

Abstract
Medulloblastomas are malignant embryonal brain tumours that may harbour mutations in histone‐modifying genes, while mutations in histone genes have not been detected to date. We here describe the first SHH medulloblastoma with H3 K27M mutation. This may have diagnostic implications as H3 K27M mutations are the hallmark of diffuse midline gliomas, H3 K27M mutant, WHO grade IV. Medulloblastomas arise in midline structures and thus must not be mistaken for DMG when using an antibody detecting the H3 K27M mutation.
Keywords: H3 K27M, H3F3A, H3K27me3, medulloblastoma, NGS, SHH
CONFLICT OF INTEREST
None to declare.
Medulloblastomas are aggressive embryonal tumours growing in the cerebellum and brainstem. They subdivide into four major molecular groups. One of these subgroups is sonic hedgehog (SHH) medulloblastoma, which typically occurs either in infants or older children/young adults, and harbours mutations in SHH pathway genes [1]. Large series of SHH medulloblastomas have been sequenced to date to analyse the mutational spectrum [1, 2]. As also discussed in a recent mini‐symposium on medulloblastomas in Brain Pathology [3], SHH medulloblastomas often have mutations in genes related to chromatin modification [1]. However, mutations in the histone proteins, which condense and structure the DNA, have not been detected in medulloblastomas so far. One subunit of the histone complexes is histone 3 (H3), which is encoded by several highly homologous genes such as H3F3A. Surprisingly, we detected an H3F3A K27M‐mutation in an SHH medulloblastoma.
A 13‐year‐old female was admitted to the A&E department with strong pain in the forehead and nape, dizziness, nausea and vomiting as well as double vision. Her mother additionally described increased sleepiness, slowness and a loss of appetite. On clinical examination, the patient was in a reduced general condition and showed signs of dehydration, a pale skin colour, a tilt of the head to the left side and slowed movements and speech. Pupils were equal, of normal size and slowly reacting to light. Apart from this, examination of the cranial nerves was inconspicuous, and no sensorimotor deficit was detected. Magnetic resonance imaging indicated a cerebellar tumour located in the right hemisphere (Figure 1A,B).
FIGURE 1.
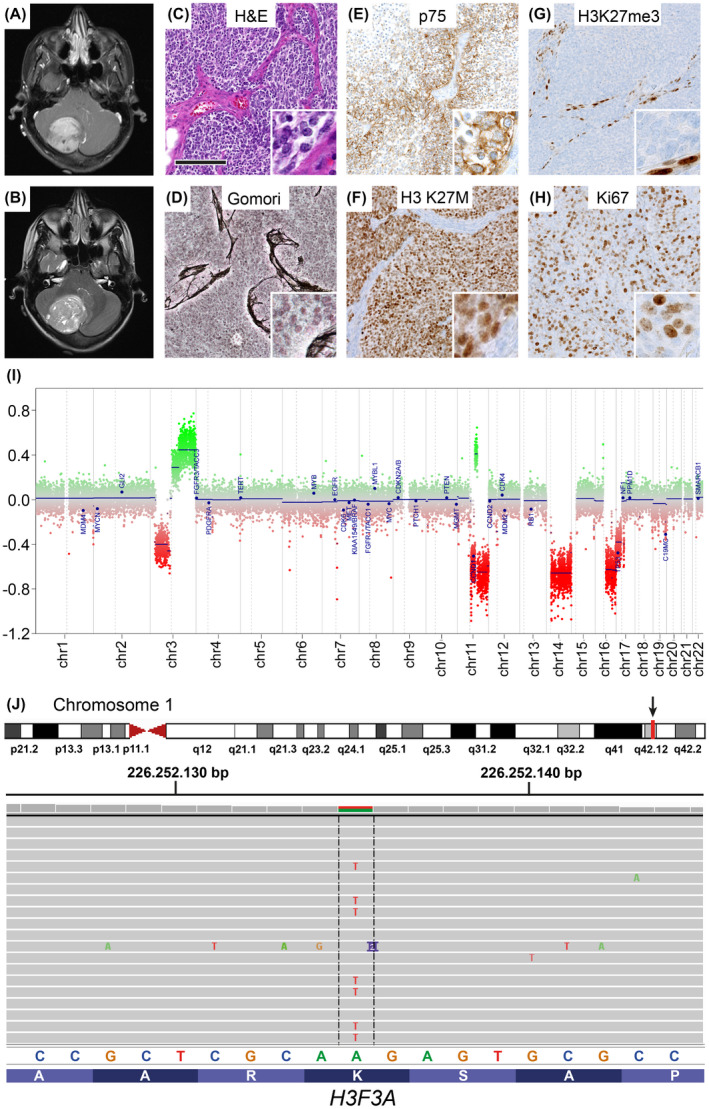
A desmoplastic/nodular SHH medulloblastoma with H3 K27M mutation. (A and B) Representative axial MRI images of the tumour showing a partial contrast enhancement in the T 1‐weighted image plus contrast medium (A) and hyperintensity in the T 2‐weighted image (B). (C–H) Histology. The H&E staining showed a small blue round cell tumour with a nodular architecture also visible in a Gomori silver impregnation (D). The internodal areas were positive for p75 (E). A mutation‐specific antibody indicated a nuclear expression of H3 K27M (F), whereas H3K27me3 trimethylation was lost in the tumour cells (G). (H) The Ki67 proliferation index amounted up to 30–60%, depending on the tumour area. Scale bar for (C–H): 150 µm. (I, J) Molecular features. (I) Copy number profile calculated from DNA methylation data. (J) H3F3A K27M mutation, detected by DNA panel sequencing. Top: genomic position on chromosome 1q, indicated by the red bar (cf. arrow). Middle: base exchange from A to T with an allele frequency of 46% (exemplary reads). Bottom: reference sequences of bases and amino acids
Histological examination of the surgical specimen revealed a small blue round cell tumour on H&E staining (Figure 1C). Tumour cells formed nodules, delineated by cell‐dense areas with a reticulin‐rich fibre network (Figure 1D). Tumour cells in the reticulin‐rich internodal areas expressed p75 (Figure 1E), whereas central areas strongly expressed NeuN (Figure S1A). Some tumour cells showed a nuclear expression of YAP1 (Figure S1B). However, tumour cells did not display nuclear expression of β‐Catenin (Figure S1C) and were negative for OTX2 (Figure S1D). Only a few scattered nuclei showed a strong nuclear accumulation of p53 (<1% of nuclei, Figure S1E). The nuclear expression of INI1 was retained (Figure S1F). The Ki67 proliferation index amounted up to 60% of the tumour cells (Figure 1H). Taken together, the histological diagnosis was desmoplastic/nodular medulloblastoma, WHO grade IV, and the immunohistochemical profile was suggestive of an SHH‐activated medulloblastoma.
To confirm this, we performed global DNA methylation profiling using an Illumina EPIC BeadChip array. We then analysed the methylation data with the DNA methylation‐based brain tumour classifier as described previously [4]. The DNA methylation profile was not classifiable but received the highest score of 0.77 for the methylation class family medulloblastoma, SHH (score 0.42 for the subclass SHH A, children and adult). A t‐SNE analysis confirmed the affiliation of this tumour with the group of SHH‐activated medulloblastomas (Figure S2). A copy number profile calculated from the DNA methylation data showed several chromosomal gains and losses that did not add diagnostic information (Figure 1I).
Next, we performed DNA panel sequencing to screen for a mutation in an SHH pathway gene and to determine the TP53 status. While a PTCH1 mutation was present (NM_000264:c.2630_2631insGGGA) and TP53 was wild‐type, we unexpectedly detected an H3F3A K27M mutation (NM_002107:c.83A>T, Figure 1J). Immunohistochemical staining for H3 K27M confirmed the mutation, as it showed a nuclear expression of the H3 K27M mutant protein (Figure 1F). Also, staining for H3K27me3 revealed a loss of the H3 K27 trimethylation (H3K27me3, Figure 1G). In sum, we diagnosed a medulloblastoma, SHH‐activated and TP53‐wild‐type, H3 K27M‐mutant, WHO grade IV.
After surgery, the patient received combined radiochemotherapy according to HIT‐MED Guidance 5.2 [radiation of the craniospinal axis with 23.4 Gy plus boost (cumulative dose in tumour area 54 Gy); additionally, six cycles of Vincristin followed by eight cycles of Cisplatin plus CCNU). Three months post‐surgery, the patient still has double vision looking into the distance but otherwise, the general condition has improved.
H3 K27M mutations have not been detected in SHH medulloblastomas so far. Thus, they seem to be very rare in these tumours. Hence, it is unclear how these mutations impact tumour biology and survival in SHH medulloblastomas. H3 K27M mutations are the genetic hallmark of a group of highly malignant gliomas that are called “diffuse midline gliomas, H3 K27M‐mutant.” These tumours typically occur in midline structures of children and young adults [5]. In these tumours, H3 K27M mutations lead to a global reduction of the immunohistochemically accessible repressive histone trimethylation H3K27me3 as well as a DNA hypomethylation phenotype. Consequently, this results in the expression of many oncogenes and in tumour growth [6, 7]. The H3 K27M mutation described here thus represents a mechanism resulting in a loss of H3K27me3. Interestingly, loss of H3K27me3 has been detected in SHH medulloblastoma previously [8]. Regulation of epigenetic histone modifications in SHH medulloblastoma is a highly complex process, as these tumours may have mutations in genes encoding different histone methyltransferases and demethylases, such as KMD3B, KDM6A and KMT2D (MLL2) [1, 8]. Also, SHH medulloblastomas may have chromosomal aberrations involving histone‐modifying genes [8]. Loss of H3K27me3 may ameliorate or worsen the prognosis of medulloblastoma, depending on the presence or the absence of other epigenetic histone modifications such as H3K4me3 [8].
Other than in diffuse midline gliomas, H3 K27M mutations have been identified in single ependymomas that were molecularly classified as subgroup ‘posterior fossa group A’ [9, 10].
In diagnostics, an antibody‐targeting H3 K27M is nowadays routinely used to screen midline tumours for the presence of an H3 K27M mutation in order to detect diffuse midline glioma, H3 K27M‐mutant. Pathologists should be aware that H3 K27M mutations rarely occur in midline tumours other than diffuse midline glioma, H3 K27M mutant, especially if the tumour has an embryonal or ependymal morphology.
Supporting information
FIGURE S1 Complementary immunohistochemical stainings. (A) The central parts of the nodules were positive for NeuN. (B) Some cells showed a nuclear expression of YAP1. (C) No nuclear expression of ß‐Catenin. (D) No expression of OTX2. (E) Only few nuclei stained for p53. (F) Nuclear expression of INI1 retained
FIGURE S2 A t‐SNE analysis of DNA methylation data from the test case and reference samples from different classes of brain tumour allocates the test case to SHH‐medulloblastoma (non‐trivial PCs 101, perplexity 55)
ACKNOWLEDGEMENTS
We thank Nicole Bernhardt and Helena Zinn (Hamburg) for technical support. Matthias Dottermusch is funded by the Erich und Gertrud Roggenbuck‐Stiftung. Annika K. Wefers is a fellow of the Mildred Scheel Cancer Career Center Hamburg/Deutsche Krebshilfe.
DATA AVAILABILITY STATEMENT
Raw data are available upon reasonable request.
REFERENCES
- 1. Kool M, Jones David TW, Jäger N, Northcott Paul A, Pugh Trevor J, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. 10.1016/j.ccr.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole‐genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–7. 10.1038/nature22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar R, Liu APY, Northcott PA. Medulloblastoma genomics in the modern molecular era. Brain Pathol. 2020;30:679–90. 10.1111/bpa.12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555:469–74. 10.1038/nature26000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 6. Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high‐grade gliomas. Cancer Cell. 2013;24:660–72. 10.1016/j.ccr.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 7. Harutyunyan AS, Chen H, Lu T, Horth C, Nikbakht H, Krug B, et al. H3K27M in gliomas causes a one‐step decrease in H3K27 methylation and reduced spreading within the constraints of H3K36 methylation. Cell Rep. 33:108390. 10.1016/j.celrep.2020.108390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez‐Lago M, et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013;125:373–84. 10.1007/s00401-012-1070-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gessi M, Capper D, Sahm F, Huang K, von Deimling A, Tippelt S, et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol. 2016;132:635–7. 10.1007/s00401-016-1608-3 [DOI] [PubMed] [Google Scholar]
- 10. Ryall S, Guzman M, Elbabaa SK, Luu B, Mack SC, Zapotocky M, et al. H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst. 2017;33:1047–51. 10.1007/s00381-017-3481-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Complementary immunohistochemical stainings. (A) The central parts of the nodules were positive for NeuN. (B) Some cells showed a nuclear expression of YAP1. (C) No nuclear expression of ß‐Catenin. (D) No expression of OTX2. (E) Only few nuclei stained for p53. (F) Nuclear expression of INI1 retained
FIGURE S2 A t‐SNE analysis of DNA methylation data from the test case and reference samples from different classes of brain tumour allocates the test case to SHH‐medulloblastoma (non‐trivial PCs 101, perplexity 55)
Data Availability Statement
Raw data are available upon reasonable request.