

Abstract
Human hypertension caused by in-frame deletion of CULLIN3 exon-9 (Cul3∆9) is driven by renal and vascular mechanisms. We bred conditionally activatable Cul3∆9 transgenic mice with tamoxifen-inducible Tie2-CREERT2 mice to test the importance of endothelial Cul3. The resultant mice (E-Cul3∆9) trended towards elevated nighttime blood pressure (BP) correlated with increased nighttime activity, but displayed no difference in daytime BP or activity. Male and female E-Cul3∆9 mice together exhibited a decline in endothelial-dependent relaxation in carotid artery. Male but not female E-Cul3∆9 mice displayed severe endothelial dysfunction in cerebral basilar artery. There was no impairment in mesenteric artery and no difference in smooth muscle function, suggesting the effects of Cul3∆9 are arterial bed-specific and sex-dependent. Expression of Cul3∆9 in primary mouse aortic endothelial cells decreased endogenous Cul3 protein, phosphorylated (S1177) endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) production. Protein phosphatase (PP) 2A, a known Cul3 substrate, dephosphorylates eNOS. Cul3∆9-induced impairment of eNOS activity was rescued by a selective PP2A inhibitor okadaic acid, but not by a PP1 inhibitor tautomycetin. Because NO deficiency contributes to salt-induced hypertension, we tested the salt-sensitivity of E-Cul3∆9 mice. While both male and female E-Cul3∆9 mice developed salt-induced hypertension and renal injury, the pressor effect of salt was greater in female mutants. The increased salt-sensitivity in female E-Cul3∆9 mice was associated with decreased renovascular relaxation and impaired natriuresis in response to a sodium load. Thus, CUL3 mutations in the endothelium may contribute to human hypertension in part through decreased endothelial NO bioavailability, renovascular dysfunction, and increased salt-sensitivity of BP.
Keywords: Endothelium, Vasodilation, Nitric Oxide, Hypertension, Salt Sensitivity
Graphical Abstract
Graphical Abstract.
Assessment of fraction new over time reveals insight into proteostatic maintenance in muscle collagen.
Introduction
Cullin3 (CUL3) provides the structural backbone for the CUL3-RING ubiquitin ligase complex (CRL3).1 While E3 ubiquitin ligase Rbx1 binds to the CUL3 C-terminus to form the catalytic core, Broad-complex, Tramtrack, and Bric-à-brac (BTB) domain-containing adaptors interact with CUL3 N-terminus to recruit substrates for ubiquitination.2 The CUL3 ubiquitin ligase regulates cardiovascular and renal function through physiological turnover of key substrates involved in redox biology, vascular tone, and renal electrolyte transport.1 Mutations in CUL3 (causing in-frame deletion of exon 9, termed CUL3∆9) cause familial hyperkalemic hypertension (FHHt) characterized by hypertension, hyperkalemia, and metabolic acidosis in humans.3
The hyperkalemia and metabolic acidosis phenotypes are caused by pathogenic impairment of CUL3-mediated ubiquitination of with-no-lysine kinases (WNKs) in the kidney.3 The resultant accumulation of WNKs upregulates two kinases, sterile 20/SPS-1 related proline/alanine-rich kinase (SPAK) and oxidative stress response kinase-1 (OSR1) that phosphorylates sodium chloride cotransporter (NCC) in the distal convoluted tubule (DCT).4,5 While the electrolyte abnormalities are primarily driven by altered pathways in the distal nephron, the hypertension in patients carrying CUL3∆9 is likely attributable to abnormalities in both renal and vascular mechanisms. For instance, we demonstrated that selective expression of CUL3∆9 or genetic deletion of CUL3 in vascular smooth muscle causes vascular dysfunction and hypertension via 1) augmented RhoA/Rho kinase signaling, and 2) impaired nitric oxide (NO)/cyclic GMP signaling.6,7 These studies provided compelling evidence that CUL3 independently regulates blood pressure (BP) through its actions in the vasculature. However, it is unclear whether endothelial CUL3∆9 contributes to the FHHt phenotypes.
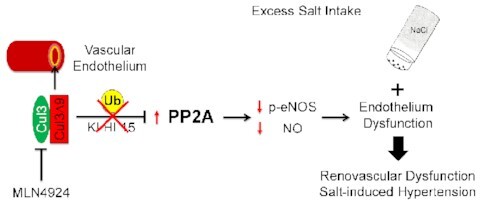
Except for nuclear factor-erythroid factor 2-related factor 2 (NRF2) dependent antioxidant mechanisms,8 where Keap1 targets NRF2 to the CRL3-complex for degradation in the absence of oxidant stress, little is known about the role of CUL3 in the endothelium. Endothelial NO synthase (eNOS) generates and releases NO to signal vasorelaxation in the vascular smooth muscle. Endothelial dysfunction caused by decreased NO bioavailability contributes to the pathogenesis of hypertension through increased total peripheral resistance (TPR), renal vascular resistance (RVR), and impaired renal blood flow (RBF).9–11 Decreased endothelial NO bioavailability also plays a key role in the pathogenesis of salt-sensitive (SS) hypertension, which accounts for over 50% of essential hypertension.12,13
The activity of eNOS is regulated transcriptionally, by post-translational modifications,14 and by the availability of its substrate L-arginine and the cofactor tetrahydrobiopterin.15,16 For example, peroxisome proliferator-activated receptor γ (PPARγ) activation promotes NO biogenesis through upregulation of eNOS expression and AMPK/Akt mediated eNOS phosphorylation (serine1177).17 Whereas phosphorylation of key serine residues promotes eNOS activation and NO production, protein phosphatase 2A (PP2A) mediated dephosphorylation of the Akt-eNOS complex decreases eNOS activity and contributes to endothelial dysfunction in obesity, diabetes, and hypertension.18–20 CUL3 regulates PP2A activity by targeting its subunits for degradation via the ubiquitin-proteasome pathway.21,22 Thus, loss of CUL3 function in the vascular endothelium may lead to accumulation of PP2A and sustained dephosphorylation of eNOS.
In the present study, we hypothesized that endothelial CUL3 plays a key role in regulating eNOS activity, endothelial function, and BP. We have made the intriguing observation that mice selectively expressing the human hypertension causing CUL3∆9 mutation in the vascular endothelium (E-CUL3∆9) exhibit impaired eNOS activation, decreased NO production, and endothelial dysfunction via PP2A-dependent mechanisms. Moreover, E-CUL3∆9 mice were susceptible to salt-induced hypertension in a sex-specific manner. To our knowledge, this is the first study to investigate the role of endothelial CUL3 in vascular function and BP regulation.
Materials and Methods
Experimental Animals
All protocols were approved by the Animal Care and Use Committees at the Medical College of Wisconsin and University of Iowa. Mouse husbandry and care in the study followed the guidelines set forth by the National Institutes of Health (NIH). Transgenic mice with a C57Bl/6 X SJL (B6SJL) background were designed to inducibly express a human hypertension-causing mutation CUL3∆9 and the tdTomato reporter in response to activation of Cre recombinase activated by tamoxifen (CAG-CUL3∆9, Figure 1A). The mice were backcrossed to C57BL/6 and most of the mice used in experiments were backcrossed for 5–10 generations. Selective expression of the transgenes in vascular endothelium were achieved by crossing the CAG-CUL3∆9 mice with transgenic mice expressing Cre recombinase under the control of Tie2 promotor (TEK-CREERT2).23 The resultant double positive mice (Cre+CUL3∆9+) termed E-CUL3∆9 were the experimental mice while the single positive and double negative littermates served as controls.
Figure 1.

Tissue-specificity of the Mouse Model. (A) Schematic illustration of constructs used to generate the inducible CAG-CUL3∆9 transgenic mice and the tamoxifen-responsive TEK-CREERT2 mice.6,30 The CAG-CUL3∆9 transgenic mice were generated with CUL3∆9 cDNA cloned by overlap splice extension PCR as previously described.6 (B) These two lines were crossed to generate four genotypes. The Cre+CUL3∆9−, Cre−CUL3∆9+, and Cre−CUL3∆9− mice served as controls, while the endothelium-specific Cre+CUL3∆9+ mice, termed E-CUL3∆9, were experimental mice. Mice at 8–10 weeks of age received five consecutive tamoxifen injections (administered via i.p. at the same time of the day). Four weeks after completion of the tamoxifen regimen, kidneys were harvested, and magnetic activated cell sorting was performed to separate non-endothelial cells (EC− fraction) from the endothelial cells (EC+ fraction). (C) Western blot analysis of the EC− and EC+ fractions. Because of auto-ubiquitination mediated self-destruction, CUL3∆9 protein was not detected. A tdTomato reporter was used as a surrogate of the CUL3∆9 transgene. ER, estrogen receptor.
Some experiments were completed at the University of Iowa and all mice were maintained on a standard laboratory rodent chow diet containing 0.8% sodium chloride or 0.3% sodium (the NIH-31 Modified Open Formula Mouse/Rat Sterilizable Diet, Teklad Catalog # 7013) at baseline. Carotid artery and basilar artery, a cerebral resistance vessel, were studied because these vessels exhibit severe impairment of vasodilation capacity in response to genetic variations and excess salt intake.6,24
Additional experiments were conducted at the Medical College of Wisconsin. All mice were weaned to a normal salt diet (NSD) containing 0.3% sodium chloride equivalent to 0.1% sodium (Teklad Catalog #2920X). All mice were fed the standard normal salt chow diet at baseline until they were provided with 4% high salt diet (HSD) (Teklad Catalog # TD. 03095) for 5 weeks. At the end of study, mice were euthanized with an overdose of a pentobarbital-based veterinary euthanasia solution (150 mg/kg, i.p.) and kidneys were harvested for renovascular function and molecular studies.
PCR Analysis of Genomic DNA Recombination
Lung tissues were harvested from tamoxifen treated control and E-CUL3∆9 mice as well as E-CUL3∆9 mice without tamoxifen treatment. Samples were frozen in liquid nitrogen and stored in −80°C until being analyzed. Genomic DNA was extracted using DNeasy blood and tissue kit (Qiagen, #69504) following the manufacturer's protocol. We designed primers to amplify the 1300bp band for the intact fragment of the transgenic construct or the 430bp band for the recombined fragment where the lox-STOP-lox sequence of the construct was excised by Cre recombinase.6 The following primer sequences were used: CUL3∆9 recombinant forward, 5’-CCTCTGCTAACCATGTTCATGCCTTCTTC-3’, and CUL3∆9 recombinant reverse, 5’-GCCTTAAGAGCTGTAATTGAACTGGGAGTG-3’. PCR was then performed using Taq DNA polymerase (Invitrogen, #18038018) following the manufacturer's protocol.
Radiotelemetry
BP, heart rate (HR), and activity was measured using radiotelemetry as previously described.6,24 Briefly, control and E-CUL3∆9 mice at 3–4 months of age were employed in age- and sex-matched cohorts, and telemetry implantation was performed under anesthesia with a mixture of ketamine (87.5 mg/kg) and xylazine (12.5 mg/kg). A catheter was inserted into the left common carotid artery, and a transmitter was subcutaneously placed along the right flank. Mice were allowed to recover for 10 d before BP, HR, and activity were recorded for 10 min every hour for a total of 7 d. Since tamoxifen was given 2 wk before telemetry implantation, baseline radiotelemetry recordings were taken 4 wk after the completion of tamoxifen treatments.
In initial cohorts of animals, data were collected and exported using the DataQuest ART Software (Data Sciences International, St Paul, MN). Due to an upgrade of hardware and software, later cohorts were measured and recorded using the Ponemah software (Data Sciences International, St Paul, MN). While the software upgrade had no impact in BP and HR recordings, the Ponemah software generated activity values that were exactly 100-fold less than those derived from the DataQuest software because of displacement of the decimal point by two digits in the activity algorithm (Figure S2A). Details of activity data derivation are provided by the manufacturer at https://support.datasci.com/hc/en-us/articles/115005030328. While the two datasets are different by 100-fold in magnitude, they showed the same circadian rhythm and inter-group trends. To combine two sets of data, activity derived from the Ponemah software were multiplied by 100 and plotted with activity data derived from the DataQuest software.
Power Spectral Analysis
Continuous radiotelemetry recording 4 weeks after tamoxifen treatment was used in power spectral analysis for HR variability and arterial pressure variability as previously described.25 Beat-by-beat HR and BP time series were derived from the BP waveforms and converted to an equidistant sampling rate using cubic spline interpolation. Those equidistant HR time series were subjected to a fast Fourier transform to calculate spectral power in the very low frequency (VLF, 0.02–0.20 Hz), low frequency (LF, 0.2–0.6 Hz, reflecting mainly sympathetic cardiac modulation) and high frequency (HF, 1.0–5.0 Hz, reflecting para-sympathetic cardiac modulation) bands. Relative LF and HF were calculated as the relative value of each power component in proportion to the total power minus the VLF component. The LF/HF ratio of HR variability and baroreflex gain was used as an indication of sympathetic nerve activity to the heart; while the LF component of the arterial pressure was affected by endothelial derived NO and sympathetic modulation of vascular tone.25
Uninephrectomy-High Salt Diet Study (UNxHSD)
Both control and E-CUL3∆9 mice received tamoxifen injections at 8–10 wk of age. Immediately after tamoxifen, uninephrectomy (UNx) was performed under anesthesia in which the right kidney was surgically removed. The right kidney was removed because its anatomy is technically more accessible for surgery. Briefly, a 1–2 cm horizontal incision was made in the right side of the mouse between the lower edge of the rib cage and the hip. Then, the skin and abdominal muscles were pulled open with rodent surgical tractors to expose the right kidney. Next, the right renal artery and vein were ligated by 6–0 suture to stop blood flow to allow the right kidney to be excised with a surgical blade. The abdominal muscle was sutured close before the skin was sutured with 6–0. A single dose of meloxicam was given intraperitoneally (i.p.) for postoperative analgesic and the surgical site was treated with a veterinary antibiotic ointment containing triple antibiotics (Bacitracin-neomycin-polymyxin) to prevent infection.
Telemetry was implanted 30 d after uninephrectomy to ensure complete recovery from the surgery and re-establishment of hemodynamic homeostasis. Mice were allowed another 10 d to recover from the telemetry surgery. BP was continuously recorded in the NSD baseline (Day −5 to Day 0, UNxNSD) and the first 4 wk of HSD (Day 0 to Day 28, UNxHSD). Because the BP of both male and female mice reached a peak by HSD week 3 and plateaued through week 4, BP in weeks 3–4 was averaged and used to compare with the NSD baseline. Two mice were excluded from the UNxHSD study because of a kidney cyst or an infarction in the remaining kidney identified post-mortem.
In the UNxHSD protocol, tGFR measurement, 48-h metabolic cage study, and the acute saline load were performed consecutively following the same time schedule for all cohorts involved in this manuscript. This was a 4-d experiment (NSD day −9 to day −5 and HSD day 29 to day 33) with tGFR at 12PM–5PM day 1, 48-h metabolic cages from 9AM day 2 through 9AM day 4 (data from the second 24-h period were used, i.e. 9AM day 3 through 9AM day 4), and acute saline load from 9AM–1PM day 4 immediately after collection of 24 h urine from the prior day. The detailed description for each of these procedures are provided below.
Metabolic Cage Studies
Mice were individually placed into metabolic cages (MMC100, Hatteras Instruments) for 3 d immediately before the NSD baseline telemetry recording and in the 5th wk of HSD. On both occasions, mice were acclimated to their cages in the first 24 h. Food intake and water intake in the second 24 h were recorded while urine was collected and measured. Sodium ingestion was calculated from food intake, and urinary sodium excretion was analyzed for the computation of apparent sodium retention as described.24 A subset of mice in the metabolic cage studies were also implanted with radiotelemetry. This provided data for the pressure-natriuresis plot.
Acute Natriuretic Challenge
Immediately after urine collection from the second 24-h time period during the metabolic cage study, natriuretic challenge studies were carried out in a subset of mice to assess renal function as described.24 In brief, mice were injected i.p. with a saline load equal to 10% of their body weight, and then, urine output in the next 4 h was collected. To assess acute natriuretic capacity, sodium excreted in the 4-h urine was calculated as percentages of the sodium injected. All urinary sodium measurements in this manuscript were analyzed using a flame photometer (Cole-Parmer, IL) by one blinded technician.
Measurement of Transdermal Glomerular Filtration Rate (tGFR)
In the UNxHSD study, tGFR was measured one day before the metabolic cage studies. Briefly, an area of 2 x 2 cm of the dorsolateral skin was shaved, and then Nair was applied topically to remove hair. The fluorescent monitor was attached to the clean prepared area and secured with tape. After 3 min of baseline transmission under isoflurane anesthesia (2%), mice received FITC-sinistrin (0.07 mg per gram body weight; MediBeacon, St. Louis, MO) via retroorbital injection. The injection was typically accomplished within 3 min of anesthesia and mice immediately returned consciousness. The clearance (removal rate) of this agent was monitored in the following 75 min using a transdermal fluorescent monitor (MediBeacon, St. Louis, MO) for the calculation of tGFR. Mice were allowed to move freely about their cages while tGFR was being measured.
Vascular Function
Wire myography studies of carotid artery was performed as previously described.24,26 Briefly, the left and right common carotid arteries were dissected free of adventitial fat and each cut into two segments. Vessel segments were then equilibrated for 45 min under a resting tension of 0.25 g, and concentration-dependent response to acetylcholine (ACh, 1 nmol/L-30 µmol/L) and sodium nitroprusside (SNP, 0.1 nmol/L-30 µmol/L) were performed after an initial submaximal precontraction (40%–60%) with a thromboxane A2 receptor agonist U46619 (60 nmol/L). Vasoconstriction was recorded in response to KCl (10–100 mmol/L), 5-hydroxytryptamine (5-HT, i.e. serotonin, 10 nmol/L–10 µmol/L), and endothelin-1 (0.1 nmol/L-0.3 µmol/L). ACh, SNP, KCl, and endothelin-1 (E7764) were obtained from Sigma Aldrich. U46619 (CAS 56985-40-1) was obtained from Cayman Chemical.
Similarly, second-order mesenteric arteries were cut into 2 mm segments and mounted on 2 tungsten wires (25 μm in diameter) on a wire myograph (DMT620M, Danish Myograph Technology). In all mesenteric rings, preload tension was set to IC90 and submaximal precontraction was induced by U46619 as previously described.27 Cumulative concentration-responsive curves were generated in response to ACh, SNP, KCl and endothelin-1. Data were recorded as percent relaxation or maximal force generation (mN).
Basilar arteries and renal interlobar arteries were isolated and cannulated onto glass micropipettes filled with oxygenated Krebs buffer (in mmol/L: 118.3 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, and 11 Glucose) in an organ chamber as we described previously.6,24,26 Arteries were transferred to a pressurized myograph system (Model 114p, Danish Myograph Technologies) and equilibrated for 30 min at 60 mmHg luminal pressure under no-flow conditions. The viability of vessels was first examined by exposure to 100 mmol/L KCl. The vessels were initially precontraction to 30% of maximal contractile capacity using U46619 for the assessment of vasodilation responses to ACh and SNP. The level of precontraction was similar in control and E-CUL3∆9 mice.
Vascular function studies in carotid artery, basilar artery, and mesenteric arteries were performed 4 weeks after completion of tamoxifen injections at 8–10 wk of age. At the time of harvesting the vessels, the mice were typically 4–5 mo of age. Because the 6-wk long UNxHSD protocol was performed after uninephrectomy, telemetry implantation, and recovery after surgeries, at the time of renal vessel harvesting, mice were typically 6–7 mo of age.
Flow Cytometry
Flow cytometry was performed in aortic tissues as previously described.28 Briefly, immediately after mice were sacrificed, 20 mL ice-cold PBS was perfused into the left ventricle to remove blood from the circulation. Aortas were harvested and minced before being incubated in a digestion buffer (phenol-free RPMI 1640 containing 1 mg/mL collagenase A, 1 mg/mL collagenase B and 0.1 mg/mL DNase I, Roche) at 37°C for 20 min. The suspension was passed through a cell strainer with a pore diameter of 70 μm to collect single cells. This single cell suspension was subsequently treated with a red blood lysis buffer (Invitrogen, #00-4333-57) to remove red blood cells. Fc receptors were blocked with anti-mouse CD16/CD32 (BD Biosciences, clone 2.4G2) for 20 min at 4°C prior to the staining of surface markers. The antibodies used were Alexa Fluor 488 anti-mouse CD45 and PE Rat anti-Mouse CD31. Each aortic cell sample was incubated in 100 μL of FACS buffer containing 1.5 μL of each antibody for 35 min. The cells were then washed twice with FACS buffer and immediately analyzed on an LSR II flow cytometer with DIVA 8.0 software (BD Biosciences). Dead cells were eliminated from analysis using Hoechst 33528 (Sigma-Aldrich). For each experiment, we performed a flow minus one (FMO) control for each fluorophore to establish the gating strategy. Data analysis was performed using FlowJo 10.2 software (Tree Star, Inc.).
Endothelial Cell Sorting
Using a method previously reported by our lab,26 we isolated endothelial cells from non-endothelial cells from the aorta and kidney in control and E-CUL3∆9 mice 4 wk after tamoxifen treatment. Mice were perfused with 20 mL heparinized DPBS (1x) through the left ventricle following euthanasia with CO2. The aorta and kidney tissues were minced in 1 mL digestion buffer containing collagenase A (1 mg/mL, Roche), collagenase B (1 mg/mL, Roche), and DNase I (100 μg/mL, Roche). Minced tissues were digested by incubation at 37°C for 20 min before being passed through a 70 μm cell strainer to yield single cell suspension (Falcon). Cells were centrifuged at 300 g at 4°C for 5 min. The cell pellets were resuspended in 80 μL magnetic activated cell sorting (MACS) buffer in 1x DPBS containing 1% bovine serum albumin (BSA), 0.5% NaN3, and 20 μL cardiac endothelial cell isolation cocktail (130-104-183, Miltenyi Biotec). Cells were incubated for 15 min at 4°C in the dark, then washed with 2 mL of MACS buffer, and centrifuged at 300 g at 4°C for 5 min. The supernatant was discarded, and the cell pellets were resuspended in 500 μL MACS buffer. Endothelial cells were isolated using positive selection method using autoMACS or OctoMACS separator (Miltenyi Biotec, Gladbach, Germany), centrifuged (300 g, 4°C, 10 min), and pellets were collected for cell culture, flow cytometry, or RNA/protein extraction.
Mouse Aortic Endothelial Cell Primary Culture
For mechanistic studies in vitro, primary mouse aortic endothelial cells (MAECs) were isolated from the inducible CAG-CUL3∆9 mice and cultured in MCDB-31 complete culture medium (Vec Technologies) up to the third passage. Third passage MAECs were planted into 12-well plates at a seeding density of 50, 000 cells/well. At 75% confluency, MAECs were treated with adenovirus expressing Cre recombinase and green fluorescence protein (Adeno-Cre-GFP) or an empty adenovirus vehicle. Viral transduction was confirmed by expression of GFP under fluorescence microscopy in cells receiving Ad-Cre-GFP. Two weeks after the viral transduction, cells were treated with the DMSO solvent, a protein phosphatase 2A inhibitor okadaic acid (4 nM), or a protein phosphatase 1 inhibitor tautomycetin (4 nM) for 24 h. Cell lysates were used for western blot analysis and measurement of NO metabolites.
Mouse Lung Endothelial Cells
The immortalized mouse lung endothelial cells (MLECs), described previously,26,29 were a kind gift from Dr. Brandon S. Davies (Department of Biochemistry, University of Iowa). MLECs were cultured in MCDB-131 complete medium with antibiotics (VEC Technologies). To determine the effects of Cullin inhibition in MLECs, the cells were seeded in 6-well cell culture plates, cultured to 90% confluency, and treated with MLN4924 (1 μM) for 24 h. Then, MLECs were collected for conducting real-time quantitative RT-PCR and western blot as described below. MLN4924 was obtained from ActiveBiochem and dissolved in DMSO.
Quantitative Real-Time PCR
To detect the gene expression in MAECs and MLECs above, cell pellets were washed with 1x DPBS twice and snap-frozen in liquid nitrogen. RNA was extracted using the TRIzol method (Invitrogen, Carlsbad, CA). Total RNA was prepared using an RNA mini kit (Invitrogen) according to the manufacturer's protocol. RNA concentration was measured using a NanoDrop spectrophotometer with an OD260/OD280 ratio of greater than 1.9. Total RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen, #18080044) and qPCR was conducted using Taqman Gene Expression Assays (Applied Biosystems). The assay numbers for TaqMan were as follows: Mm00435217_m1 (mouse NOS3), and Mm01242576_m1 (mouse CD31), Mm99999915_g1 (mouse GAPDH). The individual amount of mRNA was calculated after normalizing to its corresponding GAPDH using the 2–ΔΔCT method, as previously described.24
Western Blot
MAECs and MLECs were lysed in radioimmune precipitation assay (RIPA) buffer consisting of 1% Nonidet P-40, 0.5% deoxycholate, and 0.1% SDS in 1X PBS with proteinase and phosphatase inhibitors (Roche). Total protein lysates were collected and stored at −80°C until further analysis. Equal amounts of protein lysates were separated by SDS-PAGE and transferred overnight on ice in a cold room (Voltage = 22 mV) to a nitrocellulose membrane (GE healthcare). Membranes were blocked with 5% milk or 5% bovine serum albumin (BSA) and incubated at 4°C overnight with primary antibodies against CUL3 (Bethyl, #A301-109A), tdTomato (Gene Tex, #GTX127897), GAPDH (Santa Cruz Biotechnology, #sc-32233), eNOS (BD Bioscience, #610297), and phospho-eNOS (Cell Signaling Technology, #9571). Immunoblots were then incubated with a secondary antibody conjugated to horse radish peroxidase and developed using ECL (Amersham Biosciences, #RPN2232). Protein bands were quantified using ImageJ 1.8.0 software.
Statistics
All results were expressed as mean ± SEM. GraphPad Prism (version 9.3) was used to perform statistical analysis. Time course data of hourly or daily BP, HR, and activity, as well as dose response curves of vascular function were analyzed by two-way ANOVA with repeated measurements. Pearson's correlation was performed with SBP and activity. Cosinor analysis of BP circadian rhythm was performed with non-linear regression (least squares fit). Student's t-test was used for two-group comparison of BP, activity, circadian rhythm, vasocontraction, tissue weight, gene expression, western blot, and NO metabolites. Data from 2×2 study designs including BP, HR, activity, power spectral analysis, metabolic cage studies, and tGFR were analyzed by two-way ANOVA. Tukey's or Sidak's multiple comparison test were performed for pairwise comparisons. Because acute natriuresis data did not follow normal distribution (Anderson-Darling test, D'Agostino & Pearson test, and Shapiro-Wilk test), data were analyzed non-parametrically with Mann–Whitney U tests with Holm-Sidak multiple comparison tests. A P-value <0.05 was considered significant.
Data
The data underlying this article will be shared on reasonable request to the corresponding author.
Results
Cell-Specificity
We previously reported transgenic mice designed to inducibly express a human hypertension-causing mutation CUL3∆9 and the tdTomato reporter (CAG-CUL3∆9).6 To achieve selective expression of the transgenes in the vascular endothelium, we bred the CAG-CUL3∆9 mice with mice carrying tamoxifen-inducible CREERT2 under control of the Tie2 promoter to generate the E-CUL3∆9 mouse model (Figure 1A).30 Both E-CUL3∆9 mice and control littermates received 5 consecutive tamoxifen intraperitoneal (i.p.) injections at 8–10 weeks of age and all physiological experiments were performed 4 weeks after the last dose of tamoxifen (Figure 1B).
PCR analysis of lung genomic DNA extracted from tamoxifen-treated control mice, tamoxifen-treated E-CUL3∆9 mice, and E-CUL3∆9 mice without tamoxifen treatment was assayed for evidence of Cre-mediated recombination. Cre-dependent recombination was only identified in lung genomic DNA extracted from mice expressing both Cre and CUL3∆9 after receiving tamoxifen (Figure S1A). To enrich endothelial cells, we isolated endothelial and non-endothelial cell fractions from the kidney using magnet activated cell sorting (MACS) as previously described (Figure 1B).26 CD31 and eNOS mRNA was detected in the endothelial fractions of the kidney and aorta, while the expression of these markers was very low in the non-endothelial fractions confirming the fidelity of cell sorting (Figure S1B-C). CUL3∆9 forms an unstable heterodimer with wildtype CUL3 (CUL3WT) resulting in destruction of both proteins.5 We were unable to detect CUL3∆9 mutant protein in these samples, and thus the reporter gene was used as a surrogate for expression of the transgene. Protein expression of the embedded tdTomato reporter was identified in the endothelial fraction of E-CUL3∆9 mice, not in the non-endothelial fraction or control mice (Figure 1C). These data confirm endothelium selective expression of the transgene.
Cardiovascular Parameters
To examine the physiological significance of endothelial CUL3∆9 in vivo, we measured BP by radiotelemetry 4 weeks after completion of tamoxifen treatment. Continuous recordings over 7 d were consolidated into hourly averages and plotted using a 24-h scheme. Circadian rhythm was preserved in both male control and E-CUL3∆9 mice as evidenced by day/night variations in systolic BP, diastolic BP, HR, and activity (Figure 2A-D). While BP, HR, or activity was not different in the light phase; there was a trend towards increases in these parameters in the dark phase. Two-way ANOVA analysis detected significant interactions between genotype and time of day in BP and activity (but not HR), suggesting that the effects of genotype depend on time of day. To determine whether there was any circadian rhythm difference in arterial pressure, we performed cosinor analysis using 96-h continuous data that were representative of the 7-day telemetry recordings (Figure 2E). Both control and E-CUL3∆9 mice displayed sine waveforms with no apparent phase shift. However, there was a significant increase of SBP wave amplitude in the E-CUL3∆9 mice (P < 0.01, Student's t-test), indicating a larger magnitude of day-night arterial pressure variability.
Figure 2.

Blood Pressure, Heart Rate, Activity, and Circadian Rhythm. Four weeks after completion of tamoxifen injections, BP and activity were measured by radiotelemetry for 10 min each hour for 7 consecutive days in male control and E-CUL3∆9 mice. (A) Systolic blood pressure (SBP), (B) diastolic blood pressure (DBP), (C) HR, and (D) activity data were collapsed onto a single 24 h light/dark cycle. The time course data were analyzed using 2-way ANOVA with repeated measurements to determine the effect of genotype, time, and interaction. Averages of the 12-hour light phase (6AM-5PM) and 12-h dark phase were shown in dot plots. Despite marked circadian variations in all parameters, no effect of genotype was detected (P > 0.05, two-way ANOVA). The p values for main effects of two-way ANOVA were indicated in each panel. (E) Cosinor analysis of continuous 96-h SBP data that represented the 7-d telemetry recordings. The mean and SEM of SBP amplitude and PhaseShift generated by the cosinor analysis was tested by Student's t-test. Shaded areas indicate the 12-h dark phase (6PM–5AM or Zeitgeber time 0–12). Data were plotted as mean ± SEM. n = 9–11/genotype. *, P < 0.05.
There was a 6-hour window in the dark phase (9PM–2AM) where BP and activity were apparently different between groups. Mean values of SBP, DBP, and activity between 9PM–11PM (Figure 3A) and 9PM–2AM (Figure 3B) demonstrated a trend towards increased BP and significantly elevated activity in E-CUL3∆9 mice. Notably, there were significant correlations between BP and activity only in this 6-hour window (Figure 3C) but not in the entire 12-hour light phase or 12-hour dark phase (Figure S2B), suggesting that the nocturnal BP peaks may be driven by increased activity in the dark phase when mice were active.
Figure 3.

Nighttime Blood Pressure and Activity. Four weeks after completion of tamoxifen injections, BP and activity were recorded by radiotelemetry for 10 min each hour for 7 consecutive days in male control and E-CUL3∆9 mice. (A) Averages of blood pressure and activity between 9PM–11PM and (B) 9PM–2AM plotted and analyzed by student's t-test. Data are plotted as mean ± SEM. *, P < 0.05, two tailed. (C) Averages of SBP and activity at 9PM–11PM (left) and 9PM–2AM (right) were plotted as XY coordinates for each mouse. Pearson correlation was performed. Number of XY pairs = 20 (11 for control and 9 for E-CUL3∆9). R, Pearson correlation coefficient.
To determine whether the variations in nocturnal BP and activity were associated with changes in sympathetic nerve activity, we performed power spectral analysis of HR and arterial pressure. Low frequency (LF) component of HR variability and baroreflex gain were not different between control and E-CUL3∆9 mice in the dark or light phase, suggesting there was no change in cardiac sympathetic nerve activity (Figure S3A). Interestingly, E-CUL3∆9 mice exhibited significantly higher LF amplitude of SBP and DBP variability in the dark phase but not in the light phase (Figure S3B). Because LF arterial pressure variability is affected by both sympathetic modulation of vascular tone and endothelial-derived NO in rodents,25 we next examined the vascular function ex vivo.
Endothelial Function
Carotid artery was studied because this vessel is highly sensitive to genetic interference with peroxisome proliferator activated receptor γ (PPARγ), an upstream regulator of CUL3 activity.24,26 In an initial cohort, we studied carotid arteries of both male and female mice with an equal sex ratio. Four weeks after tamoxifen treatments, carotid arteries from E-CUL3∆9 mice exhibited mild impairment of endothelium-dependent vasodilation as evidenced by an upward shift in the ACh dose-response curve (Figure 4A left) and a statistically significant decline in Emax (Figure 4A right, ACh Emax: E-CUL3∆9 62.6 ± 2.1 vs Control 74.3 ± 2.0, p < 0.05, Student's t-test). L-NG-nitroarginine methyl ester (L-NAME, 100 μM), a NO synthase inhibitor, completely abrogated ACh-induced vasodilation, suggesting this relaxation was predominantly NO-dependent (Figure S4A). However, there was no difference in endothelium-independent vasodilation induced by an exogenous NO donor SNP (Figure 4B), or vasoconstriction responses induced by potassium chloride (KCl), serotonin (5-HT), or endothelin-1 (Figure S4B), suggesting that smooth muscle function was not affected in this vessel. These initial observations led us to confirm the effects of the transgene in resistance arteries that are more susceptible to vascular dysfunction induced by genetic perturbations.
Figure 4.

Baseline Vasodilation. A-B) Isometric tension was initially measured with wire myography in the carotid artery of control and E-CUL3∆9 mice with equal sex ratio 4 wk after completion of tamoxifen. Vessel rings were equilibrated at a resting tension of 0.25 g for 45 min before pre-contracted to submaximal levels (40%–60%) with 60 nM U46619, a thromboxane A2 receptor agonist. (A) Cumulative concentration-response curves for acetylcholine (ACh) were plotted. Curves were first analyzed using two-way ANOVA with repeated measurements and results (pgenotype = 0.06, pconcentration < 0.01, pinteraction < 0.05) indicated that the effect of genotype on ACh response depends on its concentration. Emax for ACh responses were derived from non-linear regression and were analyzed by Student's t-test (two tailed). *, P < 0.05. (B) Cumulative concentration-response curves for sodium nitroprusside (SNP). No significant genotype effect was detected (P > 0.05). (C) ACh induced vasodilation was determined by pressure myography in the basilar artery in an initial cohort of male mice (Pgenotype < 0.05; pconcentration < 0.01, Pinteraction = 0.20). (D) In separate cohorts of male and female mice, ACh- and SNP-induced vasodilation was assessed by pressure myography in the basilar artery in both male and female mice (ACh: pgenotype = 0.06; pconcentration < 0.01, pinteraction = 0.26; SNP: pgenotype = 0.32; pconcentration < 0.01, pinteraction = 0.36). Two-way ANOVA with repeated measurements was performed for statistical analysis (C-D). *, p < 0.05, control M vs E-CUL3∆9 M at 10–4 mole/L ACh, Sidak's multiple comparison tests. Data are plotted as mean ± SEM. M, male; F, female. Sample sizes were indicated in each panel.
Vasodilation properties were further evaluated in the basilar artery, a cerebral resistance vessel. In an initial cohort of male E-CUL3∆9 mice, ACh-induced endothelial-dependent vasodilation was severely impaired (Figure 4C). To assess sex differences, we repeated the basilar artery study in additional male and female animals (Figure 4D). Interestingly, impaired endothelial-dependent vasodilation was only evident in male but not female transgenic mice. While there was no genotype effect in SNP-induced endothelial-independent vasodilation, female mice seemed to be more sensitive to SNP than male mice as evidenced by the leftward shift of the curves. There was no significant genotype difference in KCl responses in the basilar artery of male or female mice (Figure S4C and D). No effect of genotype or sex was observed in vasodilation or contraction in the second order mesenteric arteries (Figure S4E and F). Together, these data indicated that selective expression of CUL3∆9 causes vessel bed-specific vascular dysfunction in a sex-dependent manner.
eNOS and NO
To investigate the molecular mechanisms underlying the endothelial dysfunction observed above, we isolated primary mouse aortic endothelial cells (MAECs) from the inducible CAG-CUL3∆9 mice using MACS as described above (Figure 5A). Flow cytometry analysis indicate that a majority of the freshly isolated MAECs (passage 0) were CD31highCD45– mature endothelial cells (Figure S5A). Two minor populations of erythro-myeloid progenitors (EMPs) that were CD31highCD45+ and CD31lowCD45+ were also present. These progenitors exist in adult tissues and can differentiate into endothelial cells.31 Passage 0 (P0) MAECs were cultured in the classical MCDB-1 endothelial cell culture medium up to three passages. The percentage of CD31high cells increased from 58.5% in passage 0 to over 90% in passage 3 (Figure S5B) and the cells exhibited typical cobble stone-like morphology (data not shown), an indication of successful enrichment of primary endothelial cells.
Figure 5.
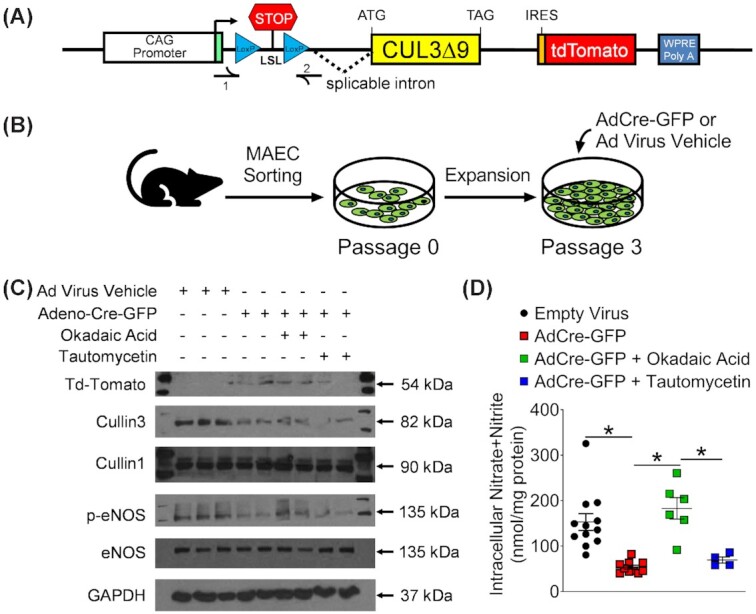
Impaired eNOS Phosphorylation and Nitric Oxide Production via Protein Phosphatase 2A. (A and B) Primary mouse aortic endothelial cells (MAECs) were isolated from the inducible CAG-CUL3∆9 mice (A) using magnet activated cell sorting and were expanded in MCDB-31 complete endothelial culture media (B). At 75% confluency in Passage 3, cells were treated with adenovirus expressing Cre recombinase and GFP (Adeno-Cre-GFP) or an empty adenovirus vehicle. (C and D) Two weeks after the viral transduction, cells were treated with the DMSO solvent, a protein phosphatase 2A inhibitor okadaic acid (4 nM), or a protein phosphatase one inhibitor tautomycetin (4 nM) for 24 hours. Cell lysates were used for western blot analysis of indicated proteins and measurement of intracellular nitric oxide (NO) metabolites nitrate/nitrite. Western blot images are representative of three independent experiments. p-eNOS represents phospho-ser1177. NO metabolites data were plotted as mean ± SEM with each symbol being an independent technical replicate (n = 4–12). *, P < 0.05, one-way ANOVA with Bonferroni's multiple comparison tests as indicated.
To induce transgene expression in vitro, third passage MAECs were treated with adenovirus expressing Cre recombinase and GFP (AdCre-GFP) or empty adenovirus vehicle (Figure 5B). TdTomato protein was only detected in cells receiving AdCre-GFP, confirming Cre recombinase activated transgene expression (Figure 5C). Like before, we were unable to detect CUL3∆9 mutant protein. Since CUL3∆9 can form unstable heterodimers with CUL3,5,32 there was a decrease in total CUL3 in AdCre-GFP treated cells. However, there was no change in the level of CUL1, an independent member of the cullin family. The decrease in CUL3 was associated with lower levels of eNOS phosphorylation (S1177, Figure 5C) and NO metabolite nitrate/nitrite in MAEC lysates (Figure 5D). In comparison, total eNOS remained unchanged suggesting that interference with CUL3 impedes eNOS activation through post-translational mechanisms.
Since CUL3-RING ubiquitin ligase (CRL) regulates the turnover of protein phosphatase 2A (PP2A),21,22 we hypothesized that decreased CUL3 causes eNOS de-phosphorylation through increased PP2A activity. In support of this hypothesis, the reduction in eNOS phosphorylation and NO metabolites were both rescued by a selective PP2A inhibitor okadaic acid (Figure 5C). In comparison, protein phosphatase 1 inhibitor tautomycetin failed to improve eNOS phosphorylation or levels of NO metabolites. These results suggest that the impairment in vasodilation caused by expression of CUL3∆9 in the endothelium is a consequence of decreased endothelial NO production.
CUL3 activation requires neddylation, covalent adduction of ubiquitin-like NEDD8 molecules (8KD) to the CUL3 C-terminus.33 To further assess whether these effects were caused by decreased CUL3 ubiquitin ligase activity, we treated immortalized MLECs with a pan Cullin inhibitor MLN4924 which inhibits the neddylation required for CRL activation. Consistent with a post-translational mechanism, the treatment did not change eNOS mRNA (Figure 6A). Whereas a slight decrease in total eNOS was observed (Figure 6B), MLN4924 caused marked suppression of eNOS phosphorylation as evidenced by dramatic decreases in p-eNOS/GAPDH and p-eNOS/eNOS ratios (Figure 6C and D). Similar to the effects observed in primary MAECs in response to the dominant negative CUL3∆9, the Cullin inhibitor also decreased intracellular NO metabolites in MLECs (Figure 6E). These data collectively support that genetic or pharmacological interference with CUL3 impairs endothelial NO biogenesis through PP2A-mediated dephosphorylation of eNOS.
Figure 6.

Pharmacological Inhibition of Cullin Decreases eNOS Phosphorylation and NO Bioavailability. Immortalized mouse lung endothelial cells (MLECs) were cultured in MCDB-131 complete medium with antibiotics (VEC Technologies) as previously described.26 To assess the effects of cullin inhibition in MLECs, the cells were seeded in 6-well cell culture plates, cultured to 90% confluency, and treated with MLN4924 (1 μM, Active Biochem) for 24 hours. MLECs lysates were collected for real-time quantitative PCR and western blot. (A) mRNA expression of eNOS. (B–D) Quantified total eNOS/GAPDH ratio, phospho-eNOS/GAPDH ratio, and phosphor-eNOS/total eNOS ratio (n = 4). p-eNOS represents phospho-ser1177. Data were plotted as mean ± SEM with each symbol being a technical replicate in two independent experiments. (E) Measurement of intracellular nitrate/nitrite (n = 6). *, P < 0.05, Student's t-test (two tailed).
Salt-Induced Hypertension
Given the endothelial dysfunction and trending BP, we asked if the E-CUL3∆9 mice were more susceptible to a hypertensive stimulus. To test this, we performed uninephrectomy (UNx) and placed the mice on a HSD containing 4% NaCl (1.57% sodium) for a total of 5 wk (Figure 7A). BP was recorded using radiotelemetry for 6 d during baseline while mice were on a normal salt chow containing 0.3% NaCl (0.1% sodium) and during the first 4 wk of HSD.
Figure 7.

Effect of High Salt Diet on Systolic Blood Pressure. (A) Protocol for the uninephrectomy high salt diet study (UNxHSD). Immediately after tamoxifen administration at 8–10 wk of age, mice received uninephrectomy (UNx) and were rested for 30 d for re-establishment of hemodynamic homeostasis. After the 30 d, telemeter devices were implanted in a second surgery and mice were allowed another 10 d for recovery. Mice consumed a normal salt diet (NSD, 0.3% NaCl) until being switched to a 4% high salt diet (HSD) for 5 wk. Blood pressure was recorded for 6 d during baseline (UNxNSD, Day −5 to Day 0) and for the first 28 d on high salt (UNxHSD). GFR and metabolic cage studies were conducted immediately before baseline blood pressure (Day −8 to Day −6) and in HSD week 5 (Day 29 to day 31). Vascular function studies were performed at the end of week 5. B-C) Nighttime SBP (6PM–5AM) during the NSD baseline and the first 4 wk of HSD in male (B) and female (C) mice. Averages of UNxNSD baseline and UNxHSD weeks 3–4 were plotted using dot/bar graphs and analyzed by two-way ANOVA with Sidak's multiple comparison tests (n = 5–8). #,P < 0.05 E-CUL3∆9 HSD vs NSD. Increases of nighttime SBP (∆SBP) from UNxNSD baseline were calculated and plotted in the panels on the right. Average ∆SBP in HSD weeks 3 and 4 were plotted using dot/bar graphs. *,P < 0.05, E-CUL3∆9 vs control by Student's t-test. Data were plotted as mean ± SEM.
Male E-CUL3∆9 and control mice exhibited similar daily SBP (24hr average) at baseline and in response to chronic HSD (Figure S6A). Interestingly, the nighttime SBP of male E-CUL3∆9 mice was significantly increased in response to chronic HSD (HSD 151.0 ± 3 mmHg vs NSD 140.7 ± 2 mmHg, P < 0.05, two-way ANOVA Sidak's multiple comparison tests), resulting in a net increase of 10.3 mmHg (Figure 7B). Surprisingly, despite the lack of endothelial dysfunction at baseline, female UNx E-CUL3∆9 mice exhibited profound 24 h SBP increases in response to high salt (HSD 143.6 ± 2 vs NSD 131.2 ± 4 mmHg, P < 0.05), resulting in a net increase of 12.4 mmHg (Figure S6B top and middle panels). This increase was primarily driven by higher nighttime BP in HSD weeks 3–4 as shown by the 24 h plot (Figure S6B bottom panels). Accordingly, the HSD-induced elevation in SBP was more pronounced at night (HSD 153.7 ± 3 mmHg vs NSD 138.3 ± 4 mmHg, P < 0.05) with a net increase of 15.3 mmHg (P < 0.05, Student's t-test) (Figure 7C).
In contrast, despite a trend towards increased 24 h and nighttime SBP in male and female controls, salt-induced SBP changes were not statistically significant (two-way ANOVA). HSD induced similar increases of DBP in both sexes, and no effect of genotype was observed (Figure S7).
Salt Sensitivity and Renal Dysfunction
To assess whether the salt-induced hypertension was associated with renal dysfunction, metabolic cage studies were performed at baseline and during the 5th week of HSD (Figure 7A). Some groups were implanted with radiotelemeters to assess natriuresis-pressure relationship. E-CUL3∆9 and control mice in response to HSD exhibited similar decreases in food intake and increases in sodium and water intake (Figure S8A). They also exhibited similar increases in urine volume and urinary sodium excretion in both sexes (Figure S8B). There were no differences in body weight gain or kidney weight/body weight ratio between genotypes (Figure S9). Baseline sodium balance was neutral in both males and females (Figure 8A and B). However, consistent with the salt-induced hypertension, both male and female E-CUL3∆9 mice exhibited positive apparent sodium retention (Na+Intake-Excretion) in response to chronic HSD. Interestingly, HSD-fed female controls also exhibited positive apparent sodium retention, although their BP did not significantly increase.
Figure 8.

Renal Function and Injury. (A-B) Apparent sodium retention (Na+In-Ex) was calculated as the difference between dietary sodium intake (Na+In) and urinary sodium excretion (Na+Ex) as measured in 24hr metabolic cages at NSD baseline and during HSD week 5 in males (A) and females (B). n = 6–13. (C-D) Natriuresis-pressure relationship in a subset of metabolic cage mice that were also implanted with radiotelemetry (n = 4–8). Six-day averages of mean arterial pressure (MAP) immediately after metabolic cages at NSD baseline (days −5 to 0) and immediately prior to metabolic cages during HSD (days 23–28) were plotted against corresponding 24 h natriuresis in the same animals. C, males. D, females. (E-F) Acute natriuretic capacity was tested in a subset of metabolic cage mice (n = 5–7). Immediately following the 24 h urine collection, male (E) and female (F) mice received a single i.p. injection of normal saline equal to 10% of their body weight. Sodium excretion in the subsequent 4 hours was collected and assayed for sodium using flame photometry. In left panels, acute natriuresis which was sodium excretion in the 4 h urine calculated as percentages of saline sodium injected was analyzed using Mann-Whitney U tests with Holms-Sidak multiple comparison tests. Right panels show HSD-induced changes in natriuresis (∆Natriuresis). * P < 0.05, control vs mutant, Mann–Whitney U test. (G) Microalbumin in 24 h urine was assayed as previously described mice.24 (H) Transcutaneous glomerular filtration rate (tGFR) was measured immediately before mice were placed into metabolic cages at NSD baseline and during HSD week 5. Data were plotted as mean ± SEM. Sample size: Male, n = 5–13; female, n = 7–10. For sodium retention, microalbumin, and tGFR, two-way ANOVA was performed. #, P < 0.05 HSD vs NSD. M, male; F, female.
The natriuresis-pressure relationship was plotted in a subset of mice in the metabolic cage studies that were also implanted with radiotelemeters (Figure 8C and D). Except for the telemetry implantation, this subset followed the same protocol in Figure 7A as the entire cohort. About 24 h sodium excretion in male mice in this subset was comparable to that of the whole dataset (Figure S8C). In female mice, 24hr sodium excretion in the subset exhibited similar trends compared to the whole dataset (Figure S8D), but the values for UNxHSD female mutants in the subset tended to be smaller perhaps due to inter-cohort variations. Nevertheless, there was no statistical difference in the 24 h sodium excretion in mice with and without telemetry implantation (data not shown). In this subset, the natriuresis-pressure relationship was similar between male control and E-CUL3∆9 mice (Figure 8C). Of note, female E-CUL3∆9 mice displayed a steeper natriuresis-pressure relationship compared to controls, indicating increased salt sensitivity of BP (Figure 8D).
To directly assess natriuresis function, we performed an acute sodium load test at NSD baseline and in HSD week 5 as previously described.24 At NSD baseline, both control and E-CUL3∆9 mice excreted ∼60% of the sodium injected. There was no significant genotype effect in either sex (Figure 8E-F left panels, P > 0.05, Mann-Whitney U tests with Holm-Sidak multiple comparison tests). In male mice, there were significant effects of diet (Figure 8E, Control: NSD vs HSD, Mann–Whitney U = 17.0, P = 0.03; E-Cul3Δ9: NSD vs HSD, Mann–Whitney U = 6.0, P = 0.03) in both genotypes. In females, high salt diet significantly increased acute natriuresis in control but not E-CUL3∆9 mice (Figure 8F, Control: NSD vs HSD, Mann–Whitney U = 7.0, P < 0.01; E-Cul3Δ9: NSD vs HSD, Mann–Whitney U = 26.0, P = 0.24). HSD induced changes in natriuresis (∆Natriuresis) were next calculated. In male mice, ∆Natriuresis was similar between control and mutant mice (Figure 8E right panel). In females, there was a positive ∆Natriuresis in controls while ∆Natriuresis was near zero in E-CUL3∆9 mice (Figure 8F right panel, P < 0.05, Mann–Whitney U test). Together, these data indicate that HSD-fed female E-CUL3∆9 mice failed to enhance their natriuretic capacities from baseline levels.
To assess hypertension-related kidney damage, we also measured urinary microalbumin and tGFR before and after HSD. Genotype-independent increases of microalbuminuria were observed in male and female mice indicating that UNxHSD induced glomerular damage regardless of hypertension status (Figure 8G). There was a trend towards increased tGFR in response to HSD in all male mice (Figure 8H, pdiet = 0.07, two-way ANOVA) but no genotype effect was observed. In female mice, while there was an overall effect of diet in tGFR (Pdiet < 0.01), Tukey's multiple comparison tests indicated that HSD significantly increased tGFR in E-CUL3∆9 mice but not in controls (Figure 8H). This is consistent with the phenomenon of salt-induced hyperfiltration in SS hypertensive humans.34
Renovascular Dysfunction
We have previously demonstrated that impaired renovascular dilation in response to salt loading restricts renal perfusion and portends SS hypertension.24,35 To assess renovascular function, we isolated third-order renal interlobar arteries at the end of the UNxHSD protocol for pressure myograph studies. These vessels are immediately upstream of the resistance interlobular arteries and have an outer diameter of ∼200 μm. Control and E-CUL3∆9 exhibited similar outer diameter, inner diameter, wall thickness, and media to lumen ratio, suggesting that CUL3∆9 did not induce remodeling in this vessel bed (Figure S10A). There was no difference in vasoconstriction in response to KCl or 5-HT (Figure S10B). Thromboxane A2 receptor agonist U46619 elicited comparable pre-contraction in control and E-CUL3∆9 vessels (Figure 9A-B). While male control and E-CUL3∆9 mice exhibited similar ACh- and SNP-induced vasodilation (Figure 9C), renal vessels from female E-CUL3∆9 mice exhibited severely impaired vasodilation in response to ACh, but not SNP (Figure 9D). These data suggest that salt-induced vasodilation impairment in the renal circulation may have contributed to the SS hypertension, particularly in the female E-CUL3∆9 mice.
Figure 9.

Renal Vasodilation. Third order renal interlobar arteries were isolated at the end of HSD week 5 and mounted on a pressure myograph chamber filled with oxygenated Kreb's physiological buffer at 37°C. Luminal pressure was maintained at 60 mmHg throughout the experiment. A-B) Pre-contraction was induced by U46619 (100 nM) in vessels from male (A) and female (B) mice. No significant difference was detected between control and mutant mice in either sex by Student's t-test (P > 0.05). n = 7–9. (C-D) Cumulative concentration-response curves for ACh- and SNP-induced relaxation were plotted as percentages of precontraction (n = 7–9) in vessels from male (C) and female (D) mice. Data were plotted as mean ± SEM. Two-way ANOVA with repeated measurements was performed for statistical analysis. *, P < 0.05, E-CUL3∆9 vs control (main effect). M, male; F, female.
Discussion
According to the 2017 American College of Cardiology/American Heart Association Guidelines, 46% of U.S. adult population has hypertension,36 and is exposed to increased risk for ischemic heart disease, stroke, and chronic kidney disease.37 SS hypertension is a major public health burden with an estimate incidence of >50% in essential hypertension and nearly 75% in African American hypertensive patients.12,13 Salt sensitivity of BP, arbitrarily defined as an increase of > 5 mmHg or 5% of mean arterial BP in response to dietary sodium load, 38 is a continuous variable that follows Gaussian distribution and is an independent cardiovascular risk factor.39 Early studies by Lewis Dahl provided the first evidence that BP elevation in response to excess dietary salt is an inheritable trait.40 Mounting evidence from human hypertension and Dahl SS rats support the genetic susceptibility to the development of SS hypertension.12,13,40
Among many genetic factors that contribute to the susceptibility to salt sensitivity, those affecting NO bioavailability and the downstream soluble guanylate cyclase (sGC)-cGMP pathway play a major role.41 While increased NO production helps to offset the pressor effect of dietary salt in salt-resistant (SR) humans and animal models, decreased NO bioavailability is a hallmark of SS hypertension.9,41,42 In Dahl rats, renal NO deficiency decreases renal blood flow and contributes to SS hypertension, while supplementation of L-arginine or stimulation of sGC promotes NO-cGMP signaling, improves renal perfusion, and prevents SS hypertension.43–46 In mice, global eNOS knockout results in profound SS hypertension.47 In keeping with a role of NO signaling in salt sensitivity, we have previously shown that mice expressing a dominant negative PPARγP467L mutation in vascular smooth muscle (S-P467L) exhibit severe impairment of smooth muscle NO responsiveness and renal vasodilation which restricts renal perfusion during excess salt intake.24 As a result, the HSD-fed S-P467L mice develop renal dysfunction and SS hypertension, which is normalized by a loop diuretic or a pharmacological vasodilator.24,35 Data in the present study extend those results and support the novel finding that genetic interference with CUL3 in the endothelium decreases NO biogenesis and causes increased salt sensitivity of BP in mice.
NO produced by any of the three isoforms of NO synthase (NOS) may contribute to the regulation of BP and hemodynamics through their activities in the cardiovascular, nervous system, and the kidney.11,47,48 However, eNOS appears to be a determinant isoform in the salt sensitivity of BP for several reasons: (i) global eNOS deletion alone is sufficient to increase salt sensitivity,47 (ii) non-selective NOS inhibition with L-NAME does not cause further salt-dependent BP increases in eNOS knockout mice which are already hypertensive;49 and (iii) ACh-induced endothelial-dependent vasodilation is reduced in SS humans even on restricted sodium diets, suggesting that preexisting impairment of eNOS activation may portend SS BP elevations through NO-deficiency.10
The activity of eNOS is regulated transcriptionally and post-transcriptionally by phosphorylation and dephosphorylation of key amino acid residues.14 In the present study, we have shown that genetic or pharmacological interference with CUL3 decreased eNOS phosphorylation without changing its mRNA levels, suggesting that CUL3 regulates eNOS activity at the post-translational level. Under physiological conditions, eNOS forms a complex with heat shock protein 90 (HSP90) and Akt.18 Formation of this complex allows Akt to phosphorylate eNOS Serine1177, which promotes eNOS activation and NO biosynthesis. In diabetes and obesity-related hypertension, lipid metabolite ceramide induces the release of PP2A, a major serine-threonine phosphatase, from the cytosolic inhibitor 2 of PP2A (I2PP2A).18–20 Free PP2A directly associates with the Hsp90-Akt-eNOS complex and dephosphorylates both Akt (T308) and eNOS (S1177) resulting in impaired endothelial-dependent vasodilation. Herein, we found that selective inhibition of PP2A, but not PP1, improved eNOS phosphorylation and NO production in CUL3∆9 endothelial cells. While the role of other phosphatases such as phosphatase and tensin homologue deleted on chromosome 10 (PTEN) cannot be ruled out,50 these data provide evidence for substrate specificity in PP2A mediated posttranslational regulation of eNOS activation.
PP2A itself is closely regulated by posttranslational mechanisms including CUL3-mediated ubiquitination that promotes the physiological turnover of PP2A in the proteasome.21,22 The PP2A heterotrimer consists of a structural A subunit, a regulatory B subunit, and a catalytic C subunit. While the A and C subunits each have two isoforms, there are over 40 isoforms for the B subunit categorized into B, B’, B’’, and B’’’ classes. Theoretically, this can give rise to hundreds of heterotrimeric combinations. Due to this variability, CUL3-mediated ubiquitination of PP2A is extremely complicated and not fully understood. Two models have been proposed. Oberg, et al identified the BTB domain-containing KLHL15 (Kelch-like 15) as the substrate adaptor that recruits PP2A B’β subunit to CUL3 ubiquitin ligase in the brain.21 CUL3 has also been shown to ubiquitinate and degrade the PP2A C subunit in breast cancer, although it is not clear whether KLHL15 acts as the substrate adaptor in this context. Thus, the cellular distribution of known PP2A isoforms appears to be tissue specific. Our data support functional involvement of PP2A in eNOS activation and endothelial NO production. However, we found that CUL3∆9 did not change the level of the PP2A C subunit, and we were unable to detect any changes in the level of the B subunit with several commercial antibodies against B subunit (data not shown). Moreover, given the spontaneous Cre recombinase activity in non-endothelial cells including the reproductive tissues, the extraembryonic vascular system, and yolk sac in the Tie2-creERT2 transgenic mice,23 we cannot rule out recombination of transgenes in other cell types and a role of non-endothelial actions of CUL3∆9. Pin-pointing the PP2A isoforms involved in eNOS dephosphorylation in vascular endothelium remains a technical challenge and may require advanced proteomic analysis followed by development of selective antibodies against a particular PP2A subunit.
Like many other behavioral and physiological processes, BP and physical activity have circadian rhythms.51 Circadian rhythm is primarily controlled by the central pacemaker in the suprachiasmatic nucleus, but clock genes expressed in the vasculature can directly regulate BP rhythm.52 Clock molecules act through direct or indirect mechanisms by binding to cell-specific enhancers to transcriptionally regulate clock-controlled genes involved in various physiological processes. Clock proteins themselves are tightly regulated by Cullin family members via ubiquitination and proteasomal degradation for periodic turnover.53 For example, CUL3 ubiquitin ligase is required for circadian control of oscillation of PERIOD and TIMELESS proteins and the rhythmicity of sleep/wakefulness in Drosophila.54,55 In the present study, E-CUL3∆9 transgenic mice exhibited an increase of activity between 9PM and 2AM in the dark phase that was correlated with a trend towards increased BP in this 6-hour time window. This raised the possibility that interference with CUL3 in the endothelium results in circadian dysregulation of locomotor activity. However, in a separate study with Promethion metabolic cages, we did not observe any differences in food intake, water intake, sleep/wakefulness, or locomotion in E-CUL3∆9 mice (data not shown). It was reported that deletion of Bmal1 gene in vascular smooth muscle resulted in altered arterial pressure amplitude without significant changes in HR or activity.52 Thus, the greater amplitude of the SBP waveform in E-CUL3∆9 mice suggests that endothelial CUL3 may directly regulate the circadian rhythm of arterial pressure through ubiquitin-mediated turnover of clock proteins.
Sex differences in vascular dysfunction and hypertension are well documented in both humans and experimental models.56 Women are protected from vascular dysfunction and hypertension before menopause but have an accelerated age-related rise in the incidence and prevalence of hypertension after menopause suggesting that the sex-specific effects may be attributable to gonadal hormones.57 Estrogen promotes eNOS-mediated NO production via both transcriptional and posttranslational mechanisms. Upon binding with 17β-estradiol in the cytoplasm, estrogen receptors (ER) α and β translocate to the nucleus and interact with the estrogen responsive DNA elements (EREs) in the promotor region of the eNOS gene, resulting in transcriptional upregulation of eNOS.58 Moreover, activation of the plasma membrane-bound ERα and G protein-coupled estrogen receptor (GPER) in response to 17β-estradiol stimulates rapid eNOS phosphorylation at the S1177 site and NO production.59,60 In healthy humans, there is a larger NO component in endothelial dependent vasodilation in men than women.61 This may contribute to the apparently larger susceptibility to disruption of eNOS activation and decreases in endothelial NO bioavailability in men. Consistent with this, only male E-CUL3∆9 mice exhibited impairment of endothelial dependent vasodilation in the basilar artery at baseline. However, this does not rule out impaired endothelial function at baseline in other vessels such as in the renal vasculature in mutant males or in mutant females. It remains to be investigated whether steroid hormone-mediated mechanisms contribute to the effect of sex in vascular function in E-CUL3∆9 mice. Furthermore, RNA sequencing in endothelial cells isolated from different vascular beds of both male and female E-CUL3∆9 mice may aid the identification of molecules mediating the sex dimorphisms in vascular function.
Multiple large population studies found salt sensitivity of BP is greater in women than in men. In the well-controlled HyperPATH consortium, women exhibited 30% higher salt sensitivity of BP than men regardless of menopause or hypertension status.62 BP responses to low-salt and high-salt interventions were greater in women than in men in the Genetic Epidemiology Network of Salt Sensitivity (GenSalt) study.39 The slope of pressor effect of increased salt intake was steeper in women than in men in the ELSA-Brasil study with 12 813 participants (35 to 64 yr).63 Consistent with this, reduction of sodium intake resulted in larger decreases in BP in women compared to men in the DASH-Sodium trial.64 Despite of these strong epidemiological evidence, the higher salt sensitivity in women is not recapitulated in animal models. In fact, female Dahl SS rats and C57Bl/6 mice have less salt sensitivity of BP compared to males.65,66 The salt resistance in female C57Bl/6 mice is attributable to more macula densa NO mediated inhibition of tubuloglomerular feedback (TGF) and the consequent facilitation of renal perfusion and sodium excretion during salt loading.66 In the present study, female but not male E-CUL3∆9 mice on C57Bl/6 background displayed increased salt sensitivity of BP as evidence by the steeper natriuresis-pressure relationship. Our data demonstrate that genetic interference with CUL3 in the vascular endothelium is sufficient to increase salt sensitivity on an otherwise salt-resistant genetic background. Thus, the E-CUL3∆9 mice may be a useful model to study the human phenotype.
HSD-fed female E-CUL3∆9 HSD developed significantly impaired endothelial-dependent vasodilation in renal microvessels. Renal vascular dysfunction plays a pivotal role in the initiation of salt-induced hypertension.67 In SR humans and Dahl SR rats, salt-induced acute increases in cardiac output (first 72 h) is simultaneously accompanied by physiological vasodilation which offsets the pressor effect of salt through a decrease in systemic vascular resistance.68,69 Lack of this compensatory vasodilation during acute salt loading portends the marked elevation of arterial pressure in SS humans and Dahl SS rats. Consistent with this, we have previously shown that salt-induced impairment of vasodilation occurred as early as day 3 of HSD, before the onset of salt-induced hypertension in a mouse model of vascular PPARγ interference.24 In the renal circulation, SR humans and rodents exhibit decreased renal vascular resistance and increased renal perfusion in response to high salt.24,70,71 The opposite occurs in SS humans and animals, where markedly increased renal vascular resistance restricts renal perfusion during excess salt. The salt induced changes in peripheral vascular resistance in SS humans is significantly correlated with changes in plasma asymmetrical dimethylarginine, an endogenous NOS inhibitor, supporting decreased endothelial NO generation being a mechanism.68 Our results indicate that interference with endothelial CUL3 increases the susceptibility to dilatory dysfunction in the renal circulation during excess salt, which may have contributed to the salt-induced hypertension through hemodynamic mechanisms.
In the present study, HSD increased the acute natriuretic capacities in control and male E-CUL3∆9 mice, but not female mutants, suggesting the salt-induced hypertension in female E-CUL3∆9 mice was accompanied by renal dysfunction. Acute diuresis/natriuresis in response to volume expansion reflects kidney function.24,72,73 In previous studies, mice with normal kidney function typically excrete 60%–80% of the volume/sodium load within 4 h following the i.p. saline injection,24,72 suggesting the majority of volume/sodium load is rapidly absorbed into intravascular compartment during the time frame of the experiment. This diuretic/natriuretic capacity is decreased in hypertension induced by angiotensin II infusion or genetic manipulations.24,72 However, results are to be interpreted with caution as this procedure extrapolates renal excretory response to an i.p. sodium load to the renal excretory response occurring with dietary increases in sodium intake. Another limitation to our studies is the lack of information about when the changes in sodium balance occurred in relation to when the differences in BP occurred. Future studies are necessary to examine whether the greater salt-induced rise of BP in the mutant animals was preceded or simultaneously accompanied by greater sodium retention.
In conclusion, we have identified novel mechanisms by which endothelial CUL3 regulates NO biogenesis, vascular function, and salt sensitivity of BP. The finding that E-CUL3∆9 mice exhibit endothelial dysfunction at baseline and salt-induced hypertension in a sex-specific manner is intriguing and clinically relevant. Opposite to humans, rodents have a rise in BP, HR, and activity at night. Thus, the nocturnal variations in E-CUL3∆9 mice that peaks at 9PM–2AM resembles the morning surge of BP in humans, which correlates with higher incidence of stroke, myocardial infarction, and sudden cardiac death than other times of the day. Our results suggest that ubiquitin-based posttranslational mechanisms are important in the regulation of vascular function and BP and may be novel targets for hypertension and cardiovascular diseases.
Sources of Funding
This work was supported through research grants from the National Institutes of Health (NIH) to CDS (HL084207, HL144807), JLG (HL134850, 18EIA33890055), and American Heart Association (15SFRN23480000) to CDS. JW was supported by an AHA postdoctoral fellowship (17POST33660685) and currently by a NIDDK K01 (DK126792). SF was supported by an AHA predoctoral fellowship (20PRE35120137).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Camille Taylor in the Biochemical Assay Core Laboratory in the Department of Physiology at Medical College of Wisconsin for measurement of urinary sodium and albumin. The Tek-CRE-ERT2 mice was the kind gift of Dr. I Grumbach from the University of Iowa.
Contributor Information
Jing Wu, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Shi Fang, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA; Department of Neuroscience and Pharmacology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, 52242 Iowa, USA.
Ko-Ting Lu, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Gaurav Kumar, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
John J Reho, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Daniel T Brozoski, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Adokole J Otanwa, Department of Neuroscience and Pharmacology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, 52242 Iowa, USA.
Chunyan Hu, Department of Neuroscience and Pharmacology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, 52242 Iowa, USA.
Anand R Nair, Department of Neuroscience and Pharmacology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, 52242 Iowa, USA.
Kelsey K Wackman, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Larry N Agbor, Department of Neuroscience and Pharmacology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, 52242 Iowa, USA.
Justin L Grobe, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Curt D Sigmund, Deparment of Physiology, Cardiovascular Center, Medical College of Wisconsin, Milwaukee, 53226 Wisconsin, USA.
Conflict of Interest/Disclosures
CDS was a member of a Scientific Advisory Board for Ionis Pharmaceuticals. His contributions to that board were unrelated to the content of this manuscript.
Data Availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
- 1. Wu J, McCormick JA, Sigmund CD. Cullin-3: Renal and Vascular Mechanisms Regulating Blood Pressure. Curr Hypertens Rep. 2020;22(9):61. doi: 10.1007/s11906-020-01076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143(6):951–965. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boyden LM, Choi M, Choate KA et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCormick JA, Yang CL, Zhang C et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J Clin Invest. 2014;124(11):4723–4736. doi: 10.1172/JCI76126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schumacher FR, Siew K, Zhang J et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7(10):1285–1306. doi: 10.15252/emmm.201505444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Agbor LN, Ibeawuchi SC, Hu C et al. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight. 2016;1(19):e91015. doi: 10.1172/jci.insight.91015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agbor LN, Nair AR, Wu J et al. Conditional deletion of smooth muscle Cullin-3 causes severe progressive hypertension. JCI Insight. 2019;4.14, doi: 10.1172/jci.insight.129793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kobayashi A, Kang MI, Okawa H et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bragulat E, de la Sierra A, Antonio MT, Coca A. Endothelial dysfunction in salt-sensitive essential hypertension. Hypertension. 2001;37(2):444–448. doi: 10.1161/01.hyp.37.2.444. [DOI] [PubMed] [Google Scholar]
- 10. Miyoshi A, Suzuki H, Fujiwara M, Masai M, Iwasaki T. Impairment of endothelial function in salt-sensitive hypertension in humans. Am J Hypertens. 1997;10(10):1083–1090. doi: 10.1016/s0895-7061(97)00226-4. [DOI] [PubMed] [Google Scholar]
- 11. Mattson DL, Meister CJ. Renal cortical and medullary blood flow responses to L-NAME and ANG II in wild-type, nNOS null mutant, and eNOS null mutant mice. Am J Physiol-Regulat Integ Comp Physiol. 2005;289(4):R991–R997. doi: 10.1152/ajpregu.00207.2005. [DOI] [PubMed] [Google Scholar]
- 12. Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986;8(6_pt_2):II127–134. doi: 10.1161/01.hyp.8.6_pt_2.ii127. [DOI] [PubMed] [Google Scholar]
- 13. Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27(3):481–490. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- 14. Kumar G, Dey SK, Kundu S. Functional implications of vascular endothelium in regulation of endothelial nitric oxide synthesis to control blood pressure and cardiac functions. Life Sci. 2020;259:118377. doi: 10.1016/j.lfs.2020.118377. [DOI] [PubMed] [Google Scholar]
- 15. Luo Z, Aslam S, Welch WJ, Wilcox CS. Activation of nuclear factor erythroid 2-related factor 2 coordinates dimethylarginine dimethylaminohydrolase/PPAR-gamma/endothelial nitric oxide synthase pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension. 2015;65(4):896–902. doi: 10.1161/HYPERTENSIONAHA.114.04760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li L, Rezvan A, Salerno JC et al. GTP cyclohydrolase I phosphorylation and interaction with GTP cyclohydrolase feedback regulatory protein provide novel regulation of endothelial tetrahydrobiopterin and nitric oxide. Circ Res. 2010;106(2):328–336. doi: 10.1161/CIRCRESAHA.109.210658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuen CY, Wong WT, Tian XY et al. Telmisartan inhibits vasoconstriction via PPARgamma-dependent expression and activation of endothelial nitric oxide synthase. Cardiovasc Res. 2011;90(1):122–129. doi: 10.1093/cvr/cvq392. [DOI] [PubMed] [Google Scholar]
- 18. Zhang QJ, Holland WL, Wilson L et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes. 2012;61(7):1848–1859. doi: 10.2337/db11-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bharath LP, Ruan T, Li Y et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes. 2015;64(11):3914–3926. doi: 10.2337/db15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li YH, Xu Q, Xu WH, Guo XH, Zhang S, Chen YD. Mechanisms of protection against diabetes-induced impairment of endothelium-dependent vasorelaxation by Tanshinone IIA. Biochimica et Biophysica Acta (BBA) – General Subjects. 2015;1850(4):813–823. doi: 10.1016/j.bbagen.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 21. Oberg EA, Nifoussi SK, Gingras AC, Strack S. Selective proteasomal degradation of the B'beta subunit of protein phosphatase 2A by the E3 ubiquitin ligase adaptor Kelch-like 15. J Biol Chem. 2012;287(52):43378–43389. doi: 10.1074/jbc.M112.420281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu J, Zhou JY, Xu Z et al. The role of Cullin3-mediated ubiquitination of the catalytic subunit of PP2A in TRAIL signaling. Cell Cycle. 2014;13(23):3750–3758. doi: 10.4161/15384101.2014.965068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Payne S, De Val S, Neal A. Endothelial-Specific Cre Mouse Models. Arterioscler Thromb Vasc Biol. 2018;38(11):2550–2561. doi: 10.1161/ATVBAHA.118.309669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu J, Agbor LN, Fang S et al. Failure to vasodilate in response to salt loading blunts renal blood flow and causes salt-sensitive hypertension. Cardiovasc Res. 2021;117(1):308–319. doi: 10.1093/cvr/cvaa147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stauss HM. Identification of blood pressure control mechanisms by power spectral analysis. Clin Exp Pharmacol Physiol. 2007;34(4):362–368. doi: 10.1111/j.1440-1681.2007.04588.x. [DOI] [PubMed] [Google Scholar]
- 26. Hu C, Keen HL, Lu KT et al. Retinol-binding protein 7 is an endothelium-specific PPARgamma cofactor mediating an antioxidant response through adiponectin. JCI Insight. 2017;2(6):e91738. doi: 10.1172/jci.insight.91738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reho JJ, Guo DF, Morgan DA, Rahmouni K. Smooth muscle cell-specific disruption of the BB some causes vascular dysfunction. Hypertension. 2019;74(4):817–825. doi: 10.1161/HYPERTENSIONAHA.119.13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu J, Saleh MA, Kirabo A et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2015;126(1):50–67. doi: 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ackah E, Yu J, Zoellner S et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115(8):2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Korhonen H, Fisslthaler B, Moers A et al. Anaphylactic shock depends on endothelial Gq/G11. J Exp Med. 2009;206(2):411–420. doi: 10.1084/jem.20082150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Monaco MCG, Maric D, Salvucci O et al. Identification of circulating CD31(+)CD45(+) cell populations with the potential to differentiate into erythroid cells. Stem Cell Res Ther. 2021;12(1):236. doi: 10.1186/s13287-021-02311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J Biol Chem. 2015;290(31):19208–19217. doi: 10.1074/jbc.M115.645358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134(6):995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rossitto G, Maiolino G, Lerco S et al. High sodium intake, glomerular hyperfiltration, and protein catabolism in patients with essential hypertension. Cardiovasc Res. 2021;117(5):1372–1381. doi: 10.1093/cvr/cvaa205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu J, Fang S, Lu KT et al. EP3 (E-Prostanoid 3) Receptor Mediates Impaired Vasodilation in a Mouse Model of Salt-Sensitive Hypertension. Hypertension. 2021;77(4):1399–1411. doi: 10.1161/HYPERTENSIONAHA.120.16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whelton PK, Carey RM, Aronow WS et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):1269–1324. doi: 10.1161/HYP.0000000000000066. [DOI] [PubMed] [Google Scholar]
- 37. Benjamin EJ, Virani SS, Callaway CW et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137(12):e67–e492. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 38. Sullivan JM. Salt sensitivity. Definition, conception, methodology, and long-term issues. Hypertension. 1991;17(1_supplement):I61–68. doi: 10.1161/01.hyp.17.1_suppl.i61. [DOI] [PubMed] [Google Scholar]
- 39. Chen J. Sodium sensitivity of blood pressure in Chinese populations. Curr Hypertens Rep. 2010;12(2):127–134. doi: 10.1007/s11906-009-0088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dahl LK, Heine M, Tassinari L. Effects of chronia excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. J Exp Med. 1962;115:6, 1173–1190. doi: 10.1084/jem.115.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Manning RD, Hu L, Tan DY, Meng S. Role of abnormal nitric oxide systems in salt-sensitive hypertension. Am J Hypertens. 2001;14(11):S68–S73. doi: 10.1016/s0895-7061(01)02072-6. [DOI] [PubMed] [Google Scholar]
- 42. Tolins JP, Shultz PJ. Endogenous nitric oxide synthesis determines sensitivity to the pressor effect of salt. Kidney Int. 1994;46(1):230–236. doi: 10.1038/ki.1994.264. [DOI] [PubMed] [Google Scholar]
- 43. Chen PY, Sanders PW. L-arginine abrogates salt-sensitive hypertension in Dahl/Rapp rats. J Clin Invest. 1991;88(5):1559–1567. doi: 10.1172/JCI115467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Geschka S, Kretschmer A, Sharkovska Y et al. Soluble guanylate cyclase stimulation prevents fibrotic tissue remodeling and improves survival in salt-sensitive Dahl rats. PLoS One. 2011;6(7):e21853. doi: 10.1371/journal.pone.0021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miyata N, Cowley AW, Jr. Renal intramedullary infusion of L-arginine prevents reduction of medullary blood flow and hypertension in Dahl salt-sensitive rats. Hypertension. 1999;33(1):446–450. doi: 10.1161/01.hyp.33.1.446. [DOI] [PubMed] [Google Scholar]
- 46. Higashi Y, Oshima T, Watanabe M, Matsuura H, Kajiyama G. Renal response to L-arginine in salt-sensitive patients with essential hypertension. Hypertension. 1996;27(3):643–648. https://www.ncbi.nlm.nih.gov/pubmed/8613217. [DOI] [PubMed] [Google Scholar]
- 47. Leonard AM, Chafe LL, Montani JP, Van Vliet BN. Increased salt-sensitivity in endothelial nitric oxide synthase-knockout mice. Am J Hypertens. 2006;19(12):1264–1269. doi: 10.1016/j.amjhyper.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 48. Rudd MA, Trolliet M, Hope S et al. Salt-induced hypertension in Dahl salt-resistant and salt-sensitive rats with NOS II inhibition. Am J Physiol-Heart Circulat Physiol. 1999;277(2):H732–H739. doi: 10.1152/ajpheart.1999.277.2.H732. [DOI] [PubMed] [Google Scholar]
- 49. Mattson DL, Meister CJ. Sodium sensitivity of arterial blood pressure in L-NAME hypertensive but not eNOS knockout mice. Am J Hypertens. 2006;19(3):327–329. doi: 10.1016/j.amjhyper.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 50. Church JE, Qian J, Kumar S et al. Inhibition of endothelial nitric oxide synthase by the lipid phosphatase PTEN. Vasc Pharmacol. 2010;52(5-6):191–198. doi: 10.1016/j.vph.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Richards J, Diaz AN, Gumz ML. Clock genes in hypertension: novel insights from rodent models. Blood Press Monit. 2014;19(5):249–254. doi: 10.1097/MBP.0000000000000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xie Z, Su W, Liu S et al. Smooth-muscle BMAL1 participates in blood pressure circadian rhythm regulation. J Clin Invest. 2015;125(1):324–336. doi: 10.1172/JCI76881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tamayo AG, Duong HA, Robles MS, Mann M, Weitz CJ. Histone monoubiquitination by Clock-Bmal1 complex marks Per1 and Per2 genes for circadian feedback. Nat Struct Mol Biol. 2015;22(10):759–766. doi: 10.1038/nsmb.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grima B, Dognon A, Lamouroux A, Chelot E, Rouyer F. CULLIN-3 controls TIMELESS oscillations in the Drosophila circadian clock. PLoS Biol. 2012;10(8):e1001367. doi: 10.1371/journal.pbio.1001367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stavropoulos N, Young MW. insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron. 2011;72(6):964–976. doi: 10.1016/j.neuron.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sabbatini AR, Kararigas G. Estrogen-related mechanisms in sex differences of hypertension and target organ damage. Biol Sex Diff. 2020;11(1):31. doi: 10.1186/s13293-020-00306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ostchega Y, Fryar CD, Nwankwo T, Nguyen DT. Hypertension prevalence among adults aged 18 and over: United States, 2017–2018. NCHS Data Brief. 2020(364):1–8. [PubMed] [Google Scholar]
- 58. MacRitchie AN, Jun SS, Chen Z et al. Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res. 1997;81(3):355–362. doi: 10.1161/01.res.81.3.355. [DOI] [PubMed] [Google Scholar]
- 59. Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103(3):401–406. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fredette NC, Meyer MR, Prossnitz ER. Role of GPER in estrogen-dependent nitric oxide formation and vasodilation. J Steroid Biochem Mol Biol. 2018;176:65–72. doi: 10.1016/j.jsbmb.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eisenach JH, Gullixson LR, Kost SL, Joyner MJ, Turner ST, Nicholson WT. Sex differences in salt sensitivity to nitric oxide dependent vasodilation in healthy young adults. J Appl Physiol. 2012;112(6):1049–1053. doi: 10.1152/japplphysiol.01197.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shukri MZ, Tan JW, Manosroi W et al. Biological Sex Modulates the Adrenal and Blood Pressure Responses to Angiotensin II. Hypertension. 2018;71(6):1083–1090. doi: 10.1161/HYPERTENSIONAHA.117.11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mill JG, Baldo MP, Molina M et al. Sex-specific patterns in the association between salt intake and blood pressure: The ELSA-Brasil study. J Clin Hypert. 2019;21(4):502–509. doi: 10.1111/jch.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vollmer WM, Sacks FM, Ard J et al. Effects of diet and sodium intake on blood pressure: subgroup analysis of the DASH-sodium trial. Ann Intern Med. 2001;135(12):1019–1028. doi: 10.7326/0003-4819-135-12-200112180-00005. [DOI] [PubMed] [Google Scholar]
- 65. Bayorh MA, Socci RR, Eatman D, Wang M, Thierry-Palmer M. The role of gender in salt-induced hypertension. Clin Exp Hypertens. 2001;23(3):241–255. doi: 10.1081/ceh-100102663. [DOI] [PubMed] [Google Scholar]
- 66. Zhang J, Zhu J, Wei J et al. New Mechanism for the Sex Differences in Salt-Sensitive Hypertension: The Role of Macula Densa NOS1beta-Mediated Tubuloglomerular Feedback. Hypertension. 2020;75(2):449–457. doi: 10.1161/HYPERTENSIONAHA.119.13822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kurtz TW, DiCarlo SE, Pravenec M, Morris RC, Jr. The pivotal role of renal vasodysfunction in salt sensitivity and the initiation of salt-induced hypertension. Curr Opin Nephrol Hypertens. 2018;27(2):83–92. doi: 10.1097/MNH.0000000000000394. [DOI] [PubMed] [Google Scholar]
- 68. Schmidlin O, Forman A, Leone A, Sebastian A, Morris RC Jr. Salt sensitivity in blacks: evidence that the initial pressor effect of NaCl involves inhibition of vasodilatation by asymmetrical dimethylarginine. Hypertension. 2011;58(3):380–385. doi: 10.1161/HYPERTENSIONAHA.111.170175. [DOI] [PubMed] [Google Scholar]
- 69. Greene AS, Yu ZY, Roman RJ, Cowley AW Jr. Role of blood volume expansion in Dahl rat model of hypertension. Am J Physiol-Heart Circul Physiol. 1990;258(2):H508–H514. doi: 10.1152/ajpheart.1990.258.2.H508. [DOI] [PubMed] [Google Scholar]
- 70. Campese VM, Parise M, Karubian F, Bigazzi R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension. 1991;18(6):805–812. doi: 10.1161/01.hyp.18.6.805. [DOI] [PubMed] [Google Scholar]
- 71. Fink GD, Takeshita A, Mark AL, Brody MJ. Determinants of renal vascular resistance in the Dahl strain of genetically hypertensive rat. Hypertension. 1980;2(3):274–280. doi: 10.1161/01.hyp.2.3.274. [DOI] [PubMed] [Google Scholar]
- 72. Trott DW, Thabet SR, Kirabo A et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. 2014;64(5):1108–1115. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang J, Rudemiller NP, Patel MB et al. Interleukin-1 Receptor Activation Potentiates Salt Reabsorption in Angiotensin II-Induced Hypertension via the NKCC2 Co-transporter in the Nephron. Cell Metab. 2016;23(2):360–368. doi: 10.1016/j.cmet.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.