

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) evolution plays a significant role in shaping the dynamics of the coronavirus disease 2019 pandemic. To monitor the evolution of SARS-CoV-2 variants, through international collaborations, we performed genomic epidemiology analyses on a weekly basis with SARS-CoV-2 samples collected from a border region between Germany, Poland, and the Czech Republic in a global background. For identified virus mutant variants, active viruses were isolated and functional evaluations were performed to test their replication fitness and neutralization sensitivity against vaccine-elicited serum neutralizing antibodies. Thereby we identified a new B.1.1.7 sub-lineage carrying additional mutations of nucleoprotein G204P and open-reading-frame-8 K68stop. Of note, this B.1.1.7 sub-lineage is the predominant B.1.1.7 variant in several European countries such as Czech Republic, Austria, and Slovakia. The earliest samples belonging to this sub-lineage were detected in November 2020 in a few countries in the European continent, but not in the UK. We have also detected its further evolution with extra spike mutations D138Y and A701V, which are signature mutations shared with the Gamma and Beta variants, respectively. Antibody neutralization assay of virus variant isolations has revealed that the variant with extra spike mutations is 3.2-fold less sensitive to vaccine-elicited antibodies as compared to the other B.1.1.7 variants tested, indicating potential for immune evasion, but it also exhibited reduced replication fitness, suggesting lower transmissibility. The wide spread of this B.1.1.7 sub-lineage was related to the pandemic waves in early 2021 in various European countries. These findings about the emergence, spread, evolution, infection, and transmission abilities of this B.1.1.7 sub-lineage add to our understanding about the pandemic development in Europe and highlight the importance of international collaboration on virus mutant surveillance.
Keywords: viral infection, pathogens, phylogeny, molecular evolution, transmission, immune evasion
1. Introduction
As one of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOCs), the Alpha variant B.1.1.7 was first detected in the UK in September 2020. This variant was shown to be more transmissible (Graham et al. 2021; Volz et al. 2021; Washington et al. 2021) compared to previously detected other variants. In Europe, B.1.1.7 accounted for the majority of coronavirus disease 2019 (COVID-19) cases from February to May in spring 2021. The originally reported B.1.1.7 was characterized by seventeen mutations including amino acid replacements and deletions on the spike protein (S), open-reading-frame-1ab (ORF1ab), open-reading-frame-8 (ORF8), and the nucleoprotein (N) (Frampton et al. 2021). These mutations might play a role in ACE2 receptor binding or neutralizing antibody escape (Greaney et al. 2021; Planas et al. 2021). One recent study has investigated the spatial invasion dynamics of B.1.1.7 in the UK, and the results of this study indicated that early B.1.1.7 growth rates were related with lineage export frequencies from a dominant source location (Kraemer et al. 2021). It remains unclear how B.1.1.7 could quickly spread to all the countries in Europe in spite of the strict lockdown including restrictions on international travels since December 2020 in almost all the European countries.
In Germany, regular large-scale genomic surveillance was initiated in early January 2021. With SARS-CoV-2 samples sequenced locally and with virus genomes shared on GISAID (Shu and McCauley 2017), we have routinely carried out genomic epidemiology analyses to investigate local, national, and international virus spreading conditions and monitor the evolution of existing variants and the emergence of new variants. Through joint efforts in genomic surveillance with Czech and Polish partners, we detected that in the B.1.1.7 samples in the Czech Republic, around 95 per cent samples carried two extra mutations: N_G204P and ORF8_68stop. In Germany, more than 30 per cent of B.1.1.7 samples showed these two extra mutations as well. More detailed analysis of international samples revealed that this B.1.1.7-N:G204P-ORF8:K68stop sub-lineage (labelled as B.1.1.7_S in the following text) has been widely distributed in Europe, among which a fraction of samples carried two additional spike mutations: D138Y and A701V (this variant is labelled as B.1.1.7_S+ in the following text). Here, we describe the detection, characterization, transmission, evolution, and functional analyses of this B.1.1.7 sub-lineage and the spreading pattern of B.1.1.7 in Europe in January 2021 when B.1.1.7 got detected in most European countries. The relevant information may help to understand why and how the B.1.1.7 waves could take place across Europe in the spring of 2021, thereby possibly promoting suitable strategies for preventing the spread of other variants of concern that evolve quickly.
2. Methods
2.1. Establishment of genome sequence data set for emerging variant monitoring
We combined SARS-CoV-2 sequences generated from samples collected in a border region between Germany, Poland, and Czech Republic, with full-length SARS-CoV-2 sequences periodically downloaded from GISAID (Shu and McCauley 2017) to build up genome sequence data set for emerging variant monitoring (locally generated sequences were shared on GISAID as well). We first performed quality check and filtered out low-quality sequences that met any of the following criteria: (1) sequences with less than 90 per cent genome coverage; (2) genomes with too many private mutations (defined as having >24 mutations relative to the closest sequence in the reference tree); (3) genomes with more than ten ambiguous bases; and (4) genomes with mutation clusters, defined as six or more private differences within a 100-nucleotide window. These are the standard quality assessment parameters utilized in NextClade (https://clades.nextstrain.org) (Aksamentov et al. 2021). The current study was based on the 2.17 million global viral genomes available as of 30 June 2021.
2.2. Lineage classification
We used the dynamic lineage classification method in this study through the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) software suite (https://github.com/hCoV-2019/pangolin) (Rambaut et al. 2020). This is intended for identifying the most epidemiologically important lineages of SARS-CoV-2 at the time of analysis (O’Toole et al. 2021b).
2.3. Phylogenetic analysis of SARS-CoV-2
Phylogenetic analysis and phylogeographical analyses were carried out to infer the transmission routes of B.1.1.7 in Europe (Dellicour, Rose, and Pybus 2016) with a custom build of the SARS-CoV-2 NextStrain build (https://github.com/nextstrain/ncov) (Hadfield et al. 2018). The pipeline includes several Python scripts that manage the analysis workflow. Briefly, it allows for the filtering of genomes, the alignment of genomes in NextClade (https://clades.nextstrain.org) (Aksamentov et al. 2021), phylogenetic tree inference in IQ-Tree (Minh et al. 2020), tree dating (Sagulenko, Puller, and Neher 2018), and ancestral state construction and annotation. The phylogeny analysis is rooted by Wuhan-Hu-1/2019 (GISAID Accession ID: EPI_ISL_402125). The subsampling of the GISAID data set for genomes was performed through a standard subsampling procedure applied in the NextStrain ncov pipeline (https://github.com/nextstrain/ncov) (Hadfield et al. 2018), which was done by specifying a subsampling scheme. In detail, for the phylogenetic analysis of B.1.1.7 in January 2021 described in results Section 3.2, we defined 150 samples from each of 10 specified European countries (Austria, Czech Republic, UK, Germany, Switzerland, Slovakia, Italy, Poland, France, and Denmark) in January 2021 should be randomly chosen from the GISAID data set. If there are fewer than 150 samples available in any country in January 2021, all of the samples from that country in January 2021 will be taken.
2.4. Epidemiology data
We analysed daily cases of SARS-CoV-2 in the Czech Republic from publicly released data provided by the Ministry of Health of the Czech Republic (https://onemocneni-aktualne.mzcr.cz/covid-19), and 7-day incidence rates per 100,000 inhabitants were calculated accordingly based on the local population. Daily cases of SARS-CoV-2 in Poland were obtained from publicly released data provided by the Service of the Republic of Poland (https://www.gov.pl/web/koronawirus/wykaz-zarazen-koronawirusem-sars-cov-2), and 7-day incidence rates per 100,000 inhabitants were calculated accordingly as well.
2.5. Viruses
All viruses used were patient isolates cultured from nasopharyngeal swabs. Virus stocks were grown on Vero E6 cells in Dulbecco’s Modified Eagle Medium (DMEM) GlutaMAX supplemented with 10 per cent fetal bovine serum (FBS), 1 per cent non-essential amino acids, and 1 per cent penicillin/streptomycin. The second passage of each virus isolate was used for experiments. The virus isolates hCoV-19/Germany/SN-RKI-I-178035/2021 (EPI_ISL_2634728, similar to originally defined B.1.1.7, labelled as B.1.1.7_O), hCoV-19/Germany/SN-RKI-I-038776/2021 (EPI_ISL_1285137, B.1.1.7-N:G204P-ORF8:K68stop sub-lineage, labelled as B.1.1.7_S), and hCoV-19/Germany/SN-UKDD-91348010S+/2021 (EPI_ISL_4932671, B.1.1.7-N:G204P-ORF8:K68stop sub-lineage with extra spike mutations D138Y and A701V, labelled as B.1.1.7_S+) were used in the virus neutralization assay and growth kinetics measurement. The representative isolate of B.1.1.7_O used here has additional mutations in addition to the characteristic mutations of the lineage: A50T in NS7a, Q72H in NS8, T140I in NSP16 as well as T1306I and V381I in NSP3 (Supplementary Material Table S1). The experimental viral cultures have been sequenced as well, and the genome sequences are deposited in the NCBI under the project number PRJNA803169.
2.6. Virus neutralization assay
All sera were derived from healthy individuals fully vaccinated with BNT162b2. A 2-fold dilution series of each serum was prepared in phosphate buffered saline (PBS)+ (supplemented with 0.3 per cent bovine albumin, 1 mM MgCl2 and 1 mM CaCl2), and each serum concentration was incubated with 50 plaque forming units (PFUs) of B.1.1.7_O, B.1.1.7_S, or B.1.1.7_S+ for 1 h at 37°C. Confluent Vero E6 cells seeded the day before were infected with the virus-containing serum dilutions for 1 h at 37°C and 5 per cent CO2 with occasional shaking. The inoculum was aspirated, cells washed with PBS, and subsequently overlaid with semi-viscous Avicel Overlay Medium (double-strength DMEM, Avicel RC-581 in H2O 0.75 per cent, 10 per cent FCS, 0.01 per cent DEAE-Dextran, and 0.05 per cent NaHCO3). After 3 days, cells were stained with 0.1 per cent crystal violet in 10 per cent formaldehyde and plaques were counted. Eleven dilutions of each serum were tested. The neutralization assay was performed in three independent experiments with each serum. Each experiment was conducted in technical duplicates of each serum. Technical duplicates were averaged before further calculations. ID50 values were calculated using 4th-order nonlinear regression curve fits with GraphPad Prism 9.
2.7. Virus growth kinetics
Calu 3 cells were seeded 3 days prior to infection. On the day of infection, cells were infected with B.1.1.7_O, B.1.1.7_S or B.1.1.7_S+ at MOI 0.1 diluted in PBS+ for 1 h at 37°C and 5 per cent CO2 with occasional shaking. Afterwards, the inoculum was aspirated, the cells were washed with PBS and fresh medium (DMEM GlutaMAX supplemented with 10 per cent FBS, 1 per cent non-essential amino acids, 1 per cent sodium pyruvate, and 1 per cent penicillin/streptomycin) was added. Supernatants were removed at 8, 16, 24, 48, 72, and 96 hours postinfection. Infectious virus particles in the supernatant were determined using plaque assay, which was performed analogously to the neutralization assay from the infection step onwards. Results are given as PFU/ml. Graphs were generated using GraphPad Prism 9.
3. Results
3.1. Identification of one specific B.1.1.7 sub-lineage with extra mutations in Europe
We evaluated 948,077 SARS-CoV-2 Alpha variant genomes available as of 30 June 2021. One specific B.1.1.7 sub-lineage with two extra mutations—nucleoprotein G204P (N_G204P) and ORF8 K68stop (Orf8_K68stop) (Fig. 1A)—was detected in thirty-five European countries with 62,225 genomes shared on GISAID. Surprisingly, this B.1.1.7 sub-lineage (referred to as B.1.1.7_S) had very unequal distribution in Europe. In the Czech Republic, Hungary, Slovakia, and Austria, B.1.1.7_S was predominant, accounting for around 60–95 per cent of local B.1.1.7-related COVID-19 cases. In Germany, Denmark, Switzerland, Poland, and France, B.1.1.7_S accounted for around 10–35 per cent of B.1.1.7-related COVID-19 cases in each country (Fig. 1B). In Belgium and Netherlands, B.1.1.7_S only accounted for less than 10 per cent of B.1.1.7-related cases. Of note, this B.1.1.7_S sub-lineage comprised only 0.4 per cent of total B.1.1.7 genomes in the UK.
Figure 1.
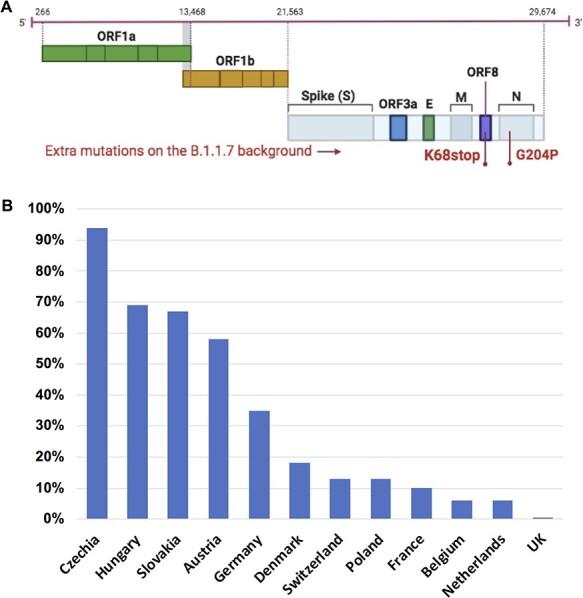
One specific SARS-CoV-2 B.1.1.7 sub-lineage with extra mutations was detected in Europe and displays unequal distribution. (A) Illustration of the genome structure of SARS-CoV-2 indicating the mutations of the B.1.1.7_S sub-lineage. This B.1.1.7_S sub-lineage was characterized by two extra mutations: nucleoprotein G204P (N_G204P) and ORF8 K68stop (Orf8_K68stop) on the originally defined SARS-CoV-2 B.1.1.7 genetic background. For visualization clearance, the background B.1.1.7 mutations are not indicated. (B) The distribution of this B.1.1.7_S sub-lineage in Europe is shown as percentage of this sub-lineage in total B.1.1.7 in each country (data collected on 30 June 2021 including all available B.1.1.7 sequences on GISAID).
3.2. Estimated transmission routes of B.1.1.7 in Europe in January 2021
Since the spreading of B.1.1.7 was one critical driving force for the February–May wave in most European countries, we performed phylogeny analysis of B.1.1.7 to estimate the early transmission routes of B.1.1.7 in Europe. In most European countries, in January 2021, the B.1.1.7 already got frequently detected (Di Domenico et al. 2021; Di Giallonardo et al. 2021; O’Toole et al. 2021a), so this analysis focused on the cross-country transmission taking place in January. Phylogeny analysis was performed with B.1.1.7 samples collected in January that are available at GISAID till 30 June 2021 from ten European countries (Austria: 202; Czech Republic: 161; UK: 58,523; Germany: 699; Switzerland: 1,118; Slovakia: 77; Italy: 373; Poland: 51; France: 1532; Denmark: 1392). In a few countries, especially in the UK, the sampling density was much higher than that of other countries, so we downsized the sample numbers to 150 randomly selected samples collected in January from each country. This condition was chosen because similar sample size from each country could largely prevent statistical errors in transmission route estimation. Figure 2A shows the phylogeny-inferred cross-country transmission routes, which indicated that in the UK and the Czech Republic there were much higher frequencies of export than import. Analysis of the transmission routes of the cluster of the B.1.1.7_S sub-lineage (Fig. 2B) revealed that the frequencies of export from the Czech Republic were much higher than that of other countries.
Figure 2.
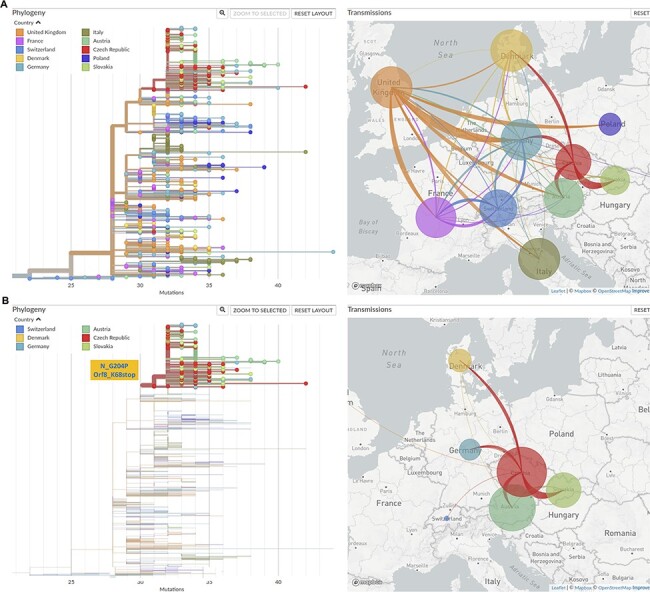
Transmission routes of B.1.1.7 in Europe in January 2021 inferred based on phylogeny analysis. The size of the circle represents the number of genomes from all B.1.1.7 (A) or from the B.1.1.7_S sub-lineage (B) in each country. The line colours correspond to the exporting locations. (A) Left: Phylogeny tree of B.1.1.7 collected in January, with branch length representing mutations; right: estimated transmission routes of B.1.1.7 in Europe in January 2021. (B) Left: only the cluster characterized by N_G204P and ORF8_K68stop is displayed; right: estimated transmission routes of B.1.1.7 samples with the two extra mutations N_G204P and ORF8_K68stop (B.1.1.7_S sub-lineage).
3.3. Exploration of the source of the B.1.1.7_S sub-lineage
As this B.1.1.7_S sub-lineage accounted for only 0.4 per cent of B.1.1.7-related cases in UK till 30 June 2021 (Fig. 1), this finding suggests the UK was possibly not the direct source of this B.1.1.7_S sub-lineage. To investigate the source and possible transmission routes of B.1.1.7_S at an early stage, we analysed all the B.1.1.7 samples belonging to this sub-lineage collected from all around Europe till end of January 2021. Earliest B.1.1.7 samples with N_G204P and Orf8_K68stop occurred in November 2020 in the following countries: Switzerland (EPI_ISL_1119202, collection date: 9 November 2020), Austria (EPI_ISL_2324194, collection date: 23 November 2020), France (EPI_ISL_1381145, collection date: 19 November 2020), Slovakia (EPI_ISL_1280136, collection date: 30 November 2020), and Denmark (EPI_ISL_1868624, collection date: 30 November 2020), with only one genome from each country being reported (Fig. 3A). Yet, till end of January 2021, it had been detected in twenty-two countries in Europe (Fig. 3B). As sample collection dates are often related to local sequencing efforts, it is not reliable to infer the source information based on the collection dates. For the ten European countries included in the B.1.1.7 phylogeny analysis in the results Section 3.2, we have provided an overview of the sequencing efforts between September 2020 and February 2021 (Supplementary Figure S1), showing the ratio of the number of sequences available per month and the number of total confirmed cases per month for each country of interest (source for the number of sequences per month: GISAID; source for the number of confirmed cases per month: Our World in Data https://ourworldindata.org/coronavirus). Except for UK, Denmark, and Switzerland, between September and December 2020, this ratio was below 0.5 per cent during most time in the other seven countries. This ratio rose sharply in early 2021 in many places, making it possible to infer more information from genome sequence phylogeny analyses. Owing to the limited number of SARS-CoV-2 genomes that were sequenced in 2020, it is difficult to analyse more details about the early transmission routes of this sub-lineage. This B.1.1.7_S sub-lineage was almost exclusive to Europe, with only 79 samples detected in Asia and 396 samples in North America.
Figure 3.
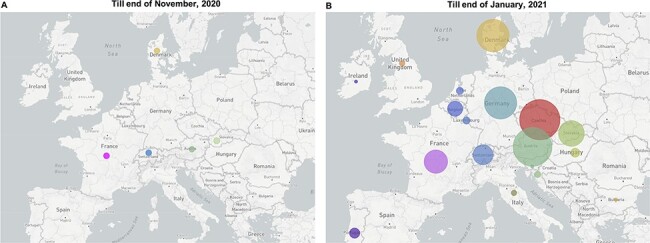
The emergence and spreading condition of this B.1.1.7_S sub-lineage at the end of November 2020 (A) and at the end of January 2021 (B). Each circle represents the number of genomes reported from each country. Each colour dot in (A) represents one genome; the circle size in (B) is proportional to the genome number in each country, e.g. Ireland: 1; Slovakia: 63; Austria: 148 (the size scale in each map is slightly adjusted based on sample size for better visualization).
In the Czech Republic, the first sample of B.1.1.7_S was reported at the beginning of January 2021 (EPI_ISL_850683, collection date: 4 January 2021), which was also the first B.1.1.7 variant being reported locally. However, it should be pointed out that in December only thirty-nine SARS-CoV-2 samples were sequenced, which means the early spreading of the B.1.1.7_S sub-lineage might be missed out owing to the low sampling density, and it could be that in late December B.1.1.7_S already started community transmission.
3.4. The COVID-19 pandemic waves in early 2021 in the Czech Republic were associated with the expansion of the B.1.1.7_S sub-lineage
Since this B.1.1.7_S sub-lineage was dominant in the Czech Republic and accounted for around 95 per cent B.1.1.7-related cases, to investigate the impact of the spreading of this B.1.1.7_S sub-lineage on the COVID-19 pandemic in the Czech Republic, we analysed the details of the COVID-19 pandemic conditions between December 2020 and April 2021 in the Czech Republic and the corresponding SARS-CoV-2 lineage development during the same period. For COVID-19 pandemic, following a second wave from September 2020 till November 2020, which was known to be caused by the spreading of other previously existing lineages (Hodcroft et al. 2021; Yi et al. 2021), a sharp wave occurred between December 2020 and January 2021 (Fig. 4A). For SARS-CoV-2 lineage development, between December 2020 and January 2021, the prevalence of B.1.1.7 (B.1.1.7 samples/all sequenced samples) increased from 0 per cent in late December 2020 to ∼60 per cent in late January 2021, replacing the majority of other lineages (Fig. 4B) (as mentioned in the results Section 3.3, the early growth of the B.1.1.7 sub-lineage might be missed out from the genome surveillance owing to the low sampling density in December), suggesting the major driving force for the sharp wave was the quick expansion of the B.1.1.7 variant, which was shown to be more transmissible than previously existing other lineages (Graham et al. 2021; Volz et al. 2021; Washington et al. 2021). Although the January wave was curbed temporarily by some countermeasures, after the January peak, the 7-day incidence rate was kept at a high level (above 400) and reached another peak in early March along with the further expansion of B.1.1.7 (Fig. 4A & B). Across the time period between January and April 2021, the ratio of the B.1.1.7_S sub-lineage to all B.1.1.7 in the Czech Republic was between 91 per cent and 100 per cent (Fig. 4C).
Figure 4.
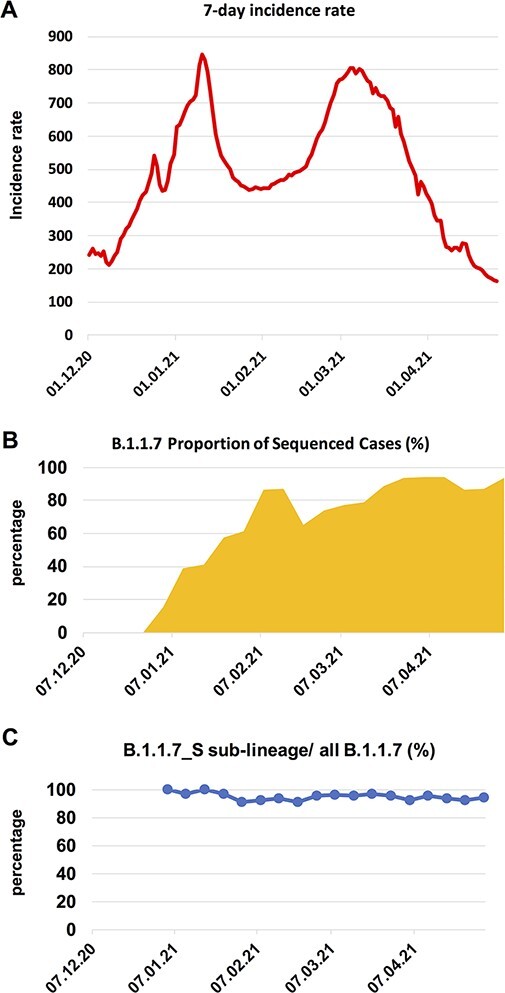
The COVID-19 pandemic waves in early 2021 and the expansion of the B.1.1.7 in the Czech Republic. (A) 7-day incidence rate per 100,000 inhabitants in the Czech Republic. (B) B.1.1.7 proportion of sequenced cases in the Czech Republic in each week, starting from the week 1st–7th December 2020. The B.1.1.7 lineage rose rapidly in early 2021, replacing the majority of other lineages (shown as the white blank space) present during this time period. (C) The ratio of the B.1.1.7_S sub-lineage to all B.1.1.7 in each week, starting from the first week when B.1.1.7 was detected.
We have also analysed lineage distribution of cases sequenced between September 2020 and February 2021 in the ten European countries included in the B.1.1.7 phylogeny analysis in the results Section 3.2 (based on GISAID data). The results are shown in the Supplementary Material Figure S2. The prevalence of B.1.1.7 in the Czech Republic in January 2021 was higher than that of most other European countries, except for UK (∼85 per cent) and Slovakia (∼75 per cent). In Slovakia, this B.1.1.7_S sub-lineage was first detected in November 2020 (Fig. 3A) and accounted for around 70 per cent B.1.1.7-related cases. In most other countries, for example, the prevalence of B.1.1.7 in Poland in late January 2021 was only around 10 per cent. Besides, in most other European countries, such as in Poland (Supplementary Material Figure S3), the B.1.1.7 wave peaked later at the end of March or the beginning of April (https://qap.ecdc.europa.eu/public/extensions/COVID-19/COVID-19.html#country comparison-tab).
3.5. Signature mutations
3.5.1. Signature mutations of the B.1.1.7_S sub-lineage
Within this sub-lineage, the most common signature mutations were the same as B.1.1.7 signature mutations, with the additional N:G204P and ORF8:K68stop mutations. As described below, there were other novel common spike mutations detected in a small portion of samples from this sub-lineage.
3.5.2. Further mutation accumulation in the B.1.1.7_S sub-lineage
Through extensive investigation of this B.1.1.7 sub-lineage, we detected one variant with further accumulated mutations in the spike protein on the genetic background of the B.1.1.7_S sub-lineage, characterized by two further extra spike mutations: D138Y and A701V (Fig. 5A). This variant (B.1.1.7_S+) was mainly detected in Germany, accounting for 0.07 per cent of all B.1.1.7 in Germany as of 30 June 2021. However, it is noteworthy that the two extra mutations carried by this B.1.1.7_S+ variant are shared with other VOCs. The spike D138Y is one signature mutation for the Gamma variant (Faria et al. 2021), and the spike A701V is one signature mutation for the Beta variant (Tegally et al. 2021). A table listing all amino acid changes defining B.1.1.7_S and B.1.1.7_S+ can be found in the supplementary material (Supplementary Material Table S1). Although B.1.1.7_S+ has a highly centralized distribution pattern (mostly in Germany), suggesting it very likely emerged at one location, we cannot completely rule out the possibility that the mutations might have appeared in a convergent way.
Figure 5.
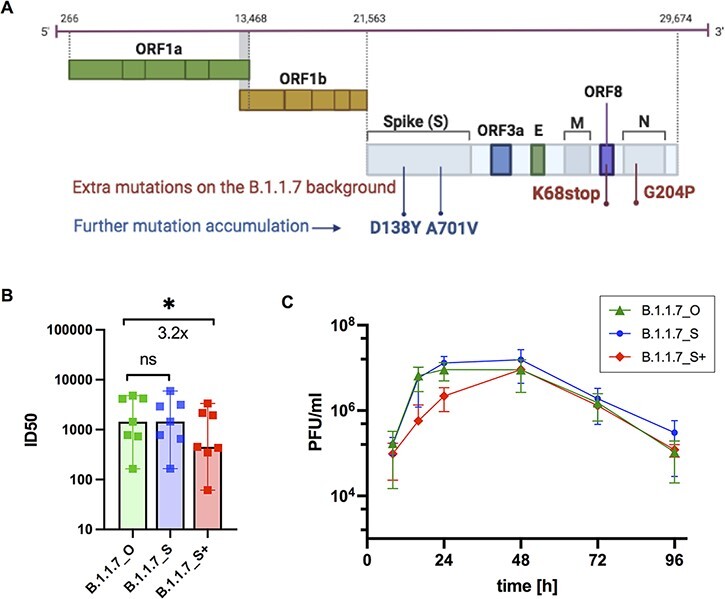
Neutralization efficacy and growth kinetics of three B.1.1.7 variants. (A) Further mutant accumulations of D138Y and A701V in the spike protein were detected within the SARS-CoV-2 B.1.1.7_S sub-lineage. (B&C) Functional evaluation of virus isolates from the three B.1.1.7 variants: the originally defined B.1.1.7 (B.1.1.7_O), B.1.1.7_S, and B.1.1.7_S sub-lineage carrying two extra spike mutations D138Y and A701V (B.1.1.7_S+). (B) Neutralization efficacy of sera from fully vaccinated individuals (n = 7, BNT162b2) against active virus of the three B.1.1.7 variants. ID50, the serum dilution required for 50 per cent virus inhibition. Bars represent the median ID50 values with 95 per cent confidence interval. *P < 0.05; Wilcoxon matched-pairs signed rank test, ns not significant. (C). Growth kinetics of B.1.1.7_O, B.1.1.7_S, and B.1.1.7_S+ on Calu 3 cells as titrated by plaque assay. All data represent three independent experiments, each with two technical replicates.
3.5.3. Impact of the mutations in the B.1.1.7 variants on virus propagation and antibody neutralization
We used virus isolates from the three variants B.1.1.7_O (similar to originally defined B.1.1.7), B.1.1.7_S, and B.1.1.7_S+ (B.1.1.7_S sub-lineage with extra spike mutations D138Y and A701V) to test their susceptibilities to vaccine-elicited serum neutralizing antibodies in individuals following vaccination with two doses BNT162b2. These experiments showed a decrease of neutralization sensitivity for B.1.1.7_S+, which carries two extra spike mutations D138Y and A701V, compared to the other two B.1.1.7 variants B.1.1.7_O and B.1.1.7_S of around 3.2-fold (Fig. 5B).
To evaluate replication abilities of these three B.1.1.7 variants, we infected a lung epithelial cell line, Calu-3, with the three B.1.1.7 variant isolates. We observed a replication disadvantage for B.1.1.7_S+ compared to B.1.1.7_O and B.1.1.7_S (Fig. 5C) in the first 24 hours after infection. These data support lower replication rate and therefore lower spread of B.1.1.7_S+ over B.1.1.7_O and B.1.1.7_S.
4. Discussion
In this article, we describe the detection, characterization, spread, evolution, and functional evaluation of the B.1.1.7_S sub-lineage that was first detected in the European continent in November 2020 and spread widely in many European countries as of 30 June 2021. In late December 2020 or early January 2021, it spread to the Czech Republic and quickly became the local predominant SARS-CoV-2 variant replacing the majority of previously existing variants. Furthermore, we detected evolution of this B.1.1.7_S sub-lineage, characterized by samples carrying two extra spike mutations A701V and D138Y on the genetic background of this B.1.1.7_S sub-lineage.
With virus isolates from the three B.1.1.7 variants (the originally defined B.1.1.7 (B.1.1.7_O), B.1.1.7_S, and the B.1.1.7_S sub-lineage carrying two extra spike mutations A701V and D138Y (B.1.1.7_S+)), we have tested among the B.1.1.7 variants if there is any difference in replication efficiency or neutralization sensitivity against vaccine-elicited serum neutralizing antibodies. The results indicated the B.1.1.7_S itself showed no replication advantage or reduced neutralization sensitivity compared to the B.1.1.7_O. This may explain the co-existence of the two variants in many European countries as shown in Fig. 1. However, for the B.1.1.7_S+, reduced neutralization sensitivity was observed, indicating potential for immune evasion (Harvey et al. 2021; Wang et al. 2021; Zhou et al. 2021).
Furthermore, using virus genome surveillance data, we have estimated the cross-country spreading pattern of B.1.1.7 in Europe in January 2021 when B.1.1.7 started being frequently detected in most European countries. We find the spreading pattern of B.1.1.7 throughout Europe was shown as high export frequencies from two locations, UK and Czech Republic, which is comparable to the early spreading pattern of B.1.1.7 in the UK (Kraemer et al. 2021) and in a few other countries at the national level (Di Domenico et al. 2021; Di Giallonardo et al. 2021; Shen et al. 2021). A limit of this study is that the transmission estimation analysis could not be optimized by taking incidence rate or other medical parameters into consideration (Dellicour et al. 2021), which is mainly due to the limited genome data.
The identification of this B.1.1.7_S sub-lineage has solved an important puzzle about the spreading of B.1.1.7 in Europe. The SARS-CoV-2 lineage B.1.1.7 was first detected in the UK in late 2020 and then spread to other countries. It is commonly acknowledged that traffic per capita as well as traffic connection to the epicentre play an important role in the spreading pattern of virus (Faria et al. 2011; Carroll et al. 2015). Usually, in the region well-connected to the epicentre, the virus would emerge and spread earlier. Among European countries, the Czech Republic is not better connected to the UK than other European countries, but in January 2021, B.1.1.7 had already become the predominant SARS-CoV-2 lineage in the Czech Republic, which was earlier than most other European countries, as shown in Supplementary Material Figure S2. It was difficult to explain the contradiction between the traffic condition and the time line of the B.1.1.7 spreading, but the spreading of this B.1.1.7_S sub-lineage in the European continent since November 2020 has provided one answer for that.
Moreover, it appeared that this B.1.1.7_S sub-lineage escaped local control measures and went on with community transmission in late December 2020 or early January 2021 in the Czech Republic. Perhaps driven by transient demographic and epidemiological factors, such as human mobilities related with vacation or family gathering, or local high incidence rate that may lead to escalating growth rate of SARS-CoV-2, this B.1.1.7_S sub-lineage rapidly increased in the Czech Republic and showed a higher export frequency than that of various other European countries.
The wide spread of this B.1.1.7_S sub-lineage was related to the pandemic waves in the Czech Republic in early 2021 and played a role in the B.1.1.7 wave in a variety of other European countries such as Austria, Slovakia, Germany, and Denmark. These findings add to the understanding about the pandemic development in Europe. Also, these findings emphasize the importance of international collaboration on virus mutant surveillance, not only for SARS-CoV-2 but also for other epidemic viruses.
Supplementary Material
Acknowledgements
We thank all researchers who are working around the clock to generate and share genome data on GISAID (http://www.gisaid.org) (a GISAID acknowledgment table is included in the Supplementary Material Table S2). We specifically thank colleagues at the Institute of Medical Microbiology and Virology, University Hospital Carl Gustav Carus, for their work in performing SARS-CoV-2 sample testing and sequencing sample preparing, and we thank the Dresden concept Genome Center for their sequencing efforts. We thank the Robert Koch Institute for the data management and sharing. We thank Dr med. Robin R. Weidemann and A. Zabzinski for help with this project. We thank all the collaboration partners who contributed to the project LüSeMut..
Contributor Information
Alexa Laubner, Institute of Medical Microbiology and Virology, University Hospital Carl Gustav Carus, Technische Universität Dresden, Fetscherstraße 74, Dresden, Saxony 01307, Germany.
Fabian Rost, DRESDEN concept Genome Center, Technische Universität Dresden, Fetscherstraße 105, Dresden, Saxony 01307, Germany; Center for Regenerative Therapies Dresden, Technische Universität Dresden, Fetscherstraße 105, Dresden, Saxony 01307, Germany.
Sylke Winkler, Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany and DRESDEN concept Genome Center, Technische Universität Dresden, Pfotenhauerstraße 108, Dresden, Saxony 01307, Germany.
Eva Patrasová, Department of Epidemiology, Regional Public Health Authority for Ustecky Kraj, Moskevská 15, Ústí nad Labem 400 01, Czech Republic; Third Faculty of Medicine, Charles University in Prague, Ruská 2411/87, Prague 100 00, Czech Republic.
Lenka Šimůnková, Department of Epidemiology, Regional Public Health Authority for Ustecky Kraj, Moskevská 15, Ústí nad Labem 400 01, Czech Republic.
Susanne Reinhardt, DRESDEN concept Genome Center, Technische Universität Dresden, Fetscherstraße 105, Dresden, Saxony 01307, Germany.
Johanna Beil, Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany and DRESDEN concept Genome Center, Technische Universität Dresden, Pfotenhauerstraße 108, Dresden, Saxony 01307, Germany.
Alexander H Dalpke, Institute of Medical Microbiology and Virology, University Hospital Carl Gustav Carus, Technische Universität Dresden, Fetscherstraße 74, Dresden, Saxony 01307, Germany.
Buqing Yi, Institute of Medical Microbiology and Virology, University Hospital Carl Gustav Carus, Technische Universität Dresden, Fetscherstraße 74, Dresden, Saxony 01307, Germany.
Data availability
All the SARS-CoV-2 genomes generated and presented in this study are publicly accessible through the GISAID platform (https://www.gisaid.org/). The processed SARS-CoV-2 genome data in the form of phylogenetic tree are available at https://github.com/genomesurveillance/alpha-variant-sublineage-revised. A list of GISAID accession ID for B.1.1.7_S+ genomes from Germany is also available at https://github.com/genomesurveillance/alpha-variant-sublineage-revised. More general information about B.1.1.7_S sub-lineage genome number in each country during certain time period can be acquired by choosing the relevant location and collection period on the GISAID database (with searching items: VOC Alpha (variants); submission date is before 30 June 2021; Substitutions: N_G204P and NS8_K68stop). To access sequence data from GISAID, registration with https://www.gisaid.org/ is necessary, which involves agreeing to GISAID’s Database Access Agreement. Biological materials (i.e. virus variant isolation) generated as a part of this study will be made available but may require execution of a materials transfer agreement.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
Parts of this study were supported by a grant from the German Ministry of Health (BMG) (project LüSeMut) to A.D. Parts of this study were supported by a grant from the State Parliament of the Free State of Saxony to A.D. B.Y. is in part supported by a funding from German Research Foundation (DFG YI 175/1-1).
Conflict of interest:
The authors declare no competing interests.
Code availability
Data processing and visualization was performed using publicly available software, primarily RStudio v1.3.1093. Code for constructing phylogenetic maximum likelihood (ML) and time trees as well as phylogeographic analyses is available at https://github.com/genomesurveillance/alpha-variant-sublineage-revised, which is modified from SARS-CoV-2-specific procedures github.com/nextstrain/ncov.
References
- Aksamentov I. et al. (2021) ‘Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes’, Journal of Open Source Software, 6: 3773. [Google Scholar]
- Carroll M. W. et al. (2015) ‘Temporal and Spatial Analysis of the 2014-2015 Ebola Virus Outbreak in West Africa’, Nature, 524: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellicour S. et al. (2021) ‘Dispersal Dynamics of SARS-CoV-2 Lineages during the First Epidemic Wave in New York City’, PLoS Pathogens, 17: e1009571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellicour S., Rose R., and Pybus O. G. (2016) ‘Explaining the Geographic Spread of Emerging Epidemics: A Framework for Comparing Viral Phylogenies and Environmental Landscape Data’, BMC Bioinformatics, 17: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico L. et al. (2021) ‘Impact of January 2021 Curfew Measures on SARS-CoV-2 B.1.1.7 Circulation in France’, Eurosurveillance, 26: 2100272.doi: 10.2807/1560-7917.ES.2021.26.15.2100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giallonardo F. et al. (2021) ‘Emergence and Spread of SARS-CoV-2 Lineages B.1.1.7 And P.1 In Italy’, Viruses, 13: 794.doi: 10.3390/v13050794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. R. et al. (2021) ‘Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil’, Science, 372: 815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2011) ‘Toward a Quantitative Understanding of Viral Phylogeography’, Current Opinion in Virology, 1: 423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton D. et al. (2021) ‘Genomic Characteristics and Clinical Effect of the Emergent SARS-CoV-2 B.1.1.7 Lineage in London, UK: A Whole-genome Sequencing and Hospital-based Cohort Study’, The Lancet Infectious Diseases, 21: 1246–56.doi: 10.1016/S1473-3099(21)00170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham M. S. et al. (2021) ‘Changes in Symptomatology, Reinfection, and Transmissibility Associated with the SARS-CoV-2 Variant B.1.1.7: An Ecological Study’, Lancet Public Health, 6: e335–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A. J. et al. (2021) ‘Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-binding Domain that Affect Recognition by Polyclonal Human Plasma Antibodies’, Cell Host & Microbe, 29: 463–76 e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘Nextstrain: Real-time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W. T. et al. (2021) ‘SARS-CoV-2 Variants, Spike Mutations and Immune Escape’, Nature Reviews. Microbiology, 19: 409–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodcroft E. B. et al. (2021) ‘Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020’, Nature, 595: 707–12. [DOI] [PubMed] [Google Scholar]
- Kraemer M. U. G. et al. (2021) ‘Spatiotemporal Invasion Dynamics of SARS-CoV-2 Lineage B.1.1.7 Emergence’, Science, 373: 889–95.doi: 10.1126/science.abj0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole A. et al. (2021a) ‘Tracking the International Spread of SARS-CoV-2 Lineages B.1.1.7 And B.1.351/501Y-V2’, Wellcome Open Research, 6: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool’, Virus Evolution, 7: veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D. et al. (2021) ‘Sensitivity of Infectious SARS-CoV-2 B.1.1.7 And B.1.351 Variants to Neutralizing Antibodies’, Nature Medicine, 27: 917–24. [DOI] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-likelihood Phylodynamic Analysis’, Virus Evolution, 4: vex042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L. et al. (2021) ‘Rapidly Emerging SARS-CoV-2 B.1.1.7 Sub-lineage in the United States of America with Spike Protein D178H and Membrane Protein V70L Mutations’, Emerging Microbes & Infections, 10: 1293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., and McCauley J. (2017) ‘GISAID: Global Initiative on Sharing All Influenza Data - from Vision to Reality’, Eurosurveillance, 22: 30494.doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H. et al. (2021) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43. [DOI] [PubMed] [Google Scholar]
- Volz E. et al. (2021) ‘Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 In England’, Nature, 593: 266–9. [DOI] [PubMed] [Google Scholar]
- Wang P. et al. (2021) ‘Increased Resistance of SARS-CoV-2 Variant P.1 To Antibody Neutralization’, Cell Host & Microbe, 29: 747–51 e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington N. L. et al. (2021) ‘Emergence and Rapid Transmission of SARS-CoV-2 B.1.1.7 In the United States’, Cell, 184: 2587–94 e2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi B. et al. (2021) ‘Phylogenetic Analysis of SARS-CoV-2 Lineage Development across the First and Second Waves in Eastern Germany in 2020: Insights into the Cause of the Second Wave’, Epidemiology and Infection, 149: e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D. et al. (2021) ‘Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-induced Sera’, Cell, 184: 2348–61 e2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the SARS-CoV-2 genomes generated and presented in this study are publicly accessible through the GISAID platform (https://www.gisaid.org/). The processed SARS-CoV-2 genome data in the form of phylogenetic tree are available at https://github.com/genomesurveillance/alpha-variant-sublineage-revised. A list of GISAID accession ID for B.1.1.7_S+ genomes from Germany is also available at https://github.com/genomesurveillance/alpha-variant-sublineage-revised. More general information about B.1.1.7_S sub-lineage genome number in each country during certain time period can be acquired by choosing the relevant location and collection period on the GISAID database (with searching items: VOC Alpha (variants); submission date is before 30 June 2021; Substitutions: N_G204P and NS8_K68stop). To access sequence data from GISAID, registration with https://www.gisaid.org/ is necessary, which involves agreeing to GISAID’s Database Access Agreement. Biological materials (i.e. virus variant isolation) generated as a part of this study will be made available but may require execution of a materials transfer agreement.