

Abstract
Pediatric bithalamic gliomas encompass several histomolecular tumoral types from benign to malignant and underlines the central role of a comprehensive neuropathological review, including immunohistochemistry, genetic, and epigenetic analyses, to achieve an accurate diagnosis.

Keywords: bithalamic, landscape, pediatric.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest directly related to the topic of this article.
ETHICS APPROVAL
This study was approved by the GHU Paris Psychiatry Neurosciences, Sainte‐Anne Hospital's local ethics committee. All the patients’ parents or legal guardians signed informed consent forms before treatment was started. We obtained human subjects’ approval from our institutional review board.
Over the past several years, based on the results of studies reported in the literature: (i) Diffuse midline gliomas (DMG) were defined as a new tumoral type in the 2016 World Health Organization (WHO) classification, and (ii) four different subtypes are now defined depending on their molecular characteristics and/or locations: DMG H3.3 K27‐mutant, DMG H3.1 or H3.2 K27‐mutant, DMG H3‐wild type with EZHIP overexpression, and DMG EGFR‐altered [1, 2, 3]. Whereas monothalamic tumors are mainly represented by H3.3 K27‐mutant, this type of tumor seems rarer in its bithalamic location [4]. Recent epigenomic analyses have pointed out the existence of a new DNA methylation class (MC) of DMG, EGFR‐altered [2, 3]. As H3K27‐mutant DMG, these tumors mainly affect children of school age and are associated with a very poor prognosis [2, 3]. While nearly all DMG are characterized by a H3K27me3 loss, DMG, EGFR‐altered are enriched for EGFR alterations (mainly mutations of exons 20 and 7, or amplification). However, in this rapidly evolving field, a more comprehensive analysis of pediatric bithalamic gliomas including clinical, radiological, histopathological, and molecular data is needed.
Here, we investigated retrospectively data from 19 pediatric bithalamic gliomas compiled consecutively from our center (18 cases diagnosed between January 1, 1988 and December 31, 2020 from Necker Hospital) and one case from Nancy hospital. They were confirmed by a central radiological review (excluding cases with obvious tumoral infiltration of another anatomic structure) by two experienced pediatric neuroradiologists in consensus, without former knowledge of the molecular data. We concluded that it was a bithalamic tumor when the epicenter of the tumor was on the thalami, with significative infiltration on both sides and not only a monothalamic mass with small infiltration of the contralateral thalamus. We also performed a comprehensive clinical, histopathological (including H3K27me3, H3K27M, and EZHIP immunoexpression), and molecular evaluation (including EGFR status by next‐generation sequencing, and FISH analyses), as well as DNA methylation profiling (using the v12.3) for those samples having sufficient material available. The immunostaining for neurofilament protein (NFP) was performed to define a circumscribed tumor (meaning the absence of or few disorganized residual axons) and a diffuse tumor (presenting numerous axons in an organoid pattern).
Clinical, histopathological, and molecular data from the cases are summarized in Table S1 and illustrated in Figures S1 and S2. The integrative histopathological, genetic, and epigenetic analyses, including t‐Distributed Stochastic Neighbor Embedding analyses (t‐SNE) (Figure 1), segregated tumors into five DMG, EGFR‐altered (26.3%); two DMG, H3K27‐altered (with H3K27M mutation) (10.5%); two DMG, H3K27‐wild type (WT) with EZHIP overexpression (10.5%); two pediatric glioblastomas, RTK2 subtype (10.5%); two pilocytic astrocytomas (PA) (10.5%); one pediatric glioblastoma, MYCN‐amplified (5.3%); one ganglioglioma (GG), grade 1, BRAF‐mutant (5.3%); one angiocentric glioma (AG), with MYB:QKI fusion (5.3%); one circumscribed glioma, BRAF and H3.3 K27‐mutant (5.3%); one high‐grade glioma, IDH‐ and histones‐WT, not elsewhere classified (NEC; 5.3%); and one high‐grade glioma, IDH‐ and histones‐WT, not otherwise specified (NOS), without sufficient material for DNA‐methylation analysis (5.3%). All DMG EGFR‐altered presented a loss of H3K27me3 immunoexpression, and three‐fifths an EZHIP overexpression (in the absence of a H3 K27 mutation). In addition, mutations in the EGFR gene, which implicated exon 7 (n = 3), exon 20 (n = 2), and exon 8 (n = 1), were detected (one case presented a mutation in exon 7 and another one in exon 20). The two DMG, H3K27‐WT with overexpression of EZHIP did not show EGFR alterations by FISH and DNA sequencing analyses as expected. Two cases presented H3K27M mutation, but no DNA‐methylation was available, and moreover, because one of them presented an EGFR amplification, we suggest the diagnosis of DMG, H3K27‐altered (with H3K27M mutation). One of the two tumors, classified as pediatric glioblastoma, RTK2 subtype by DNA methylation profiling, harbored an EGFR mutation (exon 20) and an amplification of the locus gene, but no loss of H3K27me3. The pediatric glioblastoma, MYCN‐amplified presented a retained expression of H3K27me3 and no EGFR alterations. The two last diffuse gliomas NOS/NEC presented an EGFR mutation—exon 20—without H3K27me3 loss, but one of them (case 12) did not cluster with a DNA methylation class by t‐SNE analysis.
FIGURE 1.
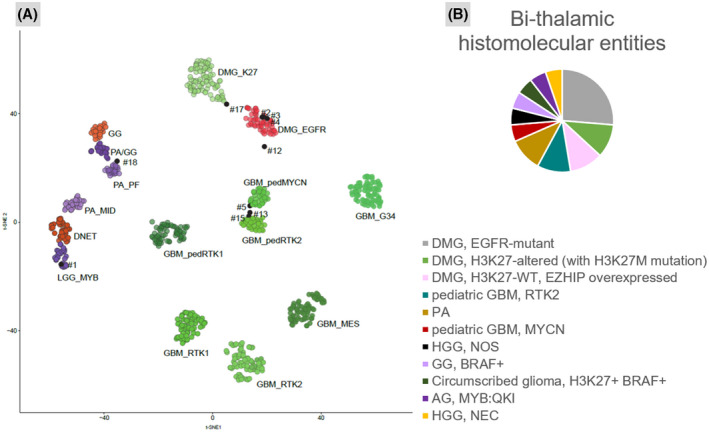
Distribution of histomolecular characteristics of the tumors within the cohort. (A) t‐distributed stochastic neighbor embedding (t‐SNE) analysis of DNA methylation profiles of nine of the investigated tumors alongside selected reference samples. Reference DNA methylation classes: low‐grade glioma, MYB (LGG_MYB); low‐grade glioma, dysembryoplastic neuroepithelial tumor (DNET); low‐grade glioma, subclass midline pilocytic astrocytoma (PA_MID); low‐grade glioma, subclass posterior fossa pilocytic astrocytoma (PA_PF); low‐grade glioma, ganglioglioma (GG); low‐grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma (PA/GG); diffuse midline glioma H3 K27 M mutant (DMG_K27); diffuse midline glioma EGFR_altered (DMG_EGFR); glioblastoma, IDH wild type, H3.3 G34 mutant (GBM_G34); pediatric glioblastoma, IDH wild type, subclass MYCN (GBM_pedMYCN); glioblastoma, IDH wild type, subclass RTK1 (GBM_RTK1); glioblastoma, IDH wild type, subclass RTK2 (GBM_RTK2); pediatric glioblastoma, IDH wild type, subclass RTK1 (GBM_pedRTK1); pediatric glioblastoma, IDH wild type, subclass RTK2 (GBM_pedRTK2); glioblastoma, IDH wild type, subclass mesenchymal (GBM_MES). (B) The integrative histopathological, genetic, and epigenetic analyses identified six diffuse midline gliomas (DMG), EGFR‐altered (26.3%); two DMG, H3K27‐altered (with H3K27 M mutation) (10.5%); two DMG, H3K27‐WT, with EZHIP overexpression (10.5%); two pediatric glioblastomas (GBM), RTK2 subtype (10.5%); two high‐grade gliomas, not elsewhere classified (NEC) IDH‐ and histones‐WT (10.5%); two pilocytic astrocytomas (PA) (10.5%); one pediatric glioblastoma, MYCN‐amplified (5.3%); one ganglioglioma (GG), grade 1, BRAF‐mutant (5.3%); one angiocentric glioma (AG), with MYB:QKI fusion (5.3%); and one circumscribed glioma, BRAF and H3K27‐mutant (5.3%).
Histopathological features of the three different subtypes of DMG were similar, corresponding to diffuse astrocytic infiltrating tumors with or without necrosis and/or neoangiogenesis. Interestingly, one (case 12) of the two diffuse gliomas NOS/NEC was different, presenting multiple calcifications, as one reported case [2]. On MRI, while most of the tumors were infiltrative with very weak contrast enhancement, and without diffusion restriction, causing volume increase of the thalami, PA, GG and the circumscribed glioma, BRAF and H3K27‐mutant, were well‐circumscribed tumors with heterogeneous contrast enhancement and microcystic components. The pediatric glioblastoma, MYCN‐amplified, presented as bulky well‐circumscribed enhancing tumor. Cerebral blood flow using arterial spin labeling was high for high‐grade gliomas, while it was low in AG (Table S1). This high blood flow in molecularly defined high‐grade tumors may appear surprising since they did not show microvascular proliferation. High tumoral metabolism may partly explain this tumoral blood flow.
As expected, progression‐free and overall survival were significantly better in PA, AG, and GG. However, due to the limited number of cases for each histomolecular subtype, no conclusion can be made and further studies addressing their prognosis are needed.
Herein, we described the histomolecular landscape of pediatric bithalamic gliomas and showed a large spectrum of different tumoral types. Moreover, we highlight that not all pediatric bithalamic gliomas are always malignant, low‐grade glioma representing 21% (4/19) of the cases in our series. To our knowledge, we present an exceptional bithalamic example of AG with an MYB:QKI fusion briefly reported in [5]. We described also for the first time a bithalamic location of a circumscribed H3K27‐ and BRAF‐mutant glioma (without neuronal differentiation in the biopsy sample), only monothalamic forms being previously reported [3, 6, 7]. This case clustered in close vicinity with DMG, H3K27‐mutant by t‐SNE analysis. Our series showed that DMG, H3K27‐WT with EZHIP overexpression (this subtype clustering in the same MC than DMG, H3K27‐mutant) are not exceptional in the bithalamic region, representing 10.5% (2/19) of cases. Our series showed that bithalamic diffuse gliomas are divided into several different histomolecular types of tumors, of which DMG, EGFR‐altered represent the most frequent but not the unique subtype. The deciphering of bithalamic tumors has been partially elucidated by the recent description of the MC DMG, EGFR‐altered [2, 3]. However, histopathological, immunophenotypical, and genetic overlaps exist between the different tumors. H3K27me3 loss represents a common characteristic of DMG, H3K27‐mutant and DMG, H3‐WT with EZHIP overexpression as well as DMG, EGFR‐altered [1, 2, 3]. H3F3A, HIST1H3B, and HIST1H3C K27M mutations are shared by DMG, H3K27‐mutant and 27% (8/30: 8/25 cases in Sievers et al., and no case in our work) of DMG, EGFR‐altered [2, 3]. Surprisingly, two cases of DMG, EGFR‐altered have been reported with histones’ genes mutations and without an EGFR alteration [3]. Consequently, for our two cases presenting H3K27M mutation without verification by DNA methylation profiling and without complete EGFR characterization (one case presented an EGFR amplification), we concluded a diagnosis of DMG, H3K27‐altered. Indeed, EGFR amplification has not been described in DMG H3K27‐mutant [8] and only six cases with concomitant H3K27M and EGFR alterations (including amplification) have been classified as DMG EGFR‐altered [3]. Further studies are needed to clarify whether or not some cases with histones’ genes mutations without EGFR alteration can be classified as DMG, EGFR‐altered. In addition, different types of tumors share EGFR alterations such as DMG EGFR‐altered, pediatric glioblastomas, RTK2 subtype (as in one of our cases and in Sievers et al. [3]), and still unclassified cases (two of our series). A subset of DMG, EGFR‐altered (22% of cases, 9/40 cases: 8/35 cases in Sievers et al., 1/5 case in Mondal et al., and no case in our work) did not harbor EGFR alterations [2, 3]. Because the loss of H3K27me3 alone may be difficult to interpret (mosaic loss described in Mondal et al. [2]) and because a single EGFR alteration is not sufficient for a diagnosis of DMG, EGFR‐altered, we suggest that a diagnosis for DMG, EGFR‐altered may be proposed when this triad is present: (i) a loss of H3K27me3, (ii) overexpression of EZHIP, and (iii) an EGFR alteration, or with a DNA‐methylation profile of DMG, EGFR‐altered. All our cases presenting this triad have always been classified as DMG, EGFR‐altered using DNA methylation analysis. If DNA‐methylation analysis is not possible, this triad can permit the suggested diagnosis of DMG, EGFR‐altered. However, in the case of the absence of this triad or if the results are unclear, DNA methylation analysis is needed to reach a conclusion. In the case of only an EGFR alteration, a DNA methylation analysis is required to distinguish DMG, EGFR‐altered from a pediatric glioblastoma, RTK2 subtype. The clinical implications for distinguishing between these different malignant tumor types need further studies.
In conclusion, our work highlights that pediatric bithalamic gliomas encompass several histomolecular tumoral types from benign to malignant and underlines the central role of a comprehensive neuropathological review, including immunohistochemistry, genetic, and epigenetic analyses, to achieve an accurate diagnosis.
Supporting information
FIGURE S1 Radiological and histomolecular features of the diffuse tumor types in the cohort. Case 3: (A) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (B) Lowly cellular astrocytic diffuse glioma (HPS x400 magnification and NFP staining, insert x400 magnification). (C) H3K27me3 loss of immunoexpression in the tumor cells (x400 magnification). (D) EZHIP overexpression by a subset of tumor cells (x400 magnification). Case 8: (E) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (F) Highly cellular astrocytic diffuse glioma (HPS x400 magnification and NFP staining, insert x400 magnification). (G) H3K27me3 loss of immunoexpression in the tumor cells (x400 magnification). (H) H3K27M expression by tumor cells (x400 magnification). Case 10: (I) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and weak heterogeneous contrast enhancement. (J) Highly cellular astrocytic diffuse glioma (HPS x400 magnification and NFP staining, insert x400 magnification). (K) H3K27me3 loss of immunoexpression in the tumor cells (x400 magnification). (L) EZHIP overexpression by a subset of tumor cells (x400 magnification). Case 5: (M) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and weak heterogeneous contrast enhancement. (N) Highly cellular astrocytic diffuse glioma with nuclear atypia and mitoses (HPS x400 magnification and NFP staining, insert x400 magnification). (O) H3K27me3 retained immunoexpression in the large part of tumor cells (x400 magnification). Case 15: (P) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and weak heterogeneous contrast enhancement. (Q) Highly cellular astrocytic diffuse glioma with nuclear atypia (HPS x400 magnification and NFP staining, insert x400 magnification). (R) H3K27me3 retained immunoexpression in the tumor cells (x400 magnification). Case 12: (S) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (T) Highly cellular astrocytic diffuse glioma with several calcifications (HPS x400 magnification and NFP staining, insert x400 magnification). (U) H3K27me3 retained immunoexpression in the tumor cells (x400 magnification). Abbreviations: DMG: diffuse midline glioma; GBM: glioblastoma; HGG: high‐grade glioma; HPS: hematoxylin, phloxine, and saffron; MRI: magnetic resonance imaging; NEC: not elsewhere classified; NFP: neurofilament protein; ped: pediatric; T2w: T2‐weigthed image. Black scale bars represent 50 μm
FIGURE S2 Radiological and histomolecular features of the circumscribed tumor types in the cohort. Case 1: (A) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (B) Astrocytic diffuse glioma of low cellularity with angiocentric features (HPS x400 magnification and NFP staining, insert x400 magnification). (C) Rearrangement of MYB gene by FISH analysis showing a signal of fusion and one isolated 5’MYB signal (3’MYB in red signals and 5’MYB in green signals) (x400 magnification). Case 19: (D) MRI showing a well‐demarcated bithalamic tumor with T2w hyperintensity, cystic component, and strong contrast enhancement. (E) A circumscribed astrocytic glioma of low cellularity with Rosenthal fibers (HPS x400 magnification and NFP staining, insert x400 magnification). (F) Rearrangement of BRAF gene by FISH analysis showing two signals of fusion associated with one extrasignal 3’BRAF (3’BRAF in red signals and 5’BRAF in green signals) (x400 magnification). Case 16: (G) MRI showing an ill‐defined bithalamic tumor with T2w hyperintensity, microcysts, and heterogeneous contrast enhancement. (H) A circumscribed glioneuronal proliferation with binucleated cells, inflammatory infiltrates, and eosinophilic granular bodies (HPS x400 magnification and NFP staining, insert x400 magnification). (I) Diffuse expression of BRAFV600E (x400 magnification). Case 17: (J) MRI showing an ill‐defined bithalamic tumor with T2w isointensity and partial heterogeneous contrast enhancement. (K) A circumscribed astrocytic diffuse glioma of low cellularity (HPS x400 magnification and NFP staining, insert x400 magnification). (L) Diffuse expression of H3K27M (x400 magnification). (M) Diffuse expression of BRAFV600E (x400 magnification). Abbreviations: AG: angiocentric glioma; GG: ganglioglioma; HPS: hematoxylin, phloxine, and saffron; MRI: magnetic resonance imaging; NFP: neurofilament protein; PA: pilocytic astrocytoma; T2w: T2‐weigthed image. Black scale bars represent 50 μm
Table S1 Summary of clinical, histopathological and molecular data of our cases
ACKNOWLEDGMENTS
We would like to thank the laboratory technicians at the GHU Paris Neurosciences, Hospital Sainte‐Anne, for their assistance.
Funding information
JG received funding from the charity “l’Etoile de Martin” and from the Carrefour Foundation “Les Boucles du Coeur” for the sequencing programme RARE.
Philipp Sievers and Pascale Varlet contributed equally to this study.
DATA AVAILABILITY STATEMENT
Data available upon request.
REFERENCES
- 1. Castel D, Kergrohen T, Tauziède‐Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild‐type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3–K27M mutation. Acta Neuropathol. 2020;139(6):1109–13. [DOI] [PubMed] [Google Scholar]
- 2. Mondal G, Lee JC, Ravindranathan A, Villanueva‐Meyer JE, Tran QT, Allen SJ, et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020;139(6):1071–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sievers P, Sill M, Schrimpf D, Stichel D, Reuss DE, Sturm D, et al. A subset of pediatric‐type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro‐Oncol. 2021;23(1):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Broniscer A, Hwang SN, Chamdine O, Lin T, Pounds S, Onar‐Thomas A, et al. Bithalamic gliomas may be molecularly distinct from their unilateral high‐grade counterparts. Brain Pathol. 2018;28(1):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wefers AK, Stichel D, Schrimpf D, Coras R, Pages M, Tauziède‐Espariat A, et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol. 2020;139(1):193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pagès M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D, et al. Co‐occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018;28(1):103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pratt D, Natarajan SK, Banda A, Giannini C, Vats P, Koschmann C, et al. Circumscribed/non‐diffuse histology confers a better prognosis in H3K27M‐mutant gliomas. Acta Neuropathol. 2018;135(2):299–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJJ, et al. Diffuse midline gliomas with histone H3–K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016;26(5):569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Radiological and histomolecular features of the diffuse tumor types in the cohort. Case 3: (A) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (B) Lowly cellular astrocytic diffuse glioma (HPS x400 magnification and NFP staining, insert x400 magnification). (C) H3K27me3 loss of immunoexpression in the tumor cells (x400 magnification). (D) EZHIP overexpression by a subset of tumor cells (x400 magnification). Case 8: (E) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (F) Highly cellular astrocytic diffuse glioma (HPS x400 magnification and NFP staining, insert x400 magnification). (G) H3K27me3 loss of immunoexpression in the tumor cells (x400 magnification). (H) H3K27M expression by tumor cells (x400 magnification). Case 10: (I) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and weak heterogeneous contrast enhancement. (J) Highly cellular astrocytic diffuse glioma (HPS x400 magnification and NFP staining, insert x400 magnification). (K) H3K27me3 loss of immunoexpression in the tumor cells (x400 magnification). (L) EZHIP overexpression by a subset of tumor cells (x400 magnification). Case 5: (M) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and weak heterogeneous contrast enhancement. (N) Highly cellular astrocytic diffuse glioma with nuclear atypia and mitoses (HPS x400 magnification and NFP staining, insert x400 magnification). (O) H3K27me3 retained immunoexpression in the large part of tumor cells (x400 magnification). Case 15: (P) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and weak heterogeneous contrast enhancement. (Q) Highly cellular astrocytic diffuse glioma with nuclear atypia (HPS x400 magnification and NFP staining, insert x400 magnification). (R) H3K27me3 retained immunoexpression in the tumor cells (x400 magnification). Case 12: (S) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (T) Highly cellular astrocytic diffuse glioma with several calcifications (HPS x400 magnification and NFP staining, insert x400 magnification). (U) H3K27me3 retained immunoexpression in the tumor cells (x400 magnification). Abbreviations: DMG: diffuse midline glioma; GBM: glioblastoma; HGG: high‐grade glioma; HPS: hematoxylin, phloxine, and saffron; MRI: magnetic resonance imaging; NEC: not elsewhere classified; NFP: neurofilament protein; ped: pediatric; T2w: T2‐weigthed image. Black scale bars represent 50 μm
FIGURE S2 Radiological and histomolecular features of the circumscribed tumor types in the cohort. Case 1: (A) MRI showing an infiltrative bithalamic tumor with T2w hyperintensity and no contrast enhancement. (B) Astrocytic diffuse glioma of low cellularity with angiocentric features (HPS x400 magnification and NFP staining, insert x400 magnification). (C) Rearrangement of MYB gene by FISH analysis showing a signal of fusion and one isolated 5’MYB signal (3’MYB in red signals and 5’MYB in green signals) (x400 magnification). Case 19: (D) MRI showing a well‐demarcated bithalamic tumor with T2w hyperintensity, cystic component, and strong contrast enhancement. (E) A circumscribed astrocytic glioma of low cellularity with Rosenthal fibers (HPS x400 magnification and NFP staining, insert x400 magnification). (F) Rearrangement of BRAF gene by FISH analysis showing two signals of fusion associated with one extrasignal 3’BRAF (3’BRAF in red signals and 5’BRAF in green signals) (x400 magnification). Case 16: (G) MRI showing an ill‐defined bithalamic tumor with T2w hyperintensity, microcysts, and heterogeneous contrast enhancement. (H) A circumscribed glioneuronal proliferation with binucleated cells, inflammatory infiltrates, and eosinophilic granular bodies (HPS x400 magnification and NFP staining, insert x400 magnification). (I) Diffuse expression of BRAFV600E (x400 magnification). Case 17: (J) MRI showing an ill‐defined bithalamic tumor with T2w isointensity and partial heterogeneous contrast enhancement. (K) A circumscribed astrocytic diffuse glioma of low cellularity (HPS x400 magnification and NFP staining, insert x400 magnification). (L) Diffuse expression of H3K27M (x400 magnification). (M) Diffuse expression of BRAFV600E (x400 magnification). Abbreviations: AG: angiocentric glioma; GG: ganglioglioma; HPS: hematoxylin, phloxine, and saffron; MRI: magnetic resonance imaging; NFP: neurofilament protein; PA: pilocytic astrocytoma; T2w: T2‐weigthed image. Black scale bars represent 50 μm
Table S1 Summary of clinical, histopathological and molecular data of our cases
Data Availability Statement
Data available upon request.