CoV nsp5. Western blotting showed that the corresponding NEMO cleavage band (1 to 152) vanished in cells expressing NEMO (E152A) and SARS-CoV nsp5 (Fig. 2H). Similar phenomena were observed in cells cotransfected with plasmids encoding NEMO (Q205A) or NEMO (Q231A) and SARS-CoV nsp5 (Fig. 2H), indicating that SARS-CoV nsp5 shares a similar recognition motif for NEMO cleavage with SARS-CoV-2 nsp5. Moreover, all three cleavage bands disappeared in cells cotransfected with plasmids encoding NEMO (E152A/Q205A/Q231A) and SARS-CoV nsp5 (Fig. 2I), suggesting that SARS-CoV nsp5 also cleaved NEMO at three sites (E152, Q205, and Q231). Taken together, these results demonstrated that SARS-CoV-2 nsp5 and SARS-CoV nsp5 cleaved NEMO at three common sites (E152, Q205, and Q231).
S46 is important for SARS-CoV-2 nsp5-mediated NEMO cleavage.
Although SARS-CoV-2 nsp5 and SARS-CoV nsp5 shared identical sites for NEMO cleavage, SARS-CoV-2 nsp5 appeared to have a stronger NEMO cleavage ability than SARS-CoV nsp5 (Fig. 1C). To explore the domain(s) within SARS-CoV-2 nsp5 and SARS-CoV nsp5 responsible for differences in catalytic activity, sequence and structural alignments of SARS-CoV-2 nsp5 and SARS-CoV nsp5 were conducted. As shown in Fig. 3A, sequence alignment showed that the sequences of SARS-CoV-2 nsp5 and SARS-CoV nsp5 were highly similar; SARS-CoV-2 nsp5 differed from SARS-CoV nsp5 at only 12 residues. We next compared the structures of SARS-CoV-2 nsp5 and SARS-CoV nsp5, especially in the region around the substrate-binding site. The two structures were highly conserved except for the domain (residues 44 to 50) near the active site. Notably, this domain (residues 44 to 50) within SARS-CoV nsp5 formed an α-helical structure, while the corresponding region in SARS-CoV-2 nsp5 adopted a loop structure (Fig. 3B). By analyzing the amino acid composition of this region (residues 44 to 50), we found that only residue 46 was polymorphic between the two coronaviruses: S46 was present in SARS-CoV-2 nsp5, while A46 was present in SARS-CoV nsp5 (Fig. 3A). Thus, the residue at position 46 could be potentially responsible for the different catalytic activities of SARS-CoV nsp5 and SARS-CoV-2 nsp5. We tested this hypothesis by constructing position 46 S/A mutants and comparing their efficiencies of NEMO cleavage efficiency. Two mutants were generated, SARS-CoV-2 nsp5 (S46A) and SARS-CoV nsp5 (A46S) (Fig. 3C). Subsequently, HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged NEMO and increasing concentrations of WT HA-tagged SARS-CoV-2 nsp5 or HA-tagged SARS-CoV-2 nsp5 (S46A). Western blotting showed that SARS-CoV-2 nsp5 (S46A) exhibited poorer cleavage of NEMO than WT SARS-CoV-2 nsp5 (Fig. 3D). Similar experiments were conducted using plasmids encoding FLAG-tagged NEMO and HA-tagged WT SARS-CoV nsp5 or SARS-CoV nsp5 (A46S). As shown in Fig. 3E, the A46S mutant of SARS-CoV nsp5 showed enhanced NEMO cleavage efficiency compared with WT SARS-CoV nsp5. These findings demonstrated that residue S46 of SARS-CoV-2 nsp5 plays an important role in its enhanced NEMO cleavage efficiency compared with SARS-CoV nsp5.
FIG 3.

S46 is important for SARS-CoV-2 nsp5-mediated NEMO cleavage. (A) Sequence alignment of SARS-CoV-2 nsp5 (GenBank accession number NC_045512) and SARS-CoV nsp5 (GenBank accession number NC_004718.3). (B) Structural alignment of SARS-CoV-2 nsp5 (PDB ID 6M2N) and SARS-CoV nsp5 (PDB ID 1UJ1). (C) Schematic representation of SARS-CoV-2 nsp5 and its S46A mutant and SARS-CoV nsp5 and its A46S mutant. (D) HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged NEMO and various amounts of expression plasmids encoding HA-tagged WT SARS-CoV-2 nsp5 (0.25 μg, 0.125 μg, 0.0625 μg, and 0.03125 μg) or SARS-CoV-2 nsp5 (S46A) (0.25 μg, 0.125 μg, 0.0625 μg, and 0.03125 μg). Cells were collected and analyzed by Western blotting. The gray value analysis of cleavage bands in Western blots was performed as described in Fig. 1C. (E) HEK-293T cells were cotransfected with expression vectors encoding FLAG-tagged NEMO and increasing amounts of plasmid encoding WT SARS-CoV nsp5 (1 μg, 0.5 μg, 0.25 μg, and 0.125 μg) or SARS-CoV nsp5 (A46S) (0.25 μg, 0.125 μg, 0.0625 μg, and 0.03125 μg). Cells were treated as described in panel D. Quantitation of bands in Western blots was performed as described in Fig. 1C. All experiments were performed at least three times, and one representative experiment is presented.
S46/A46 serves as a major strong/weak cleavage switch for the catalytic activities of SARS-CoV-2 nsp5 and SARS-CoV nsp5.
To further investigate whether the S/A polymorphism at position 46 affected the catalytic activities of SARS-CoV-2 nsp5 and SARS-CoV nsp5 in vitro, four recombinant proteins were expressed in Escherichia coli and purified, WT SARS-CoV-2 nsp5, WT SARS-CoV nsp5, SARS-CoV-2 nsp5 (S46A), and SARS-CoV nsp5 (A46S). The catalytic activities of the purified recombinant proteins were evaluated using a fluorescence resonance energy transfer assay (FRET) (36). A fluorescently labeled peptide substrate (Dabcyl-TSAVLQ↓SGFRKM-E-Edans) containing the N-terminal autocleavage site (nsp4/nsp5) of SARS-CoV-2 nsp5 derived from polyprotein was synthesized and applied in the FRET assay along with the purified proteins. SARS-CoV-2 nsp5 exhibited enhanced hydrolysis efficiency in the FRET assay compared with SARS-CoV nsp5 (Fig. 4). Similar to the findings in a cell system, SARS-CoV-2 nsp5 and SARS-CoV nsp5 (A46S) showed similar catalytic activities that were higher than those of SARS-CoV nsp5 and SARS-CoV-2 nsp5 (S46A) (Fig. 4). Thus, our data demonstrate that residue 46 is a major strong/weak cleavage switch for the catalytic activities of SARS-CoV-2 nsp5 and SARS-CoV nsp5.
FIG 4.
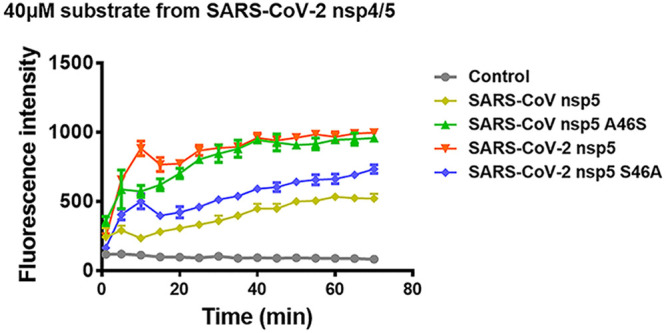
S46/A46 serves as a major strong/weak cleavage switch for the catalytic activity of SARS-CoV-2 nsp5 and SARS-CoV nsp5. Cleavage of a SARS-CoV-2 nsp5 N-terminal substrate was assessed. A fluorescence resonance energy transfer assay was conducted using 40 μM fluorogenic peptide substrate (Dabcyl-TSAVLQ↓SGFRKM-E-Edans, containing the N-terminal autocleavage site of SARS-CoV-2 3CLpro) and 50 nM SARS-CoV-2 nsp5 (WT and S46A) or SARS-CoV nsp5 (WT and A46S). All groups were performed in triplicate, and the data are represented as means ± standard deviations of three independent experiments.
SARS-CoV-2 nsp5 suppresses IFN-β production more efficiently than SARS-CoV nsp5.
NEMO is a key kinase in the RIG-I-like receptor-mediated type I IFN production pathway. Previous studies showed that NEMO cleavage mediated by viral 3CLpro led to inhibition of Sendai virus (SEV)-induced type I IFN production (28, 35, 37). To evaluate whether the cleavage products of NEMO lost the ability to induce IFN production, all cleavage products, including NEMO-K277A (1-152), NEMO-K277A (1-205), NEMO-K277A (1-231), NEMO-K277A (153-419), NEMO-K277A (206-419), and NEMO-K277A (232-419), were transfected into HEK-293T cells. As shown in Fig. 5A, all cleavage products lost the ability to induce IFN-β production, indicating that SARS-CoV-2 or SARS-CoV nsp5-mediated NEMO cleavage disrupts IFN-β induction. Because SARS-CoV-2 nsp5 and SARS-CoV nsp5 exhibited different NEMO cleavage efficiencies and different catalytic activities (Fig. 1C and Fig. 4), we speculated that the inhibitory effects of SARS-CoV-2 nsp5 and SARS-CoV nsp5 on IFN-β production might be discrepant. To confirm this hypothesis, HEK-293T cells were cotransfected with equal amounts of plasmids encoding SARS-CoV-2 nsp5 or SARS-CoV nsp5 and an IFN-β luciferase (IFN-β-Luc) reporter plasmid. Thereafter, the cells were stimulated with SEV. The dual-luciferase reporter assay showed that SARS-CoV-2 nsp5 exhibited stronger suppression of SEV-induced IFN-β promoter induction than SARS-CoV nsp5, even when the expression level of SARS-CoV nsp5 was slightly higher than that of SARS-CoV-2 nsp5 (Fig. 5B). Also, endogenous IFN expression levels in HEK-293T cells transfected with plasmids encoding SARS-CoV-2/SARS-CoV nsp5 and their mutants (S46A, A46S) were evaluated by an IFN bioassay using IFN-sensitive vesicular stomatitis virus-green fluorescent protein (VSV-GFP). The GFP-positive cells were then analyzed by flow cytometry. As shown in Fig. 5C, the percentage of GFP-positive cells in the cells treated with cell supernatants from SARS-CoV-2 nsp5-transfected cells was higher than that of SARS-CoV nsp5-transfected cells, indicating that SARS-CoV-2 nsp5 exhibited stronger inhibition ability on endogenous IFN production than SARS-CoV nsp5. The IFN bioassay was also performed and analyzed by fluorescence microscopy and Western blotting. In agreement with the data of flow cytometry, SARS-CoV-2 nsp5 more strongly recovered SEV-restricted replication of VSV-GFP than SARS-CoV nsp5 (Fig. 5D and E). These findings indicated that SARS-CoV-2 nsp5 has stronger inhibition of SEV-induced IFN production than SARS-CoV nsp5.
FIG 5.

SARS-CoV-2 nsp5 suppresses IFN-β production more efficiently than SARS-CoV nsp5. (A) We transfected 0.4 μg of plasmid expressing NEMO-truncated mutants (NEMO-K277A [1-152], NEMO-K277A [1-205], NEMO-K277A [1-231], NEMO-K277A [153-419], NEMO-K277A [206-419], and NEMO-K277A [232-419]) along with IFN-β reporter plasmid and pRL-TK plasmid into HEK-293T cells for 28 h. The cells were then lysed and evaluated by dual-luciferase assay. (B) HEK-293T cells seeded in a 24-well plate were cotransfected with equal amounts of expression vectors encoding SARS-CoV-2 nsp5 (0.015 μg) or SARS-CoV nsp5 (0.06 μg) along with IFN-β reporter plasmid and pRL-TK plasmid. At 24 h posttransfection, cells were infected with Sendai virus (SEV). The cells were collected 16 h after SEV infection and used in luciferase assays to assess the expression of the IFN-β reporter. Expression of nsp5 was analyzed by Western blotting. Bands were quantitated by densitometry using ImageJ software, and nsp5 expression was standardized to that of β-actin. (C) HEK-293T cells seeded in a 24-well plate were transfected with equal amounts of plasmid encoding SARS-CoV-2 nsp5 and SARS-CoV nsp5 for 24 h, followed by SEV infection for 12 h. The cell supernatants were then collected, treated with UV irradiation, and added to a new 24-well plate HEK-293T cells for 24 h. The IFN-treated cells were then inoculated with VSV-GFP for 12 h, followed by the detection of GFP-positive cells by flow cytometry. (D and E) Cells were treated as described in panel C. The GFP expression in cells treated with cell supernatants from cells transfected with the indicated plasmid was detected by fluorescence microscopy (D) and Western blot assay (E). The nsp5 expression of the transfected cells was also detected in Western blot assay. All groups were performed in triplicate, and the results represent means ± standard deviations of three independent experiments. *, P < 0.05; ***, P < 0.001.
S46 is responsible for the different abilities on IFN inhibition of SARS-CoV-2 nsp5 and SARS-CoV nsp5.
To further investigate whether the residue at position 46 of SARS-CoV-2 or SARS-CoV nsp5 was responsible for the differential abilities of these proteins to suppress IFN-β production, we compared the efficiency of IFN-β inhibition by WT SARS-CoV-2 nsp5 and SARS-CoV-2 nsp5 (S46A). As shown in Fig. 6A, WT SARS-CoV-2 nsp5 exhibited stronger inhibition of IFN-β promoter activity than SARS-CoV-2 nsp5 (S46A). We also compared the IFN-β inhibition efficiency of WT SARS-CoV nsp5 and SARS-CoV nsp5 (A46S). The A46S mutant more efficiently suppressed SEV-induced IFN-β promoter activation than the WT SARS-CoV nsp5 (Fig. 6B). We also compared the IFN inhibition of SARS-CoV-2/SARS-CoV nsp5 and their mutants (S46A and A46S) in an IFN bioassay and analyzed by flow cytometry. Compared with SARS-CoV-2 nsp5 mutant (S46A) and SARS-CoV nsp5, respectively, SARS-CoV-2 nsp5 and the SARS-CoV nsp5 mutant (A46S) more strongly recovered IFN-inhibited VSV replication, indicating that SARS-CoV-2 nsp5 and SARS-CoV nsp5 mutant (A46S) exhibited stronger inhibition of endogenous IFN production than the SARS-CoV-2 nsp5 mutant (S46A) and SARS-CoV nsp5 upon SEV stimulation (Fig. 6C). The IFN bioassay was also evaluated by detecting the GFP expression with fluorescence microscopy (Fig. 6D and F) and Western blotting (Fig. 6E and G). The results were consistent with the results in Fig. 6C by flow cytometry analyses. Taken together, these results indicated that the S residue at position 46 enables stronger antagonism of type I IFN production by SARS-CoV-2 nsp5 than SARS-CoV nsp5.
FIG 6.

S46 is responsible for the different abilities on IFN inhibition of SARS-CoV-2 nsp5 and SARS-CoV nsp5. (A and B) HEK-293T cells in a 24-well plate were cotransfected with expression vectors encoding SARS-CoV-2 nsp5 (WT or S46A) (A) or SARS-CoV nsp5 (WT or A46S) (B) along with an IFN-β reporter plasmid and a pRL-TK plasmid. The cells were inoculated with SEV 24 h after transfection and infected for 12 h. The cells were then lysed, and dual-luciferase assays were used to assess expression of IFN-β. Quantification of nsp5 was performed as described in Fig. 5B (C) HEK-293T cells seeded in a 24-well plate were transfected with equal amounts of plasmids encoding SARS-CoV-2 nsp5 (WT or S46A) or SARS-CoV nsp5 (WT or A46S) for 24 h and then treated as described in Fig. 5C. The GFP-positive cells were then analyzed by flow cytometry. (D to G) The cells were treated as described in panel C. The GFP expressions were detected by fluorescence microscopy (D and F) and Western blot assay (E and G). The nsp5 expression of transfected cells was also detected in Western blot assay. All groups were performed in triplicate, and the results represent means ± standard deviations of three independent experiments. **, P < 0.01.
DISCUSSION
Coronavirus nsp5 proteins have been reported to adopt similar recognition characteristics to cleave viral polyproteins or host immune-related proteins. For example, it was reported that coronavirus nsp5s targeted DCP1A at a unique site (Q343) to disrupt its antiviral activity (28), revealing a common recognition mechanism used to inhibit host innate immune responses. Our previous work demonstrated that PEDV (Alphacoronavirus) nsp5 and PDCoV (Deltacoronavirus) nsp5 cleaved NEMO at a single site, Q231 (29, 30). Additionally, feline infectious peritonitis virus (Alphacoronavirus) nsp5 was recently found to target NEMO at three sites, Q231, Q205, and Q132 (37). In this study, we found that two glutamine cleavage sites, Q205 and Q231, were involved in both SARS-CoV-2 and SARS-CoV nsp5-mediated NEMO cleavage. Surprisingly, SARS-CoV-2/SARS-CoV nsp5 cleaved NEMO at an additional site, E152, demonstrating a novel recognition characteristic used by SARS-CoV-2/SARS-CoV nsp5 to target substrates with E at the P1 position. Notably, the nsp4 protein of equine arteritis virus (EAV)—a member of the family Arteriviridae and the order Nidovirales—can also cleave NEMO by recognizing substrates with P1-E and P1-Q residues (35). Furthermore, EAV nsp4 and SARS-CoV-2 nsp5 share a similar cleavage site on NEMO (Q205) (35). Thus, we speculated that the nsp5 proteins of SARS-CoV-2 and SARS-CoV, both of which belong to lineage b of Betacoronavirus, may be more closely related to EAV nsp4 than other coronaviruses. The oxyanion hole is an important structure for proteolytic activity and substrate recognition of 3CLpro. Notably, both EAV nsp4 and SARS-CoV nsp5 possess two forms of the oxyanion hole, an active form and a collapsed one, and the signature turn in both forms is extremely similar (25, 38, 39). This finding provides a possibility of similarity of both two proteases and is consistent with our assumption of the relatedness of EAV nsp4 and SARS-CoV-2 nsp5/SARS-CoV nsp5. However, the more structural features that govern similarities in substrate recognition between EAV nsp4 and SARS-CoV-2 nsp5/SARS-CoV nsp5 need to be further investigated.
SARS-CoV-2 shows stronger transmissibility than the highly lethal SARS-CoV (40), making viral spread hard to control. Studies of innate and adaptive immune responses following viral infection will help to elucidate the mechanisms underlying discrepancies in transmission and disease process (41). IFN responses are the first line of host defense against viral infection. Coronaviruses have been reported to adopt various strategies to escape host IFN responses (42). Following the outbreak of COVID-19, many studies have been conducted on this emerging coronavirus and its IFN antagonism. To date, at least 11 SARS-CoV-2 proteins, including nsp1, nsp3, nsp6, nsp12, nsp13, nsp14, nsp15, ORF3b, ORF6, M protein, and ORF9b (31–33, 43–47), have been demonstrated to antagonize type I IFN production. In this study, we identified SARS-CoV-2 nsp5 as a novel IFN antagonist and found that both SARS-CoV-2 nsp5 and SARS-CoV nsp5 suppressed IFN-β production via NEMO cleavage, a characteristic that was previously demonstrated for other coronavirus-encoded nsp5s (29, 30, 37). Compared with the highly pathogenic SARS-CoV, SARS-CoV-2 has been shown to inhibit type I and type III IFN production more potently in human bronchial epithelial cells (48). However, which proteins encoded by SARS-CoV-2 contribute to the stronger inhibition of IFN expression remained unclear. Recently, the SARS-CoV-2-encoded accessory protein ORF3b was demonstrated to suppress IFN induction more efficiently than its SARS-CoV ortholog (44). Furthermore, the SARS-CoV-2 nonstructural proteins nsp1 and nsp6 have also been demonstrated to inhibit type I IFN signaling more efficiently than their SARS-CoV orthologs (31). Here, we found that SARS-CoV-2 nsp5 also exhibits stronger IFN antagonistic activity than SARS-CoV nsp5, which may provide an explanation of why SARS-CoV-2 poorly induces type I IFN production (48, 49) and asymptomatic transmission of COVID-19 patients. Moreover, SARS-CoV-2 nsp5 is responsible for cleaving viral polyprotein, thus playing an independent role in viral replication (20). Our finding demonstrated that SARS-CoV-2 nsp5 exhibited stronger cleavage ability on N-terminal autocleavage site of SARS-CoV-2 nsp5 from viral polyprotein than SARS-CoV nsp5, which provides evidence of stronger replication of SARS-CoV-2 than SARS-CoV. Overall, these findings on immune antagonism and viral replication help us better understand viral pathogenesis and transmission.
An interesting finding of our study was that the S46 residue of SARS-CoV-2 nsp5 was associated with its catalytic activity, cleavage efficiency, and IFN antagonism. Coronavirus nsp5 is indispensable for viral replication because of its potent cleavage of viral polypeptides (50). In this study, we demonstrated that SARS-CoV-2 nsp5 exhibited stronger cleavage efficiency on a peptide derived from the SARS-CoV-2 N-terminal autocleavage site than SARS-CoV nsp5. Thus, we speculated that SARS-CoV-2 may replicate more rapidly than SARS-CoV. Consistent with this idea, a previous study showed that SARS-CoV-2 replicated more efficiently than SARS-CoV in human lung tissues (9). Because of the high risks associated with SARS-CoV-2 and associated experimental limitations, we could not further evaluate whether SARS-CoV-2 nsp5 residue S46 plays an indispensable role in viral replication. Furthermore, we demonstrated that mutation of S46 to A weakened the catalytic activity of SARS-CoV-2 nsp5 (Fig. 4). The structure of SARS-CoV-2 nsp5 showed that its S2 pocket consists of H41, M49, Y54, and M165 (22). A recent study on the structure of SARS-CoV-2 nsp5 showed that the conformation of M49 changes along with changes in the side chain positions of S46 and L50 (51). Thus, we speculate that the S46A nsp5 mutant may result in changes to the position of the side chain at position 46 and further influence the conformation of M49 in the S2 pocket. Comparing the structures of SARS-CoV-2 nsp5 and SARS-CoV nsp5 with and without the inhibitor N3, Griffin et al. recently found that nsp5 residue 46 showed nonspecific interactions with N3 in both viruses (52). Whether S46 in SARS-CoV-2 increases catalytic activity by enforcing this interaction between protease and substrate remains to be further studied. Furthermore, we also analyzed the conservation of nsp5 amino acid sequences from SARS-CoV-2, SARS-CoV-2-related viruses, SARS-CoV, and SARS-CoV-related viruses. Position 46 in nsp5 was occupied by an S residue in all strains of SARS-CoV-2, while a conserved A residue was found at position 46 of most SARS-CoV strains. Interestingly, an S residue occupied position 46 of the nsp5s of three SARS-related isolates from bats (RaTG13, bat-SL-CoVZC45, and bat-SL-CoVZXC21; data not shown). Thus, we speculate that SARS-CoV-2 may be evolutionarily related to these three SARS-related viruses. This finding may help us better understand the origins of SARS-CoV-2.
In summary, our results demonstrated that SARS-CoV-2 nsp5 and SARS-CoV nsp5 cleaved NEMO at three sites, E152, Q205, and Q231. This recognition pattern differs from previously described patterns of coronavirus-mediated NEMO cleavage. SARS-CoV-2 nsp5 showed stronger catalytic activity and IFN antagonism than SARS-CoV nsp5. Residue S46 in SARS-CoV-2 nsp5 was responsible for stronger cleavage efficiency and IFN antagonism activity. Our findings elucidated the detailed mechanisms underlying differences in catalytic activity and IFN antagonism between SARS-CoV-2 nsp5 and SARS-CoV nsp5. These data contribute to our understanding of SARS-CoV-2 replication, transmission, and pathogenesis.
MATERIALS AND METHODS
Cells and viruses.
HEK-293T cells were obtained from the China Center for Type Culture Collection (Wuhan, China) and were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum at 37°C in a 5% CO2 incubator. SEV was acquired from the Centre for Virus Resource and Information at the Wuhan Institute of Virology, Chinese Academy of Sciences (Wuhan, China).
Plasmids.
The gene encoding SARS-CoV-2 nsp5 was amplified from the plasmid pET-32a-SARS-CoV-2 nsp5 (a kind gift from Guiqing Peng) and thereafter cloned into the pCAGGS-HA-C vector containing a C-terminal HA tag. A DNA fragment encoding SARS-CoV nsp5 was synthetized by GenScript (Nanjing, China) and cloned into the pCAGGS-HA-C vector. SARS-CoV-2 nsp5 (S46A) and SARS-CoV nsp5 (A46S) mutants were generated by overlap extension PCR using SARS-CoV-2 nsp5 or SARS-CoV nsp5 as the template. The strategy for cloning of prokaryotic expression plasmids encoding SARS-CoV-2 nsp5, SARS-CoV nsp5, and their mutants for producing recombinant nsp5 proteins was described in previous studies (20, 53). The NEMO expression plasmids were constructed as described previously (54). NEMO mutants were generated by site-directed mutagenesis using WT NEMO as the template. WT NEMO and NEMO mutants were N-terminally FLAG-tagged. The luciferase reporter plasmid, IFN-β-Luc, has previously been described (29). The sequences of all plasmids used in this study were verified.
Luciferase reporter gene assay.
HEK-293T cells were seeded in 24-well plates and cotransfected with equal amounts of plasmids encoding WT or mutant nsp5 along with IFN-β-Luc (100 ng/well) and pRL-TK (20 ng/well) plasmids. One day (24 h) after transfection, HEK-293T cells were infected with SEV for 16 h. The SEV-treated cells were lysed with 5× passive buffer for 30 min. Luciferase and Renilla luciferase activities were detected using a dual-luciferase reporter assay (Promega, Madison, WI, USA).
Western blotting.
HEK-293T cells were grown in six-well plates and treated with lysis buffer (Beyotime, Shanghai, China) 28 h after transfection. After incubating with lysis buffer for 45 min, the samples were mixed with 5× loading buffer, boiled for 10 min, and then separated by SDS-PAGE. Subsequently, proteins were transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA) and blocked with Tris-buffered saline containing 0.1% Tween 20 and 10% skim milk for 3 to 4 h. Anti-HA (1:2,000; MBL, Tokyo, Japan), anti-FLAG (1:2,000; Macgene, Beijing, China), and anti-β-actin antibodies (Beyotime), along with corresponding secondary antibodies, were used to assess the expression levels of the respective proteins.
Protein expression and purification.
To generate authentic viral proteases, the four amino acids A, V, L, and Q were added to the N terminus of nsp5, and the eight amino acids GPH6 were added to the C terminus. The resulting sequences were cloned into the pGEX-6p-1 vector to generate expression plasmids, which were then used to transform E. coli BL21(DE3) cells. The transformed cells were cultured in LB medium containing 100 μg/mL ampicillin at 37°C. When the optical density at 600 nm reached 0.8, 0.5 mM isopropyl-d-thiogalactopyranoside was added to induce protein expression. The cultures were incubated at 16°C for another 10 h. Purification of SARS-CoV-2 nsp5 (WT and S46A) and SARS-CoV nsp5 (WT and A46S) was carried out as described previously (20). Sufficient protein was obtained and stored at −80°C for detection of enzyme activity.
FRET assays.
To carry out enzyme activity assays, a fluorescent substrate (Dabcyl-TSAVLQ↓SGFRKM-E-Edans) derived from the N-terminal autocleavage site of SARS-CoV-2 nsp5 in the polyprotein was synthesized by GenScript. The FRET assays were conducted by mixing 50 nM purified SARS-CoV-2 nsp5 (WT or S46A) or SARS-CoV nsp5 (WT or A46S) proteins with 40 μM substrate in 50 mM Tris-HCl, pH 7.3, containing 1 mM EDTA. Fluorescence intensity was measured every 5 min for 1 h with excitation at 360 nm and emission at 460 nm, using a fluorescence spectrophotometer.
IFN bioassay and flow cytometry.
To evaluate the influence of SARS-CoV-2/SARS-CoV nsp5 and their mutants on the amount of SEV-induced IFN production in HEK-293T cells, a recombinant VSV-GFP was used, and the IFN bioassay was performed as described previously (55). The percentage of GFP-positive cells from IFN bioassay was calculated by an LSRFortessa X-20 cell analyzer and analyzed by FlowJo v10.
Sequence alignment.
Amino acid sequences of SARS-CoV-2 nsp5 and SARS-CoV nsp5 were collected. Multiple-sequence alignment was performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Statistical analysis.
All experiments in our study were conducted in triplicate. Differences between groups were assessed using the Student's t test. P values of <0.05 were considered statistically significant.
AKNOWLEDGMENTS
We thank Guiqing Peng for providing plasmids and Zhigao Bu for providing the VSV-GFP recombinant virus. This work was supported by the National Key Research and Development Program of China (2021YFD1801104), the National Natural Science Foundation of China (31730095), and the Emergency Project for COVID-19 from the Department of Science and Technology of Hubei Province (2020FCA003).
Contributor Information
Shaobo Xiao, Email: vet@mail.hzau.edu.cn.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang C, Horby PW, Hayden FG, Gao GF. 2020. A novel coronavirus outbreak of global health concern. Lancet 395:470–473. 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. 2020. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5:536–544. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang F, Deng L, Zhang L, Cai Y, Cheung CW, Xia Z. 2020. Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J Gen Intern Med 35:1545–1549. 10.1007/s11606-020-05762-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gates B. 2020. Responding to Covid-19 - a once-in-a-century pandemic? N Engl J Med 382:1677–1679. 10.1056/NEJMp2003762. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Zhao S, Teng T, Abdalla AE, Zhu W, Xie L, Wang Y, Guo X. 2020. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 12:244. 10.3390/v12020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen KY. 2020. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9:221–236. 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison AG, Lin T, Wang P. 2020. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol 41:1100–1115. 10.1016/j.it.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, Zhu HC, Tong YG, Shi YX, Ni XB, Liao YS, Li WJ, Jiang BG, Wei W, Yuan TT, Zheng K, Cui XM, Li J, Pei GQ, Qiang X, Cheung WY, Li LF, Sun FF, Qin S, Huang JC, Leung GM, Holmes EC, Hu YL, Guan Y, Cao WC. 2020. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583:282–285. 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- 12.Bonilla-Aldana DK, Holguin-Rivera Y, Cortes-Bonilla I, Cardona-Trujillo MC, García-Barco A, Bedoya-Arias HA, Rabaan AA, Sah R, Rodriguez-Morales AJ. 2020. Coronavirus infections reported by ProMED, February 2000-January 2020. Travel Med Infect Dis 35:101575. 10.1016/j.tmaid.2020.101575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziebuhr J. 2005. The coronavirus replicase. Curr Top Microbiol Immunol 287:57–94. 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Liu Q, Guo D. 2020. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423. 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, et al. 2003. The genome sequence of the SARS-associated coronavirus. Science 300:1399–1404. 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 16.Woo PC, Huang Y, Lau SK, Yuen KY. 2010. Coronavirus genomics and bioinformatics analysis. Viruses 2:1804–1820. 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilgenfeld R. 2014. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J 281:4085–4096. 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wojdyla JA, Manolaridis I, van Kasteren PB, Kikkert M, Snijder EJ, Gorbalenya AE, Tucker PA. 2010. Papain-like protease 1 from transmissible gastroenteritis virus: crystal structure and enzymatic activity toward viral and cellular substrates. J Virol 84:10063–10073. 10.1128/JVI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. 2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA 103:5717–5722. 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. 2020. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368:409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hegyi A, Ziebuhr J. 2002. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol 83:595–599. 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 22.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. 2020. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 23.Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. 2016. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 59:6595–6628. 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, Li C, Li Y, Bai F, Wang H, Cheng X, Cen X, Hu S, Yang X, Wang J, Liu X, Xiao G, Jiang H, Rao Z, Zhang LK, Xu Y, Yang H, Liu H. 2020. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368:1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z. 2003. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA 100:13190–13195. 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muramatsu T, Takemoto C, Kim YT, Wang H, Nishii W, Terada T, Shirouzu M, Yokoyama S. 2016. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc Natl Acad Sci USA 113:12997–13002. 10.1073/pnas.1601327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu MF, Kuo CJ, Chang KT, Chang HC, Chou CC, Ko TP, Shr HL, Chang GG, Wang AH, Liang PH. 2005. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem 280:31257–31266. 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu X, Chen J, Tian L, Zhou Y, Xu S, Long S, Wang D, Fang L, Xiao S. 2020. Porcine deltacoronavirus nsp5 cleaves DCP1A to decrease its antiviral activity. J Virol 94:e02162-19. 10.1128/JVI.02162-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, Fang L, Shi Y, Zhang H, Gao L, Peng G, Chen H, Li K, Xiao S. 2016. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J Virol 90:2090–2101. 10.1128/JVI.02514-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Fang L, Dang W, Yang Y, Chen J, Xu Y, Foda MF, Xiao S. 2017. Porcine deltacoronavirus nsp5 inhibits interferon-β production through the cleavage of NEMO. Virology 502:33–38. 10.1016/j.virol.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, Menachery VD, Rajsbaum R, Shi PY. 2020. Evasion of type 1 interferon by SARS-CoV-2. Cell Rep 33:108234. 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, Kratzat H, Hayn M, Mackens-Kiani T, Cheng J, Straub JH, Stürzel CM, Fröhlich T, Berninghausen O, Becker T, Kirchhoff F, Sparrer KMJ, Beckmann R. 2020. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369:1249–1255. 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, Cai JP, Jin DY, To KK, Chan JF, Yuen KY, Kok KH. 2020. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect 9:1418–1428. 10.1080/22221751.2020.1780953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chuck CP, Chow HF, Wan DC, Wong KB. 2011. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS One 6:e27228. 10.1371/journal.pone.0027228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Wang D, Sun Z, Gao L, Zhu X, Guo J, Xu S, Fang L, Li K, Xiao S. 2019. Arterivirus nsp4 antagonizes interferon beta production by proteolytically cleaving NEMO at multiple sites. J Virol 93:e00385-19. 10.1128/JVI.00385-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matayoshi ED, Wang GT, Krafft GA, Erickson J. 1990. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science 247:954–958. 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 37.Chen S, Tian J, Li Z, Kang H, Zhang J, Huang J, Yin H, Hu X, Qu L. 2019. Feline infectious peritonitis virus Nsp5 inhibits type 1 interferon production by cleaving NEMO at multiple sites. Viruses 12:43. 10.3390/v12010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrette-Ng IH, Ng KK, Mark BL, Van Aken D, Cherney MM, Garen C, Kolodenko Y, Gorbalenya AE, Snijder EJ, James MN. 2002. Structure of arterivirus nsp4. The smallest chymotrypsin-like proteinase with an alpha/beta C-terminal extension and alternate conformations of the oxyanion hole. J Biol Chem 277:39960–39966. 10.1074/jbc.M206978200. [DOI] [PubMed] [Google Scholar]
- 39.Tian X, Lu G, Gao F, Peng H, Feng Y, Ma G, Bartlam M, Tian K, Yan J, Hilgenfeld R, Gao GF. 2009. Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/nsp4) of porcine reproductive and respiratory syndrome virus (PRRSV). J Mol Biol 392:977–993. 10.1016/j.jmb.2009.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen E, Koopmans M, Go U, Hamer DH, Petrosillo N, Castelli F, Storgaard M, Al Khalili S, Simonsen L. 2020. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis 20:e238–e244. 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frieman M, Heise M, Baric R. 2008. SARS coronavirus and innate immunity. Virus Res 133:101–112. 10.1016/j.virusres.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fung TS, Liu DX. 2019. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol 73:529–557. 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 43.Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden van Noort GJ, Ovaa H, Müller S, Knobeloch KP, Rajalingam K, Schulman BA, Cinatl J, Hummer G, Ciesek S, Dikic I. 2020. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587:657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, Sauter D, Gifford RJ, Nakagawa S, Sato K, USFQ-COVID19 Consortium. 2020. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep 32:108185. 10.1016/j.celrep.2020.108185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, Zheng YX, Wang XN, Qi H, Zhang J, Wang PH, Han ZG, Tao SC. 2020. SARS-CoV-2 Orf9b suppresses type 1 interferon responses by targeting TOM70. Cell Mol Immunol 17:998–1000. 10.1038/s41423-020-0514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O'Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, et al. 2020. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583:459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. 2020. Activation and evasion of type 1 interferon responses by SARS-CoV-2. Nat Commun 11:3810. 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y, Shuai H, Yang D, Hu B, Huang X, Zhang X, Cai JP, Zhou J, Yuan S, Kok KH, To KK, Chan IH, Zhang AJ, Sit KY, Au WK, Yuen KY. 2020. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis 71:1400–1409. 10.1093/cid/ciaa410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045.e9. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masters PS. 2006. The molecular biology of coronaviruses. Adv Virus Res 66:193–292. 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kneller DW, Phillips G, O'Neill HM, Jedrzejczak R, Stols L, Langan P, Joachimiak A, Coates L, Kovalevsky A. 2020. Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat Commun 11:3202. 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffin JWD. 2020. SARS-CoV and SARS-CoV-2 main protease residue interaction networks change when bound to inhibitor N3. J Struct Biol 211:107575. 10.1016/j.jsb.2020.107575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xue X, Yang H, Shen W, Zhao Q, Li J, Yang K, Chen C, Jin Y, Bartlam M, Rao Z. 2007. Production of authentic SARS-CoV M(pro) with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J Mol Biol 366:965–975. 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang D, Fang L, Wei D, Zhang H, Luo R, Chen H, Li K, Xiao S. 2014. Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J Virol 88:10252–10258. 10.1128/JVI.00869-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cárdenas WB, Loo YM, Gale M, Jr., Hartman AL, Kimberlin CR, Martínez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]