

Abstract
Objective:
Smoking is associated with numerous inflammatory and autoimmune conditions. The goal of this study was to examine whether increased expression of G-protein-coupled receptor 15 (GPR15) on helper T cells in smokers could predispose to these conditions through its relationship with inflammatory biomarkers.
Methods:
We used flow cytometric measurement of GPR15+CD3+CD4+ helper T cells and serum assays for C-reactive protein (CRP) and 17 cytokines drawn from peripheral blood samples from a cohort of n = 62 primarily African American young adults (aged 27–35 years). These variables were examined cross-sectionally in conjunction with serum biomarkers of tobacco (cotinine) and cannabis (tetrahydrocannabinol) use and lifestyle factors potentially impacting immune function in correlational analyses and linear regression models.
Results:
Tobacco and cannabis smoking were strongly associated with increased GPR15 expression on helper T cells (p < 0.001), which was in turn was strongly associated with the ratio of pro-inflammatory to anti-inflammatory cytokines (p < 0.001). Mediation analyses indicated increased GPR15 expression accounted for roughly half of the relationship between smoking variables and pro-inflammatory to anti-inflammatory cytokine balance. CRP was not associated with cannabis or tobacco use or GPR15+ expression, but was associated with body mass index (p < 0.001). These relationships persisted after controlling for lifestyle and medical factors impacting immune function.
Conclusions:
Increased expression of GPR15 by helper T cells in smokers may mediate some of the relationship between smoking and a pro-inflammatory cytokine milieu. Better understanding of this relationship may help uncover how smoking increases the risk of inflammatory diseases.
Keywords: Inflammation, autoimmunity, T cells, tobacco, cannabis
1. Introduction
Smoking is responsible for over 450,000 deaths per year in the United States and is the leading cause of preventable morbidity and mortality [1, 2]. Smoking-associated diseases accounting for the majority of these deaths include cardiovascular disease (CVD), chronic obstructive pulmonary disease (COPD), stroke, and cancer [3], each of which have been linked with increased levels of systemic inflammation [4, 5]. Smokers are also at elevated risk for autoimmune diseases including psoriasis, inflammatory bowel disease, rheumatoid arthritis, and multiple sclerosis [5-10]. Lastly, smokers demonstrate increases in inflammatory biomarkers including C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-a), consistent with their increased risk of autoimmune and inflammatory conditions [4, 11, 12].
Although the mechanisms underlying this array of smoking-related inflammatory and autoimmune conditions are imperfectly understood, recent findings suggest that investigation of the role of chromosome 3 gene G-protein coupled receptor 15 (GPR15) in smokers may be a fruitful avenue of investigation. Following its discovery as an HIV co-receptor in the 1990s [13-15], GPR15’s role as regulator of T cell homing to the gut [16] was established. Its natural ligand, GPR15L, encoded by the chromosome 10 gene c10orf99, was discovered to be highly expressed in environmentally exposed epithelia including the skin, oropharynx, gut, and cervix [17, 18]. Both GPR15 and GPR15L have been independently linked with a number of smoking-associated autoimmune diseases [19-24] and cancers [25, 26].
Gene expression studies have established that GPR15 is among the most up-regulated genes in smokers [27]. In a study of 56 patients with schizophrenia, a population known to have a high rate of smoking [28], GPR15 was the single most upregulated gene in peripheral blood mononuclear cells (PBMCs) compared to controls [29]. This increase in GPR15 expression corresponds to decreased methylation of specific CpGs in the gene region, particularly cg19859270 located in the first exon of GPR15 [30-34], a finding characteristic of transcriptionally active genomic locations in general [35]. Interestingly, it appears that increased GPR15 gene expression and corresponding CpG hypomethylation occur specifically in T and B cells in peripheral blood [36, 37] rather than granulocytes or monocytes. Bauer and colleagues further demonstrated that GPR15 expression in T cells is not specific to any T cell subtype [38]. Within the T cell population, Bauer and colleagues demonstrated that 15.5% CD3+ T cells expressed GPR15 compared to only 3.7% in non-smokers, and the difference was highly significant (p = 1.8E-10).
The above findings have led to suggestions that GPR15 expression may be a highly sensitive and specific biomarker for smoking [38]. What is less clear is whether increased GPR15 expression plays a role in the development of inflammatory and autoimmune disease processes in smokers [27]. Bauer and colleagues did not find evidence of a specific association between GPR15+ expressing CD3+ T cells and the presence or absence of COPD in smokers [39], and have suggested that increased GPR15+ T cells in smokers may be an adaptive rather than pathogenic change [38]. Jhun and colleagues [40] did find associations between cg03636183, another CpG locus associated with smoking, and interleukin-18 (IL-18), but were unable examine possible associations between cg19859270 and inflammation under their Mendelian Randomization study design.
Here we seek to address the above knowledge gap by testing by conducting direct measurement of the relevant variables using serological and flow cytometry techniques, testing the following hypotheses in a sample of young African Americans with high rates of tobacco and cannabis smoking. Because anti-inflammatory effects of some cannabinoids have been reported [41] and tobacco and cannabis co-use are common, we include each substance as an independent predictor of both the proportion GPR15-expressing CD3+CD4+ Th cells (hereafter GPR15+ Th cell percent) and serum inflammatory biomarkers.
H1: Tobacco and cannabis smoking will be associated with serum inflammatory biomarkers, specifically (H1a) the ratio of pro-inflammatory to anti-inflammatory cytokines and (H1b) CRP.
H2: GPR15+ Th cell percent will mediate the relationship between tobacco and cannabis smoking and the (H2a) ratio of pro-inflammatory to anti-inflammatory cytokines and (H2b) CRP.
H3: The above relationships will persist after controlling for variables that can influence the immune system, including age, sex, race, body mass index (BMI), perceived stress, depressive symptoms, and consumption of non-steroidal anti-inflammatory drugs (NSAIDs) [42-45].
2. Methods
2.1. Participants and Recruitment
Participants were recruited from a multi-wave cohort study of African American families living in Georgia and Iowa, The Family and Community Health Study (FACHS) [46-48]. FACHS participants include a primary caretaker (“PCs”) and at least one child aged 10-12 years old at the time of initial recruitment (1995-1997). Since then, these child participants have been characterized over seven waves of data spanning a 20+ year period, with repeated interviews and biomaterial samplings. Wave 7 also included recruitment of their romantic partners to allow for examination of relationship variables. For the current study, participants in both of these groups living in Iowa who had given consent to be recontacted were recruited by phone by a trained research assistant and then provided informed consent in person.
2.2. Procedures
Approval for all study protocols and procedures was provided by the Institutional Review Board at the University of Iowa (IRB ID # 201604748). All participants provided informed consent in writing before participating in the study.
Participants provided information on their use of cannabis, tobacco, and nicotine-containing products such as e-cigarettes and pipes by means of a brief structured interview. Basic demographics including age, sex, race/ethnicity, height, and weight were also recorded. Consumption of non-steroidal anti-inflammatory drugs (NSAIDs) was assessed by a structured form listing commonly available over-the-counter formulations that asked participants to estimate their consumption over the past month. Lastly, participants provided information on their current level of depressive symptoms via the nine-item Patient Health Questionnaire depression module (PHQ-9) [49], and about their current level of stress via the Perceived Stress Scale [50].
2.3. Biomaterials and Assays
Participants were phlebotomized by a trained phlebotomist to provide whole blood for serum and flow cytometry assays, according to our previously published methods [51, 52].
2.3.1. ELISAs
Serum cotinine and tetrahydrocannabinol (THC) levels were assayed by enzyme-linked immunoassay (ELISA) using kits supplied by Abnova (Taiwan) according to the manufacturer’s recommendations. C-Reactive Protein (CRP) levels were assayed using an ELISA kit from R&D Systems (Minneapolis, MN), according to the manufacturer’s directions. A panel of 17 serum cytokines including interleukin-1 (IL-1), interleukin-2 (IL-2), interleukin-4 (IL-4), interleukin-5 (IL-5), interleukin-6 (IL-6), interleukin-7 (IL-7), interleukin-8 (IL-8), interleukin-10 (IL-10), interleukin-12 (IL-12), interleukin-13 (IL-13), interleukin-17 (IL-17), granulocyte-colony stimulating factor (G-CSF), granulocyte-monocyte colony-stimulating factor (GM-CSF), interferon-gamma (IFN-gamma), macrophage inflammatory protein (MIP-1), monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-alpha (TNF-alpha) were measured using the Bio-Plex Pro Human Cytokine 17-Plex Immunoassay by Bio-Rad (Hercules, CA), according to the manufacturer’s directions.
Finally, serum C-reactive protein was assayed using a Quantikine ELISA kit from R&D Systems, Inc. (Minneapolis, MN), according to the manufacturer’s directions.
2.3.3. PBMC preparation
Becton Dickinson (BD; Franklin Lakes, NJ) Vacutainer CPT tubes were used to isolate peripheral blood mononuclear cells (PBMCs), following the manufacturer’s protocol. Isolated PBMCs were immediately placed in a solution of 10% DMSO, 20% fetal calf serum, 70% RPMI supplemented with HEPES (1M, pH range 7.2-7.5) and L-glutamine and cryopreserved in liquid nitrogen using slow temperature-lowering method in a Mr. Frosty Nalgene polyethylene vial holder (Thermo Fisher Scientific, San Jose, CA) containing isopropyl alcohol. All PBMC samples were stored in liquid nitrogen for 7 days or longer before thawing.
For use in flow cytometry assays, cells were rapidly thawed in a 10% fetal calf serum, 90% RPMI solution supplemented with DNAse in a 37°C water bath, then washed in FACS buffer (1X PBS containing 1% BSA and 0.1% sodium azide). Viability and recovery were measured using tryptan blue exclusion.
2.3.4. Flow Cytometry
Following recovery, PBMCs were stained in 100 μL of FACS buffer with 1 μL each of anti-CD3 (clone UCHT1, BD) and anti-CD4 (monoclonal, clone Sk3, BD), and anti-GPR15 (clone SA302A10, BioLegend, San Diego, CA), according to the manufacturer’s recommendations. Mouse IgG2a kappa (clone MOPC-2710, BioLegend) was used as an isotype control. Unstained samples, fully stained (anti-CD3, anti-CD4, anti-GPR15), and control samples (anti-CD3, anti-CD4, isotype control) were acquired on a Becton Dickinson LSR II flow cytometer using FACS Diva Software and visualized with FlowJo software, version 7 (Tree Star, San Carlos, CA) to GPR15+ Th cell percent among participants.
2.4. Statistical Analyses
All statistical analyses were performed in R version 3.6.0 [53].Tobacco use intensity was coded as average cigarettes per day smoked in the last year. Due to heterogeneity in participants’ reports of their average cannabis use, which included variously “joints”, “blunts”, ounces and/or grams consumed, these reports were converted to gram equivalents according to published estimates [54]. These values for average grams used in the last year were then coded ordinally as zero (no use), one (50 grams or less), or two (more than 50 grams). Body mass index (BMI) was calculated based on self-reported weight and height. Perceived Stress Scale and PHQ-9 totals were calculated according to published instructions. NSAID consumption was calculated based on self-reported average daily dose ibuprofen, aspirin, and naproxen, which were then converted to equipotent doses of ibuprofen 200 mg [55]. Positivity for serum cotinine was defined as 1 ng/mL or greater, while serum positivity for THC was defined as 0.5 ng/mL or greater.
Cytokine assay results were subjected to quality control measures and coding as follows. First, analytes that had missing values for > 5% of the samples (n = 6) were discarded the remaining cytokines were categorized as “pro-inflammatory” (IL-1 beta, IL-5, IL-6, IL-7, IL-17, IFN-gamma, MCP-1, MIP-1b, TNF-alpha) and “anti-inflammatory” (IL-10, IL-13). For each analyte, all participants’ measured serum values were rank ordered using the rank() function, with ties coded by the “average” method in R, which awards a rank equal to the mean of the range. A the single missing “NA” cytokine value for IL-5 was imputed as the mean rank value for that participant’s remaining cytokines. Next, these rank values were summed generate “inflammatory” (IL-1 beta, IL-5, IL-6, IL-7, IL-17, IFN-gamma, MCP-1, MIP-1b, TNF-alpha) and “anti-inflammatory” (IL-10, IL-13) indices, consistent with our previously published methods [48]. Finally, the ratio of the two indices, the “cytokine ratio” was calculated. Of note, CRP values were analyzed in units of ng/mL for consistency with other analytes.
Correlational analyses were used to examine the relationship between study variables using base packages and corrplot() package [56]. Following this, linear regression models were used to examine mediation effects and control for variables that could affect immunological relationships between smoking variables, GPR15+ Th cell percent, and inflammatory biomarkers. First, cigarettes per day and scaled cannabis use were entered as linear predictors of the inflammatory biomarker. Next, GPR15+ Th cell percent was added as a linear predictor. Finally, the remaining immunological variables were entered as additional linear predictors. Moderators were not included in any regression models, and no additional mediators were examined.
Robust regression techniques including inspection of outliers and calculation of Cook’s distances were performed at each step. Each model was also examined for collinearity using the variance inflation factor function of the olsrr() package in R [57].
3. Results
3.1. Participant Characteristics and Assay Results
Interviews, measurement of GPR15+ Th cell percent by flow cytometry, and serum assays were completed for a total of 62 participants who agreed to participate in the study. Personal characteristics and assay results for these participants are provided in Table 1. The majority of participants were African American women, and their age distribution tightly distributed around age 31, consistent with the design of the cohort study. Participants’ average BMI was 34, consistent with mild obesity, and participants reported their level of depressive symptomatology on average as falling in the minimal and moderate ranges [49], whereas average stress levels were reported above average [50].
Table 1.
Means and standard deviations for study variables, all participants (n = 62).
| Variable | M or N | SD or % |
|---|---|---|
| 1. Age (years) | 30.7 | 1.2 |
| 2. Gender (male) | 23 | 37% |
| 3. Race (African American) | 53 | 85% |
| 4. Body mass index (BMI) | 34 | 8.5 |
| 5. Perceived Stress Scale | 19.3 | 4.4 |
| 6. PHQ-9 total | 4.7 | 4.1 |
| 7. NSAID daily dose | 0.43 | 1 |
| 8. Cigarettes per day (last year) | 1.8 | 4 |
| 9. Scaled cannabis use (last year) | 0.6 | 0.8 |
| 11. Cotinine positivity | 19 | 31% |
| 12. THC positivity | 20 | 32% |
| 13. Cytokine ratio | 5.3 | 3 |
| 14. CRP (mg/L) | 6.6 | 9.2 |
| 15. GPR15+ Th cell percent | 5.7 | 5 |
BMI refers to body mass index. Perceived Stress Scale refers to the total score. PHQ-9 total refers to the Patient Health Questionnaire depression module total score. NSAID daily dose refers to average daily consumption of nonsteroidal anti-inflammatory drugs in 200 mg equivalent doses of ibuprofen, naproxen, and aspirin. Cigarettes per day calculated by average consumption over the past month. Scaled cannabis use indicates average use over the past year on a zero to two scale. Cotinine positivity indicates serum values of > 1 ng/mL, while tetrahydrocannabinol (THC) positivity indicates > 0.5 ng/mL. CRP refers to C-reactive protein serum level.
Eleven percent of participants were positive for cotinine only, eleven percent positive for THC only, and twenty percent positive for both substances, while the remainder were negative for both. The distribution of GPR15+ Th cell percent by cotinine and/or THC serum positivity is provided in Figure 1. Analysis of variance indicated that GPR15+ Th cell percent varied significantly by group (F(3,58)=7.24, p < 0.001). The distribution of cytokine and CRP values are visualized in Figure 2.
Figure 1.
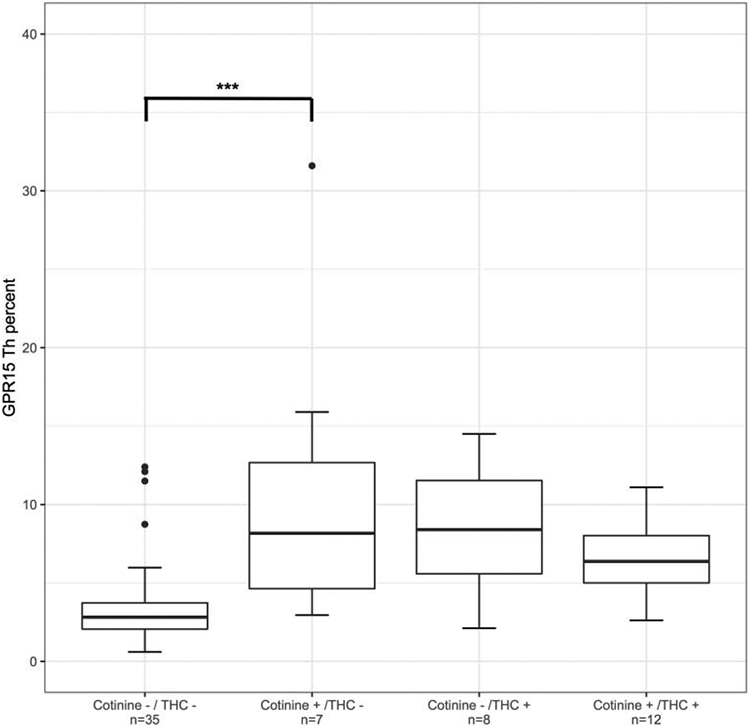
Boxplots of GPR15+ Th cell percent stratified by self-reported smoking status (tobacco and/or cannabis) and serum positivity (cotinine > 1 ng/mL and/or tetrahydrocannabinol (THC) > 0.5 ng/mL). Error bars indicate +/− 1 SD. Significance codes: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001 (Tukey’s Honest Significant Difference).
Figure 2.
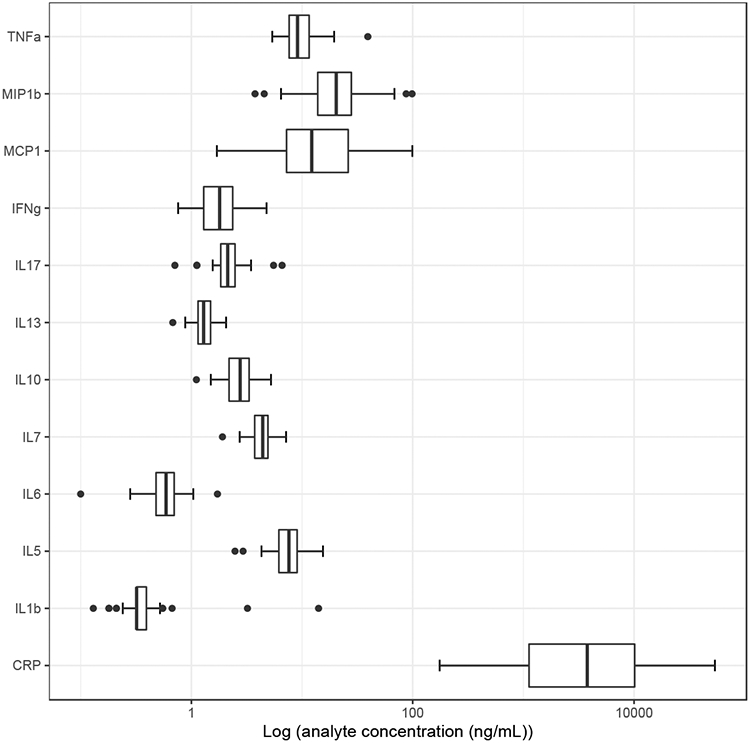
Boxplot distributions of inflammatory biomarker analytes in ng/mL. Error bars indicate +/1 SD.
3.2. Study Variable Correlations
H1: Tobacco and cannabis smoking will be associated with serum inflammatory biomarkers, specifically (H1a) the ratio of pro-inflammatory to anti-inflammatory cytokines and (H1b) CRP.
Correlation analysis of study variables (Figure 3, Supplemental Table 1) demonstrated significant correlations between GPR15+ Th cell percent and self-reported smoking intensity (r = 0.55, t = 3.66, df = 60, p < 0.001) and cannabis use intensity (r = 0.43, t = 5.11, df = 60, p < 0.001). Interestingly, GPR15+ Th cell percent was significantly correlated with serum THC positivity (r = 0.36, t = 2.96, df = 60, p < 0.001) but not serum cotinine positivity. Cotinine positivity and cigarettes per day were uncorrelated, surprisingly. GPR15+ Th cell percent was strongly correlated with the ratio of pro-inflammatory to anti-inflammatory cytokines (r = 0.43, t = 3.70, df = 60, p < 0.001) but not CRP. CRP in turn was significantly negatively correlated with male gender (r = −0.28, t = 2.30, df = 60, p < 0.05) and positively correlated with BMI (r = 0.53, t = 4.79, df = 60, p < 0.001) but no other study variables. The ratio of pro-inflammatory to anti-inflammatory cytokines was positively correlated with cigarettes per day (r = 0.39, t = 3.25, df = 60, p < 0.01) and showed a trend association with self-reported cannabis use (r = 0.22, t = 1.73, df = 60, p < 0.10), but was not correlated with serum cotinine or THC.
Figure 3.

Correlations between study variables analyzed. Ethnicity (AA) refers to American-African ethnicity. BMI refers to body mass index. Stress refers to the Perceived Stress Scale total score. PHQ refers to the Patient Health Questionnaire depression module (PHQ-9) total score. NSAIDs refers to nonsteroidal anti-inflammatory drug consumption in daily 200 mg equivalent doses of ibuprofen, naproxen, and aspirin. Cigarettes per day calculated by average consumption over the past month. Scaled cannabis use indicates average use over the past year on a zero to two scale. Serum cotinine positive indicates serum values of > 1 ng/mL, while serum tetrahydrocannabinol (THC) positive indicates > 0.5 ng/mL. Cytokine ratio refers to the ratio of pro-inflammatory and anti-inflammatory cytokine indices. Significance codes: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001.
To ensure consistency of findings, we then analyzed the correlations between each individual cytokine, CRP, and the pro-inflammatory to anti-inflammatory cytokine ratio, as well as GPR15+ Th cell percent and self-reported smoking intensity variables. As shown in Supplemental Table 2, the cytokine ratio was significantly negatively correlated with the two “anti-inflammatory” cytokines, IL-10 (r = −0.45, t = −3.94, df = 60, p < 0.001) and IL-13 (r = −0.39, t = −3.26, df = 60, p < 0.01), and positively correlated with IFN-gamma (r = 0.26, t = 2.07, df = 60, p < 0.05) and MCP-1 (r = 0.29, t = 2.38, df = 60, p < 0.05). The remaining individual cytokines failed to demonstrate significant individual correlations with the cytokine ratio and no cytokine was individually significantly correlated with GPR15+ Th cell percent.
3.3. Linear Regression Analyses
H2: GPR15+ Th cell percent will mediate the relationship between tobacco and cannabis smoking and the (H2a) ratio of pro-inflammatory to anti-inflammatory cytokines and (H2b) CRP.
H3: The above relationships will persist after controlling for variables that can influence the immune system, including age, sex, race, body mass index (BMI), perceived stress, depressive symptoms, and consumption of non-steroidal anti-inflammatory drugs (NSAIDs)
The relationship between GPR15+ Th cell percent and the ratio of pro-inflammatory to anti-inflammatory cytokines was further explored using linear regression models. First, robust regression methods were employed to examine a simple linear model relating GPR15+ Th cell percent as a predictor of cytokine ratio. Inspection indicated a single influential outlier among GPR15+ Th cell percent values (GPR15+ Th cell percent = 31.6) with high leverage and a Cook’s distance of over 1 [58]. This outlier was removed and all subsequent analyses performed on the remaining 61 participants. No further regression models demonstrated a Cook’s distance of greater than 1. No regression model demonstrated significant evidence of collinearity of predictors, with all variance inflation factors measuring between 1 and 2.
Both cigarettes per day and scaled cannabis use were entered into a linear regression model (Model 1A) as predictors of the pro-inflammatory to anti-inflammatory cytokine ratio. As shown in Table 2, cigarettes per day remained a significant predictor (b = 0.29, p < 0.01) after removal of the outlier, while cannabis use showed no independent effect. Next, to examine whether increased GPR15+ Th cell percent accounted for the relationship between increased inflammation and cigarette smoking according to hypothesis H2a, GPR15+ Th cell percent was entered as an additional predictor (Model 1B). This resulted in an improved overall model fit (R2 increased from 13.0% to 24.4%), and GPR15+ Th cell percent was a significant predictor (b = 0.33, p < 0.01), whereas cigarettes per day was no longer a significant predictor. Mediation analysis using the simple path model equation indicated that GPR15+ Th cell percent mediated 47% of the variance in the pro-inflammatory to anti-inflammatory cytokine ratio due to cigarettes per day. Finally, additional immunological predictors were entered into the model as controls. In the resulting model (Model 1C) GPR15+ Th cell percent remained a significant predictor (b = 0.34, p < 0.01) of the pro-inflammatory to anti-inflammatory cytokine ratio, whereas no other variables were significant predictors, consistent with hypothesis H3.
Table 2.
Multiple linear regression results for pro-inflammatory to anti-inflammatory cytokine ratio regressed against tobacco and cannabis smoking intensity, GPR15+ Th cell percent, and immunological variables.
| Model | Model 1A | Model 1B | Model 1C | |||
|---|---|---|---|---|---|---|
| Parameters | b | p-value | b | p-value | b | p-value |
| Cigarettes per day | 0.290 | 0.006 | 0.162 | 0.12 | 0.143 | 0.21 |
| Scaled cannabis use | 0.459 | 0.35 | 0.0004 | > 0.99 | 0.113 | 0.83 |
| GPR15+ Th cell percent | 0.333 | 0.003 | 0.337 | 0.005 | ||
| Age (years) | −0.062 | 0.84 | ||||
| Gender (male) | −0.180 | 0.83 | ||||
| Race (AA) | −0.647 | 0.53 | ||||
| BMI | 0.064 | 0.15 | ||||
| Stress | 0.058 | 0.52 | ||||
| PHQ | −0.082 | 0.41 | ||||
| NSAIDs | 0.172 | 0.65 | ||||
| R-square | 0.130 | 0.244 | 0.223 | |||
| p-value | < 0.001 | < 0.001 | < 0.001 | |||
Cigarettes per day calculated by average consumption over the past month. Scaled cannabis use indicates average use over the past year on a zero to two scale. BMI refers to body mass index. Stress refers to the Perceived Stress Scale total score. PHQ refers to the Patient Health Questionnaire depression module (PHQ-9) total score. NSAIDs refers to nonsteroidal anti-inflammatory drug consumption in daily 200 mg equivalent doses of ibuprofen, naproxen, and aspirin. Cotinine positivity indicates serum values of > 1 ng/mL, while THC positivity indicates > 0.5 ng/mL. Regression results shown for n = 61 participants.
Because of the skewed nature of the cytokine ratio (Shapiro-Wilk W = 0.785, p < 0.001), the above procedures were repeated after log transformation of this variable, which resulted in a near-normal distribution of the predictor (W 0.955, p = 0.03). In these models cigarettes per day and scaled cannabis use were no longer significant predictors, but GPR15+ Th cell percent remained a significant predictor both in conjunction with the two smoking variables (b = 0.042, p < 0.05) and in conjunction with both smoking variables and other immunological predictors (b = 0.041, p < 0.05).
Lastly, although there was no significant correlation between CRP and GPR15+ Th cell percent, in contrast to expectations (hypothesis H1b), the possibility that the strong effect of BMI and differential effects of tobacco vs. cannabis consumption might obscure such a relationship was considered and exploratory analyses performed to assess for such an impact. Similar to the mediation analyses above, BMI was first entered as a predictor, showing an R2 of 26.4%, and the model was highly significant (p < 0.001) (Model 2A). Cigarettes per day and scaled cannabis use were then entered as additional predictors of CRP (Model 2B), which did not improve model fit. Finally, the addition GPR15+ Th cell percent as a predictor (Model 2C) also did not improve model fit. Results of this analysis, provided in Supplemental Table 3, showed no significant relationships between GPR15+ Th cell percent, tobacco, or cannabis consumption and CRP after accounting for BMI, inconsistent with hypothesis H1b.
4. Conclusions
Multiple lines of evidence support a strong association between smoke exposure and increased GPR15 expression, sufficient that it has been suggested that the proportion of T cells expressing GPR15 may be a reliable biomarker for smoking [38]. What is less well understood is whether causal relationships exist between increases in GPR15-expressing T cells and increased risk of inflammatory diseases in smokers. Here, we sought to increase understanding of these relationships by direct measurement of inflammatory biomarkers and the proportion of Th cells expressing GPR15 in a young adult cohort with a high rate of tobacco and cannabis smoking.
We found a significant relationship between tobacco smoking and the ratio of pro-inflammatory to anti-inflammatory cytokines, consistent with prior studies of inflammation in smokers. In followup mediation analyses we also found tentative evidence that GPR15+ Th cell percent mediated nearly half of the relationship between smoking and a pro-inflammatory cytokine milieu. This relationship was seen even after controlling for multiple variables that could potentially impact immune function, supporting the hypothesis that GPR15+ Th cells may be drivers of inflammation in smokers.
The relationship between pro-inflammatory cytokine balance and smoking was primarily driven by cigarettes per day, with no evidence of an independent effect of cannabis use in this sample, despite evidence that some cannabinoids have an anti-inflammatory effect [41]. However, this may have been due to a number of factors, including our small sample size, less informative reporting of overall cannabis intake, or simply a preponderance of tobacco smoking in terms of total smoke exposure. Going forward, examination of inflammatory signatures in cannabis smokers who use little or no tobacco may be informative.
Against expectations, however, there was no evidence of increased CRP in smokers or any relationship between CRP and GPR15+ Th cell percent. There are multiple possible reasons for this. The first is the relatively young age of the cohort. While the high rate of tobacco and cannabis smoking, elevated BMI, and self-report measures of stress and depressive symptoms indicate this cohort is at elevated risk for inflammatory diseases, smoking-related disease processes may have not yet become sufficiently established to provoke elevations in CRP. However, given evidence that smokers in this cohort, particularly those with elevated GPR15+ Th cells, demonstrate an increased pro-inflammatory cytokine balance, and the fact that CRP production is stimulated by IL-1, IL-6, and TNF-a, all of which were included in our pro-inflammatory cytokine index, we may expect to see increased evidence of systemic inflammation with continued follow up of this cohort [59-61].
The observation that CRP was strongly associated with BMI is reassuring with respect to the above results in that it suggests the absence of association with GPR15+ Th cell percent was not due to assay failure. In addition to the liver, CRP is produced in adipocytes and its association with BMI is well documented [61, 62]. However, given the strength of this association in our sample, future examinations of the relationship between smoking-induced increases in GPR15+ Th cells may be more successful in a population with a lower rate of obesity, which could mask more subtle drivers of inflammation.
Limitations of this study stem include its cross-sectional design, possible poor performance of the cotinine assay, and its small sample size. In particular, the limited sample size may have precluded observation of significant relationships between GPR15+ Th cell percent and individual cytokine levels, which could be informative. The cross-sectional design also renders us unable to make any strong conclusions about the direction of causality. The significant correlations seen between GPR15+ Th cell percent and the cytokine ratio could be equally consistent with the hypothesis that a pro-inflammatory cytokine milieu drives an increase in GPR15+ Th cell percent in smokers. Our mediation analyses also suffer from some limitations that preclude us from drawing strong conclusions about the temporal relationships between tobacco and cannabis smoking, the balance of inflammatory and anti-inflammatory cytokines, and GPR15+ Th cell percent. In particular, cross-sectional mediational analyses are known to generate biased parameter estimates even when correctly specified [63]. Lastly, although we continued to observe a significant association between GPR15+ Th cell percent and the cytokine ratio following log transformation of the cytokine ratio due to skewness, we no longer observed a significant relationship between our smoking variables and the transformed cytokine ratio, precluding any analysis of mediation. Thus, although our findings are plausible and consistent with prior expectations, a larger, longitudinal study design would be needed to definitively establish any causal relationship between cytokine balance and GPR15+ Th cell percent in smokers. Further study is also needed to establish whether our cytokine ratio demonstrates adequate predictive validity as a derived variable in other, more well-defined diseases processes.
Strengths of this study include direct measurement of inflammatory biomarkers and GPR15+ Th cell percent in a population-based cohort, and the inclusion of appropriate health and lifestyle variables known to influence the immune system.
Going forward, further examination of the relationship between GPR15+ Th cell percent and systemic inflammation is warranted from a public health perspective because of our findings of a link between GPR15+ Th cell percent and a pro-inflammatory cytokine milieu in smokers, and because of the fact that African Americans are disproportionately affected by smoking-associated illnesses [64, 65] and have shown greater systemic inflammation compared to European American smokers [66].
Supplementary Material
GPR15 expression on helper T cells associated with smoking habits and inflammatory cytokines
GPR15 expression mediates relationship between smoking habits and inflammatory cytokine milieu
C-reactive protein associated with BMI but not smoking or GPR15 expression on helper T cells
Acknowledgments:
The data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran's Administration Medical Center.
Funding:
AA is supported by K12DA000357. RP is supported by R44 CA213507, R44AA022041 and R01DA037648. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- 1.DHHS. The Health Consequences of Smoking - 50 Yers of Progress: A Report of the Surgeon General: Office of the Surgeon General; 2014.
- 2.Jamal A, Phillips E, Gentzke AS, Homa DM, Babb SD, King BA, et al. Current Cigarette Smoking Among Adults - United States, 2016. Mmwr-Morbidity and Mortality Weekly Report. 2018;67(2):53–9. PubMed PMID: WOS:000422763400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291(10):1238–45. doi: 10.1001/jama.291.10.1238. PubMed PMID: 15010446. [DOI] [PubMed] [Google Scholar]
- 4.Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF. Systemic effects of smoking. Chest. 2007;131(5):1557–66. [DOI] [PubMed] [Google Scholar]
- 5.Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. Journal of autoimmunity. 2010;34(3):J258–J65. [DOI] [PubMed] [Google Scholar]
- 6.Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV, Tysk C, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62(4):630–49. [DOI] [PubMed] [Google Scholar]
- 7.Chang K, Yang SM, Kim SH, Han KH, Park SJ, Shin JI. Smoking and rheumatoid arthritis. International journal of molecular sciences. 2014;15(12):22279–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riise T, Nortvedt MW, Ascherio A. Smoking is a risk factor for multiple sclerosis. Neurology. 2003;61(8):1122–4. [DOI] [PubMed] [Google Scholar]
- 9.Freiman A, Bird G, Metelitsa AI, Barankin B, Lauzon GJ. Cutaneous effects of smoking. Journal of cutaneous medicine and surgery. 2004;8(6):415–23. [DOI] [PubMed] [Google Scholar]
- 10.Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S, editors. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clinic Proceedings; 2006: Elsevier. [DOI] [PubMed] [Google Scholar]
- 11.Tanni SE, Pelegrino NR, Angeleli AY, Correa C, Godoy I. Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in COPD patients. Journal of Inflammation. 2010;7(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEvoy JW, Nasir K, DeFilippis AP, Lima JA, Bluemke DA, Hundley WG, et al. Relationship of Cigarette Smoking With Inflammation and Subclinical Vascular Disease The Multi-Ethnic Study of Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2015;35(4):1002–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edinger AL, Hoffman TL, Sharron M, Lee B, O'Dowd B, Doms RW. Use of GPR1, GPR15, and STRL33 as coreceptors by diverse human immunodeficiency virus type 1 and simian immunodeficiency virus envelope proteins. Virology. 1998;249(2):367–78. [DOI] [PubMed] [Google Scholar]
- 14.Krumbiegel M, Kirchhoff F. Coreceptor usage of BOB/GPR15 and Bonzo/STRL33 by primary isolates of human immunodeficiency virus type 1. Journal of general virology. 1999;80(5):1241–51. [DOI] [PubMed] [Google Scholar]
- 15.Blaak H, Boers P, Gruters R, Schuitemaker H, Van Der Ende M, Osterhaus A. CCR5, GPR15, and CXCR6 are major coreceptors of human immunodeficiency virus type 2 variants isolated from individuals with and without plasma viremia. Journal of virology. 2005;79(3):1686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the Small Intestine and Colon. Gastroenterology. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ocon B, Pan J, Dinh TT, Chen W, Ballet R, Bscheider M, et al. A Mucosal and Cutaneous Chemokine Ligand for the Lymphocyte Chemoattractant Receptor GPR15. Front Immunol. 2017;8:1111. doi: 10.3389/fimmu.2017.01111. PubMed PMID: 28936214; PubMed Central PMCID: PMCPMC5594226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suply T, Hannedouche S, Carte N, Li J, Grosshans B, Schaefer M, et al. A natural ligand for the orphan receptor GPR15 modulates lymphocyte recruitment to epithelia. Sci Signal. 2017;10(496):eaal0180. doi: 10.1126/scisignal.aal0180. PubMed PMID: 28900043. [DOI] [PubMed] [Google Scholar]
- 19.Cartwright A, Schmutz C, Askari A, Kuiper JH, Middleton J. Orphan receptor GPR15/BOB is up-regulated in rheumatoid arthritis. Cytokine. 2014;67(2):53–9. doi: 10.1016/j.cyto.2014.02.015. PubMed PMID: WOS:000335629400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen LP, Pan J, Dinh TT, Hadeiba H, O'Hara E 3rd, Ebtikar A, et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol. 2015;16(2):207–13. doi: 10.1038/ni.3079. PubMed PMID: 25531831; PubMed Central PMCID: PMCPMC4338558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer A, Zundler S, Atreya R, Rath T, Voskens C, Hirschmann S, et al. Differential effects of α4β7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut. 2015:gutjnl-2015-310022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamczyk A, Gageik D, Frede A, Pastille E, Hansen W, Rueffer A, et al. Differential expression of GPR15 on T cells during ulcerative colitis. JCI insight. 2017;2(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ammitzbøll C, Marina R, Börnsen L, Petersen ER, McWilliam O, Ratzer R, et al. GPR15+ T cells are Th17 like, increased in smokers and associated with multiple sclerosis. Journal of Autoimmunity. 2018. [DOI] [PubMed] [Google Scholar]
- 24.Chen C, Wu N, Duan Q, Yang H, Wang X, Yang P, et al. C10orf99 contributes to the development of psoriasis by promoting the proliferation of keratinocytes. Sci Rep. 2018;8(1):8590. doi: 10.1038/s41598-018-26996-z. PubMed PMID: 29872130; PubMed Central PMCID: PMCPMC5988722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Wang X, Xiong Y, Li C-D, Xu Q, Shen L, et al. An integrated pan-cancer analysis and structure-based virtual screening of GPR15. International Journal of Molecular Sciences. 2019;20(24):6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordahl KM, Phipps AI, Randolph TW, Tindle HA, Liu S, Tinker LF, et al. Differential DNA methylation in blood as a mediator of the association between cigarette smoking and bladder cancer risk among postmenopausal women. Epigenetics. 2019;14(11):1065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koks G, Uudelepp ML, Limbach M, Peterson P, Reimann E, Koks S. Smoking-induced expression of the GPR15 gene indicates its potential role in chronic inflammatory pathologies. Am J Pathol. 2015;185(11):2898–906. doi: 10.1016/j.ajpath.2015.07.006. PubMed PMID: 26348578. [DOI] [PubMed] [Google Scholar]
- 28.De Leon J, Dadvand M, Canuso C, White AO, Stanilla JK, Simpson GM. Schizophrenia and smoking: an epidemiological survey in a state hospital. The American journal of psychiatry. 1995. [DOI] [PubMed] [Google Scholar]
- 29.Petralia MC, Ciurleo R, Saraceno A, Pennisi M, Basile MS, Fagone P, et al. Meta-Analysis of Transcriptomic Data of Dorsolateral Prefrontal Cortex and of Peripheral Blood Mononuclear Cells Identifies Altered Pathways in Schizophrenia. Genes. 2020;11(4):390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88(4):450–7. doi: 10.1016/j.ajhg.2011.03.003. PubMed PMID: 21457905; PubMed Central PMCID: PMC3071918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;21(13):3073–82. doi: 10.1093/hmg/dds135. PubMed PMID: 22492999; PubMed Central PMCID: PMC3373248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet. 2013;132(9):1027–37. doi: 10.1007/s00439-013-1311-6. PubMed PMID: 23657504; PubMed Central PMCID: PMCPMC3744600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812. doi: 10.1371/journal.pone.0063812. PubMed PMID: 23691101; PubMed Central PMCID: PMCPMC3656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clinical Epigenetics. 2015;7(1):113. doi: 10.1186/s13148-015-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Razin A CpG methylation, chromatin structure and gene silencing–a three - way connection. The EMBO journal. 1998;17(17):4905–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su D, Wang X, Campbell MR, Porter DK, Pittman GS, Bennett BD, et al. Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PloS one. 2016;11(12):e0166486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauer M, Fink B, Thürmann L, Eszlinger M, Herberth G, Lehmann I. Tobacco smoking differently influences cell types of the innate and adaptive immune system—indications from CpG site methylation. Clinical Epigenetics. 2016;8(1):83. doi: 10.1186/s13148-016-0249-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bauer M, Hackermüller J, Schor J, Schreiber S, Fink B, Pierzchalski A, et al. Specific induction of the unique GPR15 expression in heterogeneous blood lymphocytes by tobacco smoking. Biomarkers. 2019;24(3):217–24. [DOI] [PubMed] [Google Scholar]
- 39.Bauer M, Fink B, Seyfarth H-J, Wirtz H, Frille A. Tobacco-smoking induced GPR15-expressing T cells in blood do not indicate pulmonary damage. BMC pulmonary medicine. 2017;17(1):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jhun MA, Smith JA, Ware EB, Kardia SL, Mosley TH Jr, Turner ST, et al. Modeling the causal role of DNA methylation in the association between cigarette smoking and inflammation in African Americans: a 2-step epigenetic Mendelian randomization study. American journal of epidemiology. 2017;186(10):1149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russo EB, McPartland JM. Cannabis is more than simply Δ 9-tetrahydrocannabinol. Psychopharmacology. 2003;165(4):431–2. [DOI] [PubMed] [Google Scholar]
- 42.Padgett DA, Glaser R. How stress influences the immune response. Trends in immunology. 2003;24(8):444–8. [DOI] [PubMed] [Google Scholar]
- 43.Steptoe A, Kunz-Ebrecht S, Brydon L, Wardle J. Central adiposity and cortisol responses to waking in middle-aged men and women. International Journal of Obesity. 2004;28(9):1168. [DOI] [PubMed] [Google Scholar]
- 44.Cho JY. Immunomodulatory effect of nonsteroidal anti-inflammatory drugs (NSAIDs) at the clinically available doses. Archives of pharmacal research. 2007;30(1):64. [DOI] [PubMed] [Google Scholar]
- 45.Hage FG, Szalai AJ. C-reactive protein gene polymorphisms, C-reactive protein blood levels, and cardiovascular disease risk. Journal of the American College of Cardiology. 2007;50(12):1115–22. [DOI] [PubMed] [Google Scholar]
- 46.Kogan SM, Lei MK, Grange CR, Simons RL, Brody GH, Gibbons FX, et al. The contribution of community and family contexts to African American young adults' romantic relationship health: a prospective analysis. J Youth Adolesc. 2013;42(6):878–90. doi: 10.1007/s10964-013-9935-3. PubMed PMID: 23494451; PubMed Central PMCID: PMCPMC3653993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simons RL, Simons LG, Lei MK, Landor AM. Relational schemas, hostile romantic relationships, and beliefs about marriage among young African American adults. Journal of Social and Personal Relationships. 2012;29(1):77–101. doi: 10.1177/0265407511406897. PubMed PMID: WOS:000299735300004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simons RL, Lei M-K, Beach SR, Barr AB, Simons LG, Gibbons FX, et al. Discrimination, segregation, and chronic inflammation: Testing the weathering explanation for the poor health of Black Americans. Developmental psychology. 2018;54(10):1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroenke K, Spitzer RL, Williams JB. The PHQ - 9: validity of a brief depression severity measure. Journal of general internal medicine. 2001;16(9):606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen S, Kamarck T, Mermelstein R. Perceived stress scale. Measuring stress: A guide for health and social scientists. 1994:235–83. [Google Scholar]
- 51.Philibert RA, Beach SRH, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clinical Epigenetics. 2013;5(19). doi: Artn 19 10.1186/1868-7083-5-19. PubMed PMID: WOS:000329455500001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012;7(11):1331–8. doi: 10.4161/epi.22520. PubMed PMID: 23070629; PubMed Central PMCID: PMCPMC3499333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2012. ISBN 3-900051-07-0; 2014. [Google Scholar]
- 54.Mariani JJ, Brooks D, Haney M, Levin FR. Quantification and comparison of marijuana smoking practices: Blunts, joints, and pipes. Drug and alcohol dependence. 2011;113(2–3):249–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chou R MM, Nakamoto E, Griffin J. Analgesics for Osteoarthritis: An Update of the 2006 Comparative Effectiveness Review.. Rockville, MD: Agency for Healthcare Research and Quality; 2011. [PubMed] [Google Scholar]
- 56.Wei T, Simko V, Levy M, Xie Y, Jin Y, Zemla J. Package ‘corrplot’. Statistician. 2017;56:316–24. [Google Scholar]
- 57.Hebbali A. Package ‘olsrr’ 2020. [cited 2020 Nov 23]. Available from: https://cran.r-project.org/web/packages/olsrr/olsrr.pdf. [Google Scholar]
- 58.Cook RD. Detection of influential observation in linear regression. Technometrics. 1977;19(1):15–8. [Google Scholar]
- 59.Szalai AJ, van Ginkel FW, Dalrymple SA, Murray R, McGhee JR, Volanakis JE. Testosterone and IL-6 requirements for human C-reactive protein gene expression in transgenic mice. The Journal of Immunology. 1998;160(11):5294–9. [PubMed] [Google Scholar]
- 60.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. Journal of Biological Chemistry. 1996;271(16):9503–9. [DOI] [PubMed] [Google Scholar]
- 61.Sproston N, Ashworth J. Role of C-reactive protein at sites of inflammation and infection. Front Immunol 9: 754. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends in immunology. 2004;25(1):4–7. [DOI] [PubMed] [Google Scholar]
- 63.Maxwell SE, Cole DA. Bias in cross-sectional analyses of longitudinal mediation. Psychological methods. 2007;12(1):23. [DOI] [PubMed] [Google Scholar]
- 64.Control CfD, Prevention. Racial disparities in smoking-attributable mortality and years of potential life lost---Missouri, 2003-2007. MMWR Morbidity and mortality weekly report. 2010;59(46):1518. [PubMed] [Google Scholar]
- 65.Haiman CA, Stram DO, Wilkens LR, Pike MC, Kolonel LN, Henderson BE, et al. Ethnic and racial differences in the smoking-related risk of lung cancer. N Engl J Med. 2006;354(4):333–42. doi: 10.1056/NEJMoa033250. PubMed PMID: 16436765. [DOI] [PubMed] [Google Scholar]
- 66.Paalani M, Lee JW, Haddad E, Tonstad S. Determinants of inflammatory markers in a bi-ethnic population. Ethnicity & disease. 2011;21(2):142. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.