

Abstract
Development of inhibitors for histone methyllysine reader proteins is an active area of research due to the importance of reader protein-methyllysine interactions in transcriptional regulation and disease. Optimized peptide-based chemical probes targeting methyllysine readers favor larger alkyllysine residues in place of methyllysine. However, the mechanism by which these larger substituents drive tighter binding is not well understood. This study describes the development of a two-pronged approach combining genetic code expansion (GCE) and structure-activity relationships (SAR) through systematic variation of both the aromatic binding pocket in the protein and the alkyllysine residues in the peptide to probe inhibitor recognition in the CBX5 chromodomain. We demonstrate a novel change in driving force for larger alkyllysines, which weaken cation-π interactions but increases dispersion forces, resulting in tighter binding. This GCE-SAR approach establishes discrete energetic contributions to binding from both ligand and protein, providing a powerful tool to gain mechanistic understanding of SAR trends.
Keywords: post-translational modification, genetic code expansion, structure-activity relationship, chromobox protein 5, cation-π interaction
Graphical Abstract
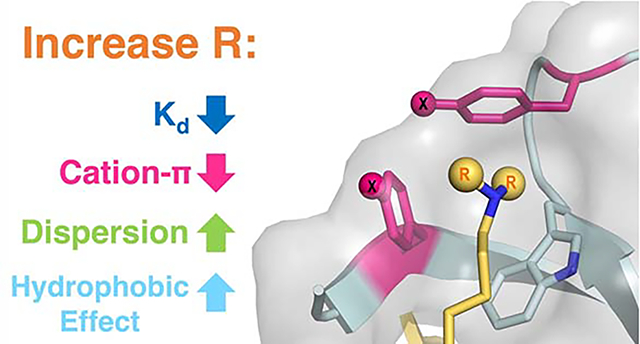
INTRODUCTION
Histone lysine methylation is a well-established molecular signal for gene expression and a validated target for many disease states.1–4 To date, more than 200 human methyllysine (Kmen, n = 0–3) reader proteins have been identified.5 Many have been deemed to be druggable,6 and a number of chemical probes and antagonists targeting these proteins have been reported to date.7–15 Trimethyllysine (Kme3) reader proteins most commonly bind their ligand via an aromatic cage consisting of 2–4 aromatic amino acid side chains. This binding event is generally driven by cation-π interactions between the methyllysine ligand (representing the cation) and the aromatic amino acids (representing the π system).16–21 Peptide-based ligands that replace the native Kme3 substituent with an unnatural alkyllysine substituent, typically a dialkyllysine (Kr2, where r is an alkyl group), have yielded molecules with improved binding affinity and selectivity in addition to enhanced cell permeability.8–9, 22–25 For example, structure-activity relationship studies have shown that increasing the size of the alkyllysine substituent in a peptide-based inhibitor from dimethyllysine (Kme2) to diethyllysine (Ket2) or ethylisopropyllysine (Kei) increases potency for Kme3 reader proteins such as chromobox protein 7 (CBX7).23 However, the energetic contributions of the alkyl group to binding have not been investigated. To this end, we aimed to determine whether the Kr2 groups in these peptidic ligands bound by the same non-covalent mechanism as the native ligand, relying on cation-π interactions, or whether higher affinity is achieved through a different mechanism. To accomplish this, we have developed a new strategy that couples typical structure-activity relationships (SAR) with a systematic variation of the protein binding site via genetic code expansion (GCE). This double systematic variation of both ligand and protein results in not only reporting on which ligand binds with highest affinity, but how the structure of the ligand influences binding. Thus, the GCE-SAR approach provides a powerful new method to gain mechanistic insight coupled with ligand development.
Cation-π interactions are driven by the interaction of a cation with the δ- charge on the face of an aromatic ring, commonly depicted by electrostatic potential maps (ESPs).18–19 The interactions are both distance- and angle-dependent, with the most favorable geometry placing the cation (or δ+) over the center of the aromatic ring at about 3.5 – 4.5 Å. Due to the electrostatic nature of the cation-π interaction, they are sensitive to the charge density of the aromatic ring, such that electron-withdrawing groups weaken cation-π interactions. Previously, our group developed a method using genetic code expansion (GCE) to probe the significance of cation-π interactions at individual amino acids to cation-π-mediated Kme3 binding in the chromodomain of the Drosophila heterochromatin protein 1 (HP1) reader protein.21 In this methodology, we used GCE to systematically tune the electrostatics of individual side chains in the aromatic binding pocket by replacing them with different non-canonical para-substituted phenylalanine derivatives. In this previous study, we found that the binding energy was strongly dependent on the electrostatics of the aromatic rings in the cage, demonstrating the role of cation-π interactions at each position.
Here, we couple this GCE approach with SAR by varying the alkyl groups on lysine to determine how the alkyl substituents influence the binding affinity of the Kme3 reader protein, CBX5 (Figure 1). CBX5 is one of the eight mammalian CBX proteins that contain a chromodomain involved in gene repression.26 CBX5 is evolutionarily related to HP1, a model Kme3 reader protein, and is capable of recognizing Kme3 at lysine 9 on histone 3 (H3K9me3).26–27 Dysregulation of CBX5 has been linked to breast,28–29 lung,30 and colon31 cancers, and as such has been identified as a relevant target for diagnostics and therapeutics.28
Figure 1.
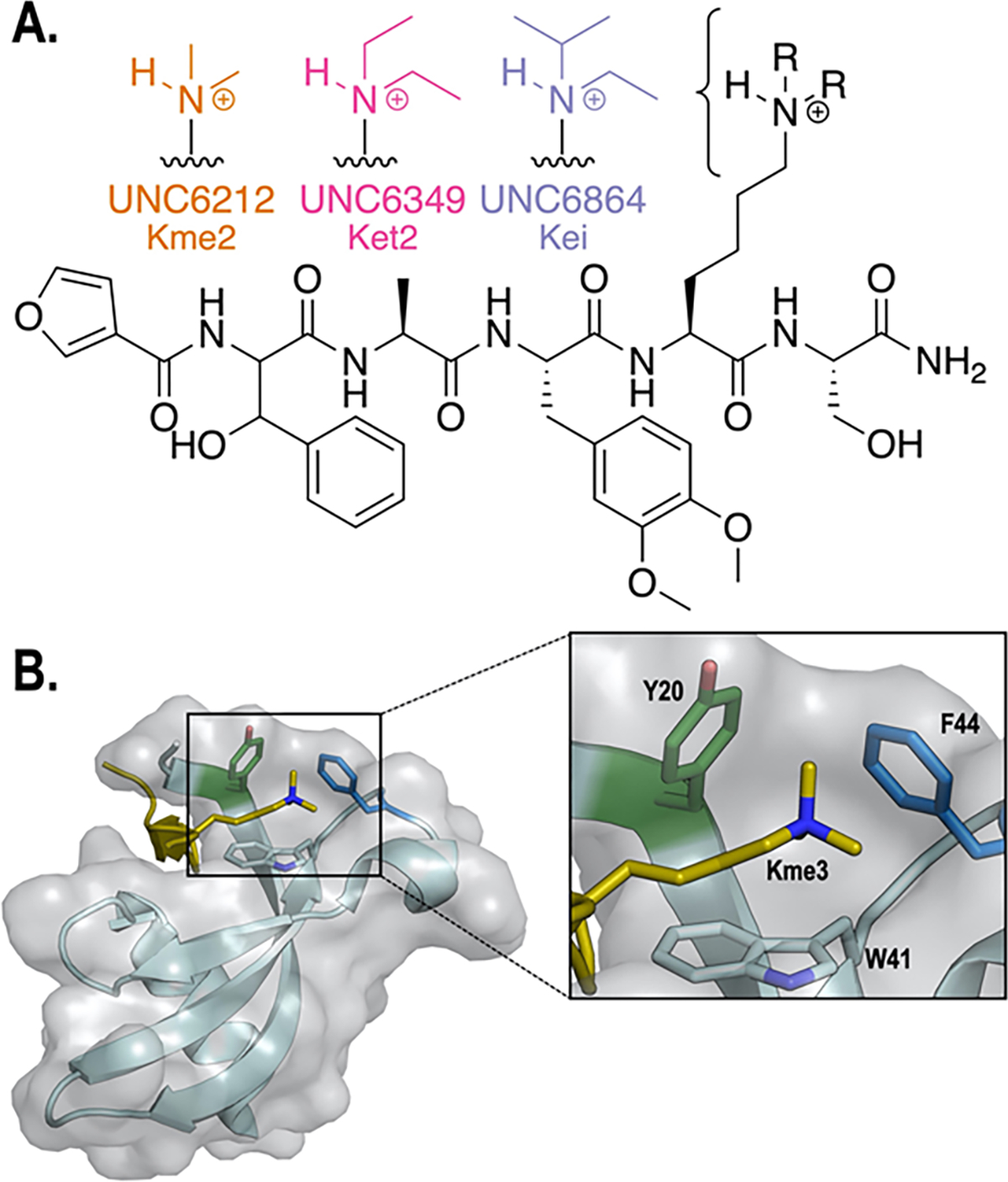
A. Structures of UNC6212 (Kme2, orange), UNC6349 ( Ket2, pink), and UNC6864 (Kei, purple) peptide-based ligands. All ligands share the same scaffold and vary only in the alkylation on lysine. B. Aromatic residues (Y20, W41, and F44) in the CBX5 chromodomain aromatic cage bound to a H3K9me3 histone peptide (PDB: 3FDT).26 Y20 (green) and F44 (blue) were targeted in this study.
We have measured the binding of non-canonical aromatic cage variants of CBX5 to a peptide-based ligand in which the alkylation of lysine was systematically varied, allowing us to probe both the participation of individual residues in the aromatic cage (the π component of the cation-π interaction) and the alkylated lysine residue (the cation component of the cation-π interaction) simultaneously. Consistent with other Kme3 reader proteins, binding is driven by the cation-π interaction. However, while larger alkyl groups generally strengthen binding overall, this study demonstrates that binding is significantly less sensitive to the electrostatics of the aromatic ring as alkyl groups on the lysine ligand increase in size. This suggests that the contribution of cation-π interactions decreases with larger alkyl groups due to dispersion of the charge. The increase in binding strength for larger alkyl groups instead arises from an increase in the contribution of dispersion forces and hydrophobic interactions. These findings provide fundamental insight into the factors that influence the strength of cation-π interactions and inform the framework for designing potent chemical probes targeting methyllysine reader proteins.
RESULTS AND DISCUSSION
Design of CBX5 peptidomimetic ligands.
We previously described a target-class approach for the discovery of novel chromodomain ligands utilizing one-bead-one-compound (OBOC) libraries stemming from hit scaffolds within the target-class.32 This approach employs negative selection steps to increase selectivity toward the chromodomain of interest, which has the added benefit of enabling novel ligand discovery efforts for other members in the target-class. As part of a chromodomain targeted OBOC library screening cascade, CBX5 was screened as a negative selection step to remove ligands with affinity for CBX5. With a growing interest in CBX5, we subsequently analyzed this pool of beads with affinity for CBX5 by MALDI mass spectrometry. This revealed a novel peptidomimetic ligand for CBX5 that incorporates 3,4-dimethoxyphenylalanine, β-phenylserine, and a furan capping residue (Figure 1A). We first synthesized the dimethyllysine (Kme2)-containing ligand, UNC6212, for comparison to the H3K9me2 peptide (ARTKQTARKme2STGGKAY). UNC6212 (Kme2) was found to have a KD for CBX5 of 5.7 μM (Figure S1), which is about 5-fold more potent than both the histone tail peptide H3K9me3, containing Kme3, (KD = 30 μM),26 and H3K9me2, containing Kme2, which we determined to have a KD of 32 μM (Figure S2). The tighter binding of UNC6212 (Kme2) provides a better starting point for the GCE studies, and so this ligand was used for subsequent studies. Next, additional ligands based on this scaffold were designed which vary only in the alkylated lysine residue, to systematically investigate the role of the N-alkyl group on binding. We synthesized a diethyllysine (Ket2)-containing ligand, UNC6349, and an ethylisopropyllysine (Kei)-containing ligand, UNC6864, (Figure 1A). Evaluation of binding to the wild-type protein indicates that the larger alkyl groups on the UNC6349 (Ket2) and UNC6864 (Kei) bind more tightly than UNC6212 (Kme2) (Figure 2, bolded values), following the same trend as has been observed previously in other methyllysine reader proteins.8, 22–25 Thus, this series of ligands was used to probe the effect of alkyl groups in a systematic way by studying the effect of variation of the electrostatics of the aromatic cage of CBX5 with respect to each ligand while keeping all other contacts constant.
Figure 2.
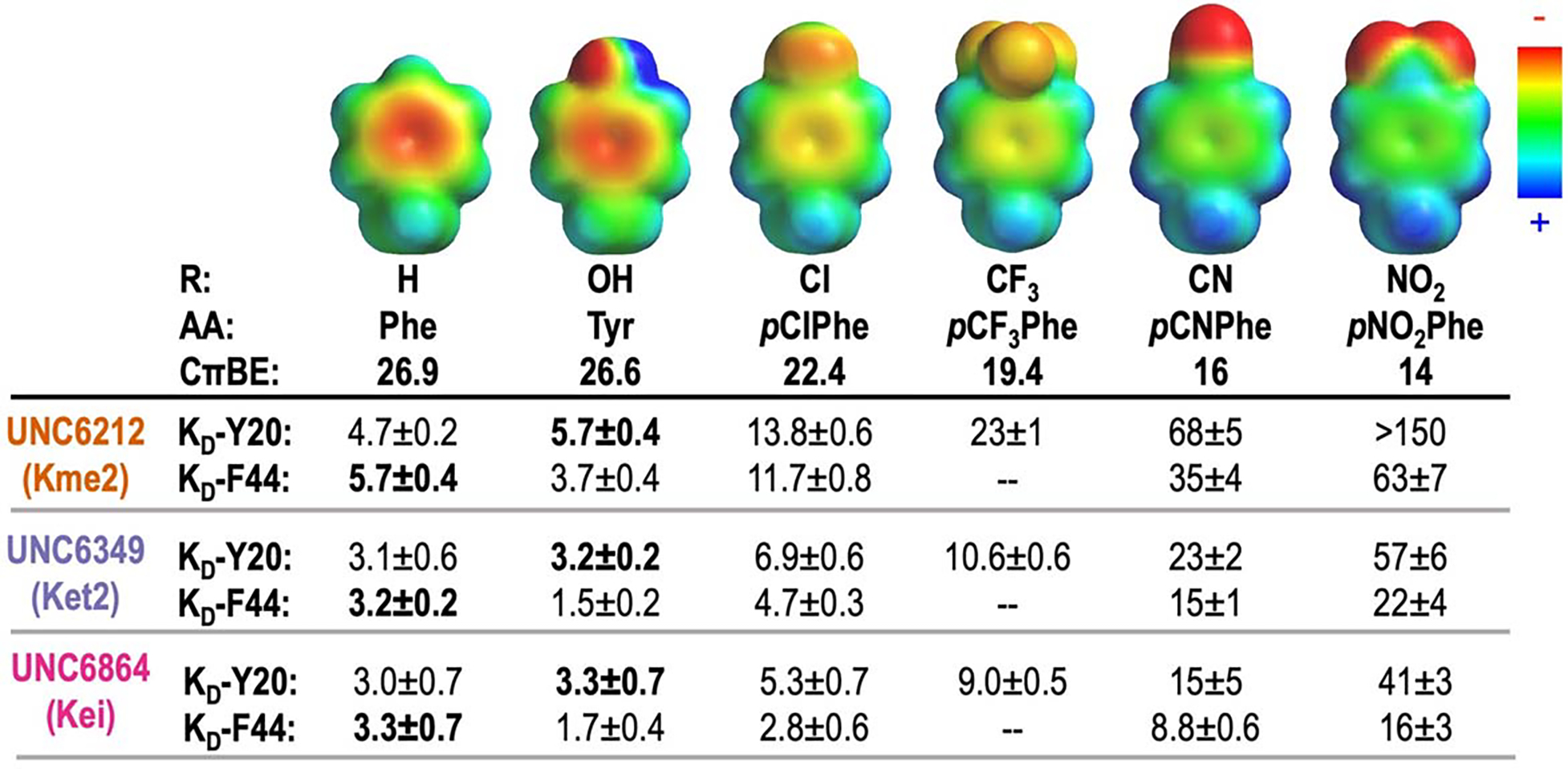
The effect of R-groups on electrostatic surface potential (ESP) maps and calculated cation-π binding energy (CπBE, kcal/mol) of amino acids tested within the aromatic cage. ESP maps were calculated in Spartan at the ωB97X-D/6–31G(d) level of theory. CπBE are calculated for substituted benzenes and Na+.20 Measured KD values (μM) for CBX5 variants at positions Y20 and F44 with peptide-based ligands, UNC6212 (Kme2), UNC6349 (Ket2), and UNC6864 (Kei). Affinities corresponding to wild-type protein are shown in bold. Binding affinity of the F44pCF3Phe variant was not determined. Errors given for KD are the standard deviation for 3 experimental replicates or the highest individual error from an individual experiment among the replicates, whichever is greater.
Development of Aromatic Cage Variants using GCE.
CBX5 binds to Kme3 at lysine 9 on histone 3 using a cage comprised of three aromatic amino acid side chains: Y20, W41, and F44 (Figure 1B). To probe the mechanism of binding to this series of alkyllysine ligands, we used GCE to systematically tune the aromatic cage of CBX5 by replacing individual residues, Y20 and F44, with different para-substituted phenylalanine derivatives, which vary the electrostatics of the aromatic residue.21 Conveniently, these two positions are solvent-exposed and can accommodate substitution at the para-position.26 While W41 also plays a role in binding, it cannot be systematically varied using current methods21 and was left constant throughout these studies. We site-specifically incorporated a series of six different para-substituted phenylalanine derivatives containing substituents with a range of electrostatic potentials (Figure 2; R = H, OH, CF3, Cl, CN, NO2) using a single permissive orthogonal tRNA/aminoacyl tRNA synthetase pair, referred to as pCNF-RS.33 pCNF-RS has been widely established to efficiently incorporate a range of para-substituted phenylalanine derivatives, but does not incorporate the canonical amino acids, tyrosine and phenylalanine. Expressions in the absence of non-canonical amino acid (ncAA) yielded no CBX5 (Figure S3). Successful ncAA incorporation in the presence of ncAA and protein purity were confirmed by LC-MS and canonical amino acid contamination was not detected (Figure S3, Table S1).
Linear Free Energy Relationships with CBX5 variants indicate that binding is driven by cation-π interactions for all dialkyllysine ligands.
To determine the effect of systematic variation of both the ligand and the aromatic cage, the binding affinity between each CBX5 variant and each ligand was measured using isothermal titration calorimetry (ITC; Figure 2, Figures S1, S4, S5, Tables S2–S4). We found that Y20pNO2Phe expressed poorly and bound UNC6212 (Kme2) weakly (>150 μM; Figure 2) and we were unable to obtain reliable binding data for this variant by ITC; therefore, it is excluded from further analysis. To account for the exclusion of Y20pNO2Phe with UNC6212 (Kme2), we included pCF3Phe at the Y20 position.
Analysis of the interactions between the CBX5 variants and ligands show measurable changes in binding affinity, with more electron-withdrawing groups weakening binding for all three ligands (Figure 2), as expected for cation-π interactions. Nonetheless, UNC6864 (Kei) remains the tightest binder regardless of the CBX5 variant. In each case, plotting ΔGbinding with respect to computationally calculated side chain cation-π binding energies (CπBE, Figure 2) results in a linear free energy relationship (LFER, Figure 3). This LFER confirms that tunable cation-π interactions underly these binding events. A LFER is also observed when plotting ΔGbinding with respect to electrostatic potential (ESP, Figure S6), indicating that the binding energy is sensitive to the electrostatics of the aromatic ring, which is also representative of cation-π interactions.
Figure 3.
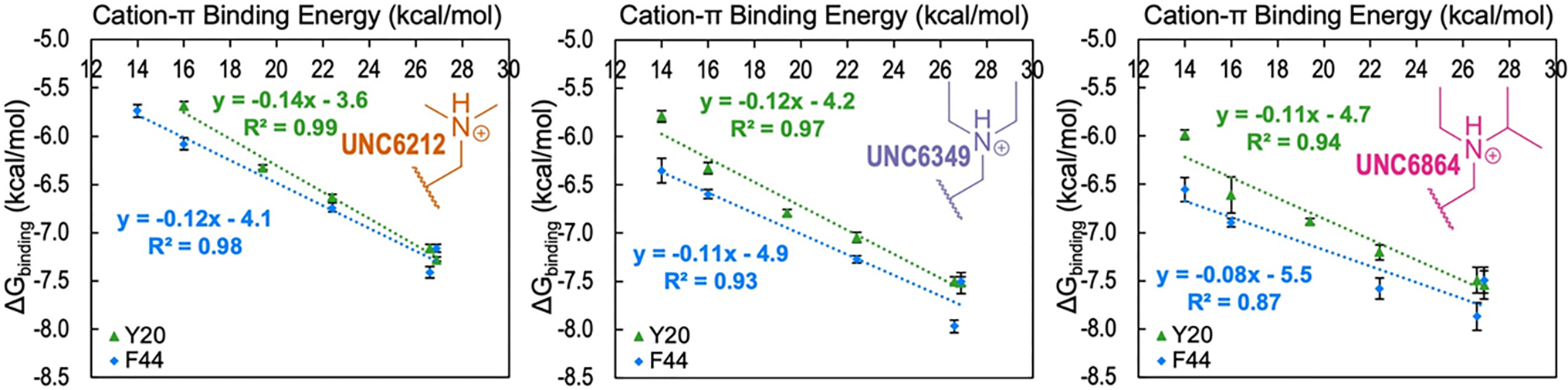
Linear free energy relationship (LFER) plots analyzing the correlation between ΔGbinding with calculated cation-π binding energies (CπBE)20 for a range of Y20 (green) and F44 (blue) variants binding peptidomimetic ligands UNC6212 (Kme2), UNC6349 (Ket2), and UNC6864 (Kei) from left to right. Error bars reflect the standard deviation for replicates or the highest individual error from an individual experiment among the replicates, whichever is greater. The error in the slopes is ±0.01 for both Y20 and F44 with UNC6212 (Kme2), ±0.01 for Y20 and ±0.02 for F44 with UNC6349 (Ket2), and ±0.01 for Y20 and ±0.02 for F44 with UNC6864 (Kei).
To evaluate the contribution of other factors that might influence binding as a result of these mutations, ΔGbinding was also plotted against Log P (a measure of hydrophobicity) and molar refractivity (MR;34 a measure of polarizability and dispersion forces), as changes in the hydrophobic effect and/or dispersion forces could also in theory contribute to the variation in binding affinity.18–19 For all three ligands (containing Kme2, Ket2, and Kei) at both protein positions (Y20 and F44), Log P exhibits no correlation (Figure S7) and molar refractivity provides a weakly negative correlation (R2 < 0.76; Figure S8). This suggests that the observed tunability of these interactions is electrostatic in nature, which is the major contributor to cation-π interactions,18–20 consistent with observations made previously in the HP1 chromodomain.21 Having confirmed that the binding of all three ligands is driven by cation-π interactions, the sections below describe additional insights gained by the SAR-GCE analysis in Figure 3, including contributions of Y20 vs F44, sensitivity to electrostatics as compared to cation-π interactions in another Kme3 reader protein, and how the size of the alkyl group influences the contribution of cation-π interactions and other non-covalent forces.
Y20 and F44 exhibit slight differences in sensitivity to the electrostatics of the ring.
Comparison of the relationship between ΔGbinding and calculated cation-π binding energy show mutations to the Y20 position appear to have a slightly larger effect on binding than at the F44 position (Figure 3, green vs blue lines in each plot). A slightly steeper slope for the LFER observed for the Y20 position relative to F44 for each compound suggests that Y20 makes a slightly stronger cation-π interaction with the ligand than F44. A similar, but more pronounced, trend was observed at the homologous positions in a series of HP1 variants binding H3K9me3.21 As with HP1, analysis of the existing crystal structure of wild-type CBX5 bound to H3K9me3 and calculated interaction energies correlate with these positional differences observed in our experimental LFER analysis. Y20 is more optimally positioned with two methyl groups and a methylene of Kme3 from H3K9me3 making van der Waals contacts (< 4.5 Å) with Y20 while only one methyl group of Kme3 from H3K9me3 makes van der Waals contact with F44 (Figure 4A). In agreement, calculated interaction energies (Eint) predict a more favorable interaction between Y20 and Kme3 (Eint = −10.9 kcal/mol, Figure 4B) than F44 with Kme3 (Eint = −8.2 kcal/mol; Figure 4C).
Figure 4. Structural analysis and computational modeling of CBX5.
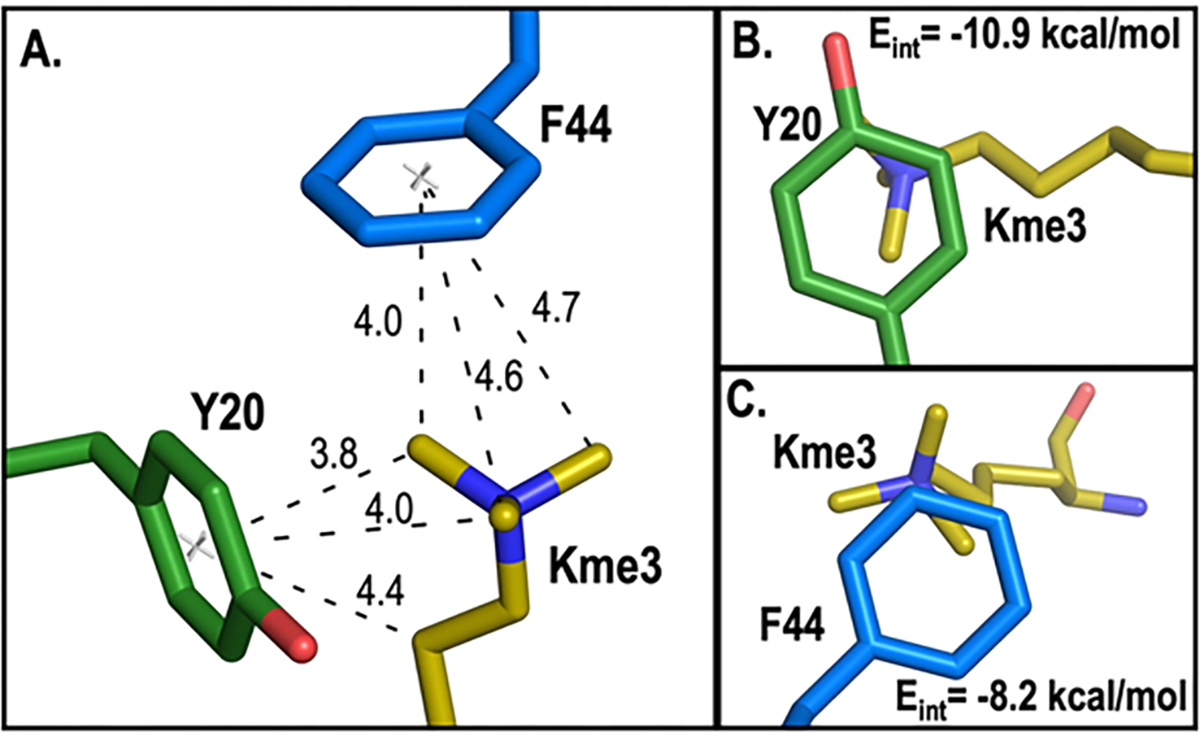
A. Measured distances (Å) between the center of Y20 (green) and F44 (blue) with Kme3 substituents from H3K9me3. B and C. Contact surface of Kme3 with Y20 (B) and F44 (C) viewed normal to the plane of the ring. Interaction energies (Eint) for Kme3 and each aromatic residue are shown and were calculated at the M06–2X/6–311+G(d,p) level of theory.35 In all panels, the structure of CBX5 bound to H3K9me3 is used (PDB: 3FDT).
CBX5 cation-π interactions are more sensitive to electrostatics than are those in HP1.
We have compared the magnitude of the electrostatic tunability of the cation-π interactions in CBX5 to our previously reported cation-π interactions in the HP1 chromodomain21 by comparing the CπBE LFER slopes of CBX5 bound to the UNC6212 (Kme2) to the slopes of the homologous positions in HP1 bound to H3K9me3 histone tail peptide (Table S5). We find that CBX5 shows greater electrostatic tunability at both positions. The magnitude of tunability is about 2-fold greater for CBX5 Y20 relative to HP1 Y24 and 2.5-fold greater for CBX5 F44 relative to HP1 Y48 (Table S5). The structures overlay very well (Figure S9), suggesting that the difference in sensitivity to electrostatics may arise from differences in hydrogen bonding of Kme2 in CBX5 versus Kme3 in HP1. The disubstituted lysines (Kme2, Ket2, and Kei) all have the capacity to form a water-bridged hydrogen bond with the backbone carbonyl of His48, a conserved interaction observed in HP136 (with Glu52) and other CBXs (e.g. CBX7 with Tyr39), that is not possible in the presence of a trimethylated lysine ligand (Figure 5A). The hydrogen bond may perturb the electrostatics of the dimethylammonium in Kme2 that results in greater tunability of the cation-π interaction. It is also worth noting that the binding affinity of CBX5 to UNC6212 (Kme2; 5.7 μM) is about 2.5-fold tighter than that of HP1 with H3K9me3 (14.4 μM),21 and that the differences in electrostatic tunability may also reflect differences in cooperativity between the alkylated lysine and other contacts between each peptide ligand and protein.
Figure 5.
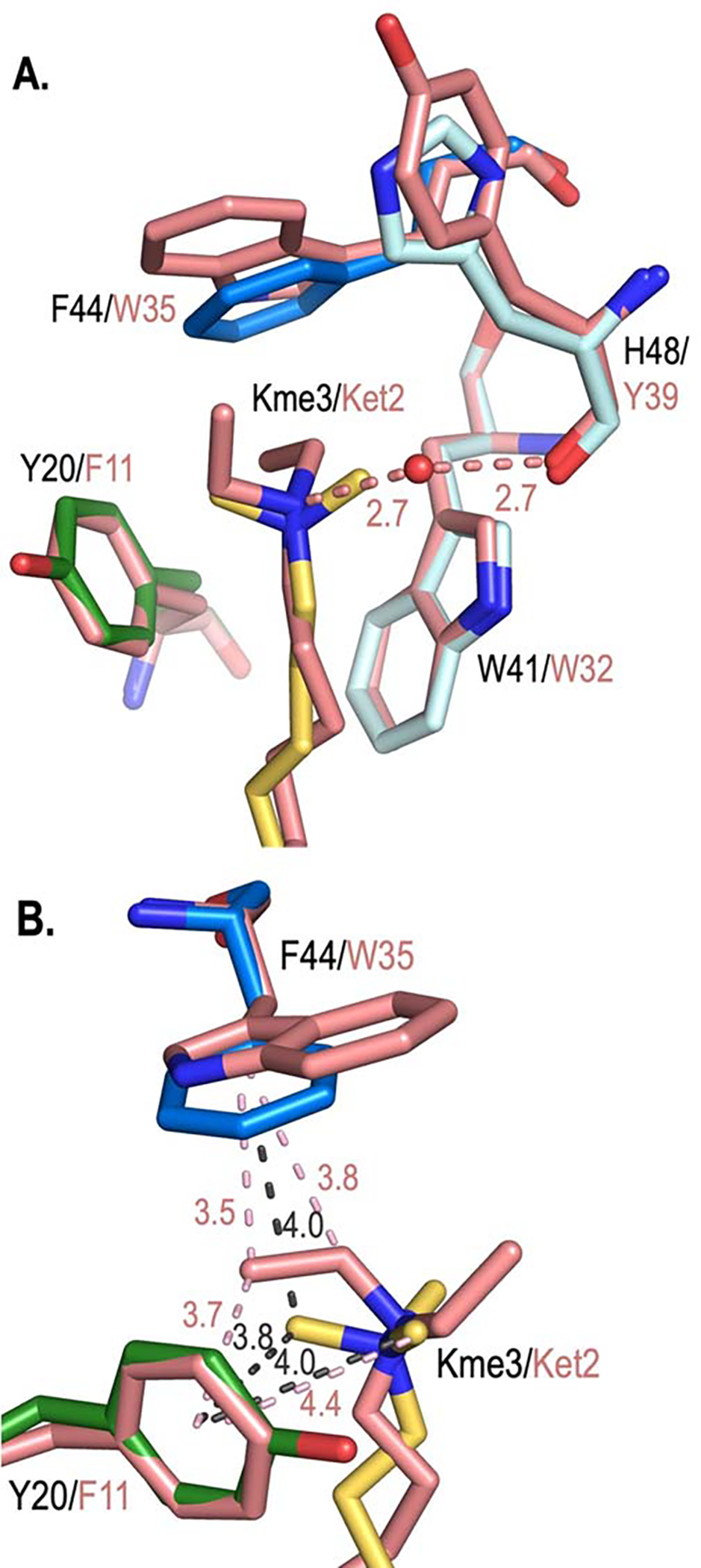
Structural comparison of CBX5 bound to Kme3 with CBX7 bound to UNC3866 containing Ket2. A. The overlay of the aromatic cages of CBX5 (PDB: 3FDT; with Y20 in green and F44 in blue) bound to H3K9me3 (yellow) and CBX7 bound to UNC3866 (containing Ket2; PDB: 5EPJ, salmon) suggest how dialkylated substrates would bind in the aromatic cage of CBX5. A conserved water-bridged hydrogen bond between di-alkylated substrate and the carbonyl of Y39 in CBX7 (H48 in CBX5) is shown with distances (Å). B. Position of Ket2 from UNC3866 bound to CBX7 as a model for binding in CBX5. Distances (Å) from ligand substituents with respect to the center of the ring of Y20 and F44 (CBX5, grey dashed lines) and F11 and W35 (CBX7, salmon dashed lines) are shown. View rotated ~90° with respect to A. W41/W32 and H48/Y39 are removed for clarity.
Cation-π interactions contribute less with larger alkyl groups.
As in a previous in vitro study of other chromodomain reader proteins,23 we find that UNC6349 (Ket2) and UNC6864 (Kei) bind with nearly equal affinity to wild-type CBX5 with binding affinities of 3.2 μM and 3.3 μM, respectively (Figure 2), which fall within error of each other, and with both binding slightly more tightly than the UNC6212 (Kme2; KD = 5.7 μM). With respect to the wild-type protein, adding a bulkier isopropyl group does not appear to improve potency compared to UNC6349 (Ket2). However, by varying the electrostatics of Y20 and F44 in the aromatic cage, we are able to determine differences in how Ket2 and Kei influence binding that are not obvious from analysis of binding to the wild-type protein alone.
Binding energies of the CBX5 variants to all three ligands show a LFER with calculated cation-π binding energies, indicating that cation-π interactions contribute to binding irrespective of the nature of the alkyl substituent (Figure 3). However, the degree of this tunability varies across compounds at both Y20 (green lines, Figure 3) and F44 (blue lines, Figure 3), as reflected by the slopes of these LFERs. A steeper slope, as observed with UNC6212 (Kme2), indicates a more consequential impact of cation-π interactions on binding, while a shallower slope, as observed with UNC6864 (Kei), indicates that cation-π interactions have less impact on binding.
The decreased contribution of cation-π interactions to binding with increase in the size of the alkyl group on lysine can be rationalized by analysis of the partial charges on each Kr2 residue. Partial charges for Kme2, Ket2, and Kei were calculated at the M06–2X/6–311+G(d,p) level of theory35 (Figure 6) and show more dispersed positive charge across larger alkyl groups. The reduction of the δ+ on the carbons adjacent the nitrogen in Ket2 and Kei is consistent with a weaker dependence on electrostatics of the aromatic residues. This phenomenon parallels observations made in gas-phase alkali metal-π binding studies showing the magnitude of a cation-π interaction with benzene varies considerably with the nature of the ion, following a classical electrostatic trend in which the smaller ions participate in stronger cation-π interactions and are more tightly bound (i.e. Li+ > Na+ > K+ > NH4+ > Rb+ > NMe4+).18 A similar observation can be made here, where cation-π interactions are most significant with the smallest alkyllysine substituent (Kme2), where charge is most localized. Another factor that may reduce the contribution of cation-π interactions to the binding energy is a change in geometry of Ket2 as suggested by X-ray structures of other CBX proteins bound to UNC3866, a Ket2-containing ligand.9 The crystal structure of CBX7 bound to UNC3866 (containing Ket2) provides a reasonable model for understanding Ket2 binding in CBX5 due to the high structural conservation of the aromatic cage as well as the water-bridged hydrogen bond observed across these CBX proteins. An overlay of CBX5 bound to H3K9me3 (PDB: 3FDT)26 and CBX7 bound to its Ket2 ligand UNC3866 (PDB: 5EPJ)9 indicate that both carbons of an ethyl group of Ket2 interact with the aromatic residue, with the terminal methyl closest to the aromatic residue (W35 in CBX7, equivalent to F44 in CBX5) in the preferred geometry to participate in cation-π interactions (Figure 5B). Since the δ+ of the terminal methyl group (+0.11) is much less than that of a Kme3 methyl (+0.33), this is also expected to reduce the contribution of cation-π interactions to binding.
Figure 6.
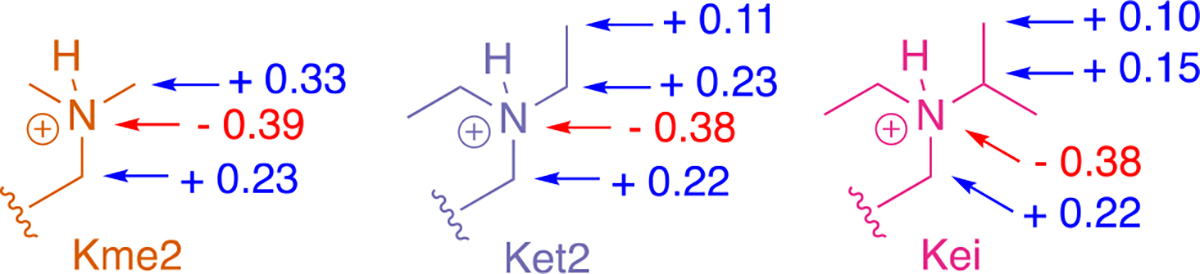
Partial charges calculated for Kme2, Ket2, and Kei lysine substituents using CM5 (charge model 5)37 charges calculated at the M06–2X/6–311+G(d,p) level of theory.
Despite the reduced dependence on cation-π interactions for UNC6349 (Ket2) and UNC6864 (Kei), CBX5 binds about 2-fold tighter to these two ligands than to UNC6212 (Kme2) (Figure 2), indicating that as the alkyllysine substituent increases in size and cation-π interactions become weaker, other non-covalent interactions that are not electrostatically tunable must contribute to this binding interaction in a more significant way. A comparison of X-ray structures of CBX5 bound to H3K9me3 relative to CBX7 bound to UNC3866 (containing Ket2) suggests that Ket2 makes more extensive van der Waals contacts with the aromatic cage (Figure 5B). The interactions between Y20 in CBX5 and the homologous position (F11) in CBX7 are similar with either ligand, both with respect to number of contacts and distances. However, additional van der Waals contacts are observed between one ethyl group of Ket2 and the 5-membered ring of W35 in CBX7, corresponding to F44 in CBX5 (Figure 5B). It has also previously been hypothesized that the isopropyl of Kei makes additional contacts within and more completely fill the aromatic cage in CBX proteins.23
To evaluate the factors that contribute to tighter binding for larger alkyl groups, we plotted ΔGbinding for a single protein variant, pCNPhe or pClPhe, with respect to ESP, Log P, and polarizability of the alkyllysine substituent of the peptidomimetic ligand (Figure 7, S10). These two protein variants were chosen because they had complete data sets and exhibited a greater variance in ΔGbinding than the more electron-rich phenylalanine derivatives. We find that an increase in size in Kr2 has a positive correlation with both hydrophobicity (Log P) and polarizability but a negative correlation with ESP. In all cases, a linear relationship is observed, unlike variation of the aromatic groups, which only exhibited a correlation with calculated cation-π binding energy and ESP. These trends are consistent with an increased contribution from both the hydrophobic effect and dispersion forces with increased size of the alkyl group, which counterbalance the weakening of the cation-π interaction.
Figure 7.
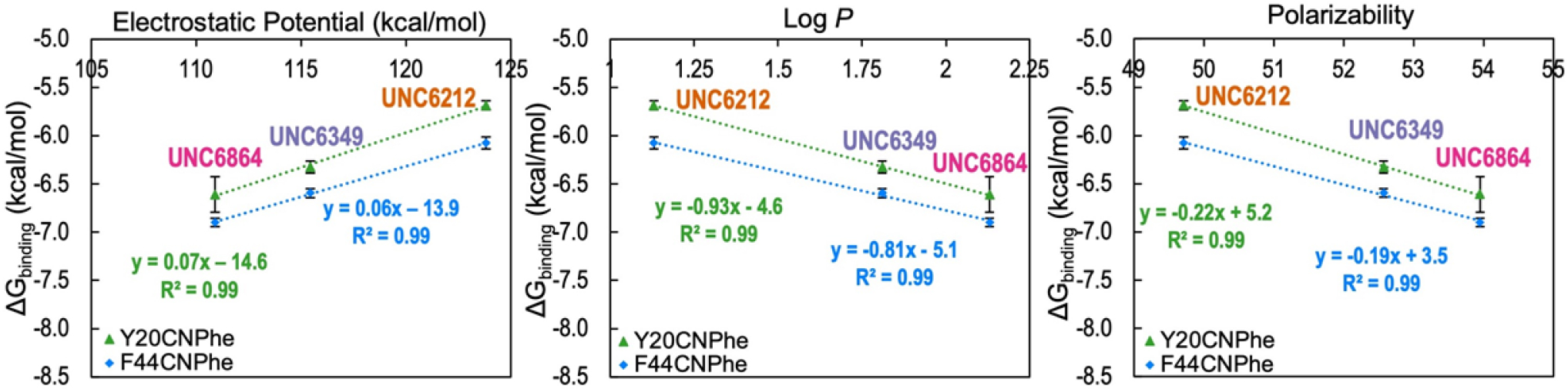
Relationship between ΔGbinding and substituent characteristics for pCNPhe variant. Plots analyzing the correlation between ΔGbinding and electrostatic potential, Log P, and polarizability for the peptidomimetic ligands UNC6212 (Kme2), UNC6349 (Ket2), and UNC6864 (Kei) for Y20 (green) and F44 (blue) positions with respect to the ΔGbinding for the pCNPhe variant are shown. Electrostatic potential, Log P, and polarizability were calculated in Spartan at the ωB97X-D/6–31G(d) level of theory. ΔGbinding error bars reflect the standard deviation for replicates or the highest individual error from an individual experiment among the replicates, whichever is greater. The same trends are observed for the pClPhe variant (Figure S10).
CONCLUSIONS
Herein we report the development of a method to couple the evaluation of structure-activity relationships with systematic variation of the protein via GCE (GCE-SAR) to gain insight into the mechanism by which structural changes to the ligand influence binding. We systematically varied the electrostatics of two residues in the aromatic cage of CBX5 as well as the size of the alkyl group on lysine in the ligand to evaluate the contribution of the larger alkyl groups to molecular recognition. In agreement with previous work,21 this study shows that the individual interactions between alkyllysine ligands and the aromatic residues in the cage are electrostatically tunable cation-π interactions where geometry and distance influence the magnitude of these non-covalent interactions. We find that not all cation-π interactions contribute equally, even across homologous Kme3 reader proteins, with greater tunability observed in CBX5 than HP1. This may arise from different contributions of Kme2 versus Kme3 due to hydrogen bonding, orientation, and/or a different degree of cooperativity.
By systematically varying the alkyl groups on lysine, we observed that larger alkyl groups at the lysine position exhibit more favorable binding, as has been seen in other reader protein-ligand interactions. However, the larger alkyl groups result in a decrease in the contribution of cation-π interactions. Calculations suggest that this is due to more disperse charge distribution on the larger alkyl groups. Tighter binding is instead due to increased dispersion forces and hydrophobic interactions, indicating a shift in forces that contribute to binding. This systematic study provides novel insights into the molecular interactions driving binding, revealing that the molecular mechanism is not wholly conserved across different size alkyllysine ligands. These fundamental insights into alkyllysine binding can be exploited for the optimization of methyllysine reader chemical probes. For example, these findings suggest that strategies that increase the alkyl group size while maintaining the cation-π interaction may further improve binding.
More broadly, the GCE-SAR strategy reported here is a powerful tool to gain mechanistic understanding of SAR trends. A single tRNA synthetase can be used to incorporate the entire series of para-substituted phenylalanine derivatives, streamlining the approach. While showcased with a methyllysine reader protein, the method can be applied broadly to protein-ligand SAR studies to gain insight that is not apparent from wild-type binding studies alone.
EXPERIMENTAL SECTION
General Methods.
Oligonucleotides were obtained from Integrated DNA Technologies. Enzymes are reagents used for cloning were obtained from New England BioLabs Inc. Non-canonical amino acids were purchased from Chem-Impex. DNA sequencing was performed by Genewiz. Protein LC-MS analysis was performed on an Agilent 6520 Accurate Mass QToF LC-MS LC-MS ESI positive in high resolution mode. The LC-MS was equipped with a Restek Viva C4 column. All other chemical reagents and solvents were obtained from chemical suppliers (Acros, Fisher Scientific, or Sigma-Aldrich) and used without further purification.
Cloning and Site-directed Mutagenesis.
pULTRA-pCNFRS was obtained from the lab of Dr. Peter Schultz and is also available from addgene (Plasmid #48215). The codon optimized human CBX5 chromodomain gene (residues 1–53) was purchased as a gBlocks Gene Fragment from IDT and cloned into a pET11a vector using NdeI and BamHI restriction sites. Mutations were generated using standard overlap PCR.
Protein Sequences.
CBX5 wild type (1–53) protein sequence: MHHHHHHSSGRENLYFQGEEYVVEKVLDRRVVKGQVEYLLKWKGFSEEHNTWEPEKNLDCPELISEFMKKYKKMKE
CBX5 Y20F (1–53) protein sequence: MHHHHHHSSGRENLYFQGEEFVVEKVLDRRVVKGQVEYLLKWKGFSEEHNTWEPEKNLDCPELISEFMKKYKKMKE
CBX5 Y20* (1–53) protein sequence (* denotes a ncAA): MHHHHHHSSGRENLYFQGEE*VVEKVLDRRVVKGQVEYLLKWKGFSEEHNTWEPEKNLDCPELISEFMKKYKKMKE
CBX5 F44Y (1–53) protein sequence: MHHHHHHSSGRENLYFQGEEYVVEKVLDRRVVKGQVEYLLKWKGYSEEHNTWEPEKNLDCPELISEFMKKYKKMKE
CBX5 F44* (1–53) protein sequence (* denotes a ncAA): MHHHHHHSSGRENLYFQGEEYVVEKVLDRRVVKGQVEYLLKWKG*SEEHNTWEPEKNLDCPELISEFMKKYKKMKE
Protein expression and purification.
For the ncAA-CBX5 variants, pET11a-CBX5-Y20TAG or pET11a-CBX5-F44TAG and pULTRA-pCNFRS were co-transformed into BL21-Gold(DE3) competent cells (Agilent Technologies). For CBX5 wild type and canonical variants, pET11a-CBX5-WT, pET11a-CBX5-Y20F, or pET11a-CBX5-F44Y were transformed into BL21-Gold(DE3) competent cells.
All proteins were expressed in LB media supplemented with 5 mM MgSO4, 5 mM MgCl2, 1% (w/v) glucose, and 1:5000 dilution of Antifoam 204 with ampicillin (100 mg/L; pET11a-CBX5) and spectinomycin (50 mg/L; pUltra-pCNFRS). After inoculation, cultures were incubated at 37°C and shaking at 210 rpm until OD600 ~0.6. For ncAA-variants, 2 mM ncAA (except for NO2Phe, 2.5 mM) was dissolved in warm water (using NaOH to dissolve if necessary) and added to cultures along with 0.5 mM IPTG. Upon induction, temperature was lowered to 18°C and cultures were left to express overnight. Cells were harvested by centrifugation and pellets either stored at −80°C until ready for use or immediately re-suspended in lysis buffer and purified.
Cell pellets were resuspended in lysis buffer (50 mM Tris pH 8, 150 mM NaCl, 30 mM imidazole, 0.25 mg/mL lysozyme) and lysed by sonication on ice. Samples were then centrifuged at 1500 rpm for 1–2 hours and supernatant was filtered to 0.45 μm. Filtered lysate was purified using an AKTAPurifier with a 5 mL HisTrap HP column (Cytiva). Using buffer A (30 mM Tris, 150 mM NaCl, 30 mM imidazole, 2 mM DTT, pH 7.4) and buffer B (30 mM Tris, 150 mM NaCl, 300 mM imidazole, 2 mM DTT, pH 7.4), samples were 6xHis-tag purified using a step gradient of 0–55% buffer B. Fractions containing desired protein were pooled and concentrated using a 3 kDa Amicon Ultra-15 centrifugal filter. Concentrated samples were further purified by size exclusion chromatography (SEC) using a Superdex 75 Increase size exclusion column (Cytiva) in SEC buffer (50 mM sodium phosphate pH 7.4, 25 mM NaCl, 2 mM DTT). Fractions containing desired protein were pooled, concentrated, and stored at 4°C for further use.
Protein characterization.
Protein purity and ncAA incorporation were confirmed by ESI-LCMS (Table S1; Figure S3). 1 mL of a 10 μM solution of each protein was exchanged into HPLC-grade water using an Amicon Ultra-15 centrifugal filter and then filtered through glass wool. The samples were run on an Agilent 6520 Accurate-Mass Q-TOF ESI positive LCMS. All LCMS chromatograms show evidence of the appropriate ncAA-incorporation with no detectible canonical amino acid contamination. In the absence of ncAA (Y20TAG and F44TAG), no background incorporation of tyrosine or phenylalanaine can be detected. Additionally, there is no evidence of tyrosine or phenylalanine incorporation detected in the presence of ncAA. Chromatograms from each LCMS can be found in the Figure S3.
Isothermal Titration Calorimetry (ITC).
Both protein and peptide-based inhibitor samples were prepared in ITC buffer (50 mM sodium phosphate pH 7.4, 25 mM NaCl, 2 mM TCEP). CBX5 protein concentration was determined by Bradford assay using bovine serum albumin as standard. Peptide-based inhibitor concentration was determined by dry mass and re-suspended in ITC buffer. ITC experiments were performed by using Malvern PEAQ ITC Automated ITC (primarily) or MicroCal Auto-ITC200 titrating peptide-based inhibitor (0.8–3 mM) into CBX5 protein (65–200 μM) at 25°C with 20 total injections (first injection 0.2 μL, followed by 19 injections of 2 μL), 4 s duration, 8 μcal/s reference power, 750 rpm stir speed, and 180 s spacing. Data were analyzed using MicroCal PEAQ-ITC analysis software (Malvern Panalytical; version 1.1.0.1262) with one-site binding model and either fitted offset or no offset (depending on saturation). Replicate ITC experiments were performed for each protein/peptide pair. Errors given for KD and ΔGbinding are the standard deviation for replicates or the highest individual error from an individual experiment among the replicates, whichever is greater.
For characterizing binding of CBX5 WT to the H3K9me2 peptide, protocols were followed as above, except H3K9me2 peptide concentration was determined by measuring absorbance at 280 nm using a Nanodrop 2000 (Thermo). ITC experiments were performed by titrating H3K9me2 peptide (3–4 mM) into CBX5 WT protein (200–230 μM). Error in slopes for further analysis of energy relationships was determined using the LINEST function in Microsoft Excel. Log P was calculated in Spartan at the ωB97X-D/6–31G(d) level of theory.
Compound Synthesis and Purification.
Fmoc-protected amino acids were purchased from Chem-Impex and Sigma-Aldrich with the exception of the Fmoc lysine derivatives which were synthesized as described previously.32 All other chemicals and solvents were purchased from TCI America and Sigma Aldrich.
Synthesis was conducted via solid-phase peptide synthesis on Fmoc Rink amide resin (50 mg, Chem-Impex). The resin was initially swollen in DCM followed by DMF (10 minutes each). Fmoc deprotection was conducted by incubation with a solution of 2.5% 1,8-diazabicycloundec-7-ene and 2.5% pyrrolidine in DMF for 10 min. The resin was filtered and washed twice with DMF, methanol, DMF, and DCM before amino acid coupling. Standard Fmoc synthesis protocols were used to generate the 6-mer peptides, UNC6212, UNC6349 and UNC6864. Briefly, Fmoc-protected amino acids (4 eq) were mixed for 5 minutes with HBTU (4 eq), HOAt (4 eq), and DIPEA (8 eq) in 1 mL of DMF and 1 mL of dichloromethane (DCM). The solution was then added to the resin and left on a shaker at room temperature for 1 hr. The resin was filtered and washed twice with DCM, DMF, methanol, and DMF again. Fmoc amino acid protecting groups were removed as described above, and then the resin was filtered and washed twice with DMF, methanol, DMF, and DCM before adding the next amino acid for coupling. Following installation of the 3-furoic acid capping residue, the resin was rinsed 6 times with DCM. Cleavage cocktail (95% trifluoroacetic acid, 2.5% triisopropylsilane, and 2.5% water) was added to the resin, the mixture was left on the shaker for 2 hours, and the filtrate was collected. The resin was rinsed twice with DCM and the filtrates were pooled and concentrated under vacuum.
The crude material was purified by preparative HPLC using an Agilent Prep 1200 series with the UV detector set to 220 nm and 254 nm. Samples were injected onto a Phenomenex Luna 75 × 30 mm, 5 μm, C18 column at 25 °C. Mobile phases of A (H2O + 0.1% TFA) and B (CH3CN) were used with a flow rate of 30 mL/min. Product fractions were pooled and concentrated to afford title compounds as TFA salts with yields as indicated in each compound characterization.
Analytical LCMS and 1H NMR were used to establish purity (>95%; Figures S11–S13). Analytical LCMS data was acquired using an Agilent 6125 Series system with the UV detector set to 220 nm and 254 nm. Samples were injected (3–5 μL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μm, C18 column at 25 °C. Mobile phases A (H2O + 0.1% acetic acid) and B (CH3CN + 1% H2O + 0.1% acetic acid) were used with a linear gradient from 10% to 100% B in 5.0 min, followed by a flush at 100% B for another 2.0 min at a flow rate of 1.0 mL/min. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6125 single quadrupole mass spectrometer with an electrospray ionization (ESI) source. Nuclear Magnetic Resonance (NMR) spectra were recorded on a Varian Mercury spectrometer at 400 MHz for proton (1H NMR); chemical shifts are reported in ppm (δ) relative to residual protons in deuterated solvent peaks. Due to intramolecular hydrogen-bonding, hydrogen-deuterium exchange between the amide protons of the molecule and the deuterated solvent is slow and requires overnight equilibration for complete exchange.
N-((7S,10S,13S,17S)-7-(((S)-1-amino-3-hydroxy-1-oxopropan-2-yl)carbamoyl)-10-(3,4-dimethoxybenzyl)-17-hydroxy-2,13-dimethyl-9,12,15-trioxo-17-phenyl-2,8,11,14-tetraazaheptadecan-16-yl)furan-3-carboxamide (UNC6212): Yield: 8.3 mg (27%) as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 8.21 – 8.05 (m, 1H), 7.60 (dt, J = 23.0, 1.7 Hz, 1H), 7.49 – 7.20 (m, 5H), 6.98 – 6.71 (m, 4H), 5.29 (d, J = 3.9 Hz, 1H), 5.01 (d, J = 7.6 Hz, 1H), 4.78 (d, J = 4.0 Hz, 1H), 4.55 – 4.19 (m, 4H), 3.90 – 3.73 (m, 8H), 3.26 – 2.92 (m, 4H), 2.90 – 2.79 (m, 6H), 1.98 – 1.20 (m, 8H), 0.93 (d, J = 7.3 Hz, 1H). MS (ESI+): 796 [M+H]+, 399 [M+2H]2+. LCMS: tR = 2.80 min.
N-((8S,11S,14S,18S)-8-(((S)-1-amino-3-hydroxy-1-oxopropan-2-yl)carbamoyl)-11-(3,4-dimethoxybenzyl)-3-ethyl-18-hydroxy-14-methyl-10,13,16-trioxo-18-phenyl-3,9,12,15-tetraazaoctadecan-17-yl)furan-3-carboxamide (UNC6349): Yield: 10.5 mg (27.6%) as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 8.21 – 8.05 (m, 1H), 7.60 (dt, J = 22.0, 1.8 Hz, 1H), 7.45 – 7.20 (m, 5H), 6.97 – 6.73 (m, 4H), 5.29 (d, J = 3.9 Hz, 1H), 5.01 (d, J = 7.6 Hz, 1H), 4.77 (d, J = 3.9 Hz, 1H), 4.56 – 4.19 (m, 4H), 3.87 – 3.75 (m, 8H), 3.25 – 2.92 (m, 8H), 2.00 – 1.17 (m, 14H), 0.93 (d, J = 7.3 Hz, 1H). MS (ESI+): 824 [M+H]+, 413 [M+2H]2+. LCMS: tR = 2.69 min.
N-((8S,11S,14S,18S)-8-(((S)-1-amino-3-hydroxy-1-oxopropan-2-yl)carbamoyl)-11-(3,4-dimethoxybenzyl)-3-ethyl-18-hydroxy-2,14-dimethyl-10,13,16-trioxo-18-phenyl-3,9,12,15-tetraazaoctadecan-17-yl)furan-3-carboxamide (UNC6864): Yield: 8.1 mg (21%) as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 8.21 – 8.03 (m, 1H), 7.59 (dt, J = 20.9, 1.8 Hz, 1H), 7.48 – 7.20 (m, 5H), 6.97 – 6.72 (m, 4H), 5.28 (d, J = 3.9 Hz, 1H), 5.00 (d, J = 7.6 Hz, 1H), 4.76 (d, J = 3.9 Hz, 1H), 4.67 – 4.17 (m, 4H), 3.90 – 3.73 (m, 8H), 3.72 – 3.57 (m, 1H), 3.28 – 2.84 (m, 6H), 2.05 – 1.14 (m, 17H), 0.92 (d, J = 7.3 Hz, 1H). MS (ESI+): 838 [M+H]+, 420 [M+2H]2+. LCMS: tR = 2.73 min.
Peptide Synthesis and Purification.
H3K9me2 (ARTKQTARKme2STGGKAY) was synthesized on a CEM Liberty Blue peptide synthesizer using standard Fmoc-protected amino acids and Rink Amide AM Resin. The amino acid residues were activated with Oxyma (Ethyl cyanohydroxyiminoacetate) and DIC (N,N’- Diisopropycarbodiimide). 5 equivalents of the amino acid, Oxyma, and 10 equivalents of DIC were used for each coupling step. Two coupling cycles of 4 minutes were performed at 90°C in DMF (dimethylformamide) for each residue. Deprotections of Fmoc were carried out in 20% piperidine in DMF, twice for 1 minute each. The resin was washed with DMF before every deprotection and coupling cycle.
K9me2 was generated by reductive amination of orthogonally protected Lys(Ivdde) on resin after peptide synthesis was complete. Ivdde deprotection was carried out in 3% hydrazine in DMF, 3 times for 3 minutes each. The resin was suspended in 50:50 DMF and ACN, then 10 equiv (1 mmol) of STAB and formaldehyde were added. The reaction was bubbled at room temperature for 24 hrs. An additional 10 equiv of STAB and formaldehyde were added and the reaction continued to bubble for another 24 hrs at room temperature. The N-terminal Fmoc deprotection was then carried out in 20% piperidine in DMF, twice for 15 minutes each.
The peptide was cleaved from the resin with 95:2.5:2.5 trifluoroacetic acid (TFA):water:triisopropylsilane for 3 hours. Crude peptide material were purified by reversed phase HPLC using a C18 S5 XBridge 5 μM column (Waters) and a gradient of 0 to 20% B in 60 minutes, where solvent A was 95:5 water:acetonitrile, 0.1% TFA and solvent B was 95:5 acetonitrile:water, 0.1% TFA. The purified peptides were lyophilized, and identity was confirmed by ESI-LCMS (Figure S14). Expected mass: 1750.98 Da; Observed mass: 1751.00 Da
Computational methods.
The geometries of the CBX5-Kme3 complex were extracted from the previously reported crystal structure (PDB: 3FDT)26 and truncated to include only the amino acid side chains. Each N and C terminus of the amino acid residues was replaced with a hydrogen atom with a C-H bond distance of 1.09 Å. Electronic interaction energies, Eint, were calculated for the Y20 and F44 positions with the lysine ammonium ion using single-point energy calculations at the M06–2X/6–311+G(d,p) level of theory with an ultrafine grid.35 The interaction energy is defined as the energy difference between the complex and each individual component: Eint = Ecomplex – (Eamino acid + EKme3). All quantum chemical calculations were performed using the Gaussian 09 software package.38
The alkyllysine substituents (Kme2, Ket2, and Kei) were optimized in the gas phase at the M06–2X/6–311+G(d,p) level of theory, and an ultrafine grid was applied to ensure calculation accuracy. Frequency calculations were carried out at the same level of theory to ensure that stationary points were truly minima or saddle points on the potential energy surface. Single point energy and CM5 charge calculations were also performed at the same level of theory. Conformational searches of the side chains were performed using the Merck molecular force field (MMFF94)39 as implemented in Spartan 18.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Katherine Albanese for synthesis of the H3K9me2 peptide and for her assistance with collecting preliminary data, Dr. Jarod Waybright for assistance with peptidomimetic ligand synthesis, and Christopher Travis for assistance with LC-MS. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM118499 to M.L.W. and K12-GM000678 to K.M.K. This work was supported by the National Institute on Drug Abuse, NIH (R61DA047023) to L.I.J. This work was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086 to support the UNC Macromolecular Interactions Facility. This work used computational and storage services associated with the Hoffman2 Shared Cluster provided by UCLA Institute for Digital Research and Education’s Research Technology Group. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding Sources
National Institutes of Health (GM118499, K12-GM000678, R61DA047023, P30CA016086).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Tables S1–S5; and Figures S1–S14. (PDF)
REFERENCES
- 1.Dawson MA; Kouzarides T; Huntly BJ, Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367 (7), 647–657. [DOI] [PubMed] [Google Scholar]
- 2.Greer EL; Shi Y, Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13 (5), 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helin K; Dhanak D, Chromatin proteins and modifications as drug targets. Nature 2013, 502 (7472), 480–488. [DOI] [PubMed] [Google Scholar]
- 4.McGrath J; Trojer P, Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 150, 1–22. [DOI] [PubMed] [Google Scholar]
- 5.Murn J; Shi Y, The winding path of protein methylation research: milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18 (8), 517–527. [DOI] [PubMed] [Google Scholar]
- 6.Santiago C; Nguyen K; Schapira M, Druggability of methyl-lysine binding sites. J. Comput. Aided Mol. Des. 2011, 25 (12), 1171–1178. [DOI] [PubMed] [Google Scholar]
- 7.James LI; Barsyte-Lovejoy D; Zhong N; Krichevsky L; Korboukh VK; Herold JM; MacNevin CJ; Norris JL; Sagum CA; Tempel W; Marcon E; Guo H; Gao C; Huang XP; Duan S; Emili A; Greenblatt JF; Kireev DB; Jin J; Janzen WP; Brown PJ; Bedford MT; Arrowsmith CH; Frye SV, Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 2013, 9 (3), 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamb KN; Bsteh D; Dishman SN; Moussa HF; Fan H; Stuckey JI; Norris JL; Cholensky SH; Li D; Wang J; Sagum C; Stanton BZ; Bedford MT; Pearce KH; Kenakin TP; Kireev DB; Wang GG; James LI; Bell O; Frye SV, Discovery and characterization of a cellular potent positive allosteric modulator of the polycomb repressive complex 1 chromodomain, CBX7. Cell Chem. Biol 2019, 26 (10), 1365–1379 e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stuckey JI; Dickson BM; Cheng N; Liu Y; Norris JL; Cholensky SH; Tempel W; Qin S; Huber KG; Sagum C; Black K; Li F; Huang XP; Roth BL; Baughman BM; Senisterra G; Pattenden SG; Vedadi M; Brown PJ; Bedford MT; Min J; Arrowsmith CH; James LI; Frye SV, A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol. 2016, 12 (3), 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simhadri C; Daze KD; Douglas SF; Quon TT; Dev A; Gignac MC; Peng F; Heller M; Boulanger MJ; Wulff JE; Hof F, Chromodomain antagonists that target the polycomb-group methyllysine reader protein chromobox homolog 7 (CBX7). J. Med. Chem. 2014, 57 (7), 2874–2883. [DOI] [PubMed] [Google Scholar]
- 11.Ren C; Smith SG; Yap K; Li S; Li J; Mezei M; Rodriguez Y; Vincek A; Aguilo F; Walsh MJ; Zhou MM, Structure-guided discovery of selective antagonists for the chromodomain of polycomb repressive protein CBX7. ACS Med. Chem. Lett. 2016, 7 (6), 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teske KA; Hadden MK, Methyllysine binding domains: Structural insight and small molecule probe development. Eur. J. Med. Chem. 2017, 136, 14–35. [DOI] [PubMed] [Google Scholar]
- 13.Xiong Y; Greschik H; Johansson C; Seifert L; Bacher J; Park KS; Babault N; Martini M; Fagan V; Li F; Chau I; Christott T; Dilworth D; Barsyte-Lovejoy D; Vedadi M; Arrowsmith CH; Brennan P; Fedorov O; Jung M; Farnie G; Liu J; Oppermann U; Schule R; Jin J, Discovery of a potent and selective fragment-like inhibitor of methyllysine reader protein spindlin 1 (SPIN1). J. Med. Chem. 2019, 62 (20), 8996–9007. [DOI] [PubMed] [Google Scholar]
- 14.He Y; Selvaraju S; Curtin ML; Jakob CG; Zhu H; Comess KM; Shaw B; The J; Lima-Fernandes E; Szewczyk MM; Cheng D; Klinge KL; Li HQ; Pliushchev M; Algire MA; Maag D; Guo J; Dietrich J; Panchal SC; Petros AM; Sweis RF; Torrent M; Bigelow LJ; Senisterra G; Li F; Kennedy S; Wu Q; Osterling DJ; Lindley DJ; Gao W; Galasinski S; Barsyte-Lovejoy D; Vedadi M; Buchanan FG; Arrowsmith CH; Chiang GG; Sun C; Pappano WN, The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017, 13 (4), 389–395. [DOI] [PubMed] [Google Scholar]
- 15.Qi W; Zhao K; Gu J; Huang Y; Wang Y; Zhang H; Zhang M; Zhang J; Yu Z; Li L; Teng L; Chuai S; Zhang C; Zhao M; Chan H; Chen Z; Fang D; Fei Q; Feng L; Feng L; Gao Y; Ge H; Ge X; Li G; Lingel A; Lin Y; Liu Y; Luo F; Shi M; Wang L; Wang Z; Yu Y; Zeng J; Zeng C; Zhang L; Zhang Q; Zhou S; Oyang C; Atadja P; Li E, An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13 (4), 381–388. [DOI] [PubMed] [Google Scholar]
- 16.Hughes RM; Wiggins KR; Khorasanizadeh S; Waters ML, Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (27), 11184–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamps JJ; Huang J; Poater J; Xu C; Pieters BJ; Dong A; Min J; Sherman W; Beuming T; Matthias Bickelhaupt F; Li H; Mecinovic J, Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 2015, 6, 8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma JC; Dougherty DA, The cation−π interaction. Chem. Rev. 1997, 97 (5), 1303–1324. [DOI] [PubMed] [Google Scholar]
- 19.Dougherty DA, The cation-pi interaction. Acc. Chem. Res. 2013, 46 (4), 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler SE; Houk KN, Substituent effects in cation/pi interactions and electrostatic potentials above the centers of substituted benzenes are due primarily to through-space effects of the substituents. J. Am. Chem. Soc. 2009, 131 (9), 3126–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baril SA; Koenig AL; Krone MW; Albanese KI; He CQ; Lee GY; Houk KN; Waters ML; Brustad EM, Investigation of trimethyllysine binding by the HP1 chromodomain via unnatural amino acid mutagenesis. J. Am. Chem. Soc. 2017, 139 (48), 17253–17256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnash KD; The J; Norris-Drouin JL; Cholensky SH; Worley BM; Li F; Stuckey JI; Brown PJ; Vedadi M; Arrowsmith CH; Frye SV; James LI, Discovery of peptidomimetic ligands of EED as allosteric inhibitors of PRC2. ACS Comb. Sci. 2017, 19 (3), 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stuckey JI; Simpson C; Norris-Drouin JL; Cholensky SH; Lee J; Pasca R; Cheng N; Dickson BM; Pearce KH; Frye SV; James LI, Structure-activity relationships and kinetic studies of peptidic antagonists of CBX chromodomains. J. Med. Chem. 2016, 59 (19), 8913–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelberg IA; Liu J; Norris-Drouin JL; Cholensky SH; Ottavi SA; Frye SV; Kutateladze TG; James LI, Discovery of an H3K36me3-derived peptidomimetic ligand with enhanced affinity for plant homeodomain finger protein 1 (PHF1). J. Med. Chem. 2021, 64 (12), 8510–8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang S; Denton KE; Hobbs KF; Weaver T; McFarlane JMB; Connelly KE; Gignac MC; Milosevich N; Hof F; Paci I; Musselman CA; Dykhuizen EC; Krusemark CJ, Optimization of ligands using focused DNA-encoded libraries to develop a selective, cell-permeable CBX8 chromodomain inhibitor. ACS Chem. Biol. 2020, 15 (1), 112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaustov L; Ouyang H; Amaya M; Lemak A; Nady N; Duan S; Wasney GA; Li Z; Vedadi M; Schapira M; Min J; Arrowsmith CH, Recognition and specificity determinants of the human cbx chromodomains. J. Biol. Chem. 2011, 286 (1), 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermeulen M; Eberl HC; Matarese F; Marks H; Denissov S; Butter F; Lee KK; Olsen JV; Hyman AA; Stunnenberg HG; Mann M, Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142 (6), 967–980. [DOI] [PubMed] [Google Scholar]
- 28.Vad-Nielsen J; Nielsen AL, Beyond the histone tale: HP1alpha deregulation in breast cancer epigenetics. Cancer Biol. Ther. 2015, 16 (2), 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Koning L; Savignoni A; Boumendil C; Rehman H; Asselain B; Sastre-Garau X; Almouzni G, Heterochromatin protein 1alpha: a hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009, 1 (3), 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu YH; Chiou GY; Huang PI; Lo WL; Wang CY; Lu KH; Yu CC; Alterovitz G; Huang WC; Lo JF; Hsu HS; Chiou SH, Network biology of tumor stem-like cells identified a regulatory role of CBX5 in lung cancer. Sci. Rep. 2012, 2, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Lange R; Burtscher H; Jarsch M; Weidle UH, Identification of metastasis-associated genes by transcriptional profiling of metastatic versus non-metastatic colon cancer cell lines. Anticancer Res. 2001, 21 (4A), 2329–2339. [PubMed] [Google Scholar]
- 32.Barnash KD; Lamb KN; Stuckey JI; Norris JL; Cholensky SH; Kireev DB; Frye SV; James LI, Chromodomain ligand optimization via target-class directed combinatorial repurposing. ACS Chem. Biol. 2016, 11 (9), 2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young DD; Young TS; Jahnz M; Ahmad I; Spraggon G; Schultz PG, An evolved aminoacyl-tRNA synthetase with atypical polysubstrate specificity. Biochemistry 2011, 50 (11), 1894–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansch C; Leo A; Unger SH; Kim KH; Nikaitani D; Lien EJ, “Aromatic” substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16 (11), 1207–1216. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Y; Truhlar DG, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120 (1), 215–241. [Google Scholar]
- 36.Jacobs SA; Khorasanizadeh S, Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2002, 295 (5562), 2080–2083. [DOI] [PubMed] [Google Scholar]
- 37.Marenich AV; Jerome SV; Cramer CJ; Truhlar DG, Charge model 5: An extension of hirshfeld population analysis for the accurate description of molecular interactions in gaseous and condensed phases. J. Chem. Theory Comput. 2012, 8 (2), 527–541. [DOI] [PubMed] [Google Scholar]
- 38.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 09 Rev. A.02, Wallingford, CT, 2016. [Google Scholar]
- 39.Halgren TA, Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry 1996, 17 (5–6), 490–519. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.