

Abstract
Familial Alzheimer's disease (FAD) is caused by autosomal dominant mutations in the PSEN1, PSEN2 or APP genes, giving rise to considerable clinical and pathological heterogeneity in FAD. Here we investigate variability in clinical data and the type and distribution of Aβ pathologies throughout the cortical layers of different FAD mutation cases. Brain tissue from 20 FAD cases [PSEN1 pre‐codon 200 (n = 10), PSEN1 post‐codon 200 (n = 6), APP (n = 4)] were investigated. Frontal cortex sections were stained immunohistochemically for Aβ, and Nissl to define the cortical layers. The frequency of different amyloid‐beta plaque types was graded for each cortical layer and the severity of cerebral amyloid angiopathy (CAA) was determined in cortical and leptomeningeal blood vessels. Comparisons were made between FAD mutations and APOE4 status, with associations between pathology, clinical and genetic data investigated. In this cohort, possession of an APOE4 allele was associated with increased disease duration but not with age at onset, after adjusting for mutation sub‐group and sex. We found Aβ pathology to be heterogeneous between cases although Aβ load was highest in cortical layer 3 for all mutation groups and a higher Aβ load was associated with APOE4. The PSEN1 post‐codon 200 group had a higher Aβ load in lower cortical layers, with a small number of this group having increased cotton wool plaque pathology in lower layers. Cotton wool plaque frequency was positively associated with the severity of CAA in the whole cohort and in the PSEN1 post‐codon 200 group. Carriers of the same PSEN1 mutation can have differing patterns of Aβ deposition, potentially because of differences in risk factors. Our results highlight possible influences of APOE4 genotype, and PSEN1 mutation type on Aβ deposition, which may have effects on the clinical heterogeneity of FAD.
Keywords: Alzheimer's disease, amyloid, familial Alzheimer’s disease
An investigation of the frequency and distribution of the Aβ pathologies throughout the cortical layers of different familial Alzheimer's disease mutations cases. The frequency of different Aβ plaque types were graded for each cortical layer and the severity of cerebral amyloid angiopathy was determined in cortical and leptomeningeal blood vessels.

1. INTRODUCTION
Familial Alzheimer's disease (FAD) is caused by dominantly inherited mutations in APP, PSEN1 and PSEN2. APP generates the amyloid precursor protein (APP), which is cleaved to produce the amyloid‐beta (Aβ) peptide. PSEN1/PSEN2 are the catalytic subunits of the ƴ‐secretase complex, which are involved in the production of Aβ (1, 2). As in sporadic AD, these Aβ peptides aggregate into extracellular deposits as amyloid plaques, a classic neuropathological hallmark of AD. Although the causative mutations all affect proteins required for Aβ production, considerable heterogeneity can be seen in the clinical and neuropathological features, which may indicate individual mutation effects (3, 4, 5).
Patients with FAD typically present with early amnestic symptoms, although atypical cognitive symptoms and additional neurological features are also seen (3, 6, 7). While FAD typically causes young onset dementia, with age at onset usually ranging between 30 and 60 years, late‐onset cases are increasingly found as older cohorts are sequenced (8, 9). In general, PSEN1 mutations cause younger‐onset than APP mutations, while PSEN2 mutations are associated with the oldest ages at onset (3, 9, 10). PSEN1 mutation site has been found to influence age at onset, with mutations before codon 200 associated with a younger onset than those located beyond codon 200 (3, 11). Although mutation type accounts for a large proportion of the variance in age at onset, substantial variation can still be observed even within carriers of the same mutation and within families (3, 6, 9). Interestingly, despite its role as a risk factor for sporadic AD, APOE genotype was not found to significantly influence age at onset in a meta‐analysis of FAD (9). In contrast, the APOE4 allele has been reported to be associated with younger ages at onset in a single large Colombian kindred harbouring the PSEN1 E280A mutation (12). Other factors, which are relevant in sporadic AD, may also play a role in clinical aspects of FAD, although they are less well understood. For example, females have a higher risk of dementia overall compared to males and in a meta‐analysis of FAD, males showed a trend to a later age at onset (9).
Although symptoms tend to begin at a younger age when individuals’ general health is better, FAD generally gives rise to similar disease durations to sporadic AD (6, 9, 13, 14). However, this too can vary within FAD, with PSEN2 cases reported to have longer disease durations than PSEN1 and APP cases (10). No difference in duration was found between APP and PSEN1 mutation carriers, nor between carriers of PSEN1 mutations located pre‐ and post‐codon 200 (15). In a meta‐analysis, shorter disease durations were observed in those with younger (<35 years) or older (>65 years) onset than in those with onset in mid‐life (9). This same ‘inverted U‐shape’ association between age at onset and disease duration was found in a recent study from our centre when all mutations were considered together. However, it resolved when the different genetic groups were examined separately as later ages at onset were associated with longer disease durations in PSEN1 mutation carriers, whereas later ages at onset were associated with shorter disease durations in the APP cohort. This study also found that survival in FAD was influenced by mutation to a much lesser extent than the age at onset and appeared to be longer in individuals with PSEN1 mutations who carried an APOE4 allele (14).
In FAD, the Aβ isoforms produced and the relative ratios of these peptide species differ between pathogenic mutations (16, 17). A study of APP mutations has demonstrated that the distinct Aβ peptides produced by different mutations have distinct aggregation conformations and so influence the type of amyloid pathology produced, resulting in histological diversity between mutations (18). Post‐mortem analysis of Aβ pathology in FAD shows considerable heterogeneity between cases, with reports suggesting total Aβ may be more severe compared to patients with sporadic AD (19). The types of plaques found in the cortex are classified by their morphological features (20, 21) and within PSEN1 cases the type and number of deposits show variability across different mutations (22). Additionally, between PSEN1 pre and post‐codon 200 mutations, distinct histological phenotypes have been noted, with mutations post‐codon 200 tending to have more severe cerebral amyloid angiopathy (CAA) (11), which is the deposition of Aβ in blood vessel walls. Severe CAA has also been observed in cases with APP mutations located within the Aβ coding domain [reviewed in Ref. (4)]. These histological differences may be underlying causes of variable disease phenotypes.
As well as morphological differences between mutations, there is also variability in Aβ distribution across the depth of the cortex. In both AD and FAD, a consistent maximal density of Aβ pathology has been found in cortical layer 3, with varying densities in other layers (23, 24, 25, 26, 27). Additionally, Aβ plaque types have differing distribution patterns over the cortical layers (28, 29). However, a previous study examining a group of 20 AD patients, including four cases with FAD mutations, reported that Aβ density and plaque‐type distribution across the cortex did not appear markedly to differ between cases, although the cortical layers were not specifically delineated (30). Yet within FAD, certain plaque types have been associated with specific clinical features, such as the frequent observation of cotton wool plaques (CWP) in patients with spastic paraparesis (31, 32). These observations are particularly associated with certain mutations, such as PSEN1 exon 9 deletions (33).
This study aimed to gain a deeper understanding of the association of genetic mutations to the morphology and distribution of Aβ pathology and their associations with disease pathogenesis in FAD. We analysed in detail the morphology and distribution of Aβ pathology, using cortical layer‐specific distributions, in a cohort of 20 FAD cases, using a range of measures and explored relationships with clinical data (age at onset and disease duration) and genotypes. Associations were examined for the cohort as a whole and also in exploratory analysis in the three mutation sub‐groups (PSEN1 pre‐codon 200, PSEN1 post‐codon 200 and APP).
Access to FAD post‐mortem tissue is limited and, as this was a study of cases donated to a single centre, our sample size was therefore small. This constrained our approaches when analysing the data and interpreting results, particularly for analysis in the mutation sub‐groups. In this context, while we report statistical significance for results in the whole cohort, our focus was on identifying trends and patterns. Our hypothesis was that a detailed examination of Aβ pathology in a variety of FAD mutations would reveal differences in Aβ plaque‐type and pattern of distribution across the cortical layers and in severity of CAA. We aimed to determine whether there are specific associations between the different types of Aβ pathology, and if these associations are observed in the FAD mutation‐specific sub‐groups.
2. MATERIALS AND METHODS
2.1. Cases
All 20 FAD cases were obtained through the brain donation program of the Queen Square Brain Bank for Neurological Disorders (QSBB), Department of Clinical and Movement Neurosciences, UCL Queen Square Institute of Neurology and all available FAD cases were investigated. The protocols used for brain donation and ethical approval for this study were approved by a London Research Ethics Committee and tissue is stored for research under a license from the Human Tissue Authority. The standard diagnostic criteria for the neuropathological diagnosis of AD and the presence of CAA were used in all cases (34, 35, 36, 37, 38).
2.2. Histological staining and Immunohistochemistry
Paraffin‐embedded serial sections (8 μm thick) were cut from the frontal cortex. For Nissl staining, deparaffinised and rehydrated sections were stained with 0.1% cresyl violet‐acetate (83860.120, Prolabo) to determine the cortical layers. Serial sections were used for Aβ immunohistochemistry (IHC), using a pan‐Aβ antibody that recognises all Aβ isoforms containing residues 8–17. Slides were pre‐treated in formic acid followed by pressure cooker in citrate buffer pH6.0. Endogenous peroxidase activity was blocked with 0.3% H202 in methanol and non‐specific binding with 10% dried milk solution. Sections were incubated with the primary Aβ antibody (1:100; M0872, DAKO) overnight at 4°C, followed by biotinylated anti‐mouse (1:200, 30 min; E0354, DAKO) and ABC complex (30 min; PK‐6100, Vector Laboratories Ltd). Colour was developed with di‐aminobenzidine/H2O2. Slides were scanned and digitised using a Leica slide scanner with a 40x objective.
2.3. Layer delineation
Digitised Nissl sections were used to delineate the cortical layers, the second frontal gyrus (Brodmann area 9) was used for analysis. Regions of analysis were used in Adobe® Photoshop® software (Adobe® Photoshop® CC 2017, Adobe Systems Incorporated) to delineate and mark the six cortical layers. The layer markings were transferred onto the corresponding area of the serial Aβ immunohistochemically stained section using multiple reference points.
2.4. Areal fraction analysis
Aβ immunohistochemically stained sections with layer overlay were analysed per cortical layer for percentage area stained (Aβ load). A 6 mm width section along the gyrus was analysed for all cases, although differences in layer depth existed between cases. For each layer and each case, the same volumetric area was analysed. Images were opened in ImageJ (39) to select individual cortical layers. For each layer, 15 randomised regions of interest were generated using Python (Python 3.6.0, Python Software Foundation) for areal fraction analysis. A pre‐defined macro was used in ImageJ for threshold analysis. For each layer, the mean value from the 15 randomised regions of area stained was generated.
2.5. Aβ plaque‐type assessment
Evaluation was conducted blinded to mutation or clinical information. Aβ pathology was evaluated semi‐quantitatively based on a four‐point grading scale of 0–3 (none, sparse, moderate and frequent). Cortical tissue was assessed at 20x magnification within the same region as areal fraction analysis, to generate scores. We used the grading scale to assess Aβ plaque frequency for each cortical layer (1–6) to produce a layer‐specific score. These scores were then combined to generate an overall total Aβ plaque score for each case. Additionally, individual plaque types (diffuse, cored or CWP – identified as large circular diffuse Aβ plaques with defined edges) were assessed by layer (1–6) using the same grading system and those scores combined to generate a total score for each plaque type. Subpial Aβ pathology was also assessed on the 0–3 scale.
2.6. Cortical and leptomeningeal vessel counts
Aβ‐positive cortical and leptomeningeal CAA pathology was determined from IHC Aβ sections. Cortical CAA was also assessed across the six layers using a similar 0–3 grading scale and a combined total generated, as previously described (40, 41, 42). The proportion of vessels affected by Aβ deposition was also analysed. Brains donated to QSBB are not stripped of the leptomeninges, and leptomeninges were visible and present in all cases. Starting in the region selected for areal fraction analysis, 100 cortical vessels were counted, and the proportion of affected vessels were calculated as a percentage. Up to 100 leptomeningeal vessels were counted along the meninges. Only vessels cut cross‐sectionally showing the full circumference of the vessel wall were included in the analysis.
2.7. Statistical analysis
Analyses were performed using all cases both for the whole cohort and by sub‐groups based on mutation (including location where relevant) or APOE4 status. The three mutation sub‐groups were PSEN1 cases with a mutation before codon 200, PSEN1 cases with a mutation after codon 200, and APP mutation cases. To classify APOE4 status, cases were separated into those without an ε4 allele, for example ε2/3 and ε3/3, and those with at least one ε4 allele, for example ε3/4 and ε4/4. For analyses incorporating APOE4 status 19 cases were used, as DNA was not available for case 9. Statistical analyses were performed using STATA 15.1 (StataCorp. 2017. Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC).
2.7.1. Clinical and genetic comparisons
Linear regression models compared both age at onset and disease duration by: (i) mutation sub‐group (adjusting for sex and APOE4 status); (ii) sex (adjusting for mutation sub‐group and APOE4 status); and (iii) APOE4 status (adjusting for mutation sub‐group and sex).
2.7.2. Aβ pathology scores
Associations between each of the four Aβ pathology scores (subpial, cored plaques, diffuse plaques, CWPs) and the two CAA scores (cortical CAA, leptomeningeal CAA) and (i) age at onset, and (ii) disease duration were investigated in the whole cohort using linear regression, adjusting for APOE4 status. Each score was assessed for evidence of differences between mutation sub‐groups (Kruskal–Wallis) and by APOE4 status (exact Mann–Whitney–Wilcoxon rank sum). All scores were then correlated pairwise against each other in the whole cohort and then in the sub‐groups in order to look for any patterns in the correlations (Kendall's tau‐b correlation coefficient, with statistical significance level determined using tables of critical values because of the small sample size). Finally, we compared individual layer Aβ pathology scores between sub‐groups (Kruskal–Wallis with Dunn's test for pairwise comparison between sub‐groups).
2.7.3. Aβ load in the cortical tissue
Associations between mean total Aβ load (measured as a percentage area stained) across the six cortical layers and (i) age at onset and (ii) disease duration were assessed using a linear regression, adjusting for APOE4 status, in the whole cohort and in sub‐groups; these analyses were repeated for each of the six cortical layers individually. Mean Aβ load was compared between mutation sub‐group (Kruskal–Wallis with Dunn's test where appropriate) and by APOE4 status (exact Mann–Whitney–Wilcoxon rank sum). Correlations between Aβ load and the frequency of the four different Aβ plaque pathologies and CAA scores were investigated in the whole cohort and in sub‐groups in order to look for any patterns in the correlations (Kendall's tau‐b as above). Finally, we assessed whether Aβ load differed between the different layers in the whole cohort (Friedman ANOVA). The Aβ load of each individual layer was then compared between the mutation sub‐groups, adjusted for APOE4 status, using linear regression.
2.7.4. Proportion of vessels affected by cortical CAA and leptomeningeal CAA
Linear regression models investigated whether in the whole cohort the proportions of vessels with CAA in the cortex and with CAA in the leptomeninges were associated with (i) age at onset and (ii) disease duration, adjusted for APOE4 status. The proportions of CAA affected vessels were then compared between mutation sub‐groups (Kruskal–Wallis and Dunn's test) and by APOE4 status (exact Mann–Whitney–Wilcoxon rank sum). Finally, we assessed whether the proportions of CAA affected vessels correlated with each of the four Aβ plaque pathologies scores and Aβ load (Kendall's tau‐b as above).
3. RESULTS
3.1. Neuropathological summary
All cases reached end‐stage AD with a score of A3B3C3 according to the current diagnostic criteria, indicating frequent neuritic plaques, neurofibrillary pathology spread to the occipital cortex reaching Braak and Braak stage 6 (except case 16 who scored 5) and Aβ plaque pathology in the cerebellum reaching Thal stage 5.
3.2. Clinical and genetic details
Subject details are summarised in Table 1. The 20 individuals with FAD in this study included: 16 subjects with PSEN1 mutations, of which 10 were located pre‐codon 200 (four intron 4, one E120K, one S132A, one M139V, one M146I and two E184D) and six were located post‐codon 200 (one I202F, one L250S, two R278I, one E280G and one double mutation A434T & T291A), and four subjects with APP mutations (one V717L and three V717I). There were no statistically significant differences between the APP and the two PSEN1 mutation sub‐groups for sex or APOE4 status (Table 1).
TABLE 1.
Study participants
| Case | Sex | Age at onset (years) | Disease duration (years)b | Mutation | APOE | Braak Tau | Thal phase | CERAD | Alpha‐syn pathology | TDP−43 pathology |
PMD (h/min) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 36 | 5 | PSEN1 Intron4 | 3/3 | 6 | 5 | Frequent | None | None | 16:15 |
| 2 | F | 35 | 16.9 | PSEN1 Intron4 | 4/4 | 6 | 5 | Frequent | Amygdala | Amygdala | 32:30 |
| 3 | F | 39 | 8.1 | PSEN1 Intron4 | 3/3 | 6 | 5 | Frequent | None | Amygdala | – |
| 4 | M | 42 | 9 | PSEN1 Intron4 | 3/3 | 6 | 5 | Frequent | Amygdala | Amygdala | 43:10 |
| 5 | F | 31 | 6 | PSEN1 E120K | 3/3 | 6 | 5 | Frequent | Amygdala | None | 24:15 |
| 6 | M | 59 | 11 | PSEN1 S132A | 3/4 | 6 | 5 | Frequent | Neocortical | None | 161:15 |
| 7 | F | 41 | 8.9 | PSEN1 M139V | 3/3 | 6 | 5 | Frequent | Amygdala | None | – |
| 8 | M | 40 | 6 | PSEN1 M146I | 2/3 | 6 | 5 | Frequent | Amygdala | None | 115:35 |
| 9 | F | 40 | 13.6 | PSEN1 E184D | ‐ | 6 | 5 | Frequent | None | None | 153:30 |
| 10 | F | 45 | 13 | PSEN1 E184D | 3/4 | 6 | 5 | Frequent | Amygdala | None | 63:25 |
| 11 | F | 48 | 11 | PSEN1 I202F | 4/4 | 6 | 5 | Frequent | Amygdala | None | 26:15 |
| 12 | M | 47 | 11 | PSEN1 L250S | 3/3 | 6 | 5 | Frequent | None | None | 32:30 |
| 13 | F | 46 | 19 | PSEN1 R278I | 3/4 | 6 | 5 | Frequent | Amygdala | None | 31:55 |
| 14 | M | 54 | 12 | PSEN1 R278I | 2/3 | 6 | 5 | Frequent | Amygdala | Amygdala | 77:45 |
| 15 | F | 42 | 11 | PSEN1 E280G | 3/4 | 6 | 5 | Frequent | Amygdala | None | 11:00 |
| 16 | M | 42 | 5 | PSEN1 A434T & T291A | 3/3 | 5 | 5 | Frequent | None | None | 43:50 |
| 17 | F | 51 | 8.7 | APP V717L | 3/3 | 6 | 5 | Frequent | None | None | 89:42 |
| 18 | M | 56 | 10.1 | APP V717I | 3/3 | 6 | 5 | Frequent | Amygdala | None | 68:05 |
| 19 | F | 50 | 6.5 | APP V717I | 3/3 | 6 | 5 | Frequent | Amygdala | None | 16:25 |
| 20 | M | 49 | 13 | APP V717I | 4/4 | 6 | 5 | Frequent | Amygdala | None | 32:10 |
| Mean | 8M:12F | 45.05 | 10.24 | ||||||||
| Whole cohort N = 20 | PSEN1 pre‐codon 200 N = 10 | PSEN1 post‐codon 200 N = 6 | APP N = 4 | p‐value * | |||||||
| APOE (% APOE4 carriera) | 37 | 33 | 50 | 25 | 0.70 | ||||||
| Sex (% female) | 60 | 70 | 50 | 50 | 0.60 |
‘–’represents missing data. No APOE data for case 9.
Abbreviations: F, female; M, male; PMD, post mortem delay.
N = 19 as PSEN1 pre‐codon 200 case 9 had no APOE genotype data available.
Disease duration accuracy based on available data.
Fisher exact test.
Regression analyses for age at onset and disease duration were conducted, with adjustments for mutation group, sex and APOE4 status, where relevant (Table 2). Case 9 has no APOE genotype data available so was not included in this analysis. The mean age at onset of the whole cohort, was lower in the PSEN1 pre‐codon 200 group (41.6 years) than the PSEN1 post‐codon 200 group (46.5 years) and the APP group (51.5 years). After adjusting for sex and APOE4 status, there was evidence of an association between age at onset and mutation sub‐group, with mean age at onset an estimated 8.8 years older (95% CI: 1.5, 16.1; p = 0.02) for APP compared with PSEN1 pre‐codon 200 mutation carriers. There were no other statistically significant associations between mutation sub‐group and age at onset or disease duration.
TABLE 2.
Clinical comparisons
| Observed mean (years) (N = 20) | Model coefficient (years) (N = 19) | 95% CI | p‐value | |
|---|---|---|---|---|
| Age at onset | ||||
| Mutation groupa | p = 0.06 | |||
| APP vs PSEN1 pre‐codon 200a | 51.5 vs 41.6 | 8.8 | 1.5, 16.1 | p = 0.02* |
| APP vs PSEN1 post‐codon 200a | 51.5 vs 46.5 | 5.9 | −2.0, 13.8 | p = 0.13 |
| PSEN1 pre‐codon 200 vs PSEN1 post‐codon 200a | 41.8 vs 46.5 | −2.9 | −9.4, 3.6 | p = 0.35 |
| Females vs malesb | 42.0 vs 49.6 | −7.3 | −13.1, −1.4 | p = 0.02* |
| APOE4 carrier vs non‐carrierc | 46.3 vs 44.8 | 3.5 | −2.5, 9.5 | p = 0.23 |
| Disease duration | ||||
| Mutation groupa | p = 0.73 | |||
| APP vs PSEN1 pre‐codon 200a | 9.6 vs 9.8 | 0.7 | −3.1, 4.4 | p = 0.71 |
| APP vs PSEN1 post‐codon 200a | 9.6 vs 11.5 | −0.6 | −4.6, 3.5 | p = 0.77 |
| PSEN1 pre‐codon 200 vs PSEN1 post‐codon 200a | 9.3 vs 11.5 | −1.2 | −4.6, 2.1 | p = 0.44 |
| Females vs malesb | 10.6 vs 9.6 | −0.2 | −3.2, 2.8 | p = 0.89 |
| APOE4 carrier vs non‐carrierc | 13.6 vs 8.0 | 5.4 | 2.4, 8.5 | p = 0.002** |
Observed mean for whole cohort (N = 20), except for APOE where N = 19. Linear regression models compared age at onset and disease duration by mutation subgroup, sex and APOE4 status (N = 19). Asterisks represent statistically significant correlations.
Adjusted for sex and APOE genotype.
Adjusted for mutation sub‐group and APOE genotype.
Adjusted for mutation sub‐group and sex.
p < 0.05
p < 0.01.
In the cohort as a whole, females had a lower observed mean age at onset than males (42.0 years vs 49.6 years). After adjusting for mutation sub‐group and APOE4 status, mean age at onset was an estimated 7.3 years younger (95% CI: 13.1, 1.4) for females compared with males (p = 0.02). No evidence of an association between disease duration and sex was found (Table 2). No statistically significant association between APOE4 status and mean age at onset was found. However, there was strong evidence of an association between disease duration and APOE4 status, after adjusting for mutation sub‐group and sex, with mean disease duration an estimated 5.4 years longer (95% CI: 2.4, 8.5) for individuals with the APOE4 allele, compared with those without (p = 0.002) (Table 2).
3.3. Aβ pathology scores
3.3.1. Associations with clinical data
The frequency of the total Aβ pathology types (representative images shown in Figure 1A–F) were analysed in relation to age at onset and disease duration. In the whole cohort, there were negative associations between leptomeningeal CAA, Aβ pathology scores (subpial pathology, diffuse plaques and total cortical pathology) and age at onset, while cortical CAA, cored plaques and CWP had little association. Negative associations between cortical CAA, Aβ pathology scores (cored and diffuse plaques and total cortical pathology) and disease duration was also seen. However, none of the adjusted associations were statistically significant, with small effect sizes and large CIs (p‐values ranged from p = 0.09 to p = 1.00, Figure 1G,H).
FIGURE 1.
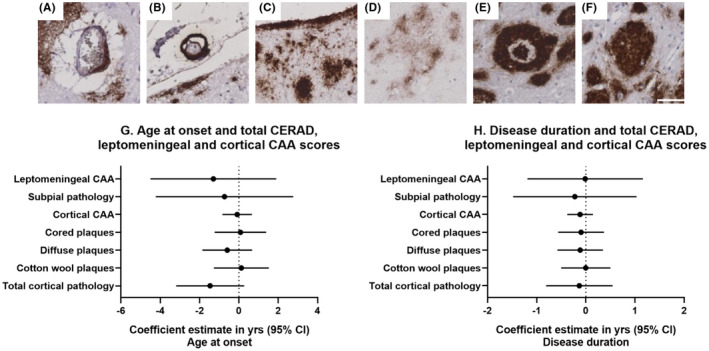
Representative images of cortical and leptomeningeal CAA (A & B), subpial pathology (C) and of Aβ plaque types (D: diffuse, E: Cored, F: CWP). White scale bar =50um, 200x objective. Linear regression adjusted for APOE4 status showing the association between total Aβ pathology scores, leptomeningeal CAA, cortical CAA and age at onset (G) and disease duration (H). Coefficient estimates and 95% confidence intervals for the whole cohort are represented for each total Aβ pathology score and the leptomeningeal and cortical CAA scores.
3.3.2. Associations by mutation sub‐groups
Microscopically, large heterogeneity in the appearance of Aβ pathology was evident throughout the cortical layers of the FAD cases (Figure 2). Table 3 shows the Aβ pathology scores for each case, giving the leptomeningeal CAA and subpial Aβ pathology, cortical CAA and plaque types in each cortical layer and the overall levels of Aβ plaque deposition. Observed leptomeningeal CAA and subpial Aβ pathology differed markedly between cases, being either absent, sparse, moderate or frequent. Based on microscopic visual inspection of the Aβ distribution throughout the cortical layers, variations were also observed between carriers of the same mutation. For example, one PSEN1 R278I mutation case (case 13) contained markedly more Aβ pathology compared with the other PSEN1 R278I case (case 14); interestingly case 13 had the genotype APOE3/4 compared to case 14 which is APOE2/3 (Figure 2). Additionally, differences within the pattern and distribution of plaques across the layers could be seen between these two cases, with case 13 having larger and greater plaque deposition particularly in layer 3, and greater cortical CAA and subpial deposition (Figure 2, Table 3E,A, and the subpial pathology row). However, similarities in layer deposition could be seen in other carriers with the same mutation, such as the PSEN1 E184D carriers, with denser deposition of Aβ across all layers (cases 9 and 10, Figure 2). Similarly, distinct distribution patterns could be seen across different cases, with APP V717L case 17 and V717I cases 18 and 20 all showing frequent subpial Aβ deposition and distinct plaque gradient from the upper to the lower of layer 3 (Figure 2). In contrast, PSEN1 E120K and S132A (cases 5 and 6) appear to have a more uniform distribution across layer 3 (Figure 2). Most cases showed the highest amount of Aβ staining within cortical layer 3, while the amount in the lower layers varied e.g. PSEN1 M139V (case 7) has very little lower layer deposition compared to the dense lower layer deposition seen in PSEN1 A434T & T291A (case 16) (Figure 2, Table 3B,D,E).
FIGURE 2.
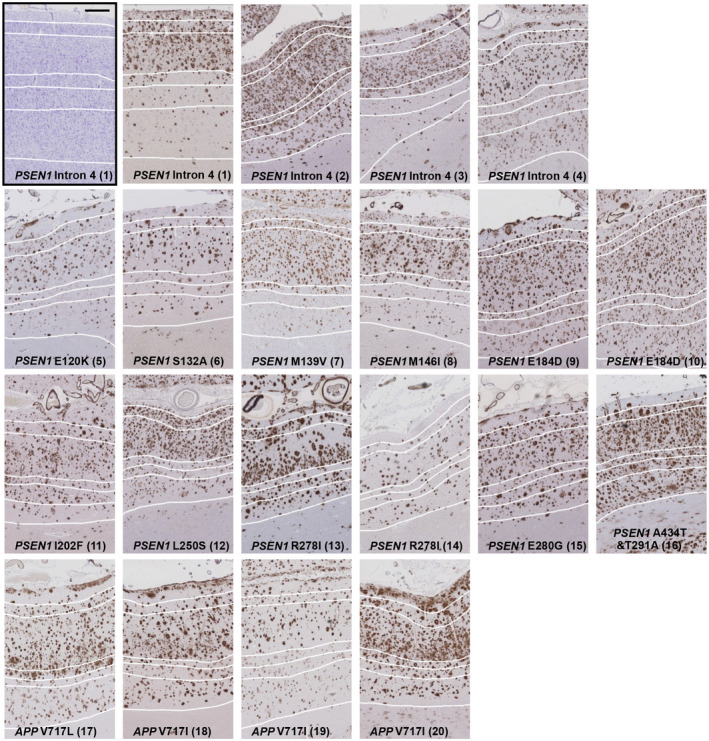
Representative images of Aβ immunohistochemically stained frontal cortex of each individual case within the mutation sub‐groups, PSEN1 pre‐codon 200 (10 cases), PSEN1 post‐codon 200 (6 cases) and APP (4 cases). One Nissl stained PSEN1 Intron 4 mutation tissue section is shown (black box) to highlight how the layers were defined based on cellular morphology and applied to the serial Aβ section. The 6 cortical layers are defined by white lines, with layer 1 at the pial surface and layer 6 adjacent to the white matter. Black scale bar =500µm. Numbers in brackets refer to case number.
TABLE 3.
Tables showing total score of Aβ pathology type and leptomeningeal and cortical CAA per cortical layer by individual case
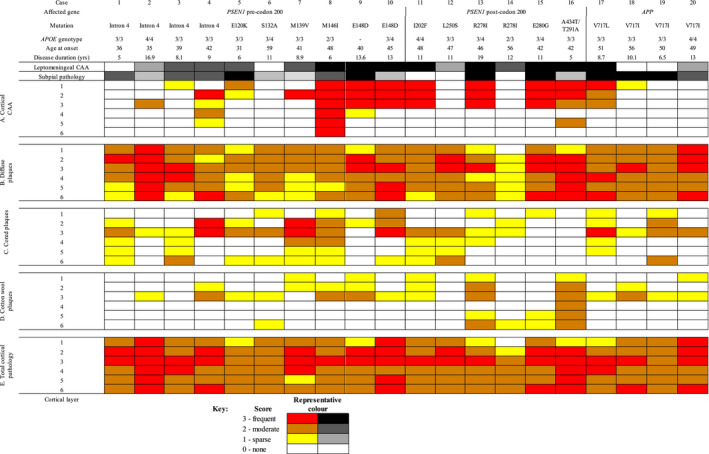
Leptomeningeal and subpial pathology varies between cases, with presence of one not indicative of the other. (A) CAA pathology scores highlight a predominance of CAA in a subset of cases, with an upper layer predisposition. (B) Diffuse type plaques are seen throughout all cases to varying degrees between cases and within individual cases. (C&D) Cored and CWP pathology scores show that there is variation in presentation between cases, and within the same cases. (E) Total cortical pathology score shows that not all cases have the same level of plaque deposition. Dashed lines divide the sub‐groups, PSEN1 pre‐codon 200, PSEN1 post‐codon 200 and APP.
Microscopically, visual inspection of the slides revealed the PSEN1 pre‐codon 200 and APP groups were more likely to have subpial pathology (presence in 100% of cases) than the PSEN1 post‐codon 200 group (presence in 50% of cases). However, comparing each total Aβ pathology score in turn formally between the three mutation sub‐groups, there were no statistically significant differences in plaque pathology (subpial p = 0.10, cored p = 0.27, diffuse p = 0.46, CWP p = 0.62 and total cortical pathology p = 0.87, global tests Kruskal–Wallis), or CAA pathology (leptomeningeal CAA p = 0.17, cortical CAA p = 0.41). None of the Aβ pathologies differed by APOE4 status.
3.3.3. Correlations between Aβ pathology scores
In the whole cohort, the frequency of leptomeningeal CAA was correlated with total cortical CAA (correlation coefficient τ b = 0.73 p ≤ 0.001, Kendall's tau‐b) (Figure 3A). When the sub‐groups were assessed separately there was a consistent positive correlation (Figure 3B–D) that remained in the two PSEN1 sub‐groups. A trend for positive correlation between both total cortical and leptomeningeal CAA with total CWP score was seen across all groups except the small APP group. Specifically, in the whole cohort, cortical and leptomeningeal CAA scores were both positively correlated to total CWP scores (τ b = 0.49 p < 0.01 and τ b = 0.37 p < 0.05, respectively) and in the sub‐group analyses this correlation was particularly evident in the PSEN1 post‐codon 200 group for cortical CAA (τ b = 0.86, p < 0.05) and leptomeningeal CAA (τ b = 0.77). In contrast, there was an observed trend for negative correlations between total cored plaque score and Aβ pathologies (subpial pathology, diffuse plaques, CWPs); this was particularly evident in the PSEN1 post‐codon 200 group, and slightly evident for diffuse plaques and CWPs in the APP group (Figure 3A–D).
FIGURE 3.
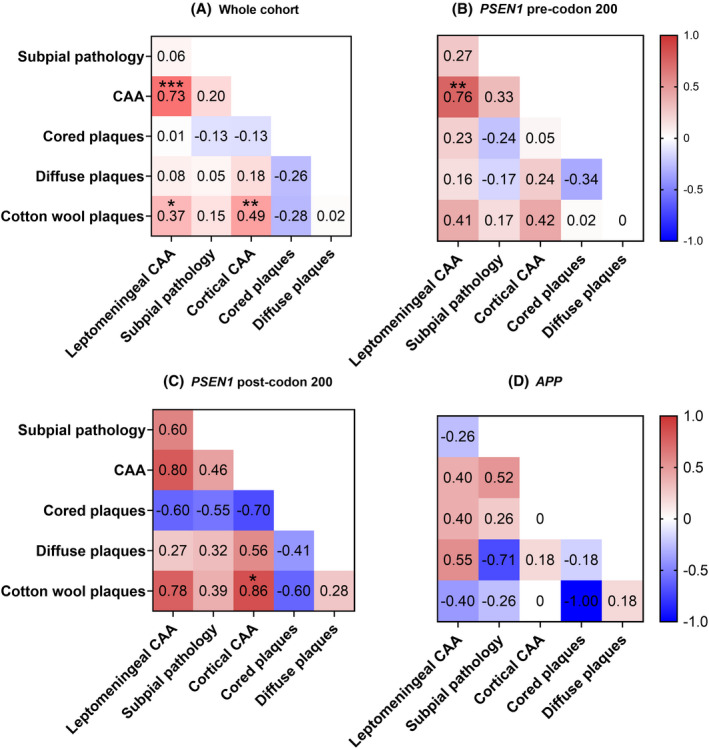
Correlation between total Aβ pathologies, leptomeningeal and cortical CAA: heat maps showing the relationship between the total Aβ pathology scores, leptomeningeal and cortical CAA ‐ A) Whole cohort, B) PSEN1 pre‐codon 200, C) PSEN1 post‐codon 200, D) APP. τ b values for Kendall's tau‐b correlation coefficients are shown. Asterisks represent statistically significant correlations: *p < 0.05, **p < 0.01, ***p < 0.001. Red =positive correlation. Blue = negative correlation
3.3.4. Differences across cortical layers
As reported above, we did not find evidence of total Aβ pathology scores differing between the sub‐groups. However, when comparing individual cortical layer scores (0–3) between sub‐groups we found CWPs in layer 5 and 6 were only seen to any measurable extent in the PSEN1 post‐codon 200 group (p = 0.02 and p = 0.02, respectively, Kruskal–Wallis). Specifically, in layer 5 the PSEN1 post‐codon 200 group had a higher CWP score (0.67 ± 0.82) compared to the PSEN1 pre‐codon 200 group (0 ± 0; p = 0.01, Dunn's test) and the APP group (0 ± 0; p = 0.04). This pattern was also seen in layer 6, with the PSEN1 post‐codon 200 group again having a higher frequency of CWPs (1.0 ± 0.89) than the PSEN1 pre‐codon 200 (0.1 ± 0.32; p = 0.01, Dunn's test) and APP group (0.0 ± 0.0; p = 0.02, Dunn's test). There was, however, considerable layer 5 and 6 variability within the sub‐groups. No differences for other plaque types were found. There were also no patterns of differences at the layer level in total Aβ pathology scores between APOE4 carriers and non‐carriers.
3.4. Aβ load in the cortical tissue
3.4.1. Associations with clinical data
Associations between mean Aβ load (measured as a percentage area stained) across the six cortical layers and age at onset and disease duration are shown in Figure 4A,B. Mean Aβ load was not statistically significantly associated with age at onset although a consistent negative association was found with mean Aβ load and Aβ load for individual cortical layers. Mean Aβ load was non‐significantly positively associated with disease duration, and this trend was seen for most cortical layers, with evidence for an association found (p = 0.0003) for layer 6 Aβ load and disease duration, (an estimated 0.21 additional years of duration for each percentage point increase in Aβ load; 95% CI: 0.004, 0.41; p = 0.05, after adjusting for APOE4 status).
FIGURE 4.
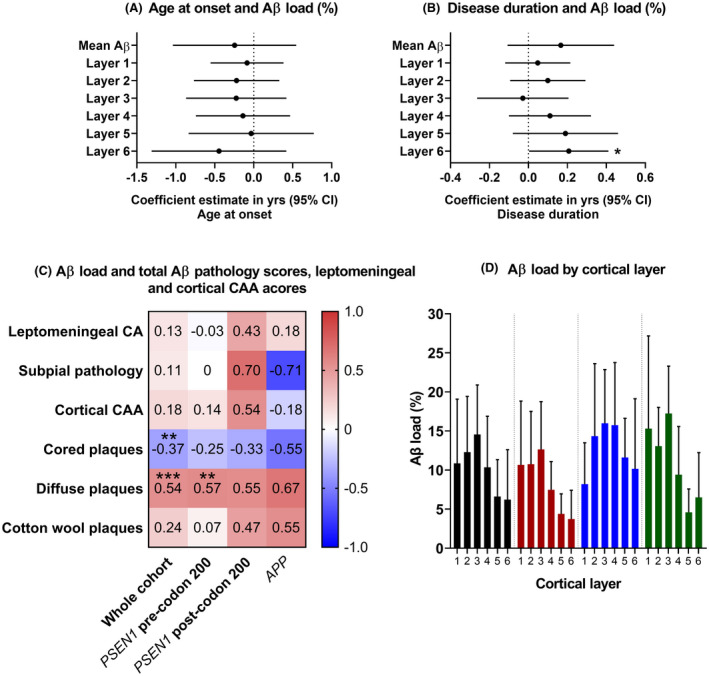
Linear regression adjusted for APOE4 status showing the association between Aβ load and age at onset (A) and disease duration (B). Coefficient estimates and 95% confidence intervals for the whole cohort are represented, for the mean Aβ load and for each cortical layer. The coefficients are the estimated difference in the outcome (age at onset or disease duration) for a one percentage point increase in the explanatory variable, after adjusting for APOE4 status. *p = 0.05. (C) Correlations between Aβ load and total Aβ pathology scores/ leptomeningeal and cortical CAA for the whole cohort and individual mutation sub‐groups. τ b values for Kendall's tau‐b correlation coefficients are shown. Asterisks represent statistically significant correlations: *p < 0.05, **p < 0.01, ***p < 0.001. Red = positive correlation. Blue = negative correlation. (D) Graphical display of the mean Aβ load (%) per cortical layer for the whole cohort and individual mutation sub‐groups. Bars and error bars represent mean and SD
3.4.2. By mutation sub‐groups
When mean Aβ load was compared between mutation sub‐groups, the PSEN1 post‐codon 200 group had the highest observed mean load, followed by the APP group, whilst the PSEN1 pre‐codon 200 group had the lowest (12.7% ± 6.4, 11% ± 5.5, 8.3% ± 3.8, respectively), although there was no statistically significant difference between the groups (p = 0.26 Kruskal–Wallis). Mean Aβ load was observed higher in APOE4 carriers than non‐carriers (13.1% ± 5.9, 8.5% ± 4.3, respectively), although with only weak evidence (p = 0.08, exact Mann–Whitney–Wilcoxon rank sum), due perhaps to the small sample size.
3.4.3. Correlations with Aβ pathology scores
Associations between Aβ load and the frequency of the different Aβ pathologies were investigated. Leptomeningeal CAA, subpial Aβ and cortical CAA did not significantly correlate with Aβ load. When the three different plaque types were examined separately, there was a clear trend of a negative correlation between cored plaque score and Aβ load, both for the whole cohort and for all sub‐groups (Figure 4C), and this was statistically significant for the whole cohort (τ b = −0.37, p < 0.01 Kendall's tau‐b). This contrasted with the consistently positive correlations seen between diffuse plaque score and Aβ load, and between CWP score and Aβ load; these associations were statistically significant for diffuse plaques in the whole cohort (p < 0.001) and in the PSEN1 pre‐codon 200 group (p < 0.01), Figure 4C.
3.4.4. Differences across cortical layers
Visual inspection of the cases revealed that Aβ deposition was not evenly distributed across the cortical layers (see Figure 2). In the whole cohort and within each mutation sub‐group, observed mean Aβ load was consistently numerically highest in layer 3 compared to the remaining layers (whole cohort 14.57% ± 6.33; PSEN1 pre‐codon 200 12.64% ± 6.12; PSEN1 post‐codon 200 15.99% ± 6.88; APP 17.24% ± 6.07).
We assessed whether Aβ load differed between the different layers. There was evidence that this was the case for the whole cohort (p < 0.0001, Friedman ANOVA), with the highest load being observed in upper layers 2 and 3, a pattern that was also generally seen across the three sub‐groups (Figures 3 and 4D). The Aβ load of each individual layer was compared between the mutation sub‐groups, adjusted for APOE4 status. We found evidence (p = 0.007) that Aβ load in layer 5 differed between sub‐groups, with mean Aβ load higher in the PSEN1 post‐codon 200 group compared to both the PSEN1 pre‐codon 200 group (7.16 percentage points higher; 95% CI: 2.9, 11.41; p = 0.003) and the APP group (6.91 percentage points higher; 95% CI: 1.68, 12.15; p = 0.01), after adjusting for APOE4 status. There was weaker evidence (p = 0.06) for differences in Aβ load in layer 4, with PSEN1 post‐codon 200 cases again having higher load than the PSEN1 pre‐codon 200 group (7.99 percentage points higher; 95% CI: 1.51, 14.46; p = 0.02). No significant differences were found for the other layers. These data suggest that mutation location may be associated with the distribution of pathology across some cortical layers but larger sub‐group sizes would be needed to investigate this further.
3.5. Proportion of cortical and leptomeningeal vessels affected by CAA
In our cohort, vessels positive for CAA were observed in the frontal cortices of the following PSEN1 mutations (three intron 4 mutation cases (1–4), E120K, M146I, both E148D, both E280G, L250S, both R278I, double mutation A434T &T291A). CAA was also present in the APP V717L and two APP V717I cases. Three cases (case 1: PSEN1 intron 4, 6: PSEN1 S132A and 19: APP V717I) had no CAA present in the frontal cortices.
3.5.1. Associations with clinical data
We assessed associations between the proportions of vessels with CAA in the cortex and leptomeninges and age at onset or disease duration. In the whole cohort, there was a trend for both older age at onset and longer disease duration to be associated with a smaller proportion of vessels with CAA, however these associations were not statistically significant (Figure S1).
3.5.2. Differences between sub‐groups
The proportion of CAA affected vessels was compared between mutation sub‐groups. PSEN1 post‐codon 200 cases had the highest observed proportion of affected vessels in both the cortex and leptomeninges compared to the other groups, with all subjects in the PSEN1 post‐codon 200 mutation sub‐group demonstrating amyloid‐beta deposition in at least 6% of their cortical vessels and 45% of their leptomeningeal vessels compared to a minimum of 2% and 0% in the PSEN1 pre‐codon 200 group and 1% and 3% in the APP group, respectively. While there was only very weak evidence for differences between the sub‐groups (p = 0.10 and p = 0.08, Kruskal–Wallis), this may have been because of small sub‐group sizes so we investigated pairwise comparisons. The proportion of vessels with cortical CAA was non‐statistically higher (p = 0.06, Dunn's test) in the PSEN1 post‐codon 200 (40.83% ± 21.43) compared to the PSEN1 pre‐codon 200 group (16.78% ± 17.56), and statistically higher (p = 0.02) compared to the APP group (10.75% ± 12.61). Additionally, the proportion of vessels with leptomeningeal CAA was higher (p = 0.07) in the PSEN1 post‐codon 200 group (71.33% ± 18.27) compared to the PSEN1 pre‐codon 200 group (45.43% ± 29.24), and higher (p = 0.01) than the APP group (30.88% ± 26.58) (Figure 5A). There were no differences in the proportions of cortical and leptomeningeal CAA by APOE4 status (p = 0.31, p = 0.47, exact Mann–Whitney–Wilcoxon rank sum).
FIGURE 5.
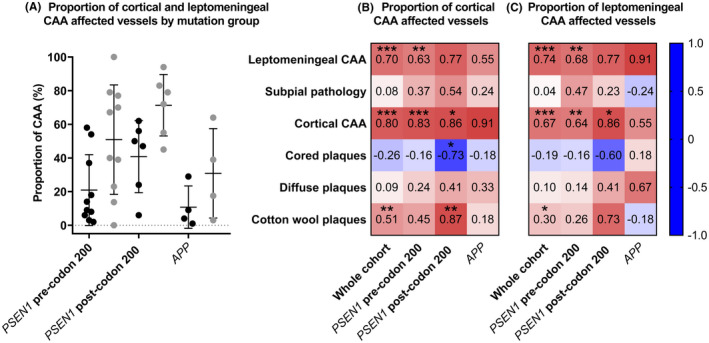
(A) Comparison of the proportion of cortical and leptomeningeal CAA between mutation groups. PSEN1 post‐codon 200 cases have significantly higher cortical and leptomeningeal CAA compared to the APP cases (p = 0.02 and p = 0.01, Dunn's test), with near significant higher proportion of cortical and leptomeningeal CAA compared to the PSEN1 post‐codon 200 group (p = 0.06 and p = 0.07). Bars represent mean and SD. Correlations between the proportion of CAA affected vessels in the cortex (B) and leptomeninges (C) and the total Aβ pathology, leptomeningeal and cortical CAA scores for the whole cohort and mutation sub‐groups. Kendall's tau‐b τ b correlation coefficients are shown. Red = positive correlation. Blue = negative correlation. Asterisks represent statistically significant correlations: *p < 0.05, **p < 0.01, ***p < 0.001
3.5.3. Correlations with Aβ pathologies
We assessed whether the proportions of cortical and leptomeningeal CAA affected vessels correlated with the total Aβ pathology scores. In contrast to the pattern seen for all other Aβ pathology scores, we found a negative correlation between cored plaque scores and the proportions of both cortical and leptomeningeal CAA affected vessels. While these relationships did not reach statistical significance, the negative correlations were seen consistently in the whole cohort, in all sub‐groups for cortical CAA, and in all but the small APP sub‐group for leptomeningeal CAA (see Figure 5B,C for the correlation coefficients). All other total Aβ pathology scores were consistently positively correlated with the proportions of both cortical and leptomeningeal CAA affected vessels in the whole cohort and also in all the sub‐groups, except again in the small APP group. As expected CAA scores and the proportions of CAA vessels were positively correlated and in the whole cohort this was statistically significant (all p < 0.001) (Figure 5B,C). Of particular interest were the results for CWPs, where in the whole cohort there was evidence for a positive correlation with the proportion of cortical CAA (τ b = 0.51, p ≤ 0.01, Kendall's tau‐b) and a consistent but weaker correlation with leptomeningeal CAA (τ b = 0.30, p < 0.05). We also assessed if the mean Aβ load for the whole cohort was significantly associated with the proportions of cortical CAA or leptomeningeal CAA, with no statistically significant correlations found (τ b = 0.17 and τ b = 0.10, respectively).
4. DISCUSSION
In this study, we investigated associations between genotype, clinical data and Aβ pathology in a cohort of FAD cases. We found no differences in the presence of Aβ pathologies and APOE status, although the overall amount of Aβ was found to be higher in APOE4 carriers than non‐carriers. In all mutation carriers, the highest amount of Aβ was found in layer 3. Aβ load also correlated with the amount of diffuse deposits found throughout the cortical layers rather than the neuritic plaques. It was also of interest that two cases with the same autosomal dominant mutations, R278I, displayed different patterns of Aβ deposition. Although one case carried an APOE4 allele, suggesting this may play a role in modifying the deposition of Aβ. We found positive associations between the frequency of CWPs and both cortical and leptomeningeal CAA (total CAA scores and proportions of CAA affected vessels). Furthermore, the presence of CWPs was observed to be higher in lower cortical layers in some PSEN1 post‐codon 200 cases, which may relate to observed clinical and pathological differences in FAD patients with mutations located post‐codon 200. In this study we used a pan‐Aβ antibody which stains all Aβ peptides and morphological conformations containing amino acid residues 8–17. It is clearly recognised within the literature that different isoforms and species are found in the different pathologies. However, in this study, we investigated the overall deposition of the Aβ peptides across the cortical layers rather than dissecting out the isoforms present in relation to the different structural forms of the peptides (soluble, oligomeric or amyloid). This could be an interesting direction for future work now that we have an overview of the total Aβ deposition.
In line with previous findings, our data show that mutation type is associated with age at onset in FAD. Specifically, PSEN1 mutations located pre‐codon 200 had a significantly earlier age at onset than APP mutations. In our cohort, females had an earlier age at onset than males, after adjusting for mutation sub‐group and APOE4 status. A trend for younger age at onset in females has also been observed in a meta‐analysis of FAD, although it did not reach statistical significance (9). Our findings highlight the importance of considering and investigating potential sex differences in disease manifestation and progression in FAD and the mechanisms that may be driving such differences. This is an area of growing interest in AD research (43), which could be informed by the insights gained from the study of FAD, where patients are typically young and lack the co‐morbidities that can confound studies of older individuals with dementia.
Similar to findings in other FAD studies, APOE4 status in our cohort did not affect age at onset (9, 44). However, we found that possession of at least one ε4 allele was associated with longer disease duration, after adjusting for mutation sub‐group and sex. Our pathological analysis did not demonstrate evidence of significant differences in the type, frequency or layer distribution of Aβ pathology between APOE4 carriers and non‐carriers, raising the question of whether the differences we observed in disease duration may be mediated by mechanisms beyond amyloid‐beta. A recent survival analysis from our centre (which included the individuals in the current study) suggested PSEN1 mutation carriers with an APOE4 allele may have longer survival (14). However, the effect of the APOE4 genotype on disease duration in FAD is not well known and in sporadic AD its effect is uncertain. In a meta‐analysis including up to 1,700 sporadic AD cases, it was shown to have no effect (45), however, in two prior studies of AD patients, a trend towards increased disease duration in APOE4 carriers was observed (46) and, separately, increased survival of APOE4 carriers was found (47). Our finding that APOE4 was associated with increased disease duration in this FAD cohort suggests that the impact of APOE4 on disease progression in AD should be explored further in future larger studies.
In the whole cohort, total pathological Aβ plaques were not significantly associated with age at onset or disease duration. Similarly, Aβ load was not significantly associated with age at onset, although Aβ load in layer 6 was positively associated with disease duration and this trend was consistent for the remaining layers. However, the associations require verifying in a larger cohort. Although differing Aβ distribution between individual cases was evident macroscopically, Aβ load was consistently higher in layer 3 for all sub‐groups, as found in previous studies of sporadic and familial AD (25, 26, 27, 30). Various hypotheses suggest an increased vulnerability to AD pathomechanisms of certain neuronal populations (48, 49, 50, 51, 52). In particular, there is some evidence that cortical–cortical connections, predominant in layer 3, may be particularly vulnerable (49). Interestingly, it has been shown in murine models that layer 3 neurons are more susceptible to Aβ toxicity than layer 5–6 neurons, indicating that there may be some intrinsic factor involved (53). A range of earlier studies has carried out investigations into amyloidogenic tau pathology in AD cortical tissue (indicated by senile plaques, neuritic plaques or NFT’s), finding this to be observed with a predominance for cortical layer 3 and 5 (24, 25, 26, 27, 54) and highlighting preferential deposition and disruption in cortico‐corticol connective regions.
Despite there being no significant differences in the total Aβ pathology scores between groups, a significantly higher frequency of CWPs in layers 5 & 6 was observed in the PSEN1 post‐codon 200 mutation carriers. This finding should be interpreted with some caution because of the small number of subjects. However, CWP pathology has often been seen in kindreds and individual cases with PSEN1 mutations post‐codon 200, such as the PSEN1 exon 9 deletion, G217R, P264L and L435F mutations (55, 56, 57, 58, 59). This suggests certain mutations post‐codon 200 support the development of CWPs; however, they can also be found in cases with PSEN1 pre‐codon 200 mutations (60). The reasons why different types of Aβ deposit are formed, and the role that these various Aβ pathologies play in AD pathogenesis, are not yet fully understood. One possibility is that differences in production of Aβ peptides (e.g., Aβ40, 42, 43) play a role in influencing plaque deposition. Different FAD mutations cause distinct alterations in the spectrum of Aβ peptides produced (16) and these peptides vary in their potential for aggregation, which is likely to influence where they deposit and how they are involved in any associated vascular or inflammatory processes. For example, two of the individuals in the post‐codon 200 group in this study carried the R278I mutation. This, and other post‐codon 200 mutations, have been found to cause a particular increase in Aβ43 together with impairment of PSEN1 autoproteolysis (16, 61). It has been speculated that altered processing of substrates other than APP, at an endopeptidase level, may also contribute to variability in the effects of PSEN1 mutations (3, 61) potentially representing an additional factor contributing to heterogeneity in Aβ pathology. Understanding the aetiology of CWPs is important because of their association with clinical phenotypes such as spastic paraparesis and other motor symptoms (31, 32, 62). Motor symptoms and spastic paraparesis are observed more frequently in PSEN1 post‐codon 200 cases (3, 63, 64, 65, 66), supporting a link between the clinical phenotype and CWP pathology. The increased frequency of CWPs in lower layers, especially layer 5, that we observed in our PSEN1 post‐codon 200 mutation cohort is perhaps consistent with this clinicopathological association as cortical layer 5 contains projections to the striatum, which is involved in motor control (67). While the evidence suggests a link, firm conclusions cannot be drawn as CWPs can exist without spastic paraparesis and CWPs can be found in sporadic AD, albeit infrequently (68, 69). Further investigation of the localisation of CWPs and their relationship with spastic paraparesis, particularly in PSEN1 post‐codon 200 cases, may provide more detail on how these observations may be linked. In future studies with larger cohorts, it would be of interest to also examine the influence of PSEN1 mutation location in more detail, moving beyond a simple codon 200 cut‐off to investigate the structural and functional consequences of mutations in different positions and how these relate to neuropathological features.
Analysis of the proportions of CAA revealed no significant correlations with Aβ load; however, positive correlations between CAA and CWPs were found in the full cohort and in the PSEN1 post‐codon 200 group separately using both the scoring system and vessel count measures. Previously, CWPs have been observed in FAD cases with severe CAA (70) and CWPs in FAD have been found to correlate with white matter hyperintensities on MRI, which are a potential imaging marker of CAA (42). However, the precise link between these pathologies is unclear and interestingly CAA is predominantly composed of Aβ40 (71) while CWPs are composed mainly of Aβ42 (69, 72, 73, 74), although Aβ40 can be present (33). Despite this, our findings relating to CWPs and CAA, especially in reference to PSEN1 post‐codon 200 mutations, supports a pathological link, which requires verification in studies with larger cohorts. Given further immunohistochemical investigations with peptide‐specific antibodies, we would expect to see predominance of Aβ40 in CAA and cored plaques, with greater abundance of Aβ42 in diffuse and CWP. The predisposition of Aβ40 to deposit in CAA or cored plaques may also account for the observed negative correlation between these two pathologies, wherein Aβ40 may be driven towards one of these main fates – depositing predominantly as either CAA or cored plaques.
In our cohort, cortical and leptomeningeal CAA were not correlated with age at onset or disease duration. There was a suggestion of differences between mutation sub‐groups but this would require larger sample sizes to investigate further. Interestingly, some of our cases had little to no CAA, suggesting it is not an inevitable consequence of Aβ pathology. Analysing a wider range of genetic cases could help decipher how different mutations are implicated in the development of CAA, for instance, because of effects on PSEN1 substrates other than APP (75) or on Aβ profiles and their aggregation potentials (11, 18). Despite the heterogeneity of CAA, understanding its role is important, especially as amyloid‐beta modifying therapies may lead to an increase in CAA and with adverse vascular events in AD, with APOE status influencing outcomes (76, 77, 78, 79).
All FAD cases in our cohort had been neuropathologically diagnosed with end‐stage severe AD. This study focussed on the distribution of Aβ throughout the cortical layers. The study did not focus on tau pathology as differences in tau deposition between cases would be difficult to observe using immunohistochemical analysis as the frontal cortex is full of neuropil threads. However, using a pan‐ Aβ immunohistochemical analysis binding the majority of Aβ species, we found that a more detailed investigation of the type and layer distribution of amyloid‐beta pathology highlighted differences between cases, with mutation location associated with certain clinical and pathological features. Broad pathological categorisations may mask more detailed mutation‐specific effects on pathogenic processes. Investigating these differences could provide better knowledge of the mechanisms of pathology and be an important tool for better understanding heterogeneity in AD. However, our cohort was small with only one case with certain mutations available. It could be misleading to generalise from one mutation case to all others, and it would be essential to have a larger number of cases to make true mutation‐based connections to pathological heterogeneity. Related to this, despite the distinct pathology patterns we observed between mutation sub‐groups, differences in pathology were also noted between cases with the same mutation indicating that other factors may influence pathological and clinical features in addition to the causative FAD mutations. We have highlighted some of those factors, with APOE4 genotype status being of importance and associated with increased disease duration. Additionally, we observed sex differences in age at onset with a younger age at onset in females, although larger studies will be needed to confirm this finding. Further investigation of the role of these factors in FAD may provide insights into how they are involved in the mechanisms underlying AD in general.
CONFLICT OF INTEREST
The authors have no conflict of interests.
AUTHOR CONTRIBUTIONS
Tammaryn Lashley and Natalie S. Ryan conceptualised the study. Material preparation, data collection and analysis were performed by Nanet Willumsen, and statistical analysis by Teresa Poole and Jennifer M. Nicholas. The first draft of the manuscript was written by Nanet Willumsen and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
ETHICAL APPROVAL
Ethical approval was obtained for the use of post‐mortem human brain tissue from the Local Research Ethics Committee of the National Hospital for Neurology and Neurosurgery. TL and NR drafted the study design, NW undertook the wet lab work and analysis. All authors read and contributed to the final manuscript.
Supporting information
FIGURE S1 Association between clinical data and the proportion of cortical and leptomeningeal CAA affected vessels
ACKNOWLEDGEMENTS
Tammaryn Lashley is supported by an Alzheimer's Research UK senior fellowship. Nanet Willumsen is supported by an Alzheimer's Research UK PhD Studentship. Natalie Ryan is supported by a University of London Chadburn Academic Clinical Lectureship in Medicine. The Queen Square Brain Bank is supported by the Reta Lila Weston Institute for Neurological Studies and the Medical Research Council. Teresa Poole and Jennifer Nicholas’ academic collaboration with the Dementia Research Centre, UCL, is supported by a grant to the DRC from Alzheimer's Research UK. This work was supported by the NIHR UCLH/UCL Biomedical Research Centre, the Rosetrees Trust, the MRC Dementia Platform UK and the UK Dementia Research Institute at UCL, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK.
Willumsen N, Poole T, Nicholas JM, Fox NC, Ryan NS, Lashley T. Variability in the type and layer distribution of cortical Aβ pathology in familial Alzheimer’s disease. Brain Pathol. 2022;32:e13009. 10.1111/bpa.13009
Natalie S. Ryan and Tammaryn Lashley are Joint senior authors.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–90. [DOI] [PubMed] [Google Scholar]
- 2. Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493(7430):56–61. [DOI] [PubMed] [Google Scholar]
- 3. Ryan NS, Nicholas JM, Weston PSJ, Liang Y, Lashley T, Guerreiro R, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer's disease: a case series. Lancet Neurol. 2016;15(13):1326–35. [DOI] [PubMed] [Google Scholar]
- 4. Ryan NS, Rossor MN. Correlating familial Alzheimer's disease gene mutations with clinical phenotype. Biomarkers in Medicine. 2010;4(1):99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shepherd C, McCann H, Halliday GM. Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathol. 2009;118(1):37–52. [DOI] [PubMed] [Google Scholar]
- 6. Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, et al. Autosomal‐dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimer's Research & Therapy. 2011;3(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joshi A, Ringman JM, Lee AS, Juarez KO, Mendez MF. Comparison of clinical characteristics between familial and non‐familial early onset Alzheimer's disease. J Neurol. 2012;259(10):2182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koriath C, Kenny J, Adamson G, Druyeh R, Taylor W, Beck J, et al. Predictors for a dementia gene mutation based on gene‐panel next‐generation sequencing of a large dementia referral series. Mol Psychiatry. 2020;25(12):3399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryman DC, Acosta‐Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2014;83(3):253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shea YF, Chu LW, Chan AOK, Ha J, Li Y, Song YQ. A systematic review of familial Alzheimer's disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J Formos Med Assoc. 2016;115(2):67–75. [DOI] [PubMed] [Google Scholar]
- 11. Mann DMA, Pickering‐Brown SM, Takeuchi A, Iwatsubo T, the members of the Familial Alzheimer’s Disease Pathology Study G . Amyloid angiopathy and variability in amyloid β deposition is determined by mutation position in presenilin‐1‐linked Alzheimer’s disease. Am J Pathol. 2001;158(6):2165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, García G, et al. Apolipoprotein Eε4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54(2):163–9. [DOI] [PubMed] [Google Scholar]
- 13. Godbolt AK, Cipolotti L, Watt H, Fox NC, Janssen JC, Rossor MN. The natural history of Alzheimer disease: A longitudinal presymptomatic and symptomatic study of a familial cohort. Arch Neurol. 2004;61(11):1743–8. [DOI] [PubMed] [Google Scholar]
- 14. Pavisic IM, Nicholas JM, O'Connor A, Rice H, Lu K, Fox NC, et al. Disease duration in autosomal dominant familial Alzheimer disease. A survival analysis. Neurology Genetics. 2020;6(5):e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ringman JM, Monsell S, Ng DW, Zhou Y, Nguyen A, Coppola G, et al. Neuropathology of autosomal dominant Alzheimer disease in the national Alzheimer coordinating center database. J Neuropathol Exp Neurol. 2016;75(3):284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arber C, Toombs J, Lovejoy C, Ryan NS, Paterson RW, Willumsen N, et al. Familial Alzheimer’s disease patient‐derived neurons reveal distinct mutation‐specific effects on amyloid beta. Mol Psychiatry. 2020;25(11):2919–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda‐Falla D, Lleo A, et al. Qualitative changes in human γ‐secretase underlie familial Alzheimer's disease. J Exp Med. 2015;212(12):2003–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hatami A, Monjazeb S, Milton S, Glabe CG. Familial Alzheimer's disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid‐ β peptide. J Biol Chem. 2017;292(8):3172–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cairns NJ, Perrin RJ, Franklin EE, Carter D, Vincent B, Xie M, et al. Neuropathologic assessment of participants in two multi‐center longitudinal observational studies: The Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology. 2015;35(4):390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dickson TC, Vickers JC. The morphological phenotype of β‐amyloid plaques and associated neuritic changes in Alzheimer's disease. Neuroscience. 2001;105(1):99–107. [DOI] [PubMed] [Google Scholar]
- 21. Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J. Spectrum of morphological appearance of amyloid deposits in Alzheimer's disease. Acta Neuropathol. 1989;78(4):337–47. [DOI] [PubMed] [Google Scholar]
- 22. Maarouf CL, Daugs ID, Spina S, Vidal R, Kokjohn TA, Patton RL, et al. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol Neurodegener. 2008;3(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colle MA, Duyckaerts C, Laquerrière A, Pradier L, Czech C, Checler F, et al. Laminar specific loss of isocortical presenilin 1 immunoreactivity in Alzheimer's disease. Correlations with the amyloid load and the density of tau‐positive neurofibrillary tangles. Neuropathol Appl Neurobiol. 2000;26(2):117–23. [DOI] [PubMed] [Google Scholar]
- 24. Dèlaere P, Duyckaerts C, Brion JP, Poulain V, Hauw JJ. Tau, paired helical filaments and amyloid in the neocortex: a morphometric study of 15 cases with graded intellectual status in aging and senile dementia of Alzheimer type. Acta Neuropathol. 1989;77(6):645–53. [DOI] [PubMed] [Google Scholar]
- 25. Duyckaerts C, Hauw J‐J, Bastenaire F, Piette F, Poulain C, Rainsard V, et al. Laminar distribution of neocortical senile plaques in senile dementia of the Alzheimer type. Acta Neuropathol. 1986;70(3):249–56. [DOI] [PubMed] [Google Scholar]
- 26. Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci USA. 1985;82(13):4531–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rogers J, Morrison J. Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer's disease. The Journal of Neuroscience. 1985;5(10):2801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delaère P, Duyckaerts C, He Y, Piette F, Hauw JJ. Subtypes and differential laminar distributions of βA4 deposits in Alzheimer's disease: relationship with the intellectual status of 26 cases. Acta Neuropathol. 1991;81(3):328–35. [DOI] [PubMed] [Google Scholar]
- 29. Majocha RE, Benes FM, Reifel JL, Rodenrys AM, Marotta CA. Laminar‐specific distribution and infrastructural detail of amyloid in the Alzheimer disease cortex visualized by computer‐enhanced imaging of epitopes recognized by monoclonal antibodies. Proc Natl Acad Sci U S A. 1988;85(16):6182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Armstrong RA. Laminar distribution of β‐amyloid (Aβ) peptide deposits in the frontal lobe in familial and sporadic Alzheimer's disease. Folia Neuropathol. 2015;53(1):15–23. [DOI] [PubMed] [Google Scholar]
- 31. Karlstrom H, Brooks WS, Kwok JBJ, Broe GA, Kril JJ, McCann H, et al. Variable phenotype of Alzheimer’s disease with spastic paraparesis. J Neurochem. 2008;104(3):573–83. [DOI] [PubMed] [Google Scholar]
- 32. Zhang S, Lei C, Liu P, Zhang M, Tao W, Liu H, et al. Association between variant amyloid deposits and motor deficits in FAD‐associated presenilin‐1 mutations: a systematic review. Neurosci Biobehav Rev. 2015;56:180–92. [DOI] [PubMed] [Google Scholar]
- 33. Mann DMA, Takeuchi A, Sato S, Cairns NJ, Lantos PL, Rossor MN, et al. Cases of Alzheimer's disease due to deletion of exon 9 of the presenilin‐1 gene show an unusual but characteristic β‐amyloid pathology known as ‘cotton wool’ plaques. Neuropathol Appl Neurobiol. 2001;27(3):189–96. [DOI] [PubMed] [Google Scholar]
- 34. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239–59. [DOI] [PubMed] [Google Scholar]
- 35. Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: The CERAD experience, part XV. Neurology. 1996;46(6):1592–6. [DOI] [PubMed] [Google Scholar]
- 36. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skrobot OA, Attems J, Esiri M, Hortobágyi T, Ironside JW, Kalaria RN, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain. 2016;139(11):2957–69. [DOI] [PubMed] [Google Scholar]
- 38. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–800. [DOI] [PubMed] [Google Scholar]
- 39. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lashley T, Holton JL, Gray E, Kirkham K, O'Sullivan SS, Hilbig A, et al. Cortical α‐synuclein load is associated with amyloid‐β plaque burden in a subset of Parkinson's disease patients. Acta Neuropathol. 2008;115(4):417–25. [DOI] [PubMed] [Google Scholar]
- 41. Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, et al. The apolipoprotein E ε4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer's disease and Lewy body variant. Neurology. 1996;47(1):190–6. [DOI] [PubMed] [Google Scholar]
- 42. Ryan NS, Biessels GJ, Kim L, Nicholas JM, Barber PA, Walsh P, et al. Genetic determinants of white matter hyperintensities and amyloid angiopathy in familial Alzheimer's disease. Neurobiol Aging. 2015;36(12):3140–51. [DOI] [PubMed] [Google Scholar]
- 43. Ferretti MT, Iulita MF, Cavedo E, Chiesa PA, Schumacher Dimech A, Santuccione Chadha A, et al. Sex differences in Alzheimer disease ‐ the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457–69. [DOI] [PubMed] [Google Scholar]
- 44. Forleo P, Nacmias B, Tedde A, Latorraca S, Piacentini S, Marcon C, et al. Presenilin genes analysis in Italian families with early‐onset Alzheimer's disease. Ital J Neurol Sci. 1997;18(4):27. [Google Scholar]
- 45. Allan CL, Ebmeier KP. The influence of ApoE4 on clinical progression of dementia: A meta‐analysis. Int J Geriatr Psychiatry. 2011;26(5):520–6. [DOI] [PubMed] [Google Scholar]
- 46. Masullo C, Daniele A, Seripa D, Filippini V, Gravina C, Carbone G, et al. Apolipoprotein E genotype in sporadic early‐ and late‐onset Alzheimer's disease. Dement Geriatr Cogn Disord. 1998;9(3):121–5. [DOI] [PubMed] [Google Scholar]
- 47. Basun H, Grut M, Winblad B, Lannfelt L. Apolipoprotein ε4 allele and disease progression in patients with late‐onset Alzheimer's disease. Neurosci Lett. 1995;183(1–2):32–4. [DOI] [PubMed] [Google Scholar]
- 48. Capetillo‐Zarate E, Staufenbiel M, Abramowski D, Haass C, Escher A, Stadelmann C, et al. Selective vulnerability of different types of commissural neurons for amyloid β‐protein‐induced neurodegeneration in APP23 mice correlates with dendritic tree morphology. Brain. 2006;129(11):2992–3005. [DOI] [PubMed] [Google Scholar]
- 49. Delatour B, Blanchard V, Pradier L, Duyckaerts C. Alzheimer pathology disorganizes cortico‐cortical circuitry: direct evidence from a transgenic animal model. Neurobiology of Disease. 2004;16(1):41–7. [DOI] [PubMed] [Google Scholar]
- 50. Hof PR, Cox K, Young WG, Celio MR, Rogers J, Morrison JH. Parvalbumin‐immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J Neuropathol Exp Neurol. 1991;50(4):451–62. [DOI] [PubMed] [Google Scholar]
- 51. Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol. 1991;111(3):293–301. [DOI] [PubMed] [Google Scholar]
- 52. Hof PR, Morrison JH, Cox K. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer's disease: I. Superior frontal and inferior temporal cortex. J Comp Neurol. 1990;301(1):44–54. [DOI] [PubMed] [Google Scholar]
- 53. Romito‐DiGiacomo RR, Menegay H, Cicero SA, Herrup K. Effects of Alzheimer's disease on different cortical layers: the role of intrinsic differences in Aβ susceptibility. J Neurosci. 2007;27(32):8496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kalimo H, Lalowski M, Bogdanovic N, Philipson O, Bird TD, Nochlin D, et al. The Arctic AβPP mutation leads to Alzheimer’s disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta Neuropathol Commun. 2013;1(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Crook R, Verkkoniemi A, Perez‐Tur J, Mehta N, Baker M, Houlden H, et al. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4(4):452–5. [DOI] [PubMed] [Google Scholar]
- 56. Heilig EA, Xia W, Shen J, Kelleher Iii RJ. A presenilin‐1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of γ‐secretase activity. J Biol Chem. 2010;285(29):22350–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martikainen P, Pikkarainen M, Pöntynen K, Hiltunen M, Lehtovirta M, Tuisku S, et al. Brain pathology in three subjects from the same pedigree with presenilin‐1 (PSEN1) P264L mutation. Neuropathol Appl Neurobiol. 2010;36(1):41–54. [DOI] [PubMed] [Google Scholar]
- 58. Norton JB, Cairns NJ, Chakraverty S, Wang J, Levitch D, Galvin JE, et al. Presenilin1 G217R mutation linked to Alzheimer disease with cotton wool plaques. Neurology. 2009;73(6):480–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smith MJ, Kwok JBJ, McLean CA, Kril JJ, Anthony Broe G, Nicholson GA, et al. Variable phenotype of Alzheimer's disease with spastic paraparesis. Ann Neurol. 2001;49(1):125–9. [DOI] [PubMed] [Google Scholar]
- 60. Sutovsky S, Smolek T, Turcani P, Petrovic R, Brandoburova P, Jadhav S, et al. Neuropathology and biochemistry of early onset familial Alzheimer’s disease caused by presenilin‐1 missense mutation Thr116Asn. J Neural Transm. 2018;125(6):965–76. [DOI] [PubMed] [Google Scholar]
- 61. Veugelen S, Saito T, Saido TC, Chávez‐Gutiérrez L, De Strooper B. Familial Alzheimer's disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron. 2016;90(2):410–6. [DOI] [PubMed] [Google Scholar]
- 62. Brooks WS, Kwok JBJ, Kril JJ, Broe GA, Blumbergs PC, Tannenberg AE, et al. Alzheimer's disease with spastic paraparesis and ‘cotton wool’ plaques: Two pedigrees with PS‐1 exon 9 deletions. Brain. 2003;126(4):783–91. [DOI] [PubMed] [Google Scholar]
- 63. Dintchov Traykov L, Mehrabian S, Van Den Broeck M, Radoslavova Raycheva M, Cruts M, Kirilova Jordanova A, et al. Novel PSEN1 mutation in a bulgarian patient with very early‐onset Alzheimer's disease, spastic paraparesis, and extrapyramidal signs. Am J Alzheimers Dis Other Demen. 2009;24(5):404–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gómez‐Tortosa E, Barquero S, Barón M, Gil‐Neciga E, Castellanos F, Zurdo M, et al. Clinical‐genetic correlations in familial Alzheimer's disease caused by presenilin 1 mutations. J Alzheimers Dis. 2010;19(3):873–84. [DOI] [PubMed] [Google Scholar]
- 65. Vöglein J, Paumier K, Jucker M, Preische O, McDade E, Hassenstab J, et al. Clinical, pathophysiological and genetic features of motor symptoms in autosomal dominant Alzheimer’s disease. Brain. 2019;142(5):1429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Raman A, Lin X, Suri M, Hewitt M, Constantinescu CS, Phillips MF. A presenilin 1 mutation (Arg278Ser) associated with early onset Alzheimer's disease and spastic paraparesis. J Neurol Sci. 2007;260(1–2):78–82. [DOI] [PubMed] [Google Scholar]
- 67. Gerfen CR, Economo MN, Chandrashekar J. Long distance projections of cortical pyramidal neurons. J Neurosci Res. 2018;96(9):1467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Le TV, Crook R, Hardy J, Dickson DW. Cotton wool plaques in non‐familial late‐onset Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(11):1051–61. [DOI] [PubMed] [Google Scholar]
- 69. Shrimpton AE, Schelper RL, Linke RP, Hardy J, Crook R, Dickson DW, et al. A presenilin 1 mutation (L420R) in a family with early onset Alzheimer disease, seizures and cotton wool plaques, but not spastic paraparesis. Neuropathology. 2007;27(3):228–32. [DOI] [PubMed] [Google Scholar]
- 70. Niwa A, Matsuo K, Shindo A, Yata K, Shiraishi T, Tomimoto H. Clinical and neuropathological findings in a patient with familial Alzheimer disease showing a mutation in the PSEN1 gene. Neuropathology. 2013;33(2):199–203. [DOI] [PubMed] [Google Scholar]
- 71. Tian J, Shi J, Mann DMA. Cerebral amyloid angiopathy and dementia. Panminerva Med. 2004;46(4):253–64. [PubMed] [Google Scholar]
- 72. Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino‐terminally truncated aβ peptide species are the main component of cotton wool plaques. Biochemistry. 2005;44(32):10810–21. [DOI] [PubMed] [Google Scholar]
- 73. Steiner H, Revesz T, Neumann M, Helmut R, Grim MG, Pesold B, et al. A pathogenic presenilin‐1 deletion causes abberrant Aβ42 production in the absence of congophilic amyloid plaques. J Biol Chem. 2001;276(10):7233–9. [DOI] [PubMed] [Google Scholar]
- 74. Verkkoniemi A, Kalimo H, Paetau A, Somer M, Iwatsubo T, Hardy J, et al. Variant Alzheimer disease with spastic paraparesis: Neuropathological phenotype. J Neuropathol Exp Neurol. 2001;60(5):483–92. [DOI] [PubMed] [Google Scholar]
- 75. Chávez‐Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of γ‐Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31(10):2261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Arrighi HM, Barakos J, Barkhof F, Tampieri D, Jack C Jr, Melançon D, et al. Amyloid‐related imaging abnormalities‐haemosiderin (ARIA‐H) in patients with Alzheimer's disease treated with bapineuzumab: a historical, prospective secondary analysis. J Neurol Neurosurg Psychiatry. 2016;87(1):106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Coric V, Van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, et al. Safety and tolerability of the γ‐secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012;69(11):1430–40. [DOI] [PubMed] [Google Scholar]
- 78. Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207. [DOI] [PubMed] [Google Scholar]
- 79. Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, et al. Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Association between clinical data and the proportion of cortical and leptomeningeal CAA affected vessels
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.