

Abstract
TGF-β/Smad signaling plays a vital role in the development of fibrosis in diabetic kidney disease (DKD). However, remedies targeting key elements in TGF-β/Smad signaling are lacking. Here, we found that TGF-β receptor 1 (TGFBR1), a key protein in TGF-β/Smad signaling, was upregulated in kidney from diabetic mice and patients with DKD. Induction of TGFBR1 was regulated by microRNA-10a and -10b (miR-10a/b) by a post-transcriptional mechanism. Furthermore, the decreased XRN2, an exoribonuclease, was identified to contribute to affecting miR-10a/b maturation in vitro. In streptozotocin (STZ)-induced DKD mice, preventing the reduction of miR-10a/b in the kidney by an in situ lentivirus-injection method attenuated collagen deposition and foot process effacement, whereas deprivation of miR-10a/b aggravated renal fibrosis. Mechanistically, manipulating miR-10a/b in the kidney influenced TGFBR1 protein expression, TGF-β/Smad signaling activation, and downstream pro-fibrotic genes expression including fibronectin (FN) and α-smooth muscle actin (α-SMA). In a cohort of patients diagnosed DKD, renal miR-10a/b expressions were downregulated, whereas both TGFBR1 and fibrosis were enhanced. Our finding suggests that overexpressing miR-10a/b in kidney may be a promising method for the treatment of fibrosis in DKD.
Keywords: MT: Non-coding RNAs, diabetic kidney disease, TGF-β/Smad signaling, TGF-β receptor 1, renal fibrosis, MicroRNA-10a/b
Graphical abstract
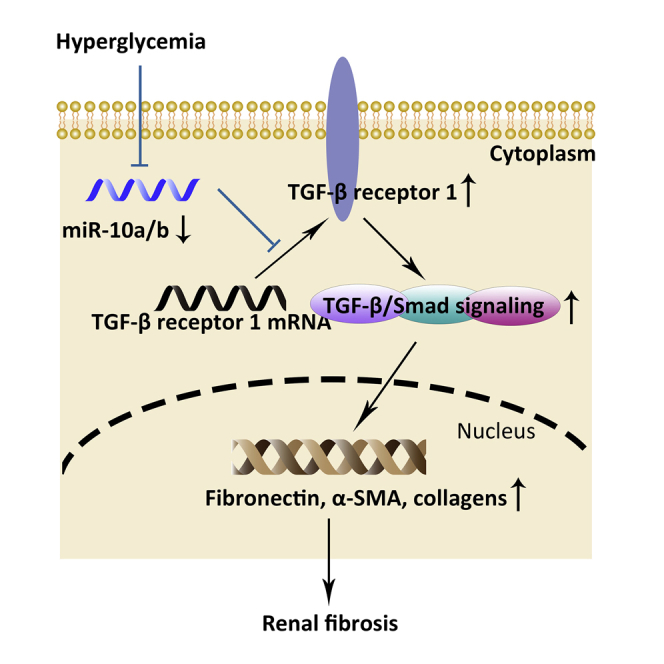
TGF-β/Smad signaling plays a vital role in the development of fibrosis in diabetic kidney disease. This study found that microRNA-10 inhibits fibrosis by suppressing TGF-β receptor 1, the key protein in TGF-β/Smad signaling, suggesting that targeting TGF-β receptor 1 by microRNA-10 is a promising method for treatment of diabetic kidney disease.
Introduction
Diabetic kidney disease (DKD) is a highly prevalent complication of diabetes mellitus, contributing to the morbidity and mortality of patients with diabetes. More than 40% of diabetic patients progress into DKD, making DKD the most common cause of end-stage renal disease (ESRD), imposing an enormous financial burden on the patient’s family and on society. However, mechanisms underlying DKD are incompletely understood.1, 2, 3 Clinically, DKD is a major microvascular complication of diabetes, with pathologic features including loss of podocytes, thickening of the glomerular basement membrane, and matrix expansion, resulting in glomerulosclerosis.4, 5, 6, 7 As for the tubular portion, recent studies showed that tubular injury in diabetic kidney occurs earlier than glomerular damage,8, 9, 10 indicating that both tubular and glomerular cells participate in the progression of DKD. Thus, it is crucial to identify a novel therapy targeting renal injury in both glomerular and tubular cells in DKD.
Renal fibrosis, one of the cardinal histological features of DKD, has been associated with transforming growth factor β (TGF-β)/Smad signaling.11, 12, 13, 14, 15 Under diabetic conditions, high glucose and its metabolites stimulate TGF-β1 expression.16,17 TGF-β1 initiates its profibrotic role by binding with TGF-β receptor 2 (TGFBR2), which activates TGFBR1,18 leading to the recruitment of Smad2/3.19,20 Then, the activated Smad2/3 complex translocates into the nucleus, where it helps to transcribe downstream genes such as fibronectin (FN), α-smooth muscle actin (α-SMA), and Snail.21, 22, 23 The accumulation of FN and α-SMA proteins in the glomerular and tubulointerstitial portion leads to glomerulosclerosis and tubulointerstitial fibrosis, respectively. Blocking TGF-β/Smad signaling has been reported to be effective for preventing the progression of DKD, proven by improved foot process effacement, alleviated renal hypertrophy, and decreased albuminuria.24,25 However, there remains a large gap in understanding the mechanism underlying the key event in TGF-β/Smad activation.
We previously reported that kidney-enriched microRNA-10a and -10b (miR-10a/b) were downregulated in high-glucose-stressed glomerular podocytes and tubular epithelial cells and in kidneys from DKD mice and patients, contributing to kidney inflammation through targeting and upregulating the NLRP3 inflammasome.26 Sustained inflammation often results in fibrosis. Results from other groups have demonstrated that in the lung tissue, declined miR-10a is associated with pulmonary fibrosis,27 making it possible that miR-10 also serves as a mediator for the fibrosis process in the kidney.
In this study, we found that the expression of miR-10a/b decreased with TGFBR1 protein increased in the kidney from both DKD mice and patients diagnosed DKD. In addition to the NLRP3-mediated inflammation pathway, the TGFBR1-mediated TGF-β/Smad pathway was also controlled by depleted miR-10. Both pathways influence renal fibrosis by regulating profibrotic genes and collagen deposition. Mechanistically, miR-10a/b exerted anti-fibrotic effect by binding to 3′ UTR of TGFBR1 mRNA. Depleted miR-10a/b lost control of TGFBR1 expression, which led to an increased amount of phosphorylated Smad3 entering the nucleus, accelerating profibrotic gene transcription. Reimbursing miR-10a/b ameliorated renal fibrosis, podocyte foot process effacement in DKD mice. Thus, our study provided a potent upstream therapeutic target for slowing the progression of fibrosis by inhibiting both TGFBR1 and inflammation pathways.
Results
TGFBR1 protein is induced in kidney from DKD mice
Uninephrectomized mice receiving 5 consecutive intraperitoneal streptozotocin (STZ) injections with a high-fat diet for 12 weeks developed albuminuria, glucosuria (Figures S1A and S1B), mesangial expansion, and renal fibrosis.28 Masson’s trichrome staining and immunohistochemical staining for TGFBR1 were employed to reveal the induction of collagen deposition and TGFBR1 protein in the kidney from DKD mice compared with the normal control (Figures 1A and 1B). Western blots for FN and α-SMA confirmed the extracellular-matrix-protein accumulation in the diabetic kidney (Figures 1C and 1D). In parallel, TGFBR1 protein, the key component in TGF-β/Smad signaling, increased in the cortex of the kidney from DKD mice (Figures 1A–1D). However, TGFBR1 mRNA was not altered (Figure 1E). Since both the glomerular and tubular regions were observed to be with TGFBR1 expression in the diabetic kidney, we selected cultured human podocytes and tubular cells to validate whether high glucose triggers TGFBR1 protein expression in vitro. Podocytes or tubular cells were treated with glucose at various concentrations for different numbers of hours. Equivalent mannitol was used as an osmotic control. Results showed that high glucose induced TGFBR1 protein expression in a time- and dose-dependent manner, with no significant alteration in TGFBR1 mRNA (Figures S2A–S2J), indicating post-transcriptional mechanisms in regulating TGFBR1 protein expression.
Figure 1.

Increase of TGFBR1 protein in mouse kidney after DKD-induced renal fibrosis
(A) Representative images of Masson’s trichrome staining and immunohistochemical staining of TGFBR1 protein (TGFBR1) in kidney sections from STZ-treated mice. Mice receiving citrate solution were used as control (Ctrl). Scale bar, 40 μm.
(B) Semi-quantification of TGFBR1 protein in (A).
(C and D) Western blot for TGFBR1, α-SMA, and FN proteins in cortex of kidney from STZ-treated or Ctrl mice (C), and quantification data (D).
(E) TGFBR1 mRNA expression by quantitative real-time PCR in kidney from STZ-treated mice.
Data were expressed as means ± SEM, n = 6 for each group. Student’s t test was used for the comparison of two groups. ∗p < 0.05, ∗∗p < 0.01.
TGFBR1 mRNA is a common target of miR-10a/b in vitro
It is acknowledged that DKD is characterized by inflammation and that sustained inflammation contributes to renal fibrosis.29 We previously reported26 that the NLRP3 inflammasome is activated in diabetic kidney and is negatively regulated by the kidney-enriched miR-10a/b. Overexpressing miR-10a/b reversed inflammation and renal fibrosis by suppressing NLRP3. Since the profibrotic role of TGFBR1 is also widely studied in other contexts, it became a doubt as to whether TGFBR1 is another independent contributor to renal fibrosis, alongside NLRP3, in DKD.
To strip the effect of NLRP3, small interfering RNA (siRNA) was used to suppress the elevation of NLRP3 expression (Figure S3A), and 25 mM high glucose was used to imitate hyperglycemia in diabetic mice. TGFBR1 was constantly upregulated after high-glucose stimulation regardless of NLRP3 expression, indicating that NLRP3 did not have a regulatory role over TGFBR1 (Figures 2A–2E). Expressions of the downstream profibrotic proteins as well as the collagen genes were partially affected by siNLRP3, but they were still increased significantly when treated with high glucose in podocytes (Figures 2A–2C) and tubular cells (Figures 2D–2F). These results coincide with previous findings that NLRP3 induces renal fibrosis by promoting inflammation30 and imply the profibrotic role of TGFBR1 in tubular cells as well as in podocytes. Interestingly, when cells were treated with siNLRP3 and miR-10 simultaneously (Figures S3B–S3E), miR-10a/b could not further lower the NLPR3 expression, possibly due to its relatively low efficiency. However, TGFBR1 expression was decreased dramatically under the miR-10a or -10b treatment. It is also noted that the same trend was observed for FN, α-SMA, and collagen genes (Figures 2A–2F). In this case, we started to investigate whether miR-10 modulates TGFBR1 expression.
Figure 2.

TGFBR1 is a direct target of miR-10a/b
(A–C) Representative immunoblotting images and quantification data of NLRP3, TGFBR1, FN, and α-SMA proteins (A and B), and quantitative real-time PCR analyzed profibrotic gene expression (C) in podocytes transfected with miR-10a or -10b mimic after knocking down NLRP3 by siRNA technology (siNLRP3). Twenty-five mM glucose was used as high-glucose (HG) stimulation, podocytes treated without high glucose. siRNA and miR-10 mimic were used as normal Ctrls.
(D–F) Representative immunoblotting images and quantification data of NLRP3, TGFBR1, FN, and α-SMA proteins (D and E), and quantitative real-time PCR analyzed profibrotic gene expression (F) in tubular cells transfected with miR-10a or -10b mimic after knocking down NLRP3 by siRNA technology (siNLRP3). Twenty-five mM glucose was used as HG stimulation. Tubular cells were treated without high glucose, and siRNA and miR-10 mimic were used as normal Ctrls.
(G) Bioinformatics prediction of miR-10a/b target site within the 3′ untranslated regions of human TGFBR1 mRNA.
(H) Relative luciferase activity in human podocytes transfected with plasmid constructed with the wild-type 3′ UTR or binding-site-mutated 3′ UTR (Mutated) of TGFBR1 mRNA, cotransfected with miR-10a or -10b mimic or antisense.
(I and J) Representative western blot for TGFBR1, FN, and α-SMA proteins in podocytes transfected with miR-10a or -10b mimic or antisense (I), and quantification data (J).
(K) Relative TGFBR1 mRNA expression by quantitative real-time PCR in podocytes transfected with miR-10a or -10b mimic or antisense.
(L) Relative luciferase activity in human tubular cells transfected with plasmid expressing wild-type or mutated 3′ UTR of TGFBR1, cotransfected with miR-10a or -10b mimic or antisense.
(M and N) Western blot for TGFBR1, FN, and α-SMA proteins in tubular cells transfected with miR-10a or -10b mimic or antisense (M), and quantification data (N).
(O) TGFBR1 mRNA expression by quantitative real-time PCR in tubular cells.
Data were expressed as means ± SEM of three independent experiments. ANOVA was used for comparison among multiple groups. Student’s t test was used for the comparison of two groups. ∗p < 0.05, ∗∗p < 0.01.
By using TargetScan and RNAhybrid,31,32 we found the binding sites for miR-10a/b in the 3′ UTR of TGFBR1 mRNA. A schematic diagram showed the species-conserved binding site within the 3′-UTR of TGFBR1 mRNA with free energies of -21.4 and -22.2 kcal/mol for miR-10a and -10b, respectively (Figure 2G). Next, the full-length 3′ UTR (wild type) or binding-site-mutated 3′ UTR (Mutated) of TGFBR1 was cloned into the luciferase-expressing plasmid. We then cotransfected luciferase plasmid and miR-10a/b mimic or antisense into podocytes or tubular cells. The results showed that overexpressing miR-10a/b decreased, whereas knocking down miR-10a/b increased, the luciferase activity (Figures 2H, 2L, and S3F–S3I). Once the putative binding sites for miR-10a/b were mutated, miR-10a/b could no longer affect luciferase activity, indicating that the putative binding sites within 3′ UTR of TGFBR1 mRNA offered a target for miR-10a/b (Figures 2H and 2L).
To evaluate whether manipulating miR-10a/b expression resulted in the alteration in TGFBR1 protein and, subsequently, the downstream genes of TGF-β/Smad signaling, we transfected podocytes (Figures 2I–2K) and tubular cells (Figures 2M–2O) with miR-10a/b mimic or antisense, and a nucleotide sequence without binding sites in cells were used as negative control (referred to as NC mimic, 10a mimic, 10b mimic, anti-NC, anti-10a, and anti-10b). The results showed that miR-10a/b significantly suppressed the protein expression of TGFBR1 as well as FN and α-SMA. In contrast, depletion of miR-10a/b stimulated protein expression of TGFBR1, FN, and α-SMA (Figures 2I, 2J, 2M, and 2N). Of note, TGFBR1 mRNA expression remained invariant (Figures 2K and 2O). These results suggested that miR-10a/b influenced fibrotic genes expression through regulating TGFBR1 protein expression at the post-transcriptional level.
XRN2 prohibits collagen deposition mainly through miR-10/TGFBR1 axis
To verify that miR-10a/b exert their anti-fibrotic function through TGFBR1 rather than other proteins in TGF-β/Smad signaling, we blocked TGFBR1 activation with TGFBR1 inhibitor. The antisense of miR-10a or -10b was administered to create miR-10a/b depletion. Twenty-four h after the miR-10a or -10b antisense transfection, podocytes or tubular cells were incubated with TGFBR1 inhibitor for 2 h to deprive its function, then glucose was introduced to stimulate TGF-β/Smad signaling. The results showed that miR-10 depletion aggravated collagen gene expression (Figures 3A, 3B, S4A, and S4B) as well as FN and α-SMA protein expression (Figures 3C, 3D, S4C, and S4D). However, TGFBR1 inhibition abolished TGF-β/Smad activation and collagen gene expression when depriving miR-10 (Figures 3A–3D and S4A–S4D), indicating that TGFBR1 is irreplaceable for miR-10 in regulating profibrotic genes expression in podocytes and tubular cells.
Figure 3.

The anti-fibrotic effect of miR-10a/b is dependent on TGFBR1 and is modulated by XRN2 in podocytes
(A and B) Relative ColIa (A) and ColIV3 (B) mRNA expression in podocytes transfected with miR-10a or -10b antisense. Twenty-five mM glucose incubation was used after TGFBR1 inhibitor treatment (TGFBR1 in).
(C and D) Western blot for TGFBR1, FN, and α-SMA proteins in podocytes transfected with miR-10a or -10b antisense in the presence of TGFBR1 inhibitor (C), and qualification data (D). DMSO was used as a dissolvent Ctrl.
(E and F) Western blot for XRN2, TGFBR1, and FN protein expression in podocytes treated with HG for 24 h (E), and quantification data (F).
(G) Relative miR-10a and -10b expression in podocytes treated with HG. siRNA target XRN2 (siXRN2) was introduced to evaluates its function in miR-10a or -10b expression. An RNA sequence without a binding site was used as a Ctrl (siNC).
(H and I) Western blot for XRN2, TGFBR1, and FN protein expression in podocytes treated with siXRN2. Twenty-four h later, miR-10a or -10b mimic was transfected into podocytes.
Data were expressed as means ± SEM. ANOVA was used for comparison among multiple groups. Student’s t test was used for the comparison of two groups. ∗p < 0.05, ∗∗p < 0.01.
It has been reported that exoribonuclease 2 (XRN2), a nuclear 5′ to 3′ exoribonuclease, is essential for miR-10a maturation in lung cancer,33 raising speculation that XRN2 may also participate in the regulation of miR-10a/b in kidney cells. Firstly, we measured XRN2 protein expression in high-glucose-treated podocytes and tubular cells. The results showed that high glucose restrained XRN2 expression (Figures 3E, 3F, S4E, and S4F) and mature miR-10a/b levels (Figures 3G and S4G), suggesting the possibility of a connection between XRN2 and miR-10a/b expression. To further investigate whether miR-10a/b levels are dictated by XRN2, we knocked down XRN2 by siRNA method. The results showed that XRN2 knockdown resulted in a significant decline of miR-10a/b, followed by intense TGFBR1 and FN activation (Figures 3G–3I and S4G–S4I). Moreover, enhanced TGFBR1 and FN expression reverted back to basal levels when miR-10a/b was restored (Figures 3H, 3I, S4H, and S4I). These results indicate that miR-10a/b regulate fibrotic gene expression mainly through inhibiting TGFBR1 and that XRN2 is required for miR-10a/b maturation.
Overexpressing miR-10a or -10b alleviates renal fibrosis in DKD mice
Based on our results, we hypothesized that high glucose reduces miR-10a/b, leading to renal fibrosis in DKD. To verify our presupposition, lentiviruses harboring miR-10a/b (referred to as 10a mimic or 10b mimic) were intrarenally injected into the kidney to restore declined miR-10a/b expression in DKD mice. A lentivirus expressing a negative-control sequence served as the control (NC mimic). One week after lentivirus injection, mice were treated with STZ injection to induce a diabetic phenotype (Figure 4A). At the end of the experiment at the 12th week, we observed that the lentivirus harboring 10a/b mimic effectively reversed the declined expression of miR-10a/b (Figures 4B and 4C). In mice overexpressing miR-10a or -10b, H&E staining suggested ameliorated kidney injury, whereas Masson trichrome and Sirius-red staining demonstrated that less collagen was deposited in both glomerular and tubular compartments of kidney (Figures 4D–4F). In addition, glomerular foot process effacement was alleviated by miR-10a/b restoration (Figures 4G and 4H).
Figure 4.

Overexpression of miR-10/b prevents renal fibrosis
(A) Schematic diagram of the animal experimental procedure.
(B and C) miR-10a (B) and -10b (C) expression by quantitative real-time PCR in kidney from STZ-treated mice. Lentivirus expressing miR-10a or -10b mimic (10a or 10b mimic) was used to overexpress miR-10a or 10b, and lentivirus expressing a nonsense sequence was used as a negative Ctrl (NC mimic) (n = 6).
(D and E) Representative images of H&E staining (D) and Masson’s trichrome and Sirius-red staining (E) in kidney sections from lentivirus-treated DKD mice. Scale bar, 40 μm.
(F) Fibrotic area was calculated in (E). n = 6 in each group.
(G and H) Representative images of transmission electron microscopy in kidney sections from lentivirus-treated DKD mice above (G), and foot-process weight was quantified (H). Scale bar, 1 μm.
Data were expressed as means ± SEM. Student’s t test was used for the comparison of two groups. ∗p < 0.05, ∗∗p < 0.01.
Abrogated miR-10a/b aggravate kidney injury in DKD mice
To further test the protective role of miR-10a/b in diabetic kidney, we knocked down miR-10a/b by a lentivirus incorporated with miR-10a/b antisense. A lentivirus expressing a negative-control sequence served as the control (referred to as anti-NC, anti-10a, and anti-10b). The results showed that intrarenal injection of lentivirus with miR-10a/b antisense effectively decreased miR-10a or -10b expression compared with anti-NC control (Figure 5A). The decline in miR-10a/b levels induced accelerated kidney injury and collagen accumulation in the glomerular and tubular interstitial regions (Figures 5B–5D). Deprived miR-10a/b induced exacerbated foot process effacement (Figures 5E and 5F). These results suggest that miR-10a/b abrogation aggravated renal fibrosis in DKD.
Figure 5.

Knocking down miR-10a/b aggravates renal fibrosis
(A) miR-10a/b expression by quantitative real-time PCR in kidney from STZ-treated mice. Lentivirus expressing miR-10a or -10b antisense (Anti-10a or Anti-10b) was used to knock down miR-10a or -10b, and lentivirus expressing a nonsense sequence was used as a negative Ctrl (anti-NC) (n = 6).
(B and C) Representative images of H&E staining (B) and Masson’s trichrome and Sirius-red staining (C) in kidney sections from lentivirus-treated DKD mice. Scale bar, 40 μm.
(D) Fibrotic area was calculated in (C). n = 6 in each group.
(E and F) Representative images of transmission electron microscopy in kidney sections from lentivirus-treated DKD mice above (E), and foot-process weight was quantified (F). Scale bar, 1 μm.
Data were expressed as means ± SEM. Student’s t test was used for the comparison of two groups. ∗p < 0.05, ∗∗p < 0.01.
miR-10a/b regulate renal fibrosis via suppressing TGF-β/Smad signaling in DKD mice
Next, we evaluated the effects of miR-10a/b on the regulation of TGFBR1 protein and the activation of TGF-β/Smad signaling in DKD mice. Immunohistochemical staining for TGFBR1 protein was increased in the kidney from DKD mice, while lentivirus expression of the NC mimic or antisense did not affect the TGFBR1 expression. Consistent with the observation in vivo, STZ-induced TGFBR1 protein in kidney was suppressed by the overexpression of miR-10a/b (Figures 6A and 6B). After TGFBR1 was induced, phosphorylated Smad3, the key mediator in TGF-β/Smad signaling, increased in the nucleus location to exert its role as a profibrotic factor (Figures 6A and 6C). Immune blotting reconfirmed that the overexpression of miR-10a/b reversed the induction of TGFBR1 protein in DKD mice, thus resulting in a reduction of fibrotic genes expression in the kidney (Figures 6D and 6E). The anti-fibrotic effect of miR-10a/b was further validated by decreased ColIa and ColIV3 gene expression in the kidney (Figure 6F). In contrast, depriving miR-10a/b enhanced the induction of TGFBR1 and phosphorylated Smad3 (Figures 6A–6C). TGFBR1, FN, and α-SMA protein expression as well as ColIa and ColIV3 gene expression was strengthened by decreased miR-10a/b levels in the cortex homogenate from miR-10a/b-antisense-treated mice (Figures 6G–6I).
Figure 6.

miR-10a/b regulate renal fibrosis in DKD mice through inhibiting TGF-β/Smad signaling pathway
(A–C) Representative images of immunohistochemical staining of TGFBR1 and phosphorylated Smad3 (A), and quantification data (B and C). Scale bar, 40 μm.
(D and E) Western blot for TGFBR1, FN, and α-SMA protein expression in kidney from DKD mice with lentivirus expressing a nonsense sequence, an miR-10a mimic, or an miR-10b mimic (D), and quantification data (E).
(F) ColIa and ColIV3 mRNA expression in kidney from mice above by quantitative real-time PCR (n = 6).
(G and H) Western blot for TGFBR1, FN, and α-SMA protein expression in kidney from mice with lentivirus expressing miR-10a or -10b antisense (G), and quantification data (H).
(I) ColIa and ColIV3 mRNA expression in kidney from STZ-induced DKD mice by quantitative real-time PCR (n = 6).
Data were expressed as means ± SEM. Student’s t test was used for the comparison of two groups. ANOVA was used for comparison among multiple groups. ∗p < 0.05, ∗∗p < 0.01.
Intrarenal miR-10a/b decrease in DKD patients with fibrosis
To measure the level of intrarenal miR-10a/b in DKD patients with fibrosis, we analyzed 19 kidney samples with biopsy-proven DKD (Table S1). Normal kidney samples were obtained from a nondiseased portion of nephrectomy specimens from patients with local renal tumors. Masson’s trichrome staining revealed renal fibrosis in DKD patients (Figures 7A and 7B). In situ hybridization of miR-10a/b showed that both miR-10a and -10b were significantly reduced compared with normal controls (Figures 7C and 7D). Immunohistochemical staining further indicated induction of TGFBR1 and FN protein expression (Figures 7E and 7F).
Figure 7.

Renal miR-10a and -10b decrease in DKD patients with fibrosis
(A and B) Representative images of Masson’s trichrome staining of renal fibrosis in patients with DKD (A), and quantification data (B). Scale bar, 40 μm.
(C and D) Representative images of in situ hybridization staining of miR-10a/b in patients with DKD (C), and quantification data (D). Scale bar, 40 μm.
(E and F) Representative images of immunohistochemical staining of TGFBR1 and FN in patients with DKD (E), and quantification data (F). Scale bar, 40 μm.
Data were expressed as means ± SEM. Student’s t test was used for the comparison of two groups. ∗∗p < 0.01.
Discussion
Our previous studies have demonstrated that overexpressing miR-10a or -10b decreased extracellular matrix deposition, whereas knocking down miR-10a or -10b enhanced kidney fibrosis.26 The altered fibrosis program may result from miR-10a/b-mediated NLRP3-inflammasome-associated sustained inflammation or from a miR-10a/b directly regulated mechanism. However, other pathways cannot be ruled out in fibrosis progression in DKD.
In this study, to uncover other contributors to renal fibrosis in DKD, we inhibited an NLRP3-mediated inflammation cascade by siRNA targeting NLRP3. The results showed that TGF-β/Smad was an independent and excessively activated pathway that contributed to profibrotic gene transcription and collagen deposition in kidney cells. In addition, TGFBR1, one of the key components of TGF-β/Smad signaling, was also directly regulated by miR-10a/b, giving rise to a very strong anti-fibrotic effect of miR-10 by suppressing the TGF-β/Smad pathway and the NLRP3-mediated inflammation pathway simultaneously.
An important finding of our study is that high-glucose levels can regulate the XRN2/miR-10a/b/TGFBR1/fibrosis axis in kidney cells. Evidence has been provided to prove that high-glucose incubation downregulated XRN2 protein, leading to decreased miR-10a/b, and thus promoting TGFBR1 protein in renal cells. Increased TGFBR1 levels in diabetic kidneys triggered the phosphorylated Smad3 translocated into the nucleus, activating TGF-β/Smad signaling. Consequently, renal fibrosis was induced in DKD mice.
Genes associated with features including extracellular-matrix accumulation and fibrosis in glomerular and tubular cells are regulated by both classical signaling pathways and epigenetic mechanisms including non-coding RNAs in DKD.3 Although several non-coding RNAs, such as miRNA-204,34 miRNA-214,35 and miRNA-29,36 have been reported to protect the kidney against injury in diabetes, there remains a need for further studies of disease etiology to develop more efficacious treatment for DKD. In this study, we demonstrated that miR-10a/b can protect both glomerular and tubular cells from fibrosis by targeting TGFBR1 at the post-transcriptional mechanism. Of note, there is a slight but not significant increase of TGFBR1 mRNA in high-glucose-stimulated tubular cells, suggesting that except for post-transcription mechanisms, there may be a possibility of an add-on or different mechanism in TGFBR1 protein in tubular cells. In both podocytes and tubular cells, overexpressing miR-10a or -10b can significantly downregulate profibrotic gene expression, including FN and α-SMA. In addition, the anti-fibrotic effects of miR-10a/b are potent enough by solely suppressing TGFBR1 (Figures 3 and S3). Delivering miR-10a or -10b into diabetic kidney prevented fibrosis and foot process effacement in DKD mice. These data suggest that in addition to inhibiting renal inflammation,26 miR-10a/b can also protect the kidney from injury by suppressing renal fibrosis, making them a promising therapeutic target for DKD.
In conclusion, our study illustrated that miR-10a/b can regulate renal fibrosis through directly targeting TGFBR1 and regulating TGF-β/Smad signaling in diabetic mellitus. These findings provide novel insight into the pathogenesis of DKD and suggest that targeting miR-10a/b may represent an effective therapeutic approach to treat DKD.
Materials and methods
Human renal biopsy samples
Six patients who underwent nephrectomy for solitary renal carcinoma without diabetes and a urine albumin-to-creatinine ratio <30 mg/g in biochemical analysis were collected as control. These control samples obtained from healthy kidney poles of solitary renal carcinoma were pathologically normal. The control kidneys used in this study were obtained from surgical nephrectomies for diagnostic purposes. Renal biopsy samples from 19 patients with biopsy-proven DKD were analyzed. Diagnosis of DKD was confirmed by both kidney histological and scanning electron microscopy examinations. The protocol regarding the use of kidney samples in this study was approved by the Human Subjects Committee of the Guangzhou Kingmed Center for Clinical Laboratory. Informed consent was obtained from all participants.
Animals and DKD models
The use of animals in this study was approved by the Institutional Animal Care and Use Committee at Nanfang Hospital. Male C57BL/6J mice (20–25 g) were used to generate a high-fat-diet- and STZ-induced DKD model with albuminuria and glomerular lesions, as previously described.37,38 Briefly, uninephrectomized mice were fasted for 4 h followed by intraperitoneal STZ injection (50 mg/kg) for 5 consecutive days. Blood glucose levels were monitored 1 week after the last STZ injection, and mice with fasting blood glucose levels ≥15 mmol/L were considered diabetic. All mice had unrestricted access to food and water for 12 weeks on the high-fat diet. Then kidneys from the control and DKD groups were harvested for further study.
Intrarenal lentivirus delivery
Seven days before STZ injection, recombinant lentivirus vector pGLV-harboring miR-10a/b mimic or antisense (referred as 10a mimic, 10b mimic, anti-10a, or anti-10b) or a nonsense sequence (referred to as NC mimic, anti-NC) were delivered into mouse kidney. In anesthetized mice, a 31G needle was inserted from the lower pole of the temporary obstructed kidney to the upper pole. As the needle was slowly withdrawn, 100 μL (105TU/μL) lentivirus was slowly injected.
Transmission electron microscopy (TEM)
Electron-microscopic sample handling and detection were performed by the Pathology Laboratory of Nephrology at Nanfang Hospital. The thickness of the glomerular basement membrane, foot process effacement, and the number of foot processes in TEM images were analyzed with ImageJ (National Institutes of Health, NIH, Bethesda, MD, USA). Five micrographs were taken in one glomerulus, and 6–8 glomeruli were randomly selected from one kidney.
Cell culture, treatment, and transfection
The human podocytes were proliferated in RPMI 1640 Media (11 mmol/L glucose, Life Technologies, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS), 1% antibiotic/antimycotic solution, and 0.1% Insulin-Transferrin-Selenium-Ethanolamine solution (ITS) (Life Technologies) at 33°C in an incubator with 5% CO2. When cells were grown to 70% confluence, they were thermoswitched to 37°C for another 10 days to induce differentiation. The human renal proximal tubular epithelial (HK2) cells were maintained in a DMEM/low-glucose (5.5 mmol/L) medium (Life Technologies) with 10% FBS and 1% antibiotics at 37°C. For high-glucose stimulation, cells were stimulated with final glucose concentration in medium at 25 mmol/L. For transient transfection, Lipofectamine 3000 was used in accordance to the manufacturer’s instructions. The miR-10a/b mimic and antisense were purchased from GenePharma (GenePharma, Shanghai, China). Twenty-four h after miR mimic or antisense transfection, cells were pretreated with TGFBR1 inhibitor SB 431542 (Sigma-Aldrich, St. Louis, MO, USA) at dosages of 10 μM to inhibit TGFBR1 activity. Two h later, indicated glucose was added into culture medium.
Histology and immunohistochemistry
Mouse kidney tissue was embedded in paraffin and cut into 2-μm-thick sections. After being rehydrated, sections were processed to the H&E, Masson’s trichrome, and Sirius-red staining. For immunohistochemistry, 8-μm-thick sections were cut. After antigen retrieval and blocking, a primary antibody against TGFBR1 (Abcam, Cambridge, UK), FN (Sigma-Aldrich, St. Louis, MO, USA), and phosphorylated Smad3 (Boster Biological Technology, Wuhan, China) was incubated at 4°C overnight. Staining was developed with DAB and counterstained with hematoxylin. Positive staining areas were quantified with Image-Pro Plus 6.0 software.
RNA isolation and quantitative real-time PCR assays
Total RNA was isolated from tissues or cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. MystiCq microRNA qPCR Assay (Sigma-Aldrich) was employed to quantify miR-10a/b expression according to the manufacturer’s instructions, and U6 was used as an internal control. For relative miR expression, miR-10a/b expression was calculated as 2-ΔCT (ΔCT = CTmiR.- CTU6).
The mRNA expression levels were determined by quantitative real-time PCR using SYBR Premix Ex Taq (Takara Bio, Shiga, Japan) on 7500 fast Real-Time PCR Systems (Applied Biosystems, Foster City, CA, USA). All reactions were run in triplicate. The sequences of specific primers are listed below.
Human
TGFBR1: GGGGCGAACGCATTACAGTGTTTCTGCCAC
TGGAATGCAGAGGAAGCAGACTGGACCAGC.
ColIa: GACTGGAAGAGCGGAGAGTACTG
CCTTGATGGCGTCCAGGTT.
ColIV3 CGGGTACCCAGGACTCATAG
GGACCTGCTTCACCCTTTTC.
Mouse
TGFBR1: CGAGAGGCAGAGATTTATCAGACTG
TCCACCAATAGAACAGCGGCG.
ColIa: TAGGCCATTGTGTATGCAGC
ACATGTTCAGCTTTGTGGACC.
ColIV3: ACCACGGCCATTCCTTCAT
CAAAAAGAAGAGAAAACCCACTATAGAGT.
Luciferase reporter assay
The 3′ UTR of TGFBR1 was amplified via PCR with human genomic DNA as the template and inserted downstream of the pGL3 promotor (Promega), referred to as wild type (WT). The binding site of the “seed sequence” for miR-10a and -10b was mutated into its complimentary sequence (ACAGGGTA into UGUCCCAU). The constructed plasmids were cotransfected with miR-10a/b mimic or nonsense RNA without a binding site in the host cells (NC) into human podocytes or HK2 using Lipofectamine 3000. Twenty-four h after transfection, cell lysates were subjected to luciferase assay with a Dual-Luciferase Reporter Assay System (Promega). Luciferase activity was normalized to Renilla luciferase activity.
Western blotting
Tissue or cell lysates were prepared with radioimmunoprecipitation assay buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA). The blots were then incubated with a primary antibody against TGFBR1 (R&D Systems, Minneapolis, MN, USA), α-SMA (Sigma-Aldrich), FN (Sigma-Aldrich), and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The relative protein expression was normalized to GAPDH.
miRNA in situ hybridization
In situ hybridization for miR-10a/b was performed with double digoxigenin-labeled locked nucleic acid (LNA) miRCURY probes (Exiqon). U6 served as the positive control, whereas a scrambled probe was conducted as the negative control. Paraffin-embedded tissue was cut into 6-μm-thick sections and incubated with indicated probes at 55°C for 2 h. An anti-digoxigenin antibody conjugated with alkaline phosphatase (Roche) was employed, followed by color development with nitroblue tetrazolium (NBT)/5-bromo-4-chloro-3-indolyl phosphate (BCIP) (Roche) as substrates. Sections were counterstained with fast red. After mounting, miR-10a/b expression was visualized under phase-contrast microscopy.
Statistical analyses
Data are representative of at least three independent experiments. Data are presented as the mean ± SEM. Differences between groups were analyzed using unpaired Student’s t test (for comparison between two samples) or analysis of variance (ANOVA) (for multiple comparisons). Statistical calculations were performed using GraphPad Prism v.6.0 (GraphPad Software, San Diego, CA, USA). A p value <0.05 was considered statistically significant.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81700640 and 81970608).
Author contributions
J.L., S.Y., J.F., J.Z., S.C., J.T., and S.N. performed the experiments. H.D. and J.L. analyzed the data. S.Y. and X.L. collected and analyzed human kidney biopsies. H.D. designed the study and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.04.002
Supplemental information
References
- 1.Thomas M.C., Cooper M.E., Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016;12:73–81. doi: 10.1038/nrneph.2015.173. [DOI] [PubMed] [Google Scholar]
- 2.Hocher B., Tsuprykov O. Diabetic nephropathy: renoprotective effects of GLP1R agonists and SGLT2 inhibitors. Nat. Rev. Nephrol. 2017;13:728–730. doi: 10.1038/nrneph.2017.140. [DOI] [PubMed] [Google Scholar]
- 3.Kato M., Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019;15:327–345. doi: 10.1038/s41581-019-0135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Declèves A.E., Sharma K. New pharmacological treatments for improving renal outcomes in diabetes. Nat. Rev. Nephrol. 2010;6:371–380. doi: 10.1038/nrneph.2010.57. [DOI] [PubMed] [Google Scholar]
- 5.Steffes M.W., Osterby R., Chavers B., Mauer S.M. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes. 1989;38:1077–1081. doi: 10.2337/diab.38.9.1077. [DOI] [PubMed] [Google Scholar]
- 6.Ziyadeh F.N. The extracellular matrix in diabetic nephropathy. Am. J. Kidney Dis. 1993;22:736–744. doi: 10.1016/s0272-6386(12)80440-9. [DOI] [PubMed] [Google Scholar]
- 7.Anders H.J., Huber T.B., Isermann B., Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018;14:361–377. doi: 10.1038/s41581-018-0001-y. [DOI] [PubMed] [Google Scholar]
- 8.Ginevri F., Piccotti E., Alinovi R., DeToni T., Biagini C., Chiggeri G.M., Gusmano R. Reversible tubular proteinuria precedes microalbuminuria and correlates with the metabolic status in diabetic children. Pediatr. Nephrol. 1993;7:23–26. doi: 10.1007/BF00861555. [DOI] [PubMed] [Google Scholar]
- 9.Magri C.J., Fava S. The role of tubular injury in diabetic nephropathy. Eur. J. Intern. Med. 2009;20:551–555. doi: 10.1016/j.ejim.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Tramonti G., Kanwar Y.S. Tubular biomarkers to assess progression of diabetic nephropathy. Kidney Int. 2011;79:1042–1044. doi: 10.1038/ki.2011.9. [DOI] [PubMed] [Google Scholar]
- 11.Reeves W.B., Andreoli T.E. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc. Natl. Acad. Sci. U S A. 2000;97:7667–7669. doi: 10.1073/pnas.97.14.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldfarb S., Ziyadeh F.N. TGF-beta: a crucial component of the pathogenesis of diabetic nephropathy. Trans. Am. Clin. Climatol Assoc. 2001;112:27–33. discussion 33. [PMC free article] [PubMed] [Google Scholar]
- 13.Huang C., Kim Y., Caramori M.L., Fish A.J., Rich S.S., Miller M.E., Russell G.B., Mauer M. Cellular basis of diabetic nephropathy: II. The transforming growth factor-beta system and diabetic nephropathy lesions in type 1 diabetes. Diabetes. 2002;51:3577–3581. doi: 10.2337/diabetes.51.12.3577. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J., Miyamoto S., You Y.H., Sharma K. AMP-activated protein kinase (AMPK) activation inhibits nuclear translocation of Smad4 in mesangial cells and diabetic kidneys. Am. J. Physiol. Renal. Physiol. 2015;308:F1167–F1177. doi: 10.1152/ajprenal.00234.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ka S.M., Yeh Y.C., Huang X.R., Chao T.K., Hung Y.J., Yu C.P., Lin T.J., Wu C.C., Lan H.Y., Chen A. Kidney-targeting Smad7 gene transfer inhibits renal TGF-beta/MAD homologue (SMAD) and nuclear factor kappaB (NF-kappaB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia. 2012;55:509–519. doi: 10.1007/s00125-011-2364-5. [DOI] [PubMed] [Google Scholar]
- 16.Xiang G., Schinzel R., Simm A., Sebekova K., Heidland A. Advanced glycation end products impair protein turnover in LLC-PK1: amelioration by trypsin. Kidney Int. Suppl. 2001;78:S53–S57. doi: 10.1046/j.1523-1755.2001.59780053.x. [DOI] [PubMed] [Google Scholar]
- 17.Yamagishi S., Inagaki Y., Okamoto T., Amano S., Koga K., Takeuchi M. Advanced glycation end products inhibit de novo protein synthesis and induce TGF-beta overexpression in proximal tubular cells. Kidney Int. 2003;63:464–473. doi: 10.1046/j.1523-1755.2003.00752.x. [DOI] [PubMed] [Google Scholar]
- 18.Heldin C.H., Miyazono K., ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 19.Tsukazaki T., Chiang T.A., Davison A.F., Attisano L., Wrana J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 20.Sato M., Muragaki Y., Saika S., Roberts A.B., Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hocevar B.A., Brown T.L., Howe P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu B., Wu Z., Phan S.H. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am. J. Respir. Cell Mol. Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 23.Peinado H., Quintanilla M., Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 24.Wang D., Zhang G., Chen X., Wei T., Liu C., Chen C., Gong Y., Wei Q. Sitagliptin ameliorates diabetic nephropathy by blocking TGF-β1/Smad signaling pathway. Int. J. Mol. Med. 2018;41:2784–2792. doi: 10.3892/ijmm.2018.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson P.G., Thompson J.C., Yoder M.H., Charnigo R., Tannock L.R. Prevention of renal apoB retention is protective against diabetic nephropathy: role of TGF-β inhibition. J. Lipid Res. 2017;58:2264–2274. doi: 10.1194/jlr.M078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding H., Li J., Li Y., Yang M., Nie S., Zhou M., Zhou Z., Yang X., Liu Y., Hou F.F. MicroRNA-10 negatively regulates inflammation in diabetic kidney via targeting activation of the NLRP3 inflammasome. Mol. Ther. 2021;29:2308–2320. doi: 10.1016/j.ymthe.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie T., Liang J., Guo R., Liu N., Noble P.W., Jiang D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol. Genomics. 2011;43:479–487. doi: 10.1152/physiolgenomics.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du P., Fan B., Han H., Zhen J., Shang J., Wang X., Li X., Shi W., Tang W., Bao C., et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013;84:265–276. doi: 10.1038/ki.2013.113. [DOI] [PubMed] [Google Scholar]
- 29.Tang S.C.W., Yiu W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020;16:206–222. doi: 10.1038/s41581-019-0234-4. [DOI] [PubMed] [Google Scholar]
- 30.Mulay S.R. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. 2019;96:58–66. doi: 10.1016/j.kint.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Grimson A., Farh K.K., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H., Lu Y., Chen E., Li X., Lv B., Vikis H.G., Liu P. XRN2 promotes EMT and metastasis through regulating maturation of miR-10a. Oncogene. 2017;36:3925–3933. doi: 10.1038/onc.2017.39. [DOI] [PubMed] [Google Scholar]
- 34.Cheng Y., Wang D., Wang F., Liu J., Huang B., Baker M.A., Yin J., Wu R., Liu X., Regner K.R., et al. Endogenous miR-204 protects the kidney against chronic injury in hypertension and diabetes. J. Am. Soc. Nephrol. 2020;31:1539–1554. doi: 10.1681/ASN.2019101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Z., Li L., Livingston M.J., Zhang D., Mi Q., Zhang M., Ding H.F., Huo Y., Mei C., Dong Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Invest. 2020;130:5011–5026. doi: 10.1172/JCI135536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H.Y., Zhong X., Huang X.R., Meng X.M., You Y., Chung A.C., Lan H.Y. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 2014;22:842–853. doi: 10.1038/mt.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu M., Liang K., Zhen J., Zhou M., Wang X., Wang Z., Wei X., Zhang Y., Sun Y., Zhou Z., et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat. Commun. 2017;8:413. doi: 10.1038/s41467-017-00498-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Azushima K., Gurley S.B., Coffman T.M. Modelling diabetic nephropathy in mice. Nat. Rev. Nephrol. 2018;14:48–56. doi: 10.1038/nrneph.2017.142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.