

Abstract
Motivation
Batch effects in omics datasets are usually a source of technical noise that masks the biological signal and hampers data analysis. Batch effect removal has been widely addressed for individual omics technologies. However, multi-omic datasets may combine data obtained in different batches where omics type and batch are often confounded. Moreover, systematic biases may be introduced without notice during data acquisition, which creates a hidden batch effect. Current methods fail to address batch effect correction in these cases.
Results
In this article, we introduce the MultiBaC R package, a tool for batch effect removal in multi-omics and hidden batch effect scenarios. The package includes a diversity of graphical outputs for model validation and assessment of the batch effect correction.
Availability and implementation
MultiBaC package is available on Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/MultiBaC.html) and GitHub (https://github.com/ConesaLab/MultiBaC.git). The data underlying this article are available in Gene Expression Omnibus repository (accession numbers GSE11521, GSE1002, GSE56622 and GSE43747).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
While omic platforms are widely accessible, data generation from a large number of samples and/or assays is still costly. For large data collections, sample acquisition may be distributed in time and space or complemented with data already available at public repositories. This results in datasets that are affected by a technical variability component associated to each acquisition event, i.e. the batch effect. Batch effects may represent the major source of variability in the combined omic dataset and compromise the detection of the underlying biological signal by standard methods (Kupfer et al., 2012).
Several Batch Effect Correction Algorithms (BECAs) have been proposed which are available as R packages (Leek et al., 2012; Risso et al., 2014; Ritchie et al., 2015). Usually, BECAs require adequate experimental designs where the batch is known and not confounded with the effects of interest (treatment, dose, time point, etc.). However, there are few BECAs providing data corrected for systematic noise coming from an unknown source. This may happen when laboratory practices introduce an un-noticed bias that affects a subset of samples. While this variability behaves as a batch effect, it is not identified as such and therefore is invisible to many traditional BECAs. Another scenario not yet covered by BECAs is multi-omic experiments. When multi-omic datasets are created by combining different omics modalities obtained asynchronously, batch effects are confounded with the ‘omic type effect’, what hampers their removal from the data.
Here, we introduce the MultiBaC R package (Fig. 1), a general tool for batch effect removal in omic data that successfully addresses these difficult cases. The MultiBaC R package integrates two different batch effect correction methods: ARSyN, a flexible approach for the correction of systematic biases in single omic datasets for both declared (batches) or hidden sources of technical noise (Nueda et al., 2012), and MultiBaC, the first batch effect correction algorithm for multi-omic data (Ugidos et al., 2020). The MultiBaC R package is available at Bioconductor.
Fig. 1.
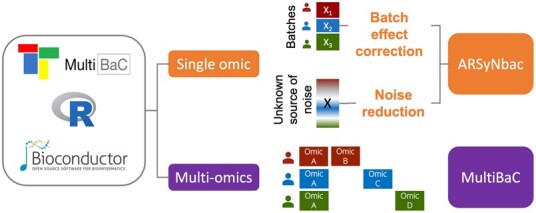
Overview of BECAs in MultiBaC R package
2 Materials and methods
Supplementary Figure S1 depicts the general scheme of ARSyNbac and MultiBaC algorithms. Basically, ARSyNbac uses ANOVA to decompose the omics signal into experimental variables and residual noise. Should the source of unwanted variation be known, the method estimates the batch effect and removes it from data. When the source of batch effect is unknown, the residual noise is analyzed by PCA to detect any systematic component which is subsequently removed from the data. It is also possible to simultaneously correct the data for both types of noise. MultiBaC operates when a multi-omic dataset is created by combining multi-omic data from different laboratories provided one omic type is shared across labs (common omic). Partial Least Squares Regression (PLS) is used to model and predict the non-common omic data as a function of the common omic type, which allows the subsequent combination of complete multi-omic data structures and removal of batch effects with ARSyNbac.
3 Implementation
The MultiBaC R package organizes all the input and output data in a MultiAssayExperiment object, a type of Bioconductor container for multi-omic studies. The createMbac() function in MultiBaC package creates a MultiAssayExperiment object for each batch from the original matrices, and generates an mbac data structure, a S3 list class of MultiAssayExperiment objects to be used as input by ARSyNbac() or MultiBaC() functions. The resulting batch effect corrected matrices are also added to the mbac structure and several plots can be generated to validate PCA or PLS models applied by MultiBaC (Fig. 2a–c), and to assess the batch effect magnitude (Fig. 2d) or the correction performance (Supplementary Fig. S2). A reduced version of a yeast multi-omic dataset described in (Ugidos et al., 2020) is included in the package. More details about the package can be found in the Bioconductor vignette and Supplementary Section S4.
Fig. 2.
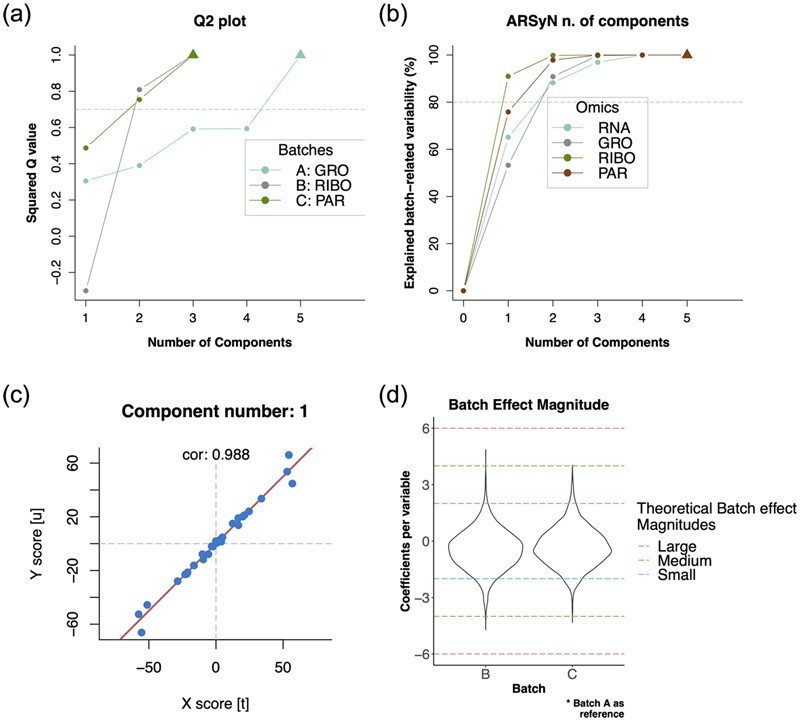
Usage of MultiBaC method on the complete yeast multiomics dataset. (a) Q2 plot shows the number of PLS components needed to reach a good predictive ability. (b) Explained variance plot shows the number of components retained by the ARSyN model. (c) Inner relation plot checks the requirement of linearity between X and Y PLS components. (d) Distribution of the magnitude of batch effects
4 Results and discussion
Supplementary Section S3.1 summarizes and discusses the qualitative comparison of methods included in the MultiBaC package to the most popular BECAs. Briefly, in the single-omic case, limma and ComBat cannot handle the unknown batches situation. ARSyN, RUV and SVA can estimate such noise effects, but SVA does not provide a corrected dataset and instead returns surrogate variables to be included in differential expression models. ARSyNbac can easily and simultaneously correct for both known and unknown sources of noise. To the best of our knowledge, MultiBaC is the only BECA for correcting multi-omic batch effects.
The ARSyN algorithm performance for noise reduction mode was validated on both real and simulated data in (Nueda et al., 2012) but not provided in a publicly available R package. The known batch option is a straightforward adaptation of the original ARSyN methodology. A very simple implementation of these two ARSyN versions was first included in the NOISeq R package (Tarazona et al., 2015) and now their functionality has been quite improved in the ARSyNbac function, with simultaneous correction of unwanted effects from both known or unknown sources, performance plots and more flexible options for PCA models. Supplementary Section S3.2 shows that ARSyNbac overperforms other popular BECAs listed in Supplementary Table S1.
The MultiBaC strategy has been also extensively validated in Ugidos et al. (2020). Figure 2 and Supplementary Figure S2 shows how MultiBaC successfully removes batch effects when four yeast omics types (RNA-seq, GRO-seq, RIBO-seq and PAR-clip) (Ugidos et al., 2020), obtained in different labs, are combined. MultiBaC returns a harmonized multi-omics dataset ready to be used in downstream analyses aiming to infer regulatory patterns across omics layers.
Funding
This work was funded by the Generalitat Valenciana through PROMETEO grants program for excellence research groups [PROMETEO 2016/093] and by the Spanish MICINN [PID2020-119537RB-I00]. Funding for open access charge: Universitat Politècnica de València.
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Manuel Ugidos, Gene Expression and RNA Metabolism Laboratory, Instituto de Biomedicina de Valencia, Consejo Superior de Investigaciones Científicas, Valencia 46010, Spain; Multivariate Statistical Engineering Group, Department of Applied Statistics, Operations Research and Quality, Universitat Politècnica de València, Valencia 46022, Spain.
María José Nueda, Department of Mathematics, Universidad de Alicante, Alicante 03690, Spain.
José M Prats-Montalbán, Multivariate Statistical Engineering Group, Department of Applied Statistics, Operations Research and Quality, Universitat Politècnica de València, Valencia 46022, Spain.
Alberto Ferrer, Multivariate Statistical Engineering Group, Department of Applied Statistics, Operations Research and Quality, Universitat Politècnica de València, Valencia 46022, Spain.
Ana Conesa, Institute for Integrative Systems Biology, Consejo Superior de Investigaciones Científicas, Valencia 46980, Spain.
Sonia Tarazona, Multivariate Statistical Engineering Group, Department of Applied Statistics, Operations Research and Quality, Universitat Politècnica de València, Valencia 46022, Spain.
References
- Kupfer P. et al. (2012) Batch correction of microarray data substantially improves the identification of genes differentially expressed in rheumatoid arthritis and osteoarthritis. BMC Med. Genomics, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T. et al. (2012) The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nueda M.J. et al. (2012) ARSyN: a method for the identification and removal of systematic noise in multifactorial time course microarray experiments. Biostatistics, 13, 553–566. [DOI] [PubMed] [Google Scholar]
- Risso D. et al. (2014) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol., 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E. et al. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. NAR, 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona S. et al. (2015) Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. NAR, 43, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugidos M. et al. (2020) MultiBaC: strategy to remove batch effects between different omic data types. Stat. Methods Med. Res., 29, 2851–2864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.