

ABSTRACT
Retroviruses are widely distributed in all vertebrates, as are their endogenous forms, endogenous retroviruses (ERV), which serve as “fossil” evidence to trace the ancient origins and history of virus-host interactions over millions of years. The retroviral envelope (Env) plays a significant role in host range determination, but major information on their genetic diversification and evolution in anamniotes is lacking. Here, by incorporating multiple-round in silico similarity search and phylogenomic analysis, more than 30,000 copies of ERV lineages with gamma-type Env (GTE), covalently associated Env, were discovered by searching against all fish and amphibian genomes and transcriptomic assemblies, but no beta-type Env (BTE), noncovalently associated Env, was found. Furthermore, a nine-type classification system of anamniote GTE was proposed by combining phylogenetic and domain/motif analyses. The elastic genomic organization and overall phylogenetic incongruence between anamniotic Env and its neighboring polymerase (Pol) implied that early retroviral diversification in anamniotic vertebrates was facilitated by frequent recombination. At last, host cellular opioid growth factor receptor (OGFr) gene capturing by anamniotic ERVs with GTE was reported for the first time. Overall, our findings overturn traditional Pol genotyping and reveal a complex evolutionary history of anamniotic retroviruses inferred by Env evolution.
IMPORTANCE Although the retroviral envelope (Env) protein in amniotes has been well studied, its evolutionary history in anamniotic vertebrates is ambiguous. By analyzing more than 30,000 copies of ERV lineages with gamma-type Env (GTE) in anamniotes, several important evolutionary features were identified. First, GTE was found to be widely distributed among different amphibians and fish. Second, nine types of GTE were discovered and defined, revealing their great genetic diversity. Third, the incongruence between the Env and Pol phylogenies suggested that frequent recombination shaped the early evolution of anamniote retroviruses. Fourth, an ancient horizontal gene transfer event was discovered from anamniotes to ERVs with GTE. These findings reveal a complex evolution pattern for retroviral Env in anamniotes.
KEYWORDS: gamma-type envelope, endogenous retrovirus, expressed retroviruses, evolution, recombination, opioid growth factor receptor, anamniote
INTRODUCTION
Retroviruses (RVs) (family Retroviridae), which were first discovered more than a hundred years ago (1), are medically and economically important because some are associated with severe infectious diseases, cancer, and immunodeficiency (2–4). RVs occasionally integrate into the germ line of the host and become endogenous retroviruses (ERVs), which serve as historical genomic “fossils” for investigating viral origins and the history of virus-host interactions over millions of years (5, 6). Due to the increasing number of sequenced vertebrate genomes, numerous ERVs have been uncovered, and some of these ERVs are distinct from the current exogenous RVs, as reflected by their genomic structure and sequence similarity, which indicates the ancient origin and long-term evolution of RVs (5).
Typical RVs contain three major protein-coding genes: group-specific antigen (gag), polymerase (pol), and envelope (env) genes. For the phylogenetic reconstruction of retroviral lineages, well-conserved Pol protein sequences, particularly the reverse transcriptase (RT) domain, are typically used, which allows the alignment of multiple ERVs or exogenous RVs from various vertebrates and can thus be used to infer the deep evolutionary history of RVs as a whole (7). Based on the RT phylogeny, ERVs can be classified into three broad classes: (i) class I, which is related to epsilonretroviruses and gammaretroviruses (GVs); (ii) class II, which is related to deltaretroviruses, lentiretroviruses, betaretroviruses, and alpharetroviruses (AVs); and (iii) class III, which is related to spumaretroviruses (8). Although an analysis based solely on Pol or RT has a distinct advantage, it also has a clear disadvantage in that it ignores many fine distinctions in other regions (such as Env) of genomes among viruses (9–11).
env, the encoded protein of which mediates entry into the host cell and thus determines the host range (11), exhibits markedly higher variability than pol. This gene encodes two subunits: the surface subunit (SU) and the transmembrane fusion subunit (TM). While SU is the most variable region of the genome, TM is relatively well conserved across many RVs (11–13). Env can be divided into two major types: covalently associated (gamma-type Env [GTE]) and noncovalently associated (beta-type Env [BTE]) (12). These types can be readily distinguished by sequence similarity and the presence or absence of two regions: the immunosuppressive domain (ISD) and the CX6CC motif. A GTE variant from an alpharetrovirus (GTE-AV), which has an internal fusion peptide (FP) flanked by a pair of cysteines, has also been well characterized (11). BTEs are typically harbored by betaretroviruses and lentiviruses and can be found only in mammals (12). In contrast, GTEs are harbored by gammaretroviruses, deltaretroviruses, alpharetroviruses, and betaretroviruses (recombinant RVs, including Mason-Pfizer monkey virus [MPMV] and simian retrovirus [SRV]) and can be found in all five major classes of vertebrates (fishes, amphibians, reptiles, birds, and mammals) (9–11, 14, 15).
Although a substantial number of RVs-GTE (retroviruses with a gamma-type Env) have been identified across the evolutionary history of vertebrates (14, 16–18), these retroviruses in anamniote vertebrates remain poorly understood. Hence, in this study, by incorporating genomic and transcriptomic data and performing comprehensive phylogenomic analyses, we were able to identify hundreds of RV-GTE lineages, which significantly increased the known set of RVs-GTE in anamniote vertebrates. Most importantly, by combining phylogenetic and genomic structure characterization, we uncovered the structural complexity and phylogenetic diversity of GTEs and redefined the classification of Env. Additionally, some unique evolutionary features were revealed, which significantly increased our understanding of the diversity and evolution of RVs.
RESULTS
Identification of ERVs-GTE, ERVs-BTE, expressed RVs-GTE, and expressed RVs-BTE in amphibians and fish.
To systematically identify ERVs-GTE (endogenous retroviruses with gamma-type Env) and ERVs-BTE (endogenous retroviruses with beta-type Env), all 974 available fish and 19 amphibian genomes (see Table S1 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]) were screened using a combined multiple-round similarity searching approach (Fig. 1). First, host genomes were screened to detect ERVs-GTE and ERVs-BTE using tBLASTn, and all Env sequences of representative RVs-GTE and RVs-BTE (see Table S2 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]), which included the vast majority of exogenous and endogenous RVs-GTE and RVs-BTE, were used as queries. Second, the significant hits were concatenated based on the host genomic location and alignment positions with reference RV proteins. These concatenated proteins were then further confirmed using phylogeny, and only sequences that clustered with RVs-GTE or RVs-BTE were included. Third, the same genomes were subjected to a second round of screening by tBLASTn using the confirmed concatenated Env protein sequences. Fourth, the significant hits that were also confirmed by phylogeny were extended to identify the full-length genome and classified into different lineages based on sequence similarity. Finally, the full-length viral genomes of each lineage were used as queries in a BLASTn search against the genome to determine the copies of each lineage (detailed in Materials and Methods). This analysis led to the discovery of 37,552 copies of ERV lineages with GTE in 37 vertebrate species, including 13 amphibians, 18 ray-finned fish, and 6 cartilaginous fish, and no ERVs-BTE were found, which was consistent with the results of previous research showing that noncovalently associated Env (beta-type Env) could be found only in mammals (12) (see Data Set S1 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). These retroviral sequences were named in accordance with a previous nomenclature proposed for ERVs (8).
FIG 1.

Schematic diagram of the pipeline to identify ERVs-GTE and ERVs-BTE. First, host genomes were screened to detect ERVs-GTE and ERVs-BTE using tBLASTn, and all reference GTE and BTE sequences were used as queries. Second, the significant hits were concatenated based on the host genomic location and alignment positions with reference proteins. Then, these concatenated proteins were further confirmed using phylogeny, and only sequences that clustered with GTE or BTE were included. Third, the same genomes were subjected to a second round of screening by tBLASTn using the confirmed concatenated protein sequences of Env. Fourth, the significant hits which were also confirmed by phylogeny were extended to identify the full-length genome and classified into different lineages based on sequence similarity. Finally, the full-length viral genomes of each lineage were used as queries for a BLASTn search against the genome to determine the copies of each lineage.
To better understand the expression of potential RVs-GTE, we also searched all available 378 fish and 61 amphibian transcriptome sequencing (RNA-seq) data sets in the transcriptome sequence assembly (TSA) database (see Table S1 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]). Notably, we found 1,093 retroviral Env contigs in 145 vertebrate species, including 29 amphibians, 113 ray-finned fish, and 3 cartilaginous fish (see Data Set S1 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). These viral sequences were named in accordance with a previous nomenclature proposed for expressed RVs (expRVs) (19). However, 133 of 145 species harboring such expRVs did not have any genomic data support. We still found that 5 amphibians (Rana catesbeiana, Rhinatrema bivittatum, Microcaecilia unicolor, Pyxicephalus adspersus, and Rhinella marina) and 7 ray-finned fish (Astyanax mexicanus, Larimichthys crocea, Oncorhynchus kisutch, Oncorhynchus tshawytscha, Salmo trutta, Salvelinus alpinus, and Seriola dumerili) harbored both endogenous and expressed forms of retroviral elements, and some of these shared high similarity (>95%) among the DNA and RNA copies in each host, which indicated the potential ability to express such viral elements. In addition, we found 4 copies of Env RNA contigs in R. catesbeiana, 1 copy in P. adspersus, 10 copies in A. mexicanus, 2 copies in O. tshawytscha, 2 copies in S. trutta, and 10 copies in S. alpinus; these copies were distantly related to the DNA copies that shared less than 50% similarity with each other, which implied the potential existence of exogenous forms of such viruses because most of these (21/29) encoded intact open reading frames (ORFs).
Characterization and classification of novel GTEs.
To better understand the relationship among all GTEs, an Env protein phylogenetic tree was generated using the reference GTEs and a representative GTE in each lineage, which is the consensus or the longest and most complete GTE (Fig. 2A; see also Fig. S1 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2] and Data Set S2 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). The phylogeny revealed the diverse evolutionary status of GTEs because they formed at least 9 major clades, including 2 well-defined GTE-C.5-AV and GTE-C.9-GV clades. Most (90.1%) of our newly identified GTEs were in the well-supported Env-C.1 clade, which was distantly related to the well-defined AV and GV clades, whereas other identified GTEs were divided into 6 novel monophyletic groups, indicating their different evolutionary statuses. Additionally, gamma-type Env previously identified in ray-finned fish (16) were clustered in Env-C.2 with newly identified GTEs in cartilaginous fish.
FIG 2.
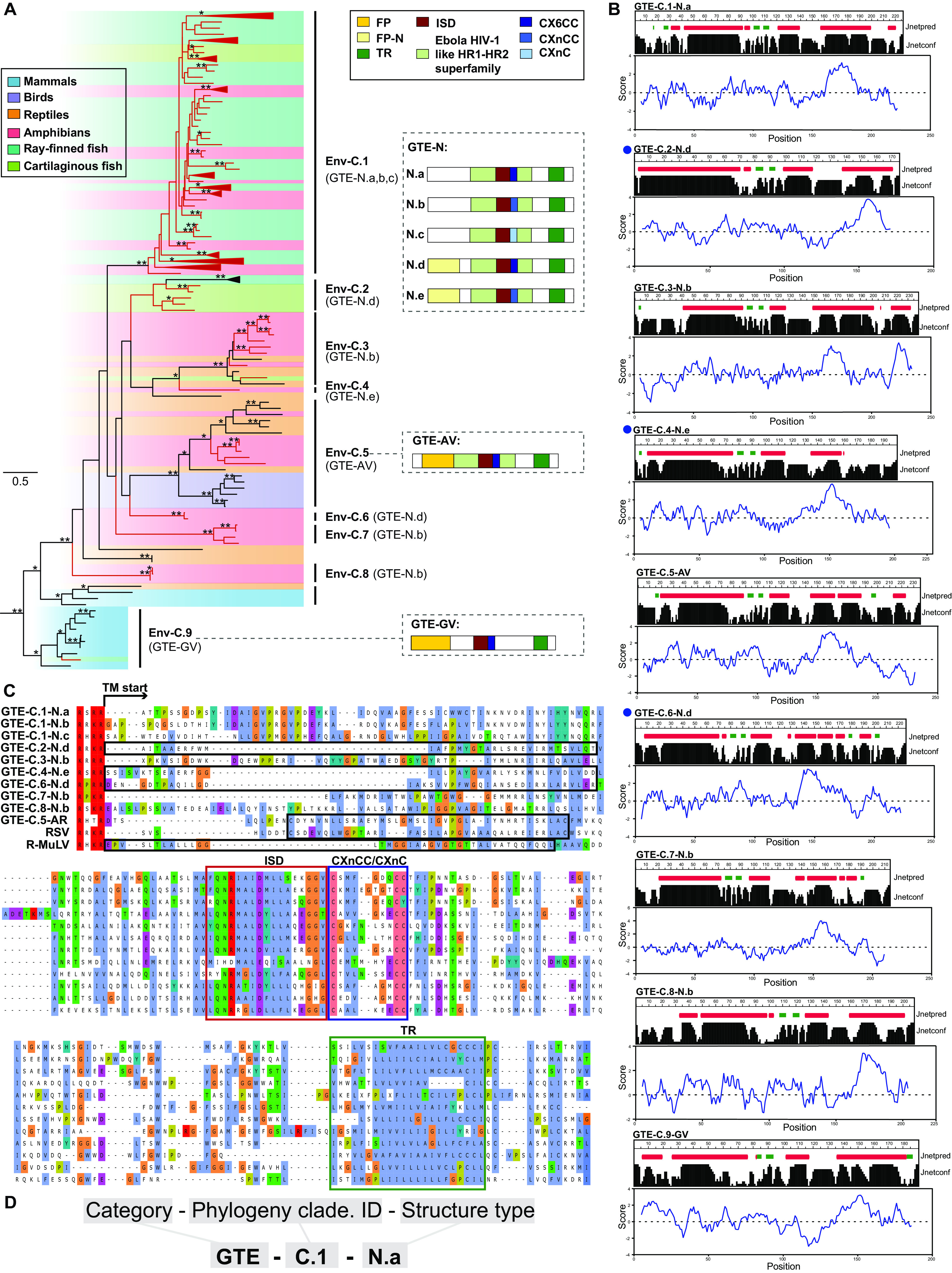
Characterization and classification of novel GTEs. (A) Phylogeny of GTEs. The left tree was inferred using amino acid sequences of the conserved region of transmembrane subunit (TM). The tree is rooted in deltaretroviruses, which are not shown in the tree. The newly identified ERVs-GTE and expRVs-GTE are labeled in red. The host information of each retrovirus is indicated using a shaded box. Bootstrap values lower than 65% are not shown. Single asterisks indicate values higher than 65%, while double asterisks indicate values higher than 80%. The scale bar indicates the number of amino acid changes per site. The typical GTE structures are shown on the right. The letters in parentheses beside the clade name indicate which GTE is found. (B) Prediction results of secondary structure and hydropathicity scores of amino acids in TM. Prediction results of all GTE types are shown here. The secondary structure is shown at the top, and the corresponding amino acid hydrophilic index is shown at the bottom. Jnetpred is secondary structure prediction for TM. The alpha helix is labeled with a red box, and the beta fold is labeled with a green box. Jnetconf shows the reliability of prediction accuracy, range from 0 to 9, and larger shapes mean more robustness. The blue curve shows the calculated hydrophilic scores of amino acids. A positive number means the protein is hydrophobic and vice versa. Sequences harboring putative FP are indicated by blue solid circles. (C) Alignment of FP, ISD, the CX6CC motif, and the flanking conserved domain of representative ERVs-GTE and other exogenous viruses. FP is labeled with black boxes. (D) The nomenclature for GTEs. GTE-N, the novel gamma-type Env; FP, fusion peptide; ISD, immunosuppressive domain; TR, transmembrane region. CX6CC represents the three-cysteine motif of GTEs. RSV, Rous sarcoma virus; R-MuLV, Rauscher murine leukemia virus.
However, the phylogeny did not show a strong classification protocol for GTEs. Therefore, we then compared the distributions of motifs and domains in different GTEs. Five major domains/motifs were found in GTEs: fusion peptide (FP), ISD, CX6CC, heptad repeat, and transmembrane region (TR). By checking the structure of all GTEs, we found that ISDs and TRs were present in all GTEs, and heptad repeats could be identified in all GTEs except GTE-C.9-GV (Fig. 2A). CX6CC had 3 subtypes, including CX6CC, CXnCC, and CXnC, which were classified according to the functional residue distribution. The first two cysteines in the CX6CC motif participate in the formation of an intramolecular loop in the TM ectodomain, whereas the third cysteine is needed for formation of a covalent bond between the TM and SU domains (20). Most (79.6%) GTEs contained CX6CC, whereas others contained CXnCC and CXnC. However, the newly identified GTEs showed no similarity to GTE-C.5-AV and GTE-C.9-GV in the N terminus of the TM, where the FP is usually located. Because the FP mostly contains an alpha-helical or beta structure and is hydrophobic (21), the secondary structure and hydrophobicity of the TM were used to predict the presence of potential novel FPs. This analysis led to the identification of 3 novel FPs that showed no sequence similarity to other viral FPs (Fig. 2B). By taking all of these findings into consideration, the structure of GTEs can be mainly classified into 7 types, as shown in Fig. 2A. To better illustrate the difference among all the GTEs, alignments of all major groups of GTEs are shown, which reconfirmed the conservation of the ISD, CX6CC, and TR and the diversity of FPs (Fig. 2C; see also Data Set S5 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). GTEs cannot be classified by phylogeny alone. Here, by combining the phylogeny and TM structure, we proposed a nomenclature for GTEs (Fig. 2D).
Extensive recombination of ERVs-GTE.
To further elucidate the relationship among ERVs-GTE, expRVs-GTE, and those from other RVs, a phylogenetic analysis based on Pol (>600 amino acids [aa]) was performed (see Fig. S2 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2] and Data Set S3 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). This phylogeny revealed that our viral elements could be divided into three different groups: (i) class I epsilon-related viruses, which can be divided into three clades, namely, Pol-LE.1, Pol-LE.2, and epsilon; (ii) class I gamma-related viruses, which can also be divided into three clades, namely, Pol-LG.1, Pol-LGE.1, and Gamma; and (iii) class III SnRV (snakehead retrovirus)-related viruses. The pol and env phylogenies of selected full-length ERVs-GTE (for selection details, see Materials and Methods) were then compared, and the results indicated that recombination was widespread and frequently occurred among different RVs over long evolutionary timescales (Fig. 3A).
FIG 3.
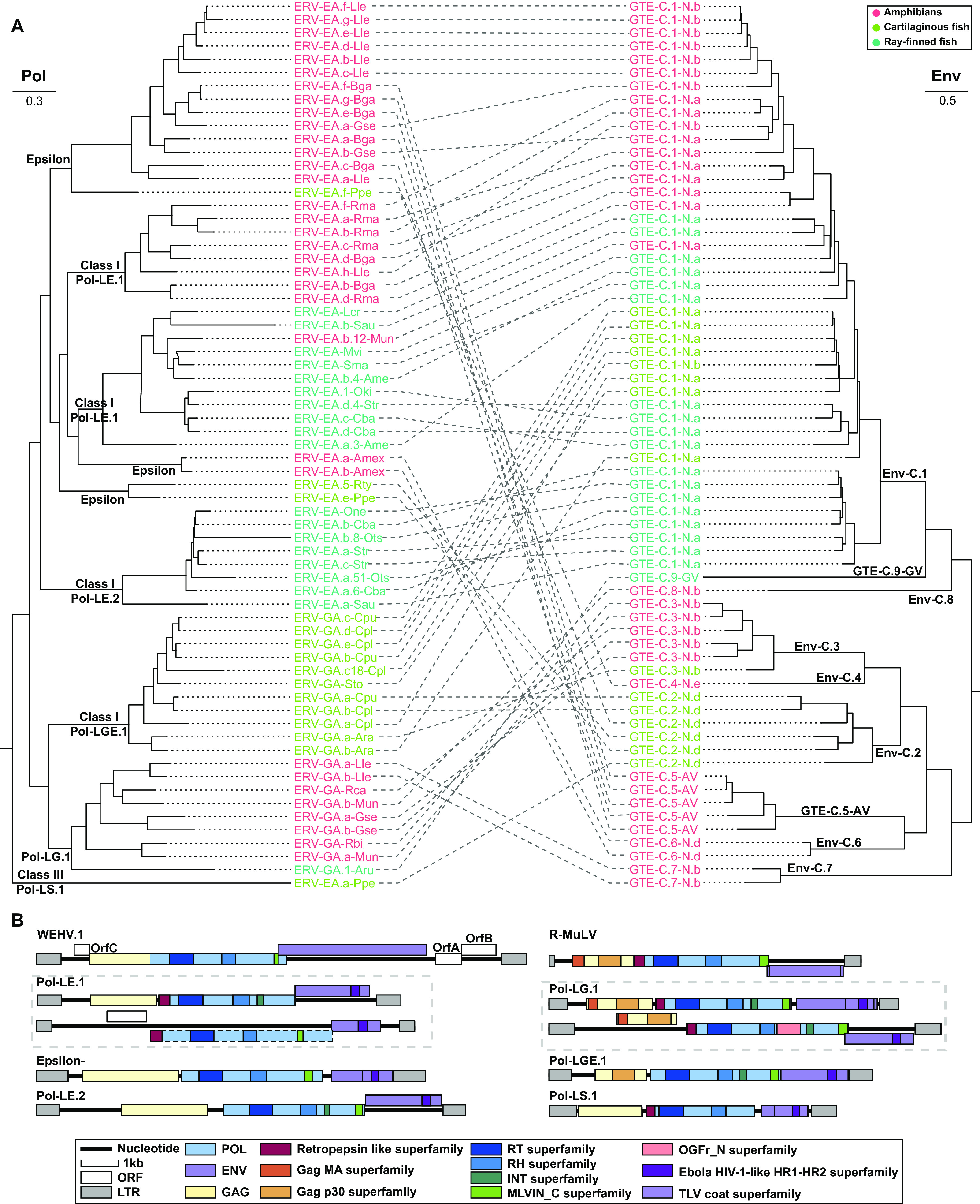
Genetic exchange of ERVs-GTE. (A) Phylogenetic incongruence between Pol and Env of ERVs-GTE. The relationship between the two phylogenies is displayed to maximize topological congruence. The classification of Pol and Env is shown at each node. (B) The comparison of genomic structure of exogenous retroviruses and representative ERVs-GTE. The walleye epidermal hyperplasia virus (WEHV) (epsilonretrovirus) and R-MuLV (gammaretrovirus) genomes are shown on the top. The predicted domains or regions that encode conserved proteins are labeled with colored boxes. The dashed boxes indicate putative open reading frames (ORFs) containing stop codons or indels.
The genomic structure also reflected recombination among viruses. Epsilonretroviruses are typical complex RVs that carry accessory genes, and their Env proteins are not GTEs or BTEs (12). However, our epsilon-related class I ERVs contained GTEs instead of epsilon Envs and did not carry any accessory gene, indicating potential recombination between epsilonretrovirus and ancient RVs-GTE (Fig. 3B). In contrast, our gamma-related class I ERVs were much more highly conserved because they exhibited a typical GV structure and contained all the conserved domains/motifs carried by exogenous RVs (e.g., Rauscher murine leukemia virus [R-MuLV]).
Conserved domain shared between GTEs and other divergent viruses.
Filoviruses {single-stranded RNA [ssRNA(−)]}, particularly Ebola virus, were previously discovered to share a similar ISD and CX6CC motif with GTEs (22). To search for other possible gene sharing events among different viruses, we used all the discovered novel GTEs to perform a search against the NCBI nr database and surprisingly found that F-env on HP35 of chelonid alphaherpesvirus 5 (a DNA virus) (23) shared 26.68% similarity and 92% coverage with ERV-GA.a-Ara and 31.75% similarity and 68% coverage with ERV-GA.a-Cpu. By searching against the conserved domain database (CDD), we found that this protein contained two conserved domains: (i) the Ebola HIV-1-like HR1-HR2 superfamily domain (cl02885), a typical retroviral domain that plays a key role in the dynamic rearrangement of the trimer during the process of fusion (11, 16), and (ii) the TLV coat superfamily domain (cl27694) (Fig. 4A). An alignment comparison indicated that chelonid alphaherpesvirus 5 also contained homologous ISD, CX6CC motif, TR, and flanking conserved regions with lengths of 164 aa (Fig. 4B and see also Data Set S6 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]) but showed no significant similarity in other regions, including the FP. However, other herpesviruses were also screened, and we found that they did not contain such domains and motifs. This finding indicated that the transfer of ISD, CX6CC, and TR could be a one-off event in reptiles.
FIG 4.
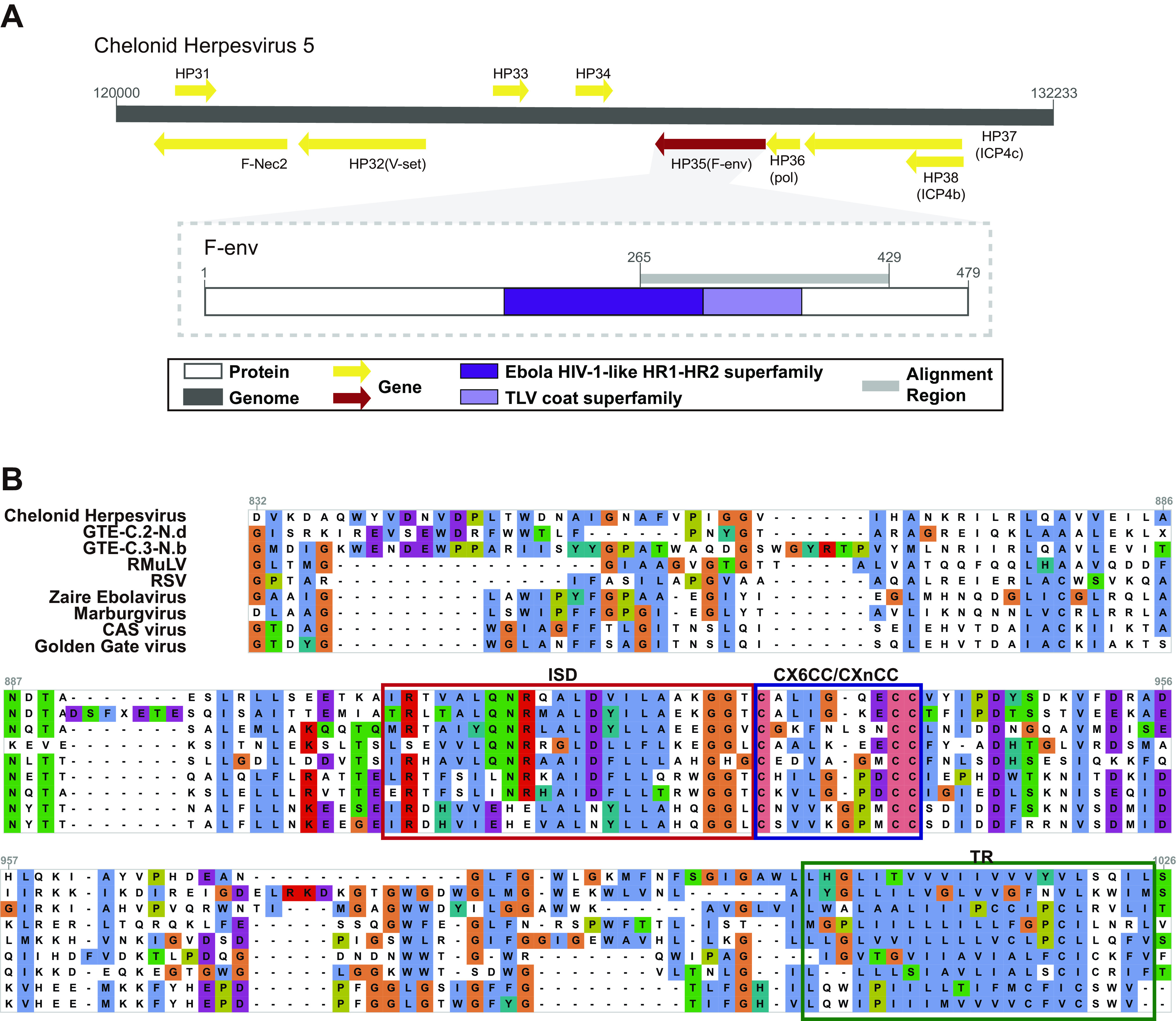
Conserved domain sharing among divergent viruses. (A) Partial genomic map of chelonid herpesvirus and genomic structure of F-env in chelonid herpesvirus. The predicted open reading frames (ORFs) or regions that encode conserved proteins are labeled with colored arrows or boxes. (B) Alignment of FP, ISD, the CX6CC motif, and the flanking conserved domain of chelonid herpesvirus, ERVs-GTE which are most homologous to chelonid herpesvirus, and other exogenous viruses.
The N terminus domain of the OGFr gene was captured by RVs-GTE in amphibians.
The opioid growth factor receptor (OGFr) gene, which plays an important role in the regulation of cell growth and embryonic development, is a typical animal cellular gene (24). Unexpectedly, by using CD-Search, we found that 51.2% of ERV-GA.a-Lle, 41.2% of ERV-GA.b-Lle, 23.1% of expRVssi-GTE, and 25% of expRVlbo-GTE sequences harbored the OGFr_N terminus domain, which was exclusively located in opioid receptors (24, 25), and all of these encoded the OGFr domain located between the RH superfamily and the INT superfamily within their Pol gene ORF (Fig. 5B). Besides, their hosts all belong to Megophryidae. By checking the structure of viral OGFrs, we found that they contained only exons (Fig. 5A and B). To further elucidate the relationship between host OGFr and viral OGFr, a phylogenetic tree of all related amphibians and viral OGFr_N terminus domain, which was the most conserved part, was constructed (Fig. 5C). This tree revealed that viral OGFrs clustered together and formed a monophyletic clade, which was distant from the OGFr of their hosts. Also worth noting was that the nucleotide identity of whole genome among ERV-GA-Lle, expRVssi, and expRVlbo is 79.2% to 86.3% (ERV-GA.a-Lle versus expRVlbo, 86.3%; ERV-GA.b-Lle versus expRVssi, 79.2% to 80.2%), which further indicated these OGFr-containing ERVs were homologous.
FIG 5.
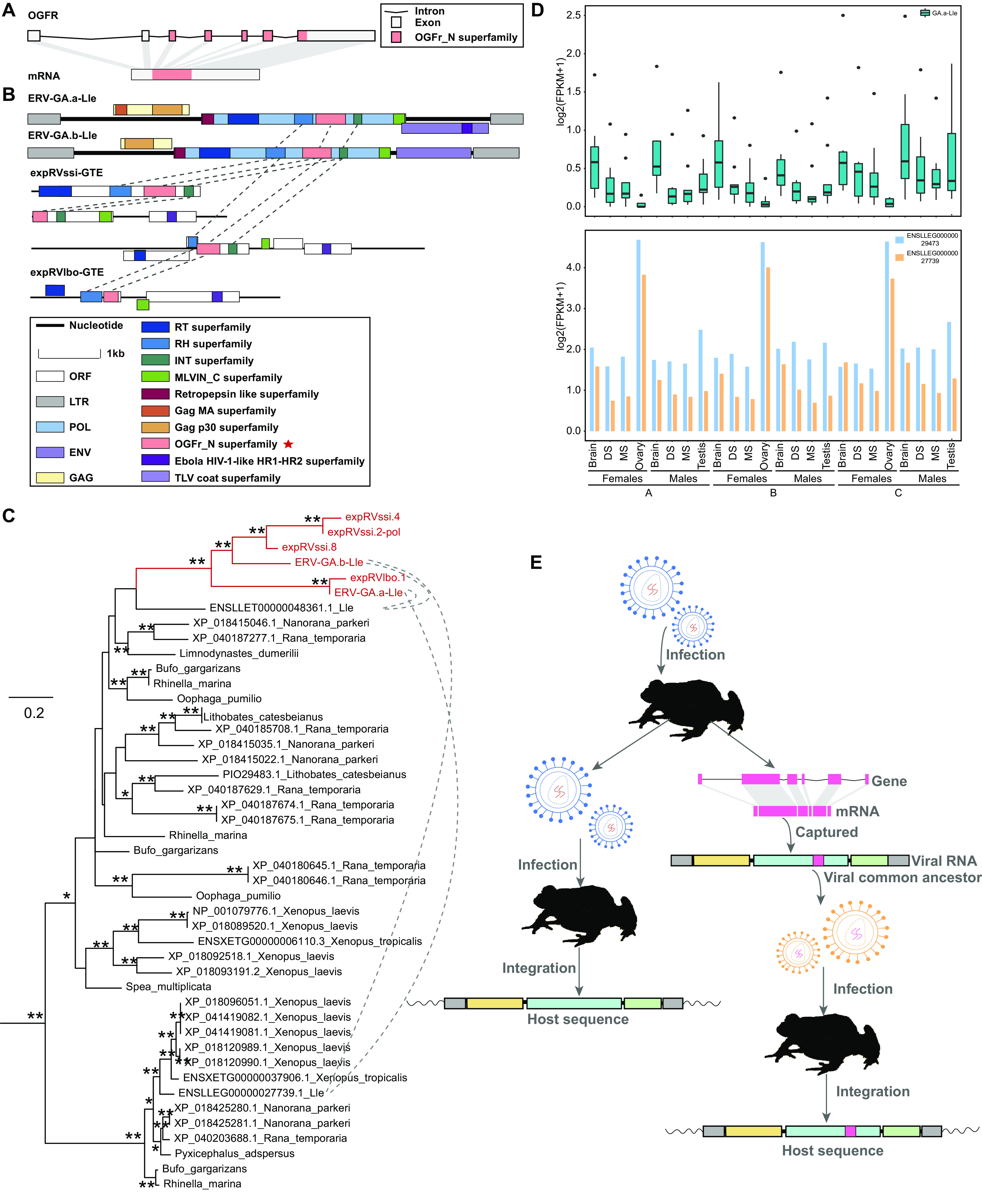
Host gene capturing by RVs-GTE. (A) Diagram of the cellular gene OGFr and its mRNA. (B) Genomic structure of ERVs-GTE and expRVs-GTE containing opioid growth factor receptor gene (OGFr). The predicted domain or region that encodes conserved proteins is labeled with a colored box. A red star indicates indicate that the motif does not have a viral origin. Homologous domains are connected with dashed lines. (C) Phylogeny of viral and frog OGFr_N terminus domain. Viral OGFr_N terminus domains are labeled in red and are linked by dashed lines with OGFr_N terminus domains of their animal hosts. Single and double asterisks denote bootstrap values 65–80% and >80% respectively. (D) The expression level of viral and host OGFrs in L. leishanense. The expression level was evaluated by log2(FPKM + 1). A box plot of the expression of viral OGFr is shown at the top, while a bar chart for the expression of host OGFr is shown at the bottom. For each box plot of viral OGFr, the locus expression values were calculated based on three biological replicates (see Data Set S7 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). The middle black box indicates the median of the expression values. The upper and lower terminal lines of a box represent the 25th and 75th percentiles, respectively. Lines extending vertically from the boxes (whiskers) indicate variability outside the upper and lower percentiles. All the sequenced transcriptomes from L. leishanense were divided into 2 genders and 3 developmental stages: A, subadult period; B, adult breeding period; and C, postbreeding stage. DS, dorsal skin; MS, maxillary skin. (E) Evolution scenario of retrovirus capturing the host OGFr_N terminus domain. The OGFr_N terminus domain is labeled with a pink box.
Furthermore, we also found that some ERVs-GA-Lle contained intact ORFs, which indicated that they had the potential to express. Accordingly, we collected data from all 72 available transcriptomes of Leptobrachium leishanense to further investigate their expression and compared them with the host OGFrs. We found that the minority (9/2,313) of the OGFrs of ERV-GA.a-Lle were expressed, whereas all OGFrs of ERV-GA.b-Lle were not expressed. Additionally, one OGFr of ERV-GA.a-Lle located on chromosome 6 was expressed at a markedly higher level than the others (see Data Set S7 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). Overall, both viral and host OGFrs were expressed in different tissues, and the expression of the latter was higher than that of the former across all tissues (Fig. 5D). Interestingly, the host OGFr was highly expressed in the ovary, whereas the viral OGFr was seldom expressed.
Taking these findings into consideration, OGFr capture more likely occurred during infection with a common exogenous ancestor RV-GTE of these viruses occasionally than during infection with these viruses independently (Fig. 5E).
Estimation of the time of ERV-GTE insertion.
We also used vertical transmission to infer the ERV-GTE insertion time. By comparing the flanking sequences and long terminal repeat (LTR) similarity, we found 46 and 2 orthologous ERV groups in the genera Chiloscyllium and Seriola, respectively (see Table S3 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]). However, no divergence time could be found for the genus Chiloscyllium. We were able to estimate the insertion of only the latter group, which occurred 4.4 to 22.2 million years ago (MYA) (Fig. 6).
FIG 6.
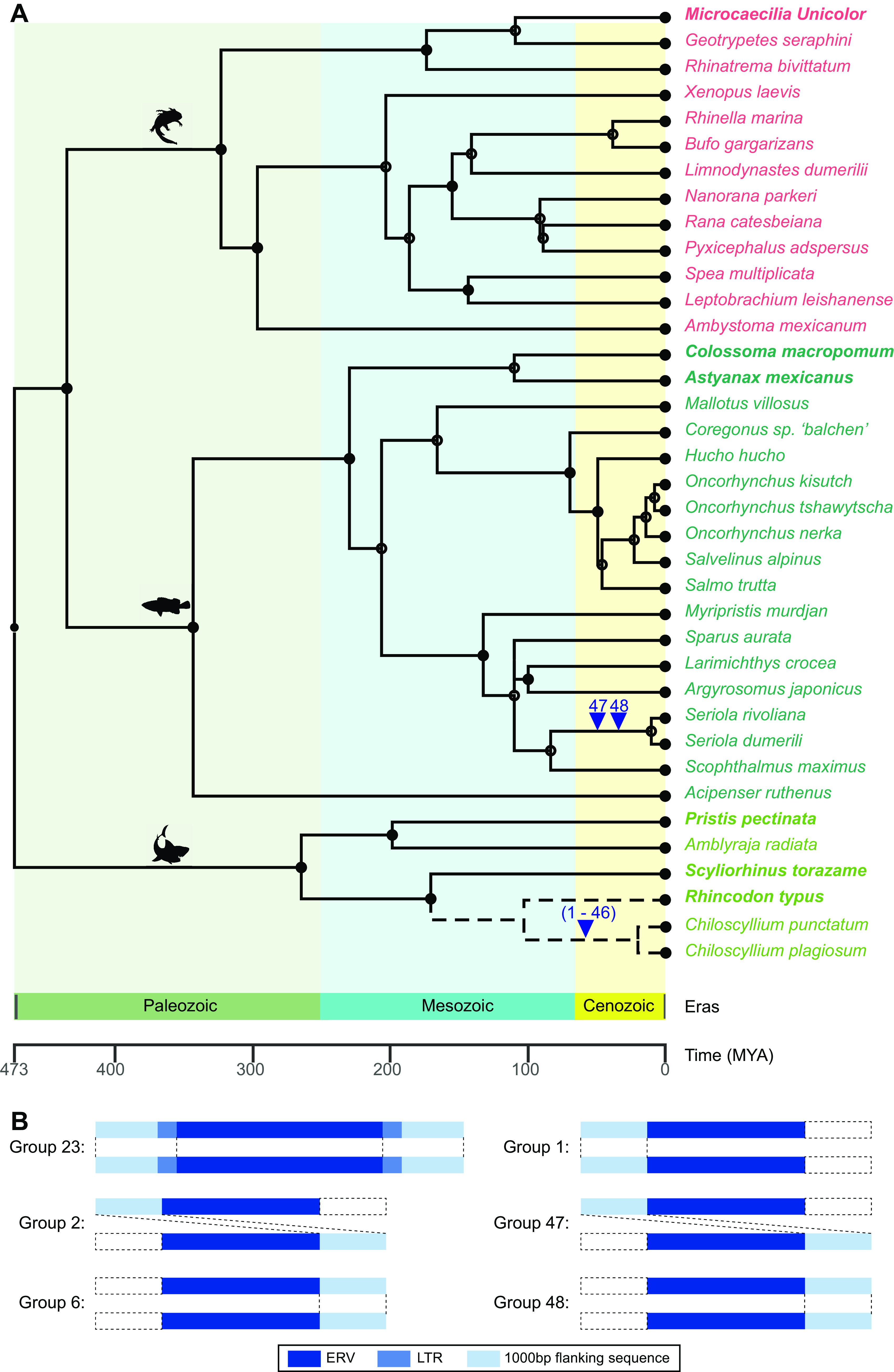
ERV-GTE orthologous insertions in Chiloscyllium and Seriola. (A) ERV-GTE integration events during fish evolution. The species tree was constructed with TimeTree. Different classes are marked with colored fonts. Arrowheads indicate the events of ERV-GTE integrations. Integration events are labeled numerically. Dotted lines indicate only the genetic relationship and do not imply the divergence time of these species. (B) Examples of orthologous insertions for ERVs-GTE. Rectangles from left to right represent 1,000-bp flanking sequence, 5′ LTR, internal genes of an ERV, 3′ LTR, and 1,000-bp flanking sequence, respectively. Dashed boxes indicate missing corresponding regions.
DISCUSSION
In this study, we developed a phylogenomic approach by regarding env genes as seeds for the detection of ERVs and expRVs. Instead of using Pol alone, we traced the evolutionary histories of RVs by combining Pol and Env, which allowed us to observe many fine distinctions and unique patterns during the macroevolution of RVs. We found that GTEs were widely distributed in vertebrates, and many of these showed diversity in well-defined GTE-C.5-AV and GTE-C.9-GV, as reflected by their phylogeny and genomic motif/domain distribution (Fig. 2). On the one hand, the majority of GTEs of anamniotic RVs were found to lie on Env-C.1, which is phylogenetically divergent from the GTEs of amniotic RVs, showing host restriction regarding the transmission of viral progenitors of RVs-GTE. On the other hand, GTEs can be placed into different phylogenetic clades, representing the dynamics of the evolutionary status of GTEs. RVs in different classes of hosts could also share similar GTEs, showing the occasional possibility of cross-class transmission. Furthermore, numerous incongruences were observed in the Env and Pol phylogenies, which implied the occurrence of frequent recombination involving various RVs (Fig. 3A). For example, ERV-SA.a-Ppe harbored Pol of class III and GTE of class I, as has also been observed previously (26). By taking these aspects into consideration, our studies suggest that the Env analyses provide additional information that cannot be obtained via Pol analyses and thus expand our understanding of the complex macroevolution of RVs.
Our studies also suggest the genomic complexity and elasticity of GTEs. First, the motifs/domains in GTEs tend to be highly variable. By combining the second structure and hydrophobicity predictions, 3 novel predicted retroviral FPs were identified in this study, and these showed no similarity to any other viral FP, as determined by BLASTp (Fig. 2). This finding could reflect the possible multiple origins of GTE segments and indicate the limitation of a similarity-based search for viral counterpart identification. Additionally, three major variants of CX6CC have also been characterized based on consensus viral GTE comparisons and the function of each cysteine. However, we admit that further experimental confirmation is needed for such identification. Second, the motif/domain distribution of GTEs is more flexible. GTE mainly has 5 major motifs/domains, including the FP, heptad repeat, ISD, CX6CC, and TR. In this study, we found that the ISD, CX6CC, and TR are relatively conserved and carried by nearly all GTEs, whereas the FP and intact heptad repeats are not necessary counterparts. However, these domains play crucial roles during the process of fusion (27–30). Overall, these findings indicate that GTEs may have their own complex evolutionary history, which involves multiple internal recombination events of different regions and results in the observed diversity.
Both GTEs and BTEs were screened in this study. However, no BTE was found, which may be due to its narrow host range (mammals) (11), whereas a large number of GTEs were found in amphibians and fish in an unbalanced manner (see Data Set S1 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). Most importantly, the majority of amphibians harbored ERVs-GTE, with considerable numbers of copies among different species. Previous research also found that many amphibians harbor other rare ERVs (19, 31). Thus, amphibians appear to be better reservoirs for diversified RVs with different evolutionary routes. However, further study is merited because only 19 amphibian genomes and 61 transcriptomes are currently available.
Horizontal gene transfer from RVs to hosts is widely distributed and well documented. However, this transfer is rare between unrelated viruses with distinct genome types. Homologous GTEs have been found in both arenaviruses and filoviruses (13, 22, 32), and herpesviruses can also acquire RV genes (superantigens [sAgs]) (33). Here, we found that chelonid alphaherpesvirus 5 can also harbor a similar partial GTE that contains intact ISD and CX6CC (Fig. 4 and see also Data Set S6 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]). However, no other herpesviruses were found to contain such domains or motifs. Herpesviruses typically infect hosts indefinitely and move from latent to lytic replication during periods of immunosuppression (33). A host infected with RVs is more likely to be in an immunocompromised state that provides an ideal environment for gene transfer in an activated and replicating herpesvirus. Because reptiles can be infected by both RVs-GTE and herpesviruses, the acquisition of ISD and CX6CC motifs could be an occasional horizontal gene transfer event that occurs during coinfection in reptiles. In addition, the results further reconfirm that gene exchange can occur among divergent viruses (33).
Numerous experimental studies have confirmed that RVs can be coopted by their host (34–36). Here, we reconfirmed that host genes could also be captured by RVs-GTE (37) and fully depicted the evolutionary scenario of such events (Fig. 5E). Four viral lineages in 3 hosts were confirmed to carry OGFrs. In regard to ERV-GA.a-Lle and ERV-GA.b, 2,331/4,552 copies and 21/51 copies contained the intronless OGFr gene, respectively, which indicates that gene capture occasionally occurred in RVs-GTE during infection rather than their endogenization. We also found that most of these had intact env genes, indicating that the proliferation of such viruses was more likely to be caused by repeated infection rather than transposons. It seems that such a cooption event occurred in a common ancestor of a single OGFr-encoding viral lineage. OGFr is thought to be related to cancer because previous studies have indicated that the upregulation of OGFr represses the growth of cancer cells in culture and in nude mice (38). On the one hand, the proliferation of OGFr could help the host repress cancer and could prolong the life span of the host. On the other hand, OGFr might help RVs produce and proliferate within hosts. It appears that the “cooption” of OGFr is a win-win situation for both hosts and viruses. Such events benefit our understanding of the interaction between the host and viruses. However, we also admit that further studies on the functional influence of viral OGFr on animal hosts is warranted.
The transcriptome data provided us with additional resources for discovering novel exogenous and endogenous viruses. Regarding their flaws, on the one hand, major genes (gag, pol, and env) of expRVs-GTE reside in different contigs and cannot be assembled at the genomic level in most cases. Therefore, it is rather difficult to characterize recombination among different viral elements. On the other hand, the efficacy of viral discovery through transcriptomic analysis is highly dependent on the sequencing depth and assembly quality. Thus, the absence of viral contigs in specific species cannot be explained by the noninfectious nature of this virus. Regardless of this fact, the pipeline presented in this work provides a refined procedure for discovering novel expRVs.
In summary, by integrating genomics and transcriptomic data and incorporating a multiple-round screening method and phylogenomic analysis, we revealed the hidden diversity and genomic elasticity of GTEs, which were found to be widely distributed in different vertebrates. Additionally, the incongruence between Env and Pol phylogenies suggested that recombination frequently occurred between different RVs across taxonomic barriers. These findings demonstrate the feasibility and practicability of using Env to perform a phylogenetic analysis and reveal hidden evolutionary features, which indicate the complex macroevolutionary history of RVs.
MATERIALS AND METHODS
Genome and transcriptome screening and identification of ERVs-GTE and expRVs-GTE.
To discover potential targeted viral elements in fish and amphibian genomes, all 974 available fish and 19 amphibian genomes (see Table S1 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]) were first screened using the tBLASTn algorithm (39), and the Env proteins of all reference RVs-GTE and their endogenous forms (see Table S2 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]) were used as probes. A 40% sequence identity over 40% of the region with an E value of 1E−5 was used to filter significant hits. Second, the significant hits confirmed by phylogeny were concatenated based on the host genomic location and alignment positions with reference RV-GTE proteins. Third, the same genomes were subjected to a second round of screening by tBLASTn using the protein sequences of concatenated Env. Finally, the flanking sequences of significant hits obtained from the second round that were confirmed by phylogeny were extended to identify viral pairwise LTRs using BLASTn (39), LTR_Finder (40), and LTR_harvest (41). Full-length ERVs and their consensus sequences were used as queries to search for ERV copies using BLASTn in each lineage. The hit parts of sequences longer than 3 kb with 80% identity were regarded as copies of each ERV-GTE lineage (see Data Set S1 at figshare [https://doi.org/10.6084/m9.figshare.19235727.v1]), and these were named in accordance with the previous nomenclature proposed for ERVs (8). The ERV copy identifier (ID) was composed of three elements, each separated by a hyphen, for example, ERV-GA.a.1-Lle. The first element indicates that the category is ERV. The second element consists of three subcomponents—the first being the taxonomic group of retroviruses which was based on Pol phylogeny (EA, epsilon-related viruses; GA, gamma-related viruses; SA, SnRV-related viruses), the second being a lineage alphabet ID, and the third being a numeric ID that uniquely identifies the ERV locus. The third element indicates the species in which the ERVs are found.
To identify potential expRVs, all 439 data sets in the transcriptome sequencing assembly database (TSA) (see Table S1 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2]) were screened using tBLASTn, and the Env proteins of the reference RVs-GTE, including the newly identified ERVs-GTE, were used as probes. A 40% sequence identity over 40% of the region with an E value of 1E−5 was used to filter significant hits. The called hit contigs were then included in the phylogenetic analysis. The viral contigs within the GTE clade were considered.
Consensus genome construction and genome annotation.
ERVs-GTE longer than 5 kb in each lineage were aligned using MAFFT 7.222 (42) and then used to construct consensus sequences for each ERV lineage by Geneious. The distributions of ORFs in copies of ERV and expERV contigs were determined using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) in the NCBI database and confirmed by BLASTp (39). Conserved domains (including RT, RH, integrase, heptad repeat, ISD, and TR) for each sequence were determined using CD-Search against the CDD (https://www.ncbi.nlm.nih.gov/cdd/) (43) and BLASTp. The TM of the Env protein of representative ERVs-GTE in each type was used to predict the secondary structure and hydropathicity scores with JPred 4 (http://www.compbio.dundee.ac.uk/jpred/) and ExPASy (https://web.expasy.org/protscale/). The region which (i) was located at the N terminus of TM, (ii) contained alpha-helical or beta structure, and (iii) was hydrophobic was considered the FP region. The predicted results are shown in Fig. 2B. The consensus sequences of ERVs-GTE can be found in Data Set S4 at figshare (https://doi.org/10.6084/m9.figshare.19235727.v1).
Phylogenetic analysis.
To investigate the evolutionary relationship between the RVs and viral elements found in this study, the protein sequences of the TM region, Env, and Pol were aligned using MAFFT 7.222 (42). The regions in the alignment that aligned poorly were removed using trimAL (44) and confirmed manually with MEGA X (45). A sequence was excluded if its length was less than 75% of the alignments. The best-fit models (Env, LG+G4; Pol, LG+F+I+G4) were selected using ProtTest (46), and phylogenetic trees for these protein sequences were inferred using the maximum likelihood (ML) method in IQ-Tree (Env and Pol phylogeny) (47) by incorporating 100 bootstrap replicates for the assessment of node robustness. The phylogenetic trees were viewed and annotated using FigTree V1.4.3 (https://github.com/rambaut/figtree/). The alignments performed in this study can be found in Data Set S2 and Data Set S3 at figshare (https://doi.org/10.6084/m9.figshare.19235727.v1). Two Pol trees in Fig. 3A and in Fig. S2 at figshare (https://doi.org/10.6084/m9.figshare.19235721.v2) are two separate similar analyses. The topologies of the two trees are the same but are presented in different manners due to the different constituent sequences (Fig. S2 at figshare [https://doi.org/10.6084/m9.figshare.19235721.v2] included viral lineages that did not contain Env) and rooted sequences. The tree in Fig. S2 at figshare (https://doi.org/10.6084/m9.figshare.19235721.v2) is composed of Pol from 7 classes of reference retroviruses and ERVs-GTE and expRVs-GTE, and is rooted to foamy retroviruses, while the Pol tree in Fig. 3A included only ERVs-GTE and is a midpoint tree.
To identify proteins harboring the OGFr_N terminus domain, we used the OGFr_N terminus domain as a probe to screen the proteome and genome of Anura. The OGFr_N terminus domain sequences of viruses and hosts were used to construct corresponding ML phylogenetic trees. The regions in the alignment that aligned poorly were removed using trimAL (44) and confirmed manually with MEGA X (45). The best-fit models (LG+G4) were selected using ProtTest (46), and the phylogenetic trees for these protein sequences were inferred using the maximum likelihood (ML) method in IQ-Tree (47) by incorporating 1,000 ultrafast bootstrap replicates for the assessment of node robustness. The phylogenetic trees were viewed and annotated using FigTree V1.4.3 (https://github.com/rambaut/figtree/). The tree is rooted in OGFrs of bony fishes which were not shown in Fig. 5C and in Data Set S8 at figshare (https://doi.org/10.6084/m9.figshare.19235727.v1).
Examination of the recombination of ERVs-GTE.
To exclude false-positive recombination events, we included only consensus viral sequences and qualified full-length ERVs-GTE, which (i) harbored pairwise similar LTRs (with divergence of <10%) and (ii) contained three complete major genes (gag, pol, and env) in the same transcriptional direction. In total, 56 consensus ERVs-GTE and 11 full-length ERVs-GTE were used to construct corresponding ML phylogenetic trees (Pol and Env). The regions in the alignment that aligned poorly were removed using trimAL (44) and confirmed manually with MEGA X (45). The best-fit models (Env, LG+I+G4; Pol, LG+F+R5) were selected using ProtTest (46), and the phylogenetic trees for these protein sequences were inferred using the maximum likelihood (ML) method in IQ-Tree (47) by incorporating 100 bootstrap replicates for the assessment of node robustness. The phylogenetic trees were viewed and annotated using FigTree V1.4.3 (https://github.com/rambaut/figtree/). The phylogenetic tree match between Pol and Env was estimated manually. Consensus sequences and representative ERVs-GTE were selected to analyze the recombinants of ERVs.
Dating analysis for determining the integration time of ERVs-GTE.
To determine the potential vertical transmission among all ERVs-GTE, we first extracted the flanking sequences of ERVs-GTE with lengths of 1 kb at both sites. The sequences on both sides of an ERV-GTE and 300 bp of its LTRs were then concatenated and compared with each other using dc-megablast with an E value of 1E−5. Significant pairwise sequences were retrieved if the alignment coverage was over 50% and longer than 800 bp.
Gene expression analysis.
Seventy-two L. leishanense RNA-seq samples, which are available in the Sequence Read Archive (SRA) database (SRR8736149 to SRR8736220), were aligned with the reference genome (ASM966780v1) using HISAT2 (48). The expression levels were then computed for each group of samples after filtering the loci with low expression (more than 20 samples had no expression). The OGFr expression level in each RNA-seq library was calculated as fragments per kilobase per million (FPKM).
Data availability.
All the data needed to support the conclusions detailed in the article are included in the article itself and the supplementary data at figshare.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31970176) and CAS Pioneer Hundred Talents Program to J.C.
Y.C. and J.C. designed research; Y.C., X.W., M.-E.L., and Y.S. performed research; Y.C., X.W., M.-E.L., and Y.S. analyzed data; and Y.C., X.W., and J.C. wrote the paper.
We declare no competing interest.
Contributor Information
Yicong Chen, Email: yc765@cornell.edu.
Jie Cui, Email: jcui@ips.ac.cn.
Frank Kirchhoff, Ulm University Medical Center.
REFERENCES
- 1.Rous P. 1911. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med 13:397–411. 10.1084/jem.13.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahn BH, Shaw GM, De Cock KM, Sharp PM. 2000. AIDS as a zoonosis: scientific and public health implications. Science 287:607–614. 10.1126/science.287.5453.607. [DOI] [PubMed] [Google Scholar]
- 3.Weiss RA. 1992. Retroviruses and human cancer. Semin Cancer Biol 3:321–328. [PubMed] [Google Scholar]
- 4.Xu W, Eiden MV. 2015. Koala retroviruses: evolution and disease dynamics. Annu Rev Virol 2:119–134. 10.1146/annurev-virology-100114-055056. [DOI] [PubMed] [Google Scholar]
- 5.Johnson WE. 2019. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol 17:355–370. 10.1038/s41579-019-0189-2. [DOI] [PubMed] [Google Scholar]
- 6.Stoye JP. 2012. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol 10:395–406. 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- 7.Xu X, Zhao H, Gong Z, Han GZ. 2018. Endogenous retroviruses of non-avian/mammalian vertebrates illuminate diversity and deep history of retroviruses. PLoS Pathog 14:e1007072. 10.1371/journal.ppat.1007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gifford RJ, Blomberg J, Coffin JM, Fan H, Heidmann T, Mayer J, Stoye J, Tristem M, Johnson WE. 2018. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 15:59. 10.1186/s12977-018-0442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henzy JE, Gifford RJ, Johnson WE, Coffin JM. 2014. A novel recombinant retrovirus in the genomes of modern birds combines features of avian and mammalian retroviruses. J Virol 88:2398–2405. 10.1128/JVI.02863-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen M, Cui J. 2019. Discovery of endogenous retroviruses with mammalian envelopes in avian genomes uncovers long-term bird-mammal interaction. Virology 530:27–31. 10.1016/j.virol.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Henzy JE, Johnson WE. 2013. Pushing the endogenous envelope. Philos Trans R Soc Lond B Biol Sci 368:20120506. 10.1098/rstb.2012.0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henzy JE, Coffin JM. 2013. Betaretroviral envelope subunits are noncovalently associated and restricted to the mammalian class. J Virol 87:1937–1946. 10.1128/JVI.01442-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bénit L, Dessen P, Heidmann T. 2001. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J Virol 75:11709–11719. 10.1128/JVI.75.23.11709-11719.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen M, Guo X, Zhang L. 2021. Unexpected discovery and expression of amphibian class II endogenous retroviruses. J Virol 95:e01806-20. 10.1128/JVI.01806-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niewiadomska AM, Gifford RJ. 2013. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol 11:e1001642. 10.1371/journal.pbio.1001642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henzy JE, Gifford RJ, Kenaley CP, Johnson WE. 2017. An intact retroviral gene conserved in spiny-rayed fishes for over 100 My. Mol Biol Evol 34:634–639. 10.1093/molbev/msw262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen CH, Steiner LA. 2004. Genome structure and thymic expression of an endogenous retrovirus in zebrafish. J Virol 78:899–911. 10.1128/jvi.78.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi J, Zhang H, Gong R, Xiao G. 2015. Characterization of the fusion core in zebrafish endogenous retroviral envelope protein. Biochem Biophys Res Commun 460:633–638. 10.1016/j.bbrc.2015.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, Zhang YY, Wei X, Cui J. 2021. Multiple infiltration and cross-species transmission of foamy viruses across the Paleozoic to the Cenozoic era. J Virol 95:e00484-21. 10.1128/JVI.00484-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenwood AD, Ishida Y, O’Brien SP, Roca AL, Eiden MV. 2018. Transmission, evolution, and endogenization: lessons learned from recent retroviral invasions. Microbiol Mol Biol Rev 82:e00044-17. 10.1128/MMBR.00044-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Angel VD, Dupuis F, Mornon JP, Callebaut I. 2002. Viral fusion peptides and identification of membrane-interacting segments. Biochem Biophys Res Commun 293:1153–1160. 10.1016/S0006-291X(02)00353-4. [DOI] [PubMed] [Google Scholar]
- 22.Gallaher WR. 1996. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell 85:477–478. 10.1016/s0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 23.Ackermann M, Koriabine M, Hartmann-Fritsch F, de Jong PJ, Lewis TD, Schetle N, Work TM, Dagenais J, Balazs GH, Leong JA. 2012. The genome of chelonid herpesvirus 5 harbors atypical genes. PLoS One 7:e46623. 10.1371/journal.pone.0046623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Y, Wang L, Zhou Z, Wang M, Liu R, Wang L, Jiang Q, Song L. 2013. An opioid growth factor receptor (OGFR) for [Met5]-enkephalin in Chlamys farreri. Fish Shellfish Immunol 34:1228–1235. 10.1016/j.fsi.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Zagon IS, Verderame MF, McLaughlin PJ. 2002. The biology of the opioid growth factor receptor (OGFr). Brain Res Rev 38:351–376. 10.1016/s0165-0173(01)00160-6. [DOI] [PubMed] [Google Scholar]
- 26.Yedavalli VRK, Patil A, Parrish J, Kozak CA. 2021. A novel class III endogenous retrovirus with a class I envelope gene in African frogs with an intact genome and developmentally regulated transcripts in Xenopus tropicalis. Retrovirology 18:20. 10.1186/s12977-021-00564-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epand RM. 2003. Fusion peptides and the mechanism of viral fusion. Biochim Biophys Acta Biomembr 1614:116–121. 10.1016/S0005-2736(03)00169-X. [DOI] [PubMed] [Google Scholar]
- 28.Apellaniz B, Huarte N, Largo E, Nieva JL. 2014. The three lives of viral fusion peptides. Chem Phys Lipids 181:40–55. 10.1016/j.chemphyslip.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mzoughi O, Teixido M, Planes R, Serrero M, Hamimed I, Zurita E, Moreno M, Granados G, Lakhdar-Ghazal F, BenMohamed L, Giralt E, Bahraoui E. 2019. Trimeric heptad repeat synthetic peptides HR1 and HR2 efficiently inhibit HIV-1 entry. Biosci Rep 39:BSR20192196. 10.1042/BSR20192196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weng Y, Weiss CD. 1998. Mutational analysis of residues in the coiled-coil domain of human immunodeficiency virus type 1 transmembrane protein gp41. J Virol 72:9676–9682. 10.1128/JVI.72.12.9676-9682.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aiewsakun P, Katzourakis A. 2017. Marine origin of retroviruses in the early Palaeozoic Era. Nat Commun 8:13954. 10.1038/ncomms13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, Reavill DR, Dunker F, Derisi JL. 2012. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. mBio 3:e00180-12. 10.1128/mBio.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aswad A, Katzourakis A. 2015. Convergent capture of retroviral superantigens by mammalian herpesviruses. Nat Commun 6:8299. 10.1038/ncomms9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chuong EB, Elde NC, Feschotte C. 2016. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351:1083–1087. 10.1126/science.aad5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feschotte C, Gilbert C. 2012. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet 13:283–296. 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- 36.Chuong EB, Rumi MA, Soares MJ, Baker JC. 2013. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet 45:325–329. 10.1038/ng.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Basta HA, Cleveland SB, Clinton RA, Dimitrov AG, McClure MA. 2009. Evolution of teleost fish retroviruses: characterization of new retroviruses with cellular genes. J Virol 83:10152–10162. 10.1128/JVI.02546-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zagon IS, McLaughlin PJ. 2014. Opioid growth factor and the treatment of human pancreatic cancer: a review. World J Gastroenterol 20:2218–2223. 10.3748/wjg.v20.i9.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 40.Xu Z, Wang H. 2007. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35:W265–W268. 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellinghaus D, Kurtz S, Willhoeft U. 2008. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9:18. 10.1186/1471-2105-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, Thanki N, Yamashita RA, Yang M, Zhang D, Zheng C, Lanczycki CJ, Marchler-Bauer A. 2020. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268. 10.1093/nar/gkz991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data needed to support the conclusions detailed in the article are included in the article itself and the supplementary data at figshare.