

Abstract
Serum concentrations of cholesterol are positively correlated with exposure to perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS) in humans. The associated change in cholesterol is small across a broad range of exposure to PFOA and PFOS. Animal studies generally have not indicated a mechanism that would account for the association in humans. The extent to which the relationship is causal is an open question. Nonetheless, the association is of particular importance because increased serum cholesterol has been considered as an endpoint to derive a point of departure in at least one recent risk assessment. To gain insight into potential mechanisms for the association, both causal and non-causal, an expert workshop was held Oct 31 and Nov 1, 2019 to discuss relevant data and propose new studies. In this report, we summarize the relevant background data, the discussion among the attendees, and their recommendations for further research.
Keywords: Fluorocarbons, Perfluoroalkyl substances, Serum cholesterol, Mechanism of action
1. Introduction
Serum concentrations of total cholesterol and related lipid measures such as low density lipoprotein (LDL) cholesterol have been reported to be positively associated with serum concentrations of perfluorinated alkyl substances (PFAS) in humans. The cholesterol-PFAS association was proposed as the point of departure for human health risk assessment, though an update of that assessment has shifted to immune effects (EFSA Panel, 2018a; EFSA Panel, 2020). As noted in a government agency critique of the cholesterol association as a point of departure (EFSA Panel, 2018b), it remains an open question whether the association is causal.
An expert workshop was conducted to provide an opportunity for cross-disciplinary discussions of the epidemiology of cholesterol and PFAS, mechanisms of PFAS sequestration and transport, their potential interactions with lipid/lipoprotein processing, and whether alterations in lipid metabolism in some way affect or correlate with PFAS disposition. This report summarizes salient background material and documents the discussions and conclusions reached at the workshop – with an emphasis on identifying data gaps regarding the interactions of PFAS with lipids – and suggests experimental, modeling, and epidemiologic studies that could further elucidate the quantitative nature of any interactions. Although exposure to PFAS other than perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS) may be related to cholesterol and related lipid measures, to keep the effort focused, the workshop addressed data on PFOA and PFOS. The background section of this report incorporated material provided in presentations from participants at the workshop.
2. Background
2.1. PFAS use, environmental contamination, and human exposure
PFAS are synthetic substances used in a variety of industrial processes and products and can also form from degradation of precursors used in manufacturing (Houde et al., 2006; Lau et al., 2007). PFAS are stable, biodegrade slowly, and persist in the environment (Fromme et al., 2009; Lau et al., 2007). PFOA and PFOS are the two PFAS most frequently detected in humans. For background-exposed adults, food is the main source of exposure (EFSA Panel, 2018). Despite the elimination of production and use of PFOA and PFOS in many countries (Sunderland et al., 2019), their widespread, global presence in human populations has raised concern over effects they might have on human health.
2.2. Epidemiology of the total cholesterol-PFAS association
We focus on the epidemiology of total serum cholesterol and PFAS in this section, because the relationships have more support than for specific lipoproteins and triglycerides. The epidemiologic data on the relation of serum cholesterol to PFOA show that the slope of the relation is relatively steep up to 50 ng/mL, and relatively modest thereafter (Fig. 1). The distribution of LDL-cholesterol among subjects in the National Health and Nutrition Examination Survey (NHANES) 2003–2016 had a higher mean and standard deviation for those in the highest compared with the lowest deciles of PFOA and PFOS (Fig. 2). Within the extremely high range of PFOA serum concentrations reached in a clinical trial to evaluate safety, dose limiting toxicity, and maximum tolerated dose, at serum concentrations > 175,000 ng/mL, a possible decrease in serum cholesterol was reported (not shown in Fig. 1) (Convertino et al., 2018). However, because all subjects in the trial had an advanced solid tumor, which may influence plasma LDL-cholesterol concentrations (Huang et al., 2020), the relevance of its results to associations in other populations is unclear. A brief review of the epidemiologic data is included in Supplementary Material Section 1. In the NHANES data, the steepest part of the curve occurred within the background range of exposure (≤ 15 ng/mL), and the change in cholesterol (mg/dl) per ng/mL PFOA was 0.77 (see Supplementary Material Section 1). The interquartile difference of PFOA in NHANES 2003–2016 was about 2.73 ng/mL, corresponding to a 2.2 mg/dl increase in cholesterol (about 1%). Thus, within the range of exposure among the general population (~2–4 ng/ml serum), the shift in cholesterol reflecting this relationship with PFOA is modest. Data for PFOS are similar, though the slope of the relationship in the general population appears lower. Most of the data are cross-sectional and use serum PFAS concentration as a measure of exposure.
Fig. 1.
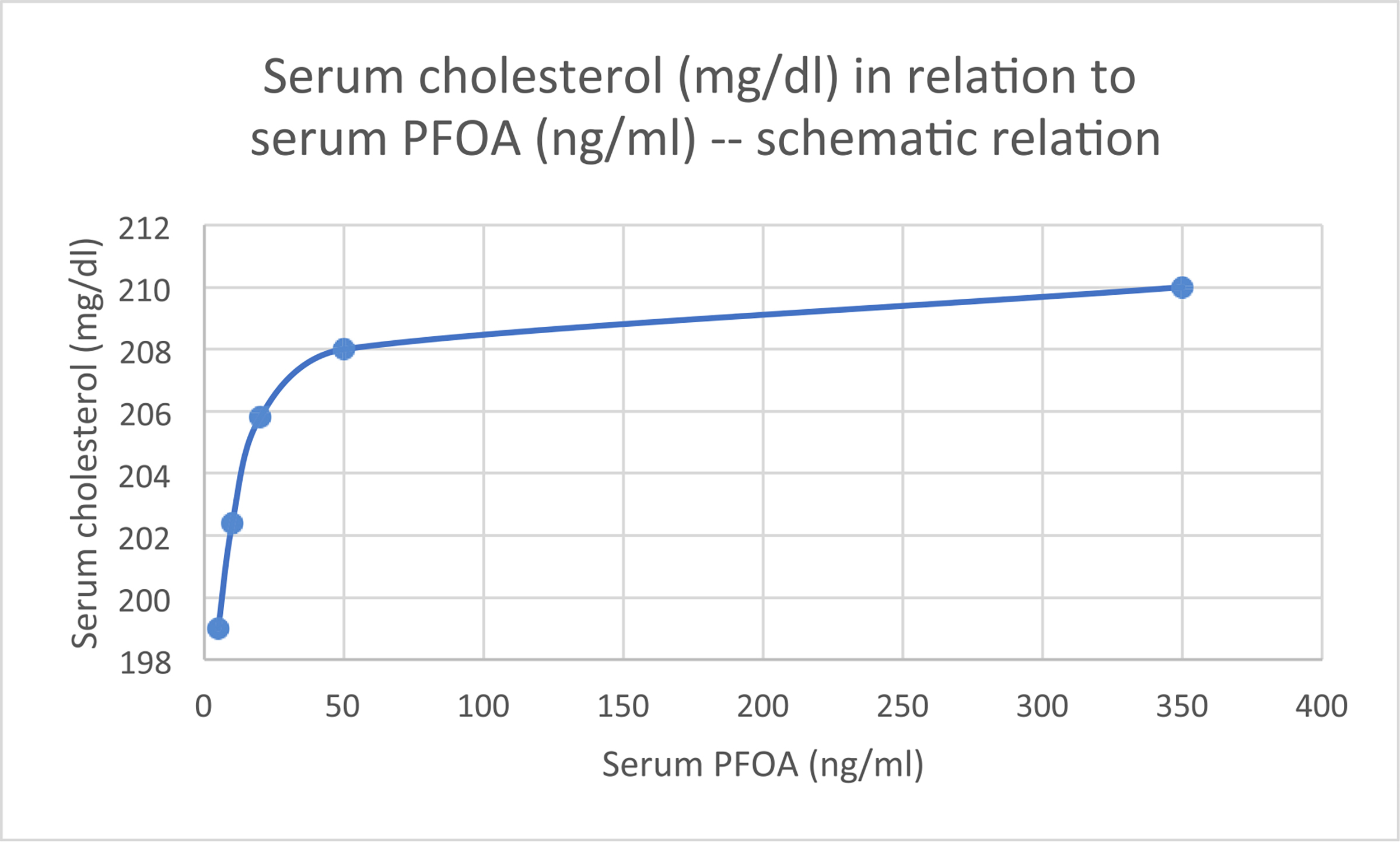
Serum cholesterol (mg/dl) in relation to serum PFOA (ng/mL) – schematic representation of the relation*.
*Based on the results of Steenland et al. (2009). Steenland et al.’s Fig. 2 was scanned and a cubic model was fit to the data. Steenland et al.’s results were adjusted for age, sex, use of cholesloterol-lowering medication, smoking, education, physical activity, alcohol consumption, and body mass index. See the original article for details about reference categories used in predicting values. The average serum cholesterol among adults in the U.S. was 202 mg/dl (Nelson et al., 2010).
Fig. 2.

Distribution of LDL cholesterol among NHANES subjects 2003–2016.
Results calculated using NHANES sampling parameters, to make them representative of the U.S. population. Those taking cholesterol-lowering drugs were excluded, as were those aged < 20 y. N = 2129 (PFOA) or 2128 (PFOS); in other words, the number of subjects under each curve shown above is roughly 1,000 (total n of subjects was 10,647).
LDL cholesterol (mg/dl) was adjusted for age, sex, ethnicity, and an index indicating survey wave. The deciles were determined for PFOA or PFOA based on the distribution of values after adjusting for age, sex, ethnicity, and an index indicating survey wave and calculated using the sampling parameters.
The smoothed curves are from a normal distribution with mean and standard deviation calculated from the observations.
As a complement to the cross-sectional studies, two studies of populations with unusually high PFOA exposure (but less than in the clinical trial of PFOA) are especially informative, because the estimate of exposure was not based on a measurement of PFAS in serum, and they were not cross-sectional. In the large and more highly exposed population in the contaminated area of Ohio and West Virginia (C8 study), when modeled exposure was examined in relation to subsequent risk of hypercholesterolemia, the hazards ratio was 1.2 in the top four quintiles of cumulative exposure (Winquist and Steenland, 2014; Geiger et al., 2014). In an occupational study where 34 workers with high exposure were compared with a matched comparison group, the serum cholesterol was about 30 mg/dl higher in the high exposure group (p = 0.003) (Costa et al., 2009). Total serum cholesterol is a risk factor in the development of cardiovascular disease (Berry et al., 2012; Peters et al., 2016). If exposure to PFAS increased serum cholesterol, then an increase in the risk of cardiovascular disease would also be expected. In the C8 study, with over 30,000 subjects of whom 2,468 developed coronary artery disease, however, no such association was found. Although the epidemiologic data are stronger for total cholesterol than for low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, or triglycerides, the majority of cholesterol in humans is present in LDL (MRFIT, 1986), and our consideration of possible mechanisms for the association will focus on LDL-cholesterol.
2.3. Toxicology and biology
PFOA and PFOS cause hepatomegaly in rodents and monkeys (ATSDR, 2018; Butenhoff et al., 2002; Quist et al., 2015; Seacat et al., 2002; Xing et al., 2016). In three mouse studies of PFOA (out of a total of 2 in rats and 7 in mice), exposure caused increased serum cholesterol (Nakamura et al., 2009; Rebholz et al., 2016; Schlezinger et al., 2020), which may have been due to use of mice with humanized PPARα, mouse strains with altered response to fat intake, higher fat diets, or a combination of these factors (Supplementary Material Table 1). The serum concentration of PFOA and PFOS in the mouse and rat studies, when measured (Supplementary Material Tables 1 and 2) were in the higher range of those reported in occupational studies (see Supplementary Material, Section 2), or greater (Loveless et al., 2006; Rebholz et al., 2016; Olsen et al., 2000, Olsen and Zobel, 2007). A hypocholesterolemic effect of PFOA and PFOS in rodents (Supplementary Material Table 1) is expected because they are HDL mammals (the majority of cholesterol carried in HDL particles (Yin et al., 2012)), and peroxisome proliferator-activated receptor alpha (PPARα) activation (described below) in rodents decreases HDL cholesterol by multiple mechanisms, whereas in humans it increases HDL cholesterol (Kersten, 2008). In monkeys, PFOA caused no changes in serum cholesterol, and PFOS caused a slight reduction, mostly from a decrease in HDL (Butenhoff et al., 2002; Chang et al., 2017). In most studies of PFOA and PFOS effects on triglycerides in rodents, higher doses caused decreased serum triglycerides (Supplementary Material Table 2). Among studies using 0.3 mg/kg/day, a relatively low dose of PFOA (Loveless et al., 2006; Loveless et al., 2008 [rats and mice]; Yan et al., 2014); the results at that dose were mixed; two showed increased triglycerides (Loveless et al., 2006 [mice], Yan et al., 2014 [mice]); two showed no statistically significant change (Loveless et al., 2006 [rats]; Loveless et al., 2008 [mice]); and one showed decreased triglycerides (Loveless et al., 2008 [rats]). Loveless et al.’s (2006) results in mice were unusual in that higher doses of PFOA caused increased triglycerides. In contrast to the general findings for serum triglycerides, PFOA and PFOS tend to increase hepatic triglyceride levels and steatosis (ATSDR, 2018; Bagley et al., 2017; Das et al., 2017; Zhang et al., 2016). In primates, ammonium perfluorooctanoate caused an increase in serum triglycerides (Butenhoff et al., 2002). In APOE*3.Leiden.CETP transgenic mice, PFOS inhibited hepatic lipoprotein production (Bijland et al., 2011). In addition, in humans and other species, PFOA and PFOS may cause immuno-and reproductive/developmental toxicities and could also affect other organ systems (ATSDR, 2018; EFSA CONTAM Panel, 2018).
In adult rodents, PFOA and PFOS activate PPARα, and currently PPARα activation is accepted as the primary mediator of the hypolipidemic effect of PFOA and PFOS (Das et al., 2017; Rosen et al., 2017). Functional PPARα levels are lower in human livers than in rodent livers (Corton et al., 2014; Rosen et al., 2017), and human PPARα is activated to lesser extents by PFAS than rodent PPARα (Maloney and Waxman, 1999; Nakamura et al., 2009; Rosen et al., 2013). Using wildtype and humanized PPARα transgenic mice, perfluorooctanoate was found to activate rodent PPARα; in comparison, PFOA activated human PPARα to lesser extents (Nakamura et al., 2009). Data from human hepatocyte cell culture models evaluating the concentration of PFOA in albumin-free media required to activate PPARα can be re-expressed to approximate the corresponding concentration in serum (see Supplemental Materials, Section 2 for an explanation and discussion). Such an approximation suggests that a serum PFOA concentration in the range of 130 μg/mL would be expected to activate human PPARα. When mice with human PPARα (hPPARα) in their liver were given PFOA resulting in a serum level of 48 μg/mL (Schlezinger et al., 2020), activation of hPPARα was demonstrated, indicating that a lower PFOA concentration may activate PPARα in humans than was suggested by the cell culture models. In similar experiments in mouse hepatocytes, the concentration of PFOA required for PPARα activation was about 20 % of that for human hepatocytes (Rosen et al., 2013). The effect of PFOA at 0.1 and 0.3 ng/kg for 2 weeks on serum cholesterol in PPARα-null 129/Sv mice was measured; the cholesterol was higher in the 0 mg/kg dose compared with wildtype mice, and a dose-response relation with PFOA was absent in the PPARα-null mice (Nakamura et al., 2009). PFAS, however, cause changes in hepatic lipid metabolism in PPARα-null mice, suggesting other mechanisms are involved (Das et al., 2017; Rosen et al., 2010).
In rodents, PFAS also activates signaling through other nuclear receptors such as liver X receptor α (LXR α), pregnane X receptor (PXR), constitutive androstane receptor (CAR), and farnesoid X receptor (FXR) (Abe et al., 2017; Bijland et al., 2011; Bjork et al., 2011), but to lesser extents than PPARα (Rosen et al., 2017). PFAS activation or inhibition of human nuclear receptors other than PPARα has been examined. Activations of CAR, PXR, LXRα, FXR, and PPARγ have been inferred from the responses of a number of target genes for each of the transcription factors (Bjork et al., 2011; Rosen et al., 2017; Wolf et al., 2012). PFAS at a concentration as low as 333 μM (approximately 130 μg/mL), a level similar to the higher range of serum concentrations in occupationally-exposed humans, decreased expression of HNF4α in primary human hepatocytes (Beggs et al., 2016) (Supplementary Materials Section 2). HNF4α is a transcription factor that regulates CYP7A1, which converts cholesterol to bile acids. Decreased CYP7A1 could lead to decreased bile acid production and increased cholesterol. This study also suggested that PFOA but not PFOS may activate PPARγ. Activation of FXR by PFAS could also decrease CYP7A1 production (Chávez-Talavera et al., 2017).
2.4. PFAS pharmacokinetic (PK) background
Although PFOA and PFOS can be absorbed dermally (Franko et al., 2012) or by inhalation (Olsen et al., 2000), for most people the majority of exposure is from the oral route (Hinderliter et al., 2006). Following oral exposure, some absorption may occur directly from the stomach (Kemper, 2003; Worley and Fisher, 2015), though absorption in the gut is facilitated by transporters (Kimura et al., 2017; Zhao et al., 2017) and probably much greater than that in the stomach. These PFAS distribute primarily to the liver, plasma, and kidney, with concentrations in other organs being considerably lower (ATSDR, 2018; Bogdanska et al., 2011, 2020). They bind extensively to fatty-acid binding protein in the liver (Khazaee et al., 2021; Luebker et al., 2002; Sheng et al., 2016) and albumin in plasma (Bischel et al., 2010) and are not metabolized (ATSDR, 2018; Andersen et al., 2008; Cui et al., 2010; Kennedy et al., 2004). Excretion in both sexes is urinary and fecal (ATSDR, 2018; Andersen et al., 2008; Cui et al., 2010; Kennedy et al., 2004). PFAS levels are lowered in females by a number of mechanisms including blood loss during menstruation (humans), transfer to the fetus during pregnancy, and though lactation and milk during breastfeeding (Brantsæter et al., 2013; Calafat et al., 2007; Chang et al., 2009; Dhingra et al., 2017; Loccisano et al., 2012, 2013; Mondal et al., 2014; Rodriguez et al., 2009; Wong et al., 2014). The rate of PFAS excretion (primarily urinary) varies greatly among species, with half-lives estimated in weeks for rodents and months for monkeys. Humans have especially long half-lives of about 2.3–3.8 years for PFOA (Bartell et al., 2010; Olsen et al., 2007) and 5.4 years for PFOS (Olsen et al., 2007). In female rats the half-life of PFOA is much shorter than in males, due to species- and sex-specific differences in renal transporters (Yang et al., 2009). The rate of excretion depends on the specific isomer, with linear isomers generally having longer half-lives than the branched compounds; perfluoro-1-methylheptaneulfonate (1 m-PFOS) may be an exception (Zhang et al., 2013).
There is some fecal excretion of PFOA and PFOS, which is better documented in animals than humans (Hundley et al., 2006). In humans, PFOA and PFOS concentrations in bile are slightly lower than in serum (Harada et al., 2007). Administration of a bile acid sequestrant reduced tissue concentrations of PFOA and PFOS in rats (Johnson et al., 1984) and humans (Genuis et al., 2010). Species-specific transporters for PFOA and PFOS involved in the enterohepatic circulation of bile acids (Zhao et al., 2015) as well as in the disposition of xenobiotics have been identified (Zhao et al., 2017). The human apical bile salt transporter (ABST) can transport both PFOS and bile salts from the gut lumen into enterocytes (Zhao et al., 2015).
2.5. Cholesterol metabolism in humans and physiologically-based pharmacokinetic models
Cholesterol is a component of cell membranes and a precursor for steroid hormones, bile acids, and other sterols. Although synthesized in all cells, dietary absorption in the small intestine and production in the liver are primary sources of plasma cholesterol (Afonso et al., 2018). In the liver cholesterol is packaged into very low-density lipoproteins (VLDL) and secreted in blood for transport throughout the body. In humans, the majority of plasma cholesterol is found esterified in the core of LDL, which are generated upon lipolysis of VLDL triglycerides. Excess cholesterol in peripheral cells can by effluxed to HDL in plasma for return to the liver, either directly or after transfer from HDL to LDL by the Cholesteryl Ester Transfer Protein (CETP). In the liver, specifically in hepatocytes, cholesterol is used as the starting material for bile acid synthesis. Production of bile acids is a major route of cholesterol utilization (Chávez-Talavera et al., 2017; Chen et al., 2002; Pullinger et al., 2002). Excretion is primarily through the gut as such or after conversion to bile acids (Reeskamp et al., 2018). Bile acids are secreted together with cholesterol and phospholipids by transporter-facilitated movement into the bile and move to the gut to aid in digestion and absorption of fatty substances from diet. About 5% of the bile acids delivered to the gut are excreted via feces, the remainder is re-absorbed and comprises the enterohepatic circulation of bile acids/salts.
Major known cholesterol homeostasis regulatory components include its intestinal absorption by the Niemann-Pick C1 Like 1 (NPC1L1) transporter, excretion by the ABCG5/8 transporter in the apical side of enterocytes, synthesis controlled by 3-hydroxy-3-methyl-glutaryl-Coenzyme A Reductase (HMGCR), and conversion to bile acids by cholesterol 7α-hydroxylase (CYP7A1) in hepatocytes (Cohen, 2008). Furthermore, plasma cholesterol levels are determined by LDL receptor activity (Goldstein and Brown, 2009), which is inhibited by proprotein convertase subtilisin/kexin type 9 (PCSK9) (Afonso et al., 2018). The genes controlling cholesterol synthesis and uptake are regulated by the intracellular free cholesterol content, which inhibits activation of sterol regulatory element-binding protein 2 (SREBP2) (Horton et al., 2002; Madison, 2016). Binding of bile acids to the FXR receptor results in decreased expression of CYP7A1 resulting in increased intra-cellular cholesterol levels (Claudel et al., 2005). In addition to genetics (Weissglas-Volkov et al., 2006; Willer et al., 2013), plasma cholesterol concentrations are affected by diet, therapeutic drugs, weight change, the microbiome, bile acid metabolism, hormones, and other factors (Alphonse and Jones, 2016). Cholesterol synthesis and absorption are reciprocal, and the proportion from each varies substantially among people (van der Wulp et al., 2013).
Insights into absorption and endogenous production of cholesterol (Gachumi and El-Aneed, 2017) can be obtained by mass spectroscopy measurement of phytosterols and cholesterol precursors in human serum and used in modeling cholesterol metabolism. Physiologically based pharmacokinetic and closely-related systems biology models of cholesterol and of bile acids have been developed (Hofmann et al., 1983; Mc Auley et al., 2012; Morgan et al., 2016; van der Wulp et al., 2013). Whole-body cholesterol metabolism in humans has been studied (Goodman et al., 1980; Grundy and Ahrens, 1969). These studies essentially have shown a 3-pool model of the turnover of the sterol which can be altered by diet and drugs.
2.6. Pharmacoepidemiology
Information about biologic mechanisms accounting for the relationship of serum concentrations of PFOA and PFOS to serum lipid concentrations may be obtained by examining the concentration of PFOA or PFOS in serum among humans taking certain drugs, compared with those not taking the drug. One man with unusually high serum concentrations of PFAS self-administered treatment with the bile acid sequestrant (BAS) cholestyramine or, at separate times, either of two herbal supplements (zeolites or saponin compounds). Cholestyramine increased fecal concentrations of PFOA and PFOS and decreased serum concentrations. With respect to herbal treatments, after treatment with zeolite PFOS was detectable in stool, and after treatment with saponins PFOA was detectable in stool (Genuis et al., 2010). Cholestyramine increased fecal elimination of PFOA and PFOS in rats, and binding of PFOA and PFOS to cholestyramine was documented (Johnson et al., 1984). In addition, oral contraceptive use has been associated with higher serum concentrations of PFOA and PFOS, probably due to the associated decrease in menstrual flow volume (Ngueta et al., 2017; Rush et al., 2018).
Due to the limited data available on PFAS concentrations in relation to use of pharmacologic agents, we analyzed U.S. NHANES data on concentrations of PFAS and prescription drug use from 2003 to 2016. The main finding was that use of bile acid sequestrants was associated with a reduction in serum PFAS concentrations (Table 1). Statins did not reduce PFAS concentrations – in fact for PFOS a small increase was observed among statin users (p = 0.001). Ezetimibe, a drug that inhibits absorption of cholesterol and related phytosterols in the brush border of the small intestine had no association. Too few people took orlistat or probenecid to have confidence in the results. Selected aspects of these results will be discussed further, below.
Table 1.
Adjusted regression coefficients and corresponding percent difference (Δ) in serum PFAS concentration (ng/mL) among those who have used prescription medications in the past 30 days, by drug or drug class, based on NHANES data from 2003–2016 (n = 14,609)*.
| PFOA |
PFOS |
||||||
|---|---|---|---|---|---|---|---|
| Drug or drug class | N used | β | %Δ | p | β | %Δ | p |
|
| |||||||
| Ezetimibe | 85 | 0.08 | 0.3 | 0.38 | 0.02 | 0.4 | 0.86 |
| Ezetimibe + Simvastatin | 71 | 0.01 | 0.0 | 0.92 | 0.12 | 2.4 | 0.17 |
| Statins | 707 | 0.07 | 0.3 | 0.09 | 0.14 | 2.9 | 0.001 |
| Bile Acid Sequestrants† | 22 | −0.36 | −1.3 | 0.009 | −1.59 | −15.1 | 0.0003 |
| Colesevelam | 10 | −0.48 | −1.6 | < 0.0001 | −2.46 | −17.4 | < 0.0001 |
| Probenecid | 5 | −0.05 | −0.2 | 0.81 | −0.26 | −0.8 | 0.37 |
Regression coefficients adjusted for age, sex, ethnicity, year, and year2 from a model of ln(PFAS). The difference in serum PFAS (ng/mL) is for a white male aged 50 y in 2010. The change in PFAS was calculated as, for PFOS, −(19−(19*eβ)), and for PFOA, −(4.2−(4.2*eβ)), where the approximate mean PFOS is 19 ng/ml and the approximate mean for PFOA is 4.2 ng/mL. NHANES sampling parameters were used in the analysis, making the results generalizable to the United States population. Statins include atorvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, simvastatin, and pitavastatin. Too few people (< 5) used orlistat for the results to be meaningful.
Bile acid sequestrants include cholestyramine, colesevelam, and colestipol.
3. Synopsis of discussions at the workshop
Multiple epidemiological studies have reported associations between PFAS and increases in plasma cholesterol. Large differences in exposure to PFAS are associated, on average, with modest increases in cholesterol (about 10 mg/dL). The relationship between serum cholesterol and PFAS body burden shows a steep slope at lower body burdens and a shallower slope at higher body burdens. The shape of the relationship is remarkably consistent across studies even though the average ranges of exposures in different populations – workers, residents in contaminated areas around production plants and the general population - vary considerably.
The expert workshop provided an opportunity for cross-disciplinary discussions on toxicokinetics of PFAS and physiological control of plasma cholesterol, including lipid/lipoprotein processing. The primary focus was to evaluate whether PFAS might affect cholesterol synthesis and metabolism or whether cholesterol metabolic processes might alter PFAS disposition. During the meeting, a number of ideas surfaced about possible mechanisms of interactions leading to discussions of the evidence for the mechanisms and whether the proposed interactions were consistent with knowledge about cholesterol disposition or of the kinetics of PFAS. The discussions highlighted the need to better understand processes affecting PFAS kinetics in humans, especially uptake from the GI tract and fecal elimination, and independent of any PFAS exposures, the processes that control plasma cholesterol in human populations. In this short overview, we note four possible types of hypotheses for the association of PFAS and LDL-cholesterol and for each we consider (1) the details of the specific hypothesis, (2) consistency of the proposed mechanism with the known biology of pathways affected by PFAS and (3) where appropriate, the consistency of expected dose-response for the hypothesized mechanism with available knowledge. There were broad discussions of possible studies/data analysis that could illuminate reasons for the observed correlations between increased cholesterol and PFAS. Here, for each of the hypotheses, we highlight key ideas developed at the workshop in relation to the hypothesis/hypotheses that would be examined by a specific study or analysis (Table 2).
Table 2.
List of studies and analyses proposed by workshop attendees, sorted by experimental vs. epidemiologic.
| 1) | Experiments that are human-relevant, either in vitro or using humanized mice (e.g., with human liver tissues), using a wide dose range relevant to humans |
| 2) | Experiments to determine whether PFAS, within the range of concentrations experienced by humans, affect the fluidity of cell membranes |
| 3) | Experiments to determine how PFAS affect the expression of genes in the human enterocyte |
| 4) | Experiments to examine the molecular targets and mechanism of action of PFAS in human enterocytes |
| 5) | Experiments to examine the activity of enzymes involved in lipid metabolism in relation to PFAS concentrations |
| 6) | Experiments to determine if PFAS displace fatty acids from fatty acid binding proteins and if this perturbs lipid metabolism |
| 7) | Experiments to examine whether transcriptional targets of PFAS in human hepatocytes vary by periportal vs centrilobular subtype |
| 8) | Experiments on single cells to evaluate effects of PFAS on transcriptomics |
| 9) | Experiments, possibly using human tissues or everted gut sacs, to examine the correlation of bile acid and PFAS transport or absorption |
| 10) | Development of a pharmacokinetic model of cholesterol metabolism that could be used to examine interactions with PFAS |
| 11) | Epidemiologic study to examine serum biomarkers of CYP7A1, such as 7α-hydroxy-4-cholesten-3-one, bile acids, and fibroblast growth factor 19 in relation to PFAS exposure (Gälman et al., 2003; Liu et al., 2020; Palmer et al., 2018) |
| 12) | Epidemiologic studies to examine dietary determinants of PFAS serum concentrations in greater detail than has been done to date, and evaluate if diet could confound the cholesterol-PFAS relationship |
| 13) | Epidemiologic study to examine the correlation of PFAS exposure and specific bile acids (preferably in serum rather than feces) |
| 14) | Epidemiologic study to evaluate whether the composition of sterols quantified as serum cholesterol changes as a function of serum PFAS concentration |
| 15) | Analyses of existing human study specimens to examine lipid profiles in relation to PFAS exposure in greater detail |
| 16) | Epidemiologic study to examine the gut microbiome in fecal samples in relation to PFAS exposure |
| 17) | Epidemiologic study to examine whether markers of steatosis (e.g., ALT, AST) are related to PFAS exposure |
| 18) | Analyses of existing human epidemiology data to see if orlistat or ezetimibe use is related to serum PFAS concentration |
| 19) | Epidemiologic study or analysis of existing data to examine if genetic variation in PFAS-sensitive receptors or transporters is related to serum cholesterol (e.g., genome-wide association study) |
3.1. Direct causality – PFAS associated biological pathways affecting cholesterol metabolism
PFAS have biological activity. They activate PPARα and increase enzymes involved in fatty acid oxidation. In four of the six statistically significant studies in rodents, doses of PFAS sufficient to activate PPARα led to decreases, not increases, in plasma cholesterol. However, in the other two studies increases in serum cholesterol were found (Rebholz et al., 2016; Schlezinger et al., 2020). In Rebholz et al. (2016) the increased cholesterol appeared to be in the “large HDL” fraction and they noted that additional studies would be needed (e.g., measurement of ApoB) to know what type of particle the excess cholesterol was in. In Schlezinger et al. (2020) the PFOA effect on total cholesterol, present in males but not females, appeared to be mostly due to increased HDL-cholesterol, though the increase in LDL-cholesterol was larger than expected. But as noted above, additional studies would be needed to confirm the type of lipoprotein affected. In non-human primates and the clinical trial (described above) such doses were associated with a decrease or no change in cholesterol. In none of the observational epidemiological cohorts were exposures considered sufficiently great to cause activation of PPARα and fatty acid oxidation pathways.
Studies using human hepatocytes suggested that exposure to PFOA at 130 μg/mL (about the highest level reported in occupational studies) decreases HNF4A expression, leading to downregulation of CYP7A1 (Beggs et al., 2016)(see Supplementary Material Section 2, which discusses the comparison of in vitro to in vivo dose metrics). In whole animal mouse studies, PFOA at 48 μg/mL serum decreased expression of hepatic CYP7A1 (Schlezinger et al., 2020). This downregulation, by slowing the conversion of cholesterol to bile acids, could cause an increase in cholesterol levels. While this mechanism could account for the small positive slope relating PFOA with serum cholesterol in occupational studies (Beggs et al., 2016), it may not explain the larger positive slope in those with background-range exposure; higher concentrations may be needed to decrease HNF4A, though additional data on the dose-response would be useful). The comparability of effective concentration in in vitro studies, however, could use further evaluation as the protein content of cell culture media can be markedly different than in serum and the exposure scenario in cell culture models (high dose, days) is different than in humans (low dose, long term) (see Supplementary Material, Section 2). Furthermore, the absence of evidence for changes in cholesterol in the clinical trial is difficult to reconcile with this potential mechanism, though the relevance of the trial results is debatable because all subjects had an advanced solid tumor refractory to standard treatment (Convertino et al., 2018).
Several studies were discussed for examining direct causality including obtaining relevant information regarding: (1) novel mechanistic responses beyond what have already been identified (e.g., PPARα, CAR, PXR), (2) possible differential regulation of cholesterol kinetics between rodents and humans, (3) aspects of PFAS disposition, and (4) possible alternative biomarkers from experimental animals using dose-response studies that could include overtly toxic doses. For instance, in animals lacking PPARα, activation of other nuclear receptors could be further studied. Could biomarkers be identified from these animals that might support follow-up examination of non-PPARα mediated responses? The utility of additional rodent studies, however, was questioned because of the fundamental differences in cholesterol metabolism noted earlier. However, a study appearing since the workshop (Schlezinger et al., 2020) suggests that experiments using mice that express hPPARα, given human-like diets, may be useful. Steatosis is also observed in animal studies. In humans with non-alcoholic fatty liver disease, an increased ratio of total to HDL cholesterol has been reported (Wu et al., 2016). A cholesterol-PFAS association among humans with no suggestion of liver disease would weigh against the “mediated-by-steatosis” hypothesis. Would there be value in examining steatosis in the epidemiological studies to see if this endpoint showed associations with PFAS levels? The challenges of such human studies were noted, due to the expense of sufficiently sensitive and specific diagnostic tests. There might also be other responses identified in rodents that might be preferentially found in the higher exposure groups in the occupational studies. Furthermore, more detailed studies of PFAS distribution could be conducted. Additional post-mortem data in humans would be useful, as would an examination of bioavailability following oral doses and the influence of dietary constituents on bioavailability in a number of species. These in-life protocols would also allow examination of metabolomics/lipidomics responses to PFAS, provide a more complete characterization of PFAS in blood and lipoprotein compartments and potentially identify new lipid biomarkers for examining possible responses to PFAS. Although the liver is probably the primary site for responses to PFAS, examination of transcriptomic enrichment and some protein-level responses of enterocytes to PFAS, either in vitro or in vivo, could help illuminate possible interactions of intestinal uptake/excretion of PFAS and bile acids. More quantitative information regarding transporter abundance in enterocytes and liver would also provide opportunities to develop PK models of PFAS with more detail regarding intestinal absorption and fecal elimination. These transporter studies in human and animal cells could be designed to assess binding affinities, maximum transport velocities and interactions of PFAS transporting proteins with bile acid transporters and improve the PK model structure for PFAS.
The detailed interactions between PFAS and human cells at human relevant exposures, i.e. concentrations consistent with those seen in high exposure occupational cohorts, could be expanded to develop refined dose-response information and to include studies in both hepatocytes and enterocytes. 3D tissue cultures might be useful if challenges associated with determining the free concentration of the chemical in the media (as opposed to the nominal concentration) can be overcome (Groothuis et al., 2015).
Measuring the specific sterols such as plant sterols that are included in estimates of serum cholesterol, and examining these in relation to PFAS exposure could be useful to understand the nature of cholesterol included in the total cholesterol measurements.
The dose-response effects of PFAS on membrane fluidity would be useful to examine, because fluidity may influence activation of SREBP family members resulting in activation of SREBP2 and increased cholesterol synthesis (Levental et al., 2020).
3.2. Reverse causality due to PFAS-carrying capacity of LDL – packaging PFAS with serum lipids
Another possible mechanism for the association could be incorporation of PFAS into cholesterol containing particles such as LDL. PFAS would then increase proportionally with the LDL across any population. This relationship could explain the similar dose responses for the various populations, though the changes in cholesterol are proportionally much smaller than the contrasts in PFAS. However, studies carried out to measure PFAS in various pools within blood lipid fractions indicated that little PFAS was carried along with lipids and instead PFAS are primarily bound to plasma proteins (Butenhoff et al., 2012). Also, medium chain-length, unesterified, saturated fatty acids, that would be most similar in size and properties to PFAS are not known constituents of LDL particles (Iglesias et al., 1996; Lecerf et al., 1993). Instead, the cholesteryl esters in LDL contain longer chain polyunsaturated (linoleic and eicopentaenoic acids) and saturated fatty acids (palmitic and stearic). Also, it was noted that use of statins to decrease cholesterol and lipoproteins was not associated with reduced serum PFAS concentrations (Table 1), weighing against this hypothesis.
Studies related to the co-distribution hypothesis: There is some information on the distribution of PFAS in plasma protein fractions after the addition of PFAS to blood in vitro. Follow-up in vivo studies with repeated PFAS dosing might show differences in this distribution in treatments where there has been time for tissue uptake, processing, export and movement through blood to both liver and kidney for biliary and urinary excretion, respectively.
3.3. Confounding by disease
If a portion of a population has a disease that is accompanied by an increase in serum cholesterol and if that disease also increases serum PFAS, this interaction could account for the cholesterol-PFAS association. For example, hypothyroidism is often accompanied by an increase in cholesterol (Elder et al., 1990) and a decrease in glomerular filtration rate (Asvold et al., 2011), which would decrease excretion of PFAS (Dzierlenga et al., 2020). Kidney failure is often accompanied by an increase in serum cholesterol (Krane and Wanner, 2007), and, with the decrease in glomerular filtration rate, serum PFAS should also increase. When a multivariate model of cholesterol in relation to PFAS was adjusted for either thyroid stimulating hormone or estimated glomerular filtration rate using multiple years of data from the National Health and Nutrition Examination Survey (NHANES), however, adjustment had no effect on the coefficients for the PFAS terms (not shown), which does not support this confounding by kidney disease hypothesis. A related issue is the adverse effect of elevated cholesterol decreasing glomerular filtration rate (Athyros et al., 2007). (This would be reverse causality rather than confounding.) In any event, analysis of NHANES data do not support this possibility (not shown).
Studies related to confounding by disease: There are studies showing associations between PFAS and uric acid (Steenland et al., 2010). There could be value in evaluating whether hyperuricemia confounds the PFAS-cholesterol association, though the PFAS-uric acid association itself may be due to confounding by glomerular filtration rate.
3.4. Confounding by common pharmacokinetic processes that alter both cholesterol and PFAS kinetics
The possibility of confounding by pharmacokinetic processes received more attention than the other three hypotheses in discussions at the meeting – emphasizing characteristics of PFAS kinetics and cholesterol disposition that might share common pathways. It was recognized that the PK models for PFAS emphasize renal excretion and tissue partitioning as the dominant factors affecting tissue time courses (Dzierlenga et al., 2020; Chou and Lin, 2019; and Worley et al., 2017). These PK models have not considered the possibility of variable percentage absorption across individuals in a population. Correlated net absorption or excretion of cholesterol or bile salts and PFAS in the gut could give rise to the association of cholesterol and PFAS in blood. A few examples are included below.
Several follow-on studies of possible confounding in the relationship of cholesterol with PFAS were discussed. Correlated absorption of bile salts or cholesterol and PFAS could occur in enterocytes. Although most cholesterol is synthesized in the body, dietary cholesterol can incrementally increase plasma levels. Ezetimibe inhibits uptake of cholesterol by enterocytes but has no effect on PFAS serum concentration (Table 1); this observation weighs against a correlation with cholesterol absorption itself. It has been demonstrated that several bile acid transporters expressed in enterocytes and hepatocytes can also transport PFAS (Zhao et al., 2015, 2017). These results suggest that PFAS could be trapped within the enterohepatic circulation, contributing in part to long half-lives. However, given the high concentrations of conjugated bile acids in the small intestine after a meal (up to 10 mM according to Hofmann and Eckmann (2006), bile acids likely affect the absorption of PFAS, at least during digestion. Potential co-modulation of the kinetics of bile acids and PFAS at these specific transporters, e.g., by cholesterol, as has been shown for rat Na+/taurocholate cotransporting polypeptide (Molina et al., 2008) needs to be examined quantitatively in hepatocytes, enterocytes or using everted gut preparations.
Correlated excretion of PFAS and bile salts or cholesterol is also conceivable. Such correlation due to agents in the gut lumen could cause the association of cholesterol and PFAS in serum. For example, bile acid sequestrants increase excretion of bile acids, and thereby decrease serum cholesterol. Bile acid sequestrants also decrease serum PFAS concentrations (Table 1) because they bind PFAS directly and then get eliminated in feces. While bile acid sequestrants do not account for the association of serum cholesterol and PFAS in epidemiologic studies, dietary constituents in the gut could act similarly. Both PFAS and cholesterol could be correlated with the same dietary constituent and have dose response curves for each that were inverse Michaelis-Menten. For instance,
A plot of cholesterol to PFAS would then have a shape similar to that observed in the various epidemiological populations and represent a confounding effect with the dietary constituent affecting both PFAS and cholesterol and this would be expected to be present in various populations regardless of the level of PFAS in plasma. There would be considerable value in developing simulation models that reflect expected dose-response in simulated epidemiological populations based on different proposed interactions of plasma PFAS and lipids.
Studies related to mechanistic investigations of confounding: Confounding occurs when a secondary variable affects both the biomarker and health outcome examined in the epidemiological study. In essence either of two scenarios – common processes that affect either biliary excretion or that affect intestinal absorption - could produce associations of the kind seen across the epidemiological studies. There was significant discussion among participants about factors that might affect absorption or elimination of cholesterol/cholesterol precursors and PFAS and how these factors might be studied. In addition to cell-based studies of interactions of transport of PFAS and bile acids/cholesterol, other work might be pursued with everted gut preparations to assess uptake from a more intact tissue. A more detailed PK model for bile acid uptake, circulation and excretion could also be developed and be designed to link bile acid disposition with key characteristics of processes affecting cholesterol and interactions between bile acid and PFAS transporters. Such models could be used to develop a more mechanistic analysis of confounding or pharmacokinetic bias. More comprehensive investigations of the relation of diet to serum PFAS concentrations should be helpful in examining possible confounding in these associations. This particular recommendation would represent a longer-term investment to bear fruit.
4. Recommendations for studies to further examine the basis for associations seen between cholesterol and PFAS in human populations
The suggested studies (Table 2) can be organized into 3- broad categories: a) biology associated with possibilities of direct causation, b) pharmacokinetic factors affecting PFAS and cholesterol levels, and c) epidemiologic evaluations. However, the information obtained from studies in any one of these categories would have broader utility. For instance, studies regarding new biology of PFAS-responsive receptors could as easily provide information on PK interactions as on direct causality or confounding. Suggested epidemiological analyses probe various possible interactions that might occur between PFAS, and various forms of cholesterol, disease states (steatosis) or genetic variability in transporters and could provide information for enhancing PK models for interactions between PFAS and cholesterol.
Pharmacokinetic factors affecting PFAS and cholesterol levels: To date most PK modeling has assumed nearly complete absorption of orally administered PFAS and excretion occurring only via urine. Persistence of these compounds has been modeled by efficient resorption of PFAS from the kidney filtrate. Discussions at the workshop, especially in relation to interactions between PFAS and cholesterol, highlighted the need to have better accounting of any role for uptake processes from the gut and biliary excretion that could jointly move some amounts of PFAS and cholesterol. A high priority need was extension of PK models to include biliary excretion and intestinal absorption of PFAS and linking them to models of cholesterol homeostasis including processing of bile acids. Further development of PK models for PFAS should be designed to account for coordinated transport of cholesterol in specific lipoproteins. Among experimental studies that would be useful for these models are studies examining transporter protein activity toward PFAS and different forms of cholesterol, including interaction studies looking at combination treatment with these two groups of compounds. Studies in hepatocytes or everted gut preparations could examine kinetic interactions between PFAS, cholesterol and bile acids. In concert with work to improve oral absorption and fecal elimination descriptions of PFAS, further work to develop/improve models for cholesterol homeostasis for bile acid excretion and resorption could aid in understanding possible interactions between bile acids and PFAS. Clearly, models for cholesterol homeostasis are considerably more complex than those for xenobiotics and the models pursued would need to be purposeful simplifications of the multiple control processes involved with cholesterol and lipoproteins.
Biology associated with possibilities of direct causation: Most of the discussion about possibilities of direct causality was targeted to increasing knowledge of the possible modes of action of PFAS on lipid metabolism, on responses in tissues other than liver and in more human-relevant models in order to determine if any particular hypotheses related to direct causality would be better supported by new data and knowledge. For instance, more detailed studies of effects on PFAS on lipid metabolism could be pursued in vitro in primary human hepatocytes or using humanized mice with human PPARα. Mechanistically, information might be gained from studies of this kind over a wide dose range but the more relevant information would be from doses consistent with exposure for the various human cohorts examined for the association between cholesterol and PFAS. While the liver has been the focus of most attention in considering the possible connections between PFAS targets and cholesterol, the workshop highlighted the need for better understanding of the responses of enterocytes to PFAS and how responses in enterocytes might alter PFAS and cholesterol transport. Enterocyte responses to PFAS could be gauged by in vitro exposures in human cells or in life exposures in animals with detailed gene expression analysis in enterocytes across a wide dose range with targeted PFAS compounds. Everted gut sacs could evaluate transport of PFAS and cholesterol and determine if there were relevant interactions in these transport processes at PFAS doses in the range of those experienced in human populations. The studies in enterocytes could also provide key information to enhance PK models for PFAS and include more detail on oral uptake/intestinal elimination of PFAS.
Other biochemical/toxicological studies were considered, including assessing effects of PFAS on activity of key enzymes in lipid metabolism, such as cholesteryl ester synthesis, effects of PFAS on membrane-fluidity at epidemiologically relevant PFAS concentrations, determining regional differences in cellular responses for centrilobular and periportal regions of the liver and assessing displacement of normal fatty acids from fatty acid binding protein by PFAS.
Evaluation of PFAS effects on liver genes expression has focused on responses in whole liver or in isolated hepatocytes. Some studies of responses in rat and human hepatocytes and rat liver have appeared for GW7647, a PPARα specific ligand (McMullen et al., 2020). While responses in specific regions of the intact liver were examined for this ligand, they have only been reported to date in a poster/abstract format (Andersen et al., 2019). No regionally specific results have been reported for PFAS. New technologies using single cell transcriptomics have measured differential expression patterns of multiple liver-resident cell types in humans and mice. These technologies could be employed to determine the effects of PFAS on other cell types or, with existing data sets, to determine the distribution of PPARα expression in these other cell types (Halpern et al., 2017; MacParland et al., 2018) and examine differences in PPARα pathway components in not just periportal and centrilobular hepatocytes but in all the liver resident cell types.
Epidemiological Evaluations: Because laboratory evidence suggests that higher concentrations of PFAS may down-regulate CYP7A1 activity (Beggs et al., 2016; Behr et al., 2020a; Schlezinger et al., 2020), it would be of interest to conduct an epidemiologic study to examine serum biomarkers of CYP7A1 (7α-hydroxy-4-cholesten-3-one (C4), specific bile acids, and fibroblast growth factor 19) in relation to PFAS exposure (Gälman et al., 2003; Liu et al., 2020; Palmer et al., 2018), especially in occupationally-exposed groups where some subjects have high serum levels, e.g., 10 μg/mL or more PFOA or PFOS.
Studies, particularly two appearing after the workshop, indicate that dietary fiber could confound the cholesterol-PFAS association (Lin et al., 2020; Dzierlenga et al., 2021). (These recent epidemiologic studies also suggest related animal experiments may be useful.) To obtain more high-quality data on the relation of diet to PFAS, and on whether uncontrolled confounding by diet might account for some of the cholesterol-PFAS association, large epidemiologic studies are needed, e. g., with 7 days of diet recalls, which may provide a better dietary measure of fiber than a food-frequency questionnaire.
An epidemiologic study to examine correlation of PFAS exposure and specific bile acids (preferably in serum rather than feces) – which is a slightly different concept than described above – could be usefully done in a background-exposed population and provide mechanistic data that could articulate with experimental data on the nature of interactions of bile acids and PFAS during transport and absorption.
If PFAS increased the absorption of plant sterols, this could result in an association of elevated total serum cholesterol and serum PFAS. Although it makes sense to verify whether the apparent PFAS-related increased in serum cholesterol quantified by standard assays is truly due to increased cholesterol, there is no evidence indicating that PFAS might alter the absorption of plant sterols.
By analyzing existing human specimens to examine lipid profiles in relation to PFAS exposure in greater detail, it may also be possible to address the odd finding that total cholesterol is more consistently related to PFAS than is LDL cholesterol. In standard epidemiologic studies, LDL cholesterol is not measured directly.
With an epidemiologic study to examine gut microbiome in relation to PFAS exposure, it may be possible to address whether PFAS affect microbial populations directly (Iszatt et al., 2019), or, if combined with a high-quality diet assessment, the microbial population merely reflects dietary factors that also affect PFAS concentrations. (Animal experiments may also be useful to address this point.)
An epidemiologic study to examine whether steatosis is related to PFAS exposure would address the possibility that increased cholesterol is secondary to PFAS-induced steatosis. However, as noted above, questions exist about the sensitivity, specificity, and practicality of diagnostic tests for steatosis, and furthermore, the inconsistent, subtle relation of liver function tests to PFAS has not strongly supported liver disease as a PFAS-related outcome in humans (EPA, 2016a,b).
Although evidence that ezetimibe is not related to serum PFAS concentration was presented (Table 1), confirmation would be useful, as would data on orlistat, to rule out an effect of these medications on PFAS absorption.
Based on the results of toxicology studies, we have a list of PFAS-sensitive nuclear receptors and PFAS transporters. To see if genetic variation in these receptors and transporters is related to serum cholesterol, the list could be compared with a list of genetic variants know to affect serum cholesterol in genome-wide association studies. A bioinformatics study might also be a useful way to systematically evaluate if the corresponding affected processes overlap.
5. Conclusion
The mechanisms underlying the association of serum cholesterol with exposure to PFAS have not been determined. Experimental studies, e.g., using human-relevant models and that include lower dose ranges could provide valuable mechanistic insights. PK modeling of both PFAS and cholesterol may also provide valuable clues. Epidemiologic studies that address mechanistic hypotheses, e.g., regarding an effect of PFAS on CYP7A1 activity, or that evaluate potential confounding by dietary factors, are among the key recommendations resulting from the workshop.
Supplementary Material
Acknowledgments
Geary Olsen and Sue Chang, both 3M employees, were present at the workshop, as observers. They neither contributed to the writing of the manuscript nor have they reviewed the finished paper.
Funding
This work was funded by 3M.
Abbreviations:
- ABST
apical bile salt transporter
- BAS
bile acid sequestrant
- CAR
constitutive androstane receptor
- CETP
cholesteryl ester transfer protein
- CYP7A1
cholesterol 7α-hydroxylase
- C4
7α-hydroxy-4-cholesten-3-one
- FXR
farnesoid X receptor
- HDL
high density lipoprotein
- HMGCR
3-hydroxy-3-methyl-glutaryl-Coenzyme A Reductase
- HNF4α
hepatic nuclear factor 4-alpha
- LDL
low density lipoprotein
- LXR α
liver X receptor alpha
- NHANES
National Health and Nutrition Examination Survey
- NPC1L1
Niemann-Pick C1 Like 1
- PCSK9
proprotein convertase subtilism/kexin type 9
- PFAS
perfluoroalkyl substances
- PFOA
perfluorooctanoic acid
- PFOS
perfluorooctane sulfonic acid
- PK
pharmacokinetic
- PPARα
peroxisome proliferator-activated receptor alpha
- PPARγ
peroxisome proliferator-activated receptor gamma
- PXR
pregnane X receptor
- SREBP2
sterol regulatory element-binding protein 2
Footnotes
Declaration of Competing Interest
3M was not involved in the preparation of the manuscript. The authors retained sole control of the manuscript content and the findings, and statements in this paper are those of the authors and not those of the author’s employer or the sponsors. No authors were directly compensated by 3 M. This project was funded through a contract between 3 M and Ramboll, an international science and engineering company that provided salary compensation to the authors. None of the authors are currently engaged to testify as experts on behalf of the sponsors in litigation related to the compound discussed in this manuscript. Dr Fletcher has served in the past as a paid consultant to law firms conducting litigation involving PFAS, for both plaintiffs and defendants. The information in this document has been subjected to review by the Center for Public Health and Environmental Assessment, Office of Research and Development, EPA and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Postscript
Since the workshop many relevant papers have appeared; a few have been cited above; the others of special note that we have come across are listed here: Behr et al. (2020b), Fragki et al. (2021), and Salihović et al. (2020).
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.tox.2021.152845.
References
- Abe T, Takahashi M, Kano M, Amaike Y, Ishii C, Maeda K, Kudoh Y, Morishita T, Hosaka T, Sasaki T, et al. , 2017. Activation of nuclear receptor CAR by an environmental pollutant perfluorooctanoic acid. Arch. Toxicol. 91 (6), 2365–2374. 10.1007/s00204-016-1888-3. [DOI] [PubMed] [Google Scholar]
- Afonso MS, Machado RM, Lavrador MS, Quintao ECR, Moore KJ, Lottenberg AM, 2018. Molecular pathways underlying cholesterol homeostasis. Nutrients 10 (6). 10.3390/nu10060760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alphonse PAS, Jones PJH, 2016. Revisiting human cholesterol synthesis and absorption: the reciprocity paradigm and its key regulators. Lipids 51 (5), 519–536. 10.1007/s11745-015-4096-7. [DOI] [PubMed] [Google Scholar]
- Andersen ME, Butenhoff JL, Chang S-C, Farrar DG, Kennedy GL, Lau C, Olsen GW, Seed J, Wallace KB, 2008. Perfluoroalkyl acids and related chemistries–toxicokinetics and modes of action. Toxicol. Sci. 102 (1), 3–14. 10.1093/toxsci/kfm270. [DOI] [PubMed] [Google Scholar]
- Andersen ME, Black MB, Pendse SN, Clewell RA, LeCluyse EL, McMullen PD, 2019. Zonal-specific transcriptional programs associated with PPARα activation in the rat liver and their role in liver cancer in rodents. Toxicologist 168, 232 (Abstract). [Google Scholar]
- ASTDR, 2018. Toxicological Profile for Perfluoroalkyls, Draft for Public Comment. Jun., p. 852. [Google Scholar]
- Asvold BO, Bjøro T, Vatten LJ, 2011. Association of thyroid function with estimated glomerular filtration rate in a population-based study: the HUNT study. Eur. J. Endocrinol. 164 (1), 101–105. 10.1530/EJE-10-0705. [DOI] [PubMed] [Google Scholar]
- Athyros VG, Kakafika AI, Papageorgiou AA, Pagourelias ED, Savvatianos SD, Elisaf M, Karagiannis A, Tziomalos K, Mikhailidis DP, 2007. Statin-induced increase in HDL-C and renal function in coronary heart disease patients. Open Cardiovasc. Med. J. 1, 8–14. 10.2174/1874192400701010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley BD, Chang S-C, Ehresman DJ, Eveland A, Zitzow JD, Parker GA, Peters JM, Wallace KB, Butenhoff JL, 2017. Perfluorooctane sulfonate-induced hepatic steatosis in male Sprague Dawley rats is not attenuated by dietary choline supplementation. Toxicol. Sci. 160 (2), 284–298. 10.1093/toxsci/kfx185. [DOI] [PubMed] [Google Scholar]
- Bartell SM, Calafat AM, Lyu C, Kato K, Ryan PB, Steenland K, 2010. Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environ. Health Perspect. 118 (2), 222–228. 10.1289/ehp.0901252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs KM, McGreal SR, McCarthy A, Gunewardena S, Lampe JN, Lau C, Apte U, 2016. The role of hepatocyte nuclear factor 4-alpha in perfluorooctanoic acid- and perfluorooctanesulfonic acid-induced hepatocellular dysfunction. Toxicol. Appl. Pharmacol. 304, 18–29. 10.1016/j.taap.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr A-C, Kwiatkowski A, Ståhlman M, Schmidt FF, Luckert C, Braeuning A, Buhrke T, 2020a. Impairment of bile acid metabolism by perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in human HepaRG hepatoma cells. Arch. Toxicol. 94 (5), 1673–1686. 10.1007/s00204-020-02732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr A-C, Plinsch C, Braeuning A, Buhrke T, 2020b. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol. In Vitro 62, 104700. 10.1016/j.tiv.2019.104700. [DOI] [PubMed] [Google Scholar]
- Berry JD, Dyer A, Cai X, Garside DB, Ning H, Thomas A, Greenland P, Van Horn L, Tracy RP, Lloyd-Jones DM, 2012. Lifetime risks of cardiovascular disease. N. Engl. J. Med. 366 (4), 321–329. 10.1056/NEJMoa1012848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijland S, Rensen PCN, Pieterman EJ, Maas ACE, van der Hoorn JW, van Erk MJ, Havekes LM, Willems van Dijk K, Chang S-C, Ehresman DJ, et al. , 2011. Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in APOE*3-Leiden CETP mice. Toxicol. Sci. 123 (1), 290–303. 10.1093/toxsci/kfr142. [DOI] [PubMed] [Google Scholar]
- Bischel HN, Macmanus-Spencer LA, Luthy RG, 2010. Noncovalent interactions of long-chain perfluoroalkyl acids with serum albumin. Environ. Sci. Technol. 44 (13), 5263–5269. 10.1021/es101334s. [DOI] [PubMed] [Google Scholar]
- Bjork JA, Butenhoff JL, Wallace KB, 2011. Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes. Toxicology 288 (1–3), 8–17. 10.1016/j.tox.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Bogdanska J, Borg D, Sundström M, Bergström U, Halldin K, Abedi-Valugerdi M, Bergman A, Nelson B, Depierre J, Nobel S, 2011. Tissue distribution of 35Slabelled perfluorooctane sulfonate in adult mice after oral exposure to a low environmentally relevant dose or a high experimental dose. Toxicology 284 (1–3), 54–62. 10.1016/j.tox.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Bogdanska J, Borg D, Bergström U, Mellring M, Bergman Å, DePierre J, Nobel S, 2020. Tissue distribution of 14C-labelled perfluorooctanoic acid in adult mice after 1–5 days of dietary exposure to an experimental dose or a lower dose that resulted in blood levels similar to those detected in exposed humans. Chemosphere 239, 124755. 10.1016/j.chemosphere.2019.124755. [DOI] [PubMed] [Google Scholar]
- Brantsæter AL, Whitworth KW, Ydersbond TA, Haug LS, Haugen M, Knutsen HK, Thomsen C, Meltzer HM, Becher G, Sabaredzovic A, et al. , 2013. Determinants of plasma concentrations of perfluoroalkyl substances in pregnant Norwegian women. Environ. Int. 54, 74–84. 10.1016/j.envint.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenhoff J, Costa G, Elcombe C, Farrar D, Hansen K, Iwai H, Jung R, Kennedy G, Lieder P, Olsen G, et al. , 2002. Toxicity of ammonium perfluorooctanoate in male cynomolgus monkeys after oral dosing for 6 months. Toxicol. Sci. 69 (1), 244–257. 10.1093/toxsci/69.1.244. [DOI] [PubMed] [Google Scholar]
- Butenhoff JL, Pieterman E, Ehresman DJ, Gorman GS, Olsen GW, Chang S-C, Princen HMG, 2012. Distribution of perfluorooctanesulfonate and perfluorooctanoate into human plasma lipoprotein fractions. Toxicol. Lett. 210 (3), 360–365. 10.1016/j.toxlet.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Calafat AM, Wong L-Y, Kuklenyik Z, Reidy JA, Needham LL, 2007. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ. Health Perspect. 115 (11), 1596–1602. 10.1289/ehp.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S-C, Ehresman DJ, Bjork JA, Wallace KB, Parker GA, Stump DG, Butenhoff JL, 2009. Gestational and lactational exposure to potassium perfluorooctanesulfonate (K+PFOS) in rats: toxicokinetics, thyroid hormone status, and related gene expression. Reprod. Toxicol. 27 (3–4), 387–399. 10.1016/j.reprotox.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Chang S, Allen BC, Andres KL, Ehresman DJ, Falvo R, Provencher A, Olsen GW, Butenhoff JL, 2017. Evaluation of serum lipid, thyroid, and hepatic clinical chemistries in association with serum perfluorooctanesulfonate (PFOS) in Cynomolgus monkeys after oral dosing with potassium PFOS. Toxicol. Sci. 156 (2), 387–401. 10.1093/toxsci/kfw267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez-Talavera O, Tailleux A, Lefebvre P, Staels B, 2017. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 152 (7). 10.1053/j.gastro.2017.01.055, 1679–1694.e3. [DOI] [PubMed] [Google Scholar]
- Chen JY, Levy-Wilson B, Goodart S, Cooper AD, 2002. Mice expressing the human CYP7A1 gene in the mouse CYP7A1 knock-out background lack induction of CYP7A1 expression by cholesterol feeding and have increased hypercholesterolemia when fed a high fat diet. J. Biol. Chem. 277 (45), 42588–42595. 10.1074/jbc.M205117200. [DOI] [PubMed] [Google Scholar]
- Chou W-C, Lin Z, 2019. Bayesian evaluation of a physiologically based pharmacokinetic (PBPK) model for perfluorooctane sulfonate (PFOS) to characterize the interspecies uncertainty between mice, rats, monkeys, and humans: development and performance verification. Environ. Int. 129, 408–422. 10.1016/j.envint.2019.03.058. [DOI] [PubMed] [Google Scholar]
- Claudel T, Staels B, Kuipers F, 2005. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler. Thromb. Vasc. Biol. 25 (10), 2020–2030. 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- Cohen DE, 2008. Balancing cholesterol synthesis and absorption in the gastrointestinal tract. J. Clin. Lipidol. 2 (2), S1–3. 10.1016/j.jacl.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertino M, Church TR, Olsen GW, Liu Y, Doyle E, Elcombe CR, Barnett AL, Samuel LM, MacPherson IR, Evans TRJ, 2018. Stochastic pharmacokinetic-pharmacodynamic modeling for assessing the systemic health risk of perfluorooctanoate (PFOA). Toxicol. Sci. 163 (1), 293–306. 10.1093/toxsci/kfy035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J, Klaunig JE, 2014. Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARa) as a case study. Crit. Rev. Toxicol. 44 (1), 1–49. 10.3109/10408444.2013.835784. [DOI] [PubMed] [Google Scholar]
- Costa G, Sartori S, Consonni D, 2009. Thirty years of medical surveillance in perfluooctanoic acid production workers. J. Occup. Environ. Med. 51 (3), 364–372. 10.1097/JOM.0b013e3181965d80. [DOI] [PubMed] [Google Scholar]
- Cui L, Liao C, Zhou Q, Xia T, Yun Z, Jiang G, 2010. Excretion of PFOA and PFOS in male rats during a subchronic exposure. Arch. Environ. Contam. Toxicol. 58 (1), 205–213. 10.1007/s00244-009-9336-5. [DOI] [PubMed] [Google Scholar]
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, Corton JC, Abbott BD, 2017. Perfluoroalkyl acids-induced liver steatosis: effects on genes controlling lipid homeostasis. Toxicology 378, 37–52. 10.1016/j.tox.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra R, Winquist A, Darrow LA, Klein M, Steenland K, 2017. A study of reverse causation: examining the associations of perfluorooctanoic acid serum levels with two outcomes. Environ. Health Perspect. 125 (3), 416–421. 10.1289/EHP273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierlenga MW, Moreau M, Song G, Mallick P, Ward PL, Campbell JL, Housand C, Yoon M, Allen BC, Clewell HJ, et al. , 2020. Quantitative bias analysis of the association between subclinical thyroid disease and two perfluoroalkyl substances in a single study. Environ. Res. 182, 109017 10.1016/j.envres.2019.109017. [DOI] [PubMed] [Google Scholar]
- Dzierlenga MW, Keast DR, Longnecker MP, 2021. The concentration of several perfluoroalkyl acids in serum appears to be reduced by dietary fiber. Environ. Int. 146, 106292 10.1016/j.envint.2020.106292. [DOI] [PubMed] [Google Scholar]
- EFSA, 2018a. Risk to Human Health Related to the Presence of Perfluorooctane Sulfonic Acid and Perfluorooctanoic Acid in Food. Dec 13 [accessed 2020 Apr 20]. European Food Safety Authority. https://www.efsa.europa.eu/en/efsajournal/pub/5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA, 2018b. Minutes of the Expert Meeting on Perfluooroctane Sulfonic Acid and Perfluorooctanoic Acid in Food Assessment. Dec 10. [Accessed 2020 Apr 20]. EFSA. http://www.efsa.europa.eu/sites/default/files/news/efsa-contam-3503.pdf. [Google Scholar]
- EFSA Panel on Contaminants in the Food Chain (EFSA CONTAM Panel), Schrenk D, Bignami M, Bodin L, Chipman JK, Del Mazo J, Grasl-Kraupp B, Hogstrand C, Hoogenboom LR, Leblanc JC, Nebbia CS, Nielsen E, Ntzani E, Petersen A, Sand S, Vleminckx C, Wallace H, Barregård L, Ceccatelli S, Cravedi JP, Halldorsson TI, Haug LS, Johansson N, Knutsen HK, Rose M, Roudot AC, Van Loveren H, Vollmer G, Mackay K, Riolo F, Schwerdtle T, 2020. Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J 18, e06223–9. 10.2903/j.efsa.2020.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder J, McLelland A, O’Reilly DS, Packard CJ, Series JJ, Shepherd J, 1990. The relationship between serum cholesterol and serum thyrotropin, thyroxine and triiodothyronine concentrations in suspected hypothyroidism. Ann. Clin. Biochem. 27 (Pt 2), 110–113. 10.1177/000456329002700204. [DOI] [PubMed] [Google Scholar]
- EPA, 2016a. Health Effects Support Document for Perfluorooctane Sulfonate (PFOS), p. 245. [Google Scholar]
- EPA, 2016b. Health Effects Support Document for Perfluorooctanoic Acid (PFOA), p. 322. [Google Scholar]
- Fragki S, Dirven H, Fletcher T, Grasl-Kraupp B, Bjerve Gutzkow K, Hoogenboom R, Kersten S, Lindeman B, Louisse J, Peijnenburg A, et al. , 2021. Systemic PFOS and PFOA exposure and disturbed lipid homeostasis in humans: what do we know and what not? Crit. Rev. Toxicol. 15, 1–24. 10.1080/10408444.2021.1888073. [DOI] [PubMed] [Google Scholar]
- Franko J, Meade BJ, Frasch HF, Barbero AM, Anderson SE, 2012. Dermal penetration potential of perfluorooctanoic acid (PFOA) in human and mouse skin. J. Toxicol. Environ. Health A 75 (1), 50–62. 10.1080/15287394.2011.615108. [DOI] [PubMed] [Google Scholar]
- Fromme H, Tittlemier SA, Völkel W, Wilhelm M, Twardella D, 2009. Perfluorinated compounds–exposure assessment for the general population in Western countries. Int. J. Hyg. Environ. Health 212 (3), 239–270. 10.1016/j.ijheh.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Gachumi G, El-Aneed A, 2017. Mass spectrometric approaches for the analysis of phytosterols in biological samples. J. Agric. Food Chem. 65 (47), 10141–10156. 10.1021/acs.jafc.7b03785. [DOI] [PubMed] [Google Scholar]
- Gälman C, Arvidsson I, Angelin B, Rudling M, 2003. Monitoring hepatic cholesterol 7alpha-hydroxylase activity by assay of the stable bile acid intermediate 7alpha-hydroxy-4-cholesten-3-one in peripheral blood. J. Lipid Res. 44 (4), 859–866. 10.1194/jlr.D200043-JLR200. [DOI] [PubMed] [Google Scholar]
- Geiger SD, Xiao J, Ducatman A, Frisbee S, Innes K, Shankar A, 2014. The association between PFOA, PFOS and serum lipid levels in adolescents. Chemosphere 98, 78–83. 10.1016/j.chemosphere.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Genuis SJ, Birkholz D, Ralitsch M, Thibault N, 2010. Human detoxification of perfluorinated compounds. Public Health 124 (7), 367–375. 10.1016/j.puhe.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS, 2009. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29 (4), 431–438. 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman DS, Smith FR, Seplowitz AH, Ramakrishnan R, Dell RB, 1980. Prediction of the parameters of whole body cholesterol metabolism in humans. J. Lipid Res. 21 (6), 699–713. [PubMed] [Google Scholar]
- Groothuis FA, Heringa MB, Nicol B, Hermens JLM, Blaauboer BJ, Kramer NI, 2015. Dose metric considerations in in vitro assays to improve quantitative in vitro-in vivo dose extrapolations. Toxicology 332, 30–40. 10.1016/j.tox.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Ahrens EH, 1969. Measurements of cholesterol turnover, synthesis, and absorption in man, carried out by isotope kinetic and sterol balance methods. J. Lipid Res. 10 (1), 91–107. [PubMed] [Google Scholar]
- Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, et al. , 2017. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542 (7641), 352–356. 10.1038/nature21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada KH, Hashida S, Kaneko T, Takenaka K, Minata M, Inoue K, Saito N, Koizumi A, 2007. Biliary excretion and cerebrospinal fluid partition of perfluorooctanoate and perfluorooctane sulfonate in humans. Environ. Toxicol. Pharmacol. 24 (2), 134–139. 10.1016/j.etap.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Hinderliter PM, Han X, Kennedy GL, Butenhoff JL, 2006. Age effect on perfluorooctanoate (PFOA) plasma concentration in post-weaning rats following oral gavage with ammonium perfluorooctanoate (APFO). Toxicology 225 (2–3), 195–203. 10.1016/j.tox.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Hofmann AF, Eckmann L, 2006. How bile acids confer gut mucosal protection against bacteria. PNAS 103 (12), 4333–4334. 10.1073/pnas.0600780103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF, Molino G, Milanese M, Belforte G, 1983. Description and simulation of a physiological pharmacokinetic model for the metabolism and enterohepatic circulation of bile acids in man. Cholic acid in healthy man. J. Clin. Invest. 71 (4), 1003–1022. 10.1172/jci110828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS, 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109 (9), 1125–1131. 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde M, Martin JW, Letcher RJ, Solomon KR, Muir DCG, 2006. Biological monitoring of polyfluoroalkyl substances: a review. Environ. Sci. Technol. 40 (11), 3463–3473. 10.1021/es052580b. [DOI] [PubMed] [Google Scholar]
- Huang B, Song B-L, Xu C, 2020. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat. Metab. 2 (2), 132–141. 10.1038/s42255-020-0174-0. [DOI] [PubMed] [Google Scholar]
- Hundley SG, Sarrif AM, Kennedy GL, 2006. Absorption, distribution, and excretion of ammonium perfluorooctanoate (APFO) after oral administration to various species. Drug Chem. Toxicol. 29 (2), 137–145. 10.1080/01480540600561361. [DOI] [PubMed] [Google Scholar]
- Iglesias A, Arranz M, Alvarez JJ, Perales J, Villar J, Herrera E, Lasunción MA, 1996. Cholesteryl ester transfer activity in liver disease and cholestasis, and its relation with fatty acid composition of lipoprotein lipids. Clin. Chim. Acta 248 (2), 157–174. 10.1016/0009-8981(95)06251-3. [DOI] [PubMed] [Google Scholar]
- Iszatt N, Janssen S, Lenters V, Dahl C, Stigum H, Knight R, Mandal S, Peddada S, González A, Midtvedt T, et al. , 2019. Environmental toxicants in breast milk of Norwegian mothers and gut bacteria composition and metabolites in their infants at 1 month. Microbiome 7 (1), 34. 10.1186/s40168-019-0645-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Gibson SJ, Ober RE, 1984. Cholestyramine-enhanced fecal elimination of carbon-14 in rats after administration of ammonium [14C]perfluorooctanoate or potassium [14C]perfluorooctanesulfonate. Fundam. Appl. Toxicol. 4 (6), 972–976. 10.1016/0272-0590(84)90235-5. [DOI] [PubMed] [Google Scholar]
- Kemper RA, 2003. Perfluorooctanoic Acid: Toxicokinetics in the Rat. DuPont Haskell Laboratories. Laboratory Project ID: DuPont-7473. USEPA Administrative Record AR-226.1499. [Google Scholar]
- Kennedy GL, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, Biegel LB, Murphy SR, Farrar DG, 2004. The toxicology of perfluorooctanoate. Crit. Rev. Toxicol. 34 (4), 351–384. 10.1080/10408440490464705. [DOI] [PubMed] [Google Scholar]
- Kersten S, 2008. Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Res. 2008, 132960 10.1155/2008/132960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazaee M, Christie E, Cheng W, Michalsen M, Field J, Ng C, 2021. Perfluoroalkyl acid binding with peroxisome proliferator-activated receptors α, γ, and δ, and fatty acid binding proteins by equilibrium dialysis with a comparison of methods. Toxics 9 (3). 10.3390/toxics9030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura O, Fujii Y, Haraguchi K, Kato Y, Ohta C, Koga N, Endo T, 2017. Uptake of perfluorooctanoic acid by Caco-2 cells: involvement of organic anion transporting polypeptides. Toxicol. Lett. 277, 18–23. 10.1016/j.toxlet.2017.05.012. [DOI] [PubMed] [Google Scholar]
- Krane V, Wanner C, 2007. Dyslipidaemia in chronic kidney disease. Minerva Urol. Nefrol. 59 (3), 299–316. [PubMed] [Google Scholar]
- Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J, 2007. Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol. Sci. 99 (2), 366–394. 10.1093/toxsci/kfm128. [DOI] [PubMed] [Google Scholar]
- Lecerf J, Rossignol A, Véricel E, Thiés F, Farnier M, Lagarde M, 1993. Variations in the fatty acid composition of lipid classes from lipoproteins in elderly women. Atherosclerosis 98 (2), 241–249. 10.1016/0021-9150(93)90133-f. [DOI] [PubMed] [Google Scholar]
- Levental KR, Malmberg E, Symons JL, Fan Y-Y, Chapkin RS, Ernst R, Levental I, 2020. Lipidomic and biophysical homeostasis of mammalian membranes counteracts dietary lipid perturbations to maintain cellular fitness. Nat. Commun. 11 (1), 1339. 10.1038/s41467-020-15203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P-ID, Cardenas A, Hauser R, Gold DR, Kleinman KP, Hivert M-F, Fleisch AF, Calafat AM, Sanchez-Guerra M, Osorio-Yáñez C, et al. , 2020. Dietary characteristics associated with plasma concentrations of per- and polyfluoroalkyl substances among adults with pre-diabetes: cross-sectional results from the Diabetes Prevention Program Trial. Environ. Int. 137, 105217 10.1016/j.envint.2019.105217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W-S, Tang M-J, Xu T-L, Su J-B, Wang X-Q, Xu F, Zhang D-M, Zhu Q, Cao J, Wang H, 2020. Association of serum fibroblast growth factor 19 levels with arteriosclerosis parameters assessed by arterial stiffness and atherogenic index of plasma in patients with type 2 diabetes. Diabetol. Metab. Syndr. 12, 44. 10.1186/s13098-020-00552-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loccisano AE, Campbell JL, Butenhoff JL, Andersen ME, Clewell HJ, 2012. Evaluation of placental and lactational pharmacokinetics of PFOA and PFOS in the pregnant, lactating, fetal and neonatal rat using a physiologically based pharmacokinetic model. Reprod. Toxicol. 33 (4), 468–490. 10.1016/j.reprotox.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Loccisano AE, Longnecker MP, Campbell JL, Andersen ME, Clewell HJ, 2013. Development of PBPK models for PFOA and PFOS for human pregnancy and lactation life stages. J. Toxicol. Environ. Health A 76 (1), 25–57. 10.1080/15287394.2012.722523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveless SE, Finlay C, Everds NE, Frame SR, Gillies PJ, O’Connor JC, Powley CR, Kennedy GL, 2006. Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (APFO). Toxicology 220 (2–3), 203–217. 10.1016/j.tox.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Loveless SE, Hoban D, Sykes G, Frame SR, Everds NE, 2008. Evaluation of the immune system in rats and mice administered linear ammonium perfluorooctanoate. Toxicol Sci. 105 (1), 86–96. 10.1093/toxsci/kfn113. [DOI] [PubMed] [Google Scholar]
- Luebker DJ, Hansen KJ, Bass NM, Butenhoff JL, Seacat AM, 2002. Interactions of fluorochemicals with rat liver fatty acid-binding protein. Toxicology 176 (3), 175–185. 10.1016/s0300-483x(02)00081-1. [DOI] [PubMed] [Google Scholar]
- MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, et al. , 2018. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9 (1), 4383. 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, 2016. Srebp2: a master regulator of sterol and fatty acid synthesis. J. Lipid Res. 57 (3), 333–335. 10.1194/jlr.C066712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney EK, Waxman DJ, 1999. Trans-activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol. Appl. Pharmacol. 161 (2), 209–218. 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- Mc Auley MT, Wilkinson DJ, Jones JJL, Kirkwood TBL, 2012. A whole-body mathematical model of cholesterol metabolism and its age-associated dysregulation. BMC Syst. Biol. 6, 130. 10.1186/1752-0509-6-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen PD, Bhattacharya S, Woods CG, Pendse SN, McBride MT, Soldatow VY, Deisenroth C, LeCluyse EL, Clewell RA, Andersen ME, 2020. Identifying qualitative differences in PPARα signaling networks in human and rat hepatocytes and their significance for next generation chemical risk assessment methods. Toxicol. In Vitro 64, 104463. 10.1016/j.tiv.2019.02.017. [DOI] [PubMed] [Google Scholar]
- Molina H, Azocar L, Ananthanarayanan M, Arrese M, Miquel JF, 2008. Localization of the Sodium-Taurocholate cotransporting polypeptide in membrane rafts and modulation of its activity by cholesterol in vitro. Biochim. Biophys. Acta 1778 (5), 1283–1291. 10.1016/j.bbamem.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Mondal D, Weldon RH, Armstrong BG, Gibson LJ, Lopez-Espinosa M-J, Shin HM, Fletcher T, 2014. Breastfeeding: a potential excretion route for mothers and implications for infant exposure to perfluoroalkyl acids. Environ. Health Perspect. 122 (2), 187–192. 10.1289/ehp.1306613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AE, Mooney KM, Wilkinson SJ, Pickles NA, Mc Auley MT, 2016. Mathematically modelling the dynamics of cholesterol metabolism and ageing. Biosystems 145, 19–32. 10.1016/j.biosystems.2016.05.001. [DOI] [PubMed] [Google Scholar]
- MRFIT, 1986. Relationship between baseline risk factors and coronary heart disease and total mortality in the Multiple Risk Factor Intervention Trial. Multiple Risk Factor Intervention Trial Research Group. Prev. Med. 15 (3), 254–273. 10.1016/0091-7435(86)90045-9. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ito Y, Yanagiba Y, Ramdhan DH, Kono Y, Naito H, Hayashi Y, Li Y, Aoyama T, Gonzalez FJ, et al. , 2009. Microgram-order ammonium perfluorooctanoate may activate mouse peroxisome proliferator-activated receptor alpha, but not human PPARalpha. Toxicology 265 (1–2), 27–33. 10.1016/j.tox.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngueta G, Longnecker MP, Yoon M, Ruark CD, Clewell HJ, Andersen ME, Verner M-A, 2017. Quantitative bias analysis of a reported association between perfluoroalkyl substances (PFAS) and endometriosis: the influence of oral contraceptive use. Environ. Int. 104, 118–121. 10.1016/j.envint.2017.03.023. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Burlew MM, Mandel JH, 2000. Plasma cholecystokinin and hepatic enzymes, cholesterol and lipoproteins in ammonium perfluorooctanoate production workers. Drug Chem. Toxicol. 23 (4), 603–620. 10.1081/dct-100101973. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR, 2007. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ. Health Perspect. 115 (9), 1298–1305. 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW, Zobel LR, 2007. Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int. Arch. Occup. Environ. Health 81 (2), 231–246. 10.1007/s00420-007-0213-0. [DOI] [PubMed] [Google Scholar]
- Palmer M, Jennings L, Silberg DG, Bliss C, Martin P, 2018. A randomised, double-blind, placebo-controlled phase 1 study of the safety, tolerability and pharmacodynamics of volixibat in overweight and obese but otherwise healthy adults: implications for treatment of non-alcoholic steatohepatitis. BMC Pharmacol. Toxicol. 19 (1), 10. 10.1186/s40360-018-0200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters SAE, Singhateh Y, Mackay D, Huxley RR, Woodward M, 2016. Total cholesterol as a risk factor for coronary heart disease and stroke in women compared with men: a systematic review and meta-analysis. Atherosclerosis 248, 123–131. 10.1016/j.atherosclerosis.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, Verhagen A, Rivera CR, Mulvihill SJ, Malloy MJ, et al. , 2002. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 110 (1), 109–117. 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist EM, Filgo AJ, Cummings CA, Kissling GE, Hoenerhoff MJ, Fenton SE, 2015. Hepatic mitochondrial alteration in CD-1 mice associated with prenatal exposures to low doses of perfluorooctanoic acid (PFOA). Toxicol. Pathol. 43 (4), 546–557. 10.1177/0192623314551841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebholz SL, Jones T, Herrick RL, Xie C, Calafat AM, Pinney SM, Woollett LA, 2016. Hypercholesterolemia with consumption of PFOA-laced Western diets is dependent on strain and sex of mice. Toxicol. Rep. 3, 46–54. 10.1016/j.toxrep.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeskamp LF, Meessen ECE, Groen AK, 2018. Transintestinal cholesterol excretion in humans. Curr. Opin. Lipidol. 29 (1), 10–17. 10.1097/MOL.0000000000000473. [DOI] [PubMed] [Google Scholar]
- Rodriguez CE, Setzer RW, Barton HA, 2009. Pharmacokinetic modeling of perfluorooctanoic acid during gestation and lactation in the mouse. Reprod. Toxicol. 27 (3–4), 373–386. 10.1016/j.reprotox.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Schmid JR, Corton JC, Zehr RD, Das KP, Abbott BD, Lau C, 2010. Gene expression profiling in wild-type and PPARα-null mice exposed to perfluorooctane sulfonate reveals PPARα-Independent effects. PPAR Res. 2010 10.1155/2010/794739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Das KP, Wood CR, Wolf CJ, Abbott BD, Lau C, 2013. Evaluation of perfluoroalkyl acid activity using primary mouse and human hepatocytes. Toxicology 308, 129–137. 10.1016/j.tox.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Das KP, Rooney J, Abbott B, Lau C, Corton JC, 2017. PPARα-independent transcriptional targets of perfluoroalkyl acids revealed by transcript profiling. Toxicology 387, 95–107. 10.1016/j.tox.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush EL, Singer AB, Longnecker MP, Haug LS, Sabaredzovic A, Symanski E, Whitworth KW, 2018. Oral contraceptive use as a determinant of plasma concentrations of perfluoroalkyl substances among women in the Norwegian Mother and Child Cohort (MoBa) study. Environ. Int. 112, 156–164. 10.1016/j.envint.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salihović S, Dickens AM, Schoultz I, Fart F, Sinisalu L, Lindeman T, Halfvarson J, Orešič M, Hyötyläinen T, 2020. Simultaneous determination of perfluoroalkyl substances and bile acids in human serum using ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 412 (10), 2251–2259. 10.1007/s00216-019-02263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlezinger JJ, Puckett H, Oliver J, Nielsen G, Heiger-Bernays W, Webster TF, 2020. Perfluorooctanoic acid activates multiple nuclear receptor pathways and skews expression of genes regulating cholesterol homeostasis in liver of humanized PPARα mice fed an American diet. Toxicol. Appl. Pharmacol. 405, 115204 10.1016/j.taap.2020.115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Olsen GW, Case MT, Butenhoff JL, 2002. Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol. Sci. 68 (1), 249–264. 10.1093/toxsci/68.1.249. [DOI] [PubMed] [Google Scholar]
- Sheng N, Li J, Liu H, Zhang A, Dai J, 2016. Interaction of perfluoroalkyl acids with human liver fatty acid-binding protein. Arch. Toxicol. 90 (1), 217–227. 10.1007/s00204-014-1391-7. [DOI] [PubMed] [Google Scholar]
- Steenland K, Tinker S, Shankar A, Ducatman A, 2010. Association of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) with uric acid among adults with elevated community exposure to PFOA. Environ. Health Perspect. 118 (2), 229–233. 10.1289/ehp.0900940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland EM, Hu XC, Dassuncao C, Tokranov AK, Wagner CC, Allen JG, 2019. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 29 (2), 131–147. 10.1038/s41370-018-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wulp MYM, Verkade HJ, Groen AK, 2013. Regulation of cholesterol homeostasis. Mol. Cell. Endocrinol. 368 (1–2), 1–16. 10.1016/j.mce.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Weissglas-Volkov D, Huertas-Vazquez A, Suviolahti E, Lee J, Plaisier C, Canizales-Quinteros S, Tusie-Luna T, Aguilar-Salinas C, Taskinen M-R, Pajukanta P, 2006. Common hepatic nuclear factor-4alpha variants are associated with high serum lipid levels and the metabolic syndrome. Diabetes 55 (7), 1970–1977. 10.2337/db06-0035. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. , 2013. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45 (11), 1274–1283. 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winquist A, Steenland K, 2014. Modeled PFOA exposure and coronary artery disease, hypertension, and high cholesterol in community and worker cohorts. Environ. Health Perspect. 122 (12), 1299–1305. 10.1289/ehp.1307943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CJ, Schmid JE, Lau C, Abbott BD, 2012. Activation of mouse and human peroxisome proliferator-activated receptor-alpha (PPARα) by perfluoroalkyl acids (PFAAs): further investigation of C4-C12 compounds. Reprod. Toxicol. 33 (4), 546–551. 10.1016/j.reprotox.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Wong F, MacLeod M, Mueller JF, Cousins IT, 2014. Enhanced elimination of perfluorooctane sulfonic acid by menstruating women: evidence from population-based pharmacokinetic modeling. Environ. Sci. Technol. 48 (15), 8807–8814. 10.1021/es500796y. [DOI] [PubMed] [Google Scholar]
- Worley RR, Fisher J, 2015. Application of physiologically-based pharmacokinetic modeling to explore the role of kidney transporters in renal reabsorption of perfluorooctanoic acid in the rat. Toxicol Appl Pharmacol 289 (3), 428–441. 10.1016/j.taap.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley RR, Yang X, Fisher J, 2017. Physiologically based pharmacokinetic modeling of human exposure to perfluorooctanoic acid suggests historical non drinking-water exposures are important for predicting current serum concentrations. Toxicol. Appl. Pharmacol. 330, 9–21. 10.1016/j.taap.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K-T, Kuo P-L, Su S-B, Chen Y-Y, Yeh M-L, Huang C-I, Yang J-F, Lin C-I, Hsieh M-H, Hsieh M-Y, et al. , 2016. Nonalcoholic fatty liver disease severity is associated with the ratios of total cholesterol and triglycerides to high-density lipoprotein cholesterol. J. Clin. Lipidol. 10 (2) 10.1016/j.jacl.2015.12.026, 420–425.e1. [DOI] [PubMed] [Google Scholar]
- Xing J, Wang G, Jichun, Zhao, Wang E, Yin B, Fang D, Jianxin, Zhao, Zhang H, Chen YQ, Chen W, 2016. Toxicity assessment of perfluorooctane sulfonate using acute and subchronic male C57BL/6J mouse models. Environ Pollut. 210, 388–396. 10.1016/j.envpol.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Yan S, Wang J, Zhang W, Dai J, 2014. Circulating microRNA profiles altered in mice after 28 d exposure to perfluorooctanoic acid. Toxicol. Lett. 224 (1), 24–31. 10.1016/j.toxlet.2013.10.017. [DOI] [PubMed] [Google Scholar]
- Yang C-H, Glover KP, Han X, 2009. Organic anion transporting polypeptide (Oatp) 1a1-mediated perfluorooctanoate transport and evidence for a renal reabsorption mechanism of Oatp1a1 in renal elimination of perfluorocarboxylates in rats. Toxicol. Lett. 190 (2), 163–171. 10.1016/j.toxlet.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, McNamara LA, Gorski JN, Eiermann GJ, Petrov A, et al. , 2012. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 53 (1), 51–65. 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Beesoon S, Zhu L, Martin JW, 2013. Biomonitoring of perfluoroalkyl acids in human urine and estimates of biological half-life. Environ. Sci. Technol. 47 (18), 10619–10627. 10.1021/es401905e. [DOI] [PubMed] [Google Scholar]
- Zhang L, Krishnan P, Ehresman DJ, Smith PB, Dutta M, Bagley BD, Chang S-C, Butenhoff JL, Patterson AD, Peters JM, 2016. Editor’s highlight: perfluorooctane sulfonate-choline ion pair formation: a potential mechanism modulating hepatic steatosis and oxidative stress in mice. Toxicol. Sci. 153 (1), 186–197. 10.1093/toxsci/kfw120. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zitzow JD, Ehresman DJ, Chang S-C, Butenhoff JL, Forster J, Hagenbuch B, 2015. Na+/taurocholate cotransporting polypeptide and apical sodium-dependent bile acid transporter are involved in the disposition of perfluoroalkyl sulfonates in humans and rats. Toxicol. Sci. 146 (2), 363–373. 10.1093/toxsci/kfv102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zitzow JD, Weaver Y, Ehresman DJ, Chang S-C, Butenhoff JL, Hagenbuch B, 2017. Organic anion transporting polypeptides contribute to the disposition of perfluoroalkyl acids in humans and rats. Toxicol. Sci. 156 (1), 84–95. 10.1093/toxsci/kfw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.