

Abstract
The contribution of domestic cattle in human societies is enormous, making cattle, along with other essential benefits, the economically most important domestic animal in the world today. To expand existing knowledge on cattle domestication and mitogenome diversity, we performed a comprehensive complete mitogenome analysis of the species (802 sequences, 114 breeds). A large sample was collected in South‐east Europe, an important agricultural gateway to Europe during Neolithization and a region rich in cattle biodiversity. We found 1725 polymorphic sites (810 singletons, 853 parsimony‐informative sites and 57 indels), 701 unique haplotypes, a haplotype diversity of 0.9995 and a nucleotide diversity of 0.0015. In addition to the dominant T3 and several rare haplogroups (Q, T5, T4, T2 and T1), we have identified maternal line in Austrian Murbodner cattle that possess surviving aurochs’ mitochondria haplotype P1 that diverged prior to the Neolithization process. This is convincing evidence for rare female‐mediated adaptive introgression of wild aurochs into domesticated cattle in Europe. We revalidated the existing haplogroup classification and provided Bayesian phylogenetic inference with a more precise estimated divergence time than previously available. Occasionally, classification based on partial mitogenomes was not reliable; for example, some individuals with haplogroups P and T5 were not recognized based on D‐loop information. Bayesian skyline plot estimates (median) show that the earliest population growth began before domestication in cattle with haplogroup T2, followed by Q (~10.0–9.5 kyBP), whereas cattle with T3 (~7.5 kyBP) and T1 (~3.0–2.5 kyBP) expanded later. Overall, our results support the existence of interactions between aurochs and cattle during domestication and dispersal of cattle in the past, contribute to the conservation of maternal cattle diversity and enable functional analyses of the surviving aurochs P1 mitogenome.
Keywords: aurochs introgression, cattle, diversity, domestication, mitogenome, phylogenetics
1. INTRODUCTION
The domestication of animals and plants is recognized as one of the most influential processes that has shaped the development and growth of human civilization (Larson et al., 2014; Zeder, 2006). The contribution of domestic cattle, which are divided into two major subspecies, the taurine or humpless cattle (Bos taurus taurus) and the indicine (Zebu) or humped cattle (Bos taurus indicus), to human societies has been enormous because cattle are able to convert large amounts of roughage into high‐quality food, while other benefits (draught power, transport, leather, manure as fertilizer, dry dung for fuel and use of other cattle products) are also substantial (Sherratt, 1983). All modern taurine and indicine cattle are descended from the extinct large‐bodied wild aurochs (Bos primigenius), which was widespread throughout much of Eurasia and northern Africa during the Pleistocene and early Holocene (Zeuner, 1963).
Taurine cattle were domesticated from Bos primigenius primigenius around 10.5 kiloyears before present (kyBP) in the Fertile Crescent (Helmer et al., 2005), while indicine cattle were domesticated from Bos primigenius namadicus about 1500 years later in the Indus Valley in present‐day Pakistan (Meadow, 1993).
In domestic animals and their close relatives, mtDNA sequences were the first source of molecular information used in the early days of ancient DNA analyses. Together with sequences obtained contemporarily, they provided seminal evidence that complemented archaeological findings and shaped our initial understanding of cattle domestication (Bailey et al., 1996; Bradley et al., 1996; Loftus et al., 1994; Mannen et al., 1998). So, the first maternal evidence from the D‐loop mtDNA showed a strong bifurcation between two geographically distinct T and I haplogroups corresponding to the two main subspecies, taurine and indicine cattle, respectively. Since the T and I haplogroups diverged long before domestication (Troy et al., 2001), this was interpreted as genetic evidence of two independent domestication events. Later analyses of complete mitogenomes in modern cattle revealed the existence of other pre‐domestication P, R, Q and C (the later present only in ancient samples from China), and significant diversity within T, so‐called ‘mega T’ haplogroup (T1, T2, T3, T4, T5 and T6), indicating the complex pattern of the domestication process (Achilli et al., 2009; Bonfiglio et al., 2010; Olivieri et al., 2015; Xia et al., 2019; Zhang et al., 2013). Interestingly, haplogroup P, found primarily in ancient bones of large European aurochs pre‐dating the Neolithic, has rarely been found in North‐east Asian cattle and never in contemporary European breeds. The absence of haplogroup P in modern European cattle has been the main argument that domesticated cattle did not interbreed with wild aurochs during Neolithization (Edwards et al., 2007). This contrasts with the pattern supported by research in pigs, sheep and horses that domestication has been gradual with continuous interbreeding between domesticated and wild forms (Frantz et al., 2020; Larson & Burger, 2013).
Turning to the Y chromosome, the presence of three different male‐specific haplogroups, the geographically distinct Y1 at North‐west Europe and Y2 at South‐east Europe, both found in taurine cattle, and the presence of the specific Y3 found in indicine cattle and crosses between African taurine and indicine cattle (Chen et al., 2018; Pérez‐Pardal et al., 2010, 2018), does not contrast with the male‐mediated introgression of northern and central European aurochs with haplogroup Y1 into domesticated cattle.
Large‐scale analyses based on high‐throughput genome‐wide nuclear markers show that modern cattle diversity is structured into three major groups representing Eurasian taurine, African taurine and Asian indicine cattle, as well as various types of crosses, all combinations and intricacies, among these three groups (Decker et al., 2014; Kim et al., 2020; McTavish et al., 2013). Chen et al. (2018) showed that indicine cattle can be further subdivided into Indian and Chinese indicines. Recently, genomic analyses of ancient bones have confirmed that interbreeding of local and geographically distinct aurochs with modern cattle has contributed to the genetic diversity of modern cattle (Park et al., 2015; Verdugo et al., 2019). Thus, all the results obtained show that the domestication of cattle is a very complex process that, although well studied, still leaves a number of questions unanswered (Larson & Burger, 2013). A good example is bovine mitogenomics, where there are still a number of open questions concerning the occurrence of rare haplogroups, their geographic and migratory origins in relation to wild ancestors and their demographic patterns.
The main objectives of this study were (i) to provide a more complete description of the classification and phylogeny of rare haplotypes, including the aurochs‐specific P haplogroup, and (ii) to reconstruct demographic events that led to the present haplogroup distributions. To achieve this, we performed a comprehensive complete mitogenome analysis of cattle (>800 mitogenomes—114 known breeds), with a large number of samples collected in South‐east Europe, an important agricultural gateway to Europe during the Neolithization and known for its biodiversity of livestock (Papachristou et al., 2020), which was under‐represented in previous complete mitogenome studies. Overall, our analyses focused (i) on estimating mitogenome diversity in modern cattle, (ii) on identifying a large number of rare haplotypes, (iii) on critically comparing the existing haplogroup classification and the classification obtained by complex Bayesian phylogenetic inference, (iv) on the construction of the Bayesian phylogenetic tree with a precise estimate of the divergence time, (v) on the evaluation of the haplogroup classification using the complete mitogenome versus partial mtDNA sequences and (vi) on the estimation of the historical maternal effective population size.
2. MATERIALS AND METHODS
2.1. Identity and origin of samples
In almost all analyses, our work was based on 809 complete mitogenomes, including five aurochs and seven bison (used only in the data set to calibrate the time clock), taken from GenBank. Of the 797 complete mitogenomes representing modern cattle, we directly sequenced 385 animals sampled mainly in central and South‐east Europe (GenBank accession numbers from MZ901375 to MZ901759). Through data mining, we also obtained 116 mitogenomes, while 296 mitogenomes were from GenBank. In some analyses, we also used an additional 189 D‐loop sequences (from np 16,042 to np 16,276) obtained from ancient carbon‐dated bones. In constructing the large median‐joining network (MJN) based on 247 mitogenomes, we also used 47 recently published mitogenomes that were not included in other analyses. To confirm maternal inheritance of the rare haplogroup found in Austrian Murbodner bull Hugo, we additionally sampled four cows related through the maternal lineage, while all living animals in this maternal lineage were identified from the pedigree data using MaGelLAn 1.0 software (Ristov et al., 2016). A T3 mitogenome obtained from 11.5 kyBP ancient bone (Lari et al., 2011), only a few mutations away from the reference sequence, was removed from the analyses because phylogenetic modelling by the algorithm BEAST was rather unstable with this sequence, indicating possible contamination. Detailed information on all mitogenome sequences used in this study, along with their corresponding accession numbers, can be found in Tables S1 and S2.
2.2. DNA extraction and library preparation for mitogenome sequencing
The commercially available reagent kit DNeasy Blood & Tissue Kit (Qiagen, Germany) was used to extract DNA from bovine samples (milk, hair roots and tissue) according to the manufacturer's instructions. The experiments reported in this work were in accordance with the ethical guidelines of the national legislation.
Amplification of the whole bovine mitogenome (~16,340 bp) was performed by polymerase chain reaction (PCR) in three overlapping fragments according to a modified protocol (Horsburgh et al., 2013). Primer pairs used are provided in Table S3, and PCR protocols applied are summarized in Table S4. The amplified three mitogenome fragments of each sample were transferred to a tube and purified according to the Wizard® SV gel and PCR Clean‐Up System Protocol (Promega).
Purified long‐range PCR products were diluted to approximately 1 ng/μl and subsequently quantified using the Qubit dsDNA HS Assay (Invitrogen).
PCR products were diluted to yield 0.2 ng/μl per sample, and a total amount of 1 ng per sample was used for library preparation. Libraries were prepared using the Nextera® XT DNA Sample Preparation Kit (Illumina) according to the manufacturer's instructions. The dsDNA was incubated with a preloaded Tn 5 transposase to generate tagged fragments and then amplified with barcoded primers to generate the final library. Amplified libraries were purified using Ampure XP beads (Beckman‐Coulter) and quantified by absorbance measurement using a NanoDrop spectrophotometer (ND‐1100; Thermo). Equal amounts of libraries were pooled, and the final pool was analysed and quantified on a Bioanalyser DNA1000 chip (Agilent). The pooled libraries were sequenced in dual‐indexed 100 bp paired‐end mode on the HiSeq 1500 System (Illumina) at Gene Center, Ludwig Maximilians University (LMU), Munich, Germany.
2.3. Primary data analysis
Primary data analysis was performed on the LMU system Galaxy Server using a workflow for demultiplexing, mapping and variant calling. Read pairs were demultiplexed using the dual indexes of Nextera XT Library Preparation Kit (Illumina). Reads were mapped to the mitochondrial sequence of the bovine reference mitogenome (GenBank accession number V00654) using the Burrows–Wheeler Alignment (BWA) tool (Li & Durbin, 2009), duplicates were removed, and variants were called using VarScan (Koboldt et al., 2012). Variants were replaced in the mitochondrial genome fasta file, and mapping and calling were repeated until no further variants could be called and no bases without coverage remained. The resulting mitochondrial genome fasta files were used for further phylogenetic analysis. The mean depth and width of mitogenome coverage was calculated using BBmap (Bushnell, 2014) and is shown in Table S5.
To confirm the results of the next‐generation sequencing (NGS) method described above and the presence of rare haplotypes, all four maternal relatives (Esta, Emma, Ella and Elke) of bull Hugo were additionally sequenced on 780‐bp‐long D‐loop sequences (Achilli et al., 2008) in both directions using the Sanger method on the ABI Prism® 310 Platform Genetic Analyzer (Applied Biosystems).
Whole‐genome sequences were downloaded from online databases and converted to Fastq files (Fastqsanger format) using NCBI Sequence Read Archive (SRA) tools (Leinonen et al., 2010). The Fastq files were mapped to a reference mitogenome (GenBank accession number V00654) using the BWA (Li & Durbin, 2009). The following workflow was set in the Galaxy platform (Afgan et al., 2018): the mapped file obtained with BWA‐MEM was processed with ‘Filter SAM or BAM’ (Galaxy vs. 1.1.29) to keep only the reads mapped to the mitochondrial genome (Li et al., 2009), duplicate molecules were removed with ‘MarkDuplicates’ (Galaxy vs. 2.7.1.1) (Broad Institute), and reads were piled up using MPileup multi‐way pileup of variants (Galaxy vs. 2.1.3) (Li, 2011a, 2011b; Li & Durbin, 2009; Li et al., 2009) and using VarScan (Koboldt et al., 2012) for variant detection (Galaxy vs. 0.1) and LMU in‐house tool Fasta SNP Replacer (Galaxy Tool vs. 1.0.0). Mapping results were reviewed using the Integrative Genomics Viewer software (Robinson et al., 2011). Consensus sequences were subjected to phylogenetic analysis. To confirm the results of the method described above, mitogenome of one animal was optioned both from whole‐genome sequencing data and from long‐range PCR products.
All mitogenomes were aligned using the Clustal Omega program (Sievers et al., 2011) imported into MEGA 7 software (Kumar et al., 2016), whereas partial mitogenome sequences were aligned using the ClustalW program (Kumar et al., 2016). The complete mitogenome data alignment was further divided into functional regions: (i) 13 regions encoding proteins, (ii) two regions encoding ribosomal RNA, (iii) one region encoding the D‐loop and (iv) one region representing encoded 22 tRNAs. Stop codons were removed from the partitions (regions) encoding proteins.
2.4. Mitogenome diversity and haplotype classification
Five ancient bovine complete mitogenome sequences were excluded from the diversity data set (797 mitogenomes) because we wanted to estimate contemporary diversity (excluding ancient diversity lost over time), while analyses were performed separately for complete mitogenomes (consensus 16,230 bp) and 17 functional regions defined as described above. Mitogenome diversity was presented by percentage of polymorphic sites to total size (S%), number of haplotypes (nH), haplotype diversity (HD) and nucleotide diversity (π), all calculated by DnaSP v6 (Rozas et al., 2017) and Arlequin v. 3.5.2.2 software packages (Excoffier & Lischer, 2010). The complete mitogenomes and D‐loop sequences (239 bp; from np 16,042 to np 16,276 plus five indels) were classified using the MitoToolPy application (Peng et al., 2015) and by the Bayesian phylogenetic inference.
To visualize the relationships among P haplotypes, after excluding sites with alignment gaps, we constructed a small MJN (Bandelt et al., 1999) by PopART (Leigh & Bryant, 2015) on a small number of representative mitogenome sequences and five ‘Hugo family’ mitogenomes. We also constructed a large MJN containing 247 mitogenomes (Figure S1) to further elucidate the mutational relationship between haplotypes and haplogroups within the entire phylogenetic network.
2.5. Phylogenetic and demography analyses
After confirming the absence of recombination using two algorithms implemented in the HyPhy software (Kosakovsky Pond et al., 2005, 2006a, 2006b), see Table S6, we proceeded with Bayesian phylogenetic inference model testing using the software package BEAST v1.4.3 (Drummond et al., 2006).
Testing of the model and calibration of the tree root was performed on the selected set with 47 mitogenomes: 36 sequences representing modern cattle haplogroups (I, R, P, Q, T1‐7), four sequences representing ancient Bison priscus, two sequences representing outlying Bison bison, and five sequences representing ancient Bos primigenius primigenius (aurochs). The program was run with Markov chain Monte Carlo length until the values of the effective sample size (ESS) exceeded 200. Calculations were performed using various substitution models: Generalize times reversible (GTR; Lanave et al., 1984), Hasegawa–Kishino–Yano (HKY; Hasegawa et al., 1985), Jukes–Cantor 1969 (JC69; Jukes & Cantor, 1969), Kimura (K80; Kimura, 1980) and Tamura–Nei (TN93; Tamura & Nei, 1993). The strict molecular clock, relaxed lognormal and exponential molecular clocks were tested (Drummond et al., 2006), and different population size priors were applied: coalescent constant, coalescent exponential and coalescent Bayesian skyline model. The different phylogenetic models were analysed in Tracer v1.6 based on a comparison of Akaike's information criterion values obtained by Markov chain Monte Carlo (AICM, see Raftery et al., 2007). The BEAST analysis was run using priors obtained from the previous analysis for the best‐fitting model: the GTR substitution model, the strict molecular clock and the coalescent Bayesian skyline tree prior (Table S7). To obtain an accurate tree topology, a total of 802 sequences (including five aurochs with information on their estimated time) were imported into BEAUti v1.4.3 (Drummond et al., 2012) and set to a best‐fitting model. The TreeHigh prior was set to 218.9 kyBP (95% HPD 316.6–136.6 kyBP) and the normal distribution with a standard deviation of 52.0 kyBP to be compatible with the calibration tree. The given trees file was compiled in TreeAnnotator v2.4.7 with burn‐in of 20%, while the most recent common ancestor tree was constructed in FigTree v1.4.3 (Drummond et al., 2012).
To estimate the population history, the Bayesian skyline plot (BSP) was calculated in Tracer v1.7.1 software (Rambaut et al., 2018). A D‐loop data set containing modern and calibrated ancient sequences was split into four different data sets representing different haplogroups Q, T2, T3 and T1. Each data set representing an individual haplogroup was run in BEAST v2.5 (Bouckaert et al., 2019) using the GTR substitution model, strict molecular clock and coalescent Bayesian skyline.
3. RESULTS
We have conducted a comprehensive, large‐scale complete mitogenome analysis in cattle that has greatly expanded the available mitogenome information by adding 501 new sequences representing 58 additional breeds. Our mitogenome information has largely focused on Europe. In addition, we obtained a large number of samples from the South‐east Europe, as this region is the main agricultural gateway to Europe during Neolithization and has not been well covered in previous analyses (Table S1). Our results are also representative of modern cattle from Africa and the West and East Asia, as mitogenomes from these regions were well represented in our data set but do not cover well indicine cattle from Asia and taurine cattle from South‐East Asia.
3.1. Mitogenome diversity and MitoToolPy haplotype classification
Mitogenome diversity (S%, nH, HD and π) for different functional regions of 797 modern cattle representing 114 breeds is shown in Figure 1. In total, we found 1725 polymorphic sites (S% = 10.6%), including 810 singletons, 853 parsimony‐informative sites and 57 indels. There were also 1563 transitions, 159 transversions and 1678 substitutions, while 701 unique haplotypes were observed with a HD of 0.9995 and a π of 0.0015.
FIGURE 1.
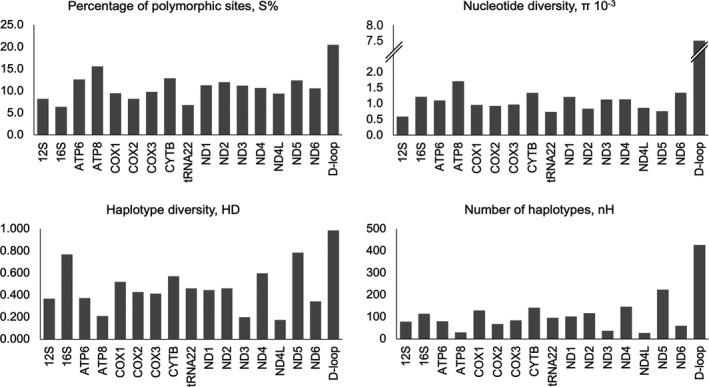
Mitogenome diversity in modern cattle (797 sequences) across different functional regions
Mitogenome diversity varied among the different functional regions (Figure 1), with the highest diversity observed in the D‐loop region (S% = 20.5, nH = 426, HD = 0.986 and π = 0.0080), followed by ATP8 (S% = 15.6), ND5 (nH = 224), ND5 (HD = 0.784) and ATP8 (π = 0.0017). In contrast, the lowest diversity was observed in 16S (S% = 6.4), ND4L (nH = 27), ATP6 (HD = 0.199) and 12S (π = 0.0006).
Mitogenomes of modern cattle (797) were classified into I (10), R (10), P‐ (6), Q (22), T5 (10), T2 (42), T3 (528), T1 (154), T4 (11), T6 (3) and T7 (1) haplogroups using the MitoToolPy algorithm. In Austrian Murbodner cattle originating from the Alpine Murtal valley, we found the aurochs‐specific haplotype P in a bull named Hugo (Figure 2a). The presence of haplotype P in Murbodner cattle is the first evidence for the survival of mitochondria of ancient northern and central European aurochs, genotyped as haplogroup P in many ancient aurochs’ bones (Ajmone‐Marsan et al., 2010; McHugo et al., 2019), in modern European cattle.
FIGURE 2.
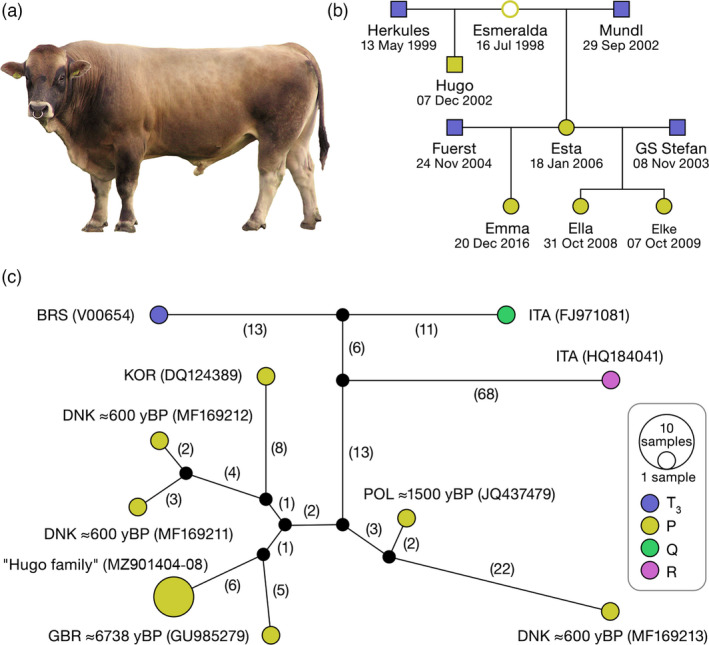
Presentation of the Murbodner cattle “Hugo family” with surviving aurochs P haplogroup mitochondria. (a) Photo of the bull named Hugo first identified with P haplotype mitogenome. (b) Reduced family tree of the members of the “Hugo family”, in which the positions of the individuals sequenced with haplotype P are marked as full yellow squares or circles, (c) Median‐joining network (mitogenome length of 16,344 bps) representing the relationship between P and few other haplogroups (R, Q and T3). Abbreviations: BRS, Bovine Reference Sequence (T3); DNK, Denmark; GBR, Great Britain; ITA, Italy; KOR, Korea; POL, Poland are associated with declared origin of sequences
This evidence, the identification of the P haplotype, was confirmed by mitogenome (NGS) and D‐loop Sanger sequencing of four cows (Esta, Ella, Emma and Elke) related to Hugo (Figure 2b). Pedigree analysis, the identification of pedigree members within the same maternal lineages, revealed that 99 animals born between 1991 and 2015 belonged to the maternal P‐lineage (Figure S2). Until recently, only several ancient or historical P haplotypes were documented in GenBank: British aurochs Pre‐Neolithic (6.7 kyBP; GU985279); historical Polish aurochs (1.5 kyBP; JQ437479); three historical ‘Danish’ aurochs (MF169211, MF169212 and MF169213; all 0.6 kyBP); and one modern cattle (Korean beef cattle sample DQ124389). After all our mitogenome analyses were completed, 14 individuals with haplotypes several mutations away from the Korean sequence DQ124389 were found in the Japanese Shorthorn (Mannen et al., 2020) and one in Chinese (Yanbian) individual (Xia et al., 2021). In the MJN (Figure 2c and Figure S1), the P haplotype found in Murbodner cattle (MZ901404‐08) was closest (11 mutations) to the reference Pre‐Neolithic aurochs (GU985279).
3.2. Bayesian phylogenetic inference and haplotype classification
Bayesian phylogenetic tree inference performed on 802 mitogenomes (modern cattle with five chronologically distant aurochs) is shown in Figure 3, while the posterior probabilities with estimated divergence time of major haplogroups (12 nodes), calculated from the calibration tree (Figure S3), are provided in Table 1. According to the results, the GTR substitution model and strict molecular clock with Bayesian skyline tree prior had the lowest AICM value, while the tree root for genus Bos was 218.9 kyBP (Figure S1). Diversification between genus Bos and Bison occurred 965.3 kyBP (95% HPD 1348.0–600.4 kyBP, posterior probability = 1). A more detailed phylogenetic tree representing the phylogenetic relationship of all individual mitogenomes is presented as a zoomable user interface (Zoom Figure S4 and its digitally formatted file, Figure SD1: Data S1, readable by the FigTree graphical viewer).
FIGURE 3.
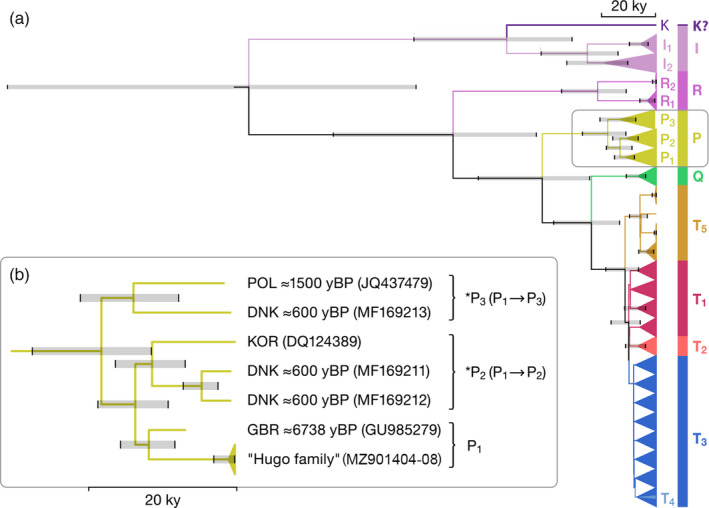
The Bayesian phylogenetic tree performed on 802 complete mitogenome cattle sequences. (a) Complete phylogenetic tree (b) Partial phylogenetic tree representing P haplotypes (DNK, Denmark; GBR, Great Britain; KOR, Korea; POL, Poland)
TABLE 1.
Haplogroup divergence time (DT) in kiloyears before present (kyBP) according to the Bayesian Markov chain Monte Carlo calibration tree with corresponding node numbers (Figure S3)
| Haplogroup divergence time | Median | 95% HPD a | Posterior probability |
|---|---|---|---|
| Node 1: I–[R–P–Q–T] | 218.9 | 316.6–136.6 | 1.00 |
| Node 2: I1–I2 | 21.2 | 46.2–10.3 | 1.00 |
| Node 3: R–[P–Q–T] | 102.7 | 149.9–63.7 | 1.00 |
| Node 4: R1–R2 | 28.5 | 46.2–14.6 | 1.00 |
| Node 5: P–[Q–T] | 56.8 | 83.9–35.0 | 1.00 |
| Node 6: P3–[P1–P2] | 21.1 | 31.4–13.2 | 1.00 |
| Node 7: P1–P2 | 15.9 | 23.6–10.3 | 0.98 |
| Node 8: Q–T | 33.5 | 51.0–20.4 | 1.00 |
| Node 9: Q1–Q2 | 11.8 | 18.2–6.4 | 1.00 |
| Node 10: T5–[T1–T2–T3–T4] | 18.2 | 27.5–11.4 | 1.00 |
| Node 11: T2–[T1–T3–T4] | 16.3 | 24.0–10.2 | 0.95 |
| Node 12: T1–T3 | 14.7 | 21.2–8.8 | 0.60 |
The highest posterior density pointing to 95% posterior credible intervals.
The divergence of haplogroups T5 and T2 from T1 to T4 was supported, whereas the divergence between haplogroups T1 and T3 was not supported by the derived Bayesian posterior probabilities (node 12 in Table 1). Thus, as a consequence of the low divergence, the assignment between T3 and T1 could be misguided, especially for the incomplete and uninformative sequences obtained from ancient samples. However, the geographically specific distribution of T1 in a large number of African cattle breeds is a solid argument to consider T1 as a separate haplogroup, while the process of its further diversification is unstoppable.
Divergence of haplogroup T4 from T3 was supported only on the hierarchically lower branch (not supported in the calibration analysis with 47 sequences), which diverged with a posterior probability of 0.95, and was positioned after several branches with very low posterior probabilities (Figure SD1: Data S1). At the same time, haplogroup T4 was clustered with a posterior probability of 1, along with individuals sampled in Korea and placed within the T4 cluster in MJN (Figure S1). The presence of a Middle Albanian Buša individual within the T4 mitogenome was therefore surprising, as T4 is considered an Asian‐specific haplotype that is most common in modern Japanese and Korean cattle (Figures S1 and S4). Recently, T4 mitogenomes were identified from ancient bones in China, which are about 3.9 kyBP old (Xia et al., 2021). In conclusion, given the geographic specificity, the frequent use in the literature and the logic followed in the case of T1, the recognition of T4 as a separate haplogroup is an acceptable decision.
On the contrary, we did not find a strong argument to support T6 and T7 as separate haplogroups, since their occurrence is extremely rare, and it is unlikely (low posterior probability) that they branch from T3 (Figure SD1: Data S1). Three sequences (AY676867, AY676870 and SRS629688) from Angus cattle classified as T6 were very similar or identical to the T6 mitogenome found in Xia et al. (2019) if we neglect the sites with alignment gaps (Figure S1). Interestingly, haplotypes T6 and T7 were located in a separate branch between haplogroups T3 and T2 along with seven haplotypes (JN817319, JN817306 and EU177841 from Italy; MZ901433, MZ901461 and MZ901447 from Croatia; and KT184470 from Egypt) classified as T1 by the MitoToolPy algorithm. For example, haplotype T7, previously found in Cabannina cattle in Italy (EU177840), was not reproduced by the diagnostic mutations (Achilli et al., 2008), where it was classified as T1′2′3 haplotype, nor by the BEAST algorithm, where the posterior probability was 0.4 (for more details, see Figure SD1: Data S1). Certainly, more information, sequences from ancient DNA and work are needed to clarify the exact classification of this entire cluster.
In addition to other known haplogroups, Bayesian inference revealed P1, P2 and P3 as separate haplogroups, with the estimated divergence time between P1–P2 and P3 ranging from 31.3 to 13.2 kyBP, and between P1 and P2 ranging from 23.6 to 10.3 kyBP (Table 1). Thus, Murbodner cattle together with British Pre‐Neolithic aurochs (GU985279) were assigned to P1, whereas Korean cattle (DQ124389) and two Scandinavian aurochs (MF169211 and MF169212) sequenced from 0.6‐kyBP‐old horns (Bro‐Jørgensen et al., 2018) were assigned to haplogroup P2 (Table 1 and Figure 3b). The Wild Polish aurochs (JQ437479), sequenced from 1.5‐kyBP‐old bone (Zeyland et al., 2013), and the Scandinavian aurochs (MF169213), sequenced from 0.6‐kyBP‐old horn (Bro‐Jørgensen et al., 2018), were assigned to haplogroup P3 (Figure 3b). Both estimated divergence times, between P1‐P2 and P3 and between P1 and P2, occurred before the domestication of cattle, as it is reasonable to assume that by 10.3 kyBP (lower 95% HPD limit of estimated divergence between P1 and P2) P aurochs were widespread, far from Fertile Crescent (Table 1).
A sequence (DQ124403), found in Korean Holstein cattle, diverged from haplogroup I and may be evidence of a new haplogroup ‘K’ descended from Bos primigenius namadicus (Figure 3a and Figure S4). Because we did not have access to sequencing quality parameters or access to their biological relatives (sequence taken from GenBank), we made no additional comments or speculations regarding this sequence.
The Bayesian phylogenetic tree obtained by the BEAST algorithms on the complete mitogenomes, even considering the ‘explanatory supported’ T4 or unsupported T6, and T7 haplogroups, was mostly consistent with that obtained by the MitoToolPy algorithms. However, we observed situations where the MitoToolPy classification was not correct, particularly in the classification of T1 and T3 (Table S1).
Even lower agreement was observed when classifications were based on partial mitogenome sequences, such as the D‐loop (Table S1 and Figure S5A). The most drastic example is the misclassification of the historical Scandinavian aurochs (MF169213 with haplotype P, which was incorrectly classified as T3 based on D‐loop information; Table S1 and Figure S3A). A very similar problem was observed for a number of T5 mitogenomes that could not be recognized at all using only the D‐loop information (Table S1 and Figure S5A). Thus, reliable haplogroup classification requires information on the complete mitogenome. Following this logic, haplogroup E, classified on the basis of the partial mitogenome from the 5.8‐kyBP‐old bone (known as EIL4) from the German Early Neolithic site of Eilsleben (Edwards et al., 2007), needs to be confirmed using the complete mitogenome information, which presents another challenge for future ancient DNA analyses.
3.3. Bayesian demographic inference
A BSP was performed for each haplogroup (Q, T2, T3 and T1) on D‐loop, from np 16,042 to np 16,276, sequence information (Figure 4). The analysis was done on 999 sequences, our mitogenomes restricted to partial D‐loop, extended by additional 189 D‐loop sequences obtained from the carbon‐dated bones as number of high‐quality complete ancient mitogenomes is very low. The inferred trend in effective female population size (N ef) for each of the four haplogroups during a period of the last 15 thousand years is shown in Figure 4 (estimates with 95% posterior credibility intervals are shown in Figure S6). The growth of N ef in T2 started before domestication and accelerates from the temperature changes in Bølling–Allerød (warm and humid interstadial period occurring in the last stages of the last ice age, from 14.7 to 12.7 kyBP). However, the steepest growth of T2 (10.0 to 7.5 kyBP) and the sudden growth of Q (from 10.0 to 8.0 kyBP) almost overlap with the timing of archaeological evidence pointing to the beginnings of cattle domestication (Helmer et al., 2005). For both T2 and Q, estimated N ef growth was slowed to ~8.0–7.0 kyBP. Around the same time, we observed a sudden and steep N ef growth in T3 that occurred around ~7.5 kyBP and then slowed down to ~2.5 kyBP. In Africa‐specific haplogroup T1, the increase in N ef growth from ~3.0 to 1.0 kyBP overlaps with the timing of the migration routes of Bantu‐speaking farmers and Cushitic‐ and Nilotic‐speaking pastoralists (Crowther et al., 2018), while it slows down with the assumed arrival date of the Zebu in Africa (Kim et al., 2020).
FIGURE 4.
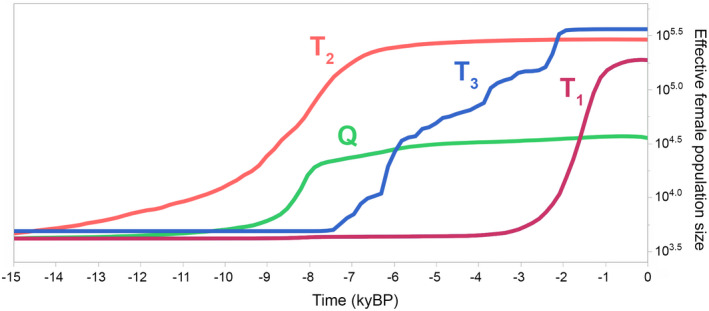
Bayesian skyline plot showing the trend in effective female population size over the last 15 thousand years for haplogroups Q, T2, T3 and T1, inferred from D‐loop sequence information (from np 16,042 to np 16,276)
Alternatively, the earliest undisputed archaeological evidence for zebu cattle in Africa comes from rock carvings of humpbacked cattle from the Horn of Africa, dated to about 1.5 kyBP, which would roughly correspond to the inflection point of the observed T1 growth function (Gifford‐Gonzalez & Hanotte, 2011). Under this assumption and the fact that all African cattle carry the T1 haplotype, it is also possible that T1 growth was promoted by zebu introgression as a result of enhanced adaptation of the paternal zebu and maternal African taurine crosses (which possess T1 haplotypes) to the local environment (high temperatures, prolonged droughts and vector‐borne diseases), which has been discussed in a number of studies related to modern cattle–zebu crosses (Kim et al., 2020; Mbole‐Kariuki et al., 2014; Mwai et al., 2015).
4. DISCUSSION
4.1. Mitogenome diversity
We estimated complete and partial mitogenome diversity in functionally distinct regions and have observed that the estimated complete mitogenome nucleotide diversity (0.0015) is comparable to the genome‐wide nucleotide diversity of 0.0010 to 0.0020 observed in other taurine breeds (Chen et al., 2018; Kim et al., 2017; Weldenegodguad et al., 2019). As expected, the D‐loop was the most diverse functional region, although notable polymorphism was observed in other functional regions. At the same time, the nucleotide diversity of the bovine mitogenome observed in this study is relatively low but within the range of values calculated for other mammals, where π ranges from 0.106 in the southern pocket gopher to 0.0004 in the grey‐browed mouse lemur (Nabholz et al., 2008).
It is worth noting that the presence of nuclear DNA sequences of historical mitochondrial origin or NUMTs (pseudogenes derived from partial mitogenome fragments historically integrated into the nuclear genome of eukaryotes by a horizontal transfer mechanism) could have an impact on the diversity estimates presented (Maude et al., 2019; Schiavo et al., 2020; Tramontin Grau et al., 2020). However, the possibility of such a bias was significantly reduced. Thus, the large number of sequences (385) was performed by the three long‐range PCRs, where the chance of narrowing down the NUMTs is almost eliminated. When controlled by other sequencing methods (Sanger sequencing), we did not detect any divergence (except for a single slip, which was corrected in further analyses). For the 116 sequences obtained by data mining, we cannot exclude the possibility of misentries in a few places caused by overlap of NUMTs and heteroplasmy calls. Such cases might have led to a slight overestimation of the number of observed mitogenomes. However, this effect and the slight overestimation of nuclear diversity could also have been caused by other sequencing errors or tissue‐specific mutations that are not inherited (very low probability). Our visual inspection of potentially suspicious ‘chains of consecutive heteroplasmy calls’ revealed no such case. Moreover, our recombination analysis found no site that would indicate the presence of recombination, a phenomenon characteristic of the nuclear genome. We provided information on the sequences obtained by data mining (Table S1) and sequence quality to inform readers of potentially suspicious sequences (Table S5).
4.2. Complexity of domestication from mitogenome perspective
With the exception of the most common T3, a large number of different haplogroups (Q, T5, T2, T1 and T4) have been identified in South‐east European breeds (Figure 5), which in Europe is only comparable to the variation found in neighbouring Italian breeds. The variation found in South‐east European cattle can be attributed to Neolithic migrations, as haplogroups T3, T2 and Q were already present in Serbia and Greece ~7.0 kyBP (Verdugo et al., 2019). Boskarin and Slavonian Syrmian Podolian cattle, two representatives of the two Podolian clusters (Senczuk et al., 2021), were predominantly assigned to the T3 haplogroup, with the exception of three T5 haplotypes in the latter breed. This result is consistent with the conclusions of Di Lorenzo et al. (2018), who found no mitogenome differences between Podolian cattle from Central Europe and the Balkans and Podolian cattle from Italy. Also, the autosomal diversity of Podolian and neighbouring shorthorn breeds does not support a clear clustering into breed groups (e.g. Papachristou et al., 2020), but rather agrees with the genetic similarity of geographic neighbours, that is the ‘isolation‐by‐distance’ model. However, two samples from Podolian cattle, one from the Boskarin breed and the other from the Calvana breed, were positioned within the ‘central’ T1‐T3 haplotype of the large MJN (Figure S1), along with three ancient samples derived from 7.6‐ to 8‐kyBP‐old bones (found in Greece and Turkey; see Verdugo et al., 2019), providing further evidence for their association with Neolithic migrations.
FIGURE 5.
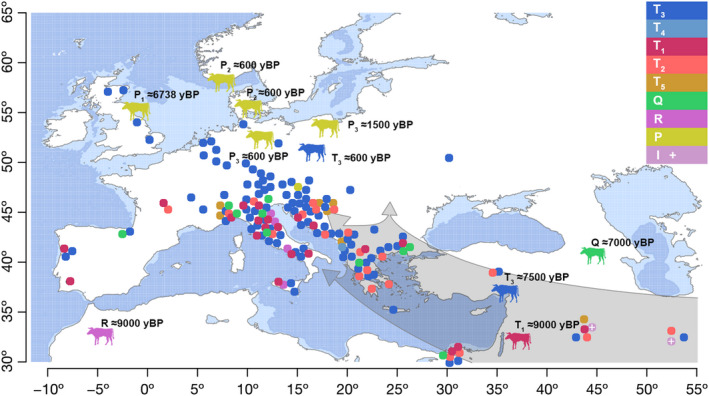
Illustration of the geographical distribution of the analysed mitogenome sequences of European modern cattle breeds with haplogroup assignment according to Bayesian phylogenetic analysis. Aurochs (silhouettes) with sequenced mitogenomes are shown with estimated time of death in years before present (kyBP). Grey shaded arrows indicate agricultural dispersal, while light blue areas show historical sections where grazing may have occurred (sea level 100 m)
The appearance of T1 in South and South‐east European breeds was not surprising, as the migratory flow from African to southern European cattle has already been estimated at the nuclear level (Decker et al., 2014; Senczuk et al., 2021; Upadhyay et al., 2019) or is evident from the presence of R and T1 haplotypes in Italian breeds (Achilli et al., 2008, 2009; Bonfiglio et al., 2010). It is also possible that the observed T1 is the result of direct migrations during Bronze Age or later, as 11 (48%) T1 haplotypes have been found in ancient bone samples from the southern Levant (Verdugo et al., 2019).
The occurrence of six T5 haplotypes in cattle from Croatia and Albania was intriguing, as only four complete mitogenomes with T5 assigned to two Italian and two Iraqi cattle were previously reported (Achilli et al., 2008). Given the divergence time of T5 from T1–T2–T3–T4 (Table 1) and the current geographic distribution of T5 in modern cattle (east and west of the centre of domestication), it is highly likely that haplogroup T5 was sampled during the domestication of the Anatolian aurochs. Note that T5 may have been missed in previous DNA analyses, especially in ancient bone samples, as the D‐loop information is usually insufficient to identify T5. However, the alternative hypothesis of a local origin from the Great Adriatic Plain (on sea‐level dynamics; see Dean et al., 2020) can be tested by analysing aurochs bones found at several interesting archaeological sites in Eastern Adriatic (Lenardić Much et al., 2018; Miracle, 2007).
The appearance of the T4 haplotype in the Middle Albanian Buša was also surprising, and its origin requires further clarification.
While the variation of diverse haplogroups was low in Central European cattle, we found a large maternal lineage of 99 individuals in the Austrian Murbodner cattle, originating from the Alpine valley Murtal, whose mitogenome is assigned to the P haplogroup specific for the large‐boned ancient European aurochs.
Murbodner P maternal lineage was assigned to the P1 haplogroup, together with the mitogenome found in 7.0‐kyBP‐old ancient bones of the Pre‐Neolithic British aurochs (Edwards et al., 2010; Park et al., 2015). This is the most convincing evidence for the sporadic taming of now extinct European wild female aurochs and the embedding of their offspring into domesticated European cattle. Unfortunately, we were unable to estimate the exact time frame, location or context of interbreeding. The taming of wild female aurochs and the inclusion of their offspring in the domesticated cattle stock are complemented by archaeozoological evidence (Schibler et al., 2014), as ancient small cattle bones found in Switzerland (5.0 kyBP), also associated with the Alps, were identified as P. Current explanations for the rare occurrence of P haplotypes in modern cattle remain speculative, for example a selective disadvantage of P mitochondria in warmer environments or a mitonuclear incompatibility (Sharbrough et al., 2017; Zaidi & Makova, 2019). The simplest and most obvious explanation, however, would be that the European and Anatolian aurochs were two well‐differentiated populations before and during the Holocene. Anatolian Neolithic Farmers selected local aurochs for domestication and tried not to lose the hard‐won domestication traits during expansion in Europe. Only in emergencies is there an increased willingness to accept a much greater effort to integrate wild or semi‐wild animals into domesticated populations. A more frequent occurrence of such emergencies is conceivable in fragmented Alpine environments.
The presence of R haplotypes in modern Italian breeds (Achilli et al., 2008) is additional evidence for the interbreeding of domesticated cattle and unidentified female aurochs with R mitogenomes, recently confirmed by the identification of R haplotypes in bones of a 9.0‐kyBP‐old aurochs, probably Bos primigenius africanus, from Morocco (Verdugo et al., 2019). However, it is difficult to speculate on the geographic distribution of R‐haplogroup aurochs on the basis of a single sample, and more information is needed to better understand the link between modern Italian breeds and R‐haplogroup aurochs.
The occurrence of P2 haplotypes in modern cattle in North‐east Asia and Japan assigned to haplogroup P2 (DQ124389 and related sequences) also provides little evidence for the pattern of cattle domestication during Neolithization in Europe. With the exception of Japanese Shorthorn, haplogroup P2 (P1a in Mannen et al., 2020) is extremely rare and has only been found in two ~0.6‐kyBP‐old aurochs’ samples (MF169211 and MF169212) in northern Europe (Figure S1). Interestingly, P2 is very similar to haplogroup T3 at the amino acid level (Figure S5B).
4.3. Bayesian demographic inference
The estimated demographic patterns of Nef obtained by the BSP for Q, T2, T3 and T1 are rather surprising. The observed trend patterns were ‘suspiciously’ consistent with archaeological evidence about the domestication process and with the growth of human populations. According to the observed trend pattern, the domestication of cattle overlaps with the accelerated rise of the T2 haplogroup and was followed by the sudden rise of the Q haplogroup between ~10.0 and ~8.0 kyBP. This is consistent with the known archaeological evidence on the location and timing of beginnings of domestication at Fertile Crescent (~10.5 kyBP) and with the geographic distribution of the T2 and Q haplogroups. Chronologically, this corresponds to general warming trend since the beginning of the Holocene (Isarin & Renssen, 1999). Interestingly, at the time when T2 and Q reached their upper growth limit, the sudden growth of T3 was initiated (by ~7.5 kyBP). We speculate that at the time T3, cattle located at South‐east Europe (see Verdugo et al., 2019), or what is colloquially referred to as the Balkans, began their growth and expanded further westwards. Chronologically, the growth of the T3 haplogroup is consistent with the estimated growth in effective female population size observed in Europeans with haplogroups of Near Eastern origin from the Holocene during the agricultural transition (Gignoux et al., 2011) and with the introduction of agriculture into Central Europe from the Balkans (Pinhasi et al., 2005). In addition, the observed growth of T1 cattle overlaps chronologically, albeit with some divergence, with human female population expansion in Africa (Gignoux et al., 2011), as well as with southward cattle herding spread in Africa (Crowther et al., 2018; Felius et al., 2014; Payne & Hodges, 1997). It is important to note that our T1 N ef trend is also based on T1 haplotypes that were not sampled in Africa and therefore reflect growth of T1 cattle outside Africa, which cannot be separated. Therefore, our discussion is based on the assumption that T1 growth was characteristic of cattle in Africa, while non‐African samples provide information on the flat T1 trend that preceded the observed growth.
Overall, the Bayesian 95% posterior credibility intervals for all N ef trends presented are wide (see Figure S6), and therefore, despite the ‘perfect’ agreement with historical demography and archaeological evidence, all our comments on the estimated N ef trends should be considered as hypotheses that need to be confirmed with additional arguments or/and evidence.
4.4. The impact of aurochs on the modern cattle diversity and adaptation
The initial evidence provided on ancient partial mitogenomes was misleading and did not support the influence of the local aurochs on the evolution and diversity of modern cattle (McHugo et al., 2019), while the presence of a divergent R mitogenome found in Italian cattle (Achilli et al., 2008) was obscured by the fact that R was not observed in any ancient aurochs until recently (Verdugo et al., 2019). Encouragingly, the presence of gene flow from aurochs to modern cattle has been estimated from the whole‐genome sequence of ancient aurochs (Park et al., 2015; Verdugo et al., 2019).
Our study provides unequivocal evidence, albeit based on a ‘single locus’ (mitogenome), for interbreeding of wild female aurochs with domesticated cattle that has been lacking for several decades, complements the findings of Park et al. (2015) and Verdugo et al. (2019), and further supports the complexity of cattle domestication, where the diversity of modern cattle reflects local adaptations of ancient aurochs populations. Thus, cattle join other domesticated species such as pigs, sheep and horses, where evidence supports gradual domestication with continuous interbreeding between domesticated and wild forms (Frantz et al., 2020; Larson & Burger, 2013). Genetic introgression within the Bos species complex appears to be a common mode of adaptation to substantially different environments (Wu et al., 2018), as introgression has been observed between taurine cattle and yak (Bos grunniens) (Medugorac et al., 2017; Xia et al., 2021) and between indicine cattle and banteng (Bos javanicus) (Chen et al., 2018). Furthermore, Medugorac et al. (2017) link periods of intense introgression to times of crisis that forced yak herders to breed all available females to restore their herds, including otherwise unwanted females obtained through backcrossing. Long‐term survival and sustainable development depend on a diversity of variants and options that can be combined to adapt to new environments. Genetic diversity represents a unique ‘information bank’ that ultimately determines the potential for life to evolve with the abiotic component of the ecosystem in the most resilient manner possible (e.g. Steffen et al., 2015). Adaptation to a changed or new environment occurs much more rapidly when an appropriate functional solution already exists in a related sympatric population. Therefore, adaptive introgression of such functional traits is a proven evolutionary solution for sustainable adaptation and long‐term survival. Given the complex and polygenic architecture of adaptation and domestication traits (Carneiro et al., 2014; Larson & Burger, 2013; Medugorac et al., 2017), we propose adaptive introgression as an important evolutionary force during the expansion of domesticated cattle into European environments.
Domestication and subsequent expansion exposed domestic animals to many new environmental conditions, including greater temperature extremes, different pathogens and nutrients. Around the time of expansion, a presumably large aurochs population existed in Europe (Wright & Viner‐Daniels, 2015). Rare intentional or unintentional hybridization thus increased the ability of domestic cattle to survive and adapt to new environmental conditions by incorporating genetic material from local species. Adaptive introgression is more likely to occur through the birth of F1 hybrids in the domestic cattle population, that is male‐mediated introgression from European aurochs into domesticated cattle (Götherström et al., 2005; Pérez‐Pardal et al., 2010) or male‐mediated introgression of indicine cattle into Middle Eastern (Verdugo et al., 2019) or African taurine cattle (Kim et al., 2020). On the contrary, female‐mediated adaptive introgression is associated with the maintenance of pure wild animals and could therefore be much more difficult. We therefore suspect that the extremely rare integration of wild female calves into domestic stock occurs only in emergencies. Of course, we cannot exclude the speculation that the introgression of the aurochs was necessary to lower the degree of inbreeding of the existing domesticated cattle or was simply a consequence of the exploitation of heterosis effects. For example, crossbreeding of yak with cattle has been traditionally practised in Asia for a long time (Zhang, 2000).
The finding of surviving aurochs’ mitochondria not only provides indisputable evidence for gene flow between European wild aurochs and domesticated cattle but also enables the protection of already endangered maternal cattle diversity. No less importantly, Murbodner cattle with the P1 haplotype are likely to be of interest for ongoing aurochs de‐extinction programmes (Sinding & Gilbert, 2016; Stokstad, 2015) and for research on the expression of mitonuclear interacting gene complexes.
CONFLICT OF INTEREST
None declared.
Supporting information
Supplementary Materials
Data S1
ACKNOWLEDGEMENT
The authors would like to thank the University of Zagreb University Computing Centre (SRCE)—Danijel Vrčić, Emir Imamagić and Tomislav Stilinović for providing computational facilities and support. We thank the anonymous reviewers for their useful comments and helpful suggestions, which greatly improved the manuscript. The authors would like to thank the members of the H2020‐TWINN‐2015 (‘Smart Integration of Genetics with Sciences of the Past in Croatia: Minding and Mending the Gap’, project number 692249, http://mendthegap.agr.hr/) for improving our understanding of the domestication process and other topics related to Sciences of the Past. Finally, the authors would like to remember Dragan Tupajić, our unforgettable friend who passed away in 2020, who designed the figures. This study was supported by two projects from Croatian Science Foundation: MITOTAUROMICS‐IP‐11‐2013‐9070 and ANAGRAMS‐IP‐2018‐01‐8708.
Cubric‐Curik, V. , Novosel, D. , Brajkovic, V. , Rota Stabelli, O. , Krebs, S. , Sölkner, J. , Šalamon, D. , Ristov, S. , Berger, B. , Trivizaki, S. , Bizelis, I. , Ferenčaković, M. , Rothammer, S. , Kunz, E. , Simčič, M. , Dovč, P. , Bunevski, G. , Bytyqi, H. , Marković, B. , … Medugorac, I. (2022). Large‐scale mitogenome sequencing reveals consecutive expansions of domestic taurine cattle and supports sporadic aurochs introgression. Evolutionary Applications, 15, 663–678. 10.1111/eva.13315
Vlatka Cubric‐Curik, Ino Curik, and Ivica Medugorac contributed equally to this work.
Contributor Information
Vlatka Cubric‐Curik, Email: vcubric@agr.hr.
Ino Curik, Email: icurik@agr.hr.
Ivica Medugorac, Email: Ivica.Medjugorac@gen.vetmed.uni-muenchen.de.
DATA AVAILABILITY STATEMENT
Mitochondrial sequences are deposited in GenBank (accession numbers from MZ901375 to MZ901759). Fasta files used in the analyses are available upon request.
REFERENCES
- Achilli, A. , Bonfiglio, S. , Olivieri, A. , Malusà, A. , Pala, M. , Kashani, B. H. , Perego, U. A. , Ajmone‐Marsan, P. , Liotta, L. , Semino, O. , Bandelt, H. J. , Ferretti, L. , & Torroni, A. (2009). The multifaceted origin of taurine cattle reflected by the mitochondrial genome. PLoS One, 4(6), e5753. 10.1371/journal.pone.0005753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achilli, A. , Olivieri, A. , Pellecchia, M. , Uboldi, C. , Colli, L. , Al‐Zahery, N. , Accetturo, M. , Pala, M. , Kashani, B. H. , Perego, U. A. , Battaglia, V. , Fornarino, S. , Kalamati, J. , Houshmand, M. , Negrini, R. , Semino, O. , Richards, M. , Macaulay, V. , Ferretti, L. , … Torroni, A. (2008). Mitochondrial genomes of extinct aurochs survive in domestic cattle. Current Biology, 18(4), R157–R158. 10.1016/j.cub.2008.01.019 [DOI] [PubMed] [Google Scholar]
- Afgan, E. , Baker, D. , Batut, B. , van den Beek, M. , Bouvier, D. , Čech, M. , Chilton, J. , Clements, D. , Coraor, N. , Grüning, B. A. , Guerler, A. , Hillman‐Jackson, J. , Hiltemann, S. , Jalili, V. , Rasche, H. , Soranzo, N. , Goecks, J. , Taylor, J. , Nekrutenko, A. , & Blankenberg, D. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research, 46(W1), W537–W544. 10.1093/nar/gky379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajmone‐Marsan, P. , Garcia, J. F. , & Lenstra, J. A. (2010). On the origin of cattle: How aurochs became cattle and colonized the world. Evolutionary Anthropology: Issues, News, and Reviews, 19(4), 148–157. 10.1002/evan.20267 [DOI] [Google Scholar]
- Bailey, J. F. , Richards, M. B. , Macaulay, V. A. , Colson, I. B. , James, I. T. , Bradley, D. G. , Hedges, R. E. M. , & Sykes, B. C. (1996). Ancient DNA suggests a recent expansion of European cattle from a diverse wild progenitor species. Proceedings of the Royal Society of London. Series B: Biological Sciences, 263(1376), 1467–1473. [DOI] [PubMed] [Google Scholar]
- Bandelt, H. J. , Forster, P. , & Röhl, A. (1999). Median‐joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37–48. 10.1093/oxfordjournals.molbev.a026036 [DOI] [PubMed] [Google Scholar]
- Bonfiglio, S. , Achilli, A. , Olivieri, A. , Negrini, R. , Colli, L. , Liotta, L. , Ajmone‐Marsan, P. , Torroni, A. , & Ferretti, L. (2010). The enigmatic origin of bovine mtDNA haplogroup R: sporadic interbreeding or an independent event of Bos primigenius domestication in Italy? PLoS One, 5(12), e15760. 10.1371/journal.pone.0015760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert, R. , Vaughan, T. G. , Barido‐Sottani, J. , Duchêne, S. , Fourment, M. , Gavryushkina, A. , Heled, J. , Jones, G. , Kühnert, D. , De Maio, N. , Matschiner, M. , Mendes, F. K. , Müller, N. F. , Ogilvie, H. A. , du Plessis, L. , Popinga, A. , Rambaut, A. , Rasmussen, D. , Siveroni, I. , … Drummond, A. J. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Computational Biology, 15(4), e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, D. G. , MacHugh, D. E. , Cunningham, P. , & Loftus, R. T. (1996). Mitochondrial diversity and the origins of African and European cattle. Proceedings of the National Academy of Sciences of the United States of America, 93(10), 5131–5135. 10.1073/pnas.93.10.5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bro‐Jørgensen, M. H. , Carøe, C. , Vieira, F. G. , Nestor, S. , Hallström, A. , Gregersen, K. M. , Etting, V. , Gilbert, T. M. P. , & Sinding, M. H. S. (2018). Ancient DNA analysis of Scandinavian medieval drinking horns and the horn of the last aurochs bull. Journal of Archaeological Science, 99, 47–54. 10.1016/j.jas.2018.09.001 [DOI] [Google Scholar]
- Bushnell, B. (2014). BBTools software package. https://sourceforge.net/projects/bbmap [Google Scholar]
- Carneiro, M. , Rubin, C. J. , Di Palma, F. , Albert, F. W. , Alföldi, J. , Barrio, A. M. , Pielberg, G. , Rafati, N. , Sayyab, S. , Turner‐Maier, J. , Younis, S. , Afonso, S. , Aken, B. , Alves, J. M. , Barrell, D. , Bolet, G. , Boucher, S. , Burbano, H. A. , Campos, R. , … Andersson, L. (2014). Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science, 345(6200), 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, N. , Cai, Y. , Chen, Q. , Li, R. , Wang, K. , Huang, Y. , Hu, S. , Huang, S. , Zhang, H. , Zheng, Z. , Song, W. , Ma, Z. , Ma, Y. , Dang, R. , Zhang, Z. , Xu, L. , Jia, Y. , Liu, S. , Yue, X. , … Lei, C. (2018). Whole‐genome resequencing reveals world‐wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nature Communications, 9(1), 2337. 10.1038/s41467-018-04737-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther, A. , Prendergast, M. E. , Fuller, D. Q. , & Boivin, N. (2018). Subsistence mosaics, forager‐farmer interactions, and the transition to food production in eastern Africa. Quaternary International, 489, 101–120. 10.1016/j.quaint.2017.01.014 [DOI] [Google Scholar]
- Dean, S. , Pappalardo, M. , Boschian, G. , Spada, G. , Forenbaher, S. , Juračić, M. , Felja, I. , Radić, D. , & Miracle, P. T. (2020). Human adaptation to changing coastal landscapes in the Eastern Adriatic: Evidence from Vela Spila cave, Croatia. Quaternary Science Reviews, 244, 106503. 10.1016/j.quascirev.2020.106503 [DOI] [Google Scholar]
- Decker, J. E. , McKay, S. D. , Rolf, M. M. , Kim, J. , Alcalá, A. M. , Sonstegard, T. S. , Hanotte, O. , Gotherstrom, A. , Seabury, C. M. , Praharani, L. , Babar, M. L. , de Almeida Regitano, L. C. , Yildiz, M. A. , Heaton, M. P. , Liu, W. S. , Lei, C. Z. , Reecy, J. M. , Saif‐Ur‐Rehman, M. , Schnabel, R. D. , & Taylor, J. F. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genetics, 10(3), e1004254. 10.1371/journal.pgen.1004254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo, P. , Lancioni, H. , Ceccobelli, S. , Colli, L. , Cardinali, I. , Karsli, T. , Capodiferro, M. R. , Sahin, E. , Ferretti, L. , Ajmone Marsan, P. , Sarti, F. M. , Lasagna, E. , Panella, F. , & Achilli, A. (2018). Mitochondrial DNA variants of Podolian cattle breeds testify for a dual maternal origin. PLoS One, 13(2), e0192567. 10.1371/journal.pone.0192567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , Ho, S. Y. , Phillips, M. J. , & Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biology, 4(5), e88. 10.1371/journal.pbio.0040088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , Suchard, M. A. , Xie, D. , & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29(8), 1969–1973. 10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, C. J. , Bollongino, R. , Scheu, A. , Chamberlain, A. , Tresset, A. , Vigne, J. D. , Baird, J. F. , Larson, G. , Ho, S. Y. W. , Heupink, T. H. , Shapiro, B. , Freeman, A. R. , Thomas, M. G. , Arbogast, R. M. , Arndt, B. , Bartosiewicz, L. , Benecke, N. , Budja, M. , Chaix, L. , … Burger, J. (2007). Mitochondrial DNA analysis shows a Near Eastern Neolithic origin for domestic cattle and no indication of domestication of European aurochs. Proceedings of the Royal Society B: Biological Sciences, 274(1616), 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, C. J. , Magee, D. A. , Park, S. D. E. , McGettigan, P. A. , Lohan, A. J. , Murphy, A. , Finlay, E. K. , Shapiro, B. , Chamberlain, A. T. , Richards, M. B. , Bradley, D. G. , Loftus, B. J. , & MacHugh, D. E. (2010). A complete mitochondrial genome sequence from a mesolithic wild aurochs (Bos primigenius). PLoS One, 5(2), e9255. 10.1371/journal.pone.0009255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Felius, M. , Beerling, M. L. , Buchanan, D. S. , Theunissen, B. , Koolmees, P. A. , & Lenstra, J. A. (2014). On the history of cattle genetic resources. Diversity, 6(4), 705–750. 10.3390/d6040705 [DOI] [Google Scholar]
- Frantz, L. A. , Bradley, D. G. , Larson, G. , & Orlando, L. (2020). Animal domestication in the era of ancient genomics. Nature Reviews Genetics, 21(8), 449–460. 10.1038/s41576-020-0225-0 [DOI] [PubMed] [Google Scholar]
- Gifford‐Gonzalez, D. , & Hanotte, O. (2011). Domesticating animals in Africa: Implications of genetic and archaeological findings. Journal of World Prehistory, 24(1), 1–23. 10.1007/s10963-010-9042-2 [DOI] [Google Scholar]
- Gignoux, C. R. , Henn, B. M. , & Mountain, J. L. (2011). Rapid, global demographic expansions after the origins of agriculture. Proceedings of the National Academy of Sciences of the United States of America, 108(15), 6044–6049. 10.1073/pnas.0914274108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götherström, A. , Anderung, C. , Hellborg, L. , Elburg, R. , Smith, C. , Bradley, D. G. , & Ellegren, H. (2005). Cattle domestication in the Near East was followed by hybridization with aurochs bulls in Europe. Proceedings of the Royal Society B: Biological Sciences, 272(1579), 2345–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau, E. T. , Charles, M. , Féménia, M. , Rebours, E. , Vaiman, A. , & Rocha, D. (2020). Survey of mitochondrial sequences integrated into the bovine nuclear genome. Scientific Reports, 10(1), 2077. 10.1038/s41598-020-59155-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, M. , Kishino, H. , & Yano, T. A. (1985). Dating of the human‐ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution, 22(2), 160–174. 10.1007/BF02101694 [DOI] [PubMed] [Google Scholar]
- Helmer, D. , Gourichon, L. , Monchot, H. , Peters, J. , & Segui, M. S. (2005). Identifying early domestic cattle from Pre‐Pottery Neolithic sites on the Middle Euphrates using sexual dimorphism. In Vigne J. D., Peters J., & Helmer D. (Eds.), The first steps of animal domestication: New archaeozoological approaches (pp. 86–95). Oxbow Books. [Google Scholar]
- Horsburgh, K. A. , Prost, S. , Gosling, A. , Stanton, J. A. , Rand, C. , & Matisoo‐Smith, E. A. (2013). The genetic diversity of the Nguni breed of African Cattle (Bos spp.): Complete mitochondrial genomes of haplogroup T1. PLoS One, 8(8), e71956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isarin, R. F. B. , & Renssen, H. (1999). Reconstructing and modelling Late Weichselian climates: The Younger Dryas in Europe as a case study. Earth‐Science Reviews, 48(1–2), 1–38. 10.1016/S0012-8252(99)00047-1 [DOI] [Google Scholar]
- Jukes, T. H. , & Cantor, C. R. (1969). Evolution of protein molecules. In Munro H. N. (Ed.), Mammalian protein metabolism, III (pp. 21–132). Academic Press. 10.1016/B978-1-4832-3211-9.50009-7 [DOI] [Google Scholar]
- Kim, J. , Hanotte, O. , Mwai, O. A. , Dessie, T. , Bashir, S. , Diallo, B. , Agaba, M. , Kim, K. , Kwak, W. , Sung, S. , Seo, M. , Jeong, H. , Kwon, T. , Taye, M. , Song, K. D. , Lim, D. , Cho, S. , Lee, H. J. , Yoon, D. , … Kim, H. (2017). The genome landscape of indigenous African cattle. Genome Biology, 18(1), 34. 10.1186/s13059-017-1153-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. , Kwon, T. , Dessie, T. , Yoo, D. A. , Mwai, O. A. , Jang, J. , Sung, S. , Lee, S. B. , Salim, B. , Jung, J. , Jeong, H. , Tarekegn, G. M. , Tijjani, A. , Lim, D. , Cho, S. , Oh, S. J. , Lee, H.‐K. , Kim, J. , Jeong, C. , … Kim, H. (2020). The mosaic genome of indigenous African cattle as a unique genetic resource for African pastoralism. Nature Genetics, 52(10), 1099–1110. 10.1038/s41588-020-0694-2 [DOI] [PubMed] [Google Scholar]
- Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution, 16(2), 111–120. 10.1007/BF01731581 [DOI] [PubMed] [Google Scholar]
- Koboldt, D. C. , Zhang, Q. , Larson, D. E. , Shen, D. , McLellan, M. D. , Lin, L. , Miller, C. A. , Mardis, E. R. , Ding, L. , & Wilson, R. K. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Research, 22(3), 568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L. , Frost, S. D. W. , & Muse, S. V. (2005). HyPhy: Hypothesis testing using phylogenies. Bioinformatics, 21(5), 676–679. 10.1093/bioinformatics/bti079 [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L. , Posada, D. , Gravenor, M. B. , Woelk, C. H. , & Frost, S. D. (2006a). Automated phylogenetic detection of recombination using a genetic algorithm. Molecular Biology and Evolution, 23(10), 1891–1901. 10.1093/molbev/msl051 [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L. , Posada, D. , Gravenor, M. B. , Woelk, C. H. , & Frost, S. D. (2006b). GARD: A genetic algorithm for recombination detection. Bioinformatics, 22(24), 3096–3098. 10.1093/bioinformatics/btl474 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanave, C. , Preparata, G. , Sacone, C. , & Serio, G. (1984). A new method for calculating evolutionary substitution rates. Journal of Molecular Evolution, 20(1), 86–93. 10.1007/BF02101990 [DOI] [PubMed] [Google Scholar]
- Lari, M. , Rizzi, E. , Mona, S. , Corti, G. , Catalano, G. , Chen, K. , Vernesi, C. , Larson, G. , Boscato, P. , De Bellis, G. , Cooper, A. , Caramelli, D. , & Bertorelle, G. (2011). The complete mitochondrial genome of an 11,450‐year‐old aurochsen (Bos primigenius) from Central Italy. BMC Evolutionary Biology, 11(1), 32. 10.1186/1471-2148-11-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, G. , & Burger, J. (2013). A population genetics view of animal domestication. Trends in Genetics, 29(4), 197–205. 10.1016/j.tig.2013.01.003 [DOI] [PubMed] [Google Scholar]
- Larson, G. , Piperno, D. R. , Allaby, R. G. , Purugganan, M. D. , Andersson, L. , Arroyo‐Kalin, M. , Barton, L. , Vigueira, C. C. , Denham, T. , Dobney, K. , Doust, A. N. , Gepts, P. , Gilbert, T. M. P. , Gremillion, K. J. , Lucas, L. , Lukens, L. , Marshall, F. B. , Olsen, K. M. , Pires, C. J. , … Fuller, D. Q. (2014). Current perspectives and the future of domestication studies. Proceedings of the National Academy of Sciences of the United States of America, 111(17), 6139–6146. 10.1073/pnas.1323964111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, J. W. , & Bryant, D. (2015). popart: Full‐feature software for haplotype network construction. Methods in Ecology and Evolution, 6(9), 1110–1116. 10.1111/2041-210X.12410 [DOI] [Google Scholar]
- Leinonen, R. , Sugawara, H. , & Shumway, M. (2011). The sequence read archive. Nucleic Acids Research, 39(Database), D19–D21. 10.1093/nar/gkq1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardić Much, J. , Sršen Oros, A. , & Radović, S. (2018). Quaternary fauna of the Eastern Adriatic (Croatia) with the special review on the Late Pleistocene sites. Quaternary International, 494, 130–151. 10.1016/j.quaint.2017.11.028 [DOI] [Google Scholar]
- Li, H. (2011a). Improving SNP discovery by base alignment quality. Bioinformatics, 27(8), 1157–1158. 10.1093/bioinformatics/btr076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2011b). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27(21), 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus, R. T. , MacHugh, D. E. , Bradley, D. G. , Sharp, P. M. , & Cunningham, P. (1994). Evidence for two independent domestications of cattle. Proceedings of the National Academy of Sciences of the United States of America, 91(7), 2757–2761. 10.1073/pnas.91.7.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannen, H. , Tsuji, S. , Loftus, R. T. , & Bradley, D. G. (1998). Mitochondrial DNA variation and evolution of Japanese black cattle (Bos taurus). Genetics, 150(3), 1169–1175. 10.1093/genetics/150.3.1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannen, H. , Yonezawa, T. , Murata, K. , Noda, A. , Kawaguchi, F. , Sasazaki, S. , Olivieri, A. , Achilli, A. , & Torroni, A. (2020). Cattle mitogenome variation reveals a post‐glacial expansion of haplogroup P and an early incorporation into northeast Asian domestic herds. Scientific Reports, 10(1), 20842. 10.1038/s41598-020-78040-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, H. , Davidson, M. , Charitakis, N. , Diaz, L. , Bowers, W. H. , Gradovich, E. , Andrew, T. , & Huntley, D. (2019). NUMT confounding biases mitochondrial heteroplasmy calls in favor of the reference allele. Frontiers in Cell and Developmental Biology, 7, 201. 10.3389/fcell.2019.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbole‐Kariuki, M. N. , Sonstegard, T. , Orth, A. , Thumbi, S. M. , Bronsvoort, B. M. D. C. , Kiara, H. , Toye, P. , Conradie, I. , Jennings, A. , Coetzer, K. , Woolhouse, M. E. J. , Hanotte, O. , & Tapio, M. (2014). Genome‐wide analysis reveals the ancient and recent admixture history of East African Shorthorn Zebu from Western Kenya. Heredity, 113(4), 297–305. 10.1038/hdy.2014.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugo, G. P. , Dover, M. J. , & MacHugh, D. E. (2019). Unlocking the origins and biology of domestic animals using ancient DNA and paleogenomics. BMC Biology, 17(1), 98. 10.1186/s12915-019-0724-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTavish, E. J. , Decker, J. E. , Schnabel, R. D. , Taylor, J. F. , & Hillis, D. M. (2013). New World cattle show ancestry from multiple independent domestication events. Proceedings of the National Academy of Sciences of the United States of America, 110(15), E1398–E1406. 10.1073/pnas.1303367110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadow, R. H. (1993). Animal domestication in the Middle East: a revised view from the eastern margin. Harappar Civilization. [Google Scholar]
- Medugorac, I. , Graf, A. , Grohs, C. , Rothammer, S. , Zagdsuren, Y. , Gladyr, E. , Zinovieva, N. , Barbieri, J. , Seichter, D. , Russ, I. , Eggen, A. , Hellenthal, G. , Brem, G. , Blum, H. , Krebs, S. , & Capitan, A. (2017). Whole‐genome analysis of introgressive hybridization and characterization of the bovine legacy of Mongolian yaks. Nature Genetics, 49(3), 470. 10.1038/ng.3775 [DOI] [PubMed] [Google Scholar]
- Miracle, P. (2007). The late glacial great adriatic plain’: Garden of Eden’ or No Man's Land' during the Epipalaeolithic? A view from Istria (Croatia). In Whallon R. (Ed.), Late paleolithic environments and cultural relations around the adriatic. BAR international series, (1716, pp.41–51). Archaeopres. [Google Scholar]
- Mwai, O. , Hanotte, O. , Kwon, Y. J. , & Cho, S. (2015). African indigenous cattle: Unique genetic resources in a rapidly changing world. Asian‐Australasian Journal of Animal Sciences, 28(7), 911–921. 10.5713/ajas.15.0002R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz, B. , Mauffrey, J. F. , Bazin, E. , Galtier, N. , & Glemin, S. (2008). Determination of mitochondrial genetic diversity in mammals. Genetics, 178(1), 351–361. 10.1534/genetics.107.073346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri, A. , Gandini, F. , Achilli, A. , Fichera, A. , Rizzi, E. , Bonfiglio, S. , Battaglia, V. , Brandini, S. , De Gaetano, A. , El‐Beltagi, A. , Lancioni, H. , Agha, S. , Semino, O. , Ferretti, L. , & Torroni, A. (2015). Mitogenomes from Egyptian cattle breeds: new clues on the origin of haplogroup Q and the early spread of Bos taurus from the Near East. PLoS One, 10(10), e0141170. 10.1371/journal.pone.0141170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papachristou, D. , Koutsouli, P. , Laliotis, G. P. , Kunz, E. , Upadhyay, M. , Seichter, D. , Russ, I. , Gjoko, B. , Kostaras, N. , Bizelis, I. , & Medugorac, I. (2020). Genomic diversity and population structure of the indigenous Greek and Cypriot cattle populations. Genetics Selection Evolution, 52(1), 43. 10.1186/s12711-020-00560-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. D. , Magee, D. A. , McGettigan, P. A. , Teasdale, M. D. , Edwards, C. J. , Lohan, A. J. , Murphy, A. , Braud, M. , Donoghue, M. T. , Liu, Y. , Chamberlain, A. T. , Rue‐Albrecht, K. , Schroeder, S. , Spillane, C. , Tai, S. , Bradley, D. G. , Sonstegard, T. S. , Loftus, J. B. , & MacHugh, D. E. (2015). Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biology, 16(1), 171. 10.1186/s13059-015-0790-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, W. J. A. , & Hodges, J. (1997). Tropical cattle: origins, breeds and breeding policies. Blackwell Science Ltd. [Google Scholar]
- Peng, M. S. , Fan, L. , Shi, N. N. , Ning, T. , Yao, Y. G. , Murphy, R. W. , Wang, W. Z. , & Zhang, Y. P. (2015). DomeTree: A canonical toolkit for mitochondrial DNA analyses in domesticated animals. Molecular Ecology Resources, 15(5), 1238–1242. 10.1111/1755-0998.12386 [DOI] [PubMed] [Google Scholar]
- Pérez‐Pardal, L. , Royo, L. J. , Beja‐Pereira, A. , Chen, S. , Cantet, R. J. , Traoré, A. , Curik, I. , Solkner, J. , Bozzi, R. , Fernandez, I. , Alvarez, I. , Gutierrez, J. P. , Gomez, E. , Ponce de Leon, F. A. , & Goyache, F. (2010). Multiple paternal origins of domestic cattle revealed by Y‐specific interspersed multilocus microsatellites. Heredity, 105(6), 511–519. 10.1038/hdy.2010.30 [DOI] [PubMed] [Google Scholar]
- Pérez‐Pardal, L. , Sánchez‐Gracia, A. , Álvarez, I. , Traoré, A. , Ferraz, J. B. S. , Fernández, I. , Costa, V. , Chen, S. , Tapio, M. , Cantet, R. J. C. , Patel, A. , Meadow, R. H. , Marshall, F. B. , Beja‐Pereira, A. , & Goyache, F. (2018). Legacies of domestication, trade and herder mobility shape extant male zebu cattle diversity in South Asia and Africa. Scientific Reports, 8(1), 18027. 10.1038/s41598-018-36444-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhasi, R. , Fort, J. , & Ammerman, A. J. (2005). Tracing the origin and spread of agriculture in Europe. PLoS Biology, 3(12), e410. 10.1371/journal.pbio.0030410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery, A. E. , Newton, M. A. , Satagopan, J. M. , & Krivitsky, P. N. (2007). Estimating the integrated likelihood via posterior simulation using the harmonic mean identity. In Bernardo J. M., Bayarri M. J., & Berger J. O. (Eds.), Bayesian statistics (pp. 1–45). Oxford University Press. [Google Scholar]
- Rambaut, A. , Drummond, A. J. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristov, S. , Brajkovic, V. , Cubric‐Curik, V. , Michieli, I. , & Curik, I. (2016). MaGelLAn 1.0: a software to facilitate quantitative and population genetic analysis of maternal inheritance by combination of molecular and pedigree information. Genetics Selection Evolution, 48(1), 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J. , Ferrer‐Mata, A. , Sánchez‐DelBarrio, J. C. , Guirao‐Rico, S. , Librado, P. , Ramos‐Onsins, S. E. , & Sánchez‐Gracia, A. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34(12), 3299–3302. 10.1093/molbev/msx248 [DOI] [PubMed] [Google Scholar]
- Schiavo, G. , Bovo, S. , Ribani, A. , Kazemi, H. , & Fontanesi, L. (2020). A comparative genome landscape of mitochondrial DNA insertions into two cattle nuclear genome versions. Animal Genetics, 51(1), 149–151. 10.1111/age.12889 [DOI] [PubMed] [Google Scholar]
- Schibler, J. , Elsner, J. , & Schlumbaum, A. (2014). Incorporation of aurochs into a cattle herd in Neolithic Europe: Single event or breeding? Scientific Reports, 4(1), 5798. 10.1038/srep05798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senczuk, G. , Mastrangelo, S. , Ajmone‐Marsan, P. , Becskei, Z. , Colangelo, P. , Colli, L. , Ferretti, L. , Karsli, T. , Lancioni, H. , Lasagna, E. , Marletta, D. , Persichilli, C. , Portolano, B. , Sarti, F. M. , Ciani, E. , & Pilla, F. (2021). On the origin and diversification of Podolian cattle breeds: testing scenarios of European colonization using genome‐wide SNP data. Genetics Selection Evolution, 53(1), 48. 10.1186/s12711-021-00639-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharbrough, J. , Havird, J. C. , Noe, G. R. , Warren, J. M. , & Sloan, D. B. (2017). The mitonuclear dimension of Neanderthal and Denisovan ancestry in modern human genomes. Genome Biology and Evolution, 9(6), 1567–1581. 10.1093/gbe/evx114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherratt, A. (1983). The secondary exploitation of animals in the Old World. World Archaeology, 15(1), 90–104. 10.1080/00438243.1983.9979887 [DOI] [Google Scholar]
- Sievers, F. , Wilm, A. , Dineen, D. , Gibson, T. J. , Karplus, K. , Li, W. , Lopez, R. , McWilliam, H. , Remmert, M. , Söding, J. , Thompson, J. D. , & Higgins, D. G. (2011). Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 7(1), 539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinding, M. H. S. , & Gilbert, M. T. P. (2016). The draft genome of extinct European aurochs and its implications for de‐extinction. Open Quaternary, 2, 7. 10.5334/oq.25 [DOI] [Google Scholar]
- Steffen, W. , Richardson, K. , Rockström, J. , Cornell, S. E. , Fetzer, I. , Bennett, E. M. , Biggs, R. , Carpenter, S. R. , de Vries, W. , de Wit, C. A. , Folke, C. , Gerten, D. , Heinke, J. , Mace, G. M. , Persson, L. M. , Ramanathan, V. , Reyers, B. , & Sörlin, S. (2015). Planetary boundaries: Guiding human development on a changing planet. Science, 347(6223), 1259855. 10.1126/science.1259855 [DOI] [PubMed] [Google Scholar]
- Stokstad, E. (2015). Bringing back the aurochs. Science, 350(6265), 1144–1147. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , & Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution, 10(3), 512–526. [DOI] [PubMed] [Google Scholar]
- Troy, C. S. , MacHugh, D. E. , Bailey, J. F. , Magee, D. A. , Loftus, R. T. , Cunningham, P. , Chamberlain, A. T. , Sykes, B. C. , & Bradley, D. G. (2001). Genetic evidence for Near‐Eastern origins of European cattle. Nature, 410(6832), 1088–1091. [DOI] [PubMed] [Google Scholar]
- Upadhyay, M. , Bortoluzzi, C. , Barbato, M. , Ajmone‐Marsan, P. , Colli, L. , Ginja, C. , Sonstegard, T. S. , Bosse, M. , Lenstra, J. A. , Groenen, M. A. M. , & Crooijmans, R. P. (2019). Deciphering the patterns of genetic admixture and diversity in southern European cattle using genome‐wide SNPs. Evolutionary Applications, 12(5), 951–963. 10.1111/eva.12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdugo, M. P. , Mullin, V. E. , Scheu, A. , Mattiangeli, V. , Daly, K. G. , Delser, P. M. , Hare, A. J. , Burger, J. , Collins, M. J. , Kehati, R. , Hesse, P. , Fulton, D. , Sauer, E. W. , Mohaseb, F. A. , Davoudi, H. , Khazaeli, R. , Lhuillier, J. , Rapin, C. , Ebrahimi, S. , … Bradley, D. G. (2019). Ancient cattle genomics, origins, and rapid turnover in the Fertile Crescent. Science, 365(6449), 173–176. [DOI] [PubMed] [Google Scholar]
- Weldenegodguad, M. , Popov, R. , Pokharel, K. , Ammosov, I. , Ming, Y. , Ivanova, Z. , & Kantanen, J. (2019). Whole‐genome sequencing of three native cattle breeds originating from the northernmost cattle farming regions. Frontiers in Genetics, 9, 728. 10.3389/fgene.2018.00728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, E. , & Viner‐Daniels, S. (2015). Geographical variation in the size and shape of the European aurochs (Bos primigenius). Journal of Archaeological Science, 54, 8–22. 10.1016/j.jas.2014.11.021 [DOI] [Google Scholar]
- Wu, D. D. , Ding, X. D. , Wang, S. , Wójcik, J. M. , Zhang, Y. I. , Tokarska, M. , Yan, L. , Wang, M. S. , Faruque, O. , Nielsen, R. , Zhang, Q. , & Zhang, Y. P. (2018). Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nature Ecology & Evolution, 2(7), 1139–1145. 10.1038/s41559-018-0562-y [DOI] [PubMed] [Google Scholar]
- Xia, X. T. , Achilli, A. , Lenstra, J. A. , Tong, B. , Ma, Y. , Huang, Y. Z. , Han, J. L. , Sun, Z. Y. , Chen, H. , Lei, C. Z. , Hu, S. M. , & Chen, N. B. (2021). Mitochondrial genomes from modern and ancient Turano‐Mongolian cattle reveal an ancient diversity of taurine maternal lineages in East Asia. Heredity, 126(6), 1000–1008. 10.1038/s41437-021-00428-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X. , Qu, K. , Li, F. , Jia, P. , Chen, Q. , Chen, N. , Zhang, J. , Chen, H. , Huang, B. , & Lei, C. (2019). Abundant genetic diversity of Yunling Cattle based on mitochondrial genome. Animals, 9(9), 641. 10.3390/ani9090641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi, A. A. , & Makova, K. D. (2019). Investigating mitonuclear interactions in human admixed populations. Nature Ecology & Evolution, 3(2), 213–222. 10.1038/s41559-018-0766-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeder, M. A. (2006). Central questions in the domestication of plants and animals. Evolutionary Anthropology: Issues, News, and Reviews, 15(3), 105–117. 10.1002/evan.20101 [DOI] [Google Scholar]
- Zeuner, F. E. (1963). A history of domesticated animals. Hutchinson. [Google Scholar]
- Zeyland, J. , Wolko, L. , Bocianowski, J. , Szalata, M. , Slomski, R. , Dzieduszycki, A. M. , Ryba, M. , Przystalowska, H. , & Lipinski, D. (2013). Complete mitochondrial genome of wild aurochs (Bos primigenius) reconstructed from ancient DNA. Polish Journal of Veterinary Sciences, 16(2), 265–273. 10.2478/pjvs-2013-0037 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Paijmans, J. L. , Chang, F. , Wu, X. , Chen, G. , Lei, C. , Yang, X. , Wei, Z. , Bradley, D. G. , Orlando, L. , O’Connor, T. , & Hofreiter, M. (2013). Morphological and genetic evidence for early Holocene cattle management in northeastern China. Nature Communications, 4(1), 2755. 10.1038/ncomms3755 [DOI] [PubMed] [Google Scholar]
- Zhang, R. C. (2000). Interspecies hybridization between yak, Bos taurus and Bos indicus and reproduction of the hybrids. In Zhao X. X. & Zhang R. C. (Eds.), Recent advances in yak reproduction. International Veterinary Information Service; (http://www.ivis.org). Paper No. A1304.0900. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials
Data S1
Data Availability Statement
Mitochondrial sequences are deposited in GenBank (accession numbers from MZ901375 to MZ901759). Fasta files used in the analyses are available upon request.