

Abstract
目的
分析17例软骨发育不全(achondroplasia,ACH)患儿的临床特征及成纤维细胞生长因子受体3(fibroblast growth factor receptor 3,FGFR3)基因变异情况。
方法
回顾性分析2009年1月至2021年10月确诊的17例ACH患儿临床资料及FGFR3基因检测结果。
结果
ACH最常见的临床表现是不匀称型身材矮小(100%,17/17)、大头畸形(100%,17/17)、三叉戟手畸形(82%,14/17)、膝内翻(88%,15/17)。最普遍的影像学是根茎状长骨缩短(100%,17/17)和腰椎椎间距变窄(88%,15/17)。主要并发症有骨骼异常(100%,17/17)、中耳功能障碍(82%,14/17)、运动及语言发育迟缓(88%,15/17)、慢性疼痛(59%,10/17)、睡眠呼吸暂停(53%,9/17)、肥胖(41%,7/17)、枕骨大孔缩小(35%,6/17)、脑积水(24%,4/17)。17例(100%)均存在FGFR3基因变异,13例为FGFR3基因c.1138G>A的热点突变;2例FGFR3基因c.1138G>C变异;2例为未报道的变异,其中1例FGFR3基因c.1252C>T变异,1例FGFR3基因c.445+2_445+5delTAGG变异。
结论
该研究检出FGFR3基因未报道变异位点,扩展了ACH基因变异谱。ACH是一种进行性发展的疾病,其相关并发症由多学科团队协作进行终身管理。
Keywords: 软骨发育不全, 成纤维细胞生长因子受体3基因, 基因变异, 儿童
Abstract
Objective
To study the clinical features and fibroblast growth factor receptor 3 (FGFR3) gene mutations of children with achondroplasia (ACH) through an analysis of 17 cases.
Methods
A retrospective analysis was performed on the clinical data and FGFR3 gene detection results of 17 children with ACH who were diagnosed from January 2009 to October 2021.
Results
Of the 17 children with ACH, common clinical manifestations included disproportionate short stature (100%, 17/17), macrocephaly (100%, 17/17), trident hand (82%, 14/17), and genu varum (88%, 15/17). The common imaging findings were rhizomelic shortening of the long bones (100%, 17/17) and narrowing of the lumbar intervertebral space (88%, 15/17). Major complications included skeletal dysplasia (100%, 17/17), middle ear dysfunction (82%, 14/17), motor/language developmental delay (88%, 15/17), chronic pain (59%, 10/17), sleep apnea (53%, 9/17), obesity (41%, 7/17), foramen magnum stenosis (35%, 6/17), and hydrocephalus (24%, 4/17). All 17 children (100%) had FGFR3 mutations, among whom 13 had c.1138G>A hotspot mutations of the FGFR3 gene, 2 had c.1138G>C mutations of the FGFR3 gene, and 2 had unreported mutations, with c.1252C>T mutations of the FGFR3 gene in one child and c.445+2_445+5delTAGG mutations of the FGFR3 gene in the other child.
Conclusions
This study identifies the unreported mutation sites of the FGFR3 gene, which extends the gene mutation spectrum of ACH. ACH is a progressive disease requiring lifelong management through multidisciplinary collaboration.
Keywords: Achondroplasia, Fibroblast growth factor receptor 3 gene, Gene mutation, Child
软骨发育不全(achondroplasia,ACH;OMIM #100800)是人类最常见的侏儒症,临床特征包括不成比例的身材矮小、大头畸形与前额突出、面中部发育不全、胫骨弯曲等[1]。ACH发病率为1/15 000~1/25 000[2],据估计会影响全球超过250 000人[3]。1994年ACH致病基因确定为成纤维细胞生长因子受体3(fibroblast growth factor receptor 3,FGFR3)基因[4]。ACH是一种常染色体显性遗传病,具有100%的外显率。国外研究显示超过97%的病例是由位于4号染色体短臂上FGFR3基因跨膜结构域的错义突变(p.G380R)引起的,约80%的病例是由于健康父母所生患者的新发突变所致,与父系年龄效应相关,另外约20%是由家族遗传所致[1]。国内研究显示99%(208/210)基因变异为热点突变,仅有2例为新发未报道变异[5],且大部分研究为个案报道,样本量小。本研究对17例ACH的临床特征、并发症及基因变异进行总结,发现了2例未报道基因变异,对扩充ACH基因库意义重大。现总结如下。
1. 资料与方法
1.1. 研究对象
回顾性分析2009年1月至2021年10月在空军军医大学第一附属医院儿科诊断的17例ACH患儿的临床资料。ACH诊断标准[1]:(1)临床表现:非匀称性矮小及四肢短小(身高<-3 SD,臂展/身高<0.96);双上肢或下肢肢根缩短;大头畸形伴前额突出(头围>1 SD);面中部发育不良,鼻梁扁平、鼻棘短小及鼻前倾;手指短小和三叉戟形结构。(2)影像学检查:骨盆和股骨的单次前后位X线片提示方形髂翼;扁平、水平移位的髋臼;坐骨切迹明显变窄;典型的股骨近端透亮度增加,腰椎椎弓根间距变窄(在新生儿中罕见)。(3)基因检测:FGFR3基因存在c.1138G>A(p.G380R)或c.1138G>C(p.G380R)变异。
本研究已获得患儿父母知情同意,并通过我院医学伦理委员会批准(KY20212126-F-1号)。
1.2. 临床资料收集
收集患儿临床资料,包括患儿性别、出生史、生长发育史、起病年龄、首发症状、临床表现、家族史等;测量患儿身高、坐高、指间距、体重、头围;实验室检查、影像学检查等。
1.3. 基因检测
FGFR3基因热点突变检测:采集患儿及父母外周血2 mL,使用DNA试剂盒提取基因组DNA,对患儿及父母的FGFR3基因第9号外显子和邻近内含子区域进行聚合酶链式反应扩增,引物序列为正向5'-GGTCTGGCCCTCTAGACTCA-3'和反向5'-CGTGCCCCAAAGTACCCTAG-3'。聚合酶链式反应产物经1%琼脂糖凝胶电泳,获得目的带后,交由基因公司测序。突变位点采用Chromos与Mutation Surveyor V4.0.6软件分析。
全外显子基因测序分析:采集患儿及父母外周血2 mL。使用Gen Exome Research Panel v1.0(Integrated DNA Technologies,美国)试剂盒进行外显子捕获实验,Illumina HiSeq 4000测序仪双端150 bp测序模式测序。测序原始数据经过BWA和SAMtools软件分析,使用GATK V3.70对变异进行识别。全外显子设计针对检测出变异的Sanger测序扩增引物,PCR扩增,通过Sanger测序在患儿及父母中验证外显子测序发现的变异。
1.4. 临床管理及治疗
随访需监测头围及囟门闭合情况、体重、身高(<2岁测卧位身长,≥2岁测立位身高)[5]、发育测评(根据情况进行)、听力测试、脊柱影像学、颅颈交界区CT或MRI检查、多导睡眠监测等。通过多学科团队协作(multidisciplinary team,MDT)对并发症及时干预,包括对症处理、手术干预和终身随访护理[1]。
2. 结果
2.1. 临床特征
17例ACH患儿中,男性10例(59%),女性7例(41%)。患儿均来自非近亲婚配家庭。14例(82%)患儿为剖宫产出生,3例(18%)顺产出生,均为足月儿。均无基础疾病。就诊年龄范围1 d至11.3岁。14例(82%)以身材矮小为主诉就诊,2例(12%)以发育落后、抽搐为主诉就诊,1例(6%)因产前超声异常就诊。
17例患儿中,10例(59%)为散发病例,7例(41%)由家族遗传,其中4例变异遗传来自母亲,3例来自父亲。所有产妇平均年龄27.3岁。散发病例的父亲平均生育年龄38.8岁。15例(88%)患儿存在产前超声异常,其中13例发现长骨缩短。1例患儿在新生儿期基因诊断。病例4母亲再次怀孕后B超显示股骨长度缩短,小于同孕周胎儿第3个百分数,羊水穿刺基因确诊ACH后终止妊娠。
仅病例4使用重组人生长激素(recombinant human growth hormone,rhGH)治疗3年,身高由<P 3提升至P 10~P 25,见表1(表中身高百分位数均参照ACH特定的生长曲线[5])。
表 1.
17例ACH患儿的临床资料及基因结果
| 病例 | 性别 | 就诊年龄 | 就诊生长或身高(cm)/百分位数 | 主诉 | FGFR3基因变异位点 | 遗传来源 | 父亲受孕年龄 (岁) | 目前年龄(岁) | 目前身高(cm)/百分位数 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 男 | 11.3岁 | 103.1/P 3~P 10 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 35 | 18 | 130 |
| 2 | 女 | 8.5岁 | 93.6/P 3~P 10 | 身材矮小 | c.1138G>C(p.G380R) | 新发突变 | 39 | 16 | 120 |
| 3 | 女 | 2.3岁 | 71.7/P 3~P 10 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 42 | 11 | 100/<P 3 |
| 4 | 男 | 1.5岁 | 63.6/<P 3 | 身材矮小 | c.1138G>C(p.G380R) | 母源 | 23 | 5.6 | 87/P 10~P 25 |
| 5 | 女 | 6.5岁 | 88/P 10 | 身材矮小 | c.1138G>A(p.G380R) | 母源 | 26 | 18.4 | 120 |
| 6 | 男 | 2.1岁 | 70.6/P 10 | 身材矮小 | c.1138G>A(p.G380R) | 母源 | 30 | 13.8 | 117 |
| 7 | 女 | 1.2岁 | 64.4/P 25 | 身材矮小 | c.1138G>A(p.G380R) | 母源 | 27 | 2.5 | 67/<P 3 |
| 8 | 男 | 3岁 | 71.8/<P 3 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 46 | 15.3 | 118 |
| 9 | 男 | 1.3岁 | 63/P 3~P 10 | 发育落后、抽搐 | c.1258C>T(p.P420S) | 父源 | 36 | 3.5 | 70/<P 3 |
| 10 | 男 | 0.75岁 | 60/P 3~P 10 | 发育落后、抽搐 | c.1144G>A(p.G382R) | 新发突变 | 41 | 3.7 | 66/<P 3 |
| 11 | 男 | 0.7岁 | 65/P 25 | 身材矮小 | c.1138G>A(p.G380R) | 父源 | 36 | 5.1 | 82/P 3~P 10 |
| 12 | 女 | 1 d | 48/P 25~P 50 | 胎儿期股骨短小、头颅大 | c.1138G>A(p.G380R) | 新发突变 | 35 | 0.5 | 57/P 10~P 25 |
| 13 | 男 | 1.6岁 | 68/P 25 | 身材矮小 | c.445+2_445+5delTAGG | 父源 | 27 | 3.8 | 77/P 3~P 10 |
| 14 | 男 | 3.2岁 | 74.7/P 3 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 32 | 17.3 | 131 |
| 15 | 女 | 8.3岁 | 95.8/P 10 ~P 25 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 29 | 18.6 | 121 |
| 16 | 女 | 3.8岁 | 77/P 10 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 50 | 15.3 | 102 |
| 17 | 男 | 10.1岁 | 98.9/P 3~P 10 | 身材矮小 | c.1138G>A(p.G380R) | 新发突变 | 39 | 11.8 | 103/P 3~P 10 |
主要临床表现:17例(100%)患儿表现为根茎状短肢矮小身材(长骨短及四肢近端骨骼节段不成比例缩短)及特征性的面貌(颅骨较大、前额突出、鼻梁扁平、中面部发育不全、下颌骨相对突出);14例(82%)为三叉戟手畸形及短指。
主要并发症:17例(100%)患儿均有骨骼异常;15例(88%)有膝内翻畸形;10例(59%)有脊柱畸形;9例(53%)有肘关节伸展受限;7例(41%)有髋关节和膝关节过度活动。14例(82%)有中耳功能障碍,均曾患有中耳炎,其中4例听力下降(1例安放中耳压力平衡管)。9例(53%)报告了睡眠障碍,进行了多导睡眠监测,均显示睡眠呼吸暂停,其中4例进行扁桃体切除术和/或腺样体切除术,有1例重度阻塞性睡眠呼吸暂停低通气综合征(obstructive sleep apnea hypopnea syndrome,OSAHS),患儿接受了2次手术,第1次行腺样体切除术,第2次行气管切开术,扁桃体切除术和悬雍垂-腭-咽成形术,鼻甲消融术,舌根消融术,术后睡眠呼吸暂停均得到缓解。7例(41%)有肥胖。15例(88%)有发育迟缓,其中13例运动发育落后,8例语言发育迟缓。6例(35%)有枕骨大孔缩小,无明显脊髓压迫症状,动态随访观察。4例(24%)有脑积水,1例脑室扩大并脑积水,有颅高压表现,行脑室-腹腔分流术,余3例予随访观察。2例(12%)癫痫。10例(59%)有出现慢性疼痛(发生时间9~18岁),其中7例关节疼痛,5例背痛、腰痛、臀部疼痛,有1例佩戴改良胸腰骶部支具。
放射学表现:17例(100%)患儿为长骨根茎缩短;15例(88%)为腰尾椎体椎弓根间距变窄;14例(82%)为方形髂骨和水平髋臼;12例(71%)为全身干骺端改变;11例(65%)为股骨近端射线可透;10例(59%)为坐骨切迹变窄;6例(35%)为颅底及枕骨大孔缩小(诊断年龄范围1.5~5.4岁,平均年龄3.8岁)。
2.2. 遗传学特征
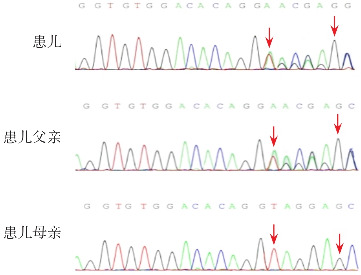
本研究中12例(71%)携带FGFR3基因c.1138G>A(p.G380R)杂合变异;2例(12%)携带FGFR3基因c.1138G>C(p.G380R)杂合变异;1例(6%)携带FGFR3基因c.1144G>A(p.G382R)杂合变异(转录本为NM_001163213,将其转换为常用转录本NM_000142,此变异位点与c.1138G>A是同一位点),参照美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)相关指南[6],以上15例FGFR3基因变异评定为致病性变异(PS1+PM1+PM2+PP3)。2例(12%)为FGFR3基因未报道的杂合变异,1例携带FGFR3基因c.1258C>T(p.P420S)变异(转录本NM_001163213,将其转换为常见转录本NM_000142,此变异位点为c.1252C>T(p.P418S),依据ACMG指南,该变异评定为可能致病(PM1+PM2+PS3+PP3),见图1。1例携带FGFR3基因c.445+2_445+5delTAGG缺失变异(转录本NM_000142),依据ACMG指南,该变异评定为可能致病(PVS1+PM2),见图2。此2例患儿临床表型与15例FGFR3基因热点突变患儿的临床表型无差异。
图1. 病例9患儿及父母FGFR3基因Sanger测序图 患儿所携带的FGFR3基因c.1258C>T(p.P420S)变异(转录本为NM_001163213),转换为常见转录本NM_000142,此变异为c.1252C>T(p.P418S)杂合变异,遗传自父亲。变异位点如箭头所示。.
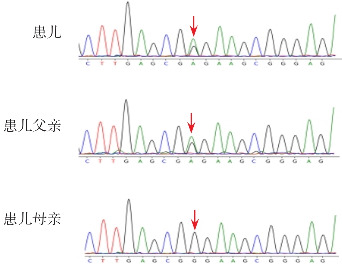
图2. 病例13患儿及父母FGFR3基因Sanger测序图 患儿所携带的FGFR3基因c.445+2_445+5delTAGG缺失变异,遗传自父亲。变异区域如箭头所示。.
3. 讨论
FGFR3基因在软骨细胞和成熟的成骨细胞中表达,作为一种负性调节因子抑制软骨的生长发育。FGFR3基因突变不是失活突变,而是功能获得性突变[3]。这种突变的受体通过多步骤的受体后级联反应,对所有生长板中的软骨细胞增殖造成持续的永久性中断。ACH根茎外观表明,增殖速度较快的生长板(股骨、肱骨)往往比增殖速度较慢的其他部位生长板受到更大的影响[7]。ACH几乎所有临床特征和并发症都是由软骨及骨生长异常而产生的,无论是直接的,还是因为与来自附近其他组织的结构相比,软骨及骨的生长不成比例。ACH是一种进行性发展的疾病,表型的严重程度与年龄相关,临床表现在新生儿中已经很明显,随着年龄增长,骨骼生长板会逐渐瓦解,临床特征加重,并发症逐渐显现[8]。ACH的并发症贯穿整个生命阶段[9],会影响多系统多器官,其治疗也需多专业共同协作完成[1]。
散发病例的发生与父系年龄效应相关,归因于精原干细胞随年龄增长的阳性选择和克隆扩增[10]。本研究10例散发病例的父亲受孕平均年龄38.8岁,有研究显示当父亲超过35岁时,这种突变的频率显著增加[11]。国外文献报道超过60%的ACH患儿有睡眠呼吸暂停[12],可能是由于胸廓及面中部发育不全、上呼吸道阻塞、颈髓过渡区受压等原因[13],其中45%接受了扁桃体切除术和/或腺样体切除术[14]。本研究中53%有睡眠呼吸暂停,仅24%接受了手术治疗。其中1例诊断为重度OSAHS,由MDT进行临床管理,防止危及生命的意外事件发生。总之,临床建议通过多导睡眠图定期评估,以免错过手术干预的时机[13]。
最近美国对114例ACH成年人进行自然史研究发现,17.4%的病例诊断为脑积水[14],意识到脑脊液空间扩大属于ACH的自然病程,并不意味着一定需要分流[15]。本研究仅1例因颅压增高患儿行脑室-腹腔分流术。ACH偶尔会出现癫痫发作[16],本研究中有2例伴发癫痫,具体原因尚不明确。颅颈交界处异常很常见,有症状的脊髓受压不多见[17],但临床表现严重,已经观察到中枢性睡眠呼吸暂停、脑干受压导致呼吸困难、脊髓型颈椎病和猝死等症状[18]。因此临床上在婴儿早期应及时评估与颅颈交界处受压相关的解剖学、祌经学和呼吸学表现,预测不良事件的风险。据报道,6.7%~13.3% ACH在2岁时需要枕骨大孔减压术[19]。本研究中6例枕骨大孔缩小,平均诊断年龄3.8岁,目前均无明显脊髓压迫症状,动态随访中。
ACH伴发的发育迟缓包括运动发育里程碑延迟、语言发育落后。本研究17例均无认知问题,智力正常,这与Bodensteiner[18]文献报道一致,但也有报道[20]显示在中枢神经系统残疾的情况下,智力水平下降。中耳功能障碍在ACH中极为常见[8]。据报道,几乎90%的ACH儿童在2岁前都曾患过中耳炎,至少有25%的儿童患有慢性和复发性中耳炎[13],这与面中部发育不全、咽鼓管缩短、咽小、腺样体或扁桃体相对肥大的有关。本研究中82%的病例患有中耳炎,与Ceroni等[21]文献报道大致接近。复发性中耳炎是传导性听力损失的危险因素[22],听力损失会导致语言延迟,因此建议尽早进行听力评估和鼓室测压[13]。
慢性疼痛是ACH一个未充分诊断且治疗不足的方面[9],其始于儿童期[23],随着年龄的增长,骨骼畸形进行性加重,引发持续疼痛,导致身体机能下降[24]。据统计,高达74.5%的成人慢性疼痛,但很少有人去寻求支持[25]。本研究中59%患儿存在慢性疼痛,后续骨科和康复科管理可以减轻疼痛[3]。鉴于慢性疼痛的高患病率以及缺乏针对ACH疼痛管理的循证指南,故仍需进一步探索获得有效疼痛管理的途经。
肥胖是ACH的常见并发症,用体重指数评估肥胖时应使用特定的标准[26],因身体比例的显著差异,普通人的标准会将大多数ACH患者错误地定义为肥胖。根据特定的体重指数标准[26],本研究中7例诊断为肥胖。虽然ACH患者脂肪组织优先积聚在腹部,不会导致典型的代谢并发症[27],但肥胖会增加阻塞性睡眠呼吸暂停、心血管疾病、腰椎管狭窄或关节超负荷风险[8]。因此参考ACH特有的生长曲线适当控制体重很重要[28]。
Miccoli等[29]报道,短期rhGH治疗后生长速度增加,但长期治疗没有明确益处,对身体比例的影响不确定,因此不常规推荐使用rhGH治疗ACH。目前的药理学方法旨在直接阻断FGFR3激活或调节其他控制软骨细胞增殖和分化的信号通路[1,7-8]。如C型利钠肽类似物,但分子药物需要在多大的生命窗口期使用它才能对健康产生有益的影响及有无不良反应等,这些都仍然未知,需进一步深入研究。
本研究存在一定局限性,随着分子生物学技术的日渐成熟和基因数据库的不断更新,对基因变异的解读可能会发生改变;本研究对FGFR3基因变异未进一步行功能研究,对其真正的危害性和意义尚待了解,未来将会着眼于此方向研究。
总之,ACH的管理是终身的,需要在整个生命过程中涉及多个专业,旨在应用内、外科手段改善其身高问题,并对不同并发症进行积极处理,以提高ACH患者的生活质量,并积极开展遗传咨询,应用产前超声和羊水穿刺基因检测手段,尽可能避免患儿出生。
参 考 文 献
- 1. 中国医师协会医学遗传医师分会, 中华医学会儿科学分会内分泌遗传代谢学组, 中华医学会儿科学分会罕见病学组, 等. 软骨发育不全诊断及治疗专家共识[J]. 中华儿科杂志, 2021, 59(7): 545-550. DOI: 10.3760/cma.j.cn112140-20201229-01142. [DOI] [PubMed] [Google Scholar]
- 2. Unger S, Bonafé L, Gouze E. Current care and investigational therapies in achondroplasia[J]. Curr Osteoporos Rep, 2017, 15(2): 53-60. DOI: 10.1007/s11914-017-0347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pauli RM. Achondroplasia: a comprehensive clinical review[J]. Orphanet J Rare Dis, 2019, 14(1): 1. DOI: 10.1186/s13023-018-0972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le Merrer M, Rousseau F, Legeai-Mallet L, et al. . A gene for achondroplasia-hypochondroplasia maps to chromosome 4p[J]. Nat Genet, 1994, 6(3): 318-321. DOI: 10.1038/ng0394-318. [DOI] [PubMed] [Google Scholar]
- 5. 代伟倩, 顾学范, 余永国. 中国210例软骨发育不全患儿临床遗传特征分析及生长发育曲线探索[J]. 中华儿科杂志, 2020, 58(6): 461-467. DOI: 10.3760/cma.j.cn112140-20200217-00096. [DOI] [PubMed] [Google Scholar]
- 6. Richards S, Aziz N, Bale S, et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Högler W, Ward LM. New developments in the management of achondroplasia[J]. Wien Med Wochenschr, 2020, 170(5-6): 104-111. DOI: 10.1007/s10354-020-00741-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wrobel W, Pach E, Ben-Skowronek I. Advantages and disadvantages of different treatment methods in achondroplasia: a review[J]. Int J Mol Sci, 2021, 22(11): 5573. DOI: 10.3390/ijms22115573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cormier-Daire V, AlSayed M, Ben-Omran T, et al. . The first European consensus on principles of management for achondroplasia[J]. Orphanet J Rare Dis, 2021, 16(1): 333. DOI: 10.1186/s13023-021-01971-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ornitz DM, Legeai-Mallet L. Achondroplasia: development, pathogenesis, and therapy[J]. Dev Dyn, 2017, 246(4): 291-309. DOI: 10.1002/dvdy.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coi A, Santoro M, Garne E, et al. . Epidemiology of achondroplasia: a population-based study in Europe[J]. Am J Med Genet A, 2019, 179(9): 1791-1798. DOI: 10.1002/ajmg.a.61289. [DOI] [PubMed] [Google Scholar]
- 12. Tenconi R, Khirani S, Amaddeo A, et al. . Sleep-disordered breathing and its management in children with achondroplasia[J]. Am J Med Genet A, 2017, 173(4): 868-878. DOI: 10.1002/ajmg.a.38130. [DOI] [PubMed] [Google Scholar]
- 13. Kubota T, Adachi M, Kitaoka T, et al. . Clinical practice guidelines for achondroplasia[J]. Clin Pediatr Endocrinol, 2020, 29(1): 25-42. DOI: 10.1297/cpe.29.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okenfuss E, Moghaddam B, Avins AL. Natural history of achondroplasia: a retrospective review of longitudinal clinical data[J]. Am J Med Genet A, 2020, 182(11): 2540-2551. DOI: 10.1002/ajmg.a.61825. [DOI] [PubMed] [Google Scholar]
- 15. Hoover-Fong J, Cheung MS, Fano V, et al. . Lifetime impact of achondroplasia: current evidence and perspectives on the natural history[J]. Bone, 2021, 146(5): 115872. DOI: 10.1016/j.bone.2021.115872. [DOI] [PubMed] [Google Scholar]
- 16. Manikkam SA, Chetcuti K, Howell KB, et al. . Temporal lobe malformations in achondroplasia: expanding the brain imaging phenotype associated with FGFR3-related skeletal dysplasias[J]. AJNR Am J Neuroradiol, 2018, 39(2): 380-384. DOI: 10.3174/ajnr.A5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ireland PJ, Pacey V, Zankl A, et al. . Optimal management of complications associated with achondroplasia[J]. Appl Clin Genet, 2014, 7: 117-125. DOI: 10.2147/TACG.S51485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bodensteiner JB. Neurological manifestations of achondroplasia[J]. Curr Neurol Neurosci Rep, 2019, 19(12): 105. DOI: 10.1007/s11910-019-1008-x. [DOI] [PubMed] [Google Scholar]
- 19. King JAJ, Vachhrajani S, Drake JM, et al. . Neurosurgical implications of achondroplasia[J]. J Neurosurg Pediatr, 2009, 4(4): 297-306. DOI: 10.3171/2009.3.PEDS08344. [DOI] [PubMed] [Google Scholar]
- 20. Ireland PJ, Donaghey S, McGill J, et al. . Development in children with achondroplasia: a prospective clinical cohort study[J]. Dev Med Child Neurol, 2012, 54(6): 532-537. DOI: 10.1111/j.1469-8749.2012.04234.x. [DOI] [PubMed] [Google Scholar]
- 21. Ceroni JRM, DCDQ Soares, Testai LDC, et al. . Natural history of 39 patients with achondroplasia[J]. Clinics (Sao Paulo), 2018, 73: e324. DOI: 10.6061/clinics/2018/e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tunkel D, Alade Y, Kerbavaz R, et al. . Hearing loss in skeletal dysplasia patients[J]. Am J Med Genet A, 2012, 158A(7): 1551-1555. DOI: 10.1002/ajmg.a.35373. [DOI] [PubMed] [Google Scholar]
- 23. Hoover-Fong J, Scott CI, Jones MC, et al. . Health supervision for people with achondroplasia[J]. Pediatrics, 2020, 145(6): e20201010. DOI: 10.1542/peds.2020-1010. [DOI] [PubMed] [Google Scholar]
- 24. Fredwall SO, Steen U, de Vries O, et al. . High prevalence of symptomatic spinal stenosis in Norwegian adults with achondroplasia: a population-based study[J]. Orphanet J Rare Dis, 2020, 15(1): 123. DOI: 10.1186/s13023-020-01397-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dhiman N, Albaghdadi A, Zogg CK, et al. . Factors associated with health-related quality of life (HRQOL) in adults with short stature skeletal dysplasias[J]. Qual Life Res, 2017, 26(5): 1337-1348. DOI: 10.1007/s11136-016-1455-7. [DOI] [PubMed] [Google Scholar]
- 26. Hoover-Fong JE, Schulze KJ, McGready J, et al. . Age-appropriate body mass index in children with achondroplasia: interpretation in relation to indexes of height[J]. Am J Clin Nutr, 2008, 88(2): 364-371. DOI: 10.1093/ajcn/88.2.364. [DOI] [PubMed] [Google Scholar]
- 27. Saint-Laurent C, Garcia S, Sarrazy V, et al. . Early postnatal soluble FGFR3 therapy prevents the atypical development of obesity in achondroplasia[J]. PLoS One, 2018, 13(4): e0195876. DOI: 10.1371/journal.pone.0195876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoover-Fong J, McGready J, Schulze K, et al. . A height-for-age growth reference for children with achondroplasia: expanded applications and comparison with original reference data[J]. Am J Med Genet A, 2017, 173(5): 1226-1230. DOI: 10.1002/ajmg.a.38150. [DOI] [PubMed] [Google Scholar]
- 29. Miccoli M, Bertelloni S, Massart F. Height outcome of recombinant human growth hormone treatment in achondroplasia children: a meta-analysis[J]. Horm Res Paediatr, 2016, 86(1): 27-34. DOI: 10.1159/000446958. [DOI] [PubMed] [Google Scholar]