

Abstract
Age is one of the strongest risk factors for the development of neurodegenerative diseases, the majority of which involve misfolded protein aggregates in the brain. These protein aggregates are thought to drive pathology and are attractive targets for the development of new therapies. However, it is unclear how age influences the onset of pathology and the accompanying molecular response. To address this knowledge gap, we used a model of seeded tau pathology to profile the transcriptomic changes in 3 and 12 month old mice in response to developing tau hyperphosphorylation and aggregation. First, we found the burden of hyperphosphorylated tau pathology in mice injected at 12 months of age was moderately reduced compared to animals injected at 3 months. On a molecular level, we found an inflammation‐related subset of genes, including C3 and the disease‐associated microglia genes Ctsd, Cst7, and Clec7a, were more expressed early in disease in 12 but not 3 month old mice. These findings provide evidence of an early, age‐specific response to tau pathology, which could serve as a marker for the severity of downstream pathology.
Keywords: aging, RNA‐seq, tauopathy
Aging is the strongest risk factor for developing neuropathology, however the underlying molecular reason for this is unclear. In this study we find that there is an age‐specific inflammatory response to tau pathology that precedes an altered pathology burden. This emphasises that age is a critical factor in not only the risk of disease but also the course of its development.
1. INTRODUCTION
Neurodegenerative conditions typically harbor proteinaceous deposits that are considered clinical hallmarks of disease [1, 2]. These pathologies are not only diagnostic markers of brain dysfunction, but are also linked to disease progression and remain a target for therapeutics [3]. The tau protein, which is involved in microtubule dynamics under normal physiological conditions, can misfold and deposit in neurons and glia in a number of neurodegenerative diseases, including Alzheimer's disease (AD) [4]. In AD, as with other tauopathies, the disease course is believed to begin decades prior to the onset of cognitive decline [5]. As such, it is important to consider that, while idiopathic tauopathies are thought of as diseases of the elderly, the onset of disease occurs much earlier in life. This early stage of critical changes in the brain remains poorly understood at the molecular level and further investigation of this period is still needed.
While tau pathology is a central player in many neurodegenerative diseases, the strongest risk factor for the development of these conditions is advanced age [6]. Numerous aspects of aging are hypothesized to contribute to brain deterioration, including cellular senescence and genome instability [7]. The influence of host age on the onset of pathology is difficult to investigate with traditional transgenic models where the pathology begins at a specific age and accumulates with time. A model for sporadic tau pathology was recently described that utilizes injection of AD human brain extracts into wild‐type (WT) mice to induce pathology [8]. This was further expanded to investigate other tauopathies and the interplay between tau and beta‐amyloid pathology [9, 10, 11]. This model relies on the ability of disease‐associated misfolded proteins to spur misfolding and aggregation of naïve proteins in a process termed seeding [1, 12]. The development of hyperphosphorylated, insoluble tau deposits in WT mouse tissue provides an opportunity to investigate (i) the early stages of nascent pathology in the absence of overt effects caused by transgene overexpression and (ii) the influence of age on the cellular response to pathogenesis.
In the present study, we hypothesized that age would modify the burden of pathology in a seeding model of tauopathy in WT mice. Additionally, we projected that this age‐specific susceptibility to pathology would be accompanied by a unique transcriptional response. Our results provide evidence that age does indeed alter the burden of pathology and, intriguingly, that an early, age‐specific inflammatory gene expression profile is linked to reduced downstream pathology.
2. METHODS
2.1. Extract preparation
Human brain extracts were prepared following a previously published protocol [8]. Briefly, frozen frontal cortex tissue from healthy control (Ctrl; Braak score 0‐I; Table S1) or AD (Braak stage VI, ABC score “high”; Table S1; [13]) patients was homogenized using a glass dounce tissue grinder (10% w/v) in high salt buffer (10 mM Tris‐HCl pH 7.4, 0.8 M NaCl, 1 mM EDTA, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.1% sarkosyl, 10% sucrose, protease/phosphatase inhibitor [ThermoFisher Mini tablets]) and centrifuged at 10,000 g for 10 min (all steps were carried out at 4℃ unless otherwise specified). The pellet was re‐homogenized as above and supernatants pooled prior to adjusting the sarkosyl concentration to 1% and incubating at room temperature for 60 min. Samples were centrifuged at 300,000 g for 60 min and the resulting pellet was washed with ice‐cold PBS prior to resuspension in PBS using probe sonication (20, 1 s pulses at 80% amplitude). Samples were centrifuged at 100,000 g for 30 min and the pellet was resuspended in PBS with sonication (60, 1 s pulses at 80% amplitude) prior to a clarifying centrifugation step of 10,000 g for 10 min. The remaining supernatant was used as the extract for injection experiments and was aliquoted and frozen for storage at −80℃. Extracts from patients Ctrl1‐Ctrl5 and AD1‐AD5 were pooled for injections in a mouse cohort used for AT8 immunohistochemistry and RNA‐seq experiments (Table S1). Extracts from patients Ctrl5‐Ctrl9 and AD1‐AD5 were pooled for injections in a mouse cohort used for sarkosyl insoluble and soluble western blots and qPCR experiments (Table S1). Use of de‐identified human brain material was approved by the University of Queensland human research ethics committee (HREC/18/MHS/60).
2.2. Stereotactic injections
Wildtype C57BL/6 (WT) mice at either 3 or 12 months old (3 mo or 12 mo) were anaesthetized with ketamine/xylazine (100/10, mg/kg) and placed in a stereotactic frame (Kopf Instruments) after losing reflexes. Mice were bilaterally injected with either AD or control extracts (2.5 µl with a Hamilton Syringe) into the hippocampus (Bregma: −2.5 mm rostral/caudal, ±2.0 mm left/right, −1.8 mm dorsal/ventral). After closely monitored recovery, animals were incubated for 1, 3, or 5 months (n = 4–7, sex balanced). Mice were sacrificed using overdose of anaesthesia (250 ketamine/25 xylazine, mg/kg) and perfusion with 20 ml of ice‐cold PBS. Brains were removed and separated into hemispheres for downstream processing. For immunohistochemistry, brains were immersion fixed in 4% paraformaldehyde (PBS) for 48 h then cryo‐protected in 30% sucrose (PBS) and snap frozen in 2‐methylbutane on dry ice for storage at −80℃. For RNA‐sequencing or sarkosyl extraction, brains were dissected to isolate the hippocampus and snap frozen on dry ice and stored at −80℃. All animal experiments were approved by the University of Queensland animal ethics committee (QBI/226/18).
2.3. Pathology staining and quantification
Brains were sectioned on a freeze‐sliding microtome (Leica Biosystems, 25 μm) and collected in a 12‐well plate containing cryoprotectant solution (35% ethylene glycol, 25% glycerol, PBS) so that each well contained every 12th section. Sections were quenched for endogenous peroxidase activity using 0.3% hydrogen peroxide and blocked for 60 min with 5% normal horse serum (Vector labs) and 1% bovine serum albumin (Sigma Aldrich, A7906‐100G) containing 0.3% triton X‐100 (TBST). Staining with AT8 (1:1000, ThermoFisher, MN1020) to label hyperphosphorylated tau was carried out overnight at 4℃ in 2% normal horse serum and 1% bovine serum albumin in TBST. The remainder of the staining protocol followed the manufacturer's specification for the Vectastain ABC kit and DAB reagent (Vector labs). Counterstaining was achieved using hematoxylin. Slides were dehydrated and cover‐slipped with Eukitt quick‐hardening mounting media (Sigma Aldrich). Stereological quantification of AT8‐positive granular puncta and inclusions were carried out on an Axio Imager Z2 microscope (Zeiss) equipped with a motorized stage and Stereo Investigator software (MBF Bioscience). Contours outlining the dentate gyrus of the hippocampus were drawn using the 5x objective and counting was performed with the 20x objective. Counting was performed using the fractionator protocol with a 200 µm × 200 µm grid size and 100 µm × 100 µm counting frame. Calculations of puncta per area were carried out using Neurolucida Explorer (MBF Bioscience).
2.4. Sarkosyl‐insoluble tau extraction and western blotting
Frozen hippocampus tissue was homogenized (10% w/v) with a pestle and 25G syringe in RIPA buffer, without SDS (50 mM Tris‐HCl pH 7.6, 150 mM NaCl, 5 mM EDTA, 1% Igepal CA‐630, 0.5% sodium deoxycholate, protease/phosphatase inhibitor [ThermoFisher Mini tablets]) and centrifuged at 18,000 g for 20 min at 4℃. The supernatant was collected and sarkosyl was added to a final concentration of 1% prior to incubation at room temperature for 60 min in a thermomixer at 600 rpm (Eppendorf). The samples were centrifuged at 120,000 g for 60 min at room temperature and the pellet was resuspended in RIPA buffer without SDS using probe sonication (three 10 s pulses at 30% amplitude). The sarkosyl soluble supernatant and insoluble pellet were used in downstream western blotting.
Protein concentrations were determined using a BCA Protein assay kit (ThermoFisher) and absorbance readings were collected at 562 nm on a spectrophotometer. Samples were heated at 95℃ for 10 min in sample buffer (63 mM Tris‐HCl pH 6.8, 10% glycerol, 2% SDS) prior to running on a 10% Tris‐Glycine polyacrylamide gel (4% stacking gel) using the SureCast gel system or 4%–12% Tris‐Glycine polyacrylamide gel (ThermoFisher, XV04125PK20) in Tris‐Glycine running buffer (25 mM Tris‐base, 192mM Glycine, 0.1% SDS) at 125 V for 60–80 min. Samples were transferred to a nitrocellulose membrane at 25 V for 60 min in Tris‐Glycine buffer (12 mM Tris‐base, 96 mM Glycine, 20% methanol). Revert 700 total protein stain (Licor, LCR‐926‐11011) was used for downstream loading control normalization and was followed by blocking with 5% milk in TBS with 0.1% Tween‐20 or Intercept (TBS) Blocking buffer (Licor, LCR‐927‐80001) for 60 min at room temperature. Membranes were probed with Tau46 (1:2500, ThermoFisher, 13‐6400), pS396 (1:10,000, Abcam, ab109390), TAU‐13 (1:1000, BioLegend, 835201), AT8 (1:1000, ThermoFisher, MN1020), C3 (1:2000, ThermoFisher, PA5‐21349), Ctsd (1:5000, ThermoFisher, MA5‐17236), Cst7/Cystatin F (1:3000, ThermoFisher, PA5‐103772), or Clec7a/Dectin‐1 (1:1000, ThermoFisher, PA5‐34382) overnight at 4℃. After washing, membranes were incubated in mouse‐IR680 (1:10,000, Licor, 926‐68070) and rabbit‐IR800 (1:10,000, Licor, 926‐32211) and imaged with a Licor Fc Imager (Licor). Quantification was carried out using Licor Image Studio and the relative signal to 3 mo AD samples were calculated followed by normalization for total protein using Revert 700 signal.
2.5. RNA‐sequencing and analysis
RNA extraction of frozen hippocampus tissue was carried out using the Qiagen RNeasy Mini Kit (Qiagen, 74104) and followed manufacturer's specifications. RNA quality was assessed using an Agilent 2100 Bioanalyzer, with all samples having a RIN value greater than 8.3. Mature messenger RNA was isolated using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB) followed by fragmentation and priming with NEBNext First Strand Synthesis Reaction buffer (NEB) and NEBNext Random Primers. First and second strand synthesis were completed using ProtoScript II Reverse Transcriptase and Second Strand Synthesis Enzyme Mix (NEB), respectively. Double‐stranded cDNA was purified with AxyPrep Mag PCR Clean‐up (Axygen) and End Prep Enzyme Mix was used to repair ends and A‐tail followed by addition of adaptors with T‐A ligation (NEB). AxyPrep Mag PCR Clean‐up was used for selection of ~360 bp fragments and these were amplified with 11 cycles of PCR using P5 and P7 primers. Libraries were purified with AxyPrep Mag PCR Clean‐up and quantified using an Agilent 2100 Bioanalyzer and Qubit 2.0 Fluorometer (Invitrogen). RNA‐sequencing was carried out on an Illumina HiSeq platform in a 2x150bp configuration. Data are available from the GEO archive under accession number GSE169686.
RNA‐seq reads were aligned to the Ensembl mouse genome build GRCm38 using STAR 2.7.3a with default parameters [14]. Results were examined for 3ʹ bias, leading to one sample from the 3 mo late AD group being excluded (n = 3 after removal). Duplicate reads were marked with picard 2.18.0 (Broad Institute) and reads were counted against Ensembl genes GRCm38 build 98. Gene expression results were analyzed in edgeR version 3.24.3 [15]. Gene expression normalization factors were calculated via the TMM method. The edgeR plotMDS function was used to screen for outliers and the desmear function was used to assess the relationship between fold change and expression level. Differential expression was assessed using the glmLRT function in edgeR. Network analysis was performed on differentially expressed genes using STRING [16]. The expression of genes in different cell types was determined using a previously published dataset of single nucleus RNA‐sequencing on the mouse hippocampus [17]
To quantify transposable element (TE) subfamily expression, RNA‐seq reads were again aligned to the reference genome assembly with STAR (parameters ‐‐twopassMode Basic ‐‐outSAMprimaryFlag AllBestScore ‐‐winAnchorMultimapNmax 1000 ‐‐outFilterMultimapNmax 1000) and duplicate reads marked with Picard. An established strategy was then followed to, where possible, assign multi‐map reads a weighting at each genomic alignment based on the relative abundance of uniquely mapping reads nearby [18, 19, 20]. Specifically, for each multi‐map read, the number of uniquely mapped reads within 100bp of the aligned multi‐map read at each of its potential best map genomic locations was counted. A weighting was then assigned to each multi‐map position proportionate to the fraction of uniquely mapped reads found at that position out of the total number of uniquely mapped reads found at any position for the given multi‐map read. If no uniquely mapped reads were found at any of the n multi‐map positions, each position was assigned a weighting of 1/n. Uniquely mapped reads were assigned a weighting of 1. To produce estimates of transcript abundance for TE subfamilies, weighted alignments were intersected with RepeatMasker coordinates to yield read count totals for each individual TE [21], and were then summed to produce a value for each TE subfamily genome‐wide. Values were normalized by the total number of weighted mapped reads (tags per million). Animals were grouped by age or treatment and differential TE subfamily expression assessed via edgeR.
2.6. qPCR
RNA extraction of hippocampus tissue from different biological replicates was completed as for RNA‐sequencing experiments. cDNA synthesis was achieved using High‐Capacity cDNA Reverse Transcription Kit while following manufacturers specifications (ThermoFisher, 4368814). Primers were designed using Primer‐BLAST (NCBI). The LightCycler 480 SYBR Green I Master kit was used for qPCR on a LightCycler 480 Instrument following manufacturer's specifications and an annealing temperature of 56℃. Samples were ran in triplicate. The delta‐delta Ct method of calculating fold change relative to 3 mo control samples normalized to beta‐actin primers was used. Primer sequences: C3‐fwd GCTTCAGGGTCCCAGCTACT; C3‐rev AGCCGTAGGACATTGGGAGT; Ctsd‐fwd CT TCGTCCTCCTTCGCGATT; Ctsd‐rev GGGCC TTTGAGGATCAGGTC; Cst7‐fwd GGAGCTGT ACTTGCCGAGC; Cst7‐rev CATGGGTGTCAG AAGTTAGGC; Clec7a‐fwd GCTCCCAGCTAG GTGCTCATC; Clec7a‐rev TGTTTGGCTTTCA ATGAACTCAA; beta‐actin‐fwd CGCAGCCA CTGTCGAGTC; beta‐actin‐rev GTCATCCATG GCGAACTGGT.
2.7. Data analysis
Heatmaps (heatmap.2) and data scaling for z‐score calculations were completed using RStudio (version 1.2.5042; R version 4.0.0). Graphs and statistical tests (pathology, western blotting and qPCR) were performed with Prism 8.0.2 (GraphPad).
3. RESULTS
The goal of our study was to investigate the influence of age on the molecular response to developing tau pathology. To accomplish this, we utilized a tau seeding model where the intrahippocampal injection of pathological tau isolated from human AD brains into WT C57BL/6 mice leads to tau pathology [8]. In order to more closely model the middle‐aged onset of tauopathies, we used 12 month old (mo) WT mice for our injection paradigm and compared this with young, 3 mo WT mice (Figure 1A). Animals were incubated for one, three, or five months post injection (mpi), which we termed early, middle, and late time points. Human control (Ctrl) and AD frontal cortex brain material was processed using an established sarkosyl insolubility fractionation method to isolate pathological tau ([8]; see Section 2). Western blot analysis showed that AD extracts contained substantial amounts of insoluble, hyperphosphorylated tau (pS396) compared to the Ctrl (Figure 1B).
FIGURE 1.
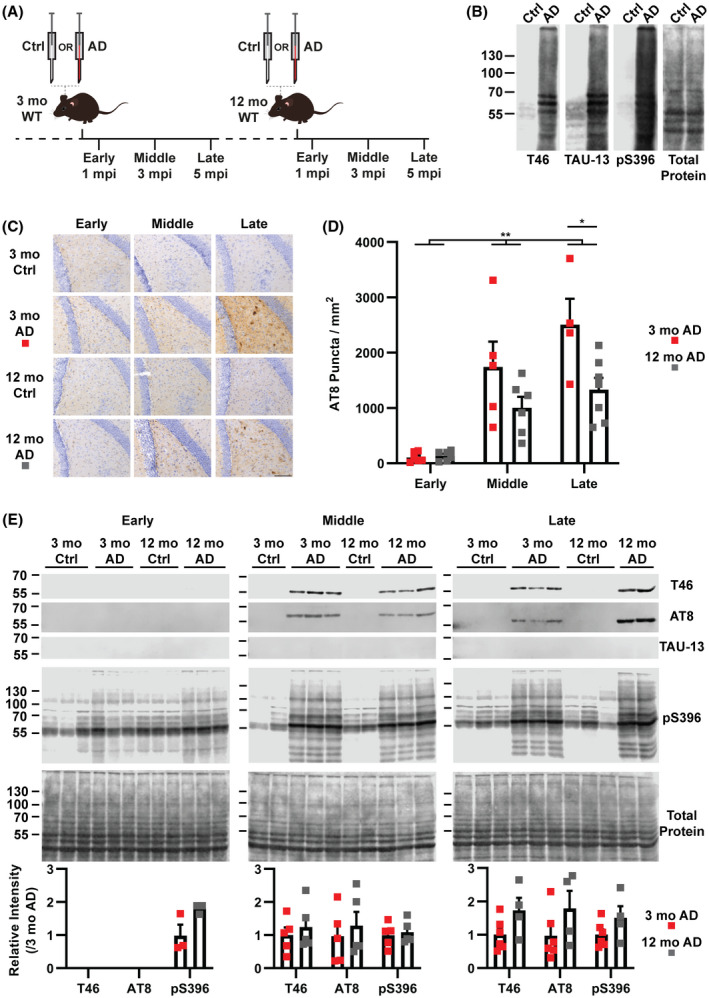
Host age influences the burden of tau pathology in seeded WT mice. (A) WT mice at three (3 mo) or twelve months old (12 mo) were injected in the dentate gyrus with control (Ctrl) or Alzheimer's disease (AD) human brain extracts and were then incubated for three different times termed early (1 mpi; mpi, months postinjection), middle (3 mpi), and late (5 mpi). (B) Western blot analysis of Ctrl and AD extracts showing total (Tau46), human (TAU‐13), and phosphorylated tau (pS396). Extracts from five patients were pooled for injections (see methods). Total protein staining was completed with Revert 700 (Licor). (C) Representative images of hyperphosphorylated tau (AT8) immunohistochemistry staining in the dentate gyrus of injected mice (n = 4–7, Scale bar = 100 μm). (D) Quantification of AT8‐positive puncta in the dentate gyrus of injected mice (Two‐way ANOVA, Age: F 1,28 = 9.144, p = 0.0053 (**); Incubation: F 2,28 = 27.01, p < 0.0001, no significant interaction; Bonferroni correction, late 3 mo vs. 12 mo p = 0.0117 (*)). Error bars represent the standard error of the mean. (E) Western blot analysis of sarkosyl insoluble extracts from the hippocampus of injected mice (n = 3–6). Antibodies used are the same as in (B) and include AT8 (used in C). Signal intensity relative to the 3 mo AD sample was calculated and then normalized to total protein. No significant differences were identified between 3 and 12 mo AD samples for pS396, AT8 or total tau for any of the stages (Mann–Whitney Test). Error bars represent the standard error of the mean
Immunohistochemistry analysis of injected mice identified hyperphosphorylated tau in AD injected animals but not Ctrl injected animals (Figure 1C). While we did identify pathology in regions functionally connected to the hippocampus, namely the mammillary body and entorhinal cortex (Figure S1A), the primary site of pathology was the hippocampus where extracts were injected. The injection of AD extracts into WT mice gave rise to hyperphosphorylated tau pathology that we classified into two categories, puncta and inclusions (Figure S1B). These two pathologies are similar in morphology to neuropil pathology and somatic inclusions, respectively [22]. Inclusions were only identified at the middle (3 mpi) and late (5 mpi) time points but no differences were identified between 3 and 12 mo animals in the dentate gyrus (Figure S1C, two‐way ANOVA). Puncta were seen at all time points in the dentate gyrus and the effect of host age was significant, with 12 mo animals having less pathology than the 3 mo group (two‐way ANOVA, age p = 0.0053, incubation p < 0.0001, no interaction) (Figure 1C,D). Specifically, the late stage showed the strongest reduction of punctate pathology in 12 compared to 3 mo animals (Bonferroni correction, p = 0.01) (Figure 1C,D).
In order to determine whether the hyperphosphorylated tau identified with immunohistochemistry was also insoluble, as seen in other models of tauopathy [23], we processed the hippocampus of a new animal cohort, distinct to those used in immunohistochemistry, with a sarkosyl insolubility extraction protocol. Sarkosyl‐insoluble tau isolated from injected animals were not reactive for a human tau‐specific antibody (TAU‐13), suggesting no residual human tau from the extracts was present (Figure 1E). A dilution series of the human AD extract used for injections shows that the TAU‐13 western blot can detect a 1600‐fold reduction in the extract, underlining the sensitivity of this method to detect residual extract in the mouse brain following injection (Figure S2). Insoluble and hyperphosphorylated mouse tau (T46 and pS396) could be detected at the early time point (Figure 1E, Figure S2), but at the middle and late time points, hyperphosphorylated, insoluble mouse tau was abundant in AD injected animals as shown by the antibodies T46 (total tau), AT8, and pS396 (hyperphosphorylated tau) (Figure 1E, Figure S3). No differences were found between 3 and 12 mo AD injected animals for either total or hyperphosphorylated tau in any of the stages (Figure 1E, Figure S3, Mann–Whitney test). The differential pathology burden of 12 and 3 mo mice paired with the presence of insoluble tau was intriguing and suggested a unique underlying molecular response.
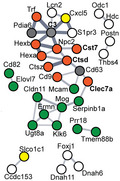
We next performed gene expression analysis of the mature polyadenylated hippocampus transcriptome of injected mice to identify molecular changes accompanying pathology. Our analysis of protein‐coding genes identified few instances of robust differential expression when comparing Ctrl to AD animals for the middle and late time points; and, specifically, no changes for the 12 mo middle time point (Tables [Link], [Link], [Link]). In contrast, the early time point had 63 differentially expressed transcripts (Benjamini–Hochberg, FDR < 0.05) in AD compared to Ctrl for 12 mo, but none for 3 mo animals (Figure 2A, Table S5). Network analysis using STRING [16] showed a number of interactions between the proteins encoded by the differentially expressed transcripts (Figure 2B). A subset of these transcripts (25 of 63 genes) were expressed in microglia, astrocytes, and oligodendrocytes, as informed by a prior single nucleus RNA‐sequencing analysis of the mouse hippocampus [17]. An analysis of transcription emanating from transposable elements (TEs) identified no examples of significant differential TE expression (Benjamini–Hochberg, p < 0.001, >twofold change) associated with tau pathology or age (Figure S4).
FIGURE 2.
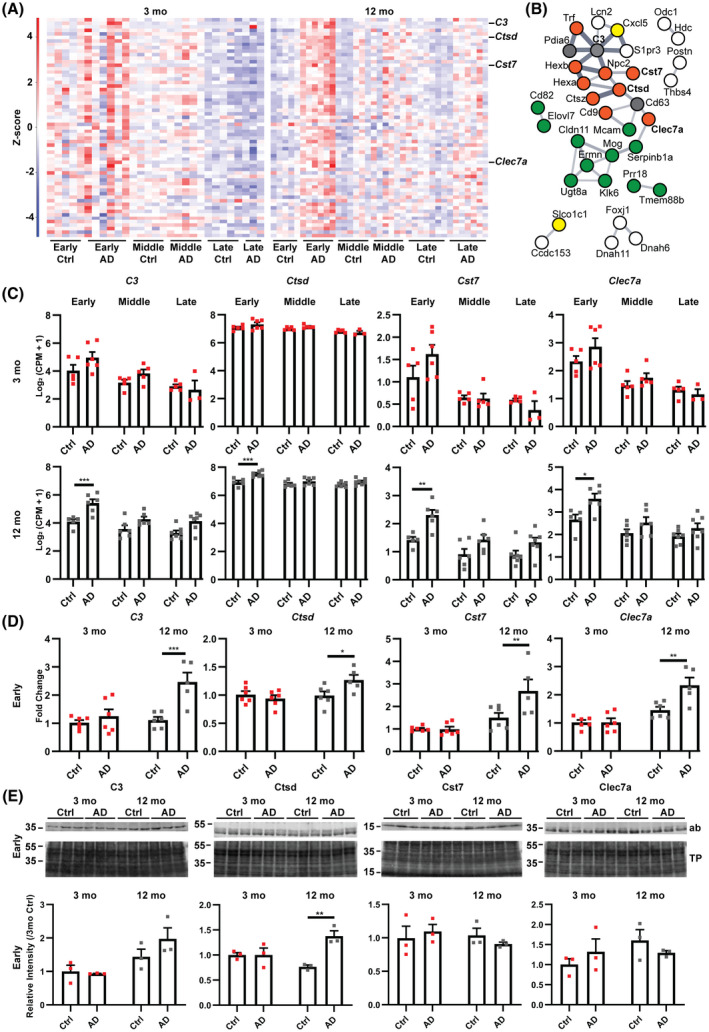
A unique gene expression signature that includes a subset of glial genes is found in 12 mo mice at the early time point in AD injected mice. (A) Heatmap of RNA‐seq analysis showing z‐scores of log2(CPM + 1) values for all injected mice of differentially expressed genes (DEGs, EdgeR glm, Benjamini–Hochberg FDR < 0.05) at the early time point of 12 mo AD versus Ctrl. Red and blue colors represent up and downregulated genes, respectively. Sixty‐three DEGs in total were found in 12 mo mice and these were not significant in 3 mo mice at the early time point (n = 3–7). (B) STRING network analysis (v11.0) of DEGs identified in (A) showing relations between a subset of 35 related genes and their protein products. Line thickness indicates the strength of supporting data in STRING. The glial cell type where transcripts are expressed was determined using the Broad Institute Single Cell portal (Oligodendrocyte‐green; Microglia‐orange; Astrocyte‐yellow; Multiple‐gray; White‐undefined) [17]. (C) RNA‐seq gene plots for a selection of glial genes (EdgeR, glm, Benjamini–Hochberg). Error bars represent the standard error of the mean (n = 3–7). (*FDR < 0.05; **FDR < 0.01; ***FDR < 0.001) (D) Validation of differentially expressed glial genes using qPCR on additional biological replicates. Gene expression is presented as fold change relative to 3 mo Ctrl after normalizing to beta‐actin. (n = 5–6, Two‐way ANOVA [C3 Age: F 1,19 = 10.16, p = 0.0048; Treatment: F 1,19 = 14.80, p = 0.0011; Interaction: F 1,19 = 7.52, p = 0.013; Bonferroni correction 12 mo Ctrl vs. AD p = 0.0004 (***)] [Ctsd Age: F 1,19 = 4.92, p = 0.0389; Treatment: F 1,19 = 2.15, p = 0.1588; Interaction: F 1,19 = 6.11, p = 0.0231; Bonferroni correction 12 mo Ctrl vs. AD p = 0.0271 (*)] [Cst7 Age: F 1,19 = 18.82, p = 0.0004; Treatment: F 1,19 = 5.36, p = 0.0319; Interaction: F 1,19 = 5.66, p = 0.028; Bonferroni correction 12 mo Ctrl vs. AD p = 0.0086 (**)] [Clec7a Age: F 1,19 = 30.75, p < 0.0001; Treatment: F 1,19 = 7.86, p = 0.0114; Interaction: F 1,19 = 7.76, p = 0.0118; Bonferroni correction 12 mo Ctrl vs. AD p = 0.0021 (**)]). Error bars represent the standard error of the mean. (E) Western blot analysis of protein products from differentially expressed glial genes. Sarkosyl soluble fractions were analyzed with the signal normalized to total protein (Revert 700) and the abundance relative to 3 mo Ctrl. Ab, antibody probed blot image; TP, total protein stain (Revert 700). (n = 3, Two‐way ANOVA [C3 Age: F 1,8 = 11.14, p = 0.01; Treatment: F 1,8 = 1.13, p = 0.32; Interaction: F 1,8 = 1.84, p = 0.21] [Ctsd Age: F 1,8 = 0.61, p = 0.46; Treatment: F 1,8 = 11.38, p = 0.0097; Interaction: F 1,8 = 11.31, p = 0.0099; Bonferroni correction 12 mo Ctrl vs. AD p = 0.0028 (**)] [Cst7 Age: F 1,8 = 0.39, p = 0.55; Treatment: F 1,8 = 0.01, p = 0.91; Interaction: F 1,8 = 0.96, p = 0.36] [Clec7a Age: F 1,8 = 1.69, p = 0.23; Treatment: F 1,8 = 0.0008, p = 0.98; Interaction: F 1,8 = 2.04, p = 0.19]). Error bars represent the standard error of the mean
In particular amongst protein‐coding genes, Ctsd, Cst7, and Clec7a were differentially expressed in 12 mo animals and have been identified as transcripts specific to disease‐associated microglia (Figure 2B, C; [24]). We therefore validated the differential expression of Ctsd, Cst7, and Clec7a and the complement gene C3 (two‐way ANOVA, Bonferroni correction, p = 0.0271, p = 0.0086, p = 0.0021, and p = 0.0004, respectively) in 12 mo compared to 3 mo mice using quantitative PCR on biological replicates obtained from an additional animal cohort (Figure 2D). Given the confirmed change in transcript level, we next investigated the protein products of these genes with western blotting (Figure 2E, Figure S5). The abundance of Ctsd was significantly increased in AD compared to Ctrl samples in 12 mo but not 3 mo animals at the early time point (two‐way ANOVA, Bonferroni correction, p = 0.0028) while the levels of other proteins remained unchanged (Figure 2E, Figure S5). Taken together, our analysis suggests that protein‐coding genes, but not TEs, provide an age‐specific proinflammatory response to pathology in 12 mo animals which precedes a reduced pathology burden.
4. DISCUSSION
Advanced age is a near ubiquitous feature of neurodegenerative conditions, but has an unclear influence on disease at the molecular level. In this study, we found that age produces a unique proinflammatory gene expression pattern at the early stages of nascent tau pathology initiated in WT mice by intracranial injection with human AD extracts. This molecular phenotype preceded a reduced level of pathology in 12 mo animals. This is interesting given that this age of mouse was chosen to investigate the paradigm proposed in humans where disease begins in middle‐aged people, decades prior to cognitive decline [5]. In tau seeded mice, we identified stereotypical spreading of tau pathology to anatomically connected brain regions, a feature that validates the relevance of this model to human disease ([8]; Figure S1A). Additionally, the most abundant hyperphosphorylated tau feature in our mice were small punctate foci of staining (Figure S1B), similar to an earlier study which investigated the injection of pathological tau into WT mice [25]. These puncta are similar to neuropil staining seen in AD and other tauopathies [22]. Tau inclusions, a pathological feature that resembles somatic neurofibrillary tangles, did not show any clear association with host age in our study (Figure S1C). This finding was surprising given that puncta were significantly reduced in 12 compared to 3 mo animals (Figure 1D). It is possible that puncta and inclusions represent different levels of tau deposit maturation given that puncta were found at all stages (Figure 1D) and, the less abundant inclusions, were only found after 3 months of incubation (Figure S1C). Our results point to a scenario where the differences in puncta may lead to a difference in inclusion numbers between host ages at later time points with longer incubation times than used in our study. The lack of any significant difference in insoluble tau from different aged mice may suggest a similar explanation; with longer incubation times, insoluble tau levels may have reached significantly different levels (Figure 1E). It is worth noting that the immunohistochemistry analysis (Figure 1D) does not probe the exact same population of tau as the sarkosyl extraction followed by western blotting (Figure 1E). This methodological difference likely explains how a modest significant change in AT8‐positive tau deposits was not also observed in sarkosyl insoluble tau levels between age groups.
The influence of advanced age on tau pathology has been investigated experimentally in various mouse models [26]. For example, in a prior study of the same seeding model of sporadic tauopathy, older WT mice (15–19 mo) had a moderately increased level of pathology when compared to younger animals in the entorhinal cortex but not other regions [8]. Another study using an AAV system to express tau in WT mice similarly showed that in aged animals (22–24 mo) there was a greater level of pathology compared to young (3 mo) animals [27]. It is important to note that the older animals used in our study (12 mo) were younger than those mentioned above. This could explain why we found moderately less punctate pathology in 12 compared to 3 mo mice as opposed to the increase seen with older animals in these other experiments (Figure 1D). Additionally, when compared to the study using the same seeding model of pathology [8], we quantified specific pathology morphologies in a different region, namely the dentate gyrus.
TEs have been associated with tauopathies and aging in animal models and human autopsy tissue [28, 29]. More specifically, these studies identified that certain endogenous retroviruses (ERVs) and long interspersed element‐1 (LINE‐1, L1) transcripts were enriched in AD and progressive supranuclear palsy autopsy material [30, 31]. The involvement of ERVs has also been investigated in other neurodegenerative diseases, including amyotrophic lateral sclerosis [32, 33, 34]. It is therefore somewhat surprising that our RNA‐seq analysis did not yield any significant dysregulation of TEs in the presence of tauopathy, nor an association with advancing age (Figure S4). However, differences between the model used here and those mentioned above are important to consider, most importantly the model of early pathology used here compared to end‐stage human material that would have more severe pathology and dysfunction.
In numerous neurodegenerative diseases, inflammation has been identified as a key player in the progression of disease [35]. More specifically, microglia have been increasingly singled out as a main effector of pathology‐related deterioration [36]. Several studies have identified a disease‐specific class of microglia in distinct pathology models, and in humans [24, 37, 38]. In our study, the differentially expressed genes in 12 mo animals at the early stage were enriched for a set of genes that are related to the inflammatory response (Figure 2A,B; [17]). Interestingly, this transcriptional signature in 12 mo animals preceded a reduced pathology burden, suggesting this may trigger an initial protective response (Figures 1 and 2). The validated genes Cst7, Ctsd, and Clec7a, of which Ctsd protein was also significantly increased, have been identified as differentially regulated in a disease‐specific class of microglia [24]. This suggests that microglial response in the nascent pathology stage of 12 mo mice may be similar to that identified in previous publications [24, 37, 38]. Additionally, C3 has been linked to AD and a tauopathy mouse model, where increased levels are associated with the disease state [39]. As indicated above, it is expected that inflammatory cells would be the first to respond to a neurological insult, however the age‐specific nature of the response encountered here is interesting. Given that few gene expression changes were seen at the later stages of pathology, we interpret our finding as an early marker of altered pathology burden. The lack of a similar gene expression response at later time points when pathology is more pronounced was unexpected, yet could be explained by the nature of this tauopathy model not being driven by a human transgene. Furthermore, given the moderate severity of pathology in this model, it could be that in the later stages of incubation where pathology has increased, the cellular milieu may have entered an immune‐tolerant state with few transcriptional alterations compared to controls [40]. Notwithstanding, the validation of gene expression changes during a period of nascent pathology, which remains an understudied phase of disease, strongly indicates that host age dictates the molecular response to tau pathology.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
All animal experiments were approved by the University of Queensland animal ethics committee (QBI/226/18). Use of de‐identified human brain material was approved by the University of Queensland human research ethics committee (HREC/18/MHS/60).
AUTHOR CONTRIBUTIONS
Jay Rasmussen performed all experiments and analyzed data. Adam D. Ewing performed bioinformatics analyses. Liviu‐Gabriel Bodea and Gabriela O. Bodea contributed expertise and resources. Marla Gearing provided human tissue and data. Geoffrey J. Faulkner contributed resources and conceptualized the project with Jay Rasmussen, who wrote the manuscript with input from all authors.
Supporting information
FIGURE S1 Features of pathology in injected mice. (A) AT8 staining of the mammillary body and entorhinal cortex of 3 and 12 mo WT AD injected mice at the late incubation time point (scale bar = 100 μm). (B) Higher magnification image of the hippocampus showing hyperphosphorylated (AT8) puncta and inclusion pathology in a 3 mo WT AD injected mouse (scale bar = 50 μm). (C) Stereological quantification of AT8‐positive inclusions in the dentate gyrus of injected mice. No inclusion pathology was seen at the early time point so it was omitted from the figure. No significant differences were observed (n = 4–7, Two‐way ANOVA). Error bars represent the standard error of the mean
{kind=link}
FIGURE S2 Western blotting images for T46 and TAU‐13. (A) From Figure 1E, a long exposure image of the T46 total tau western blot. Small amounts of mouse tau can be found in control animals. Quantification of the tau band at >55 kDa in Figure 1E focussed on a comparison between 3 and 12 mo AD animals. (B) From Figure 1E, showing a transgenic mouse (a 5‐month‐old female rTg4510 brain extract) positive control lane next to experimental samples in the TAU‐13 human tau western blot. Short and long exposures are shown. (C) A dilution series of the human AD brain extract injected into mice probed with TAU‐13. The TAU‐13 blot can detect 1600‐fold less tau than what was injected into mice (2.5 μl)
{kind=link}
FIGURE S3 Western blotting images used for quantification of insoluble tau in injected animals. Blotting as in Figure 1E but with all AD injected samples on a single blot in order to complete signal intensity analysis
{kind=link}
FIGURE S4 Expression of repetitive element families in injected mice. Read counts from bulk RNA‐seq data is presented for the most highly expressed families for SINE (B1 and B3), LINE (L1Md_F and L1Md_T), and ERV superfamilies (MuLV and IAPEz). Two‐way ANOVA with Bonferroni correction was performed (n = 3–7) and no significant differences were observed. Error bars represent the standard error of the mean
{kind=link}
FIGURE S5 Western blotting images used for quantification of protein products of differentially expressed glial genes. Blotting as in Figure 2E with a red arrow indicating the band that was quantified. For C3, a fragment of the alpha chain was quantified (~40 kDa). For Ctsd, Cst7 (CstF), and Clec7a (Dectin‐1) the monomeric band (~50, ~20, and ~30 kDa, respectively) was quantified
{kind=link}
TABLE S1 Human patient data used for extract preparation
TABLE S2 3 mo middle time point differentially expressed genes (AD vs Ctrl)
TABLE S3 3 mo late time point differentially expressed genes (AD vs Ctrl)
TABLE S4 12 mo late time point differentially expressed genes (AD vs Ctrl)
TABLE S5 12 mo early time point differentially expressed genes (AD vs Ctrl)
ACKNOWLEDGEMENTS
This work was supported by the Mater Foundation (GJF, AEW), an NHMRC Investigator Grant (GNT1173711, GJF), a CSL Behring Fellowship (GJF) and an NHMRC‐ARC Dementia Research Development Fellowship (GNT1108258, GOB). Microscopy analysis was possible due to Linkage Infrastructure, Equipment and Facilities funding (LIEF LE100100074). We would like to thank the Queensland Brain Institute Animal and Behaviour Facility for access to animal surgery facilities and also all animal care technicians for assistance with the project.
Rasmussen J, Ewing AD, Bodea L‐G, Bodea GO, Gearing M, Faulkner GJ. An early proinflammatory transcriptional response to tau pathology is age‐specific and foreshadows reduced tau burden. Brain Pathol. 2022;32:e13018. 10.1111/bpa.13018
Contributor Information
Jay Rasmussen, Email: jdoug.rasmussen@gmail.com.
Geoffrey J. Faulkner, Email: faulknergj@gmail.com.
DATA AVAILABILITY STATEMENT
Sequencing data is available from GEO under the accession number GSE169686. Other Supporting Information is available upon request from JR or GJF.
REFERENCES
- 1. Jucker M, Walker LC. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci. 2018;21:1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovacs GG. Molecular pathology of neurodegenerative diseases: principles and practice. J Clin Pathol. 2019;72:725–35. [DOI] [PubMed] [Google Scholar]
- 3. Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov. 2015;14:759–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Götz J, Halliday G, Nisbet RM. Molecular pathogenesis of the tauopathies. Ann Rev Pathol. 2019;14:239–61. [DOI] [PubMed] [Google Scholar]
- 5. McDade E, Bateman RJ. Stop Alzheimer's before it starts. Nature. 2017;547:153–5. [DOI] [PubMed] [Google Scholar]
- 6. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–81. [DOI] [PubMed] [Google Scholar]
- 7. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213:2635–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, et al. Amyloid‐beta plaques enhance Alzheimer's brain tau‐seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018;24:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci. 2017;37:11406–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Narasimhan S, Changolkar L, Riddle DM, Kats A, Stieber A, Weitzman SA, et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J Exp Med. 2020;217:e20190783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peng C, Trojanowski JQ, Lee VM. Protein transmission in neurodegenerative disease. Nat Rev Neurol. 2020;16:199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging‐Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2012;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Habib N, Li Y, Heidenreich M, Swiech L, Avraham‐Davidi I, Trombetta JJ, et al. Div‐Seq: single‐nucleus RNA‐Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353:925–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Faulkner GJ, Forrest AR, Chalk AM, Schroder K, Hayashizaki Y, Carninci P, et al. A rescue strategy for multimapping short sequence tags refines surveys of transcriptional activity by CAGE. Genomics. 2008;91:281–8. [DOI] [PubMed] [Google Scholar]
- 19. Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–71. [DOI] [PubMed] [Google Scholar]
- 20. Hashimoto T, de Hoon MJ, Grimmond SM, Daub CO, Hayashizaki Y, Faulkner GJ. Probabilistic resolution of multi‐mapping reads in massively parallel sequencing data using MuMRescueLite. Bioinformatics. 2009;25:2613–4. [DOI] [PubMed] [Google Scholar]
- 21. Smit AFA, Hubley R, Green P. RepeatMasker Open‐3.0. 1996. [Google Scholar]
- 22. Gibbons GS, Lee VMY, Trojanowski JQ. Mechanisms of cell‐to‐cell transmission of pathological tau: a review. JAMA Neurol. 2019;76:101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Julien C, Bretteville A, Planel E. Biochemical isolation of insoluble tau in transgenic mouse models of tauopathies. Methods Mol Biol. 2012;849:473–91. [DOI] [PubMed] [Google Scholar]
- 24. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169:1276–90. [DOI] [PubMed] [Google Scholar]
- 25. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bodea LG, Evans HT, Van der Jeugd A, Ittner LM, Delerue F, Kril J, et al. Accelerated aging exacerbates a pre‐existing pathology in a tau transgenic mouse model. Aging Cell. 2017;16:377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wegmann S, Bennett RE, Delorme L, Robbins AB, Hu M, McKenzie D, et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci Adv. 2019;5:eaaw6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bodea GO, McKelvey EGZ, Faulkner GJ. Retrotransposon‐induced mosaicism in the neural genome. Open Biol. 2018;8:180074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dubnau J. The Retrotransposon storm and the dangers of a Collyer's genome. Curr Opin Genet Dev. 2018;49:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo C, Jeong HH, Hsieh YC, Klein HU, Bennett DA, De Jager PL, et al. Tau activates transposable elements in Alzheimer's disease. Cell Rep. 2018;23:2874–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun W, Samimi H, Gamez M, Zare H, Frost B. Pathogenic tau‐induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci. 2018;21:1038–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krug L, Chatterjee N, Borges‐Monroy R, Hearn S, Liao WW, Morrill K, et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP‐43 model of ALS. PLoS Genet. 2017;13:e1006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, et al. Human endogenous retrovirus‐K contributes to motor neuron disease. Sci Transl Med. 2015;7:307ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tam OH, Ostrow LW, Gale Hammell M. Diseases of the nERVous system: retrotransposon activity in neurodegenerative disease. Mob DNA. 2019;10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scheiblich H, Trombly M, Ramirez A, Heneka MT. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 2020;41:300–12. [DOI] [PubMed] [Google Scholar]
- 36. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21:1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep. 2019;27:1293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019;28:2111–23. [DOI] [PubMed] [Google Scholar]
- 40. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Features of pathology in injected mice. (A) AT8 staining of the mammillary body and entorhinal cortex of 3 and 12 mo WT AD injected mice at the late incubation time point (scale bar = 100 μm). (B) Higher magnification image of the hippocampus showing hyperphosphorylated (AT8) puncta and inclusion pathology in a 3 mo WT AD injected mouse (scale bar = 50 μm). (C) Stereological quantification of AT8‐positive inclusions in the dentate gyrus of injected mice. No inclusion pathology was seen at the early time point so it was omitted from the figure. No significant differences were observed (n = 4–7, Two‐way ANOVA). Error bars represent the standard error of the mean
FIGURE S2 Western blotting images for T46 and TAU‐13. (A) From Figure 1E, a long exposure image of the T46 total tau western blot. Small amounts of mouse tau can be found in control animals. Quantification of the tau band at >55 kDa in Figure 1E focussed on a comparison between 3 and 12 mo AD animals. (B) From Figure 1E, showing a transgenic mouse (a 5‐month‐old female rTg4510 brain extract) positive control lane next to experimental samples in the TAU‐13 human tau western blot. Short and long exposures are shown. (C) A dilution series of the human AD brain extract injected into mice probed with TAU‐13. The TAU‐13 blot can detect 1600‐fold less tau than what was injected into mice (2.5 μl)
FIGURE S3 Western blotting images used for quantification of insoluble tau in injected animals. Blotting as in Figure 1E but with all AD injected samples on a single blot in order to complete signal intensity analysis
FIGURE S4 Expression of repetitive element families in injected mice. Read counts from bulk RNA‐seq data is presented for the most highly expressed families for SINE (B1 and B3), LINE (L1Md_F and L1Md_T), and ERV superfamilies (MuLV and IAPEz). Two‐way ANOVA with Bonferroni correction was performed (n = 3–7) and no significant differences were observed. Error bars represent the standard error of the mean
FIGURE S5 Western blotting images used for quantification of protein products of differentially expressed glial genes. Blotting as in Figure 2E with a red arrow indicating the band that was quantified. For C3, a fragment of the alpha chain was quantified (~40 kDa). For Ctsd, Cst7 (CstF), and Clec7a (Dectin‐1) the monomeric band (~50, ~20, and ~30 kDa, respectively) was quantified
TABLE S1 Human patient data used for extract preparation
TABLE S2 3 mo middle time point differentially expressed genes (AD vs Ctrl)
TABLE S3 3 mo late time point differentially expressed genes (AD vs Ctrl)
TABLE S4 12 mo late time point differentially expressed genes (AD vs Ctrl)
TABLE S5 12 mo early time point differentially expressed genes (AD vs Ctrl)
Data Availability Statement
Sequencing data is available from GEO under the accession number GSE169686. Other Supporting Information is available upon request from JR or GJF.