

Abstract
Targeted therapy is a groundbreaking innovation for cancer treatment. Among the receptor tyrosine kinases, the fibroblast growth factor receptors (FGFRs) garnered substantial attention as promising therapeutic targets due to their fundamental biological functions and frequently observed abnormality in tumors. In the past 2 decades, several generations of FGFR kinase inhibitors have been developed. This review starts by introducing the biological basis of FGF/FGFR signaling. It then gives a detailed description of different types of small-molecule FGFR inhibitors according to modes of action, followed by a systematic overview of small-molecule-based therapies of different modalities. It ends with our perspectives for the development of novel FGFR inhibitors.
Keywords: FGFR, tyrosine kinase, small-molecule inhibitors, targeted therapy, cancer
1 Introduction
Currently, one of the most important modalities for cancer treatment is the targeted therapy which hampers the growth of cancer cells by chemical intervention against specific target biomolecules known to be essential for tumorigenesis and proliferation. A number of protein kinases in the human body are associated with cancer initiation and progression, and small molecules that inhibit these kinases have thus far gained notable achievement manifested by ∼70 FDA-approved small molecule kinase inhibitor drugs for the treatment of a variety of malignancies (Ayala-Aguilera et al., 2022). FGFRs are a family of receptor tyrosine kinases that have been successfully targeted by three approved small-molecule inhibitors. Due to their functional versatility and frequent alterations in different cancers, FGFRs are considered to be a promising target, and more inhibitors are expected to be translated from bench to bedside in the near future.
Small-molecule FGFR inhibitors have been reviewed by others in the past several years, but these papers mainly focused on small molecules targeting the kinase domain. Herein, we make a systematic and comprehensive description on FGF/FGFR signaling, their role in cancer development, and drug resistance. We also update the development of different modalities targeting FGF-FGFR axis with a detailed discussion of their advantages and future trend.
2 Fibroblast Growth Factors
The mammalian fibroblast growth factors (FGFs) are a family of 23 proteins, which exert a wide variety of biological effects on different types of cells. Based on their sequence homology and mode of action, these proteins are classified as secreted FGFs and intracellular FGFs (iFGFs). The iFGFs (FGF11-14) are non-signaling factors that do not bind to any cell surface receptor. Instead, they function as cofactors for voltage-gated sodium channels (Goldfarb et al., 2007). In contrast, all secreted FGFs signal to a class of receptor tyrosine kinases named fibroblast growth factor receptors (FGFRs). In general, secreted FGFs are produced intracellularly and secreted to extracellular matrix (ECM) and eventually bind to FGFRs to initiate signal transduction.
Depending on how far they can travel, secreted FGFs are further classified into two subfamilies: canonical FGFs and endocrine FGFs. In the ECM, canonical FGFs (FGF1-10,16–18, 20, and 22) interact with copious cofactors named heparan sulfate proteoglycans (HSPGs), which limit diffusion of FGFs and regulate their specificity toward FGFRs (Ornitz, 2000; Matsuo et al., 2013). Hence, canonical FGFs function as autocrine or paracrine factors, traveling merely a short distance before binding to the FGFRs on the cells of their origin or adjacent cells (Belov et al., 2013). The binding of canonical FGFs to FGFRs triggers a series of cellular processes related to cellular survival, metabolism, proliferation and differentiation, and consequently mediates organogenesis, tissue metabolism, repair, regeneration and inflammatory response (Belov et al., 2013; Powers et al., 2000; N.; Wang et al., 2018).
Due to the reduced affinity for HSPGs, endocrine FGFs (FGF15/19, 21 and 23) often permeate through the HSPGs-rich extracellular matrix into the circulatory system, and subsequently reach all parts of the body like endocrine hormones (Fernandes-Freitas et al., 2015). Instead of HSPGs, endocrine FGFs require members of Klotho family, including αKlotho, βKlotho, and Klotho-LPH related protein (KLPH), to generate FGF-FGFR-Klotho ternary complex (Angelin et al., 2012; Ding et al., 2012). As a result of their hormone-like features, endocrine FGFs play important roles in the metabolism of bile acid, glucose and lipid in addition to the canonical FGF functions.
Therefore, dysregulation of expression, secretion, and degradation of FGFs lead to aberrations in the metabolism, organogenesis (Dorey et al., 2010; Yu et al., 2017), wound healing (Müller et al., 2012), and are responsible for many cancers (Brooks et al., 2012).
3 Fibroblast Growth Factor Receptors
The human fibroblast growth factor receptors belong to receptor tyrosine kinases (RTKs), consisting of FGFR1, FGFR2, FGFR3, FGFR4, and FGFR5. Although FGFR1-4 are encoded by four distinct genes, they share great sequence homology with an identity of 56–71%. The FGFR5, also called FGFR-like 1 (FGFRL1), possesses structural similarity with FGFR1-4 but lacks an intracellular kinase domain (Wiedemann et al., 2000). Activated FGFRs participate in multiple cell processes through intervening several signaling pathways.
3.1 Structure of FGFR
FGFRs are single-pass transmembrane proteins containing approximately 800 amino acids, which are composed of several domains: an extracellular ligand binding domain, a transmembrane domain, and an intracellular domain with kinase activity (Figure 1).
FIGURE 1.

Structure of FGFR. The upper panel shows the main domains of FGFR; the bottom panel demonstrates their corresponding sequences (UniProtKB: P11362). Three dimensional structures (brown) and functional regions (cyan) involved in the kinase domain of FGFR are marked.
The extracellular ligand binding domain of FGFRs is composed of three immunoglobulin-like subdomains (IgI, IgII and IgIII) and an acidic-residues-rich sequence termed acid box (Itoh et al., 2004). IgI and acid box have been demonstrated to play a key role in autoinhibition of FGFRs in the absence of FGFs (Sanchez-Heras et al., 2006). IgII and IgIII form the FGF-binding pocket, thus are responsible for the binding specificity between FGFRs and FGFs. There are two isoforms of IgIII (b/c) in FGFR1-3 that result from alternative splicing, while this is not observed for FGFR4.
The single-pass transmembrane domain (TM) is embedded in the cell membrane, functioning as an anchor of FGFR. The TM also supports the dimerization of cytoplasmic kinase domains of two FGFRs which leads to activation of FGFR (Itoh et al., 2004).
The intracellular tyrosine kinase domain of FGFR1-4 (∼300 amino acids) is the most extensively investigated part, which possesses a classical kinase architecture (Figure 2) (Itoh et al., 2004; Mohammadi, Schlessinger, et al., 1996; Sanchez-Heras et al., 2006). The small N-terminal lobe (N-lobe, ∼100 amino acids) is composed of a five-stranded anti-parallel β-sheet (β1-β5) and an αC-helix that resides between β3 and β4 in sequence and flanks the β-sheet spatially. A highly-flexible glycine-rich loop between β1 and β2 termed P-loop is able to swing downward in the presence of ATP to create the nucleotide binding site (Guimarães et al., 2011). In contrast, seven α-helices (αD, αE, αEF, αF, αG, αH and αI) gather to form the main part of the larger C-terminal lobe (C-lobe, ∼200 amino acids).
FIGURE 2.

FGFR1 kinase domain structure (PDB: 4UWY). Basic secondary structures and critical regions described in this review are highlighted in (A). The critical DFG, HRD motif and molecular brake are highlighted by close-up in (B–D), respectively.
The N-lobe and C-lobe are connected by a hinge region containing conserved residues which could offer critical contacts with the adenine moiety of an ATP molecule. In addition, a triad of residues around the hinge region (e.g., N549, E565, and K641 in FGFR2) acts as the “molecular brake” of FGFRs to regulate their autoinhibition (Chen et al., 2007). The C-lobe also contains two long loops and some short strands at the interface of the two lobes, all of which contribute to the exquisite machinery for the activation and functioning of the kinase.
In between β8 and αEF, 20–30 amino acids round up to form the activation loop (A-loop), which encompasses tyrosine phosphorylation sites (Webster et al., 1996). At the beginning of the A-loop, an Asp-Phe-Gly triad constitutes the highly conserved DFG-motif, which is indicative of the active/inactive states of kinase. Located between αE and β7 is another important loop named catalytic loop, which contains the His-Arg-Asp (HRD) motif. The Asp of HRD-motif interacts with the hydroxyl group of the substrate tyrosine and therefore contributes to the phosphorylation (Vijayan et al., 2015).
3.2 FGF/FGFR Signaling
3.2.1 Activation of FGFRs
Once FGFs bind to the extracellular domains of FGFRs, the dimerization of transmembrane and intracellular domains takes place along with a series of conformational changes that lead to trans-phosphorylation of dimerized kinase domains for activation.
The activation of kinase domain is a fined-tuned process (Furdui et al., 2006). Several critical tyrosine residues including Y463, Y583, Y585, Y653, Y654, Y730, and Y766 are autophosphorylated by precisely ordered reactions while ATP binds to the highly conserved pocket located in the hinge region during the activation of FGFR1 (Mohammadi, Dikic, et al., 1996). The autophosphorylations of Y653 and Y654 in the A-loop, which appear to induce the binding of substrate but not ATP, have increased by 50–100 fold and 500–1,000 fold in the rate of substrate phosphorylation, respectively. This suggested that these autophosphorylations have an indispensable role in kinase activation. The function of other tyrosine autophosphorylation sites contributes to the activation of FGFRs and downstream signal transduction through diverse biochemical reactions yet to be discovered.
In addition to the phosphorylation of critical tyrosine residues, the DFG motif of kinase domain toggles between two different conformations in line with the state of FGFR (active or inactive). When the motif adopts a DFG-in conformation, its Asp coordinates with phosphate groups of ATP and/or magnesium ion and causes the A-loop to display an open conformation, rendering the kinase an active state. Conversely, a DFG-out conformation, where the Asp and Phe point away from and toward the ATP binding pocket, respectively, is an indicator of inactive state of kinase. Noticeably, the flipped conformation of the DFG motif results in the formation of a large adjacent hydrophobic pocket (Hu et al., 2015; Vijayan et al., 2015). A valine in the ATP binding pocket (V561/564/555/550 in FGFR1/2/3/4), which is highly conserved in a variety of kinases and known as a “gatekeeper” residue, is the switch of the large hydrophobic pocket. The gatekeeper mutations give rise to many drug resistances due to hampered drug binding (Azam et al., 2008; Roskoski, 2010; Vijayan et al., 2015).
3.2.2 FGFR Signaling Pathways
The autophosphorylated kinase domain can recruit and phosphorylate multiple downstream effector molecules to initiate several signaling pathways (Figure 3).
FIGURE 3.

The FGF/FGFR signaling pathways. The binding of FGFs with FGFRs initiates a series of conformational changes, which consequently result in phosphorylation of tyrosine residues in the kinase domain. The phosphorylated tyrosine triggers cascaded docking and phosphorylation of downstream molecules including SHP2, GRB2, GAB1 and SOS, forms a multi-complex, and subsequently activates RAS-MAPK-ERK and PI3K-AKT pathways. Activated FGFRs are also involved in JAK-STAT and PLCγ-PKC pathways. The Cbl, SPRY, MKP3 negatively regulate FGF/FGFR signaling by ubiquitination, docking prevention and dephosphorylation, respectively.
The Fibroblast Growth Factor Receptor Substrate 2 (FRS2), a major FGFR substrate, binds to the juxtamembrane region of FGFR via its phospho-tyrosine binding domain (PTB) in a constitutive manner, regardless of the activation and phosphorylation state of the kinase domain. Following the activation of FGFR, multiple tyrosine residues of FRS2 are subject to phosphorylation and serve as docking sites for subsequent molecules.
The RAS-MAPK-ERK signaling pathway is activated by a serial docking of FRS2 with multiple proteins, including SH2-containing protein tyrosine phosphatase (SHP2), growth factor receptor-bound protein 2 (GRB2), Son of Sevenless (SOS) and noted RAS. Both SHP2 and GRB2 contain a Src homology domain (SH2 domain), which can recognize and bind the phosphorylated tyrosine residues of FRS2 and GRB2. Therefore, the GRB2-SOS complex is recruited to FRS2 directly or through the formation of the SHP2-GRB2-SOS complex (Hadari et al., 1998; Ong et al., 2000). The complex, in turn, initiates a phosphorylation cascade in the RAS-MAPK-ERK signaling pathway. Upon activation, ERK1/2 is translocated from cytoplasm into nucleus and regulates the activity of diverse transcription factors to influence cell proliferation, differentiation and signal transduction, which makes it the most persuasive signaling molecules in this pathway for the evaluation of FGFR inhibitors (Guo et al., 2020).
The docking protein GRB2 associated binding protein 1 (GAB1) is recruited to the complex via binding to the SH3 domain of GRB2, which enables tyrosine phosphorylation on itself. Similarly, the phosphorylated tyrosine residues of GRB2 are captured by the phosphoinositide 3 kinase (PI3K) containing a SH2 domain, thus initiating the activation of PI3K-AKT signaling pathway. The downstream effector molecules of AKT vary, including the well-known mTOR, which is closely related to cell metabolism, transcription and so forth (Quan et al., 2020).
Besides FRS2, the phospholipase Cγ (PLCγ) binds to a phosphorylated tyrosine in the C-terminal of phosphorylated kinase domain, and hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into two secondary messengers, inositol triphosphate (IP3) and diacyl glycerol (DAG). The binding between IP3 and its receptor on the endoplasmic reticulum leads to the release of Ca2+ from intracellular stores and thus increases Ca2+ concentration (Mikoshiba, 2007). When coordinated with Ca2+, DAG activates PKC signaling pathway, which causes crosstalk with RAS-MAPK pathway due to the competition between GRB2 and PLCγ to bind with FGFR (Fearon et al., 2014).
In addition, FGFR can activate the signal transducer and activator of transcription (STAT) proteins to partially mediate cell transformation (Hart et al., 2000).
The negative regulation of FGF/FGFR signaling includes dephosphorylation, ubiquitination and obstruction in a serial of docking. In response to FGF stimulation, an ubiquitin ligase called Casitas B-lineage lymphoma (Cbl) is recruited to the FRS2 (-SHP2)-GRB2-SOS complex and induces ubiquitination and subsequent degradation of FGFR and FRS2α (Wong et al., 2002). In addition, the binding of Sprouty to GRB2 can block the interaction between GRB2 and FRS2 or SHP2 so as to exert an inhibitory effect on downstream RAS-MAPK signaling (Hanafusa et al., 2002). The dual-specificity MAPK phosphatases 3 (MKP3) also inhibits RAS-MAPK signaling by dephosphorylating activated MAPK (Farooq et al., 2004).
The activations of these FGFR-dependent or related signaling pathways converge into the regulation of diverse cellular events and physical functions.
3.3 FGF/FGFR Signaling in Cancer
FGFR genetic alterations have been involved in the development and progression of a variety of diseases, particularly cancers (Figure 4). The majority of FGFR aberrations are gene amplifications (66%), followed by gene mutations (26%) and, less frequently, rearrangements (8%), according to a recent sequencing study involving 4,853 patients with various types of cancers (Helsten et al., 2016). FGFR amplification leads to enhanced level of FGF binding. Generally, extracellular mutations increase binding affinity and disturb specificity between FGFs and FGFRs (Ibrahimi et al., 2001; Ibrahimi et al., 2004), or increase receptor dimerization by forming unexpected disulfide bridge (Plotnikov et al., 2000); while kinase domain mutations directly induce a higher level of intracellular phosphorylation. Despite the low incidence, chromosome rearrangements usually cause ligand-independent dimerization. However, most FGFR aberrations are oncogenic drivers, whereas prognostic indicators or “passenger co-aberrations” in different cancers remain ambiguous.
FIGURE 4.

Main abnormalities in FGFRs and their frequency in related cancers.
3.3.1 FGFR1
As the most commonly altered FGFR subtype, FGFR1 aberrations account for 49% of all cases with FGFR aberrations according to the sequencing analysis (Helsten et al., 2016). The most frequent type of FGFR1 aberrations is gene amplification, which is reported in 8.7–20.0% of non-small cell lung carcinoma (NSCLC) cases (Miao et al., 2016; Miao et al., 2020) and involved in several acquired resistances against NSCLC therapeutics (Gammelgaard et al., 2019; Zhang et al., 2019). FGFR1 amplification is common in breast cancer (10%), predominantly in estrogen receptor-positive breast cancer, and harmful to the survival of patients (Gelsi-Boyer et al., 2005; Wang et al., 2017). FGFR1 amplification is also seen in prostate cancer (15%) (Edwards et al., 2003), bladder cancer (9%) (Ross et al., 2014), and other cancers (ovarian cancer, colorectal carcinoma, and squamous non-lung tumors). FGFR amplification (mainly in FGFR1 and 2) causes overexpression of proteins and increases the FGFR-dependency of cancer cells. Therefore, it is regarded as a biomarker for the efficacy of some FGFR inhibitors (Weiss et al., 2010). FGFR1 mutation has been detected in several tpyes of cancers, including midline gliomas (18%), glioblastoma and melanoma, whereas FGFR1 fusion is rare.
3.3.2 FGFR2
Amplification (predominantly observed in triple-negative breast cancer, 4%) and mutation (e.g., K660N) of FGFR2 occur frequently in breast cancer. Besides, FGFR2 also amplifies in gastric cancer (4.5–9%) and is associated with its venous and lymphatic invasion (Jung et al., 2012; Cancer Genome Atlas Research Network, 2014). Apart from breast cancer, 10–12% of endometrial carcinoma and 4–5% of NSCLCs bear FGFR2 mutations (Mohammadi et al., 1998; Dutt et al., 2008; Kandoth et al., 2013). FGFR2 mutants are infrequently reported in urothelial cancers (1.2%). Several FGFR2 fusions have been reported including FGFR2-AFF3, FGFR2-CASP7 and FGFR2-CCDC6 (Turner et al., 2010; Reintjes et al., 2013; Wu et al., 2013). In addition, FGFR2 fusions occur in cholangiocarcinoma, lung squamous cell carcinoma (LSCC), thyroid cancer, prostate cancer, according to a study of FGFR targetable gene fusions (Wu et al., 2013). Notably, a FGFR2-PPHLN1 fusion generated by the chromosomal translocation t (10; 12) (q26; q12) is identified to possess oncogenic and transforming activity in 16% of intrahepatic cholangiocarcinoma (iCCA).
3.3.3 FGFR3
FGFR3 aberrations are predominantly implicated in bladder cancer (Baldia et al., 2016; Helsten et al., 2016). The incidence of FGFR3 mutations in non-muscle-invasive bladder cancer is as high as 75%, as determined by the presence of mutations in the p53 suppressor gene (Zhang et al., 2015), whereas it is relatively low (20%) in muscle-invasive bladder cancer (Couffignal et al., 2015; Solomon et al., 2016; Siracusano et al., 2020). Suppression of FGFR3 activation is sufficient to reduce the survival and proliferation of carcinoma cells harboring FGFR3 mutations (Markham, 2019; Montazeri et al., 2020). Furthermore, FGFR3 mutations are found in uterine cervical cancer (16.3%) (Yoshimoto et al., 2020), including invasive cervical cancer (5%) (Rosty et al., 2005), myeloma (2.16%) (Walker et al., 2015), and spermatocytic seminoma (6.66%) (Goriely et al., 2009). FGFR3 amplification is not frequent in cancers, but is sporadically reported in bladder cancer and adenoid cystic carcinomas (Vékony et al., 2007). Translocations at the t (4; 14), in which FGFR3 is significantly mutated, occurs in multiple myeloma (15%) frequently (Walker et al., 2018). Fusions of BAIAP2L1 or TACC3 to 5’ terminal of FGFR3 can also cause aberrant activation of FGFR3 by inducing oligomerization of fusion proteins even in the absence of FGFs. These fusions are reported in a variety of cancers including bladder cancer, LSCC, NLSCC, glioblastoma and oral cancer.
3.3.4 FGFR4
Amplification or mutation of FGFR4 is rarely perceived as an oncogene except in rhabdomyosarcoma (7.5%). It is confirmed that kinase inhibitor treatment increased cell apoptosis in FGFR4-mutant rhabdomyosarcoma (RMS) cell lines, which is consistent with increased SubG1 fraction and high level of activated caspase-3, suggesting the strong dependency of RMS on FGFR4 (Taylor et al., 2009).
4 Small-Molecule FGFR Inhibitors
To fight against FGFR-driven abnormalities in various cancers, continuous efforts are devoted to various types of therapeutics, including monoclonal antibodies interacting with extracellular domain of FGFR, ligand traps restricting FGF, and small-molecule inhibitors targeting the kinase domain. During the past decade, we have witnessed multiple preclinical and clinical breakthroughs of FGFR inhibitors. To help developing novel therapeutics, we reviewed the current status of discovery of small-molecule FGFR inhibitors as well as other small molecule-based modalities from the standing point of medicinal chemists.
Although the development of tyrosine kinase inhibitors started in the 20th century (Porta et al., 2017), targeting FGFR was validated as a therapeutic strategy for cancer treatment only recently, when FDA approved the use of erdafitinib (JNJ-42756493) in 2019 (Markham, 2019), pemigatinib (INCB054828) in 2020 (Qu et al., 2022) and infigratinib (BGJ-398) in 2021 (Yu et al., 2021) for the treatment of FGFR-altered cancers. In addition, a larger number of inhibitors are in clinical trial or preclinical investigation, such as LY2874455, ARQ-087, AZD4547, FGF401, BLU9931, and H3B6527s (Supplementary Table S1). The following part will elaborate the discovery of small-molecule FGFR inhibitors in structure-based fashion.
4.1 The First-Generation: Non-Selective FGFR Kinase Inhibitors
The FGFR kinase domain share high homology with other receptor tyrosine kinases. The first-generation FGFR inhibitors are non-selective tyrosine kinase inhibitors (TKIs) that compete with ATP for ATP-binding site. As a result, these inhibitors inhibit not only FGFR but also a variety of other tyrosine kinases, such as vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fms-like tyrosine kinase 3 (FLT-3), c-Kit and BCR-ABL (Huang et al., 2020).
Many approved TKIs show mild to strong activity for FGFR, and some of them are being (or have been) assessed in clinical trials for diseases where FGFR alterations are implicated, including nintedanib, dovitinib, ponatinib, lucitanib, derazantinib, anlotinib, and so on. Nintedanib (BIBF1120), first discovered in 2009 by Roth et al. (2009), is an inhibitor targeting VEGFR, FGFR and PDGFR (Capdevila et al., 2014). Nintedanib was approved for the treatment of idiopathic pulmonary fibrosis and interstitial lung diseases (ILD) by FDA in 2014 and 2020, respectively. It is currently under active clinical trials, including the treatment of FGFR3 mutated urothelial carcinoma (Phase 2, NCT02278978), and the treatment of SARS-CoV-2 induced pulmonary fibrosis (Phase 3, NCT04541680). This inhibitor resulted from the optimization of a hit compound 1 bearing a 5-substituted indolinone core that was initially identified as a VEGFR-2 inhibitor (Figure 5). The computational modeling of hit compound 1 with VEGFR-2 suggested that the carbonyl oxygen of the amide group can form a hydrogen bond with Lys868. The hydrophobic region flanked by Val916 indicated that replacing the amide moiety with a more lipophilic substituent (e.g., methoxycarbonyl) could improve potency and maintain selectivity. Meanwhile, the basic side chain pointing toward the solvent was further modified with additional polar fragments, resulting in two compounds BIBF1000 and BIBF1120. The latter compound exhibited a favorable IC50 values for VEGFR, FGFR, and PDGFR within nanomolar range and showed selectivity over other homologous kinases. The indolinone scaffold formed two hydrogen bonds with Cys919 and Glu917 in the hinge region. The methyl piperazinyl group directed into the solvent region, and its 4-nitrogen atom formed a bidentate ionic interaction with the carboxylate oxygens of Glu850 based on a published X-ray crystal structure in complex with VEGFR-2 (PDB: 3C7Q) (Hilberg et al., 2008).
FIGURE 5.

Discovery of BIBF1120. H-bonds are outlined as blue hashed lines. The methyl-piperazinyl moiety is involved in the ionic interaction with the side chains of Glu850 (Black). Hydrophobic interaction is outlined by hashed brown rectangle. Solvent-exposed region is highlighted using green rectangle. Hinge region is indicated by pink arc.
Dovitinib (TKI258) inhibits VEGFR-2, FGFR-1, and PDGFR with IC50 values below 0.1 μM, and several clinical trials for advanced solid tumors have been conducted (Angevin et al., 2013; Taeger et al., 2011). Dovitinib contains a benzimidazole core, which makes critical contacts with the hinge region and also binds to FGFR1 and FGFR4 in a DFG-in mode as usually observed for this type of inhibitors (Bunney et al., 2015; Lesca et al., 2014). Ponatinib, which targets PDGFR, VEGFR and FGFR, was initially approved for the treatment of refractory chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph + ALL) in 2012 (Pao et al., 2004; Cortes et al., 2018), then it entered two clinical trials in 2014 for the treatment of malignant neoplasm with FGFR fusions or activating mutations (NCT02265341, NCT02272998). Structural study revealed that ponatinib bound to either FGFR1 or FGFR4 in a unique DFG-out mode, which is distinct from most of reported FGFR inhibitors (Lesca et al., 2014). Liu et al. (2017) conducted extensive SAR study of ponatinib and obtained optimized analogs with improved activity and selectivity. Anlotinib, a quinoline-based inhibitor of VEGFR, FGFR, PDGFR and c-kit (Shen et al., 2018), is being investigated for treatment of advanced solid tumors with FGFR alterations (NCT03929965). Lucitanib (E3810) is also a TKI that targets VEGFR1/2/3, FGFR1/2 and PDGFR (Babina et al., 2017), and a phase 2 trial (NCT02053636) for testing Lucitanib in patients with FGFR1-amplified or non-amplified ER + metastatic breast cancer was completed. Derazantinib (ARQ087) inhibits multiple kinases including RET, DDR2, PDGFR, VEGFR, KIT and FGFR, and its phase 1/2 study in FGFR-altered patients was recently completed as well. Representative kinase small-molecule inhibitors in this category are shown in Supplementary Figure S1 .
Although moderate suppression on tumors harboring FGFR aberrations was observed, these non-selective inhibitors still brought some issues of therapeutic regimen. The human kinome comprises ∼535 protein kinases (Zhong et al., 2021). A wide range of off-target effects attributed to their poor selectivity leads to blockage of multiple signaling pathways and causes a multiplicity of related side effects such as diarrhea, vomiting and nausea (Konecny et al., 2015). Albeit these factors have restricted the broad application of multi-target TKIs, they are widely recognized as a decent treatment for tumors in absence or unawareness of the “oncogenic driver,” and have provided the impetus to the development of on target FGFR inhibitors.
4.2 The Second-Generation: Selective FGFR Kinase Inhibitors
Thanks to the rapidly evolving high throughput screening methods and structure-based strategies, a number of second-generation FGFR inhibitors have been discovered with higher potency, selectivity, safety as well as novel modality. Three inhibitors in this category, namely erdafitinib, pemigatinib, and infigratinib, have been approved by FDA, and a lot more compounds are being evaluated in preclinical and clinical investigations. These second-generation inhibitors were tentatively divided into several subclasses on the basis of different modes of action. Chemical structures of these reported selective FGFR small molecule inhibitors are shown in Supplementary Figure S2 .
4.2.1 Non-Covalent Pan-FGFR Inhibitors
The three approved FGFR inhibitors and quite a few candidates are all non-covalent inhibitors with pan-FGFR inhibitory activity, although some of them showed reduced, yet still considerable, potency for FGFR4 because of its relatively notable difference from FGFR1-3.
Erdafitinib (JNJ-42756493) is the first approved FGFR inhibitor for treatment of adult patients with locally advanced or metastatic urothelial carcinoma. It is an orally active and selective pan-FGFR inhibitor (Perera et al., 2017) that inhibits the kinase activity of FGFR1-4 with similar potency (Markham, 2019). Erdafitinib features quinoxaline element, which was first identified through virtual screening based on the crystal structure with FGFR1. The compound 2 was next generated through fragment growing approach. Removal of the methylene group in compound 2 produced compound 3, which has shown much improvement in activity due to better shape complementarity with the hydrophobic pocket. An additional substitution on the secondary nitrogen occupied the ribose-binding region, leading to the discovery of erdafitinib with increased affinity, better physicochemical and pharmacokinetic properties (Figure 6 ) (Murray et al., 2019).
FIGURE 6.

Discovery of erdafitinib.
In order to replace the pyrido [2,3-d]pyrimidin-7-one core, which was a common feature in a well-established class of protein kinase inhibitors (e.g., PD166285), Guagnano et al. (2011) developed a pseudo six-membered ring structure stabilized by an intramolecular hydrogen bond (e.g., prototype compound) (Figure 7) (Furet et al., 2008). Using the same strategy, infigratinib was eventually discovered through a lead optimization of a known FGFR inhibitor PD173074. Structural studies confirmed that hydrogen bonds with hinge region were retained and both chlorines and methoxy groups would form favorable hydrophobic contacts with the deep pocket inside ATP binding site. It was also observed that the phenyl ring at the entrance of the pocket was hydrophobically sandwiched between Leu478 and Gly561. These hydrophobic effects contributed to the selectivity of infigratinib for FGFR, especially FGFR1-3, over other tyrosine kinases. Infigratinib was approved by FDA for the treatment of cholangiocarcinoma patients with FGFR2 fusion in 2021 (Botrus et al., 2021; Javle et al., 2021).
FIGURE 7.

Discovery of infigratinib (BGJ-398) and summary of interactions of infigratinib and FGFR3 ATP binding site. H-bonds with hinge region are indicated by blue hashed lines. Hydrophobic interactions are outlined by hashed brown rectangle (PDB: 3TT0).
Pemigatinib is another FGFR inhibitor featuring a tricyclic urea scaffold for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement. Like infigratinib, pemigatinib contains a 3,5-dimethoxyphenyl for the high affinity and selectivity by filling the hydrophobic pocket, and two fluorine atoms, which led to further improvements in potency (Figure 8) (Wu et al., 2021). Many rounds of optimization also demonstrated that the ethyl group on the N-1 position of the cyclic urea and the pyrrole ring were of great importance to the potency and PK profile.
FIGURE 8.

Discovery of pemigatinib and summary of interactions of pemigatinib and FGFR1 ATP binding site. H-bonds are indicated by blue hashed lines. Hydrophobic interactions are outlined by hashed brown arc. Solvent-exposed region is highlighted using green arc.
Starting from erdafitinib and its quinazolinone analogue, a series of pyrido [1,2-a] pyrimidinone derivatives were designed as novel selective FGFR inhibitors through scaffold hopping (Figure 9A) (Ran et al., 2021). Molecular docking suggested an overall similar binding mode with erdafitinib, while the rotatable pyrazole ring could lead to increased potency. The rotation also disrupted the planarity, which might enhance the aqueous solubility owing to reduced crystal-stacking.
FIGURE 9.

(A) Discovery of pyrido [1,2-a] pyrimidinone derivatives as non-covalent pan-FGFR inhibitor. (B) Discovery of 1H-Pyrazolo [3,4-b]pyridine derivatives from AZD4547 and summary of its interactions with FGFR1 ATP binding site. Hydrophobic interactions are outlined by hashed brown arc (PDB: 4V05). (C) Summary of CH5183284 and designed fragment.
AZD4547 is another pan-FGFR inhibitor bearing the pyrazole scaffold and just completed phase 2 clinical trial (NCT04439240) in multiple cancers with FGFR alterations. Based on the structure of AZD4547, Zhao et al. developed 1H-Pyrazolo [3,4-b]pyridine derivatives using scaffold hopping strategy, and incorporated two chlorines at the dimethoxyphenyl ring (Figure 9B) (Zhao et al., 2016). Notably, AZD4547 maintains efficacy to FGFR1 harboring gatekeeper mutation V561M as the flexible linker between dimethoxyphenyl and pyrazole allows conformational adaption within the hydrophobic region (Sohl et al., 2015). However, it remains unknown whether 1H-Pyrazolo [3,4-b]pyridine derivative (a) has compromised its activity for mutated FGFRs.
CH5183284 (Debio1374) is a potent pan-inhibitor of FGFR1-3 with IC50 values of 9.3, 7.6 and 22 nM, respectively. It was discovered through a conventional high throughput screening and its interactions with the hinge region, hydrogen bonding pattern and an additional π-π interaction were also identified (Ebiike et al., 2016). CH5183284 is under clinical investigation for the treatment of cancer patients with FGFR genetic alterations (Nakanishi et al., 2014). Turner et al. (2017) described the application of SPROUT, a de novo computation program, to develop the active indazole-based pharmacophore for the inhibition of FGFR kinases ( Figure 9C ). Beginning with the co-crystal structure of CH5183284-FGFR1, they modified the indole moiety and obtained a small library of 23 indazole derivatives. Subsequent biological evaluation indicated that these indazole-containing fragments inhibited FGFR1-3 with IC50 values of 0.8–90 µM, suggesting that structure-based drug discovery (SBDD) is becoming an essential tool for identifying potent and selective FGFR inhibitors.
There are numerous other non-covalent pan-FGFR inhibitors in clinical trial or development. For example, ASP5878 inhibited cell proliferation of urothelial cancer cell lines harboring FGFR3 point mutation or fusion and has completed phase 1 clinical trial in 2017 (NCT02038673) (Kikuchi et al., 2017). LY2874455 is a phase 1 orally available inhibitor with IC50 values of 2.8, 2.6, 6.4, and 6.0 nM against FGFR1-4, respectively (Michael et al., 2017). Rogaratinib (BAY 1163877) is another potent and selective FGFR1-4 inhibitor (Collin et al., 2018). Rogaratinib alone or in combination with other agents have been in a few clinical trials. 3D185, a highly selective FGFR1-3 inhibitor, was approved for investigational new drug by the NMPA in 2018 and followed by a phase 1 study in patients with the advanced solid tumors (NCT04221204). ICP-192 is a pan-inhibitor against FGFR1-4 and entered the phase 1/2 clinical trial for the treatment of advanced solid tumors, urothelial carcinoma, and cholangiocarcinoma (NCT04565275). E7090 has favorable pharmacokinetic profiles and sub-nanomolar inhibitory activity against FGFR1-3 with IC50 values of 0.71, 0.50, and 1.2 nM, respectively (Watanabe Miyano et al., 2016). The phase 2 study of E7090 in participants with unresectable advanced or metastatic cholangiocarcinoma with FGFR2 fusion is recruiting (NCT04238715).
4.2.2 Covalent Pan-FGFR Inhibitors
Covalent inhibition is a re-emerging strategy especially in the development of kinase inhibitors, which can make a big difference in binding affinity and selectivity. A covalent inhibitor typically consists of a drug-like scaffold offering noncovalent interactions and an appropriate electrophilic warhead to react with nucleophilic residues of target proteins. Cysteine represents the most targeted amino acid in kinases, due to its non-catalytic roles, low abundance, high reactivity and chemical plasticity of the anionic thiolate (Abdeldayem et al., 2020; Galbiati et al., 2020). For FGFRs, the conserved cysteine in the P-loop (C488 in FGFR1, C491 in FGFR2, C482 in FGFR3 and C477 in FGFR4) and the unique C552 in FGFR4 in the hinge region are sites for covalent attachment (Dai et al., 2019). This section focuses on recent publications with regard to the discovery of covalent pan-FGFR covalent inhibitors.
Zhou et al. (2010) discovered FIIN-1 as the first irreversible inhibitor of FGFR1−4 in 2010. The acrylamide of FIIN-1 formed covalent bond with a conserved cysteine (Cys488 of FGFR1) located at the rim of the P-loop. Replacing the acrylamide with a propylamide led to the failure of covalent bond formation. In addition, its selectivity over some other kinases (e.g., c-Src, TNK1, and YES) bearing the P-loop cysteine at the same position as FGFRs was also confirmed. Tan et al. (2014) developed FIIN-2 and FIIN-3 as irreversible inhibitors with potent in vitro inhibitory activity against FGFR1 and FGFR2 gatekeeper mutants, which conferred resistance to first-generation FGFR inhibitors. The acrylamide moiety in both molecules was installed on the 4-poistion of their phenyl rings in contrast to 3-acrylamide as found in FIIN-1, which still maintained the bond formation with P-loop cysteine while changed the selectivity over other kinases (PDB 4QQC, 4R5S, 4R6V).
Brameld et al. (2017) developed another irreversible inhibitor, PRN-1371, which shared a common core with FIIN-1. PRN-1371 was proven to be highly selective for FGFR1-4 over other kinases, including KDR, FLT-4, etc., and showed high FGFR1 occupancy and ideal PK profile. Ding et al. focused on the modification of the acrylamide-containing side chain of FIIN-1 and obtained the promising lead compound showing inhibitory effect in FGFR-amplified cancer cell lines ( Figure 10A) (Li et al., 2017). The crystal structure of FGFR1 C488A in complex with lead compound revealed that the acrylamide side chain was located in the solvent accessible space and easily performed nucleophilic attack by the target cysteine.
FIGURE 10.

(A) Structure optimization of FIIN-1. H-bonds are indicated by blue hashed lines. Hydrophobic interactions are outlined by hashed brown arc (PDB: 5B7V). The covalent bond-forming Michael acceptor carbon of the acrylamide is indicated by red arrow. (B) Discovery of irreversible inhibitors bearing novel pyrrolopyrimidine scaffold. (C) Discovery of 2H-pyrazolo [3,4-d]pyrimidine derivatives. Hydrophobic interaction region is outlined by hashed brown arc. Covalent binding region is highlighted by dashed red arc. (D) Covalent FGFR inhibitors bearing pyrazolo [3,4-d]pyridazinone.
Goyal et al. (2019) demonstrated that Futibatinib (TAS-120), a highly selective and irreversible pan-FGFR inhibitor, exhibited in vitro potency against wild-type FGFR1-4, as well as some FGFR2 kinase domain mutants. Hiroshi Sootome et al. (2020) reported the preclinical profile of futibatinib and suggested that it is an orally available, potent pan-FGFR inhibitor. Futibatinib is the most advanced candidate in the category of covalent pan-FGFR inhibitors because it is in phase 3 clinical trial to evaluate the treatment of metastatic and recurrent unresectable cholangiocarcinoma harboring FGFR2 gene rearrangements.
Wang et al. (2018) designed and discovered a set of irreversible inhibitors bearing novel pyrrolopyrimidine scaffold. By analyzing the ATP binding pocket, they introduced a substituted phenyl moiety at the C-3 position of pyrrolopyrimidine to interact with hydrophobic region I ( Figure 10B ). Then the electrophilic warhead attached to N-1 via a pyrrolidine linker led to the identification of lead compound that showed excellent potencies against FGFR1−4 and acceptable selectivity over VEGFR2.
Wei et al. (2021) recently reported a class of 2H-pyrazolo [3,4-d]pyrimidine derivatives as a potent irreversible pan-FGFR inhibitor (Figure 10C). The lead compound was derived from BTK inhibitor ibrutinib and also shares a similar core with the above-mentioned lead compound. Interestingly, an electron-withdrawing terminal-CF3 substituted acrylamide group provided the most potent inhibition against FGFRs.
There are several other covalent pan-FGFR inhibitors in development. For example, Yamani et al. (2021) discovered a pyrazole-benzimidazole CPL304110 as a pan-FGFR inhibitor for the treatment of bladder, gastric and squamous cell lung cancer, which also showed favorable pharmacokinetic properties after oral administration. Dai et al. (2020) reported that DW14383 simultaneously inhibited tumor proliferation and angiogenesis via inhibition of FGFR1–4 with similar potency. In addition, they claimed that its pan-tumor spectrum potential might overcome compensatory activation among FGFR1–4. Wang et al. (2019) developed a series of compounds featuring pyrazolo [3,4-d] pyridazinone as covalent FGFR inhibitors. Their structural optimization resulted in more analogues that could remarkably inhibit proliferation of various FGFR-dysregulated cancer cells and display potent antitumor efficacy in xenograft model as well (Figure 10D) (Xie et al., 2020).
4.2.3 FGFR4-Specific Covalent Inhibitors
The kinase domains of FGFR1–3 share high structural similarity, whereas FGFR4 is relatively distinct from FGFR1-3 (Babina et al., 2017), which is consistent with the fact that many foregoing pan-FGFR inhibitors show strong inhibition of FGFR1-3 but reduced potency for FGFR4. Detailed comparison of the active sites of FGFR1-3 and FGFR4 revealed a key difference in the hinge region: Tyr563 in FGFR1-3 versus the Cys552 in FGFR4 ( Figure 11 ) (Tucker et al., 2014). This unique Cys552 provides great opportunity for the design of highly selective, covalent inhibitors of FGFR4. Although there is no approved FGFR4-specific drug, the past years have witnessed growing numbers of promising compounds as discussed below.
FIGURE 11.

Critical Cys552 in FGFR4 and replacement by tyrosine in FGFR1-3 (PDB: 4QRC, the crystal structure of FGFR4 in complex with Ponatinib).
Hagel et al. (2015) discovered BLU9931 as the first selective FGFR4 inhibitor for the treatment of hepatocellular carcinomas (HCC) with aberrant FGFR4 signaling. The dimethoxyphenyl group of BLU9931 occupied the hydrophobic pocket located near the gatekeeper valine of all FGFRs. In addition, an acrylamide at the ortho-position of the aniline can form the covalent bond with Cys552 in the hinge region of FGFR4. To achieve a better selectivity, the rotation of the phenyl ring was also taken into consideration because it could cause steric clash with the corresponding tyrosine in FGFR1-3 hinge region. Moreover, the addition of 3-methyl group on the aniline ring rendered BLU9931 with high selectivity for FGFR4 over FGFR1-3 (Figure 12A). Unfortunately, BLU9931 failed to enter clinical stage, presumably due to its rapid metabolism in liver microsomes. BLU554 (fisogatinib), an orally available analog of BLU9931, is now in phase 1 clinical trial to treat patients with HCC (NCT02508467) and in phase 1b/2 clinical trial in combination with CS1001.
FIGURE 12.

(A) Structures of FGFR4 covalent inhibitors BLU9931 and its analogs. (B) Discovery of a covalent inhibitor of FGFR4 from BGJ-398 and summary of its interactions with FGFR 4 kinase domain. H-bonds are outlined by hashed blue line. Covalent binding contact is highlighted using red (PDB: 6JPE). (C) Discovery of dipyridylamine. The novel warhead is highlighted by red color and H-bonds are outlined by hashed blue line.
H3B-6527 (Joshi et al., 2017) is another highly selective covalent FGFR4 inhibitor and is currently undergoing phase 1 study for the treatment of advanced HCC, liver neoplasms, hepatic carcinoma and so on. Co-crystal structure of FGFR4 in complex with H3B-6527 illustrated the covalent bond formation between Cys552 and the acrylamide at the ortho-position of the N-aryl substituent.
Fairhurst et al. (2020) reported the discovery of FGF401 (roblitinib) as a potent, selective FGFR4 inhibitor. Through high throughput screening, 2-formylquinoline amide (2-FQA) derivatives were identified as the starting hits. After optimization of the 2-FQA and the substituent groups on the hinge-binding pyridyl ring, roblitinib was eventually obtained with outstanding selectivity for FGFR4. Being assessed in phase 2 clinical trial, roblitinib is the most advanced covalent FGFR4-specific inhibitor. This inhibitor features a covalent yet rapidly reversible mode of action that may reduce off-target related toxicity. Therefore, roblitinib is regarded as a promising next-generation drug to offer a new approach to target FGFR covalently.
Starting from BGJ-398 (infigratinib), Miranda et al. developed a novel covalent inhibitor of FGFR4 for the treatment of HCC (Rezende Miranda et al., 2020). According to the crystal structure of FGFR1-BGJ-398, the urea group and aminopyridine group should exhibit similar geometrical and electronic properties ( Figure 12B). An acrylamide group was also attached to the ortho position of a 2-methylaniline ring for targeting Cys552 of FGFR4. Interestingly, crystallographic study revealed that the introduction of the methyl group into aniline phenyl rings facilitates the covalent reaction from a conformational perspective (PDB: 6JPE). As expected, its exceptional selectivity among the FGFR family is due to the fact that the acrylamide group formed steric clash with the bulky tyrosine residue present in FGFR1-3 hinge region.
Fairhurst et al. (2017) reported the discovery of dipyridylamine through high throughput screening (HTS). Dipyridylamine contains a novel warhead 3-nitro-6-chloropyridyl, which was designed to achieve covalent binding with Cys552 of FGFR4 (Figure 12C ). The 6-chloro substituent in this warhead is positioned for attack by the Cys552 thiolmethyl group through a nucleophilic aromatic substitution (SNAr). Dipyridylamine demonstrated high potency against FGFR4 with IC50 value of 53 nM while sparing the FGFR1-3 with IC50 values higher than 10 μM. Besides, each nitrogen atom in pyridyl ring formed a hydrogen bond with hinge residue Ala553 (Lu et al., 2019). Dipyridylamine features a relatively low molecular weight and novel mechanism of covalent binding that may serve as a promising lead compound for future discovery of FGFR4-specific covalent inhibitors.
Several candidates have entered clinical stages without full disclosure of chemical structures. For example, INCB-62079 entered phase 1 trial in 2017 but was terminated for business strategic consideration. ICP-105, a selective FGFR4 inhibitor, is now in phase 1 clinical trial for the treatment of solid tumor (NCT03642834). Other clinical trials involving inhibitors including ZSP-1241 and ABSK-011 are actively recruiting patients.
4.2.4 Selective FGFR2 Inhibitors
Unlike FGFR4, few FGFR1-3 subtype-specific kinase inhibitors have been reported to date, mainly aiming at FGFR2. Casaletto et al. (2021) recently reported RLY-4008 as a highly selective inhibitor of FGFR2 WT/mutant, which exhibited >200-fold higher potency than FGFR1. Although the structure has not been disclosed, RLY-4008 showed no difference in binding modes with FGFR1 or FGFR2. Instead, a flexible loop in FGFR1/2 validated from MD simulation might be the cause of the selectivity. It is encouraging that a recent de novo design by Turner et al. (2021) has provided a paradigm for perhaps the next-generation member of FGFR-specific inhibitors. They started from a fragment with moderate potency and carried out iterative rounds of de novo design as well as a classical SAR study to generate compound 31. Interestingly, compound 31 specifically inhibited FGFR2 with an IC50 of 29 nM, whereas 389 nM for FGFR1 and 758 nM for FGFR3, suggesting at least 10-fold selectivity for FGFR2 over FGFR1 (Figure 13).
FIGURE 13.

Structure-guided approach for the development of selective FGFR2 inhibitors. H-bonds are outlined as blue hashed lines. Hydrophobic region is indicated by brown arc.
4.3 Miscellaneous Types
4.3.1 Extracellular Allosteric Inhibitors
An extracellular allosteric inhibitor of FGFR can bind to its extracellular domain and modulate the receptor conformation, thus blocking the signaling pathway. The extracellular domains of FGFR1-4 exhibit discernible structural differences compared to their kinase domains, therefore it is possible to achieve selective inhibition within FGFR members. Alofanib (RPT835) belongs to this type of inhibitors and has entered a phase 1 clinical trial in Russia. By specifically binding to IgIII of FGFR2, alofanib is able to inhibit the FGF2-induced phosphorylation of FRS2α with nanomolar activity in cancer cells expressing different FGFR2 isoforms (Tyulyandina et al., 2017). In addition, Tsimafeyeu et al. (2016) performed in vivo experiments to demonstrate that alofanib could ablate FGF-induced angiogenesis. Another example is SSR128129E, an orally-active, allosteric inhibitor of FGFR1 (Herbert et al., 2013). It interacts with extracellular part of FGFR without interfering with FGF binding or receptor dimerization. Critical conformational changes were observed in IgIII subdomain when treated with SSR128129E, resulting in defective internalization of FGFRs.
4.3.2 Natural Products
Efforts have been made to identify natural products that act on FGFRs. These include a few phenolic compounds (resveratrol, caffeic acid phenethyl ester, kaempferol, etc.), stilbene glycosides, and sesterterpenes compounds (leucosesterterpenone, leucosterlactone, etc.), and a detailed review has been completed by Yin et al. (2019). Recently, Pagano et al. (2021) reported rosmarinic acid (RA), a natural phenolic compound, could induce FGF2/FGFR complex dissociation as verified by experimental mechanistic study. In addition, gambogenic acid and ferulic acid, originated from natural plants, exhibited inhibitory effect of FGFR autophosphorylation. Taken together, natural products stand for a prime source of FGFR inhibitors, while more studies are needed to improve their activities and elucidate the molecular mechanisms. Chemical structures of these represent natural products are shown in Supplementary Figure S3 .
5 Other Small Molecule-Based Therapeutic Modalities
5.1 Combination Therapy
Due to the extensive crosstalk between FGF/FGFR and other signaling pathways, the inhibition of FGF/FGFR signaling can be rescued by activation or upregulation of multiple signaling pathways. The most involved proteins are among the receptor tyrosine kinase family, such as c-Met, EGFR, ErbB2/3 or even among four members of FGFR. The compensatory activation of alternative receptors or/and signaling pathways occurs frequently while a receptor function is suppressed specifically, and consequently the resistance to FGFR inhibitor is developed. Therefore, a combination of FGFR inhibitor with other types of drugs is a promising avenue to improve clinical efficacy of available therapeutics and overcome drug resistance.
Fischer et al. (2008) reported that cotreatment with FGFR inhibitors (SU5404/PD166866) and EGFR-targeting drugs (erlotinib/lapatinib) improved in vivo antiproliferative effects, indicating its potential as combination therapy in NSCLC. In addition to EGFR, other RTKs were also involved in the combination therapy with FGFR. The combined treatment of RET inhibitor (ST1571) and FGFR inhibitor (PD173074) significantly suppressed tumor growth of medullary thyroid cancer, which is intractable by surgery and has no widely accepted treatment (Ezzat et al., 2005). The addition of VEGFR1 inhibitors solved the limited practical effects of FGFR inhibitors in FGFR1-amplified breast cancers through blocking the contribution of FGFR1 to VEGF secretion (Golfmann et al., 2018).
Besides RTKs, many kinases are attractive targets in the combination therapy with FGFR inhibitors. The phosphoinositide 3-kinase (PI3K) inhibitor is used to achieve superior antitumor effect in FGFR2 mutant endometrial cancer cell lines (Packer et al., 2017). PI3K also mediates resistance to FGFR inhibitors in urothelial cell carcinomas harboring alterations of FGFR3 gene, which both highlight the prospect of combination of their inhibitors (Wang et al., 2017). FGFR was also identified as a promoter to induce resistance to CDK4/6 inhibitors, which was diminished by complementary inhibition of FGFR in ER+/FGFR1-amplified breast cancers (Formisano et al., 2019). Similar antitumor effects were observed in synergism of mTOR and FGFR inhibitors, which resulted in significantly arrested cell cycle in G1 phase in AN3CA-derived endometrial tumor models (Gozgit et al., 2013). Krook et al. (2020) further implemented a combination therapy using an mTOR inhibitor (INK258) and demonstrated that this strategy may overcome the resistance to FGFR inhibitor like infigratinib. Moreover, through a kinome-wide CRISPR-based screening, Yang et al. identified PLK1 and FGFR as promising synthetic lethal targets for treating FGFR1-amplified lung cancer (Yang et al., 2021).
Overexpression of FGFs may also cause hyperactivated FGF/FGFR signaling, which is present in some tumors and can be co-targeted accordingly. The frequent presence of both BRAF mutations and FGF2 overexpression in melanomas, which lack a recognized systematic therapy so far, leads to the combination of FGFR inhibitor PD166866 and BRAF V600E inhibitor, consequently increased cell apoptosis and restricted tumor growth (Metzner et al., 2011). Wang et al. (2019) further revealed that upregulated secretion of FGF1 gave rise to resistance to the combined therapy of RAF inhibitor vemurafenib and MEK inhibitor cobimetinib in BRAF V600E-driven tumors, which was abrogated by addition of FGFR inhibitors to achieve a triple BRAF/MEK/FGFR inhibition.
Tremendous potential also lies in cooperation of FGFR inhibitors with immune checkpoint inhibitors (Qin et al., 2020). For instance, Palakurthi et al. demonstrated the combination of erdafitinib and PD-1 blockade RMP1-14 could achieve remarkable tumor regression and significantly improve survival in mice with a FGFR2-driven lung tumor harboring dual mutations on FGFR2 and P53 genes (Palakurthi et al., 2019).
Despite a number of successful attempts of combination therapy, the drug-drug interactions may cause unpredictable toxicity and should be assessed with meticulousness. For example, the combination of infigratinib with imatinib encountered higher toxicity and frequent adverse effects, including CPK elevation, lipase elevation, hyperphosphatemia, anemia, and peripheral edema (NCT02257541).
5.2 Dual/Multi-Target Inhibitors
Dual/multi-target inhibitors have several potential advantages over combination therapy, such as more predictable pharmacokinetics, better patient compliance, reduced administration dosage and toxicities (Duan et al., 2021). Previous advances in the field of TKIs validated a diversity of promising and well-tolerated targets, including EGFR, ALK, ROS1, HER2, NTRK, VEGFR, RET, MET, MEK, FGFR, PDGFR, PI3K and KIT, which have inspired the discovery and rational design of dual/multi-target inhibitors.
From the perspective of medicinal chemistry, most of the present dual/multi-target inhibitors can be assigned to the first- or second-generation FGFR TKIs. In some cases, their low selectivity over aforementioned targets has in turn created a synergistic inhibitory effect in diseases involving abnormal FGFR and the other target(s). MPT0L145, an alleged dual-target inhibitor of PIK3C3 and FGFR, not only increased autophagosome formation due to FGFR inhibition but also interfered with autophagic flux via PIK3C3 inhibition, It synergistically sensitized anticancer effects of targeted- or chemo-therapy in different cancer cell lines (Chen et al., 2018). Besides, FGFR/EGFR and FGFR/VEGFR dual inhibition strategies are also frequently reported. FGFR/EGFR dual inhibitors can be exemplified by FIIN3, while FGFR/VEGFR dual inhibitors include PD173074, AZD2171 (cediranib), BMS-540215 (brivanib), ODM-203, and so on (Chen et al., 2019).
Recently, several publications have paved the way for the rational design of dual/multi-target FGFR inhibitors. Chen et al. applied SVM machine learning algorithm to establish QSAR models for FGFR4 and EGFR, which led to the identification of Cpd 34 as a potent inhibitor of FGFR and EGFR with similar IC50 values but distinct binding modes (Figure 14) (Chen et al., 2020). Xie et al. developed a series of 4,6-pyrimidinediamine derivatives through incorporation of key scaffolds from FGFR inhibitors (FIIN3 and infigratinib) and EGFR inhibitors (Figure 14, Cpd 32) (Sacks et al., 2018). The most promising compound, BZF2 ( Figure 14, Cpd 33), potently inhibited cell proliferation and cell migration, and induced apoptosis in NSCLC cell lines with FGF2-FGFR1 autocrine loop. Moreover, it exhibited outstanding in vivo anti-tumor activity. Apart from FGFR/EGFR dual inhibitors, the FGFR/HDAC dual inhibitors were also reported by Liu et al. (2018). The 1-H-indazol-3-amine-derived FGFR/HDAC dual inhibitor ( Figure 14, Cpd 35) exhibited HDAC6 and FGFR1 dual inhibitions with IC50 values of 34 and 9 nM, respectively.
FIGURE 14.

Discovery of representative dual/multi-target inhibitors. H-bonds are outlined as blue hashed lines. Hydrophobic region is indicated by brown arc.
5.3 FGFR Degraders
Proteolysis targeting chimera (PROTAC) was first reported by the Crews and Deshaies laboratories in 2001 (Hu et al., 2020; Pettersson et al., 2019). Rather than acting as conventional inhibitors, PROTACs induce selective intracellular proteolysis of target proteins. This novel strategy is likely to circumvent the common disadvantages of traditional occupancy-driven inhibitors such as the toxicity due to off-target and drug resistance caused by compensatory feedback activation of alternative kinases (Paiva et al., 2019). Several kinase targets employing the PROTAC strategy have been explored, including EGFR, HER2, c-Met, ALK, Akt, CK2, ERK1/2, FLT3, PI3K, BTK, RIPK2, and BCR-ABL, most of which are cytosol- or nuclei-located proteins. As for membrane-associated tyrosine kinase receptors like EGFR, Burslem et al. (2018) conjugated a kinase inhibitor Lapatinib to a VHL ligand for degradation of EGFR, HER2, and c-Met. Interestingly, the PROTAC mediated the internalization of EGFR and sorted to lysosomal degradation, although the RTKs usually prefer to be internalized into a recycling endosome (Zou et al., 2019). Du et al. (2021) recently reported a bivalent degrader DGY-09-192, which coupled pan-FGFR inhibitor BGJ-398 to a CRL2VHL E3 ligase (Figure 15). Surprisingly, DGY-09-192 preferentially induced FGFR1 and FGFR2 degradation while largely sparing FGFR3 and FGFR4. Despite multiple concerns regarding cellular permeability, the feasibility of scalable synthesis, and so on (Goracci et al., 2020; Liu et al., 2020), these pioneering studies have demonstrated that the PROTAC approach has a great potential to expand the arsenal against a variety of FGFRs-altered cancers.
FIGURE 15.
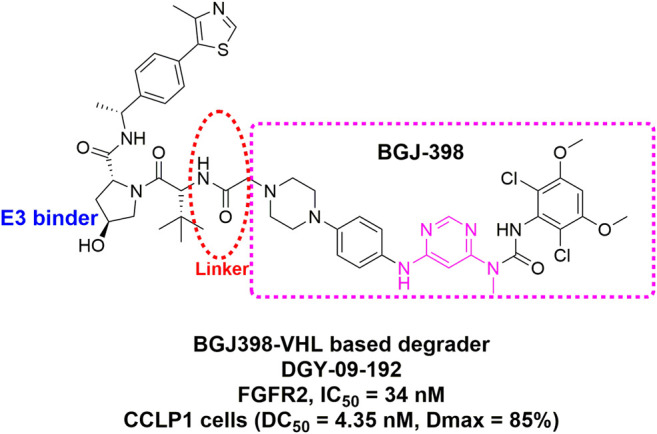
Chemical structure of potent FGFR 1/2 degrader DGY-09–192.
6 Concluding Remarks
As a type of membrane receptor, FGFR plays a critical role in cell signal transduction and mediates diverse cellular events and processes through a ligand-dependent characteristic. Genetic amplification, mutation, and/or fusion of FGFR occurring frequently in various kinds of cancers, can over-activate downstream signaling pathways and cause excessive common oncogenic inducements, such as cell proliferation, inadequate cell apoptosis and cell transformation.
Due to the increasing incidence of abnormal FGF/FGFR signaling axis in various malignancy, such as breast cancers, lung cancers and bladder cancers, FGFRs have been recognized as attractive therapeutic targets. A great number of FGFR inhibitors have been developed in the past decades. Erdafitinib, Pemigatinib and Infigratinib were approved by FDA in 2019, 2020 and 2021, respectively, to treat limited subsets of bladder cancer and cholangiocarcinoma patients with corresponding FGFR alterations, while dozens of other inhibitors are racing in preclinical and clinical development.
TKIs make up a major portion of FGFR-targeting small molecules. The first-generation FGFR inhibitors are general TKIs with a wide spectrum of inhibitory effects for multiple kinases. In contrast, second-generation FGFR inhibitors have improved selectivity, potency and lowered drug resistance, as a result of delicate structure-based design focusing on either optimizing non-covalent interactions with ATP-binding site or the use of covalent warheads to modify critical Cys residues.
The FGFR2 selective inhibitors generated by de novo design hold great promise for targeting specific members of FGFR1-3. These inhibitors may possess higher safety, as pan-FGFR inhibitors often display “FGFR1-specific” toxicity that leads to adverse side effects which are presumably originated from abnormal signaling of FGF23 (Chae et al., 2017).
Macrocyclization may serve as another potential strategy for novel FGFR inhibitors. Generally, macrocyclic molecules offer superior binding affinity with targets bearing large and featureless pockets. As seen in other kinase inhibitors, macrocyclic inhibitors also exhibit improved cell permeability, plasma stability and oral absorption when compared to traditional small molecules (Engelhardt et al., 2019; Begnini et al., 2021).
Although there is no allosteric TKI reported for FGFR, this strategy was applied to overcome the drug-resistant EGFR T790M mutant. Jia et al. (2016) described the rational discovery of EAI045, a fourth-generation EGFR inhibitor that targeted an allosteric pocket of the EGFR mutant but not the wild-type kinase. Given the structural similarity between EGFR and FGFR, this finding could give a hint about the design of allosteric TKIs for the latter.
Compared with conventional CADD, it is believed that artificial intelligence (AI) can expand chemical space in a more comprehensive way. Zhavoronkov et al. (2019) established the first deep-learning-based de novo design method (GENTRL) to discover inhibitors for a receptor tyrosine kinase called DDR1. In addition, the aforementioned work regarding FGFR4/EGFR dual inhibitor by Chen et al. (2020) also demonstrated the great potential of AI for the discovery of novel FGFR inhibitors with different modes of action.
Small molecules other than TKIs, such as extracellular domain binders or natural products, is another potential source of novel modalities. Many of these molecules show selectivity towards certain subtype of FGFR, probably because the extracellular domains of FGFR1-4 are structurally more distinct than kinase domains. The rapidly emerging resistance to current TKIs may also be overcome by these novel types of molecules. Additionally, it seems unnecessary to impose harsh criteria for cell permeability of these molecules as they typically function outside cells.
Combination therapy and dual/multi-target inhibitor are conceptually similar, while the latter apparently has more advantages since it circumvents any drug-drug interactions. While the rational design of dual inhibitors of FGFR/EGFR, FGFR/VEGFR, and FGFR/HDAC may inspire rapid discovery of more inhibitors simultaneously acting on FGFR and another target, identification of more genes that can robustly cause synthetic lethality with FGFR is the central problem and requires extensive in-depth research.
The thriving techniques of targeted protein degradation including PROTAC, molecular glue, as well as other TACs (AUTAC, LYTAC, ATTEC, etc.), have demonstrated broad applicability. PROTACs for RTKs including EGFR and FGFR, have been recently reported, all of which utilized current kinase inhibitors as the RTK binders. It can be envisaged that small molecules occupying a pocket of the kinase beyond ATP-binding site are especially suitable for designing new PROTACs. Such PROTACs are supposed to improve the selectivity and offer solutions to combat drug resistance.
This review encompassed most of the existing FGFR inhibitors and elaborated important structures from a medicinal chemistry perspective. We anticipate that more and more tailor-made novel small molecules of different types and modalities will be developed to improve future targeted therapy with higher efficacy and lower toxicity.
Author Contributions
JZ and WZ conducted the literature review and wrote the draft. LL, SN and ZG edited the draft and made significant revisions. All authors read and approved the final.
Funding
This work was supported by the research foundation of talented scholars from Chongqing Medical University, the National Natural Science Foundation of China (22177017) and Innovative Group of Natural Science Foundation of Chongqing (CXQT21016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.860985/full#supplementary-material
References
- Abdeldayem A., Raouf Y. S., Constantinescu S. N., Moriggl R., Gunning P. T. (2020). Advances in Covalent Kinase Inhibitors. Chem. Soc. Rev., 49, 2617–2687. 10.1039/c9cs00720b [DOI] [PubMed] [Google Scholar]
- Akbulut O., Lengerli D., Saatci O., Duman E., Seker U. O. S., Isik A., et al. (2020). A Highly Potent TACC3 Inhibitor as a Novel Anticancer Drug Candidate. Mol. Cancer Ther. 19, 1243–1254. 10.1158/1535-7163.MCT-19-0957 [DOI] [PubMed] [Google Scholar]
- Angelin B., Larsson T. E., Rudling M. (2012). Circulating Fibroblast Growth Factors as Metabolic Regulators-A Critical Appraisal. Cel Metab., 16, 693–705. 10.1016/j.cmet.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Angevin E., Lopez-Martin J. A., Lin C.-C., Gschwend J. E., Harzstark A., Castellano D., et al. (2013). Phase I Study of Dovitinib (TKI258), an Oral FGFR, VEGFR, and PDGFR Inhibitor, in Advanced or Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 19, 1257–1268. 10.1158/1078-0432.CCR-12-2885 [DOI] [PubMed] [Google Scholar]
- Ayala-Aguilera C. C., Valero T., Lorente-Macías Á., Baillache D. J., Croke S., Unciti-Broceta A. (2022). Small Molecule Kinase Inhibitor Drugs (1995-2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem., 65, 1047–1131. 10.1021/acs.jmedchem.1c00963 [DOI] [PubMed] [Google Scholar]
- Azam M., Seeliger M. A., Gray N. S., Kuriyan J., Daley G. Q. (2008). Activation of Tyrosine Kinases by Mutation of the Gatekeeper Threonine. Nat. Struct. Mol. Biol., 15, 1109–1118. 10.1038/nsmb.1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babina I. S., Turner N. C. (2017). Advances and Challenges in Targeting FGFR Signalling in Cancer. Nat. Rev. Cancer, 17, 318–332. 10.1038/nrc.2017.8 [DOI] [PubMed] [Google Scholar]
- Baldia P. H., Maurer A., Heide T., Rose M., Stoehr R., Hartmann A., et al. (2016). Fibroblast Growth Factor Receptor (FGFR) Alterations in Squamous Differentiated Bladder Cancer: a Putative Therapeutic Target for a Small Subgroup. Oncotarget 7, 71429–71439. 10.18632/oncotarget.12198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begnini F., Poongavanam V., Over B., Castaldo M., Geschwindner S., Johansson P., et al. (2021). Mining Natural Products for Macrocycles to Drug Difficult Targets. J. Med. Chem. 64, 1054–1072. 10.1021/acs.jmedchem.0c01569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello E., Colella G., Scarlato V., Oliva P., Berndt A., Valbusa G., et al. (2011). E-3810 Is a Potent Dual Inhibitor of VEGFR and FGFR that Exerts Antitumor Activity in Multiple Preclinical Models. Cancer Res. 71, 1396–1405. 10.1158/0008-5472.CAN-10-2700 [DOI] [PubMed] [Google Scholar]
- Belov A. A., Mohammadi M. (2013). Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harbor Perspect. Biol., 5, a015958. 10.1101/cshperspect.a015958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide R. S., Cai Z.-W., Zhang Y.-Z., Qian L., Wei D., Barbosa S., et al. (2006). Discovery and Preclinical Studies of (R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5- Methylpyrrolo[2,1-F][1,2,4]triazin-6-Yloxy)propan- 2-ol (BMS-540215), an In Vivo Active Potent VEGFR-2 Inhibitor. J. Med. Chem. 49, 2143–2146. 10.1021/jm051106d [DOI] [PubMed] [Google Scholar]
- Bono F., De Smet F., Herbert C., De Bock K., Georgiadou M., Fons P., et al. (2013). Inhibition of Tumor Angiogenesis and Growth by a Small-Molecule Multi-FGF Receptor Blocker with Allosteric Properties. Cancer Cell 23, 477–488. 10.1016/j.ccr.2013.02.019 [DOI] [PubMed] [Google Scholar]
- Botrus G., Raman P., Oliver T., Bekaii-Saab T. (2021). Infigratinib (BGJ398): an Investigational Agent for the Treatment of FGFR-Altered Intrahepatic Cholangiocarcinoma. Expert Opin. Investig. Drugs, 30, 309–316. 10.1080/13543784.2021.1864320 [DOI] [PubMed] [Google Scholar]
- Brameld K. A., Owens T. D., Verner E., Venetsanakos E., Bradshaw J. M., Phan V. T., et al. (2017). Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-Yl)propyl)-6-(2,6-Dichloro-3,5-Dimethoxyphenyl)-2-(methylamino)pyrido[2,3-D]pyrimidin-7(8h)-One (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 60, 6516–6527. 10.1021/acs.jmedchem.7b00360 [DOI] [PubMed] [Google Scholar]
- Brooks A. N., Kilgour E., Smith P. D. (2012). Molecular Pathways: Fibroblast Growth Factor Signaling: a New Therapeutic Opportunity in Cancer. Clin. Cancer Res., 18, 1855–1862. 10.1158/1078-0432.Ccr-11-0699 [DOI] [PubMed] [Google Scholar]
- Bunney T. D., Wan S., Thiyagarajan N., Sutto L., Williams S. V., Ashford P., et al. (2015). The Effect of Mutations on Drug Sensitivity and Kinase Activity of Fibroblast Growth Factor Receptors: A Combined Experimental and Theoretical Study. EBioMedicine 2, 194–204. 10.1016/j.ebiom.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbridge M. F., Bossard C. J., Saunier C., Fejes I., Bruno A., Léonce S., et al. (2013). S49076 Is a Novel Kinase Inhibitor of MET, AXL, and FGFR with strong Preclinical Activity Alone and in Association with Bevacizumab. Mol. Cancer Ther. 12, 1749–1762. 10.1158/1535-7163.MCT-13-0075 [DOI] [PubMed] [Google Scholar]
- Burslem G. M., Smith B. E., Lai A. C., Jaime-Figueroa S., McQuaid D. C., Bondeson D. P., et al. (2018). The Advantages of Targeted Protein Degradation over Inhibition: An RTK Case Study. Cel Chem. Biol. 25, 67–77. 10.1016/j.chembiol.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 513, 202–209. 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.-X., Zheng R.-L., Lin H.-J., Luo S.-D., Zhou Y., Xu Y.-Z., et al. (2011). SKLB610: a Novel Potential Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases Inhibits Angiogenesis and Tumor Growth In Vivo . Cell Physiol Biochem 27, 565–574. 10.1159/000329978 [DOI] [PubMed] [Google Scholar]
- Capdevila J., Carrato A., Tabernero J., Grande E. (2014). What Could Nintedanib (BIBF 1120), a Triple Inhibitor of VEGFR, PDGFR, and FGFR, Add to the Current Treatment Options for Patients with Metastatic Colorectal Cancer? Crit. Rev. Oncology/Hematology, 92, 83–106. 10.1016/j.critrevonc.2014.05.004 [DOI] [PubMed] [Google Scholar]
- Carpinelli P., Ceruti R., Giorgini M. L., Cappella P., Gianellini L., Croci V., et al. (2007). PHA-739358, a Potent Inhibitor of Aurora Kinases with a Selective Target Inhibition Profile Relevant to Cancer. Mol. Cancer Ther. 6, 3158–3168. 10.1158/1535-7163.MCT-07-0444 [DOI] [PubMed] [Google Scholar]
- Casaletto J., Maglic D., Toure B. B., Taylor A., Schoenherr H., Hudson B., et al. (2021). Abstract 1455: RLY-4008, a Novel Precision Therapy for FGFR2-Driven Cancers Designed to Potently and Selectively Inhibit FGFR2 and FGFR2 Resistance Mutations. Cancer Res. 81, 1455. 10.1158/1538-7445.AM2021-1455 [DOI] [Google Scholar]
- Chae Y. K., Ranganath K., Hammerman P. S., Vaklavas C., Mohindra N., Kalyan A., et al. (2017). Inhibition of the Fibroblast Growth Factor Receptor (FGFR) Pathway: the Current Landscape and Barriers to Clinical Application. Oncotarget 8, 16052–16074. 10.18632/oncotarget.14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-H., Changou C. A., Hsieh T.-H., Lee Y.-C., Chu C.-Y., Hsu K.-C., et al. (2018). Dual Inhibition of PIK3C3 and FGFR as a New Therapeutic Approach to Treat Bladder Cancer. Clin. Cancer Res. 24, 1176–1189. 10.1158/1078-0432.CCR-17-2066 [DOI] [PubMed] [Google Scholar]
- Chen C.-H., Liu Y.-M., Pan S.-L., Liu Y.-R., Liou J.-P., Yen Y. (2016). Trichlorobenzene-substituted Azaaryl Compounds as Novel FGFR Inhibitors Exhibiting Potent Antitumor Activity in Bladder Cancer Cells In Vitro and In Vivo . Oncotarget, 7, 26374–26387. 10.18632/oncotarget.8380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Bao Y., Weng Q., Zhao Y., Lu X., Fu L., et al. (2019). Compound 15c, a Novel Dual Inhibitor of EGFRL858R/T790M and FGFR1, Efficiently Overcomes Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor Resistance of Non-small-cell Lung Cancers. Front. Pharmacol. 10, 1533. 10.3389/fphar.2019.01533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Ma J., Li W., Eliseenkova A. V., Xu C., Neubert T. A., et al. (2007). A Molecular Brake in the Kinase Hinge Region Regulates the Activity of Receptor Tyrosine Kinases. Mol. Cel 27, 717–730. 10.1016/j.molcel.2007.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Xie W., Yang Y., Hua Y., Xing G., Liang L., et al. (2020). Discovery of Dual FGFR4 and EGFR Inhibitors by Machine Learning and Biological Evaluation. J. Chem. Inf. Model. 60, 4640–4652. 10.1021/acs.jcim.0c00652 [DOI] [PubMed] [Google Scholar]
- Collin M.-P., Lobell M., Hübsch W., Brohm D., Schirok H., Jautelat R., et al. (2018). Discovery of Rogaratinib (BAY 1163877): a Pan-FGFR Inhibitor. ChemMedChem 13, 437–445. 10.1002/cmdc.201700718 [DOI] [PubMed] [Google Scholar]
- Cortes J. E., Kim D.-W., Pinilla-Ibarz J., le Coutre P. D., Paquette R., Chuah C., et al. (2018). Ponatinib Efficacy and Safety in Philadelphia Chromosome-Positive Leukemia: Final 5-year Results of the Phase 2 PACE Trial. Blood 132, 393–404. 10.1182/blood-2016-09-739086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couffignal C., Desgrandchamps F., Mongiat-Artus P., Ravery V., Ouzaid I., Roupret M., et al. (2015). The Diagnostic and Prognostic Performance of Urinary FGFR3 Mutation Analysis in Bladder Cancer Surveillance: A Prospective Multicenter Study. Urology 86, 1185–1191. 10.1016/j.urology.2015.07.036 [DOI] [PubMed] [Google Scholar]
- Dai M.-d., Wang Y.-l., Fan J., Dai Y., Ji Y.-c., Sun Y.-m., et al. (2020). DW14383 Is an Irreversible Pan-FGFR Inhibitor that Suppresses FGFR-dependent Tumor Growth In Vitro and In Vivo . Acta Pharmacol. Sin 42, 1498–1506. 10.1038/s41401-020-00567-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S., Zhou Z., Chen Z., Xu G., Chen Y. (2019). Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells, 8, 614. 10.3390/cells8060614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X., Boney-Montoya J., Owen B. M., Bookout A. L., Coate K. C., Mangelsdorf D. J., et al. (2012). βKlotho Is Required for Fibroblast Growth Factor 21 Effects on Growth and Metabolism. Cel Metab. 16, 387–393. 10.1016/j.cmet.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorey K., Amaya E. (2010). FGF Signalling: Diverse Roles during Early Vertebrate Embryogenesis. Development, 137, 3731–3742. 10.1242/dev.037689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G., Jiang J., Wu Q., Henning N. J., Donovan K. A., Yue H., et al. (2021). Discovery of a Potent Degrader for Fibroblast Growth Factor Receptor 1/2. Angew. Chem. Int. Ed. 60, 15905–15911. 10.1002/anie.202101328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y.-C., Zhang S.-J., Shi X.-J., Jin L.-F., Yu T., Song Y., et al. (2021). Research Progress of Dual Inhibitors Targeting Crosstalk between Histone Epigenetic Modulators for Cancer Therapy. Eur. J. Med. Chem. 222, 113588. 10.1016/j.ejmech.2021.113588 [DOI] [PubMed] [Google Scholar]
- Dubreuil P., Letard S., Ciufolini M., Gros L., Humbert M., Castéran N., et al. (2009). Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS One 4, e7258. 10.1371/journal.pone.0007258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt A., Salvesen H. B., Chen T.-H., Ramos A. H., Onofrio R. C., Hatton C., et al. (2008). Drug-sensitive FGFR2 Mutations in Endometrial Carcinoma. Proc. Natl. Acad. Sci. U.S.A. 105, 8713–8717. 10.1073/pnas.0803379105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebiike H., Taka N., Matsushita M., Ohmori M., Takami K., Hyohdoh I., et al. (2016). Discovery of [5-Amino-1-(2-Methyl-3h-Benzimidazol-5-Yl)pyrazol-4-Yl]-(1h-Indol-2-Yl)methanone (CH5183284/Debio 1347), an Orally Available and Selective Fibroblast Growth Factor Receptor (FGFR) Inhibitor. J. Med. Chem. 59, 10586–10600. 10.1021/acs.jmedchem.6b01156 [DOI] [PubMed] [Google Scholar]
- Edwards J., Krishna N. S., Witton C. J., Bartlett J. M. (2003). Gene Amplifications Associated with the Development of Hormone-Resistant Prostate Cancer. Clin. Cancer Res., 9, 5271–5281. [PubMed] [Google Scholar]
- Engelhardt H., Böse D., Petronczki M., Scharn D., Bader G., Baum A., et al. (2019). Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 62, 10272–10293. 10.1021/acs.jmedchem.9b01169 [DOI] [PubMed] [Google Scholar]
- Ezzat S., Huang P., Dackiw A., Asa S. L. (2005). Dual Inhibition of RET and FGFR4 Restrains Medullary Thyroid Cancer Cell Growth. Clin. Cancer Res., 11, 1336–1341. [PubMed] [Google Scholar]
- Fairhurst R. A., Knoepfel T., Buschmann N., Leblanc C., Mah R., Todorov M., et al. (2020). Discovery of Roblitinib (FGF401) as a Reversible-Covalent Inhibitor of the Kinase Activity of Fibroblast Growth Factor Receptor 4. J. Med. Chem. 63, 12542–12573. 10.1021/acs.jmedchem.0c01019 [DOI] [PubMed] [Google Scholar]
- Fairhurst R. A., Knoepfel T., Leblanc C., Buschmann N., Gaul C., Blank J., et al. (2017). Approaches to Selective Fibroblast Growth Factor Receptor 4 Inhibition through Targeting the ATP-Pocket Middle-Hinge Region. Med. Chem. Commun. 8, 1604–1613. 10.1039/c7md00213k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq A., Zhou M.-M. (2004). Structure and Regulation of MAPK Phosphatases. Cell Signal., 16, 769–779. 10.1016/j.cellsig.2003.12.008 [DOI] [PubMed] [Google Scholar]
- Fearon A. E., Grose R. P. (2014). Grb-ing Receptor Activation by the Tail. Nat. Struct. Mol. Biol., 21, 113–114. 10.1038/nsmb.2767 [DOI] [PubMed] [Google Scholar]
- Fernandes-Freitas I., Owen B. M. (2015). Metabolic Roles of Endocrine Fibroblast Growth Factors. Curr. Opin. Pharmacol., 25, 30–35. 10.1016/j.coph.2015.09.014 [DOI] [PubMed] [Google Scholar]
- Fischer H., Taylor N., Allerstorfer S., Grusch M., Sonvilla G., Holzmann K., et al. (2008). Fibroblast Growth Factor Receptor-Mediated Signals Contribute to the Malignant Phenotype of Non-small Cell Lung Cancer Cells: Therapeutic Implications and Synergism with Epidermal Growth Factor Receptor Inhibition. Mol. Cancer Ther. 7, 3408–3419. 10.1158/1535-7163.Mct-08-0444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher G. C., Brokx R. D., Denny T. A., Hembrough T. A., Plum S. M., Fogler W. E., et al. (2011). ENMD-2076 Is an Orally Active Kinase Inhibitor with Antiangiogenic and Antiproliferative Mechanisms of Action. Mol. Cancer Ther. 10, 126–137. 10.1158/1535-7163.MCT-10-0574 [DOI] [PubMed] [Google Scholar]
- Formisano L., Lu Y., Servetto A., Hanker A. B., Jansen V. M., Bauer J. A., et al. (2019). Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat. Commun. 10, 1373. 10.1038/s41467-019-09068-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furdui C. M., Lew E. D., Schlessinger J., Anderson K. S. (2006). Autophosphorylation of FGFR1 Kinase Is Mediated by a Sequential and Precisely Ordered Reaction. Mol. Cel, 21, 711–717. 10.1016/j.molcel.2006.01.022 [DOI] [PubMed] [Google Scholar]
- Furet P., Caravatti G., Guagnano V., Lang M., Meyer T., Schoepfer J. (2008). Entry into a New Class of Protein Kinase Inhibitors by Pseudo Ring Design. Bioorg. Med. Chem. Lett., 18, 897–900. 10.1016/j.bmcl.2007.12.041 [DOI] [PubMed] [Google Scholar]
- Futami T., Okada H., Kihara R., Kawase T., Nakayama A., Suzuki T., et al. (2017). ASP5878, a Novel Inhibitor of FGFR1, 2, 3, and 4, Inhibits the Growth of FGF19-Expressing Hepatocellular Carcinoma. Mol. Cancer Ther. 16, 68–75. 10.1158/1535-7163.MCT-16-0188 [DOI] [PubMed] [Google Scholar]
- Galbiati A., Zana A., Conti P. (2020). Covalent Inhibitors of GAPDH: From Unspecific Warheads to Selective Compounds. Eur. J. Med. Chem., 207, 112740. 10.1016/j.ejmech.2020.112740 [DOI] [PubMed] [Google Scholar]
- Gammelgaard K. R., Vad-Nielsen J., Clement M. S., Weiss S., Daugaard T. F., Dagnæs-Hansen F., et al. (2019). Up-Regulated FGFR1 Expression as a Mediator of Intrinsic TKI Resistance in EGFR-Mutated NSCLC. Translational Oncol. 12, 432–440. 10.1016/j.tranon.2018.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavine P. R., Mooney L., Kilgour E., Thomas A. P., Al-Kadhimi K., Beck S., et al. (2012). AZD4547: an Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family. Cancer Res. 72, 2045–2056. 10.1158/0008-5472.CAN-11-3034 [DOI] [PubMed] [Google Scholar]
- Gelsi-Boyer V., Orsetti B., Cervera N., Finetti P., Sircoulomb F., Rougé C., et al. (2005). Comprehensive Profiling of 8p11-12 Amplification in Breast Cancer. Mol. Cancer Res. 3, 655–667. 10.1158/1541-7786.Mcr-05-0128 [DOI] [PubMed] [Google Scholar]
- Goldfarb M., Schoorlemmer J., Williams A., Diwakar S., Wang Q., Huang X., et al. (2007). Fibroblast Growth Factor Homologous Factors Control Neuronal Excitability through Modulation of Voltage-Gated Sodium Channels. Neuron 55, 449–463. 10.1016/j.neuron.2007.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golfmann K., Meder L., Koker M., Volz C., Borchmann S., Tharun L., et al. (2018). Synergistic Anti-angiogenic Treatment Effects by Dual FGFR1 and VEGFR1 Inhibition in FGFR1-Amplified Breast Cancer. Oncogene 37, 5682–5693. 10.1038/s41388-018-0380-3 [DOI] [PubMed] [Google Scholar]
- Goracci L., Desantis J., Valeri A., Castellani B., Eleuteri M., Cruciani G. (2020). Understanding the Metabolism of Proteolysis Targeting Chimeras (PROTACs): The Next Step toward Pharmaceutical Applications. J. Med. Chem., 63, 11615–11638. 10.1021/acs.jmedchem.0c00793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A., Hansen R. M. S., Taylor I. B., Olesen I. A., Jacobsen G. K., McGowan S. J., et al. (2009). Activating Mutations in FGFR3 and HRAS Reveal a Shared Genetic Origin for Congenital Disorders and Testicular Tumors. Nat. Genet. 41, 1247–1252. 10.1038/ng.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L., Shi L., Liu L. Y., Fece de la Cruz F., Lennerz J. K., Raghavan S., et al. (2019). TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 9, 1064–1079. 10.1158/2159-8290.CD-19-0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozgit J. M., Squillace R. M., Wongchenko M. J., Miller D., Wardwell S., Mohemmad Q., et al. (2013). Combined Targeting of FGFR2 and mTOR by Ponatinib and Ridaforolimus Results in Synergistic Antitumor Activity in FGFR2 Mutant Endometrial Cancer Models. Cancer Chemother. Pharmacol. 71, 1315–1323. 10.1007/s00280-013-2131-z [DOI] [PubMed] [Google Scholar]
- Grünewald S., Politz O., Bender S., Héroult M., Lustig K., Thuss U., et al. (2019). Rogaratinib: A Potent and Selective pan‐FGFR Inhibitor with Broad Antitumor Activity in FGFR‐overexpressing Preclinical Cancer Models. Int. J. Cancer 145, 1346–1357. 10.1002/ijc.32224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guagnano V., Furet P., Spanka C., Bordas V., Le Douget M., Stamm C., et al. (2011). Discovery of 3-(2,6-Dichloro-3,5-Dimethoxy-Phenyl)-1-{6-[4-(4-Ethyl-Piperazin-1-Yl)-Phenylamino]-Pyrimidin-4-Yl}-1-Methyl-Urea (NVP-Bgj398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 54, 7066–7083. 10.1021/jm2006222 [DOI] [PubMed] [Google Scholar]
- Guimarães C. R. W., Rai B. K., Munchhof M. J., Liu S., Wang J., Bhattacharya S. K., et al. (2011). Understanding the Impact of the P-Loop Conformation on Kinase Selectivity. J. Chem. Inf. Model. 51, 1199–1204. 10.1021/ci200153c [DOI] [PubMed] [Google Scholar]
- Guo Y. J., Pan W. W., Liu S. B., Shen Z. F., Xu Y., Hu L. L. (2020). ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med., 19, 1997–2007. 10.3892/etm.2020.8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadari Y. R., Kouhara H., Lax I., Schlessinger J. (1998). Binding of Shp2 Tyrosine Phosphatase to FRS2 Is Essential for Fibroblast Growth Factor-Induced PC12 Cell Differentiation. Mol. Cel Biol, 18, 3966–3973. 10.1128/mcb.18.7.3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagel M., Miduturu C., Sheets M., Rubin N., Weng W., Stransky N., et al. (2015). First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 5, 424–437. 10.1158/2159-8290.CD-14-1029 [DOI] [PubMed] [Google Scholar]
- Hall T. G., Yu Y., Eathiraj S., Wang Y., Savage R. E., Lapierre J.-M., et al. (2016). Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS One 11, e0162594. 10.1371/journal.pone.0162594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H., Torii S., Yasunaga T., Nishida E. (2002). Sprouty1 and Sprouty2 Provide a Control Mechanism for the Ras/MAPK Signalling Pathway. Nat. Cel Biol, 4, 850–858. 10.1038/ncb867 [DOI] [PubMed] [Google Scholar]
- Harris P. A., Boloor A., Cheung M., Kumar R., Crosby R. M., Davis-Ward R. G., et al. (2008). Discovery of 5-[[4-[(2,3-Dimethyl-2h-Indazol-6-Yl)methylamino]-2-Pyrimidinyl]amino]-2-Methyl-Benzenesulfonamide (Pazopanib), a Novel and Potent Vascular Endothelial Growth Factor Receptor Inhibitor. J. Med. Chem. 51, 4632–4640. 10.1021/jm800566m [DOI] [PubMed] [Google Scholar]
- Hart K. C., Robertson S. C., Kanemitsu M. Y., Meyer A. N., Tynan J. A., Donoghue D. J. (2000). Transformation and Stat Activation by Derivatives of FGFR1, FGFR3, and FGFR4. Oncogene, 19, 3309–3320. 10.1038/sj.onc.1203650 [DOI] [PubMed] [Google Scholar]
- Helsten T., Elkin S., Arthur E., Tomson B. N., Carter J., Kurzrock R. (2016). The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res., 22, 259–267. 10.1158/1078-0432.Ccr-14-3212 [DOI] [PubMed] [Google Scholar]
- Herbert C., Schieborr U., Saxena K., Juraszek J., De Smet F., Alcouffe C., et al. (2013). Molecular Mechanism of SSR128129E, an Extracellularly Acting, Small-Molecule, Allosteric Inhibitor of FGF Receptor Signaling. Cancer Cell 23, 489–501. 10.1016/j.ccr.2013.02.018 [DOI] [PubMed] [Google Scholar]
- Hilberg F., Roth G. J., Krssak M., Kautschitsch S., Sommergruber W., Tontsch-Grunt U., et al. (2008). BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 68, 4774–4782. 10.1158/0008-5472.CAN-07-6307 [DOI] [PubMed] [Google Scholar]
- Holmström T. H., Moilanen A.-M., Ikonen T., Björkman M. L., Linnanen T., Wohlfahrt G., et al. (2019). ODM-203, a Selective Inhibitor of FGFR and VEGFR, Shows Strong Antitumor Activity, and Induces Antitumor Immunity. Mol. Cancer Ther. 18, 28–38. 10.1158/1535-7163.Mct-18-0204 [DOI] [PubMed] [Google Scholar]
- Howard S., Berdini V., Boulstridge J. A., Carr M. G., Cross D. M., Curry J., et al. (2009). Fragment-based Discovery of the Pyrazol-4-Yl Urea (AT9283), a Multitargeted Kinase Inhibitor with Potent aurora Kinase Activity. J. Med. Chem. 52, 379–388. 10.1021/jm800984v [DOI] [PubMed] [Google Scholar]
- Hu B., Zhou Y., Sun D., Yang Y., Liu Y., Li X., et al. (2020). PROTACs: New Method to Degrade Transcription Regulating Proteins. Eur. J. Med. Chem. 207, 112698. 10.1016/j.ejmech.2020.112698 [DOI] [PubMed] [Google Scholar]
- Hu J., Ahuja L. G., Meharena H. S., Kannan N., Kornev A. P., Taylor S. S., et al. (2015). Kinase Regulation by Hydrophobic Spine Assembly in Cancer. Mol. Cel Biol 35, 264–276. 10.1128/mcb.00943-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Jiang S., Shi Y. (2020). Tyrosine Kinase Inhibitors for Solid Tumors in the Past 20 Years (2001-2020). J. Hematol. Oncol., 13, 143. 10.1186/s13045-020-00977-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi O. A., Eliseenkova A. V., Plotnikov A. N., Yu K., Ornitz D. M., Mohammadi M. (2001). Structural Basis for Fibroblast Growth Factor Receptor 2 Activation in Apert Syndrome. Proc. Natl. Acad. Sci. U.S.A., 98, 7182–7187. 10.1073/pnas.121183798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi O. A., Zhang F., Eliseenkova A. V., Itoh N., Linhardt R. J., Mohammadi M. (2004). Biochemical Analysis of Pathogenic Ligand-dependent FGFR2 Mutations Suggests Distinct Pathophysiological Mechanisms for Craniofacial and Limb Abnormalities. Hum. Mol. Genet., 13, 2313–2324. 10.1093/hmg/ddh235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N., Ornitz D. M. (2004). Evolution of the Fgf and Fgfr Gene Families. Trends Genet., 20, 563–569. 10.1016/j.tig.2004.08.007 [DOI] [PubMed] [Google Scholar]
- Javle M., Roychowdhury S., Kelley R. K., Sadeghi S., Macarulla T., Weiss K. H., et al. (2021). Infigratinib (BGJ398) in Previously Treated Patients with Advanced or Metastatic Cholangiocarcinoma with FGFR2 Fusions or Rearrangements: Mature Results from a Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Gastroenterol. Hepatol. 6, 803–815. 10.1016/s2468-1253(21)00196-5 [DOI] [PubMed] [Google Scholar]
- Jia Y., Yun C.-H., Park E., Ercan D., Manuia M., Juarez J., et al. (2016). Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 534, 129–132. 10.1038/nature17960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.-f., Dai Y., Peng X., Shen Y.-y., Su Y., Wei M.-m., et al. (2018). SOMCL-085, a Novel Multi-Targeted FGFR Inhibitor, Displays Potent Anticancer Activity in FGFR-Addicted Human Cancer Models. Acta Pharmacol. Sin 39, 243–250. 10.1038/aps.2017.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi J. J., Coffey H., Corcoran E., Tsai J., Huang C.-L., Ichikawa K., et al. (2017). H3B-6527 Is a Potent and Selective Inhibitor of FGFR4 in FGF19-Driven Hepatocellular Carcinoma. Cancer Res. 77, 6999–7013. 10.1158/0008-5472.CAN-17-1865 [DOI] [PubMed] [Google Scholar]
- Jung E.-J., Jung E.-J., Min S. Y., Kim M. A., Kim W. H. (2012). Fibroblast Growth Factor Receptor 2 Gene Amplification Status and its Clinicopathologic Significance in Gastric Carcinoma. Hum. Pathol., 43, 1559–1566. 10.1016/j.humpath.2011.12.002 [DOI] [PubMed] [Google Scholar]
- Kikuchi A., Suzuki T., Nakazawa T., Iizuka M., Nakayama A., Ozawa T., et al. (2017). ASP5878, a Selective FGFR Inhibitor, to Treat FGFR3-dependent Urothelial Cancer with or without Chemoresistance. Cancer Sci. 108, 236–242. 10.1111/cas.13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konecny G. E., Finkler N., Garcia A. A., Lorusso D., Lee P. S., Rocconi R. P., et al. (2015). Second-line Dovitinib (TKI258) in Patients with FGFR2-Mutated or FGFR2-Non-Mutated Advanced or Metastatic Endometrial Cancer: a Non-randomised, Open-Label, Two-Group, Two-Stage, Phase 2 Study. Lancet Oncol. 16, 686–694. 10.1016/s1470-2045(15)70159-2 [DOI] [PubMed] [Google Scholar]
- Kovalenko M., Gazit A., Böhmer A., Rorsman C., Rönnstrand L., Heldin C. H., et al. (1994). Selective Platelet-Derived Growth Factor Receptor Kinase Blockers Reverse Sis-Transformation. Cancer Res. 54, 6106–6114. [PubMed] [Google Scholar]
- Krook M. A., Lenyo A., Wilberding M., Barker H., Dantuono M., Bailey K. M., et al. (2020). Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Mol. Cancer Ther. 19, 847–857. 10.1158/1535-7163.MCT-19-0631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird A. D., Vajkoczy P., Shawver L. K., Thurnher A., Liang C., Mohammadi M., et al. (2000). SU6668 Is a Potent Antiangiogenic and Antitumor Agent that Induces Regression of Established Tumors. Cancer Res. 60, 4152–4160. [PubMed] [Google Scholar]
- Lesca E., Lammens A., Huber R., Augustin M. (2014). Structural Analysis of the Human Fibroblast Growth Factor Receptor 4 Kinase. J. Mol. Biol., 426, 3744–3756. 10.1016/j.jmb.2014.09.004 [DOI] [PubMed] [Google Scholar]
- Levine D. A., Schultz N., Cherniack A. D., Akbani R., Liu Y., Shen H., et al. (2013). Integrated Genomic Characterization of Endometrial Carcinoma. Nature 497, 67–73. 10.1038/nature12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Guise C. P., Taghipouran R., Yosaatmadja Y., Ashoorzadeh A., Paik W.-K., et al. (2017). 2-Oxo-3, 4-dihydropyrimido[4, 5- D ]pyrimidinyl Derivatives as New Irreversible pan Fibroblast Growth Factor Receptor (FGFR) Inhibitors. Eur. J. Med. Chem. 135, 531–543. 10.1016/j.ejmech.2017.04.049 [DOI] [PubMed] [Google Scholar]
- Liu J., Ma J., Liu Y., Xia J., Li Y., Wang Z. P., et al. (2020). PROTACs: A Novel Strategy for Cancer Therapy. Semin. Cancer Biol. 67, 171–179. 10.1016/j.semcancer.2020.02.006 [DOI] [PubMed] [Google Scholar]
- Liu J., Qian C., Zhu Y., Cai J., He Y., Li J., et al. (2018). Design, Synthesis and Evaluate of Novel Dual FGFR1 and HDAC Inhibitors Bearing an Indazole Scaffold. Bioorg. Med. Chem. 26, 747–757. 10.1016/j.bmc.2017.12.041 [DOI] [PubMed] [Google Scholar]
- Liu Y., Peng X., Guan X., Lu D., Xi Y., Jin S., et al. (2017). Discovery of Novel Ponatinib Analogues for Reducing KDR Activity as Potent FGFRs Inhibitors. Eur. J. Med. Chem. 126, 122–132. 10.1016/j.ejmech.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Lu X., Chen H., Patterson A. V., Smaill J. B., Ding K. (2019). Fibroblast Growth Factor Receptor 4 (FGFR4) Selective Inhibitors as Hepatocellular Carcinoma Therapy: Advances and Prospects. J. Med. Chem., 62, 2905–2915. 10.1021/acs.jmedchem.8b01531 [DOI] [PubMed] [Google Scholar]
- Müller A.-K., Meyer M., Werner S. (2012). The Roles of Receptor Tyrosine Kinases and Their Ligands in the Wound Repair Process. Semin. Cel Develop. Biol., 23, 963–970. 10.1016/j.semcdb.2012.09.015 [DOI] [PubMed] [Google Scholar]
- Markham A. (2019). Erdafitinib: First Global Approval. Drugs 79, 1017–1021. 10.1007/s40265-019-01142-9 [DOI] [PubMed] [Google Scholar]
- Matsui J., Yamamoto Y., Funahashi Y., Tsuruoka A., Watanabe T., Wakabayashi T., et al. (2008). E7080, a Novel Inhibitor that Targets Multiple Kinases, Has Potent Antitumor Activities against Stem Cell Factor Producing Human Small Cell Lung Cancer H146, Based on Angiogenesis Inhibition. Int. J. Cancer 122, 664–671. 10.1002/ijc.23131 [DOI] [PubMed] [Google Scholar]
- Matsuo I., Kimura-Yoshida C. (2013). Extracellular Modulation of Fibroblast Growth Factor Signaling through Heparan Sulfate Proteoglycans in Mammalian Development. Curr. Opin. Genet. Develop., 23, 399–407. 10.1016/j.gde.2013.02.004 [DOI] [PubMed] [Google Scholar]
- Metzner T., Bedeir A., Held G., Peter-Vörösmarty B., Ghassemi S., Heinzle C., et al. (2011). Fibroblast Growth Factor Receptors as Therapeutic Targets in Human Melanoma: Synergism with BRAF Inhibition. J. Invest. Dermatol. 131, 2087–2095. 10.1038/jid.2011.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J.-L., Liu R.-J., Zhou J.-H., Meng S.-H. (2016). Fibroblast Growth Factor Receptor 1 Gene Amplification in Nonsmall Cell Lung Cancer. Chin. Med. J. (Engl), 129, 2868–2872. 10.4103/0366-6999.194649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J.-L., Zhou J.-H., Cai J.-J., Liu R.-J. (2020). The Association between Fibroblast Growth Factor Receptor 1 Gene Amplification and Lung Cancer: a Meta-Analysis. Arch. Med. Sci., 16, 16–26. 10.5114/aoms.2020.91284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael M., Bang Y.-J., Park Y. S., Kang Y.-K., Kim T. M., Hamid O., et al. (2017). A Phase 1 Study of LY2874455, an Oral Selective Pan-FGFR Inhibitor, in Patients with Advanced Cancer. Targ Oncol. 12, 463–474. 10.1007/s11523-017-0502-9 [DOI] [PubMed] [Google Scholar]
- Mikoshiba K. (2007). The IP3 receptor/Ca2+ Channel and its Cellular Function. Biochem. Soc. Symp. 74, 9–22. 10.1042/bss0740009 [DOI] [PubMed] [Google Scholar]
- Mohammadi M., Dikic I., Sorokin A., Burgess W. H., Jaye M., Schlessinger J. (1996). Identification of Six Novel Autophosphorylation Sites on Fibroblast Growth Factor Receptor 1 and Elucidation of Their Importance in Receptor Activation and Signal Transduction. Mol. Cel Biol, 16, 977–989. 10.1128/mcb.16.3.977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M., Froum S., Hamby J. M., Schroeder M. C., Panek R. L., Lu G. H., et al. (1998). Crystal Structure of an Angiogenesis Inhibitor Bound to the FGF Receptor Tyrosine Kinase Domain. Embo j 17, 5896–5904. 10.1093/emboj/17.20.5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M., McMahon G., Sun L., Tang C., Hirth P., Yeh B. K., et al. (1997). Structures of the Tyrosine Kinase Domain of Fibroblast Growth Factor Receptor in Complex with Inhibitors. Science 276, 955–960. 10.1126/science.276.5314.955 [DOI] [PubMed] [Google Scholar]
- Mohammadi M., Schlessinger J., Hubbard S. R. (1996). Structure of the FGF Receptor Tyrosine Kinase Domain Reveals a Novel Autoinhibitory Mechanism. Cell, 86, 577–587. 10.1016/s0092-8674(00)80131-2 [DOI] [PubMed] [Google Scholar]
- Montazeri K., Bellmunt J. (2020). Erdafitinib for the Treatment of Metastatic Bladder Cancer. Expert Rev. Clin. Pharmacol., 13, 1–6. 10.1080/17512433.2020.1702025 [DOI] [PubMed] [Google Scholar]
- Murray C. W., Newell D. R., Angibaud P. (2019). A Successful Collaboration between Academia, Biotech and Pharma Led to Discovery of Erdafitinib, a Selective FGFR Inhibitor Recently Approved by the FDA. Med. Chem. Commun., 10, 1509–1511. 10.1039/c9md90044f [DOI] [Google Scholar]
- Nakanishi Y., Akiyama N., Tsukaguchi T., Fujii T., Sakata K., Sase H., et al. (2014). The Fibroblast Growth Factor Receptor Genetic Status as a Potential Predictor of the Sensitivity to CH5183284/Debio 1347, a Novel Selective FGFR Inhibitor. Mol. Cancer Ther. 13, 2547–2558. 10.1158/1535-7163.MCT-14-0248 [DOI] [PubMed] [Google Scholar]
- O'Hare T., Shakespeare W. C., Zhu X., Eide C. A., Rivera V. M., Wang F., et al. (2009). AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 16, 401–412. 10.1016/j.ccr.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S. H., Guy G. R., Hadari Y. R., Laks S., Gotoh N., Schlessinger J., et al. (2000). FRS2 Proteins Recruit Intracellular Signaling Pathways by Binding to Diverse Targets on Fibroblast Growth Factor and Nerve Growth Factor Receptors. Mol. Cel Biol 20, 979–989. 10.1128/mcb.20.3.979-989.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M. (2000). FGFs, Heparan Sulfate and FGFRs: Complex Interactions Essential for Development. Bioessays 22, 108–112. [DOI] [PubMed] [Google Scholar]
- Packer L. M., Geng X., Bonazzi V. F., Ju R. J., Mahon C. E., Cummings M. C., et al. (2017). PI3K Inhibitors Synergize with FGFR Inhibitors to Enhance Antitumor Responses in FGFR2mutant Endometrial Cancers. Mol. Cancer Ther. 16, 637–648. 10.1158/1535-7163.MCT-16-0415 [DOI] [PubMed] [Google Scholar]
- Pagano K., Carminati L., Tomaselli S., Molinari H., Taraboletti G., Ragona L. (2021). Molecular Basis of the Antiangiogenic Action of Rosmarinic Acid, a Natural Compound Targeting Fibroblast Growth Factor‐2/FGFR Interactions. Chembiochem, 22, 160–169. 10.1002/cbic.202000610 [DOI] [PubMed] [Google Scholar]
- Paiva S.-L., Crews C. M. (2019). Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol., 50, 111–119. 10.1016/j.cbpa.2019.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palakurthi S., Kuraguchi M., Zacharek S. J., Zudaire E., Huang W., Bonal D. M., et al. (2019). The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunol. Res. 7, 1457–1471. 10.1158/2326-6066.Cir-18-0595 [DOI] [PubMed] [Google Scholar]
- Pan B.-S., Chan G. K. Y., Chenard M., Chi A., Davis L. J., Deshmukh S. V., et al. (2010). MK-2461, a Novel Multitargeted Kinase Inhibitor, Preferentially Inhibits the Activated C-Met Receptor. Cancer Res. 70, 1524–1533. 10.1158/0008-5472.CAN-09-2541 [DOI] [PubMed] [Google Scholar]
- Pao W., Miller V. A., Kris M. G. (2004). ‘Targeting’ the Epidermal Growth Factor Receptor Tyrosine Kinase with Gefitinib (Iressa) in Non-small Cell Lung Cancer (NSCLC). Semin. Cancer Biol., 14, 33–40. 10.1016/j.semcancer.2003.11.005 [DOI] [PubMed] [Google Scholar]
- Peng X., Hou P., Chen Y., Dai Y., Ji Y., Shen Y., et al. (2019). Preclinical Evaluation of 3D185, a Novel Potent Inhibitor of FGFR1/2/3 and CSF-1R, in FGFR-dependent and Macrophage-Dominant Cancer Models. J. Exp. Clin. Cancer Res. 38, 372. 10.1186/s13046-019-1357-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera T. P. S., Jovcheva E., Mevellec L., Vialard J., De Lange D., Verhulst T., et al. (2017). Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 16, 1010–1020. 10.1158/1535-7163.MCT-16-0589 [DOI] [PubMed] [Google Scholar]
- Pettersson M., Crews C. M. (2019). PROteolysis TArgeting Chimeras (PROTACs) - Past, Present and Future. Drug Discov. Today Tech., 31, 15–27. 10.1016/j.ddtec.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov A. N., Hubbard S. R., Schlessinger J., Mohammadi M. (2000). Crystal Structures of Two FGF-FGFR Complexes Reveal the Determinants of Ligand-Receptor Specificity. Cell, 101, 413–424. 10.1016/s0092-8674(00)80851-x [DOI] [PubMed] [Google Scholar]
- Porta R., Borea R., Coelho A., Khan S., Araújo A., Reclusa P., et al. (2017). FGFR a Promising Druggable Target in Cancer: Molecular Biology and New Drugs. Crit. Rev. Oncology/Hematology 113, 256–267. 10.1016/j.critrevonc.2017.02.018 [DOI] [PubMed] [Google Scholar]
- Powers C. J., McLeskey S. W., Wellstein A. (2000). Fibroblast Growth Factors, Their Receptors and Signaling. Endocr. Relat. Cancer, 7, 165–197. 10.1677/erc.0.0070165 [DOI] [PubMed] [Google Scholar]
- Qin Q., Patel V., Galsky M. D. (2020). Urothelial Carcinoma: the Development of FGFR Inhibitors in Combination with Immune Checkpoint Inhibitors. Expert Rev. Anticancer Ther., 20, 503–512. 10.1080/14737140.2020.1770600 [DOI] [PubMed] [Google Scholar]
- Qiu H., Yashiro M., Zhang X., Miwa A., Hirakawa K. (2011). A FGFR2 Inhibitor, Ki23057, Enhances the Chemosensitivity of Drug-Resistant Gastric Cancer Cells. Cancer Lett., 307, 47–52. 10.1016/j.canlet.2011.03.015 [DOI] [PubMed] [Google Scholar]
- Qu L., Chen X., Wei H., Guo M., Dai S., Jiang L., et al. (2022). Structural Insights into the Potency and Selectivity of Covalent Pan-FGFR Inhibitors. Commun. Chem. 5. 10.1038/s42004-021-00623-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan M.-Y., Guo Q., Liu J., Yang R., Bai J., Wang W., et al. (2020). Corrigendum: An FGFR/AKT/SOX2 Signaling Axis Controls Pancreatic Cancer Stemness. Front. Cel Dev. Biol. 8, 594589. 10.3389/fcell.2020.594589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran K., Zeng J., Wan G., He X., Feng Z., Xiang W., et al. (2021). Design, Synthesis and Biological Evaluations of a Series of Pyrido[1,2-A]pyrimidinone Derivatives as Novel Selective FGFR Inhibitors. Eur. J. Med. Chem. 220, 113499. 10.1016/j.ejmech.2021.113499 [DOI] [PubMed] [Google Scholar]
- Reintjes N., Li Y., Becker A., Rohmann E., Schmutzler R., Wollnik B. (2013). Activating Somatic FGFR2 Mutations in Breast Cancer. PLoS One, 8, e60264. 10.1371/journal.pone.0060264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezende Miranda R., Fu Y., Chen X., Perino J., Cao P., Carpten J., et al. (2020). Development of a Potent and Specific FGFR4 Inhibitor for the Treatment of Hepatocellular Carcinoma. J. Med. Chem. 63, 11484–11497. 10.1021/acs.jmedchem.0c00044 [DOI] [PubMed] [Google Scholar]
- Risuleo G., Ciacciarelli M., Castelli M., Galati G. (2009). The Synthetic Inhibitor of Fibroblast Growth Factor Receptor PD166866 Controls Negatively the Growth of Tumor Cells in Culture. J. Exp. Clin. Cancer Res., 28, 151. 10.1186/1756-9966-28-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronca R., Giacomini A., Di Salle E., Coltrini D., Pagano K., Ragona L., et al. (2015). Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 28, 225–239. 10.1016/j.ccell.2015.07.002 [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr. (2010). RAF Protein-Serine/threonine Kinases: Structure and Regulation. Biochem. Biophysical Res. Commun. 399, 313–317. 10.1016/j.bbrc.2010.07.092 [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr. (2020). The Role of Fibroblast Growth Factor Receptor (FGFR) Protein-Tyrosine Kinase Inhibitors in the Treatment of Cancers Including Those of the Urinary Bladder. Pharmacol. Res. 151, 104567. 10.1016/j.phrs.2019.104567 [DOI] [PubMed] [Google Scholar]
- Ross J. S., Wang K., Al-Rohil R. N., Nazeer T., Sheehan C. E., Otto G. A., et al. (2014). Advanced Urothelial Carcinoma: Next-Generation Sequencing Reveals Diverse Genomic Alterations and Targets of Therapy. Mod. Pathol. 27, 271–280. 10.1038/modpathol.2013.135 [DOI] [PubMed] [Google Scholar]
- Rosty C., Aubriot M.-H., Cappellen D., Bourdin J., Cartier I., Thiery J., et al. (2005). Clinical and Biological Characteristics of Cervical Neoplasias with FGFR3 Mutation. Mol. Cancer 4, 15. 10.1186/1476-4598-4-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth G. J., Heckel A., Colbatzky F., Handschuh S., Kley J., Lehmann-Lintz T., et al. (2009). Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J. Med. Chem. 52, 4466–4480. 10.1021/jm900431g [DOI] [PubMed] [Google Scholar]
- Sacks D., Sacks D., Baxter B., Campbell B. C. V., Carpenter J. S., Cognard C., et al. (2018). Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. AJNR Am. J. Neuroradiol 39, E61–E632. 10.3174/ajnr.A5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Heras E., Howell F. V., Williams G., Doherty P. (2006). The Fibroblast Growth Factor Receptor Acid Box Is Essential for Interactions with N-Cadherin and All of the Major Isoforms of Neural Cell Adhesion Molecule. J. Biol. Chem., 281, 35208–35216. 10.1074/jbc.M608655200 [DOI] [PubMed] [Google Scholar]
- Shen G., Zheng F., Ren D., Du F., Dong Q., Wang Z., et al. (2018). Anlotinib: a Novel Multi-Targeting Tyrosine Kinase Inhibitor in Clinical Development. J. Hematol. Oncol. 11, 120. 10.1186/s13045-018-0664-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siracusano S., Rizzetto R., Porcaro A. B. (2020). Bladder Cancer Genomics. Urologia, 87, 49–56. 10.1177/0391560319899011 [DOI] [PubMed] [Google Scholar]
- Sohl C. D., Ryan M. R., Luo B., Frey K. M., Anderson K. S. (2015). Illuminating the Molecular Mechanisms of Tyrosine Kinase Inhibitor Resistance for the FGFR1 Gatekeeper Mutation: the Achilles' Heel of Targeted Therapy. ACS Chem. Biol., 10, 1319–1329. 10.1021/acschembio.5b00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon J. P., Hansel D. E. (2016). The Emerging Molecular Landscape of Urothelial Carcinoma. Surg. Pathol. Clin., 9, 391–404. 10.1016/j.path.2016.04.004 [DOI] [PubMed] [Google Scholar]
- Sootome H., Fujita H., Ito K., Ochiiwa H., Fujioka Y., Ito K., et al. (2020). Futibatinib Is a Novel Irreversible FGFR 1-4 Inhibitor that Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res. 80, 4986–4997. 10.1158/0008-5472.CAN-19-2568 [DOI] [PubMed] [Google Scholar]
- Sun L., Tran N., Liang C., Tang F., Rice A., Schreck R., et al. (1999). Design, Synthesis, and Evaluations of Substituted 3-[(3- or 4-Carboxyethylpyrrol-2-Yl)methylidenyl]indolin-2-Ones as Inhibitors of VEGF, FGF, and PDGF Receptor Tyrosine Kinases. J. Med. Chem. 42, 5120–5130. 10.1021/jm9904295 [DOI] [PubMed] [Google Scholar]
- Taeger J., Moser C., Hellerbrand C., Mycielska M. E., Glockzin G., Schlitt H. J., et al. (2011). Targeting FGFR/PDGFR/VEGFR Impairs Tumor Growth, Angiogenesis, and Metastasis by Effects on Tumor Cells, Endothelial Cells, and Pericytes in Pancreatic Cancer. Mol. Cancer Ther. 10, 2157–2167. 10.1158/1535-7163.MCT-11-0312 [DOI] [PubMed] [Google Scholar]
- Tan L., Wang J., Tanizaki J., Huang Z., Aref A. R., Rusan M., et al. (2014). Development of Covalent Inhibitors that Can Overcome Resistance to First-Generation FGFR Kinase Inhibitors. Proc. Natl. Acad. Sci. U.S.A. 111, E4869–E4877. 10.1073/pnas.1403438111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel S., Li Z. H., Wei E., Wiesmann M., Chang H., Chen C., et al. (2005). CHIR-258, a Novel, Multitargeted Tyrosine Kinase Inhibitor for the Potential Treatment of T(4;14) Multiple Myeloma. Blood 105, 2941–2948. 10.1182/blood-2004-10-3913 [DOI] [PubMed] [Google Scholar]
- Tsimafeyeu I., Ludes-Meyers J., Stepanova E., Daeyaert F., Khochenkov D., Joose J.-B., et al. (2016). Targeting FGFR2 with Alofanib (RPT835) Shows Potent Activity in Tumour Models. Eur. J. Cancer 61, 20–28. 10.1016/j.ejca.2016.03.068 [DOI] [PubMed] [Google Scholar]
- Tucker J. A., Klein T., Breed J., Breeze A. L., Overman R., Phillips C., et al. (2014). Structural Insights into FGFR Kinase Isoform Selectivity: Diverse Binding Modes of AZD4547 and Ponatinib in Complex with FGFR1 and FGFR4. Structure 22, 1764–1774. 10.1016/j.str.2014.09.019 [DOI] [PubMed] [Google Scholar]
- Turner L. D., Summers A. J., Johnson L. O., Knowles M. A., Fishwick C. W. G. (2017). Identification of an Indazole-Based Pharmacophore for the Inhibition of FGFR Kinases Using Fragment-Led De Novo Design. ACS Med. Chem. Lett., 8, 1264–1268. 10.1021/acsmedchemlett.7b00349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner L. D., Trinh C. H., Hubball R. A., Orritt K. M., Lin C.-C., Burns J. E., et al. (2021). From Fragment to Lead: De Novo Design and Development toward a Selective FGFR2 Inhibitor. J. Med. Chem. 65, 1481–1504. 10.1021/acs.jmedchem.1c01163 [DOI] [PubMed] [Google Scholar]
- Turner N., Lambros M. B., Horlings H. M., Pearson A., Sharpe R., Natrajan R., et al. (2010). Integrative Molecular Profiling of Triple Negative Breast Cancers Identifies Amplicon Drivers and Potential Therapeutic Targets. Oncogene 29, 2013–2023. 10.1038/onc.2009.489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyulyandina A., Harrison D., Yin W., Stepanova E., Kochenkov D., Solomko E., et al. (2017). Alofanib, an Allosteric FGFR2 Inhibitor, Has Potent Effects on Ovarian Cancer Growth in Preclinical Studies. Invest. New Drugs 35, 127–133. 10.1007/s10637-016-0404-1 [DOI] [PubMed] [Google Scholar]
- Vékony H., Ylstra B., Wilting S. M., Meijer G. A., van de Wiel M. A., Leemans C. R., et al. (2007). DNA Copy Number Gains at Loci of Growth Factors and Their Receptors in Salivary Gland Adenoid Cystic Carcinoma. Clin. Cancer Res. 13, 3133–3139. 10.1158/1078-0432.Ccr-06-2555 [DOI] [PubMed] [Google Scholar]
- Vi J. G. t., Cheuk A. T., Tsang P. S., Chung J.-Y., Song Y. K., Desai K., et al. (2009). Identification of FGFR4-Activating Mutations in Human Rhabdomyosarcomas that Promote Metastasis in Xenotransplanted Models. J. Clin. Invest. 119, 3395–3407. 10.1172/jci39703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan R. S. K., He P., Modi V., Duong-Ly K. C., Ma H., Peterson J. R., et al. (2015). Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 58, 466–479. 10.1021/jm501603h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B. A., Boyle E. M., Wardell C. P., Murison A., Begum D. B., Dahir N. M., et al. (2015). Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 33, 3911–3920. 10.1200/jco.2014.59.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B. A., Mavrommatis K., Wardell C. P., Ashby T. C., Bauer M., Davies F. E., et al. (2018). Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 132, 587–597. 10.1182/blood-2018-03-840132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Šuštić T., Leite de Oliveira R., Lieftink C., Halonen P., van de Ven M., et al. (2017). A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur. Urol. 71, 858–862. 10.1016/j.eururo.2017.01.021 [DOI] [PubMed] [Google Scholar]
- Wang N., Li J.-y., Li S., Guo X.-c., Wu T., Wang W.-f., et al. (2018). Fibroblast Growth Factor 21 Regulates Foam Cells Formation and Inflammatory Response in Ox-LDL-Induced THP-1 Macrophages. Biomed. Pharmacother. 108, 1825–1834. 10.1016/j.biopha.2018.09.143 [DOI] [PubMed] [Google Scholar]
- Wang S., Ding Z. (2017). Fibroblast Growth Factor Receptors in Breast Cancer. Tumour Biol., 39, 101042831769837. 10.1177/1010428317698370 [DOI] [PubMed] [Google Scholar]
- Wang V. E., Xue J. Y., Frederick D. T., Cao Y., Lin E., Wilson C., et al. (2019). Adaptive Resistance to Dual BRAF/MEK Inhibition in BRAF-Driven Tumors through Autocrine FGFR Pathway Activation. Clin. Cancer Res. 25, 7202–7217. 10.1158/1078-0432.Ccr-18-2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Dai Y., Wu X., Li F., Liu B., Li C., et al. (2019). Discovery and Development of a Series of Pyrazolo[3,4-D]pyridazinone Compounds as the Novel Covalent Fibroblast Growth Factor Receptor Inhibitors by the Rational Drug Design. J. Med. Chem. 62, 7473–7488. 10.1021/acs.jmedchem.9b00510 [DOI] [PubMed] [Google Scholar]
- Wang Y., Li L., Fan J., Dai Y., Jiang A., Geng M., et al. (2018). Discovery of Potent Irreversible Pan-Fibroblast Growth Factor Receptor (FGFR) Inhibitors. J. Med. Chem. 61, 9085–9104. 10.1021/acs.jmedchem.7b01843 [DOI] [PubMed] [Google Scholar]
- Watanabe Miyano S., Yamamoto Y., Kodama K., Miyajima Y., Mikamoto M., Nakagawa T., et al. (2016). E7090, a Novel Selective Inhibitor of Fibroblast Growth Factor Receptors, Displays Potent Antitumor Activity and Prolongs Survival in Preclinical Models. Mol. Cancer Ther. 15, 2630–2639. 10.1158/1535-7163.MCT-16-0261 [DOI] [PubMed] [Google Scholar]
- Webster M. K., D'Avis P. Y., Robertson S. C., Donoghue D. J. (1996). Profound Ligand-independent Kinase Activation of Fibroblast Growth Factor Receptor 3 by the Activation Loop Mutation Responsible for a Lethal Skeletal Dysplasia, Thanatophoric Dysplasia Type II. Mol. Cel Biol, 16, 4081–4087. 10.1128/mcb.16.8.4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedge S. R., Kendrew J., Hennequin L. F., Valentine P. J., Barry S. T., Brave S. R., et al. (2005). AZD2171: a Highly Potent, Orally Bioavailable, Vascular Endothelial Growth Factor Receptor-2 Tyrosine Kinase Inhibitor for the Treatment of Cancer. Cancer Res. 65, 4389–4400. 10.1158/0008-5472.Can-04-4409 [DOI] [PubMed] [Google Scholar]
- Wei M., Peng X., Xing L., Dai Y., Huang R., Geng M., et al. (2018). Design, Synthesis and Biological Evaluation of a Series of Novel 2-Benzamide-4-(6-Oxy-N-Methyl-1-Naphthamide)-Pyridine Derivatives as Potent Fibroblast Growth Factor Receptor (FGFR) Inhibitors. Eur. J. Med. Chem. 154, 9–28. 10.1016/j.ejmech.2018.05.005 [DOI] [PubMed] [Google Scholar]
- Wei Y., Tang Y., Zhou Y., Yang Y., Cui Y., Wang X., et al. (2021). Discovery and Optimization of a Novel 2H-Pyrazolo[3,4-D]pyrimidine Derivative as a Potent Irreversible Pan-Fibroblast Growth Factor Receptor Inhibitor. J. Med. Chem. 64, 9078–9099. 10.1021/acs.jmedchem.1c00174 [DOI] [PubMed] [Google Scholar]
- Weiss J., Sos M. L., Seidel D., Peifer M., Zander T., Heuckmann J. M., et al. (2010). Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci. Transl. Med. 2, 62ra93. 10.1126/scitranslmed.3001451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann M., Trueb B. (2000). Characterization of a Novel Protein (FGFRL1) from Human Cartilage Related to FGF Receptors. Genomics, 69, 275–279. 10.1006/geno.2000.6332 [DOI] [PubMed] [Google Scholar]
- Wong A., Lamothe B., Lee A., Schlessinger J., Lax I. (2002). FRS2α Attenuates FGF Receptor Signaling by Grb2- Mediated Recruitment of the Ubiquitin Ligase Cbl. Proc. Natl. Acad. Sci. U.S.A., 99, 6684–6689. 10.1073/pnas.052138899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Zhang C., He C., Qian D., Lu L., Sun Y., et al. (2021). Discovery of Pemigatinib: A Potent and Selective Fibroblast Growth Factor Receptor (FGFR) Inhibitor. J. Med. Chem. 64, 10666–10679. 10.1021/acs.jmedchem.1c00713 [DOI] [PubMed] [Google Scholar]
- Wu Y.-M., Su F., Kalyana-Sundaram S., Khazanov N., Ateeq B., Cao X., et al. (2013). Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 3, 636–647. 10.1158/2159-8290.Cd-13-0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Wu K., Wang Y., Pan Y., Chen B., Cheng D., et al. (2020). Discovery of 4,6-pyrimidinediamine Derivatives as Novel Dual EGFR/FGFR Inhibitors Aimed EGFR/FGFR1-positive NSCLC. Eur. J. Med. Chem. 187, 111943. 10.1016/j.ejmech.2019.111943 [DOI] [PubMed] [Google Scholar]
- Xu J. M., Wang Y., Chen Y. L., Jia R., Li J., Gong J. F., et al. (2017). Sulfatinib, a Novel Kinase Inhibitor, in Patients with Advanced Solid Tumors: Results from a Phase I Study. Oncotarget 8, 42076–42086. 10.18632/oncotarget.14942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Meng X., Xu N., Fu W., Tan H., Zhang L., et al. (2018). Gambogenic Acid Inhibits Fibroblast Growth Factor Receptor Signaling Pathway in Erlotinib-Resistant Non-small-cell Lung Cancer and Suppresses Patient-Derived Xenograft Growth. Cell Death Dis 9, 262. 10.1038/s41419-018-0314-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamani A., Zdżalik-Bielecka D., Lipner J., Stańczak A., Piórkowska N., Stańczak P. S., et al. (2021). Discovery and Optimization of Novel Pyrazole-Benzimidazole CPL304110, as a Potent and Selective Inhibitor of Fibroblast Growth Factor Receptors FGFR (1-3). Eur. J. Med. Chem. 210, 112990. 10.1016/j.ejmech.2020.112990 [DOI] [PubMed] [Google Scholar]
- Yang G.-W., Jiang J.-S., Lu W.-Q. (2015). Ferulic Acid Exerts Anti-angiogenic and Anti-tumor Activity by Targeting Fibroblast Growth Factor Receptor 1-Mediated Angiogenesis. Int. J. Mol. Sci., 16, 24011–24031. 10.3390/ijms161024011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Liang S.-Q., Yang H., Xu D., Bruggmann R., Gao Y., et al. (2021). CRISPR-mediated Kinome Editing Prioritizes a Synergistic Combination Therapy for FGFR1-Amplified Lung Cancer. Cancer Res. 81, 3121–3133. 10.1158/0008-5472.Can-20-2276 [DOI] [PubMed] [Google Scholar]
- Yin B., Fang D.-M., Zhou X.-L., Gao F. (2019). Natural Products as Important Tyrosine Kinase Inhibitors. Eur. J. Med. Chem., 182, 111664. 10.1016/j.ejmech.2019.111664 [DOI] [PubMed] [Google Scholar]
- Yoshimoto Y., Sasaki Y., Murata K., Noda S.-e., Miyasaka Y., Hamamoto J., et al. (2020). Mutation Profiling of Uterine Cervical Cancer Patients Treated with Definitive Radiotherapy. Gynecol. Oncol. 159, 546–553. 10.1016/j.ygyno.2020.08.020 [DOI] [PubMed] [Google Scholar]
- Yu J., Mahipal A., Kim R. (2021). Targeted Therapy for Advanced or Metastatic Cholangiocarcinoma: Focus on the Clinical Potential of Infigratinib. Onco Targets Ther., 14, 5145–5160. 10.2147/OTT.S272208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P., Wilhelm K., Dubrac A., Tung J. K., Alves T. C., Fang J. S., et al. (2017). FGF-dependent Metabolic Control of Vascular Development. Nature 545, 224–228. 10.1038/nature22322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Han L.-l., Du F., Liu X.-m., Li J., Wang H.-h., et al. (2019). FGFR1 Induces Acquired Resistance against Gefitinib by Activating AKT/mTOR Pathway in NSCLC. Onco Targets Ther. 12, 9809–9816. 10.2147/ott.S220462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lv H., Zhou Q., Elkholi R., Chipuk J. E., Reddy M. V. R., et al. (2014). Preclinical Pharmacological Evaluation of a Novel Multiple Kinase Inhibitor, ON123300, in Brain Tumor Models. Mol. Cancer Ther. 13, 1105–1116. 10.1158/1535-7163.MCT-13-0847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhang Y. (2015). Bladder Cancer and Genetic Mutations. Cell Biochem Biophys, 73, 65–69. 10.1007/s12013-015-0574-z [DOI] [PubMed] [Google Scholar]
- Zhao B., Li Y., Xu P., Dai Y., Luo C., Sun Y., et al. (2016). Discovery of Substituted 1H-Pyrazolo[3,4-B]pyridine Derivatives as Potent and Selective FGFR Kinase Inhibitors. ACS Med. Chem. Lett. 7, 629–634. 10.1021/acsmedchemlett.6b00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G., Li W.-Y., Chen D., Henry J. R., Li H.-Y., Chen Z., et al. (2011). A Novel, Selective Inhibitor of Fibroblast Growth Factor Receptors that Shows a Potent Broad Spectrum of Antitumor Activity in Several Tumor Xenograft Models. Mol. Cancer Ther. 10, 2200–2210. 10.1158/1535-7163.MCT-11-0306 [DOI] [PubMed] [Google Scholar]
- Zhavoronkov A., Ivanenkov Y. A., Aliper A., Veselov M. S., Aladinskiy V. A., Aladinskaya A. V., et al. (2019). Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 37, 1038–1040. 10.1038/s41587-019-0224-x [DOI] [PubMed] [Google Scholar]
- Zhong L., Li Y., Xiong L., Wang W., Wu M., Yuan T., et al. (2021). Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Sig Transduct Target. Ther. 6, 201. 10.1038/s41392-021-00572-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Hur W., McDermott U., Dutt A., Xian W., Ficarro S. B., et al. (2010). A Structure-Guided Approach to Creating Covalent FGFR Inhibitors. Chem. Biol. 17, 285–295. 10.1016/j.chembiol.2010.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Ma D., Wang Y. (2019). The PROTAC Technology in Drug Development. Cell Biochem Funct, 37, 21–30. 10.1002/cbf.3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.