

Summary
Understanding macrophage heterogeneity in tissue repair is a major challenge. Here, we describe a protocol that combines isolation of immune cells from skin wounds with subsequent flow-cytometry-based sorting of wound macrophages and single-cell RNA sequencing. We use a modified version of the original Smart-seq2 protocol to increase speed and accuracy. This protocol is useful for analyzing the pronounced heterogeneity of activation phenotypes in wound macrophages and might be adapted to other experimental models of skin inflammation.
For complete details on the use and execution of this protocol, please refer to Willenborg et al. (2021).
Subject areas: Cell Biology, Cell isolation, Single Cell, Flow Cytometry/Mass Cytometry, Sequencing, RNAseq, Immunology, Metabolism, Molecular Biology
Graphical abstract
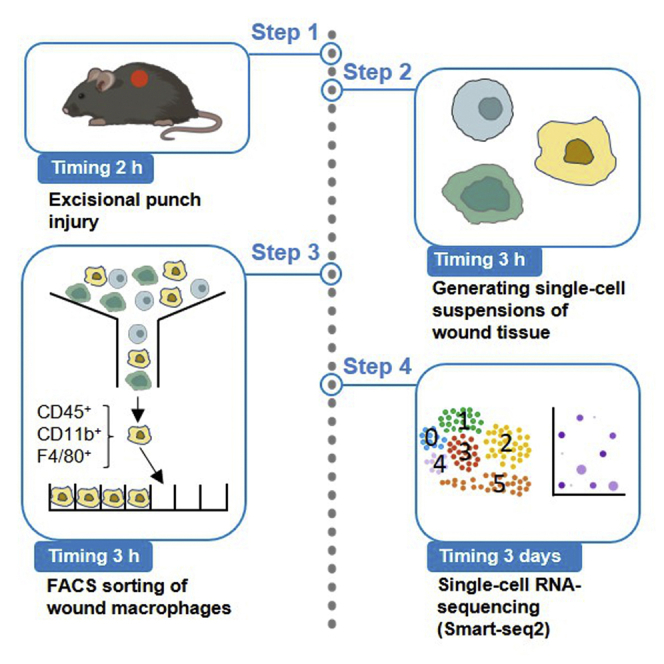
Highlights
-
•
Procedure for excisional punch injury in mouse skin
-
•
Isolation of macrophages from skin wounds suitable for single-cell RNA sequencing
-
•
FACS-based sorting of macrophages into 384-well plates
-
•
Single-cell RNA sequencing following the Smart-seq2 workflow
Understanding macrophage heterogeneity in tissue repair is a major challenge. Here, we describe a protocol that combines isolation of immune cells from skin wounds with subsequent flow-cytometry-based sorting of wound macrophages and single-cell RNA sequencing. We use a modified version of the original Smart-seq2 protocol to increase speed and accuracy. This protocol is useful for analyzing the pronounced heterogeneity of activation phenotypes in wound macrophages and might be adapted to other experimental models of skin inflammation.
Before you begin
The protocol below describes the specific steps for single-cell RNA sequencing in wild-type macrophages isolated from mouse skin wounds. Sequencing the mRNA content of single wound macrophages with the Smart-seq2 workflow allows an accurate and comprehensive profiling of the genes expressed at the single-cell level. We have also used parts of the protocol for the isolation of skin macrophages in mouse models of skin inflammation (Knuever et al., 2015; Ding et al., 2020).
Institutional permission for the animal study should be obtained. All animal experiments in this study were in accordance with the German Law for Protection of Animals and approved by the Landesdirektion Dresden (approval number TVV 62/2015).
Preparation on the day of excisional punch injury
Timing: 1 h
-
1.
Prepare Anesthesia Solution in a sterile environment as described in the materials and equipment section.
-
2.
Prepare 70% Ethanol solution in sterile H2O.
-
3.
Set the heating plate at 37°C.
-
4.
Prepare clean autoclaved mouse cages. Use one cage for each mouse because mice should be kept separately after injury.
-
5.
Monitor the health status of the mice before injury.
Preparation on the day before wound excision
-
6.Prepare buffers and solutions as described in the materials and equipment section.
-
a.Liberase Stock Solution.
-
b.FACS Buffer 1.
-
c.FACS Buffer 2.
-
a.
-
7.Make sure that centrifuge and ThermoMixer have reached the required temperature before starting.
-
a.Centrifuge at 4°C.
-
b.ThermoMixer at 37°C.
-
a.
Preparation on the day of wound excision
-
8.
Prepare Red Blood Cell Lysis Solution as described in the materials and equipment section.
Preparation right before FACS sorting
-
9.
Prepare Lysis Buffer as described in the materials and equipment section.
CRITICAL: The Lysis Buffer is stable for a maximum of 4 h if stored properly at 4°C. Do not freeze this buffer as it reduces the RNase inhibitor activity. We recommend always preparing it fresh right before FACS sorting.
-
10.
Preload the Lysis Buffer onto the 384-well plate.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Rat anti-mouse Trem2 (237920), 100 tests, dilution 1:10 | R&D Systems | Cat# FAB17291A; RRID: AB_884527 |
| APC-eFluor780 Rat anti-mouse CD11b (M1/70), 0.2 mg/mL, dilution 1:400 | Thermo Fisher Scientific | Cat# 47-0112-80; RRID: AB_1603195 |
| eFluor450 Rat anti-mouse MHC II (M5/114.15.2), 0.2 mg/mL, dilution 1:200 | Thermo Fisher Scientific | Cat# 48-5321-82; RRID: AB_1272204 |
| FITC Rat anti-mouse CD45 (30-F11), 0.2 mg/mL, dilution 1:200 | Thermo Fisher Scientific | Cat# 11-0451-82; RRID: AB_465050 |
| PE Rat anti-mouse F4/80 (CI:A3-1), 100 tests, dilution 1:10 | Bio-Rad Laboratories | Cat# MCA497PE; RRID: 2098196 |
| PerCP/Cy5.5 Rat anti mouse CD301b (URA-1), 0.2 mg/mL, dilution 1:50 | BioLegend | Cat# 146809; RRID: AB_2563391 |
| Rat anti-mouse CD16/CD32, 0.5 mg/mL, dilution 1:50 | Thermo Fisher Scientific | Cat# 14-0161-82; RRID: AB_467133 |
| Chemicals, peptides, and recombinant proteins | ||
| Betaine (5 M) | Thermo Fisher Scientific | Cat# J77507.AB |
| dNTP | Thermo Fisher Scientific (Life Technologies) | Cat# R0192 |
| EDTA (0.5 M, pH 8.0) solution | Thermo Fisher Scientific (Ambion) | Cat# AM9261 |
| Ethanol | Merck | Cat# 1009835000 |
| H2O, sterile | Fresenius Kabi | 40676.00.00 |
| Liberase TM Research Grade | Roche | Cat# 05401127001 |
| MgCl2 (1 M) | Thermo Fisher Scientific (Invitrogen) | Cat# AM9530G |
| Murine RNase Inhibitor | New England Biolabs | Cat# M0314 S |
| NaCl (5 M) | Thermo Fisher Scientific (Invitrogen) | Cat# AM9759 |
| Poly(ethylene glycol) avg mol wt 8,000 | Sigma-Aldrich | Cat# P5413-500G |
| Red Blood Cell Lysis Solution | Miltenyi Biotec | Cat# 130-094-183 |
| TE buffer (1×) | Thermo Fisher Scientific (Invitrogen) | Cat# AM9858 |
| Tris-Buffer pH 8.0 (1 M) | VWR (AppliChem) | Cat# A4577.1000 |
| Triton-X 100 | Merck | Cat# T8787-50ML |
| Critical commercial assays | ||
| AccuBlue Broad Range ds DNA Kit, incl 9 standards | VWR/Biotium | Cat# 31007 |
| BD CaliBRITE™ beads | BD Biosciences | Cat# 340486 |
| ERCC spike-in | Thermo Fisher Scientific (Invitrogen/Ambion) | Cat# 4456740 |
| HS NGS Fragment Kit (1–6,000 bp) | Agilent | Cat# NF-474-0500 |
| Illumina® DNA Prep, (M) Tagmentation (96 Samples) | Illumina | Cat# 20018705 |
| Kapa HiFi HotStart Readymix (2×) | Roche | Cat# 7958935001 |
| Magnetic beads (GE; Sera-Mag SpeedBead Carboxylate-Modified Magnetic Particles (Hydrophobic)) | GE Healthcare | Cat# 65152105050250 |
| SuperScript II Reverse Transcriptase, incl. First Strand Buffer of Superscript II (5×), DTT (100 mM) | Thermo Fisher Scientific (Invitrogen) | Cat# 18064071 |
| TruePrep DNA Library Prep Kit V2 for Illumina® (50 ng input/96rxn) | Vazyme | Cat# TD501-02 |
| Deposited data | ||
| Single-cell RNA-seq wound macrophages | Willenborg et al. (2021) | GSE183489; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183489 |
| Experimental models: Organisms/strains | ||
| C57BL/6J wild-type mice, sex: male or female, age: 8–12 weeks | Charles River | Strain code: 632 |
| Oligonucleotides | ||
| Poly-dT-UP (10 μM); 5′-C6-Amino | metabion | C6-aminolinker-AAGCAGTGGTATC AACGCAGAGTCGAC TTTTTTTTTT TTTTTTTTTTTTTTTTTTTTVN |
| Universal amp primer (10 μM); 5′-C6-Amino | metabion | C6-aminolinker-AAGCAGTGGTA TCAACGCAGAGT |
| TSO (100 μM) | metabion | AAGCAGTGGTATCAA CGCAGAGTACATggg |
| IDT® for Illumina Nextera DNA Unique Dual Indexes Set D (96 Indexes, 96 Samples) or equivalent | Illumina | Cat# 20027216 |
| Software and algorithms | ||
| FACSDiva Version 7.0 | BD Biosciences | https://www.bdbiosciences.com/en-de/products/software/instrument-software/bd-facsdiva-software#Overview Cat# 659528 |
| FlowJo Version 10.7.1 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| Other | ||
| Biopsy punch | Stiefel Laboratorium | Cat# 270036 |
| Tissue for moue cages, MARACEL light | Loftex | Cat# 1136010 |
| Eye ointment | Bayer | 6029009.00.00 |
| Ketavet 100 mg/mL | Pfizer | 6187926.00.00 |
| NaCl 0.9% | Fresenius Kabi | 6096595.00.00 |
| Tramadol | Grünenthal | Cat# 95005446 |
| Xylazin 20 mg/mL | Serumwerk Bernburg AG | 3100265.00.00 |
| Adhesive PCR Plate Foils (Silver) | Thermo Fisher Scientific | Cat# AB0626 |
| Aluminium Foil Heat Seal | Brooks (4titude) | Cat# 4ti-0536 |
| 384-well Magnet Plate | Alpaqua | Cat# A001222 |
| Nanodispenser: Mantis v3.2 | FORMULATRIX | Cat# MANTV3.2 |
| Liquid handling device: Biomek FXP | Beckman Coulter | Cat# A31844 |
| Fluorometer, Infinite M Nano+ (Series M200) | Tecan | Cat# 30111058 |
| Bluewasher (automated plate washer) | BlueCatBio | Cat# 1573-L |
| Semiautomated sheet heat sealer 4s3 | 4titude | Cat# 4ti-0655 |
| Centrifuge | Thermo Fisher Scientific | Cat# 75004230 |
| ThermoMixer | Eppendorf | Cat# EP5387000013 |
| Fragment Analyzer | Agilent | Cat# 2621 (successor instruments like Fragment Analyzer 5200 with Cat# M5310AA are available) |
| Echo 525: Nanodispenser with ultrasound | Beckman Coulter | Cat# 001-10080 |
| Electric shaver for animals (Aesculap ISIS GT420 Type HS61) | Braun | Cat# 90200714 |
Materials and equipment
Anesthesia Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Ketavet 100 mg/mL | 10 mg/mL | 20 μL |
| Xylazin 20 mg/mL | 1 mg/mL | 10 μL |
| 0.9% NaCl | N/A | 170 μL |
| Total | N/A | 200 μL |
Note: We recommend preparing the Anesthesia Solution in a sterile environment and using it on the same day. Keep it at 18°C–25°C.
Liberase Stock Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| H2O | N/A | 2 mL |
| LiberaseTM TM Research Grade | 2.5 mg/mL | 5 mg |
| Total | N/A | 2 mL |
Note: Dissolve LiberaseTM TM Research Grade on ice. Store at −20°C in 100–200 μL aliquots for a maximum of 6 months.
Digestion Medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM | N/A | 1.2 mL |
| Liberase Stock Solution | 30 μg/mL | 14.4 μL |
| Total | N/A | 1.214 mL |
Note: Thaw Liberase Stock Solution on ice. Avoid repeated freeze and thaw cycles. Use digestion medium on the same day of preparation.
FACS Buffer 1 (FB1)
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS (1×) | N/A | 448 mL |
| Fetal calf serum | 10% | 50 mL |
| EDTA (500 mM) | 2 mM | 2 mL |
| Total | N/A | 500 mL |
Note: Prepare FB1 one day before the experiment. Store FB1 at 4°C for a maximum of 4 weeks.
Note: FB1 with high FCS content is used during the cell isolation procedure after the enzymatic digestion and the mechanical homogenization to promote cell survival.
Red Blood Cell Lysis Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Red Blood Cell Lysis Solution (10×) | 1× | 100 μL |
| H2O | N/A | 900 μL |
| Total | N/A | 1 mL |
Note: Prepare Red Blood Cell Lysis Solution on the day of the experiment for single use and keep it at 18°C–25°C.
FACS Buffer 2 (FB2)
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS (1×) | N/A | 488 mL |
| Fetal calf serum | 2% | 10 mL |
| EDTA (500 mM) | 2 mM | 2 mL |
| Total | N/A | 500 mL |
Note: Prepare FB2 one day before the experiment. Store FB1 at 4°C for a maximum of 4 weeks.
Note: FB2 with low FCS content is used after step 34 to avoid clumping during FACS sorting.
Note: To prepare the necessary buffers and mastermixes for Smart-seq2, please combine and mix the reagents as specified in the tables below. With the exception of the Hypotonic Buffer, all other mixes should be prepared freshly before each step. All reaction volumes were carefully optimized for a miniaturized version of the Smart-seq2 protocol, and are to be used with cell sorting into 384-well plates. Dispensing of small volumes and handling of samples were also optimized to be carried out on (semi-)automated nanodispenser and liquid handlers.
Hypotonic Buffer (HB)
| Reagent | Final concentration | Amount |
|---|---|---|
| 10% Triton-X 100 | 0.2% | 20 μL |
| Nuclease-free water | N/A | 980 μL |
| Total | N/A | 1 mL |
Note: Store HB at 18°C–25°C for a maximum of 6 month.
Lysis Buffer (LB)
| Reagent | Final concentration | Amount |
|---|---|---|
| Hypotonic Buffer | N/A | 0.45 μL |
| Murine RNase Inhibitor (40 U/μL) | 4 U/μL | 0.05 μL |
| Total | N/A | 0.50 μL |
Note: Store LB at 4°C for a maximum of 4 h. Do not freeze this buffer as it reduces the RNase Inhibitor activity. We recommend to always preparing it fresh right before FACS sorting.
dT-Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Hypotonic Buffer | N/A | 0.124 μL |
| dNTPs (10 mM) | 5 mM | 0.249 μL |
| Poly-dT-UP (10 μM) | 0.5 μM | 0.0244 μL |
| Murine RNase Inhibitor (40 U/μL) | 4 U/μL | 0.0488 μL |
| ERCC spikes 1:2∗106 | N/A | 0.0532 μL |
| Total | N/A | 0.5 μL |
Note: ERCC spikes are optional. To prepare the ERCC dilution, first prepare a 1:100 dilution by mixing 1 μL of ERCC with 9 μL TE buffer (10 mM Tris + 1 mM EDTA, pH 8). This dilution is stable for about 2 weeks at −80°C. Further dilute the 1:100 dilution with nuclease free water until the 2∗106 dilution is reached and use it right away. The final concentration of ERCC may be adjusted for different cell types, but a general rule when working with viable cells is to aim for 0.5%–5% ERCC reads in the sequencing data.
Note: Dispense volume with a nanodispenser for high accuracy and speed.
SMART Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 5× First Strand Buffer of Superscript II | N/A | 2 μL |
| Betaine (5 M) | 1.9 M | 2 μL |
| DTT (100 mM) | 9.6 mM | 0.5 μL |
| MgCl2 (1 M) | 0.012 M | 0.06 μL |
| Nuclease-free water | N/A | 0.64 μL |
| Total | N/A | 5.2 μL |
Note: Store SMART Buffer at −20°C for a maximum of 3 months.
Reverse Transcription Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| SMART Buffer | N/A | 1.3 μL |
| Murine RNase Inhibitor (40 U/μL) | 1.6 U/μL | 0.059 μL |
| TSO (100 μM) | 1.7 μM | 0.025 μL |
| Superscript II Reverse Transcriptase (200 U/μL) | 15.2 U/μL | 0.114 μL |
| Total | N/A | 1.5 μL |
Note: Add the Reverse Transcriptase immediately before use. Dispense volume with a nanodispenser for high accuracy and speed. Prepare this buffer fresh before use and do not store.
cDNA Amplification Master Mix
| Reagent | Final concentration | Amount |
|---|---|---|
| Universal amplification primer (10 μM) | 0.17 μM | 0.05 μL |
| Kapa HiFi HotStart Ready mix (2×) | 1.8× | 2.75 μL |
| Nuclease-free water | N/A | 0.2 μL |
| Total | N/A | 3 μL |
Note: Prepare cDNA Amplification Master Mix fresh before use and do not store.
Tagmentation Master mix A (Vazyme TruePrepTM DNA Library Prep Kit V2 for Illumina)
| Reagent | Final concentration | Amount |
|---|---|---|
| 5× TTBL Buffer | 1× | 0.2 μL |
| TTE Mix V50 | N/A | 0.1 μL |
| Nuclease-free water | N/A | 0.7 μL |
| Total | N/A | 1.0 μL |
Note: Prepare Tagmentation Master mix A fresh before use and do not store.
Tagmentation Master Mix B (Illumina DNA prep BLT Tagmentation)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tagmentation Buffer 1 (TB1) | N/A | 0.2 μL |
| Bead-linked Transposome (BLT) | N/A | 0.2 μL |
| Nuclease-free water | N/A | 0.6 μL |
| Total | N/A | 1.0 μL |
Note: Prepare Tagmentation Master mix B fresh before use and do not store.
AccuBlue BR dsDNA Quantitation Mix
| Reagent | Final concentration | Amount |
|---|---|---|
| BR Buffer | N/A | 20 μL |
| BR Dye | N/A | 0.2 μL |
| BR Enhancer | N/A | 0.2 μL |
| Total | N/A | 20.4 μL |
Note: Prepare AccuBlue BR dsDNA Quantitation Mix fresh before use and do not store.
Step-by-step method details
Excisional punch injury
This step describes how to induce a tissue repair response in mice by performing full-thickness punch injury in the skin.
Note: This protocol describes a model of skin wound healing in which the wounds are left open after injury. No wound dressing is used.
Note: The same protocol can be used for male and female wound cell isolation.
-
1.
Anesthetize a mouse (8–12 weeks old, male or female) by intraperitoneal injection of 100 mg/kg body weight Ketavet and 10 mg/kg bodyweight Xylazin in sterile 0.9% NaCl solution.
-
2.
Cover the eyes of the mouse with eye ointment to avoid drying-out.
-
3.
Shave the back skin of the mouse with an electric shaver for animals.
-
4.
Disinfect the back skin with a paper towel soaked in 70% ethanol.
-
5.
Perform 4 full thickness punch injuries in the back skin left and right of the spine using a biopsy puncher (4 mm in diameter).
-
6.
Keep the mouse on a heating plate at 37°C in a laminar flow hood until it wakes up.
-
7.
After the mouse wakes up, place it into a clean autoclaved cage which should contain autoclaved tissue instead of beddings.
Note: Cage embeddings should not be used up to day 3 after injury as they may contaminate the wound. After this, cages may contain embeddings as a dry scab will cover the wound tissue and protect it from dirt.
-
8.
Exchange the autoclaved tissues daily up to day 3 after injury.
-
9.
Keep the wounded mouse separated from other mice until the wound tissue is excised.
-
10.
To alleviate pain after injury, add 1 mg/mL tramadol to the drinking water up to day 3 after injury.
Note: We recommend tramadol as painkiller because, in our experience, it does not interfere with inflammation and other wound healing mechanisms. To ensure sufficient oral uptake of tramadol we recommend measuring the daily water uptake per mouse (4–7 mL is sufficient).
Generating single-cell suspensions of wound tissue
This step describes how to digest wound tissue in order to generate a single-cell suspension for FACS staining.
Note: The day of wound excision after skin injury depends on the scientific question. To compare gene expression in inflammatory macrophages in the early phase of healing with the resolution phenotype of macrophages in the late phase of healing, we recommend isolating macrophages at day 2–4 after injury (inflammatory phase) and day 12–14 after injury (resolution phase).
-
11.
Sacrifice the mice according to institutional guidelines.
-
12.
Excise the wound tissues with forceps and scissor. Place the wound tissue onto a wooden spatula (Figure 1A).
-
13.
Remove the skin surrounding the wound tissue. Leave 0.5–1.0 mm of normal skin around the wound tissue because the wound edge region contains a high number of inflammatory cells (can be seen histologically) that may be lost if the normal skin is completely removed (Figure 1B).
-
14.
Cut the wound tissue with a scalpel into small pieces (∼ 1 mm edge length) (Figure 1C).
-
15.
Transfer the cut wound tissue into a 1.5 mL tube filled with 1.2 mL of DMEM medium.
-
16.
Add 30 μg/mL Liberase TM Research Grade into the DMEM medium to digest the wound tissue enzymatically.
-
17.
Incubate the wound tissue in the digestion medium for 1 h at 37°C in a ThermoMixer (shaking at 400/min).
Pause point: The incubation time can be used for a break. However, it is critical to mix the wound tissue manually every 15 min by taking the tube out of the ThermoMixer and shaking it.
-
18.
Open the lid of a Medicon.
-
19.
Aspirate the digested wound tissue together with the digestion medium and transfer it into a Medicon using a 1 mL pipette tip (Figure 1D).
Note: Truncating the pipette tip with a scissor in order to widen the opening makes it easier to aspirate the pieces of digested wound tissue.
-
20.
Close the lid of the Medicon and homogenize the tissue mechanically for 5 min (Figure 1E).
-
21.
Aspirate wound cell suspension with a 1 mL syringe and pour it through a 70 μM cell strainer into a 50 mL tube placed on ice (Figure 1F).
-
22.
Rinse the Medicon with 2 mL DMEM and pour it through the 70 μM cell strainer into the 50 mL tube.
-
23.
Centrifuge cell suspension at 300 × g for 8 min at 4°C.
-
24.
Aspirate supernatant carefully and discard it.
-
25.
Resuspend the cell pellet in 2 mL of FB1.
-
26.
Aspirate wound cell suspension and pour it through a 40 μM cell strainer into a 50 mL tube.
-
27.
Rinse the 40 μM cell strainer with 3 mL of FB1.
-
28.
Centrifuge cell suspension at 300 × g for 8 min at 4°C.
-
29.
Aspirate supernatant carefully and discard it.
-
30.
Add 1 mL of Red Blood Cell Lysis Solution and incubate at 18°C–25°C for 2 min.
-
31.
Centrifuge cell suspension at 300 × g for 8 min at 4°C.
-
32.
Aspirate supernatant carefully and discard it. Resuspend the cell pellet in 200 μL of FB1.
-
33.
Transfer cell suspension into a 96 well conical (V) bottom plate for FACS staining.
Figure 1.

Generating single-cell suspensions of wound tissue
(A) Representative photographs of wound tissue excised at 4 days after injury.
(B) Removal of skin surrounding the wound tissue with a scalpel.
(C) Mincing the wound tissue with a scalpel for enzymatic digestion.
(D and E) (D) Transfer of wound tissue after enzymatic digestion into a Medicon and (E) homogenization for 5 min.
(F) Aspiration of single-cell suspension with a 1 mL syringe.
FACS sorting of wound macrophages
This step describes how to stain wound macrophage populations and how to index FACS-sort single macrophages into 384-well plates.
-
34.
Centrifuge 96-well plate at 300 × g for 5 min at 4°C.
-
35.
Aspirate supernatant and discard it. Resuspend the cell pellet in 100 μL of FB2 with anti-CD16/CD32 and incubate for 10 min at 4°C.
-
36.
Centrifuge 96-well plate at 300 × g for 5 min at 4°C.
-
37.
Aspirate supernatant and discard it. Resuspend the cell pellet in 100 μL of antibody mix (see table below) and incubate for 20 min at 4°C in the dark.
Antibody Mix
| Antigen | Fluorophore | Clone | Stock concentration | Final concentration |
|---|---|---|---|---|
| CD45 | FITC | 30-F11 | 0.2 mg/mL | 1.0 μg/mL |
| CD11b | APC-eFluor780 | M1/70 | 0.2 mg/mL | 0.5 μg/mL |
| F4/80 | PE | Cl:A3-1 | 100 tests | 10 μL/test |
| Trem2 | APC | 237920 | 100 tests | 10 μL/test |
| CD301b | PerCP/Cy5.5 | URA-1 | 0.2 mg/mL | 4 μg/mL |
| MHC II | eFluor450 | M5/114.15.2 | 0.2 mg/mL | 1 μg/mL |
-
38.
Centrifuge 96-well plate at 300 × g for 5 min at 4°C.
-
39.
Aspirate supernatant and discard it. Resuspend the cell pellet in 200 μL of FB2.
-
40.
Centrifuge 96-well plate at 300 × g for 5 min at 4°C.
-
41.
Aspirate supernatant and discard it. Resuspend the cell pellet in 200 μL of FB2.
-
42.
Centrifuge 96-well plate at 300 × g for 5 min at 4°C.
-
43.
Aspirate supernatant and discard it. Resuspend the cell pellet in 100 μL of FB2.
-
44.
Prior to FACS-sorting add 50 ng/mL DAPI and pour the cell suspension through a 40 μM cell strainer into a FACS tube. Keep the sample tubes on ice and in the dark.
Note: To excite DAPI fluorescence a flow cytometer equipped with a violet (405 nm)/UV (355 nm) laser is required. In case the flow cytometer has only a 405 nm laser line, either eFluor450/Brilliant Violet 421/Pacific Blue coupled antibodies or DAPI can be used. Alternatively, propidium iodide (1 μg/mL final concentration) can be used.
Note: The following procedure is established for the use of a BD FACS ARIA II/III cell sorter equipped with automated cell deposition unit (ACDU) and BD FACS Diva software version 7 or higher.
-
45.
Setup your flow cytometer: activate appropriate laser lines, make sure that filters and mirrors match your fluorochromes, activate desired parameters, setup PMT voltages and perform compensation with single stain controls. Fluorochrome spill-over should be minimized to avoid high compensation. Appropriate compensation as well as autofluorescence for each fluorescence parameter can be determined with a set of FMO (fluorescence minus one) controls (i.e., prepare a set of control stainings with leaving out each antibody once) (Herzenberg et al., 2006; Roederer 2002).
-
46.
Install a 100 μm nozzle and the splash shield.
-
47.
Bring the ACDU to the front.
-
48.
Optimize drop delay (see BD FACS Diva reference manual).
-
49.
Install collection plate (384-well plate preloaded with 0.5 μL of Lysis Buffer per well, leave the plate sealed/covered during setup) and choose the appropriate device layout.
-
50.
Switch on plate voltage and activate “test sort”.
-
51.
Optimize far left side stream to pass through splash shield with minimal deflection.
-
52.
Bring ACDU to home position, deposit drop by shortly activating “test sort” button and inspect deposition of the drop. Adjust home position to hit the center of well A1.
-
53.
To validate accurate set up of the sorting device and cell sorter, sort single polystyrene beads (e.g., BD CaliBRITE™ beads) premixed in PBS with horse radish peroxidase solution (100 μg/mL) into individual wells of plates containing 1 μL of 3,3′,5,5′-Tetramethylbenzidine (TMB) solution (BioLegend, see Rodrigues and Monard, 2016). Wells that were correctly sorted with a single bead will turn blue within minutes.
-
54.
Set sort precision “single cell” and target event = 1.
-
55.
Choose as target cell population gate DAPI- single CD45+CD11b+F4/80+ macrophages.
Note: To account for the heterogeneity of wound macrophages, our protocol is designed to broadly identify wound macrophages based on expression of CD45, CD11b, F4/80. However, additional markers such as MHC II, Trem2, CD301b, Ly6C, CCR2, and CD64 (which has been used to identify dermal macrophages), can be added for sorting and/or as an index marker (Joshi et al., 2020; Kolter et al., 2019; Tamoutounour et al., 2013; Willenborg et al., 2012). Furthermore, lineage markers, e.g., CD3ε, NK1.1, B220, Siglec F and Ly6G can be used to further help exclusion of non-macrophage cell populations.
Note: Signal intensities of FSC, SSC and fluorescence parameters are relative and depend on the individual settings of the flow cytometer (e.g., PMT voltages, filters and mirrors). Before starting the sorting, we recommend backgating CD45+CD11b+F4/80+ cells to ensure no macrophage subpopulation is missed and gates are appropriately placed. Typically, we recover about 35%–40% (4 days post injury) and 80%–90% (day 14 post injury) of CD45+CD11b+F4/80+ cells among total CD45+ cells. In addition, FMO controls can help in determining appropriate gating thresholds for populations with dim separation.
-
56.
Check “Index Sorting” option.
-
57.
Begin with index sorting of single macrophages.
-
58.
Record for each sort run a data file that will contain all recorded events. In addition, record an index sort file, containing the well ID and the corresponding optical parameters (i.e., FSC, SSC and fluorescence intensity) of the sorted event. This data can be merged with gene expression data.
Since a high abundance of debris and red blood cells can massively increase the sorting time, we strongly recommend to use cell strainers to remove aggregates (steps 21 and 26) and to perform lysis of red blood cells (step 30). Adjust the cell suspension to a cell density of approximately 5 × 106/mL. A higher dilution of the cell suspension increases the sorting time; at lower dilutions, target cell events are more likely to be missed. Leave the sorting plate sealed as long as possible to prevent evaporation of the Lysis Buffer and uncover the plate immediately before sorting.
-
59.
Seal plates with adhesive foil and centrifuge plates at minimum 100 × g for 1 min at 4°C to collect all liquid at the bottom. Freeze the plates at −80°C for at least 1 h or not longer than 4 months.
Single-cell RNA sequencing following the Smart-seq2 workflow
This step describes cDNA synthesis, cDNA amplification, cDNA purification, and library preparation after wound macrophages were FACS-sorted and lysed in 384-well plates.
Note: The workflow described below is a modified version of the original Smart-seq2 protocol (Picelli et al., 2014). Adaptations include miniaturization of reaction volumes (thus changing enzyme ratios) and implementing automation in certain steps of the protocol for increased speed and accuracy, and to minimize costs. Additionally, we use blocked poly-dT and amplification primers to reduce background coming from, e.g., primer dimers, and added control samples (positive control of isolated RNA and bulk RNA) for troubleshooting. Importantly, here we describe two methods for library preparation. Although both are based on tagmentation, they differ from what was presented in the original method.
-
60.
Thaw the plates on ice.
-
61.
Add 0.5 μL of dT-Buffer (as described in the materials and equipment section) to each well of the 384-well plate with a nanodispenser. Heat-seal the plate (1 s at 166°C), mix well and spin it down.
Note: The ERCC in the dT-Buffer mix is optional. It supports the detection of low-quality cells (which will show a significant ERCC proportion) or differences in overall RNA content (inactive cells with low RNA content will show higher ERCC proportions than active cells) during the subsequent analysis. Further, the ERCC can be used to estimate the detection sensitivity on levels of expressed genes, as the mix contains 92 different constructs with varying length, GC content and concentration.
Note: We prefer heat-sealing over adhesive foils as we often observed evaporation at the wells located in the corners of the plate.
Note: The optimal sealing temperature and time can vary for different plate types, foils and/or heat-sealing systems. Please ensure that the plate is properly sealed by (carefully) lifting the foil at the corners.
-
62.
Denature RNA by incubating the plate for 3 min at 72°C.
-
63.
Prepare the Reverse Transcription Buffer on ice as described in the materials and equipment section.
-
64.
Add 1.5 μL to each well of the 384-well plate using a nanodispenser, heat-seal the plate (1 s at 166°C) and spin it down. Vortex thoroughly and spin it down once more.
-
65.
Perform Reverse Transcription as follows.
Reverse Transcription
| Temperature | Time |
|---|---|
| 42°C | 90 min |
| 70°C | 15 min |
| 4°C | hold |
Note: Plates can be kept at 4°C for 8–16 h in the thermocycler or frozen at −80°C for 8–16 h (preferred).
-
66.
Prepare cDNA Amplification Master Mix as described in the materials and equipment section.
-
67.
Add 3 μL of the cDNA Amplification Master Mix to each well with a nanodispenser, heat-seal the plate (1 s at 166°C), vortex the plate and spin it down.
-
68.
Amplify cDNA with the following cycling conditions.
cDNA amplification
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 3 min | 1 |
| Denaturation | 98°C | 20 s | |
| Annealing | 67°C | 15 s | 23 |
| Extension | 72°C | 6 min | |
| Final extension | 72°C | 5 min | 1 |
| Hold | 4°C | forever | |
Note: Amplified cDNA can be kept at 4°C for 8–16 h or at −20°C for a few days. The purification of the reaction should take place within 72 h after cDNA amplification. The purified cDNA can then be stored for longer.
-
69.Purify the amplified cDNA on a liquid handling robot with magnetic bead purification as follows:
-
a.Add 9 μL of magnetic beads to each well, mix well, and incubate for 5 min at 18°C–25°C followed by 3 min incubation on the magnet.
-
b.Wash the beads twice with 30 μL of 80% ethanol.
-
c.If necessary, dry bead pellet by vacuum centrifuge for 8 min at 30°C to remove any remaining ethanol.
-
d.Elute beads in 12 μL nuclease-free water.
-
e.Let beads and cDNA rehydrate at 18°C–25°C for approximately 1 h.
-
a.
Note: Samples can be stored at 4°C for 8–16 h or at −20°C for a maximum of 6 months.
-
70.
Analyze fragment size distribution and measure concentration of the cDNA of few randomly picked samples using a fragment analyzer. Measure the concentration of all samples in a microplate reader by loading 20 μL of the AccuBlue BR dsDNA Quantitation Mix into each well of a 384-well black LV plate and adding 1.5 μL of the purified cDNA. In parallel, always prepare a standard row (in triplicates) covering the concentration range of the samples. We recommend at least eight measure points: 10, 8, 6, 4, 2, 1, 0.5, 0 ng/μL of standard. Make sure to use appropriate excitation and emission parameters for the measurements.
Note: Below, we describe two protocols for library preparation. The first protocol, described in step 71, uses a library preparation kit that is currently not manufactured, but we opt for detailing this workflow because it was used in the study by Willenborg et al. (2021). Since then, we optimized the library preparation to use the currently available library prep chemistry from Illumina, using the bead linked transposome (BLT). This second protocol is described in step 72.
-
71.Library preparation using the Vazyme TruePrep DNA Library Prep Kit V2:Note: This kit is currently not available.
-
a.For library preparation, scale all samples to a concentration of 350 pg/μL (for lower concentrated samples, use 2 μL).Note: Some samples will likely have concentrations below the detection limit because they are low in RNA content and/or the sort failed.
-
b.Transfer 2 μL (700 pg) of the amplified cDNA in an empty 384-well plate.
-
c.Desiccate the liquid completely by vacuum centrifuge for 15 min at 30°C.
-
d.In the meantime, thaw all reagents from the Tagmentation Master Mix A on ice and, once thawed, prepare the mix as described in the materials and equipment section.
-
e.Add 1 μL of Tagmentation Master Mix A with a nanodispenser to each well, heat-seal the plate for 1 s at 166°C, vortex vigorously and spin it down.
-
f.Incubate the plate in a pre-heated thermal cycler using the following program.
Step Temperature Time 1 20°C 5 min 2 55°C 5 min 3 10°C hold -
g.To amplify the tagmented DNA, add 3 μL of the KAPA High-Fidelity Hot Start ReadyMix (2×) and 2 μL of P5 and P7 barcoded Nextera amplification primers (1 μM of each) to a total reaction volume of 6 μL.Note: Barcode sets can be prepared in advance by combining P5 and P7 primers to generate a dual-indexing primer set, for example by distributing each P7 barcode row-wise and each P5 column-wise. The barcode plate can be stored at −20°C and thawed shortly before use.
-
h.Heat-seal the plate, vortex and spin it down.
-
i.Amplify tagmented DNA with the following cycling conditions (pre-heat lid to 105°C).Tagmented DNA amplification
Steps Temperature Time Cycles 1 72°C 3 min 1 2 98°C 30 s 1 98°C 10 s 3 63°C 30 s 13 72°C 1 min 4 72°C 5 min 1 5 4°C Hold 1 Note: Samples can be kept at 2°C–8°C for up to 24 h or frozen at −20°C for longer. -
j.Add 10 μL of nuclease-free water to each sample to reach a total volume of 16 μL.
-
k.Heat-seal the plate at 166°C for 1 s, vortex thoroughly and spin it down.
-
a.
-
72.
Library preparation using the Illumina DNA prep Tagmentation kit:
Note: This is the currently established protocol. Users should refer to step 72 for library preparation.
-
73.Quantification of libraries and pooling.
-
a.Measure the concentration of the unpurified libraries in a microplate reader by loading 20 μL of the AccuBlue BR dsDNA Quantitation Mix into each well of a 384-well black LV plate and adding 1.5 μL of the unpurified library. In parallel, always prepare a standard row (in triplicates) covering the concentration range of the libraries. We recommend 12 measure points: 80, 60, 40, 20, 10, 8, 6, 4, 2, 1, 0.5 of standard and a blank value.
-
b.Seal the plate with a foil, vortex, spin it down and incubate for at least 10 min at 18°C–25°C in the dark.
-
c.Before measurement, remove foil and quickly spin down the plate. Measure with a plate reader using appropriate excitation and emission parameters.
-
d.Evaluate library concentration according to the values measured in the standard row.
-
e.Pool libraries equimolarly according to reader measurements and desired sequencing output. It is advisable to pool libraries with similar concentrations.
-
a.
-
74.Library purification and size selection.
-
a.For purification of library pools, add 0.9× of Agencourt XP beads (or comparable) and pipette up and down to mix well. Quickly spin down the tubes and incubate at 18°C–25°C for 5 min.
-
b.Place tubes on a magnetic rack and discard supernatant after beads attach to the magnet.
-
c.Keep tubes on magnetic rack and wash library pools twice with 200 μL of freshly-prepared 80% EtOH. For each wash, wait for 30 s before removing the EtOH. Pulse-spin the tubes, place them back in the magnetic rack and remove any residual EtOH.
-
d.Air-dry the beads at 18°C–25°C for approximately 5 min.
-
e.For size selection, first hydrate the beads with nuclease-free water and add 0.6× volume of magnetic beads to each library pool. Incubate at 18°C–25°C for 5 min.
-
f.Place tubes on the magnetic rack to separate beads from buffer, and transfer the supernatants (that contain the fragments of interest) into a new tube.
-
g.Add 0.9× of magnetic beads to each library pool and incubate at 18°C–25°C for 5 min. Place tubes on the magnetic rack to separate beads from buffer, and discard all supernatant.
-
h.Keep tubes on magnetic rack and wash library pools twice with 200 μL of freshly-prepared 80% EtOH. For each wash, wait for 30 s before removing the EtOH. Pulse-spin the tubes, place them back in the magnetic rack and remove any residual EtOH.
-
i.Air-dry the beads at 18°C–25°C for approximately 2 min. Beads should have a rusty color when dry.Note: Avoid excessive drying of the beads (cracks will form) to minimize the loss of DNA.
-
j.Dissolve the beads with appropriate volume of nuclease-free water.Note: Expected concentrations will be ∼50% lower than those determined by the microplate reader measurements for each library pool. This is mainly due to loss of material during size selection and depletion of primer dimers.
-
k.Place the tubes back on the magnetic rack. When solution has cleared, transfer all supernatant to a clean PCR tube.
-
l.Analyze library size distribution and concentration using a fragment analyzer.Optional: If necessary, an additional 0.9× purification may follow to avoid fragments smaller than 200 bp.
-
a.
-
75.
RNA sequencing can be performed as desired, choosing read sizes and sequencing depths that better address the research question. We find that 0.5 mio to 1.5 mio reads per cell (paired-end 50–100 bp) is a good setting for most experiments.
Expected outcomes
This protocol is intended for the isolation of macrophages from mouse skin wounds for scRNA-seq using the Smart-seq2 workflow. Depending on the time-point after injury, approximately 50,000 (day 14 after injury) – 200,000 (day 4 after injury) CD45+CD11b+F4/80+ wound macrophages can be obtained from 4 wounds of a single mouse. Cell viability ranges from approximately 75 - 85% when determined by DAPI staining and FACS analysis (Figures 2A and 2B). FACS analysis generates highly reproducible results for surface markers (e.g., CD301b, Trem2, MHC II) that can be used to differentiate macrophage subpopulations and that can be combined with the gene expression data after RNA sequencing.
Figure 2.

Gating strategy for FACS-sorting of wound macrophages
(A and B) Representative flow cytometry plots at (A) 4 and (B) 14 days post injury. DAPI- single cells were gated (Gates G1-G4) and expression of CD45 was analyzed. CD45+ cells were gated (G5) and analyzed for expression of CD11b and F4/80. Single CD45+CD11b+F4/80+ wound macrophages (G6) were sorted in 384-well plates for RNA sequencing. CD45+CD11b+F4/80+ wound macrophages can further be analyzed for expression of Trem2, CD301b, and MHC II. Index sorting allows merging FACS data with gene expression data at single cell level.
The Smart-seq2 approach, coupled with careful dissection of skin wounds and subsequent FACS-sorting of macrophages, makes it possible to sequence and study macrophage heterogeneity over the time course of the healing response with great specificity. This specificity comes at the cost of a limited number of cells that can be processed, both in terms of experimental capacity and time. This is contrasted by other single-cell sequencing approaches based on microfluidics that allow for a much higher capture and sequencing throughput of thousands of cells. In contrast to high-throughput single-cell methods, however, the gene detection sensitivity achieved with the Smart-seq2 workflow is generally higher, mostly because the lower number of cells per experiment allows for each cell to be sequenced more deeply while keeping sequencing costs to a reasonable amount.
The Smart-seq2 protocol is designed to amplify and sequence the full transcript, unlike other methods which capture only 3′ or 5′ ends of the mRNA. This is advantageous not only because it allows for the detection of a higher number of genes, but also for downstream quantitative and qualitative analysis, giving deeper insights into the heterogeneity of expression both within and between cells. Details of scRNA-seq analysis can be found in our recent publication (Willenborg et al., 2021).
Limitations
To ensure proper cell viability before cell sorting and high RNA quality, it is critical to proceed as fast as possible after wound excision. Therefore, the number of individual mice in one experiment should be limited in order to avoid delays.
The experimental workflow that follows FACS sorting, from the Reverse Transcription reaction until library preparation, was designed to the 384-well plate format. Reagents and reaction mixes were carefully optimized for a miniaturized version of the Smart-seq2 protocol, and do not necessarily scale to other plate formats (e.g., 96-well plates) or tubes.
Given the low volumes used for almost all steps from Reverse Transcription reaction to library prep, access to nanodispensing and liquid handling robots is essential. This ensures i) high precision in volume dispensing, necessary for miniaturized reactions, and ii) speed in processing of the plates, necessary to avoid evaporation.
The miniaturized reaction volume also limits sorting times and conditions. In order to avoid evaporation of the Lysis Buffer, sorting should be done in a cooled system and should last no longer than 1 h.
A precise set up of cell sorting is critical, in order to ensure that the cell lands directly in the middle of the well – and thus directly into the Lysis Buffer. Inaccurate dispensing with the FACS instrument may lead to cells being stuck on the walls of the wells, which results in RNA degradation. We recommend the following colorimetric assay to ensure that cells are dispensed correctly into the center of the well: https://www.well.ox.ac.uk/ogc/a-colorimetric-method-to-determine-efficiency-of-facs-sorting/.
Tagmentation of the cDNA for library preparation was done using the Vazyme TruePrepTM DNA Library Prep Kit V2 for Illumina, which is currently not available. Thus, we offer an alternative protocol for library preparation that uses the Illumina DNA Prep Tagmentation chemistry, based on bead-linked transposase, in parallel. The workflow has been carefully optimized for such low-volume reactions, and is described in step 72.
As with the original protocol (Picelli et al., 2014), the Smart-seq2 workflow described here is limited to sequencing of polyadenylated RNAs and lacks the strand specificity of the mRNA transcripts.
Troubleshooting
Problem 1
The viability of the cells as determined by FACS in gate 3 is lower than 70% (step 55 and Figure 2).
Potential solution
After wound excision, cell viability decreases with time. We recommend preparing all buffers and reagents prior to each step to avoid delays. It is important that the centrifuges have reached 4°C before starting the experiment. Make sure that the incubation time in digestion medium (with Liberase) does not exceed 1 h and that from step 21 on the cells and all buffers are kept on ice or at 4°C. Keep the incubation time with Red Blood Cell Lysis Buffer no longer than 2 min.
Problem 2
The proportion of macrophages as determined by FACS analysis in gate 6 is too low (lower than 25%) (step 55 and Figure 2). Macrophage populations cannot be gated properly (step 55 and Figure 2).
Potential solution
The experimental procedure described in this protocol requires in-depth expertise in multicolor flow cytometry. We recommend titrating all the antibodies used in step 37 carefully in your own lab. Make sure that the instrument setup (e.g., voltage, compensation) of the flow cytometer is correct. To maximize the total cell number, make sure to excise the granulation tissue of the wound (which contains the immune cells) properly (step 12). Furthermore, make sure that the excised wound tissue is constantly shaking at 400/min at 37°C and that the tubes are shacked manually every 15 min (step 17). Be careful when aspirating and discarding the supernatant in steps 24, 29, 32, 35, 37, 39, 41, and 43.
Problem 3
cDNA and library yields are low (steps 57, 60–65, 70, 71a, 72a, 73a).
Potential solution
Low cDNA and library yields can be due to many different factors affecting the integrity of the RNA. To troubleshoot for low yields, first make sure the cell sorting set up is precisely adjusted for the plate format so that cells land directly to the bottom of the wells and into the Lysis Buffer. To avoid RNA degradation, make sure working benches are properly cleaned to avoid unwanted and accidental contamination with environmental RNases. It is also important to work with plates on ice until step 68, where double-stranded cDNA is generated.
We noted that increasing the amount of RNase inhibitor to the sorting buffer actually led to a decrease in yield, likely due to a reduction of the efficiency of the Reverse Transcription reaction and/or amplification. Thus, we advise to follow the concentration specified in this protocol. Low yields can also happen when working with cells that are too small or silenced. In this case, it is possible to increase the number of cycles for cDNA amplification – but be careful as to not over-amplify the material, which can lead to preferential amplification of certain fragments and, consequently, to biased libraries.
We recommend using a few different controls during the process to check the optimal conditions of instrument set up, reagents, and input material. First, include one well with a bulk control to the experiment with 10–20 cells, to check and compare fragment size distribution. Second, use a positive control of isolated total RNA (40 pg) to check the quality of reagents and make sure cycling programs went through as expected.
A thorough mix of all reagents prior to preparing buffers and master mixes and after adding it to the samples is critical to ensure the quality of the reaction. Failure to do so, especially with enzymes, can lead to a failed experiment.
Problem 4
Fragment sizes of sequencing library out of optimal range (200–700 bp) (steps 71a, 72a, 73a).
Potential solution
The optimal library size distribution ranges from 200–700 bp with a peak at 300–400 bp (Figure 3), when libraries are sequenced on an Illumina platform with short reads (for example up to 150 bp). If libraries are too short (peak smaller than 250 bp) or too long (peak larger than 600 bp), we recommend starting the library preparation again with different amounts of the cDNA and/or of the tagmentation enzyme to increase/decrease the efficiency of the tagmentation reaction. For example, for libraries with short fragment sizes, we recommend increasing the amount of cDNA and/or decreasing the amount of tagmentation enzyme. For libraries with longer fragments, the opposite should be done.
Figure 3.

Examples of libraries with optimal and suboptimal fragment size distributions
An optimal library (black line) has fragments ranging from 200–700 bp with a peak at around 400 bp. Examples of suboptimal, short and long libraries are depicted by yellow and red lines.
Problem 5
Unusually high ERCC content in sequencing data (step 61).
Potential solution
A too high ERCC spike-in content in the sequencing data can be due to low mRNA content in the cells (for example when working with small cells). However, if cells are damaged the ratio between mRNA and ERCC will be lower, which will lead to a higher amplification of the ERCC spike-in. If unusual high ERCC percentages are observed in the data, the percentage of mitochondrial reads should be monitored to additionally check cell quality and integrity: a mitochondrial content much higher than 20% is a sign of a damaged cell for most cell types – thus, indicates low quality input.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sabine Eming (sabine.eming@uni-koeln.de).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation): CRC829 (project number 73111208 to S.A.E.), the CRC1218 (project number 269925409 to S.A.E.), the Research Unit FOR2599 (project number 3927 49992 to S.A.E. and A.R.), the CRC1403 (project number 414786233 to S.A.E.), Germany’s Excellence Strategy – CECAD, EXC 2030 – 390661388 to S.A.E.), the Research Unit FOR2240 (EM48/5-2 to S.A.E.), Center for Molecular Medicine Cologne (to S.A.E.), and the DEBRA International Foundation (to S.A.E.). DcGC is supported by the Deutsche Forschungsgemeinschaft (DFG), Sächsisches Staatsministerium für Wissenschaft, Kultur und Tourismus (SMWK), Bundesministerium für Bildung und Forschung (BMBF), Center for Molecular and Cellular Bioengineering (CMCB), Max Planck Institute of Molecular Cell Biology and Genetics (MPI-CBG), and Paul Langerhans Institut Dresden (PLID).
Author contributions
S.W., J.G.R., A.G., and S.R. wrote the manuscript. S.R. developed and implemented the Smart-seq2 protocol described here and performed the experimental part corresponding to this method. A.D. supervised the developmental work at the DcGC. A.R., A.D., and S.A.E. supervised the study. All authors commented on and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Sabine A. Eming, Email: sabine.eming@uni-koeln.de.
Susanne Reinhardt, Email: susanne.reinhardt@tu-dresden.de.
Data and code availability
This study did not generate datasets. The single-cell RNA sequencing dataset mentioned in this protocol has been described in the study by Willenborg et al. (2021).
References
- Ding X., Willenborg S., Bloch W., Wickström S.A., Wagle P., Brodesser S., Roers A., Jais A., Brüning J.C., Hall M.N., et al. Epidermal mammalian target of rapamycin complex 2 controls lipid synthesis and filaggrin processing in epidermal barrier formation. J. Allergy Clin. Immunol. 2020;145:283–300.e8. doi: 10.1016/j.jaci.2019.07.033. [DOI] [PubMed] [Google Scholar]
- Herzenberg L.A., Tung J., Moore W.A., Herzenberg L.A., Parks D.R. Interpreting flow cytometry data: a guide for the perplexed. Nat. Immunol. 2006;7:681–685. doi: 10.1038/ni0706-681. [DOI] [PubMed] [Google Scholar]
- Joshi N., Pohlmeier L., Greenwald M.B.Y., Haertel E., Hiebert P., Kopf M., Werner S. Comprehensive characterization of myeloid cells during wound healing in healthy and healing-impaired diabetic mice. Eur. J. Immunol. 2020;50:1335–1349. doi: 10.1002/eji.201948438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuever J., Willenborg S., Ding X., Akyüz M.D., Partridge L., Niessen C.M., Brüning J.C., Eming S.A. Myeloid cell-restricted insulin/IGF-1 receptor deficiency protects against skin inflammation. J. Immunol. 2015;195:5296–5308. doi: 10.4049/jimmunol.1501237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter J., Feuerstein R., Zeis P., Hagemeyer N., Paterson N., d'Errico P., Baasch S., Amann L., Masuda T., Lösslein A., et al. A subset of skin macrophages contributes to the surveillance and regeneration of local nerves. Immunity. 2019;50:1482–1497.e7. doi: 10.1016/j.immuni.2019.05.009. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Rodrigues O.R., Monard S. A rapid method to verify single-cell deposition setup for cell sorters. Cytom. A. 2016;89:594–600. doi: 10.1002/cyto.a.22865. [DOI] [PubMed] [Google Scholar]
- Roederer M. Compensation in flow cytometry. Curr. Protoc. Cytom. 2002;Chapter 1:Unit 1.14. doi: 10.1002/0471142956.cy0114s22. [DOI] [PubMed] [Google Scholar]
- Tamoutounour S., Guilliams M., Sanchis F.M., Liu H., Terhorst D., Malosse C., Pollet E., Ardouin L., Luche H., Sanchez C., et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. 2013;39:925–938. doi: 10.1016/j.immuni.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Willenborg S., Lucas T., van Loo G., Knipper J.A., Krieg T., Haase I., Brachvogel B., Hammerschmidt M., Nagy A., Ferrara N., et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood. 2012;120:613–625. doi: 10.1182/blood-2012-01-403386. [DOI] [PubMed] [Google Scholar]
- Willenborg S., Sanin D.E., Jais A., Ding X., Ulas T., Nüchel J., Popović M., MacVicar T., Langer T., Schultze J.L., et al. Mitochondrial metabolism coordinates stage-specific repair processes in macrophages during wound healing. Cell Metab. 2021;33:2398–2414.e9. doi: 10.1016/j.cmet.2021.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate datasets. The single-cell RNA sequencing dataset mentioned in this protocol has been described in the study by Willenborg et al. (2021).