

Abstract
Diabetes is one of the most common disorders that substantially contributes to an increase in global health burden. As a metabolic disorder, diabetes is associated with various medical conditions and diseases such as obesity, hypertension, cardiovascular diseases, and atherosclerosis. In this review, we cover the scientific studies on sodium/glucose cotransporter (SGLT) inhibitors published during the last decade. Our focus on providing an exhaustive overview of SGLT inhibitors enabled us to present their chemical classification for the first time.
Diabetes is one of the most common disorders that substantially contributes to an increase in global health burden.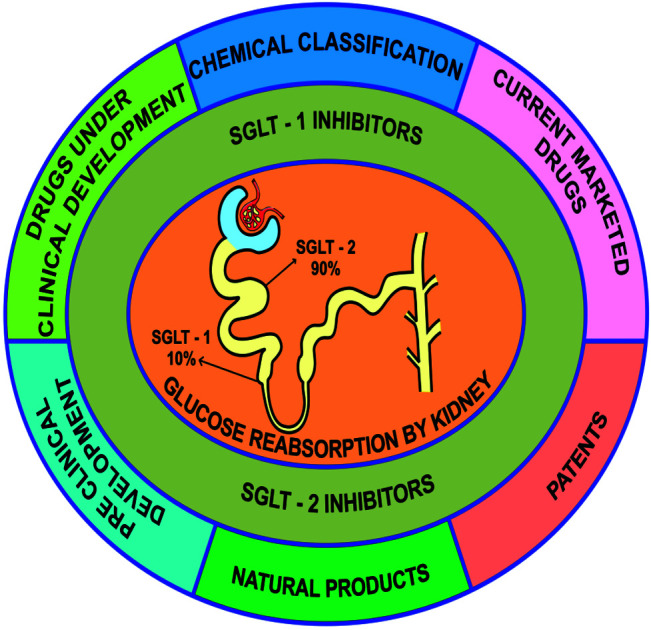
1. Introduction
Diabetes mellitus (DM) is a common disorder associated with metabolic dysfunction that affects people all around the world. In 2011, the estimated prevalence of DM was 366 million cases, which are predicted to increase to approximately 552 million by 2030.1 The growing pervasiveness of diabetes has been linked to an increasing global health burden over the last several decades.2 DM is often linked to various other chronic conditions and disorders such as obesity, hypertension, cardiovascular diseases or atherosclerosis, resulting in a significant decrease in life expectancy. It also increases the associated healthcare expenditure. The etiology of DM is multifaceted, hence, its management is also a challenging task.1,2
Existing glucose-reducing treatments mostly target skeletal muscles, liver, pancreas, adipose tissues, and the small intestine. This glucose-lowering action is mediated by the direct or indirect regulation of endogenous insulin, which further regulates the utilization of tissue glucose in the patient.3 Retinopathy, nephropathy, and neuropathy are associated complications (secondary microvascular complications) that can also be avoided with these treatments. A change in lifestyle along with a healthy and restricted diet is beneficial for long-term metabolic control in diabetic patients, but pharmacological assistance is frequently required for early management in the majority of patients.4–6 Selection of the appropriate drug mostly depends on biochemical and clinical factors. Anti-hyperglycemic therapy is initiated only after a careful consideration of the potential risks and benefits of the treatment for the individual patient.7
2. Current therapy and its limitations
Many studies have shown that lifestyle modification along with a restricted healthy diet can control the glycemic index in diabetes patients, but lifestyle modification on its own is rarely adequate enough to achieve the required glycemic goal. However, along with modification in lifestyle, single drug therapy is not an assured beneficial treatment for attaining long-term glycemic control.6,7 Currently, DM treatment includes more than 10 classes of molecules and several new ones that are being developed. For about 40 years (from the mid-1950s to mid-1990s), only insulin, sulfonylureas, and biguanides were available for treatment. Other classes of DM therapeutics were discovered and developed after 1995.7,8
Improvement in glycemic control is the principal goal of all antidiabetic agents. Similar to agents used in other diseases, the mechanism of action is typically used as a criterion for categorizing antidiabetic agents. Some agents such as metformin, sulfonylureas or insulin principally lower the fasting glucose level, while other agents such as α-glucosidase inhibitors, meglitinides or prandial insulins mainly the lower postprandial glucose level, but the remaining antidiabetics such as thiazolidinediones, dipeptidyl peptidase 4 inhibitors or mixed insulins can perform both functions.8–16
Currently available pharmacological agents can neither control the gradual reduction in the functioning of pancreatic β-cells nor the secretion of insulin.4,6 Accordingly, single and combination therapy can exert the required glycemic control for a short duration and only in a subset of patients associated with type-2 diabetes mellitus (T2DM).5,17–19 Clinical trial programs such as ACCORD are recommended for the most intensive glycemic control, but some patients with T2DM are still unable to attain a target HbA1c level of ≤7%. The reduction rate of the HbA1c level by pharmacological agents depends on the patient's baseline.19 In patients with a baseline HbA1c level of more than 9%, sulfonylureas or metformin can lower the fasting glucose level along with an approximately 2.0% decline in the level of HbA1c. The HbA1c level is reduced by approximately 0.5% in patients with a baseline HbA1c level of less than 7.5%. The agents (e.g. nateglinide or α-glucosidase inhibitors) that can reduce changes in postprandial glucose are also able to decrease the HbA1c level by about 0.4–0.8%. However, the efficiency of these agents and the associated adverse effects limit their use for pharmacological management in T2DM patients.18
Theoretically, insulin preparations are capable of reducing the HbA1c level in T2DM patients to any preferred level. However, when the insulin treatment reduces the HbA1c level from 7.5% to 6.0%, there is also a gradual rise in the occurrence of moderate to severe hypoglycemia. Moreover, consistent weight gain is associated with gradually rigorous insulin therapy using gradually increased dosing.5,19,20
Thiazolidinediones have been shown to reduce insulin resistance. They also improve HbA1c levels when administered in combination with other agents. However, some patients have developed congestive heart failure (CHF) due to fluid retention caused by thiazolidinediones. Also, an increase in the frequency of bone fractures in women is another adverse effect that is associated with long-term treatment using thiazolidinediones.21,22
The incidence of insulin resistance is increased in overweight or obese T2DM patients, thereby complicating disease management. Persistent hyperglycemia, which is associated with hypertension and dyslipidemia, increases the risk of macrovascular complications (peripheral arterial disease, myocardial infarction, and stroke). Hence, the treatment of hypertension and dyslipidemia along with the consequences associated with glucose-lowering therapies has a major effect on cardiovascular morbidity and mortality.
3. New targets of antidiabetic therapeutics explored over the last two decades
Over the last two decades, numerous orally administered compounds such as sulfonylureas and biguanides have been developed to control the plasma glucose level in T2DM patients. These agents target various pathways and have different adverse effects.
3.1. The GLP1 (glucagon-like peptide 1) receptor
The GLP1 (glucagon-like peptide 1) receptor is targeted by agonists such as liraglutide and exenatide that are distinct from other antidiabetic agents. When the plasma glucose levels are elevated, the GLP1 receptor agonists increase insulin and reduce glucagon secretion. These agents are known to delay the gastric emptying time and increase satiety, which reduces the intake of food and thereby causes a modest loss in weight. Like other agents, GLP1 receptor agonists are also associated with unwanted side effects such as nausea and vomiting. Although these symptoms generally diminish with continued treatment, approximately 5–15% of patients have to stop using these agents. According to the US Food and Drug Administration (US FDA), GLP1 receptor agonists increase the risk for acute pancreatitis and thyroid medullary carcinoma.14,23,24
3.2. Alpha-glucosidase
Alpha-glucosidase, a crucial enzyme involved in carbohydrate metabolism, is targeted by agents such as acarbose, miglitol, and voglibose that competitively inhibit this enzyme located in the small intestine, thereby delaying carbohydrate absorption. The main effect of these inhibitors is a reduction in the postprandial glucose level. Because the alpha-glucosidase inhibitors have been found to alter only the postprandial glucose level but not the fasting plasma glucose level, the treatment results in a mild reduction in HbA1c. Their known side effects include gastric discomfort such as flatulence, diarrhea, bloating, and abdominal cramps.25,26
3.3. Dipeptidyl peptidase 4 (DPP-4)
Dipeptidyl peptidase 4 (DPP-4) is located in the cell membrane and functions to reduce GLP1 rapidly. Thus, suppression of DPP-4 can be a beneficial treatment. This effect is glucose-dependent, i.e., it increases insulin secretion and simultaneously suppresses glucagon secretion. Sitagliptin, vildagliptin, linagliptin, saxagliptin, and alogliptin are FDA-approved DPP-4 inhibitors.24,25,27
3.4. Peroxisome proliferator-activated receptor (PPAR-γ)
Peroxisome proliferator-activated receptor (PPAR-γ) is another target for diabetes treatment. By a corresponding agonist effect on muscles; PPAR-γ activation enhances insulin sensitivity and increases glucose uptake because glucose homeostasis is maintained by adipose tissues in the human body. After attaching with a ligand or an agonist like thiazolidinediones, PPAR-γ becomes activated and forms a retinoid X-receptor (RXR) complex. The PPAR-γ–RXR complex then binds to a specific DNA sequence motif, the peroxisome proliferator response element, of certain target genes to regulate their expression. Thiazolidinediones (or glitazones) induce insulin sensitivity mainly by acting on PPAR-γ.28
3.5. 11β-Hydroxysteroid dehydrogenase-1 (11β HSD-1)
11β-Hydroxysteroid dehydrogenase-1 (11β HSD-1) converts cortisone to cortisol. Cortisol stimulates glucocorticoid receptors and plays an important role in diabetes. An elevated blood level of cortisol results in a decrease in glucose and fat metabolism and, hence, escalates blood glucose and fat levels, contributing to insulin resistance. Inhibition of 11β HSD-1 by carbenoxolone, a peptic ulcer drug, has been experimentally demonstrated. According to one study, 11β HSD-1 activity was found to be higher in T2DM subjects.29,30
3.6. Glutamine fructose-6-phosphate amidotransferase (GFAT)
Glutamine fructose-6-phosphate amidotransferase (GFAT) is an enzyme that mainly functions in the hexosamine biosynthetic pathway (HBP). Two important effects in diabetes, the development of glucose-induced insulin resistance and vascular complications, are linked to the HBP. GFAT activity in the HBP induces growth factor synthesis.31 GFAT affects uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) synthesis as the product of HBP. Hence, GFAT has attracted attention as a therapeutic target for T2DM treatment.32
3.7. Protein tyrosine phosphatase 1B (PTP1B)
Protein tyrosine phosphatase 1B (PTP1B) is involved in cellular tyrosine phosphorylation. PTP1B inhibitors may act as a new type of agent that sensitize insulin as a treatment in diseases such as diabetes, obesity, and cardiovascular disorders.33
3.8. G-protein coupled receptor (GPR)
G-protein coupled receptor (GPR) represents another antidiabetic target; activation of GPR120 and subsequent GPRs mediates GLP1 secretion, which generally stimulates insulin secretion. Thus, targeting the GPR is a possibility to inhibit glucagon secretion by stimulating insulin secretion; therefore, GPR is considered as one of the possible drug targets for diabetes treatment.34
3.9. Glucose transporter type 4 (GLUT4)
Glucose transporter type 4 (GLUT4) facilitates the transport of glucose into the cells in response to insulin, thereby contributing to the homeostasis of glucose. Importantly, mutations in GLUT4 are associated with T2DM. Hence, there is also an increased interest in exploring GLUT4 as a therapeutic target for diabetes treatment.35
3.10. Sodium/glucose cotransporters (SGLTs)
Sodium/glucose cotransporters (SGLTs) belongs to a membrane protein family that facilitates the transport of compounds such as glucose, amino acids, and vitamins, as well as various ions through the proximal convoluted tubules (PCT) and intestinal epithelium. The sodium/glucose cotransporter 2 (SGLT2) is mainly expressed in the kidneys. Nearly 90% of glucose transported by SGLTs gets reabsorbed in the kidneys, and, therefore, SGLT2 is of great interest in the field of diabetes treatment. Inhibition of these transporters blocks glucose reabsorption, leading to glycosuria. This mechanism is a promising therapeutic approach to improve glycemic control in DM patients.36
4. Need for new targets
Even with the impressive developments in effective therapeutic methods and modern medicines, the search for promising diabetes therapies is still a major ongoing task. Many efforts have been made to improve insulin action on its target tissue and to identify lead compounds that effectively enhance insulin secretion by β cells. Research groups are searching for safer, more convenient, and economical methods to treat diabetes by assessing the antidiabetic activity of natural products and synthetic derivatives on various proteins as novel therapeutic targets.37,38
To improve diabetes management, the discovery and development of new antidiabetic therapeutics should be pursued. Currently, with the exception of α-glucosidase inhibitors, most antidiabetic agents employ an insulin-dependent mechanism. Importantly, sodium/glucose cotransporter-2 (SGLT2) inhibitors have been identified as novel agents for diabetes treatment. They improve the elimination of glucose through urine (urinary glucose excretion, UGE), which decreases renal glucose reabsorption inside the nephron. Because the mechanism of action employed by SGLT2 inhibitors does not involve insulin, these inhibitors can be administered at any stage of T2DM. SGLT2 inhibitors have also been found to exert a promising efficacy in insulin-resistance and β-cell-dysfunction cases, providing an exceptional treatment option in diabetes management.39
5. Sodium/glucose cotransporter (SGLT)
Crane was the first scientist to present the active cotransport concept by describing that the deposition of glucose at the brush border of epithelial cells of the intestine is associated with the transport of sodium ions downward of their gradient.38,40 The first identified cotransporter proteins mediating sodium/glucose and sodium/proline transport were found on the intestinal brush border in rabbits.41
In animal and bacterial cells, more than 220 members of the SGLT family (gene symbol, SLC5A) have been identified to date. In humans, from the epithelial cells to the nerve cells of the central nervous system (CNS), eleven genes encoding members of the SGLT family have been identified. From these eleven genes, studies using heterologous expression systems have revealed the function of nine:
• Sodium/chloride/choline cotransporter.
• Anion transporter.
• Glucose-activated ion channel.
• Six (tightly coupled) sodium/substrate cotransporters42
5.1. SGLT1
The membrane-bound protein SGLT1 has a mass of 75 kDa and is encoded by the SLC5A1 gene. It is mainly found in the cell membranes in the small intestine, kidney (renal proximal tubule), and heart. The transport of sodium and glucose by this protein occurs at a ratio of 2 : 1. SGLT1 has a high affinity for glucose and galactose, but possesses a low capacity to transport them. In the gastrointestinal tract (GIT), SGLT1 serves as a vital transporter for glucose uptake, but its effect on the kidneys has been found to be less significant (approximately 10% reabsorption of glucose has been observed). Mutations in the SGLT1 gene cause a genetic disorder known as glucose-galactose malabsorption (GGM). In newborn infants, this rare autosomal recessive disease leads to life-threatening diarrhea. However, the withdrawal of dietary sugars such as glucose, galactose, and lactose typically stops diarrhea, which rebounds if these dietary sugars are reintroduced in the diet. Researchers have developed an interest in SGLT1 because it has been hypothesized that the inhibition of this transporter could decrease the gastrointestinal glucose uptake and may also induce weight loss or reduce postprandial hyperglycemia.42,43
5.2. SGLT2
SGLT2 as a second membrane-bound SGLT family member is encoded by SLC5A2 in humans. It has a high capacity but low affinity for glucose and galactose; the transport occurs at a stoichiometric ratio of 1 : 1 of the sodium ion to the sugar molecule, which differentiates SGLT2 from SGLT1. It has been shown that SGLT2 is mostly expressed in the kidneys where it functions as the most important transporter (reabsorbs about 90% glucose). In the initial segment of the proximal tubule in the kidneys, the glucose transport is driven during tubular filtration by the reabsorption of sodium ions along their electrochemical gradient via a mechanism that is called secondary active transport. Accordingly, high levels of SGLT2 have been found in the initial segment of the proximal tubule. This transporter has gained much interest in the diabetes field because of its much involvement in the glucose reabsorption process inside the kidneys.42,44,61
5.3. SGLT3
SGLT3 is the third membrane-bound protein, which is encoded by the SLC5A4 gene. The glucose transport occurs at the same sodium to glucose stoichiometry as that of SGLT1. This protein is mostly found in skeletal muscles, the small intestine, the kidneys, and cholinergic neurons. Human SGLT3 is a basic glucose-gated ion channel present in the cell membranes of the muscles and the neuronal membranes but it generally does not act as a sodium/glucose transporter.42,44
The available information on the SGLT4, SGLT5, and SGLT6 proteins is still insufficient. The tissue distribution, substrates, and encoding genes of the discussed SGLT proteins are provided in Table 1.42
Genes, substrates and tissue distribution of SGLT proteins42.
| Human gene | Protein | Substrates | Tissue distribution |
|---|---|---|---|
| SLC5A1 | SGLT1 | Glucose and galactose | Small intestine, kidney and heart |
| SLC5A2 | SGLT2 | Glucose | Kidney |
| SLC5A4 | SGLT3 | Na+ (H+) | Small intestine, skeletal muscle, kidney, uterus and testis |
| SLC5A8 | SGLT4 | — | Small intestine, kidney, liver, lung and brain |
| SLC5A9 | SGLT5 | — | Kidney |
| SLC5A10 | SGLT6 | Chiro-inositol, myo-inositol, glucose and xylose | Small intestine, kidney, liver, heart, lung and brain |
5.4. Pathology
GGM is a principal genetic disorder that is linked to the SGLT family of genes. It is a rare autosomal recessive disease that occurs in newborn infants, where mutations in the SGLT1 gene cause life-threatening diarrhea. However, diarrhea can be managed by the withdrawal of dietary sugars, such as glucose, galactose, and lactose, but it can quickly recur after the reintroduction of these sugars.43 SGLT2 is a principal transporter found in the kidneys, which are associated with glucose reabsorption, hence, confirming why autosomal recessive renal glycosuria occurs in diabetes patients. DNA sequencing was conducted on two younger brothers and their parents to investigate the formation of two mutations, a homozygous nonsense mutation and a heterozygous mutation, which both occurred in exon 11 of SGLT2.42,44
5.5. Structure and functions of SGLT
5.5.1. Crystal structure
The crystal structure of SGLT was derived from Vibrio parahaemolyticus as it shows 60% similarity (32% sequence identity) to human SGLT1 (Fig. 1). The model of the SGLT structure predicts 14 transmembrane α-helices (TMH) and features NH2-terminal and a COOH-terminal halves. Studies on this type of transporters indicate that the NH2-terminal half carries the sodium ion whereas the COOH-terminal half carries the sugar moiety. NH2-terminal binding of a sodium ion induces a conformational change in the protein to permit sugar binding and translocation. Furthermore, these studies postulated that the TMHs numbered 10–13 are critical for maintaining the sugar translocation pathway in SGLT1.44,45
Fig. 1. Crystal structure of the SGLT protein.

Researchers identified two important binding sites in the crystal structure, i.e. the galactose and Na+ binding sites. The function of the galactose binding site is to identify the precise point of reference and location of the sugar molecule. The galactose binding site is located in between hydrophobic residues of the intracellular and extracellular gates. Owing to the densitometric similarity between Na+ and water, it is difficult to locate the sodium binding site precisely on vSGLT. Researchers have claimed that the sodium binding site is located at the crossing of the TM2 and TM9 helices and is 10 Å away from the substrate-binding site on the basis of sequence alignment studies on the LeuT structure.45b
5.5.2. Functions
In a mutagenesis analysis of SGLT1, the residues at positions 457, 468, and 499 were replaced by cysteines, which make the transporter susceptible to methanethiosulfonate (MTS), but only when the transporter is in the C2 conformation. If the MTS reaction is performed either with glucose/phlorizin in the presence/absence of sodium ions or in the presence of sodium ions alone (when there is a depolarization of the membrane potential), the accessibility of the cysteine residues to MTS is found to be blocked. The probability of the protein to adopt the C2 conformation is directly proportional to the accessibility of these residues (457, 468, and 499) to MTS. The main conclusion of these experiments was that the transporter residues 457, 468, and 499 are critical for maintaining the translocation pathway, which is accessible in C2 conformation.44,46,47 MTS reagents can react with these critical residues, but phlorizin can block the entry of sugar derivatives towards them. It is suggested that the sodium ion creates a large conformational change by rotating (tilting) one or more TMHs in the 10–13 helical bundle. In the cell, the translocation of sugar generally needs a substantial change in the helical rotation, which results in the cotransportation of water and urea. Under these conditions, the stoichiometry of sodium ions : glucose : water in such transport has been found to be 2 : 1 : 250.44,47,48
5.5.3. Role in diabetes
Although the kidneys employ various mechanisms to maintain glucose levels, this primarily occurs by glomerular filtration and reabsorption mechanisms in the kidneys.44,49 To determine urinary glucose excretion (UGE), the reabsorbed glucose is subtracted from the total amount of filtered glucose. It has been observed that in a healthy adult, approximately 180 g per day of glucose is generally filtered. However, after filtration, the glucose gets reabsorbed almost completely and only a very low amount (below 1%) gets eliminated in the urine.50,51
Glucose reabsorption consists of several steps, involving various mechanisms. Tubular epithelial cells must carry filtered glucose from the tubule. This filtered glucose is further carried into the peritubular capillary through basolateral membranes. Loss of glucose does not normally occur through the urine under non-diabetic conditions (when the glucose load is normal). But, whenever the renal glucose threshold is exceeded, UGE occurs. However, to trigger this process, there is no set threshold value for plasma glucose which in most individuals found at approximately 200 mg dL−1.52
During the reuptake from urine (governed by the SGLTs), glucose is transported to tubular or luminal epithelial cells. Numerous members of the SGLT protein family that are present in the cell membrane facilitate the movement of several vital constituents such as glucose, amino acids, ions or vitamins through renal tubules and luminal epithelial cells.42,53 The SGLT1 and SGLT2 transporters are important members of this family. SGLT1 is the principal transporter in the GIT responsible for glucose absorption. However, in the kidneys (S3 segment of proximal tubule), SGLT1 reabsorbs only about 10% of the glucose.
SGLT2 as another transporter has a high capacity and a low affinity for the substrate. SGLT2 is mostly found in the kidneys where most of the glucose (approximately 90%) gets reabsorbed. SGLT2 interacts and binds both glucose molecules and sodium ions of the tubular filtrate to facilitate secondary active transport. This type of glucose transport across the membrane is coupled to the movement of sodium ions, which drive the transport along the electrochemical sodium ion gradient between the tubular filtrate and the cell.51
Once SGLT2 has mediated the glucose transport into the luminal epithelial cells, the glucose molecules are then passed through the basolateral cell membrane (facilitated diffusion) and again back into the peritubular capillary. Glucose transporters (GLUTs) such as GLUT1 (present in the late proximal tubule) and GLUT2 (present in the early proximal tubule) are the main transporters involved in this facilitated diffusion.54 It has been previously reported that glucose reabsorption is higher in insulin-treated diabetic patients than in healthy subjects.55 In addition, diabetic patients show increased SGLT2 expression in renal proximal tubular cells.56
6. SGLT inhibitors
Phlorizin, a chalcone compound, was originally identified as a natural nonspecific inhibitor of SGLT1 and SGLT2. It was first isolated from apple tree bark by French researchers before the 1850s. Research on phlorizin has shown that the inhibition of SGLT may have an application as a possible strategy for treating diabetes. Because of its major effects, such as decreasing urinary glucose reuptake and increasing the renal elimination of glucose, phlorizin lowers the risks of hyperglycemia and glucotoxicity. However, although phlorizin causes nonselective inhibition of SGLT1 (at the intestinal brush border), it was not found to be an appropriate antidiabetic agent because it induces GGM as a side effect on the GIT and has poor oral bioavailability.57–59
Moreover, a hydrolyzed phlorizin metabolite, phloretin, was found to block GLUT1 and interfere with glucose uptake in various tissues. Inhibitors help to reveal the role of SGLTs in certain inherited and acquired disorders, such as GGM in the small intestine (involves SGLT1) and glycosuria in the renal system (involves SGLT2). Various studies on these defects led to the discovery of the molecular mechanism employed by the SGLTs and their function in the pathophysiology of diabetes.52,58,60
Before the mid-20th century, phlorizin was considered to be responsible for blocking the glucose uptake of red blood cells (RBCs) in the kidneys and small intestine. However, in the late 20th century, after the studies on SGLT2, newer reports described the impact of phlorizin-generated glycosuria on the renal system and its mechanism.39,61 Also, after the discovery of T-1095 in the 1990s, the pioneering work by Tanabe Seiyaku inspired by the natural SGLT inhibitor phlorizin, multiple classes of the scaffolds were extensively explored, and huge numbers of the SGLT inhibitors have since been reported by many pharmaceutical companies.61
6.1. Current drugs in the market
Recently, the FDA approved several SGLT2 inhibitors, such as canagliflozin, empagliflozin dapagliflozin and more recently, ertugliflozin. Treatment with these therapeutics should be accompanied by an improved diet and exercise regimen to reduce the blood sugar levels in T2DM patients. These medicines are available as a single therapy or in combination with other antidiabetic agents such as metformin.62
6.1.1. Canagliflozin
Janssen, a division of Johnson & Johnson, is licensed for marketing the first FDA-approved SGLT2 inhibitor called canagliflozin (Mitsubishi Tanabe Pharma), which was approved in 2013. Canagliflozin is categorized under the gliflozin class, the mechanism of action of which is based on the inhibition of glucose reuptake inside the proximal tubules of the kidneys.63
In extensive placebo-controlled trials, canagliflozin doses of 100 and 300 mg per day were evaluated as a single therapy and also in combination with other antidiabetic agents. At both doses, canagliflozin caused statistically significant declines in HbA1c from the baseline relative to the placebo when used as a single therapy and also in combination with sulfonylureas, metformin, metformin plus pioglitazone, metformin plus sulfonylurea, and insulin. When used as a single therapy, the reductions from the HbA1c baseline at a dose of 100 and 300 mg were −0.91 and −1.16, respectively, as placebo-subtracted HbA1c. When canagliflozin was administered in combination with other antidiabetic therapeutics, the placebo-subtracted difference ranged from −0.62 to −0.92 (excluding special populations such as elderly and renally impaired subjects). An average 0.4% to 3.3% reduction in weight across multiple trials of 100 and 300 mg canagliflozin treated patients was another observation recorded. Average reductions in systolic blood pressure (SBP) by 0.1 to 7.9 mmHg along with increased HDL-C relative to the placebo were also observed across trials. Inconsistent changes in triglycerides were modest. Any effect on these two lipid parameters of canagliflozin was countered by the elevation of the LDL-C level ranging from 4.6–12% for a dose of 300 mg and 2–8% for a dose of 100 mg.62,64
Treatment with canagliflozin and other SGLT-2 inhibitors has some adverse effects, such as elevation in thirst, urination, and LDL cholesterol, as well as episodes of low blood pressure and the increased occurrence of urogenital infections. Hence, canagliflozin is contraindicated in people with renal insufficiency, decreased glomerular filtration rate (GFR) or renal problems.64
The FDA had additional concerns about the cardiovascular safety of canagliflozin. More cardiovascular events were observed in canagliflozin-receiving patients (0.45%) during the initial 30 days of study compared to those in placebo-receiving patients (0.07%), suggesting an increased cardiovascular risk during the early treatment period. Furthermore, subjects receiving canagliflozin appeared to have an increased risk of stroke. However, this risk was not statistically significant.62
The FDA issued a warning in May 2015 that canagliflozin and certain SGLT2 inhibitors may induce the development of ketoacidosis (increased acid levels in the blood). In September 2015, the FDA also issued a drug safety communication for canagliflozin stating that decreased bone density can occur, resulting in probable risks of bone fracture. This fact was already included in the adverse reactions of the drug and had been provided to health care professionals (to assess this issue before prescribing) and to patients (to immediately report any fractures that occurred during therapy).65
In December 2015, the FDA also gave information on the addition of new warnings on the canagliflozin label about urinary tract infections and elevated blood acid levels due to the risk of dehydration with diuretic drugs.62,65 In 2017, canagliflozin showed favourable cardiovascular effects in a clinical study consisting of 666 T2DM patients.62
6.1.2. Empagliflozin (EMPA)
Boehringer Ingelheim and Eli Lilly and Company together developed empagliflozin (Jardiance), another SGLT2 inhibitor belonging to the gliflozin class, which was approved in 2014.66
Empagliflozin contains a C-glucoside with similarity to that in phlorizin (the glucose and aglycone groups are linked by a carbon–carbon bond ensuring efficient oral administration and the bypassing of GIT degradation). In addition, the drug is more selective for SGLT2 (by more than 25 × 102-fold) along with it having only minimal adverse reactions on the GIT.66
According to statistical data obtained from empagliflozin combination studies, a marked decline in systolic blood pressure (SBP) and body weight was observed as compared to those of the placebo treatment. Different combinations such as metformin, metformin plus sulfonylurea, pioglitazone, and pioglitazone plus metformin were included in these combination studies. When empagliflozin with or without metformin at doses of 10 or 25 mg were included in a multidose regimen of insulin, it showed a marked loss in body weight and a nonsignificant decrease in SBP. In combination with metformin, a dose of 25 mg of empagliflozin showed a marked loss in body weight and SBP compared to those in glimepiride-treated subjects. However, in these combination studies, empagliflozin was also linked to a minor elevation in LDL-C and HDL-C levels.62,66
The post-marketing clinical trial “EMPA-REG OUTCOME” released in August 2015 and in January 2019 indicated that the inclusion of empagliflozin in standard care can reduce the risk of stroke, myocardial infarction, and cardiovascular death. Urogenital infections were the common adverse reactions that were found to be associated with the use of empagliflozin in the treatment of DM.66
6.1.3. Dapagliflozin
Dapagliflozin as the next member of the gliflozin class used to treat T2DM was developed by Bristol-Myers Squibb under the trade names Farxiga™ and Forxiga™ (in partnership with AstraZeneca). Initially, in July 2011, the FDA rejected the approval until more data were available. And later, on January 8, 2014, the FDA approved dapagliflozin for glycemic control in diabetes patient, along with diet and exercise. In October 2014, the FDA also approved a combination therapy of dapagliflozin and metformin hydrochloride (Xigduo™).67
Adverse effects associated with the treatment of dapagliflozin include heavy glycosuria, which may be up to 70 g per day occasionally, tiredness with rapid weight loss (leading to dehydration), and an increased risk of diabetic ketoacidosis (DKA). Existing infections due to diabetes, mostly urinary tract infections and candidiasis, become worse upon an increase in glucose in the urine.67,68
Canagliflozin, empagliflozin, dapagliflozin and ertugliflozin are now on the market in the US and EU. Ipragliflozin, tofogliflozin, and luseogliflozin were approved in Japan as the first national approval granted in the world. Other drug candidates such as sotagliflozin had successfully completed the phase III clinical trial, but in early 2019, was rejected as an adjunct to insulin for T1DM (type 1 diabetes mellitus). In addition, various drug combinations containing SGLT2 inhibitors along with other oral antidiabetic agents are currently in clinical development and are being rigorously tested in clinical trials.62,66,68
6.1.4. Ipragliflozin
In January 2014, ipragliflozin was approved first in Japan as an orally administered SGLT2 inhibitor under the brand name Suglat. Ipragliflozin is being used as a treatment for T2DM either alone or as an add-on to various other anti-hyperglycemic agents such as metformin (sulfonylurea), DPP-4 inhibitors or nateglinide (α-glucosidase inhibitor) and pioglitazone.69,70 25 and 50 mg tablets of ipragliflozin have been approved, administered as a dose of 50 mg per day either before or after breakfast. The ipragliflozin dose can be increased up to 100 mg per day if 50 mg is found to be insufficient. Ipragliflozin approval was based on the results of various trials that were conducted in Japan. One such trial was a pivotal phase III clinical trial, which was carried out in patients with insufficient glycemic control. It was included in a total of six trials of ipragliflozin in combination with other anti-hyperglycemic agents (listed below).67,69
(1) Ipragliflozin + metformin (randomized, placebo-controlled trial).
(2) Ipragliflozin + α-glucosidase inhibitors, such as voglibose, miglitol or acarbose (uncontrolled 52 week open-label trial).
(3) Ipragliflozin + pioglitazone (randomized, double-blind, placebo-controlled trial).
(4) Ipragliflozin + nateglinide (uncontrolled open-label trial).
(5) Ipragliflozin + sulfonylurea, such as gliclazide, glibenclamide or glimepiride (randomized, double-blind, placebo-controlled trial).
(6) Ipragliflozin + DPP-4 inhibitors, such as vildagliptin, saxagliptin, sitagliptin or alogliptin.
Ipragliflozin was also explored in phase III monotherapy studies in Japanese patients with T2DM, in:
(1) An uncontrolled, open-label, six-month trial.
(2) Randomized, double-blind, placebo-controlled trials.
(3) A 52 week study comparing the effects of pre-meal and post-meal administration along with evaluating its long-term safety and efficacy.
In the US and EU, as well as in other countries, ipragliflozin passed phase II clinical trials for the treatment of T2DM, in combination with metformin or administered alone. Very recently, in December 2018, Astellas Pharma and Kotobuki pharmaceutical Co. Ltd. got approval for Suglat for the additional indication of T1DM in Japan.69,70 After in vitro pharmacological studies, ipragliflozin was found to inhibit SGLT2 competitively and also have high selectivity for SGLT2 over other members of the SGLT family.69
6.1.5. Tofogliflozin
Tofogliflozin, an orally administered SGLT2 inhibitor, was developed by Chugai Pharmaceuticals (Japan) and approved in 2014 for the treatment of adult patients suffering from T2DM. Chugai Pharmaceuticals is tied up to a tofogliflozin co-development license agreement (2012) with Sanofi K. K. and Kowa. Tofogliflozin will be marketed in Japan under the trade name Apleway by Sanofi and as Deberza by Kowa. It can be administered as a single drug therapy or as an add-on with various anti-hyperglycemic drugs. A single dose of 20 mg per day has been approved and it should be taken either before or after breakfast.71,72
Tofogliflozin was approved after phase III trials in a total of approximately 800 Japanese T2DM patients. In the first study, a single daily dose of 20 or 40 mg was administered to 194 patients with an improper diet and poor exercise response. In the second study, the same tofogliflozin doses in addition to various orally administered anti-hyperglycemic agents (sulfonylurea, biguanide, thiazolidinedione, α-glucosidase inhibitor or DPP4 inhibitor) were administered to around 600 patients with insufficient/poor glycemic control along with an improper diet and poor exercise habits. Japan also conducted a six-month phase II/III clinical trial (randomized and double-blind, placebo-controlled) of single tofogliflozin therapy in 10, 20, and 40 mg doses accompanied by an appropriate diet and exercise plan. The trial was conducted on 230 T2DM patients with insufficient glycemic control.
Tofogliflozin was initially licensed to Roche in 2007, but Roche returned its research and development rights to Chugai in 2011.71,73 The former licensee, i.e., Roche, had also contributed to the development of tofogliflozin. Roche conducted three phase I clinical trials along with a three-month multinational phase II clinical trial on around 400 patients with T2DM.72
6.1.6. Luseogliflozin
Japan also developed luseogliflozin (Lusefi), a selective SGLT2 inhibitor. It was the first SGLT2 inhibitor to get approval for marketing as oral tablets in 2014. Although the approved doses of luseogliflozin are 2.5 and 5 mg per day, a starting dose of 2.5 mg per day is generally recommended. This dose may be increased to a daily single dose of 5 mg if optimal results are not achieved with the former dose. Luseogliflozin was approved after conducting a series of four phase III clinical trials and evaluating its efficacy in Japanese patients with T2DM. In two clinical trials, luseogliflozin was administered with other anti-hyperglycemic agents (in patients, initially treated with luseogliflozin alone, who had suboptimal glycemic control); in two other trials, luseogliflozin was evaluated as a single therapy.
In Japan, Novartis filed a marketing license agreement for luseogliflozin (2012). According to the terms and conditions of this agreement, Taisho Pharmaceutical is authorized to manufacture luseogliflozin. However, it will be marketed by Taisho as well as by Novartis.73–75 Luseogliflozin was found to decrease HbA1c, fasting plasma glucose levels, and body weight when administered long-term both as a single therapy and as an add-on with other oral anti-hyperglycemic drugs in patients with T2DM.73
6.1.7. Ertugliflozin
Phase III studies of ertugliflozin conducted by Merck in partnership with Pfizer met the initial endpoints for the development of an oral SGLT2 inhibitor in T2DM treatment. When compared to placebo, significant HbA1C reductions of 0.69% and 0.76% were observed using doses of 5 and 15 mg, respectively. Researchers also observed that ertugliflozin in combination with sitagliptin reduced HbA1C to less than 7.0% compared to that of ertugliflozin or sitagliptin administrated individually. In 2017, ertugliflozin (Steglatro) got its approval from the FDA.62,76,77
6.1.8. Sotagliflozin and sergliflozin
In 2015, Lexicon Pharmaceuticals and Sanofi collaborated and signed a license agreement according to which Sanofi obtained the rights to have worldwide royalties and also be licensed to develop, manufacture, and commercialize sotagliflozin. Under this collaboration, all clinical development activities related to T1DM will be conducted by Lexicon, whereas Sanofi will perform all clinical developments related to T2DM and will commercialize sotagliflozin worldwide for DM treatment.78,79
In 2016, Lexicon Pharmaceuticals reported that the phase III clinical trial of sotagliflozin in patients with T1DM demonstrated a marked decrease in HbA1C. The results of a 24 week treatment suggested that a single dose of 200 mg per day of sotagliflozin reduces HbA1C from the baseline by up to 0.43%, while a single dose of 400 mg per day reduces HbA1C by up to 0.49%, compared to the placebo that reduced HbA1C by only 0.08%. Importantly, this improvement in HbA1C in T1DM patients is statistically significant and clinically meaningful, and is not associated with severe hypoglycemia. Glucose absorption in the GIT occurs through SGLT1 and reuptake of glucose in the kidneys is facilitated by SGLT2. Sotagliflozin was also the first in its class to inhibit both SGLT1 and SGLT2.78,79 In 2019, sotagliflozin was rejected by the US FDA for the treatment of T1DM.68 Another candidate, sergliflozin, did not undergo further development after phase II.

More recently, due to cases for risk of DKA observed in studies on T1DM patients receiving SGLT inhibitors, an international consensus report was published in which recommendations were provided so as to safeguard the use of SGLT inhibitors mainly with respect to DKA in these patients. Goldenberg et al. have also suggested a “STOP DKA protocol” in which a risk overcoming strategy is given to reduce DKA in patients receiving SGLT inhibitors (Fig. 2).68
Fig. 2. Chemical classification of SGLT1/SGLT2 inhibitors.

6.2. Chemical classification of SGLT1/SGLT2 inhibitors
6.2.1. Glucosides (including C, O, N glucosides)
6.2.1.1. Triazoles
Substituted 1,2,3- and 1,2,4-triazoles are core structures found in various therapeutics such as antiviral, antiallergic, anti-HIV, and antibacterial agents.
Banerjee et al. synthesized an l-rhamnose-derived glucoside series with aliphatic C-nucleosides coupled to the 1,2,3-triazole nucleus. Step-by-step synthesis of l-rhamnose (a β-ketoester) was carried out under treatment with a variety of aromatic azides to produce 1,2,3-triazole derivatives with better yields. A screen of these derivatives using a glucose-uptake assay (for assessing SGLT1 and SGLT2 inhibition) identified a p-nitrophenyl derivative (1) as a highly potent SGLT2 inhibitor with an IC50 value of 125.9 nM.80–82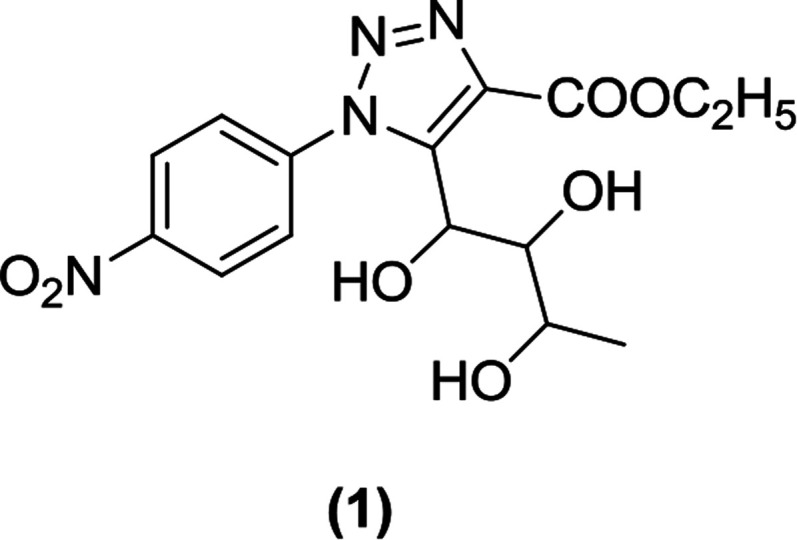
Wang et al. used ‘click chemistry’ to synthesize triazolylmethylaryl glucosides and assessed their effect on glucosuria. They discovered C-glucosides (2) containing a triazole aglycon core as novel SGLT2 inhibitors. These compounds were synthesized via cycloaddition reactions involving azides and alkynes in the presence of copper as a catalyst. The newly synthesized triazole C-glucosides were tested for their effect on UGE. Nearly all of the synthesized compounds were found to increase UGE when administered to Sprague Dawley (SD) rats, and they also enlarged the urine volume to a lesser extent than dapagliflozin.82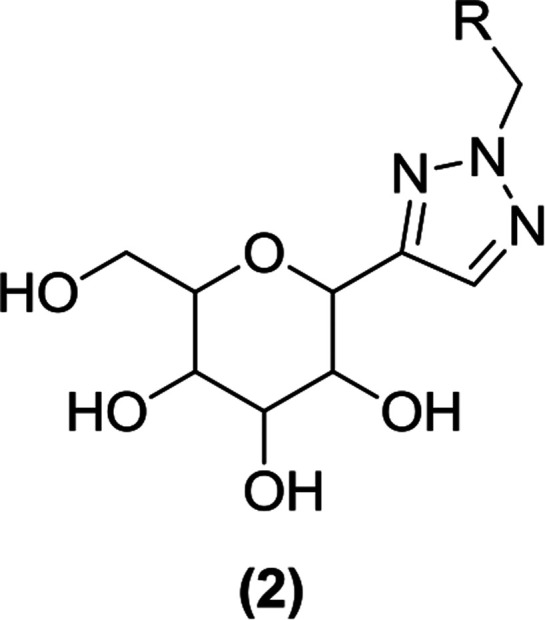
Wu and Ye et al. carried out reactions between triazoles and carbohydrates in the presence of copper thiocyanide as a catalyst to prepare a novel N-glycoside series. The synthesis of these N-glycoside derivatives was done via click reactions and chemistry similar to the Ullmann reaction. The new derivatives were tested in a cell-based assay for glucose transport inhibition using the drug dapagliflozin as a control. The analysis demonstrated that compounds containing a triazole moiety combined with N-glucosides or N-galactosides (3 and 4) exerted a strong inhibitory activity in this assay (at a concentration of 100 nM).83
Du et al. synthesized novel, non-phlorizin-based SGLT2 inhibitors and tested them in a glucose transporter assay. Compound (5) was found to be the most potent inhibitor (IC50 = 10 nM) when tested against human SGLT2. Compound (5) also displayed selectivity against hSGLT1 (IC50 = 9 μM). When the compound was tested in rats, low microsomal stability and increased clearance was observed.84
The SAR analysis of this series indicated that compounds containing benzooxazinone attached to an aromatic group were potent hSGLT2 inhibitors; cyanide or pyridine (polar groups) linked to the C-8 position of benzooxazinone were tolerated by this moiety. Most of the inhibitors such as compound (6) (IC50 = 3 nM; solubility = 1.1 μg mL−1) and compound (7) (IC50 = 12 nM; solubility = 64 μg mL−1) exhibited improved efficacy compared to that of the early lead compound (5).84
6.2.1.2. Indole derivatives
Substitution of d-xylose by glucose joined with an indole at the third position resulted in N-indolyl xylosides, which exhibited potent SGLT2 inhibitor activity. Moreover, C-indolyl glucoside derivatives were also found to be good SGLT2 inhibitors.85
Zhang et al. prepared indole-O- and C-glucosides by modifying previously reported blood-glucose lowering compound (8). In vitro testing of the new compounds for inhibitory activity against SGLT1 and SGLT2 using cell-based assays identified compounds (9) and (10) as the most potent SGLT2 inhibitor and SGLT1/SGLT2-mixed inhibitor, respectively. Both compounds were further tested for efficacy in ZDF (Zucker diabetic fatty) male rats. The pharmacokinetic parameters of the two new inhibitors were found to be similar to those of compound (8) but neither was as effective as the original compound. Compounds (9) and (10) were also found to have similar UGE efficacy. The SAR analysis showed that shortening the linker to the 2,3-dihydrobenzofuran moiety within compound (9) to create compound (11) abolished the SGLT2 inhibitory activity, whereas a modification of the 2,3-dihydrobenzofuran moiety with another aromatic group typically generated compounds with potent SGLT2 inhibitory activity. The study also indicated that anomeric oxygen (glycosidic oxygen) is important for SGLT inhibitory activity and its loss creates C-glucosides with reduced biological activity.85
Chu et al. synthesized N-indolyl glucoside compounds and tested them in vitro for inhibitory activity against human SGLT2 expressed in Chinese hamster ovary cells using phlorizin as a reference compound. The SAR analysis revealed that varying the C6-glucose substituent improves the antidiabetic activity. Among the synthesized compounds, compound (12) with C6-amide substitution was found to be a good inhibitor of hSGLT2 (compound 12, EC50 ∼ 42 nM; phlorizin, EC50 ∼ 123 nM) but with a reduced selectivity towards hSGLT1 (compound 12, EC50 ∼ 1412 nM; phlorizin, EC50 ∼ 153 nM). This compound also displayed efficacy in vivo.86
Li et al. synthesized an N-glycoside indole (13) using a different route as described by the Mitsubishi Tanabe Pharma Corporation, where the original compound was discovered. The steps of this new route included the Vilsmeier alkylation and coupling via the Grignard reaction. Compound (13) was obtained in a good yield (more than 70%) and further derivatized using the Friedel–Crafts reaction to create compound (14). Both compounds were found to be potent SGLT inhibitors.87
Lee et al. synthesized and developed a series of indolyl xyloside derivatives. The researchers developed this concept from lead C-glucosides containing a C-glycosidic linkage. They observed that the C-glycosidic linkage is critical for the biological activity of these compounds because it cannot be hydrolyzed by gastrointestinal glucosidase, which eliminated the process of conversion of these inhibitors in the GIT. To synthesize novel SGLT inhibitors, potent indolyl xyloside derivatives were created by substituting the glucose of C-glucosides with d-xylose joined to the 3rd position of the indolyl moiety. Among the newly synthesized derivatives, compound (15) was the most potent SGLT2 inhibitor (EC50 = 47 ± 3 nM against SGLT2 and EC50 = 282 ± 11 nM against SGLT1), whereas compound (16) was the most potent SGLT1 inhibitor (EC50 = 50 ± 11 nM against SGLT2 and EC50 = 55 ± 5 nM against SGLT1). Compound (16) was also more potent than reference compound dapagliflozin (EC50 = 2.4 ± 0.6 nM against SGLT2 and EC50 = 593 ± 176 nM against SGLT1). The SAR study indicated that the ‘N’ of the indolyl moiety attached to the para position of the cyclopropylphenyl group and a substitution at the 7th position of the indolyl moiety (compounds 15 and 16) were important for optimal inhibition. An in vivo study showed that compound (15) is metabolically stable with a low clearance rate along with a good anti-hyperglycemic activity (>35% reduction in the blood glucose level in diabetic rats by compound 15 as compared to that by a reference drug).88
6.2.1.3. Thiophene derivatives
Lee et al. designed and synthesized two different types of C-aryl glucoside derivatives containing a thiophene ring and evaluated them as inhibitors of SGLT2. Among these derivatives, compound (17), a 5-chloro-substituted 4-ethylbenzylthiophene, was found to be the most potent inhibitor with an IC50 value of 4.47 nM. This result indicates that an alkyl chain (with an appropriate number of carbon atoms) attached at the 4th position of the benzyl group helped to achieve good in vitro activity against hSGLT2 when tested.89
Nomura et al. synthesized various derivatives by replacing the benzene ring in canagliflozin with different moieties such as pyridine, pyrimidine, and five-membered thiophene-coupled heteroarenes. The researchers found that most of the 3-pyridyl, 2-pyrimidyl five-membered thiophene-coupled heteroarenes exhibited good antidiabetic activity against SGLT2. The most potent derivative was compound (18), which showed strong inhibitory activity against SGLT2 (IC50 = 2 nM) that was similar to that exhibited by canagliflozin (IC50 = 2.2 nM). When tested in vivo at a dose of 30 mg per kg in male rats, after one day of treatment, compound (18) had a UGE of 2666 mg per 200 g whereas canagliflozin had a UGE of 3696 mg per kg at the same dose.90
6.2.1.4. Pyrazole glucopyranoside
Fushimi et al. synthesized a novel series of β-d-glucopyranoside derivatives containing hydrophilic substitutions at the phenyl ring and assayed them for SGLT1 inhibitory activity. Among the synthesized derivatives, compound (19) displayed significantly stronger activity (IC50 = 50 nM against SGLT1) and low permeability compared to those of the initial lead compound reported by the authors. The in vivo efficacy of compound (19) was demonstrated by its improved carbohydrate tolerance in diabetic rats compared to that of the clinically used reference compound acarbose, an α-glucosidase inhibitor.91
6.2.1.5. Pyridazine
Lee et al. designed and synthesized C-glucosides with a pyridazine ring attached as an aromatic substitution at a carbon position of the glucoside. Among the evaluated derivatives, compounds (20) and (21) displayed the most potent activity against SGLT2 with IC50 values of 11.4 and 13.4 nM, respectively. Moreover, compound (21), containing a methylthio moiety, exhibited significant in vivo activity in normal SD rats.92
The researchers also designed and synthesized novel pyridazinyl derivatives. They reported the attachment of a pyridazine ring at the 1st carbon (anomeric carbon) of the d-glucopyranose moiety using a stereoselective approach. The resulting compound (22) was examined for biological activity using an in vitro SGLT2 inhibition assay, where (22) had an IC50 value of 610 nM.93
6.2.1.6. Thiazole and thiadiazole
Lee et al. worked on a lead optimization that resulted in the synthesis of a new thiazole core bearing a furanyl moiety. All compounds of this series exhibited in vitro antidiabetic activity against SGLT2 in the nanomolar range from which compounds (23) and (24), featuring a benzene ring with alkoxy and hydroxy substitutions at the ortho position, respectively, were found to be the most potent inhibitors. The IC50 value was 0.934 nM for compound (23) and 0.797 nM for compound (24).94
Lee et al. also coupled perbenzyl gluconolactone 9 with 2-lithiothiazole and therefore synthesized a novel series of thiazole analogues. During synthesis, the thiazole ring is linked to the C-glucoside (using 2-lithiated thiazole) and the C-4 or C-5 position of the thiazole gains a halogen attachment. After in vitro evaluation of these derivatives, using the SGLT2 inhibition assay, the authors reported that the novel benzylthiazolyl derivatives can be tested as potential SGLT2 inhibitors useful for pharmacotherapy. Compound (25) was found to be the most potent inhibitor of the series with an IC50 value of 121 nM.93
In addition, the researchers designed and synthesized 1,3,4-thiadiazole coupled C-aryl glucosides from commercially available perbenzylated gluconolactone and evaluated them as SGLT2 inhibitors. Among the heterocycle-containing derivatives, the compounds (26), (27), and (28) exhibited significant in vitro inhibitory activity against SGLT2 with IC50 values of 4.06, 7.03, and 3.51 nM, respectively.96
The SAR analysis indicated that replacement of the benzene ring attached at the distal end of dapagliflozin by 1,3,4-oxadiazole decreased the antidiabetic activity, whereas the 1,3,4-thiadiazole ring increased it. Furthermore, the substitution of an aliphatic linear chain on the thiadiazole ring decreased the activity and a bulkier group resulted in a complete loss of the activity. If halogen-like small lipophilic substituents were introduced at the C-4′ position of the benzene ring (directly attached to C-glucoside), the antidiabetic activity was improved. Because the most potent inhibitors contain a pyrazine, furan or thiophene ring [compound (26) contains a pyrazine ring, compound (27) contains a 2-furan ring, and compound (28) contains a 3-thiophene ring], these heterocycles are important for strong inhibitory activity against SGLT2.95,96
6.2.1.7. Spirocyclic derivatives
Lv et al. prepared a novel series of C-arylglucosides with a spiroketal ring attached at the ortho position. This series was tested in vitro against SGLT1 and SGLT2 using a cell based assay. Among the evaluated derivatives, compounds (29) and (30) were identified as the most effective inhibitors with IC50 values of 0.3 and 3.8 nM against SGLT2 and IC50 values of 3.1 and 0.7 μM against SGLT1, respectively. Compound (29) was found to be the more potent compound against SGLT2, whereas compound (30) was more potent against SGLT1 compared to that of the reference compound dapagliflozin (IC50 = 6.7 nM and 0.89 μM against SGLT2 and SGLT1, respectively). According to this report, the spiro[isobenzofuran-1,20-pyran] core with a chlorine substitution at the C-4 position of the B ring displayed significant antidiabetic activity. Administration of compound (30) in mice elevated glucosuria and reduced the blood glucose levels.97
Xu and Chen et al. synthesized two series of O-spiro-C-aryl glucosides where the spiro group was attached to the 2′ and 6′ positions of the C-aryl glucosides. The compounds (1 μM) were tested against SGLT1 and SGLT2 in vitro; the C-aryl glycoside derivatives containing the 6′-O-spiro substitution showed the highest inhibitory activity against SGLT2. However, the 2′-O-spiro derivatives were not effective against the same transporter. Two 6′-O-spiro derivatives, compounds (31) and (32), were identified as potent SGLT2 inhibitors, with IC50 values of 71 and 6.6 nM, respectively. Furthermore, compound (32) was found to be as effective as the reference compound dapagliflozin (IC50 = 6.7 nM). The SAR showed that the substituent at the 4′ position of the first phenyl ring was crucial for potent SGLT2 inhibitory activity; specifically, compound (31) without this substitution is 10 times less potent than compound (32) with a chlorine substituent at the 4′ position of the first phenyl ring.98
Robinson et al. prepared a series of C-5 spiro derivatives. By assessing their in vitro activity, some compounds, specifically (33), (34), (35), (36), and (37), were found to possess potent inhibitory activity against SGLT2 (Table 2).99 Moreover, two compounds (35), and (37) also showed good in vivo efficacy in rats. Interestingly, the 5th position is the only position at which removal of the hydroxyl group does not cause any significant loss of SGLT2 inhibitory activity.99
In vitro evaluation results of compounds 33–37 against SGLT2 (ref. 99).
| Compound | IC50 (nm) |
|---|---|
| 33 | 2.4 ± 0.5 |
| 34 | 5.1 |
| 35 | 3.0 ± 0.5 |
| 36 | 7.9 ± 3.5 |
| 37 | 6.6 ± 2.5 |
Mascitti et al. also synthesized C-glycoside derivatives with spirocycle substituents at the 5th position of the pyranose ring using a one-pot aldol-Cannizzaro type reaction and spiro oxetane production (using pentaol C-glycoside as a substrate). Evaluation of these derivatives for inhibitory activity and selectivity against SGLT1 and SGLT2 identified compounds (37), (38) and (39) as promising SGLT2 inhibitor candidates with IC50 values of 6.6 ± 2.5, 14 and 3.4 ± 0.8 nM, respectively. Also, it was observed that removal of the H-bond-donor at the C-5 position does not affect the potency and selectivity of candidates towards SGLT2.
However, in the case of azetidine, substituting an H-bond-donor at the C-5 position was not tolerated since this substitution resulted in a decrease in the SGLT2-inhibitory activity.100
6.2.1.8. C-aryl glucoside derivatives
Xu and Chen et al. synthesized a series of C-aryl glucosides with substitutions at the second carbon. These glucoside derivatives were tested in vitro for inhibitory activity against SGLT1 and SGLT2 using a cell-based assay. From the evaluated derivatives, the compounds (40) and (41) exhibited strong inhibitory activity against SGLT1 and SGLT2. Their IC50 values were 0.9 nM (compound 40) and 1.2 nM (compound 41) against SGLT2 and >2 nM (compound 40) and >4.2 nM (compound 41) against SGLT1. According to the in vitro tests, compound (40) exhibited 7 times stronger inhibitory activity and 11 times better selectivity against SGLT2 compared to those of the reference compound dapagliflozin and one compound that had been synthesized previously by the authors. Furthermore, the authors reported that introducing a substituent at the 2nd carbon (ortho position) of the benzene ring adjacent to the glycosidic linkage not only increased the SGLT2-inhibitory activity but also improved the selectivity of the respective compounds for SGLT2 over SGLT1.101
Lee et al. designed and synthesized C-aryl glucoside derivatives of dapagliflozin containing a macrocycle with the goal of creating new SGLT2 inhibitors. Two diverse synthetic schemes, intramolecular alkylation (OH = nucleophile and R from R–X = electrophile) and olefin metathesis (involving ring closure in the presence of a Grubbs catalyst), were implemented to achieve macrocyclization of dapagliflozin between the 6th position of the pyranose ring and the ortho position of the benzene ring (which was directly attached to a sugar moiety). The derivatives were tested in vitro for inhibitory activity against SGLT2 using a cell-based assay with dapagliflozin as a reference compound (IC50 = 1.35 ± 0.15 nM). In this assay, compound (42) was the most potent compound with an IC50 value of 0.778 nM against SGLT2. Compound (43) also exhibited strong anti-SGLT2 activity (IC50 = 0.899 nM) in the in vitro assay, but it was only moderately active when tested in SD rats. However, the macrocyclization between the sugar and the proximal phenyl ring was crucial for the inhibitory activity against SGLT2. Also, a methylthiophene or ethoxybenzene substituent at the central ring of the dioxacyclopentadecine macrocycle contributed to the best in vitro inhibition of SGLT2.102
Li et al. prepared dioxa-bicyclo C-aryl glucoside derivatives, which were analyzed in vitro for inhibitory activity against hSGLT2 using a cell-based assay. Among the newly synthesized derivatives, only compound (44) was found to be effective against SGLT2 with an IC50 value of 700 nM. The authors further reported that changing the oxygen position resulted in a loss in the SGLT2 inhibitory activity.103
6.2.1.9. Azuline derivatives
Ikegai and Imamura et al. evaluated the in vitro anti-hSGLT1 and anti-hSGLT2 activities (Chinese hamster ovary cells) of a newly synthesized series of C-glucoside derivatives containing azulene rings attached to the benzene ring present on the sugar moiety. Among the new derivatives, compounds (45) and (46) were active against hSGLT2 (IC50 = 22 and 8.9 nM, respectively) and more selective towards hSGLT2 with a selectivity index over hSGLT1 of 590 and 280 times, respectively. The most potent compound (46) also showed good in vivo activity when tested in diabetic rats and mice. The SAR analysis indicated that attaching a hydroxyl group at the 6th position of the benzene ring linked to the pyranose ring (ortho position) resulted in a more active compound (46), indicating that even the pyranose ring with an ortho substitution can improve the anti-SGLT2 activity. As a result of the SAR, the monocholine compound (47), which was derived from compound (46), was selected for further clinical development.104
6.2.1.10. Isoquinoline derivatives
Liu et al. synthesized C-aryl glucoside derivatives containing a tetrahydroisoquinoline ring attached to the ortho position of the sugar moiety. The authors examined these derivatives in vitro for their inhibitory activity against SGLT2 using a cell-based assay. Among the evaluated derivatives, compound (48) was found to be the most potent inhibitor, reducing the SGLT2 activity by 81.7%, which was similar to the inhibition determined for the reference compound dapagliflozin (85.4% inhibition of SGLT2). The SAR study indicated that the presence of electron-withdrawing or -donating groups at the 4th position of the aromatic ring attached to the indolyl core further increased the in vitro anti-SGLT2 activity.105
6.2.2. Non-glucosides
Benzothiazinone and benzooxazinone derivatives were synthesized and evaluated by Li et al. for their potential inhibitory activity against SGLT2. The authors identified compound (49) as a target molecule in high throughput screening (HTS). Compound (49) showed an IC50 value of 1.4 μM against hSGLT2 (when tested in vitro using a Chinese hamster ovary cell-based assay). Derivatization of compound (49) resulted in a series of compounds containing benzothiazinone and benzooxazinone cores. From these compounds, compounds (50) and (51) possessed the highest activity. Compound (50) had IC50 values of 0.009 and 9.14 μM and compound (51) had IC50 values of 0.010 and 6.29 μM against SGLT2 and SGLT1, respectively. The researchers further reported that N-substituents on triazole and chloro substitutions at the 6th and 8th position of the benzooxazinone and benzothiazinone rings were found to be important for anti-SGLT2 activity.106
6.2.3. Miscellaneous
Qing et al. described the design and synthesis of dapagliflozin derivatives with a geminal difluoro substitution at the C-4 position. In vitro evaluation of these compounds for inhibitory activity against SGLT2 resulted into identification of certain compounds with very potent inhibitory activity (IC50 < 0.60 nM) when compared with the activity of the reference compound dapagliflozin (IC50 = 1.98 nM). The most potent derivative was compound (52) with an IC50 value of 0.35 nM. The SAR analysis indicated that the presence of a strong electron withdrawing group such as trifluoromethyl results in a decrease in the activity, whereas geminal difluoro substituents at the C-4 position of dapagliflozin resulted in potent anti-SGLT2 activity.107
Zhao and Wang et al. reported the synthesis of C-glucosides with geminal dimethyl substitution and their evaluation as SGLT2 inhibitors (53). The synthesis of these compounds included a Friedel–Crafts alkylation (as the main step) reaction using anhydrous AlCl3 as a catalyst. When tested in mice, these compounds showed good anti-hyperglycemic efficacy but they were less potent than the reference compound dapagliflozin.108
Kakinuma et al. prepared derivatives of d-glucitol linked to C-phenyl rings and a pyranose-like ring with the oxygen atom replaced by a sulfur atom. These compounds were tested in vitro against SGLT1 and SGLT2 using a cell-based assay. From these compounds, compound (54) was found to be the most potent with an IC50 value of 2.26 nM along with a 1650 times stronger selectivity over SGLT1. The in vivo results of this compound were also promising, which initiated the testing of this compound in clinical trials (it reached Phase II clinical trials).109
Lansdell et al. were the first to work on the design and synthesis of fluorophore-conjugated SGLT2 basic pharmacophore molecules as promising SGLT2 inhibitors. They tested these compounds in vitro for inhibitory activity against SGLT2 and identified compound (55) as the most potent inhibitor in this series, with an IC50 value of 55 nM, which was ten times more potent than the reference compound phlorizin (IC50 = 752 nM). According to this report, for optimum potency of the synthesized molecules, the mode by which the fluorophore binds with the SGLT2 basic pharmacophore was identified as the most essential aspect. In addition, the report indicated that alkyl substituents, such as propyl (–C3H7) and butyl (–C4H9) when introduced on nitrogen, increased the size of the amide, which resulted in a reduction in potency.110
Wang and Shen et al. reported on nine compounds derived from sergliflozin-A. After performing alterations to the O-glucoside chain, a 4-O methyl derivative (56) was found to be pharmacokinetically stable with good pharmacodynamic potency.111
Tsujihara and Saito et al. synthesized and evaluated derivatives of 4′-dehydroxyphlorizin substituted on the B ring. The researchers indicated that a small alkyl group substitution at the 4′ position increases the biological activity of these derivatives. Thus, compound (57) was found to be the most potent SGLT inhibitor among the new series. The researchers also prepared prodrugs that were susceptible to hydrolysis by a β-glucosidase in the GIT. Among the prodrugs, compound (58) had very beneficial features when tested in mice, such as a decreased hyperglycemia and the inhibition of blood sugar level increase. Finally, the report indicated that compound (57) displayed therapeutic efficacy in the treatment of non-insulin-dependent DM.112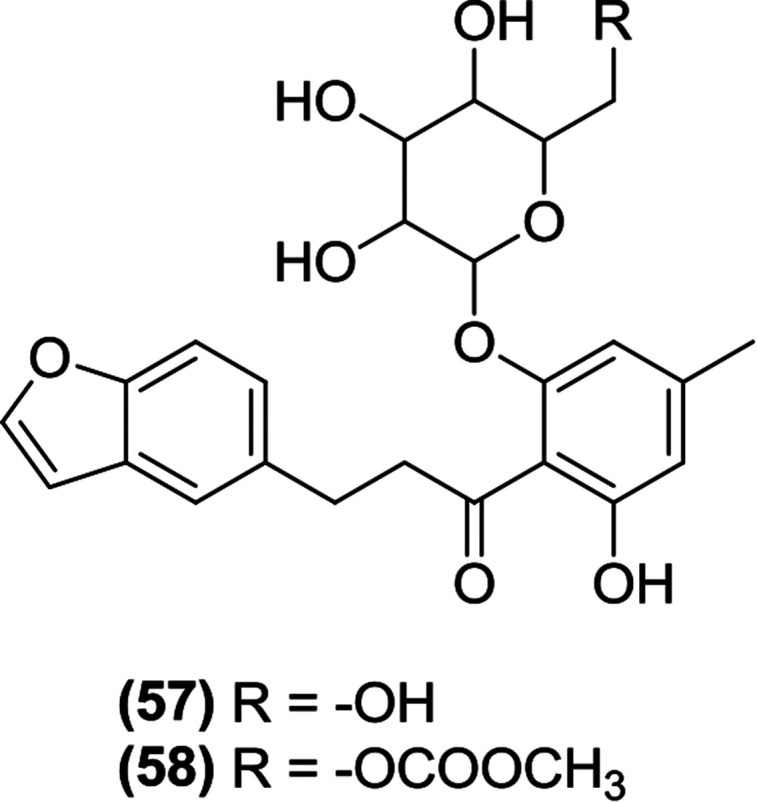
After synthesizing novel N-glucoside derivatives, Nomura et al. evaluated them for their inhibitory activity against hSGLT2. Among the evaluated compounds, compound (59) containing an N-glucoside attached to the indolyl core exhibited good in vitro activity with an IC50 value of 7.1 nM.113
6.3. SGLT inhibitors from natural sources
As described earlier, phlorizin was the first natural substance that displayed SGLT inhibitory activity.57,59 Among the compounds that Sato et al. identified in the methanol extract prepared from dried roots of the Chinese plant Sophora flavescens, three isoflavonoids, formononetin (60), maackiain (61) and variabilin (62), exhibited selective activity against SGLT 2, whereas another flavone, l-kurarinone (63), was the most potent inhibitor of both SGLT1 (IC50 = 10.4 μM) and SGLT2 (IC50 = 1.7 μM).114 Furthermore, Yang et al. also isolated several compounds from the same plant species and found that compound (64) possessed the most potent SGLT2 inhibitory activity (IC50 = 2.6 ± 0.18 μM).115
Compound (65) was identified as the most potent compound among the constituents of the bark of the tree Acer nikoense, isolated by Morita et al. Compound (65) showed IC50 values of 26 and 43 μM against SGLT1 and SGLT2, respectively.116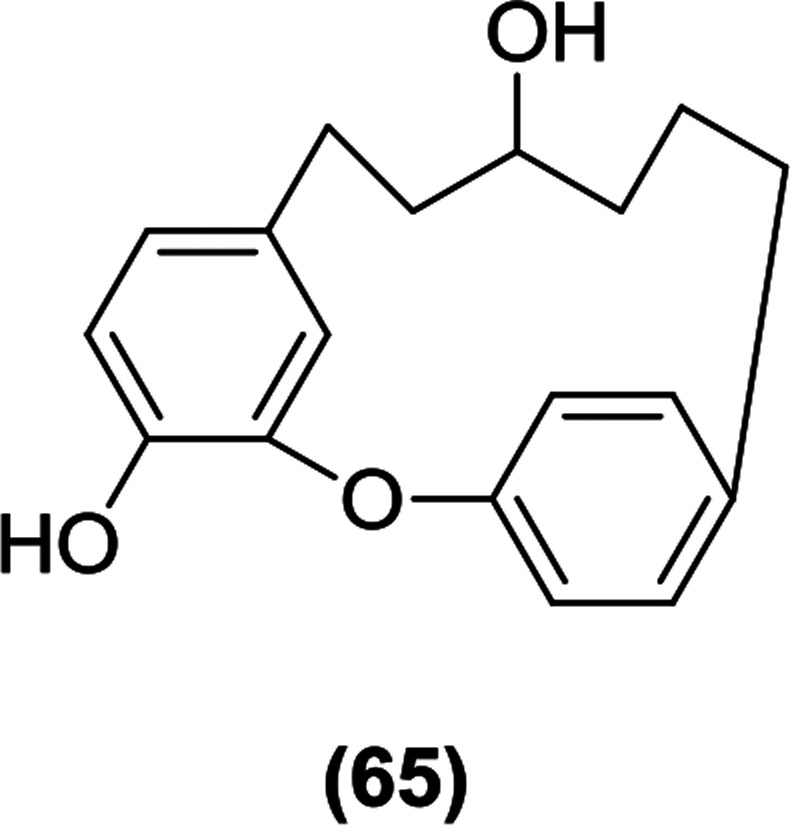
Natural stilbenes were isolated from the dried bark of the tree Gnetum gnemonoides by Shimokawa et al. Among these compounds, gneyulin A (66) was found to be the most active inhibitor against both SGLT1 and SGLT2, with IC50 values of 27 and 25 μM, respectively (Table 3).116
Patents on SGLT inhibitors.
| Sr No. | Patent no. (patent family) | Applicant/assignee | Filling year | Title | Country |
|---|---|---|---|---|---|
| 1 | WO2016041470A1 (CN107108539A) | National Institute of Biological Sciences, Beijing | 2015 | SGLT2 inhibitors117 | China |
| 2 | WO2015173584A1 (EP3142661A1a, US20170135981A1b, JP2017515908Aa, TW201607540Aa, CN106714796Aa) | AstraZeneca AB | 2015 | Method for suppressing glucagon secretion of an SGLT2 inhibitor118 | Europe, United States, Japan, Taiwan, China |
| 3 | WO2014159151A1 (US9573959B2, EP3466431A1a, EP2968375B1) | MSD International GmbH | 2014 | Methods for preparing SGLT2 inhibitors119 | United States, Europe |
| 4 | WO2012163990A1 (EP2714052B1, US20180177794A1a) | Boehringer Ingelheim International GmbH | 2012 | SGLT2 inhibitors for treating metabolic disorders in patients treated with neuroleptic agents120 | Europe, United States |
| 5 | WO2010048358A2 (US20100167988A1b) | Auspex Pharmaceutical, Inc. | 2010 | Ethoxyphenylmethyl inhibitors of SGLT2 (ref. 121 and 122) | United States |
| 6 | WO2010012153A1 (CN101638423B) | Changzhou Multiple Dimension Institute of Industry Technology | 2009 | New phlorizin derivatives having SGLT2 inhibitory activity: useful for treating metabolic diseases such as diabetes and diabetic complications122,123 | China |
Under prosecution.
Abandoned.
7. Conclusions
DM is a major chronic disease associated with metabolism that affects a large proportion of the world population. It is linked to other medical conditions and disorders such as obesity, hypertension, cardiovascular diseases, and atherosclerosis. In most cases, the DM management period includes the entire lifespan of a diabetic person, requiring a multidisciplinary approach that involves lifestyle changes, medical treatments such as antidiabetic therapies, and input from various persons with different backgrounds (e.g., health-related professionals, dieticians, exercise instructors, awareness educators, and support from family and society). We intended to cover all published information on SGLTs as targets for DM treatment and the compounds with inhibitory activity against the SGLTs, including current drugs, drugs in the pipeline, and drugs in clinical development. We sorted the current and potential therapeutics into chemical classes that we identified from the various reports. In this review, a systematic overview of the research published over the last 10 years in the area of SGLT inhibition as an antidiabetic target is provided to serve as a comprehensive resource for the scientific community.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
The authors are thankful to Padmashri Mrs Fatma Rafiq Zakaria, Chairman, Maulana Azad Educational Trust and Dr Zahid Zaheer, Principal, Y. B. Chavan College of Pharmacy, Dr Rafiq Zakaria Campus, Aurangabad 431 001 (M.S.), India for support and encouragement.
Biographies
Biography
Rahul P. Kshirsagar.

Rahul Kshirsagar is currently working as a Professor and Principal at the Yashodeep Institute of Pharmacy, Aurangabad, Maharashtra, India. He completed his M. Pharmacy in Pharmacology and submitted his Ph.D. in Pharmaceutical Sciences. He has 20 peer reviewed research publications in various national and international journals to his credit. He received a Young Investigator Grant in 2014 from the International Atherosclerosis Society Asia-Pacific Federation and a Young Investigator Fellowship Award in 2015 from the European Atherosclerosis Society. His area of research is diabetes and cardiovascular complications. Up until now he has guided 20 postgraduate students.
Biography
Abhishek A. Kulkarni.

Abhishek A. Kulkarni graduated from Shri Bhagwan College of Pharmacy, Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India in 2013 with distinction. He received his master's degree in Pharmaceutical Chemistry with distinction from the Y. B. Chavan College of Pharmacy, Aurangabad (MS), India under the guidance of Dr Jaiprakash N. Sangshetti in 2015. He has presented various scientific posters on topics related to Pharmaceutical Chemistry at both state and national level. His area of research includes the design and synthesis of novel heterocyclic coupled bioactive compounds and evaluation of their antileishmanial, antimicrobial and antioxidant activities. He is also working on the one-pot multicomponent synthesis of various bioactive molecules using various reusable catalysts.
Biography
Rashmi S. Chouthe.

Rashmi S. Chouthe is Assistant Professor in the BGPS groups, Srinath College of Pharmacy, Aurangabad Campus. She completed her M. Pharm in 2012 at the Dr Babasaheb Ambedkar Marathwada University, Aurangabad and is currently pursuing her PhD in pharmaceuticals and fine chemicals. She has been involved in teaching and research for seven years. Rashmi has 15 peer reviewed research publications in various national and international journals to her credit. She received the ‘Best paper award 2015’ for the best paper published in 2014 in the journal Chinese Chemical Letters.
Biography
Hemant D. Une.

Hemant Devidas Une works as an Associate Professor and Head at the Department of Pharmacology, Y. B. Chavan College of Pharmacy, Rouza Baugh, Aurangabad, Maharashtra, India. His research areas include preclinical evaluations for CNS acting agents, anti-diabetics and immunomodulators, herbal extraction and isolation. He has received two research grants from government funding agencies.
Biography
Jaiprakash N. Sangshetti.

Dr Jaiprakash N. Sangshetti graduated from Y. B. Chavan College of Pharmacy, Aurangabad (MS), India, in 1999. He completed an M. Pharmacy at the Prin. K. M. Kundnani College of Pharmacy, Mumbai. He obtained his PhD degree from Dr Babasaheb Ambedkar Marathwada University, Aurangabad under the supervision of Dr Devanand B. Shinde in 2009. Since 2010, he has been working as an Associate Professor at Y. B. Chavan College of Pharmacy, Aurangabad (MS), India. He has four patents to his credit and has published more than 200 research papers in international journals. He has authored two books, one on pharmaceutical validation and the other on quality by design. He was a recipient of a Young Scientist Award from the Department of Science and Technology, India. He has received research grants from different funding agencies such as DST, UGC and AICTE. His major area of research includes the design and synthesis of bioactive molecules, the development and validation of analytical methods for drugs and pharmaceutical dosage forms. He is currently working on different targets in antibacterial drug discovery.
References
- (a) Leung M. Y. M. Pollack L. M. Colditz G. A. Chang S. H. Diabetes Care. 2015;38:460–468. doi: 10.2337/dc14-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whiting D. R. Guariguata L. Weil C. Shaw J. E. Diabetes Res. Clin. Pract. 2011;94:311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Guariguata L. Whiting D. R. Hambleton I. Beagley J. Linnenkamp U. Shaw J. E. Diabetes Res. Clin. Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- (a) Inzucchi S. E. Bergenstal R. M. Buse J. B. Diamant M. Ferrannini E. Nauck M. Peters A. L. Tsapas A. Wender R. Matthews D. R. Diabetes Care. 2012;35:1364–1379. doi: 10.2337/dc12-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Groop L. C. Ferrannini E. Bailliere. Clin. Endocrinol. Metab. 1993;7:1007–1032. doi: 10.1016/S0950-351X(05)80243-5. [DOI] [PubMed] [Google Scholar]
- Turner R. C. Cull C. A. Frighi V. Holman R. R. J. Am. Med. Assoc. 1999;281:2005–2012. doi: 10.1001/jama.281.21.2005. [DOI] [PubMed] [Google Scholar]
- Nathan D. M. Buse J. B. Davidson M. B. Ferrannini E. Holman R. R. Sherwin R. Zinman B. Diabetes Care. 2009;32:193–203. doi: 10.2337/dc08-9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kahn S. E. Haffner S. M. Heise M. A. Herman W. H. Holman R. R. Jones N. P. Kravitz B. G. Lachin J. M. O'Neill M. C. Zinman B. Viberti G. N. Engl. J. Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]; (b) Nathan D. M. Genuth S. Lachin J. Cleary P. Crofford O. Davis M. Rand L. Siebert C. N. Engl. J. Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]; (c) Reichard P. Nilsson B.-Y. Rosenqvist U. N. Engl. J. Med. 1993;329:304–309. doi: 10.1056/NEJM199307293290502. [DOI] [PubMed] [Google Scholar]
- Lebovitz H. E. Nat. Rev. Endocrinol. 2011;7:408–419. doi: 10.1038/nrendo.2011.10. [DOI] [PubMed] [Google Scholar]
- Lebovitz H. E., in Diabetes mellitus: A fundamental and clinical text, ed. D. LeRoith, S. I. Taylor and J. M. Olefsky, Lippincott Williams and Wilkins, Philadelphia, 3rd edn, 2004, ch. 76, pp. 1107–1122 [Google Scholar]
- Hundal R. S. Inzucchi S. E. Drugs. 2003;63:1879–1894. doi: 10.2165/00003495-200363180-00001. [DOI] [PubMed] [Google Scholar]
- (a) Lebovitz H. E. Endocrinol. Metab. Clin. North Am. 1997;26:539–551. doi: 10.1016/S0889-8529(05)70266-8. [DOI] [PubMed] [Google Scholar]; (b) Lebovitz H. E. Nat. Rev. Endocrinol. 2010;6:326–334. doi: 10.1038/nrendo.2010.49. [DOI] [PubMed] [Google Scholar]
- Hanefeld M. Cardiovasc. Diabetol. 2007;6:1–7. doi: 10.1186/1475-2840-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman R. R. Thorne K. I. Farmer A. J. Davies M. J. Keenan J. F. Paul S. Levy J. C. N. Engl. J. Med. 2007;357:1716–1730. doi: 10.1056/NEJMoa075392. [DOI] [PubMed] [Google Scholar]
- Heine R. J. Van Gaal L. F. Johns D. Mihm M. J. Widel M. H. Brodows R. G. Ann. Intern. Med. 2013;143:559–569. doi: 10.7326/0003-4819-143-8-200510180-00006. [DOI] [PubMed] [Google Scholar]
- Drucker D. Diabetes Care. 2007;30:1335–1343. doi: 10.2337/dc07-0228. [DOI] [PubMed] [Google Scholar]
- Lebovitz H. E. Banerji M. A. Eur. J. Pharmacol. 2004;19:135–146. doi: 10.1016/j.ejphar.2004.02.051. [DOI] [PubMed] [Google Scholar]
- Buse J. B. Rosenstock J. Sesti G. Schmidt W. E. Montanya E. Brett J. H. Zychma M. Blonde L. Lancet. 2009;374:39–47. doi: 10.1016/S0140-6736(09)60659-0. [DOI] [PubMed] [Google Scholar]
- Riddle M. C. Ambrosius W. T. Brillon D. J. Buse J. B. Byington R. P. Cohen R. M. Goff D. C. Malozowski S. Margolis K. L. Probstfield J. L. Schnall A. Seaquist E. R. Diabetes Care. 2010;33:983–990. doi: 10.2337/dc09-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolen S. Feldman L. Vassy J. Wilson L. Yeh H. Marinopoulos S. Wiley C. Selvin E. Wilson R. Bass E. B. Brancati F. L. Ann. Intern. Med. 2007;147:386–399. doi: 10.7326/0003-4819-147-6-200709180-00178. [DOI] [PubMed] [Google Scholar]
- (a) Rodbard H. W. Blonde L. Braithwaite S. S. Brett E. M. Cobin R. H. Handelsman Y. Hellman R. Jellinger P. S. Jovanovic L. G. Levy P. Mechanick J. I. Zangeneh F. AACE diabetes mellitus clinical practice guidelines task force. Endocr. Pract. 2007;13:1–68. doi: 10.4158/EP.13.S1.1. [DOI] [PubMed] [Google Scholar]; (b) DeFronzo R. A. Stonehouse A. H. Han J. Wintle M. E. Diabetic Med. 2010;27:309–317. doi: 10.1111/j.1464-5491.2010.02941.x. [DOI] [PubMed] [Google Scholar]
- Purnell J. Q. Hokanson J. E. Marcovina S. M. Steffes M. W. Cleary P. A. Brunzell J. D. J. Am. Med. Assoc. 1998;280:140–146. doi: 10.1001/jama.280.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Nesto R. W. Bell D. Bonow R. O. Fonseca V. Grundy S. M. Horton E. S. Winter M. L. Porte D. Semenkovich C. F. Smith S. Young L. H. Kahn R. Circulation. 2009;108:2941–2948. doi: 10.1161/01.CIR.0000103683.99399.7E. [DOI] [PubMed] [Google Scholar]; (b) Home P. D. Pocock S. J. Beck-Nielsen H. Curtis P. S. Gomis R. Hanefeld M. Jones N. P. Komajda M. McMurray J. J. Lancet. 2009;373:2125–2135. doi: 10.1016/S0140-6736(09)60953-3. [DOI] [PubMed] [Google Scholar]
- Meier C. Kraenzlin M. E. Bodmer M. Jick S. S. Jick H. Meier C. R. Arch. Intern. Med. 2008;168:820–825. doi: 10.1001/archinte.168.8.820. [DOI] [PubMed] [Google Scholar]
- Holst J. J. Physiol. Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- (a) Drucker D. J. Sherman S. I. Gorelick F. S. Bergenstal R. M. Sherwin R. S. Buse J. B. Diabetes Care. 2010;33:428–433. doi: 10.2337/dc09-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ahmad S. R. Swann J. N. Engl. J. Med. 2008;358:1970–1971. [PubMed] [Google Scholar]; (c) FDA drug safety communication: FDA investigating reports of possible increased risk of pancreatitis and pre-cancerous findings of the pancreas from incretin mimetic drugs for type 2 diabetes, https://www.fda.gov/Drugs/DrugSafety/ucm343187.htm, accessed April, 2019
- (a) Van de Laar F. A. Lucassen P. L. Akkermans R. P. Van de Lisdonk E. H. Rutten G. E. Van Weel C. Cochrane Database Syst. Rev. 2005;CD003639:1–6. doi: 10.1002/14651858.CD003639.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) American Diabetes Association Diabetes Care. 2009;32:S13–S61. doi: 10.2337/dc09-S013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D. M. Buse J. B. Davidson M. B. Heine R. J. Holman R. R. Sherwin R. Zinman B. Diabetes Care. 2006;29:1963–1972. doi: 10.2337/dc06-9912. [DOI] [PubMed] [Google Scholar]
- (a) Drucker D. J. Nauck M. A. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]; (b) Drugs@FDA: FDA approved drug products, https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm, accessed April, 2019
- (a) Osada S. Tsukamoto T. Takiguchi M. Mori M. Osumi T. Genes Cells. 1997;2:315–327. doi: 10.1046/j.1365-2443.1997.1220319.x. [DOI] [PubMed] [Google Scholar]; (b) Balasubramanyam M. Mohan V. Curr. Sci. 2000;79:1440–1446. [Google Scholar]
- (a) Chiodini I. Adda G. Scillitani A. Coletti F. Morelli V. Di Lembo S. Epaminonda P. Masserini B. Beck-Peccoz P. Orsi E. Ambrosi B. Arosio M. Diabetes Care. 2007;30:83–88. doi: 10.2337/dc06-1267. [DOI] [PubMed] [Google Scholar]; (b) Andrews R. C. Rooyackers O. Walker B. R. J. Clin. Endocrinol. Metab. 2003;88:285–391. doi: 10.1210/jc.2002-021194. [DOI] [PubMed] [Google Scholar]; (c) Jellinck P. H. Monder C. McEwen B. S. Sakai R. R. J. Steroid Biochem. Mol. Biol. 1993;46:209–213. doi: 10.1016/0960-0760(93)90296-9. [DOI] [PubMed] [Google Scholar]
- Shukla R. Basu A. K. Mandal B. Mukhopadhyay P. Maity A. Chakraborty S. Devrabhai P. K. BMC Endocr. Disord. 2019;19:1–9. doi: 10.1186/s12902-018-0327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buse M. G. Am. J. Physiol. Endocrinol. Metabol. 2005;290:E1–E8. doi: 10.1152/ajpendo.00329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K. C. J. Proteome Res. 2004;3:1284–1288. doi: 10.1021/pr049849v. [DOI] [PubMed] [Google Scholar]
- Goldstein B. J. Curr. Drug Targets: Immune, Endocr. Metab. Disord. 2001;1:265–275. doi: 10.2174/1568008013341163. [DOI] [PubMed] [Google Scholar]
- (a) creutzfeldt W. Ebert R. Diabetologia. 1985;28:565–573. doi: 10.1007/BF00281990. [DOI] [PubMed] [Google Scholar]; (b) Kieffer T. J. Habener J. F. Endocr. Rev. 1999;20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]; (c) Kreymann B. Williams G. Ghatei M. A. Bloom S. R. Lancet. 1987;2:1300–1304. doi: 10.1016/S0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]; (d) Ineedi S. Kandasamy A. D. Veeranjaneyulu A. Kumar V. Pharmacologyonline. 2009;2:17–28. [Google Scholar]; (e) Hirasawa A. Tsumaya K. Awaji T. Katsuma S. Adachi T. Yamada M. Sugimoto Y. Miyazaki S. Tsujimoto G. Nat. Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- Huang S. Czech M. P. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Neumiller J. J. White J. R. Campbell R. K. Drugs. 2010;70:377–385. doi: 10.2165/11318680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Misra M. J. Pharm. Pharmacol. 2013:317–327. doi: 10.1111/j.2042-7158.2012.01574.x. [DOI] [PubMed] [Google Scholar]
- Rhodes C. J. White M. F. Eur. J. Clin. Invest. 2002;32:3–13. doi: 10.1046/j.1365-2362.32.s3.2.x. [DOI] [PubMed] [Google Scholar]
- (a) Idris I. Donnelly R. Diabetes, Obes. Metab. 2009;11:79–88. doi: 10.1111/j.1463-1326.2008.00982.x. [DOI] [PubMed] [Google Scholar]; (b) Keller D. M. Lotspeich W. D. J. Biol. Chem. 1959;234:991–994. [PubMed] [Google Scholar]; (c) Alvarado F. C. Crane R. K. Biochim. Biophys. Acta. 1962;56:170–172. doi: 10.1016/0006-3002(62)90543-7. [DOI] [PubMed] [Google Scholar]; (d) Lee W.-S. Kanai Y. Wells R. G. Hediger M. A. J. Biol. Chem. 1994;269:12032–12039. [PubMed] [Google Scholar]
- Crane R. K., Miller D. and Bihler I., in Membrane transport and metabolism, ed. A. Kleinzeller and A. Kotyk, Academic Press, New York, 1961, pp. 439–449 [Google Scholar]
- (a) Schultz S. G. Curran P. F. Physiol. Rev. 1970;50:637–718. doi: 10.1152/physrev.1970.50.4.637. [DOI] [PubMed] [Google Scholar]; (b) Wright E. M. Peerce B. E. J. Biol. Chem. 1984;259:14993–14996. [PubMed] [Google Scholar]
- (a) OMIM182380, Solute carrier family-5 (sodium/glucose cotransporter), member1, SLC5A1, https://www.omim.org/entry/182380, accessed April, 2019; (b) Wright E. M. Turk E. Eur. J. Physiol. 2004;447:510–518. doi: 10.1007/s00424-003-1063-6. [DOI] [PubMed] [Google Scholar]; (c) Diez-Sampedro A. Hirayama B. A. Osswald C. Gorboulev V. Baumgarten K. Volk C. Wright E. M. Koepsell H. Proc. Natl. Acad. Sci. U. S. A. 2003;100:11753–11758. doi: 10.1073/pnas.1733027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Wright E. M. Am. J. Physiol. 1998;275:G879–G882. doi: 10.1152/ajpgi.1998.275.5.G879. [DOI] [PubMed] [Google Scholar]; (b) Kasahara M. Maeda M. Hayashi S. Mori Y. Abe T. Biochim. Biophys. Acta, Mol. Basis Dis. 2001;1536:141–147. doi: 10.1016/S0925-4439(01)00043-6. [DOI] [PubMed] [Google Scholar]
- (a) Turk E. Wright E. M. J. Membr. Biol. 1997;159:1–20. doi: 10.1007/s002329900264. [DOI] [PubMed] [Google Scholar]; (b) Wright E. M. Am. J. Physiol. Renal. Physiol. 2001;280:F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]; (c) Wright E. M., Martin G. M. and Turk E., in Familial glucose–galactose malabsorption and hereditary renal glycosuria, ed. C. R. Scriver, A. L. Beaudet, W. S. Sly and D. Valle, McGrow-Hill, New York, 8th edn, 2001, Metabolic basis of inherited disease, pp. 4891–4908 [Google Scholar]
- (a) Panayotova-Heiermann M. Loo D. D. F. Klong C. T. Lever J. E. Wright E. M. J. Biol. Chem. 1996;271:10029–10034. doi: 10.1074/jbc.271.17.10029. [DOI] [PubMed] [Google Scholar]; (b) Faham S. Watanabe A. Besserer G. M. Cascio D. Specht A. Hirayama B. A. Wright E. M. Abramson J. Science. 2008;321:810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayotova-Heiermann M. Eskandari S. Zampighi G. A. Wright E. M. J. Biol. Chem. 1997;272:20324–20327. doi: 10.1074/jbc.272.33.20324. [DOI] [PubMed] [Google Scholar]
- Xie Z. Turk E. Wright E. M. J. Biol. Chem. 2000;275:25959–25964. doi: 10.1074/jbc.M002687200. [DOI] [PubMed] [Google Scholar]
- (a) Loo D. D. F. Hirayama B. A. Meinild A. K. Chandy G. Zeuthen Z. Wright E. M. J. Physiol. 1999;518:195–202. doi: 10.1111/j.1469-7793.1999.0195r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schoolwerth A. C. Smith B. C. Culpepper R. M. Miner. Electrolyte Metab. 1988;14:347–361. [PubMed] [Google Scholar]
- (a) Brown G. K. J. Inherited Metab. Dis. 2000;23:237–246. doi: 10.1023/A:1005632012591. [DOI] [PubMed] [Google Scholar]; (b) Mitrakou A. Diabetes Res. Clin. Pract. 2011;93:S66–S72. doi: 10.1016/S0168-8227(11)70016-X. [DOI] [PubMed] [Google Scholar]
- (a) Guyton A. C. and Hall J. E., in Textbook of medical physiology, ed. W. B. Saunders, Philadelphia, PA, 9th edn, 1996, Urine formation and the kidneys, pp. 332–335 [Google Scholar]; (b) White J. R. Clin. Diabetes. 2010;28:5–10. doi: 10.2337/diaclin.28.1.5. [DOI] [Google Scholar]
- (a) Quamme G. A. Freeman H. J. Am. J. Physiol. 1987;253:F151–F157. doi: 10.1152/ajpcell.1987.253.1.C151. [DOI] [PubMed] [Google Scholar]; (b) Mather A. Pollock C. Kidney Int. 2011;79:S1–S6. doi: 10.1038/ki.2010.509. [DOI] [PubMed] [Google Scholar]
- (a) Wright E. M. Loo D. D. F. Hirayama B. A. Physiol. Rev. 2011;91:733–794. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]; (b) Chao E. C. Clin. Diabetes. 2014;32:4–11. doi: 10.2337/diaclin.32.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakris G. L. Fonseca V. A. Sharma K. Wright E. M. Kidney Int. 2009;75:1272–1277. doi: 10.1038/ki.2009.87. [DOI] [PubMed] [Google Scholar]
- (a) Gerich J. E. Meyer C. Woerle H. J. Stumvoll M. Diabetes Care. 2001;24:382–391. doi: 10.2337/diacare.24.2.382. [DOI] [PubMed] [Google Scholar]; (b) Thorens B. Invited review. Am. Physiol. Soc. 1996:G541–G553. doi: 10.1152/ajpgi.1996.270.4.G541. [DOI] [PubMed] [Google Scholar]
- Farber S. Berger E. Earle D. J. Clin. Invest. 1951;30:125–129. doi: 10.1172/JCI102424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmoune H. Thompson P. W. Ward J. M. Smith C. D. Hong G. Brown J. Diabetes. 2005;54:3427–3434. doi: 10.2337/diabetes.54.12.3427. [DOI] [PubMed] [Google Scholar]
- (a) Petersen C. Ann. Pharm. 1835;15:178. doi: 10.1002/jlac.18350150210. [DOI] [Google Scholar]; (b) Dardi I. Kouvatsos T. Jabbour S. A. Biochem. Pharmacol. 2016;101:27–39. doi: 10.1016/j.bcp.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Ehrenkranz J. R. L. Lewis N. G. Kahn C. R. Roth J. Diabetes/Metab. Res. Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- Nair S. Wilding J. P. H. J. Clin. Endocrinol. Metab. 2010;95:34–42. doi: 10.1210/jc.2009-0473. [DOI] [PubMed] [Google Scholar]
- Gallo L. A. Wright E. M. Vallon V. Diabetes Vasc. Dis. Res. 2015;12:78–89. doi: 10.1177/1479164114561992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ferrannini E. Muscelli E. Frascerra S. Baldi S. Mari A. Heise T. Broedl U. C. Woerle H. J. J. Clin. Invest. 2014;124:499–508. doi: 10.1172/JCI72227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gouvea W. L. Alpert H. C. Kelley J. Pardo V. Vaamonde C. A. Kidney Int. 1989;35:1041–1048. doi: 10.1038/ki.1989.88. [DOI] [PubMed] [Google Scholar]; (c) Oku A. Ueta K. Arakawa K. Ishihara T. Nawano M. Kuronuma Y. Matsumoto M. Saito A. Tsujihara K. Anai M. Asano T. Kanai Y. Endau H. Diabetes. 1999;48:1794–1800. doi: 10.2337/diabetes.48.9.1794. [DOI] [PubMed] [Google Scholar]
- (a) Fujita Y. Inagaki N. J. Diabetes Invest. 2014;5:265–275. doi: 10.1111/jdi.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Drugs@FDA: FDA approved drug products, https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=209803, accessed April 2019; (c) Januzzi J. L. Butler J. Jarolim P. Sattar N. Vijapurkar U. Desai M. Davis M. J. J. Am. Coll. Cardiol. 2017;70:704–712. doi: 10.1016/j.jacc.2017.06.016. [DOI] [PubMed] [Google Scholar]
- (a) Johnson and Johnson, homepage on internet, https://www.jnj.com/media-center/press-releases/fda-advisory-committee-recommends-approval-of-canagliflozin-for-treatment-of-adults-with-type-2-diabetes, accessed April 2019; (b) Rosenwasser R. F. Sultan S. Sutton D. Choksi R. Epstein B. J. Diabetes, Metab. Syndr. Obes.: Targets Ther. 2013;6:453–467. doi: 10.2147/DMSO.S34416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Wilding J. P. H. Charpentier G. Hollander P. Gonzalez-Galvez G. Mathieu C. Vercruysse F. Usiskin K. Law G. Black S. Canovatchel W. Meininger G. Int. J. Clin. Pract. 2013;67:1267–1282. doi: 10.1111/ijcp.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Halimi S. Verges B. Diabetes Metab. 2014;40:S28–S34. doi: 10.1016/S1262-3636(14)72693-X. [DOI] [PubMed] [Google Scholar]
- (a) FDA drug safety communication: FDA confirms increased risk of leg and foot amputations with the diabetes medicine canagliflozin (Invokana, Invokamet, Invokamet XR), https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-confirms-increased-risk-leg-and-foot-amputations-diabetes-medicine, accessed May 2019; (b) FDA drug safety communication: FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood, http://wayback.archive-it.org/7993/20170112031553/http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm, accessed May 2019; (c) FDA drug safety communication: FDA revises label of diabetes drug canagliflozin (Invokana, Invokamet) to include updates on bone fracture risk and new information on decreased bone mineral density, https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-revises-label-diabetes-drug-canagliflozin-invokana-invokamet, accessed May 2019; (d) Taylor S. I. Blau J. E. Rother K. I. J. Clin. Endocrinol. Metab. 2015;100:2849–2852. doi: 10.1210/jc.2015-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Drugs@FDA: FDA approved drug products, https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=204629, accessed May 2019; (b) Grempler R. Thomas L. Eckhardt M. Himmelsbach F. Sauer A. Sharp D. E. Bakker R. A. Mark M. Klein T. Eickelmann P. Diabetes, Obes. Metab. 2012;14:83–90. doi: 10.1111/j.1463-1326.2011.01517.x. [DOI] [PubMed] [Google Scholar]; (c) Shubrook J. H. Bokaie B. B. Adkins S. E. Drug Des., Dev. Ther. 2015;9:5793–5803. doi: 10.2147/DDDT.S69926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rosenstock J. Jelaska A. Zellar C. Kim G. Broedl U. C. Woerle H. J. On behalf of the EMPA-REG BASAL™ trial investigators. Diabetes, Obes. Metab. 2015;17:936–948. doi: 10.1111/dom.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) American College of Cardiology, empagliflozin cardiovascular outcome event trial in type 2 diabetes mellitus patients-EMPA-REG OUTCOME, https://www.acc.org/latest-in-cardiology/clinical-trials/2015/09/17/10/11/empa-reg-outcome, accessed May 2019
- (a) Drugs@FDA: FDA approved drug products, https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=202293, accessed May 2019; (b) Drugs@FDA: FDA approved drug products,https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=205649, accessed May 2019; (c) Bailey C. J. Gross J. L. Pieters A. Bastien A. List J. F. Lancet. 2010;375:2223–2233. doi: 10.1016/S0140-6736(10)60407-2. [DOI] [PubMed] [Google Scholar]; (d) Kim G. W. Chung S. H. Arch. Pharmacal Res. 2014;37:957–966. doi: 10.1007/s12272-014-0419-0. [DOI] [PubMed] [Google Scholar]; (e) Madaan T. Akhtar M. Najmi A. K. Eur. J. Pharm. Sci. 2016;93:244–252. doi: 10.1016/j.ejps.2016.08.025. [DOI] [PubMed] [Google Scholar]
- (a) Ferrannini E. Ramos S. J. Salsali A. Tang W. List J. F. Diabetes Care. 2010;33:2217–2224. doi: 10.2337/dc10-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sanofi press release, http://www.news.sanofi.us/2019-01-17-FDA-advisory-committee-votes-on-Zynquista-TM-sotagliflozin-as-treatment-for-adults-with-type-1-diabetes, accessed May 2019; (c) Danne T. Garg S. Peters A. L. Buse J. B. Mathieu C. Pettus J. H. Alexander C. M. Battelino T. Javier Ampudia-Blasco F. Bode B. W. Cariou B. Close K. L. Dondona P. Dutta S. Ferannini E. Fourlanos S. Grunberger G. Heller S. R. Henry R. R. Kurian M. J. Kushner J. A. Oron T. Parkin C. G. Pieber T. R. Rodbard H. W. Schatz D. Skyler J. S. Tamborlane W. V. Yokote K. Phillip M. Diabetes Care. 2019;42:1147–1154. doi: 10.2337/dc18-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Goldenberg R. M. Gilbert J. D. Hramiak I. M. Woo V. C. Zinman B. Diabetes, Obes. Metab. 2019:1–11. doi: 10.1111/dom.13811. [DOI] [PubMed] [Google Scholar]
- (a) Poole R. M. Dungo R. T. Drugs. 2014;74:611–617. doi: 10.1007/s40265-014-0204-x. [DOI] [PubMed] [Google Scholar]; (b) Takasu T. Yokono M. Tahara A. Takakura S. Biol. Pharm. Bull. 2019;42:507–511. doi: 10.1248/bpb.b18-00728. [DOI] [PubMed] [Google Scholar]
- (a) Astellas Pharma Inc., Approval of Suglat Tablets, a Selective SGLT2 Inhibitor for Treatment of Type 2 Diabetes, in Japan, https://www.astellas.com/en/news/11351, accessed May 2019; (b) Astelas Pharma Inc., Approval of Suglat tablets, a selective SGLT2 inhibitor, for additional indication of type 1 diabetes mellitus and additional dosage and administration, in Japan, https://www.astellas.com/en/news/14481, accessed May 2019
- Poole R. M. Prossler J. E. Drugs. 2014;74:939–944. doi: 10.1007/s40265-014-0229-1. [DOI] [PubMed] [Google Scholar]
- Suzuki M. Honda K. Fukazawa M. Ozawa K. Hagita H. Kawai T. Takeda M. Yata T. Kawai M. Fukuzawa T. Kobayashi T. Sato T. Kawabe Y. Ikeda S. J. Pharmacol. Exp. Ther. 2012;341:692–701. doi: 10.1124/jpet.112.191593. [DOI] [PubMed] [Google Scholar]
- (a) Kaku K. Watada H. Iwamoto Y. Utsunomiya K. Terauchi Y. Tobe K. Tanizawa Y. Araki E. Ueda M. Suganami H. Watanabe D. Cardiovasc. Diabetol. 2014;13:1–15. doi: 10.1186/1475-2840-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chugai Pharmaceutical Co. License agreement of SGLT2 inhibitor ‘‘CSG452’’ in Japan, https://www.kowa.co.jp/eng/news/press12102602.pdf, accessed February 2019
- Seino Y. Sasaki T. Fukatsu A. Ubukata M. Sakai S. Samukawa Y. Curr. Med. Res. Opin. 2014;30:1245–1255. doi: 10.1185/03007995.2014.912983. [DOI] [PubMed] [Google Scholar]
- (a) Taisho Pharmaceutical Co. LTD., News release, https://www.taisho.co.jp/en/company/release/2012/index.html, accessed May 2019; (b) Markham A. Elkinson S. Drugs. 2014;74:945–950. doi: 10.1007/s40265-014-0230-8. [DOI] [PubMed] [Google Scholar]
- Amin N. B. Wang X. Jain S. M. Lee D. S. Nucci G. Rusnak J. M. Diabetes, Obes. Metab. 2015;17:591–598. doi: 10.1111/dom.12460. [DOI] [PubMed] [Google Scholar]
- (a) Amin N. B. Wang X. Mitchell J. R. Lee D. S. Nucci G. Rusnak J. M. Diabetes, Obes. Metab. 2015;17:805–808. doi: 10.1111/dom.12486. [DOI] [PubMed] [Google Scholar]; (b) Miller S. Krumins T. Zhou H. Huyck S. Johnson J. Golm G. Terra S. G. Mancuso J. P. Engel S. S. Lauring B. Diabetes Ther. 2018;9:253–268. doi: 10.1007/s13300-017-0358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands A. T. Zambrowicz B. P. Rosenstock J. Lapuerta P. Bode B. W. Garg S. K. Buse J. B. Banks P. Heptulla R. Rendell M. Cefalu W. T. Strumph P. Diabetes Care. 2015;38:1181–1188. doi: 10.2337/dc14-2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lapuerta P. Zambrowicz B. Strumph P. Sands A. Diabetes Vasc. Dis. Res. 2015;12:101–110. doi: 10.1177/1479164114563304. [DOI] [PubMed] [Google Scholar]; (b) Finucane F. M. Med. Hypotheses. 2018;114:11–12. doi: 10.1016/j.mehy.2018.02.025. [DOI] [PubMed] [Google Scholar]; (c) Collaboration and licence agreement between Lexicon Pharmaceuticals, Inc. and Sanofi, 2015, https://www.sec.gov/Archives/edgar/data/1062822/000106282216000068/exh1014collaborationandlic.htm, accessed February 2019; (d) 2015 Annual report, Lexicon pharmaceuticals, 2015, 1
- (a) Makabe O. Suzuki H. Umezawa S. Bull. Chem. Soc. Jpn. 1977;50:2689–2693. doi: 10.1246/bcsj.50.2689. [DOI] [Google Scholar]; (b) whiting M. Tripp J. C. Lin Y.-C. Lindstorm W. Olson A. J. Elder J. H. Sharpless K. B. Fokin V. V. J. Med. Chem. 2006;49:7697–7710. doi: 10.1021/jm060754+. [DOI] [PubMed] [Google Scholar]; (c) Kume M. Kubota T. Kimura Y. Nakashimizu H. Motokawa K. Nakano M. J. Antibiot. 1993;46:177–192. doi: 10.7164/antibiotics.46.177. [DOI] [PubMed] [Google Scholar]; (d) Buckle D. R. Outred D. J. Rockell C. J. M. Smith H. Spicer B. A. J. Med. Chem. 1983;26:251–254. doi: 10.1021/jm00356a025. [DOI] [PubMed] [Google Scholar]
- Putapatri S. R. Kanwal A. Sridhar B. Banerjee S. K. Kantevari S. Org. Biomol. Chem. 2014;12:8415–8421. doi: 10.1039/C4OB01319K. [DOI] [PubMed] [Google Scholar]
- Li L.-T. Zhou L.-F. Li Y.-J. Huang J. Liu R.-H. Wang B. Wang P. Bioorg. Med. Chem. Lett. 2012;22:642–644. doi: 10.1016/j.bmcl.2011.10.062. [DOI] [PubMed] [Google Scholar]
- Bai S.-T. Xiong D.-C. Niu Y. Wu Y.-F. Ye X.-S. Tetrahedron. 2015;71:4909–4919. doi: 10.1016/j.tet.2015.05.108. [DOI] [Google Scholar]
- Du X. Lizarzaburu M. Turcotte S. Lee T. Greenberg J. Shan B. Fan P. Ling Y. Medina J. C. Houze J. Bioorg. Med. Chem. Lett. 2011;21:3774–3779. doi: 10.1016/j.bmcl.2011.04.053. [DOI] [PubMed] [Google Scholar]
- Zhang X. Urbanski M. Patel M. Cox G. G. Zeck R. E. Bian H. Conway B. R. Beavers M. P. Rybczynski P. J. Demarest K. T. Bioorg. Med. Chem. Lett. 2006;16:1696–1701. doi: 10.1016/j.bmcl.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Chu K.-F. Yao C.-H. Song J.-S. Chen C.-T. Yeh T.-K. Hsieh T.-C. Huang C.-Y. Wang M.-H. Wu S.-H. Chang W.-E. Chao Y.-S. Lee J.-C. Bioorg. Med. Chem. 2016;24:2242–2250. doi: 10.1016/j.bmc.2016.03.058. [DOI] [PubMed] [Google Scholar]
- (a) Nomura S. and Sakamaki S., WO Pat., 2008/013322 A1, 2008; (b) Nomura S., Kawanishi E. and Ueta K., Novel compounds having inhibitory activity against sodium-dependent transporter, WO/2005/012326, 2005; (c) Li X. Wells K. M. Branum S. Damon S. Youells S. Beauchamp D. A. Palmer D. Stefanick S. Russell R. K. Murray W. Org. Process Res. Dev. 2012;16:1727–1732. doi: 10.1021/op3001355. [DOI] [Google Scholar]
- Yao C.-H. Song J.-S. Chen C.-T. Yeh T.-K. Hsieh T.-C. Wu S.-H. Huang C.-Y. Huang Y.-L. Wang M.-H. Liu Y.-W. Tsai C.-H. Kumar C. R. Lee J.-C. Eur. J. Med. Chem. 2012;55:32–38. doi: 10.1016/j.ejmech.2012.06.053. [DOI] [PubMed] [Google Scholar]
- Lee S. H. Song K.-S. Kim J. Y. Kang M. Lee J. S. Cho S.-H. Park H.-J. Kim J. Lee J. Bioorg. Med. Chem. 2011;19:5813–5832. doi: 10.1016/j.bmc.2011.08.014. [DOI] [PubMed] [Google Scholar]
- Koga Y. Sakamaki S. Hongu M. Kawanishi E. Sakamoto T. Yamamoto Y. Kimata H. Nakayama K. Kuriyama C. Matsushita Y. Ueta K. Tsuda-Tsukimoto M. Nomura S. Bioorg. Med. Chem. 2013;21:5561–5572. doi: 10.1016/j.bmc.2013.05.048. [DOI] [PubMed] [Google Scholar]
- Fushimi N. Teranishi H. Shimizu K. Yonekubo S. Ohno K. Miyagi T. Itoh F. Shibazaki T. Tomae M. Ishikawa-Takemura Y. Nakabayashi T. Kamada N. Yamauchi Y. Kobayashi S. Isaji M. Bioorg. Med. Chem. 2013;21:748–765. doi: 10.1016/j.bmc.2012.11.041. [DOI] [PubMed] [Google Scholar]
- Kim M. J. Lee J. Kang S. Y. Lee S. H. Son E. J. Jung M. E. Lee S. H. Song K. S. Lee M. Han H. K. Kim J. Lee J. Bioorg. Med. Chem. Lett. 2010;20:3420–3425. doi: 10.1016/j.bmcl.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Kang S. Y. Lee J. Song K.-S. Lee J. Lee S.-H. Lee J. Bioorg. Med. Chem. 2010;18:6069–6079. doi: 10.1016/j.bmc.2010.06.076. [DOI] [PubMed] [Google Scholar]
- Lee S. H. Kim M. J. Lee S.-H. Kim J. Park H.-J. Lee J. Eur. J. Med. Chem. 2011;46:2662–2675. doi: 10.1016/j.ejmech.2011.03.052. [DOI] [PubMed] [Google Scholar]
- Xie J. Durrat F. Valery J.-M. J. Org. Chem. 2003;68:7896–7898. doi: 10.1021/jo034795e. [DOI] [PubMed] [Google Scholar]
- Lee J. Lee S.-H. Seo H.-J. Son E.-J. Lee S. H. Jung M. E. Lee M. Han H.-K. Kim J. Kang J. Lee J. Bioorg. Med. Chem. 2010;18:2178–2194. doi: 10.1016/j.bmc.2010.01.073. [DOI] [PubMed] [Google Scholar]
- Lv B. Xu B. Feng Y. Peng K. Xu G. Du J. Zhang L. Zhang W. Zhang T. Zhu L. Ding H. Sheng Z. Welihinda A. Seed B. Chen Y. Bioorg. Med. Chem. Lett. 2009;19:6877–6881. doi: 10.1016/j.bmcl.2009.10.088. [DOI] [PubMed] [Google Scholar]
- Xu B. Lv B. Feng Y. Xu G. Du J. Welihinda A. Sheng Z. Seed B. Chen Y. Bioorg. Med. Chem. Lett. 2009;19:5632–5635. doi: 10.1016/j.bmcl.2009.08.030. [DOI] [PubMed] [Google Scholar]
- Robinson R. P. Mascitti V. Boustany-Kari C. M. Carr C. L. Foley P. M. Kimoto E. Leininger M. T. Lowe A. Klenotic M. K. MacDonald J. I. Maguire R. J. Masterson V. M. Maurer T. S. Miao Z. Patel J. D. Preville C. Reese M. R. She L. Steppan C. M. Thuma B. A. Zhu T. Bioorg. Med. Chem. Lett. 2010;20:1569–1572. doi: 10.1016/j.bmcl.2010.01.075. [DOI] [PubMed] [Google Scholar]
- Mascitti V. Robinson R. P. Preville C. Thuma B. A. Carr C. L. Reese M. R. Maguire R. J. Leininger M. T. Lowe A. Steppan C. M. Tetrahedron Lett. 2010;51:1880–1883. doi: 10.1016/j.tetlet.2010.02.024. [DOI] [Google Scholar]
- Xu B. Feng Y. Lv B. Xu G. Zhang L. Du J. Peng K. Xu M. Dong J. Zhang W. Zhang T. Zhu L. Ding H. Sheng Z. Welihinda A. Seed B. Chen Y. Bioorg. Med. Chem. 2010;18:4422–4432. doi: 10.1016/j.bmc.2010.04.088. [DOI] [PubMed] [Google Scholar]
- Kim M. J. Lee S. H. Park S. O. Kang H. Lee J. S. Lee K. N. Jung M. E. Kim J. Lee J. Bioorg. Med. Chem. 2011;19:5468–5479. doi: 10.1016/j.bmc.2011.07.045. [DOI] [PubMed] [Google Scholar]
- (a) Yan Q. Ding N. Li Y. Carbohydr. Res. 2016;421:1–8. doi: 10.1016/j.carres.2015.10.011. [DOI] [PubMed] [Google Scholar]; (b) Nomura S. and Sakamaki S., WO/2008/013322, 2008; (c) Nomura S., Kawanishi E. and Ueta K., WO/2005/012326, 2005
- Ikegai K. Imamura M. Suzuki T. Nakanishi K. Murakami T. Kurosaki E. Noda A. Kobayashi Y. Yokota M. Koide T. Kosakai K. Ohkura Y. Takeuchi M. Tomiyama H. Ohta M. Bioorg. Med. Chem. 2013;21:3934–3948. doi: 10.1016/j.bmc.2013.03.067. [DOI] [PubMed] [Google Scholar]
- Pan X. Huan Y. Shen Z. Liu Z. Eur. J. Med. Chem. 2016;114:89–100. doi: 10.1016/j.ejmech.2016.02.053. [DOI] [PubMed] [Google Scholar]
- Li A.-R. Zhang J. Greenberg J. Lee T. Liu J. Bioorg. Med. Chem. Lett. 2011;21:2472–2475. doi: 10.1016/j.bmcl.2011.02.056. [DOI] [PubMed] [Google Scholar]
- Chen Z.-H. Wang R.-W. Qing F.-L. Tetrahedron Lett. 2012;53:2171–2176. doi: 10.1016/j.tetlet.2012.02.062. [DOI] [Google Scholar]
- Zhao W. J. Shi Y. H. Zhao G. L. Wang Y. L. Shao H. Da Tang L. Wang J. W. Chin. Chem. Lett. 2011;22:1215–1218. [Google Scholar]
- Kakinuma H. Oi T. Hashimoto-Tsuchiya Y. Arai M. Kawakita Y. Fukasawa Y. Iida I. Hagima N. Takeuchi H. Chino Y. Asami J. Okumura-Kitajima L. Io F. Yamamoto D. Miyata N. Takahashi T. Uchida S. Yamamoto K. J. Med. Chem. 2010;53:3247–3261. doi: 10.1021/jm901893x. [DOI] [PubMed] [Google Scholar]
- Lansdell M. I. Burring D. J. Hepworth D. Strawbridge M. Graham E. Guyot T. Betson M. S. Hart J. D. Bioorg. Med. Chem. Lett. 2008;18:4944–4947. doi: 10.1016/j.bmcl.2008.08.036. [DOI] [PubMed] [Google Scholar]
- Cao X. Zhang W. Yan X. Huang Z. Zhang Z. Wang P. Shen J. Bioorg. Med. Chem. Lett. 2016;26:2170–2173. doi: 10.1016/j.bmcl.2016.03.065. [DOI] [PubMed] [Google Scholar]
- Tsujihara K. Hongu M. Saito K. Kawanishi H. Kuriyama K. Matsumoto M. Oku A. Ueta K. Tsuda M. Saito A. J. Med. Chem. 1999;42:5311–5324. doi: 10.1021/jm990175n. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y. Kawanishi E. Koga Y. Sakamaki S. Sakamoto T. Ueta K. Matsushita Y. Kuriyama C. Tsuda-tsukimoto M. Nomura S. Bioorg. Med. Chem. Lett. 2013;23:5641–5645. doi: 10.1016/j.bmcl.2013.08.042. [DOI] [PubMed] [Google Scholar]
- Sato S. Takeo J. Aoyama C. Kawahara H. Bioorg. Med. Chem. 2007;15:3445–3449. doi: 10.1016/j.bmc.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Yang J. Yang X. Wang C. Lin Q. Mei Z. Zhao P. Med. Chem. Res. 2015;24:1265–1271. doi: 10.1007/s00044-014-1200-0. [DOI] [Google Scholar]
- Shimokawa Y. Akao Y. Hirasawa Y. Awang K. Hadi A. H. Sato S. Aoyama C. Takeo J. Shiro M. Morita H. J. Nat. Prod. 2010;73:763–767. doi: 10.1021/np9007987. [DOI] [PubMed] [Google Scholar]
- (a) Zhang Z., Huang S., Zhang C., Su Y. and Ren Y., WO Pat., WO2016041470A1, 2015; (b) Zhang Z., Huang S., Zhang C., Su Y. and Ren Y., CN Pat., CN107108539A, 2015
- (a) Hirshberg B. and Iqbal N., WO Pat., WO2015173584A1, 2015; (b) Hirshberg B. and Iqbal N., EP Pat., EP3142661A1, 2015; (c) Hirshberg B. and Iqbal N., US Pat., US20170135981A1, 2015; (d) Hirshberg B. and Iqbal N., JP Pat., JP2017515908A, 2015; (e) Hirshberg B. and Iqbal N., TW Pat., TW201607540A, 2015; (f) Hirshberg B. and Iqbal N., CN Pat., CN106714796A, 2015
- (a) Brenek S. J., Caron S., Nelson J. D., Webster M. E. and Weekly R. M., WO Pat., WO2014159151A1, 2014; (b) Brenek S. J., Caron S., Nelson J. D., Webster M. E. and Weekly R. M., US Pat., US9573959B2, 2014; (c) Brenek S. J., Caron S., Nelson J. D., Webster M. E. and Weekly R. M., EP Pat., EP3466431A1, 2014; (d) Brenek S. J., Caron S., Nelson J. D., Webster M. E. and Weekly R. M., EP Pat., EP2968375B1, 2014
- (a) Wienrich M. and Mayoux E., WO Pat., WO2012163990A1, 2012; (b) Wienrich M. and Mayoux E., EP Pat., EP2714052B1, 2012; (c) Wienrich M. and Mayoux E., US Pat., US20180177794A1, 2018
- (a) Gant T. G. and Manoucherhr S., WO Pat., WO2010048358A2, 2010; (b) Gant T. G. and Manoucherhr S., US Pat., US20100167988A1, 2010
- Harrison C. Patent watch. Nat. Rev. Drug Discovery. 2010;9:508. doi: 10.1038/nrd3218. [DOI] [PubMed] [Google Scholar]
- (a) Weixin S., Weimin Z. and Shuhua G., WO Pat., WO2010012153A1, 2009; (b) Weixin S., Weimin Z. and Shuhua G., CN Pat., CN101638423B, 2009