

Abstract
Lignin, the second most abundant biopolymer, is a promising renewable energy source and chemical feedstock. A key element of lignin biosynthesis is unknown: how do lignin precursors (monolignols) get from inside the cell out to the cell wall where they are polymerized? Modeling indicates that monolignols can passively diffuse through lipid bilayers, but this has not been tested experimentally. We demonstrate significant monolignol diffusion occurs when laccases, which consume monolignols, are present on one side of the membrane. We hypothesize that lignin polymerization could deplete monomers in the wall, creating a concentration gradient driving monolignol diffusion. We developed a two-photon microscopy approach to visualize lignifying Arabidopsis thaliana root cells. Laccase mutants with reduced ability to form lignin polymer in the wall accumulated monolignols inside cells. In contrast, active transport inhibitors did not decrease lignin in the wall and scant intracellular phenolics were observed. Synthetic liposomes were engineered to encapsulate laccases, and monolignols crossed these pure lipid bilayers to form polymer within. A sink-driven diffusion mechanism explains why it has been difficult to identify genes encoding monolignol transporters and why the export of varied phenylpropanoids occurs without specificity. It also highlights an important role for cell wall oxidative enzymes in monolignol export.
The lignin polymerizing laccase enzymes in cell walls establish an energetically favorable concentration gradient that drives diffusion of lignin monomers from inside the cell to the wall outside.
Introduction
Annually, plants deposit billions of tons of lignin mostly in the form of woody biomass (Balat and Ayar, 2005). Lignin is essential to the integrity of the vascular system of the majority of land plants and a potentially valuable bioproduct to replace fossil fuel-derived products. All vascular plants utilize lignin to strengthen their secondary cell walls in cell types ranging from water-conducting vessels to supportive fibers (Boerjan et al., 2003). Lignin is a phenolic polymer composed primarily of monolignols, although other constituents can be incorporated readily (Mottiar et al., 2016). The core monolignols are synthesized by the phenylpropanoid and monolignol biosynthetic pathways (Dixon and Barros, 2019). The enzymes catalyzing these reactions are both soluble in the cytoplasm and associated with the cytoplasmic face of the endoplasmic reticulum (Gou et al., 2018). Following export across the plasma membrane, lignin polymerization occurs in cell walls where the oxidative enzymes, peroxidases, and laccases, produce monolignol radicals that couple into lignin polymer (Freudenberg, 1959; Ralph et al., 2004). The oxidative enzymes are embedded into the cell walls by vesicle-mediated secretion with the carbohydrate components in advance of the synthesis of lignin monomers (Chou et al., 2018). Laccases, which are multicopper oxidoreductases, couple monolignol oxidation to reduction of oxygen, while class III peroxidases require hydrogen peroxide to oxidize monolignols (Tobimatsu and Schuetz, 2019). Laccases are required for lignification in Arabidopsis thaliana stems (Berthet et al., 2011; Zhao et al., 2013). They are abundant in lignifying cell wall domains and determine where lignin is deposited in the cell wall (Schuetz et al., 2014; Chou et al., 2018; Hoffmann et al., 2020). Both the laccase substrate specificity for monolignols and the localization of laccases in cell wall domains contribute to the unique lignin compositions found in different cell types and wall domains (Hiraide et al., 2021).
As lignin represents ∼25% of woody biomass (Novaes et al., 2010), the spatial separation between the intracellular site of monolignol synthesis and the extracellular polymerization in the cell wall means that significant amounts of monolignols are exported across cellular membranes. Despite the physiological and economic importance of lignin, it is remarkable that the mechanism of monolignol export from cells to cell walls is still unknown (Sibout and Höfte, 2012; Barros et al., 2015; Perkins et al., 2019). While several mechanisms for monolignol export from their site of synthesis to the cell wall have been proposed, none of these explain the export of the most abundant monolignols (coniferyl alcohol [CA] and sinapyl alcohol) and no gene products encoding transporters for these monolignols have been identified. Inhibitor studies implicated ATP-binding cassette (ABC) transporters in transport activity in isolated membrane vesicles (Miao and Liu, 2010); however, other studies have not found evidence for active transport of monolignols (Väisänen et al., 2020). The Arabidopsis ABCG29 transporter showed transport activity for p-coumaryl alcohol, but not coniferyl or sinapyl, when heterologously expressed in yeast (Alejandro et al., 2012). During wood formation, gymnosperms accumulate monolignol glucosides in the vacuoles of tracheids, and following programmed cell death, these are deglucosylated in the wall and incorporated into lignin (Aoki et al., 2016). Proton-dependent transport activity for monolignol glucosides has been reported in secondary xylem from gymnosperms (Tsuyama et al., 2013, 2019; Väisänen et al., 2020). In contrast to gymnosperms, flowering plants do not accumulate the glucoside conjugates of monolignols in lignifying cells, so monolignol transport in angiosperms is likely not dependent on monolignol glucosides (LeRoy et al., 2016). Any transport mechanism must account for the wide range of structurally diverse nontraditional monolignols that can be incorporated into lignin either due to biological variation or intentional manipulation (reviewed in Mottiar et al., 2016)).
In addition to their roles in gymnosperm wood formation, monolignol glucosides are part of a complex homeostatic network that can buffer monolignol cytotoxicity, solubility, and stability (LeRoy et al., 2016; Speeckaert et al., 2020). If monolignol export is blocked, the resulting back-up of cytoplasmic monolignols could trigger monolignol glucosylation as a detoxification mechanism, as demonstrated in cell cultures (Väisänen et al., 2015) or Arabidopsis engineered to overproduce monolignols (Perkins et al., 2020). Therefore, the phenotype of a plant with impaired monolignol export is predicted to include the accumulation of monolignol glucosides in vacuoles.
Molecular dynamics modeling indicates that simple diffusion of monolignols across lipid bilayers is an energetically feasible route for not only monolignols, but dimers as well (Vermaas et al., 2019). The computed permeability coefficients of monolignol glucosides indicated that they would not readily diffuse across membranes (Vermaas et al., 2019). This modeling predicts that monolignols will move across lipid bilayers, e.g. out of cells, in the presence of a concentration gradient. While net movement of monolignols has not been observed into purified microsomal vesicles (Miao and Liu, 2010; Alejandro et al., 2012), model monolignol-like compounds could associate with lipid disks and liposomes (Boija and Johansson, 2006; Boija et al., 2007). Importantly, these experimental approaches did not consider that monolignol export occurs in the presence of oxidative enzymes, which are abundant in cell walls of lignifying cells. Thus, the role of oxidative enzymes consuming monolignols in the cell wall creating a concentration gradient to drive diffusion, has been predicted (Vermaas et al., 2019), but it has not been experimentally tested.
Here, we test the hypothesis that the oxidation and subsequent polymerization of monolignols by oxidative enzymes removes them from solution, forming a concentration gradient that drives monolignol diffusion across membranes. Using lignifying xylem vessels of Arabidopsis roots, we developed a two-photon microscopy approach to quantify lignin and soluble phenolics’ intrinsic fluorescence in cell walls and inside living cells. Using this system, we investigated the effect of the loss of oxidative enzymes LACCASE4 and LACCASE17 (lac4 lac17 double mutants) on both cell wall polymer and soluble phenolics in double mutants (Berthet et al., 2011). lac4 lac17 mutants had decreased lignin fluorescence in the cell wall and increased intracellular fluorescent phenolics, suggesting monolignol export was impaired. This was not the case when lignifying xylem cells were treated with an inhibitor of P-type ATPases, such as ABC transporters. We also created an in vitro system where laccases were encapsulated inside of synthetic lipid bilayers (liposomes), and monolignols in the surrounding solution declined as polymer accumulated inside the liposome. Overall, these data highlight the important role of lignin polymerization in the cell wall, creating concentration gradients that lead to monolignol diffusion out of the cell.
Results
Loss of laccases increases intracellular phenolics and decreases extracellular lignin polymer
We hypothesized that the loss of lignin-associated oxidative enzymes would lead to impaired monolignol transport and monolignol accumulation inside of cells. It has not been possible to localize monolignols inside living cells, as lignifying cells are typically buried deep inside plant organs, e.g. tracheary elements in Arabidopsis primary roots (Figure 1A). An additional complexity is that monolignols are exported not only from the lignifying cells themselves, but also by “good neighbors” in the immediate vicinity, e.g. pericycle and endodermal cells that export monolignols into the cell wall of xylem vessels in roots (Smith et al., 2013). To overcome these challenges, a two-photon microscopy approach was developed, where a pulsed, red-shifted laser is used to penetrate deep into an organ. Direct visualization of phenolic autofluorescence showed strong lignin polymer autofluorescence in protoxylem tracheary elements of Arabidopsis roots, with scant soluble phenolic autofluorescence in the surrounding cells (Figure 1B). To validate that this fluorescence is phenolic in nature, roots were treated with piperonylic acid (PA) to inhibit cinnamate 4-hydroxylase (C4H), a critical enzyme in the synthesis of all phenylpropanoids (Schalk et al., 1998). PA extinguished the lignin autofluorescence seen in the spiral wall thickenings of the first lignified protoxylem cells, verifying that the fluorescent signal is due to phenolic compounds downstream of C4H. PA had no effect on the deposition of the polysaccharide component of the spiral secondary cell wall, which was visualized with propidium iodide (Figure 1C). Previous studies found that light exposure induced significant accumulation of monolignol glucosides, coniferin, and syringin, in Arabidopsis roots (Hemm et al., 2004). The two-photon microscope detected high levels of auto-fluorescent material in the vacuoles of light-grown, but not dark-grown, roots (Figure 1D). This accumulation of fluorescent intracellular signal could also be blocked by treatment with PA (Figure 1D). These signals were localized to the central portion of the root cells, consistent with localization in the vacuole. To test if this signal was in the vacuole or cytoplasm, fluorescein diacetate (FDA) was used to stain living cytoplasm (Supplemental Figure S1A), which did not overlap with the autofluorescence, but formed a peripheral signal surrounding it. FM4–64 was used to stain membranes including the tonoplast membrane (Scheuring et al., 2015; Supplemental Figure S1B). The staining was observed at the perimeter of the vacuole, encircling the autofluorescent signal. These colocalizations demonstrate that the autofluorescent signal was entirely within the tonoplast, consistent with vacuolar localization. Thus, both soluble phenolics inside the vacuoles of light-grown root cells and lignin polymer in the cell walls of xylem cells were well-resolved in living roots using the two-photon microscopy system.
Figure 1.
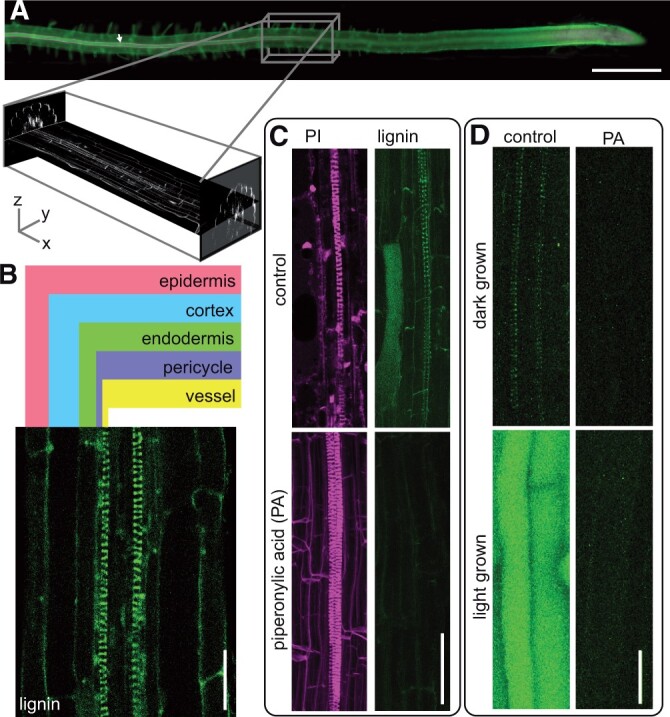
Multiphoton autofluorescence microscopy simultaneously reveals Arabidopsis root protoxylem lignin and soluble phenolics. A, Conventional epifluorescence imaging of Arabidopsis root stained with basic fuchsin (magenta) (white arrow) and calcofluor (green). Scale bar 500 µm. Inset represents the equivalent portion of the root imaged using multiphoton autofluorescence microscopy shown in (B)–(D). XY and YZ planes represent the 3D multiphoton data captured in a z stack. B, Cross-section of a representative Col-0 Arabidopsis root with cell types labeled. C, Control and 24 h 5-µM piperonylic acid (PA)-treated dark-grown roots, lignin imaged by two-photon excitation at 730 nm and emission filter 520–560 nm (green) and propidium iodide (PI) imaged by two-photon excitation at 1,000 nm and emission filter 630 nm (magenta). Images are maximum intensity Z-projections. D, Control and 24 h 5-µM PA-treated roots grown in continuous light or continuous dark conditions imaged by two-photon excitation at 730 nm and emission filter 520–560 nm. Images are maximum intensity Z-projections. Scale bars in (B)–(D) are 25 µm.
If lignin polymerization in the cell wall produces a concentration gradient required for net monolignol diffusion, then the loss of important oxidative enzymes is predicted to impair monolignol export. In contrast, if membrane transporters export monolignols, the monolignols should accumulate in the cell wall, even in the absence of oxidative enzymes. To decrease the amounts of oxidative enzymes in the lignifying cells of these roots, lac4 lac17 double mutants were chosen. These lac4 lac17 mutants (lac4-2 lac17) have reduced lignin levels in stems and altered soluble phenolics in leaves (Berthet et al., 2011), and both LAC4 and LAC17 are expressed in experimentally induced protoxylem cells (Schuetz et al., 2014; Chou et al., 2018). To test if these oxidative enzymes are expressed in native root protoxylem cells, LAC4 and LAC17 were tagged with an mCherry fluorescent tag and expressed under their native promoters (pLAC4:LAC4-mCherry and pLAC17:LAC17-mCherry). Previous work demonstrated that these fluorescent protein fusions are functional in the double mutant (Schuetz et al., 2014; Hoffmann et al., 2020). Both LAC4-mCherry and LAC17-mCherry signals were found near or in advance of the first appearance of lignin in the developing strand of protoxylem (Figure 2, A and B). The lac4 lac17 mutants were then assessed with two-photon microscopy to determine the distribution of phenolics inside and outside of the cells, compared to wild-type (WT) roots. Consistent with the decreased stem lignin in these mutants (Berthet et al., 2011), autofluorescent signal for lignin in the root vessel cell wall was significantly reduced in the lac4 lac17 double mutant compared to WT (Figure 2, D and E). In contrast to the low intracellular phenolic signal observed in the WT stele, the neighboring cells surrounding the lignifying tracheary elements in lac4 lac17 had significantly elevated fluorescent signals inside both pericycle and endodermal cells (Figure 2, D and F).
Figure 2.

Intrinsic fluorescence shows that lac4 lac17 mutant roots have increased intracellular phenolics. A, Schematic of an Arabidopsis root with the region of cell wall lignification highlighted by the black box. B, pLAC4:LAC4-mCherry and pLAC17:LAC17-mCherry in young root xylem tracheary elements imaged for mCherry fluorescence and lignin autofluorescence. Two-photon excitation at 730 nm and emission filter 520–560 nm for the lignin/phenolic channel. C, Coniferin levels in Col WT roots compared to lac4 lac17 measured by HPLC/QTOF. n = 5 experimental replicates of two to three plates of seedlings containing approximately 200 seedlings each, two-sample t test, P = 0.000045. D, Representative images of lignin/phenolic autofluorescence of Col-0 WT controls and lac4 lac17 double mutants. White rectangles indicate representative portion of tracheary element sampled for quantification. Gray rectangles indicate region of neighbor cells sampled for vacuolar fluorescence quantification. Scale bar 25 µm. E(i), Quantification of lignin autofluorescence intensity in WT Col-0 compared to lac4 lac17 double mutants. n = 17 roots from a total of four experimental replicates consisting of two to three plates of seedlings grown and images on separate occasions. Two-sample t test, P<0.01. E(ii), Quantification of lignin autofluorescence intensity in WT Col-0 compared to lac4 lac17 double mutant, lac4 lac17 LAC4-mCherry, and lac4 lac17 LAC17-mCherry lines. ANOVA and Tukey’s post hoc test, P <0.05. n = 7 roots from three experimental replicates. Letters indicate statistical significance. F(i), Quantification of neighboring cell autofluorescence intensity in WT Col-0 compared to lac4 lac17 double mutants. n = 17 roots from a total of four experimental replicates. Two-sample t test, P<0.01. F(ii), Quantification of neighboring cell autofluorescence intensity in WT Col-0 compared to lac4 lac17 double mutant, lac4 lac17 LAC4-mCherry, and lac4 lac17 LAC17-mCherry lines. ANOVA and Tukey’s post hoc test, P <0.05. n = 7 roots from three experimental replicates consisting of two to three plates of seedlings grown and images on separate occasions. Letters indicate statistical significance. Box and whiskers in C, E, and F represent median values and quartiles.
If monolignols are accumulating inside the vacuoles of neighboring cells, we predicted that they would be in the form of monolignol glucosides. High-pressure liquid chromatography (HPLC) and quadrupole time-of-flight mass spectrometry (Q-TOF MS) of soluble phenolics revealed that whole dark-grown lac4 lac17 roots had a statistically significant increase in the monolignol glucoside coniferin compared to the WT (Figure 2C). In the original characterization of the soluble stem or leaf phenolics of lac mutants, both sinapoyl esters and flavonol glycosides were elevated (Berthet et al., 2011; Zhao et al., 2013). In these dark-grown lac4lac17 roots, flavonol glycosides were unchanged while there were significant increases in two feruloyl derivatives and N′,N′′-disinapoylspermidine (Supplemental Figures S2 and S3). Coniferin was the most elevated phenolic compound, supporting our interpretation that the intracellular signal that we observe in the two-photon microscopy studies is related to impaired monolignol export.
To test if these changes in lignin and intracellular phenolics were the result of a developmental defect, the growth of lac4 lac17 mutant lines was characterized; however, lac4 lac17 mutants did not differ from Col-0 WT in epidermal cell length or width or the overall root diameter (Supplemental Figure S4). When WT LAC4 or LAC17 (pLAC4:LAC4-mCherry or pLAC17:LAC17-mCherry) was expressed in the lac4 lac17 double mutants, the mutant phenotype of increased intracellular phenolics was complemented to WT levels (Figure 2, E and F). The mutant phenotype demonstrates that in the absence of laccase-dependent lignin deposition, monolignols accumulate inside of cells and that monolignol export is deficient.
Monolignol transport in roots is not inhibited by vanadate
To test the alternative hypothesis that ABC transporters are transporting lignin monomers, both the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and vanadate, the well-characterized P-type ATPase inhibitor, were tested (Rea, 2007). CCCP was lethal, even at low concentrations, following 24-h incubation (Supplemental Figure S5). Roots were exposed to various concentrations of vanadate for 24 h, and the lethal concentration was above 100 µM (Supplemental Figure S5). Following 50-µM vanadate inhibition, there was no significant changes in the lignin signal in the cell walls of the protoxylem compared to controls, suggesting that monolignol export was not affected (Figure 3, A and B). Similarly, there was no strong vacuolar accumulations inside cells with the vanadate treatment (Figure 3C). A lower autofluorescent signal was observed in the periphery of some vanadate-treated endodermal cells, but this was distinct from the vacuolar fluorescence in the lac4lac17 mutants (Figure 3C). This may reflect a role for ABC transporters in the complex phenolic metabolism of endodermal cells, which are producing their own lignin barrier as Casparian strip formation occurs (Lee et al., 2013).
Figure 3.

Vanadate treatment does not change lignification in root protoxylem. A, Representative lignin signals from control and 24 h 50-µM vanadate-treated roots. White rectangles indicate representative portion of tracheary element sampled for quantification. Two-photon excitation at 730 nm and emission filter 520–560 nm. Scale bar 25 µm. B, Quantification of lignin signal from two-photon microscopy in control and 24 h 50-µM vanadate-treated roots. n = 14 roots from a total of three experimental replicates consisting of independently grown plates of plants. Two-sample t test indicated no significant difference (n.d.). C, Comparison of representative two-photon autofluorescent signals Col-0 WT roots, lac4lac17 mutant roots, and vanadate-treated roots. te indicates xylem tracheary elements. en indicates endodermal cells neighboring xylem vessels. co indicates cortical cells. ep indicates epidermal cells. Scale bar indicates 25μm.
As vanadate affects multiple classes of transporters and biological processes (Palmgren and Morsomme, 2019), we did a series of control experiments to ensure the conditions of vanadate treatment was not stopping xylem development, but was effective in inhibiting known ABC transporters. To test if 50-mM vanadate for 24 h permitted new tracheary elements to differentiate, but was not long enough to arrest cell division or growth, roots were dipped in fluorescent beads at the beginning of the inhibition period. Beads adhered to the entire root surface (Supplemental Figure S6A). In both control roots and vanadate treatments, new growth, including areas containing newly differentiated and lignifying protoxylem, was detected by the presence of root segments without beads (Supplemental Figure S6, B and C). To ensure that 50-μM vanadate was sufficient to inhibit root ABC transporters, we tested known ABC transporter-mediated processes, such as the gravity-induced auxin asymmetry mediated by the MDR4/ABCB4 transporter, using the protocol used to phenotype the mdr4-1 (abcb4) mutant (Lewis et al., 2007). Vanadate phenocopied the mdr4-1 mutant phenotype, with more auxin (detected by the ProDR5:GFP signal, Benková et al., 2003) in the lower root epidermis, suggesting that 50-µM vanadate was sufficient to inhibit known ABC transporter activity in the roots (Supplemental Figure S7A). Vanadate treatment during continued root growth also elicited a characteristic abundant growth of root hairs (Supplemental Figure S7B) as previously reported (Lin et al., 2015). These controls establish that the 50-µM vanadate treatment was effective in its well-established role as an ABC transporter inhibitor, but that it did not interfere with protoxylem development. Lignin was still deposited in growing regions of the roots, despite the inhibition of transporters, including ABC transporters.
Net flux of monolignols across a synthetic lipid bilayer driven by a sink
If monolignols can diffuse across lipid bilayers, without transporter proteins and driven only by concentration gradients, then they should move across any membrane with a sink on one side and a source on the other. In lignifying cells, the presence of oxidative enzymes in the cell wall creates the sink, while monolignol production provides the source (Figure 4A). Spheres of synthetic lipid bilayers (liposomes) were prepared with laccase enzyme in the interior volume as a sink, with the surrounding solution representing the monolignol source (Figure 4A). Artificial liposomes were generated using a lipid composition that balanced the practical consideration of preparing stable unilamellar liposomes with the need to realistically and conservatively mimic real plant plasma membranes. Plant plasma membranes contain large proportions of sterols (Bohn et al., 2001), which are known to increase membrane ordering and packing while reducing partitioning of solutes into the membrane (Wennberg et al., 2012). For this reason, 45% cholesterol was included. The composition of fatty acyl tails on membrane lipids varies considerably across plant species and tissues (Reszczyńska and Hanaka, 2020). For this experiment, we selected a saturated fatty acid, palmitic acid (16:0) that would pack tightly and conservatively model membrane permeability (Wennberg et al., 2012). Laccase extracted from Trametes versicolor was used as the source of laccase, as it was not possible to extract mCherry-tagged lignin-associated laccases from Arabidopsis in pull-down experiments. Laccase from another Trametes, Trametes hirsute, produced dehydrogenation polymer (DHP) with good structural similarity to native lignins, when incubated with CA in vitro (Warinowski et al., 2016). Further, comparative modeling of fungal, plant, and bacterial laccases demonstrates that T. versicolor and plant (poplar) laccases share common 3D structures consisting of three cupredoxin-like domains, with high amino acid conservation at copper binding sites (Dwivedi et al., 2011).
Figure 4.

Monolignols cross pure lipid bilayers to encounter laccase inside. A, Schematic representation of the lignifying thickened secondary wall of a root protoxylem vessel and the in vitro laccase-containing liposome system. Top row: laccase (black bean) has been exported to the cell wall in advance of monolignol production; in liposomes, lipid bilayer (magenta) encapsulates laccase proteins. Middle: CA monomers (blue) cross the lipid bilayer. Bottom: Monolignols are oxidized by laccase to form monolignol radicals (blue with highlight and dot) that form dimers of CA. B, Cryo-transmission electron microscopy of liposomes before and after incubation with CA. Scale bar 100 nm. C, CA levels over time with liposomes containing laccase, BSA, or buffer. n = 3 replicated independent reactions. Bars indicate standard deviation. D, CA incubated with liposomes containing laccase generate β-β-, β-O-4-, and β-5-linked dimers over time. Bars indicate standard deviation. CA and dimer analysis by HPLC was replicated three times. Bars indicate standard deviation.
The laccase-containing liposomes incubated with the monolignol (CA) provided a simplified system to test if monolignols would cross the pure lipid bilayer in the presence of a sink. An additional control experiment using cryogenic transmission electron microscopy (cryo-TEM) of liposomes before and after incubation with the CA demonstrated that the liposomes retained their size and integrity throughout the course of the assays (Figure 4B). To assess if monolignols crossed the bilayer and were consumed, the total amount of CA present (both inside and outside of the liposome) was determined with HPLC at different time points. When CA was added to media surrounding laccase-containing liposomes, the monolignol concentration declined, while CA concentrations were stable in the controls of empty liposomes or liposomes encapsulating a control protein of bovine serum albumin (BSA) (Figure 4C). As monolignol levels declined, dimers accumulated, indicating that the CA was crossing the membrane to reach the laccase, which was able to facilitate radical coupling (Figure 4D).
To ensure that all laccases were contained within the liposomes during these experiments, the solution surrounding the liposomes was tested for free laccase activity, which would be predicted if liposomes had broken open (Supplemental Figure S8A). A preparation of laccase-containing liposomes was incubated with CA as above, and then the liposomes were separated from the surrounding solution using a size exclusion filter with a cut-off larger than the size of laccase proteins. If free laccase were present, then the filtrate would contain laccase activity; however, no laccase activity, in either decreased CA levels (Supplemental Figure S8A) or increases of dimers, was found in the liposome-free filtrate (flow through) samples (Supplemental Figure S8B). This indicates that all the laccase activity was inside the liposomes for the duration of the assays. Another possibility was that laccases might have adhered to the exterior surface of the liposomes, so a parallel liposome preparation was made by mixing empty liposomes with free laccase in the surrounding solution (Supplemental Figure S9A). Following the size exclusion chromatography purification step, no laccase activity remained associated with these empty liposomes (Supplemental Figure S9B). Thus, all laccase activity was contained within the liposomes, and monolignol oxidation and dimerization is evidence that the monolignols crossed the pure phospholipid bilayer to encounter the laccase within.
The disappearance of monolignols from the solution suggested that they were crossing the lipid bilayer and encountering the laccase, leading to polymerization. To assess the formation of higher molecular weight products by laccase-containing liposomes, gel permeation chromatography was performed (Figure 5A). When laccase-containing liposomes were incubated with CA, higher molecular weight (shorter retention time) compounds appeared over time from time zero (light gray) to 24 and 48 h (medium gray) and finally 96 h (black). From 24 to 96 h, products consistent with polymeric phenolics, as determined by comparison with a standard DHP, were formed (Figure 5A).
Figure 5.

Stable laccase-containing liposomes accumulate phenolic polymer. A(i), Gel permeation chromatographic analysis of products formed by laccase-containing liposomes fed CA. A(ii), GPC standards of laccase DHP, pure CA, and blank solvent control. GPC analysis was replicated four times by preparing liposomes in four separate tubes. B, Super-resolution total internal reflection micrographs of immobilized laccase-containing liposomes (arrows) following 24 h 1-mM CA incubation. Autofluorescence of phenolic material (405 nm) green in merged image, signal from the DiI (DiIC18(3)) lipid dye (561 nm) magenta in merged images. Representative images of laccase-containing liposomes with CA (LAC+CA) and without CA (LAC−CA), and DHP formed by T. versicolor laccase incubated directly with CA (no liposomes). ≥20 fields of view from three replicated experiments, consisting of independent reactions with liposomes, analyzed per treatment, a representative image is shown for each treatment. C, Quantification of (i) density and (ii) size of autofluorescent puncta in laccase-contain liposomes with (LAC + CA) or without CA (LAC − CA). Control liposomes contained BSA with (BSA + CA) or without CA (BSA − CA). More than 20 fields of view from three replicated experiments per treatment. ***Indicates Dunn’s pairwise comparison, P < 0.001. D, Schematic representation of the end point of the liposome reaction in Figure 5A (96 h). Laccase-containing liposomes with polymer in the interior and no monolignols remaining in the surrounding solution.
To test if the polymer was inside the laccase-containing liposomes, super-resolution total internal reflection fluorescence (TIRF) microscopy was employed. Liposomes were imaged using the red fluorescent dye incorporated into their lipids, and the polymeric lignin was visualized by its characteristic UV-autofluorescence. Autofluorescent puncta were visible in laccase-containing liposomes incubated with CA (Figure 5B, top row), while control laccase-containing liposomes without CA had background levels (Figure 5B, middle row). The size, shape, and distribution of fluorescent puncta were notably different than the DHP made by T. versicolor laccase in solution (Figure 5B, bottom row). Lignin signal was measured as the number of fluorescent puncta per square micrometer, and liposomes containing laccase (LAC + CA, Figure 5C) had statistically significantly more signal than controls. Lignin autofluorescence was minimal in controls of BSA-containing liposomes fed with CA or in laccase-containing or BSA-containing liposomes incubated without CA (Figure 5C). While only a subset of liposomes contained the lignin signal, the lignin signals were highly co-localized with the liposome fluorescence (Manders overlap co-efficient 0.85 ± 0.19 [standard deviation], n = 10). The particles observed differed significantly in size from background signals seen in controls. The uniformity of the size of the lignin signals in the laccase-containing liposomes was distinct from the large and irregular autofluorescence seen in DHP controls. Thus, the monolignol depletion observed in Figure 4 was accompanied by lignin formation inside the liposomes (Figure 5D). This experimental system demonstrates that laccase oxidation of monolignols and polymer formation is capable of driving significant monolignol movement across a pure lipid bilayer in the absence of protein transporters.
Discussion
The presence of a sink, e.g. laccase-mediated oxidation of monolignols, is a previously unappreciated factor in monolignol export. The consequence of monolignol consumption by oxidative coupling/polymerization is that a concentration gradient of monolignols occurs across the membrane that can drive the net diffusion out of cells (or into liposomes). Decreased monolignol levels were observed as CA diffused through the synthetic lipid bilayers of liposomes to form polymeric lignin structures. Importantly, net movement was found only in the presence of an oxidative enzyme, laccase, which promotes polymer formation. The presence of dimers and polymers are consistent with the conclusion that monolignols have diffused across the lipid bilayer to encounter the laccase inside the liposome. The encapsulated laccase in the liposome experiments is analogous to LAC4 and LAC17 in the cell walls of xylem cells, which provide a sink sufficiently strong to drive significant flux out of cells. The loss of these oxidative enzymes led to intracellular accumulations of monolignol glucosides, which could be detected inside vacuoles by live-cell imaging with two-photon microscopy. These data validate the prediction that simple diffusion is sufficient to transport monolignols across lipid bilayers, based on in silico analysis of the physico-chemical properties of lignin-related compounds (Vermaas et al., 2019). While that modeling indicates that diffusion is energetically feasible, this work reveals that oxidative enzymes are a key component of the export pathway, creating the conditions to drive monolignol diffusion into the wall.
Monolignol oxidation in the wall is linked to export from the cells
In the present experiments, we developed an application of two-photon microscopy to directly visualize the intrinsic fluorescence of lignin and intracellular phenolics (Figure 1), and examined phenolic localizations in lac4 lac17 double mutant (Figure 2). The reduced phenolic cell wall signal showed that, with the loss of oxidative laccases in the cell wall, lignin polymerization declines. The corresponding increased intracellular fluorescence in lac4 lac17 double mutants support the view that oxidation in the wall is linked to export from the cells. The intracellular elevated fluorescence in the vacuoles of lac4 lac17 mutants was correlated with increased levels of monolignol glucosides detected by liquid chromatography–mass spectrometry, consistent with the paradigm that plant cells deal with elevated levels of monolignols in the cytoplasm by glucosylation and storage in the vacuole (LeRoy et al., 2016; Speeckaert et al., 2020). This is also consistent with protoplast feeding experiments, where mono- or dilignols supplied from the media moved into cells and were subsequently oxidatively coupled intracellularly and glucosylated (Dima et al., 2015). Leaf tissues that hyperaccumulate monolignol glucosides due to the overexpression of the phenylpropanoid pathway also exhibit a high vacuolar autofluorescence (Perkins et al., 2020). In lac4 lac17 double mutants, there was slight downregulation of some monolignol biosynthetic pathway genes (Berthet et al., 2011), which could contribute to the decreased lignin levels seen in the xylem cell walls. This decrease does not explain why there is a strong intracellular soluble phenolic signal in these mutants that was not found in WT roots. The accumulation of soluble phenolics in the vacuoles of lac4 lac17 mutants, as well as their WT growth, suggest that the slight downregulation detected by Berthet et al. (2011) is not enough to halt monolignol production. Instead, monolignol production continues, but the monomers do not move directionally out of the cell without the oxidative activity in the wall and accumulate as monolignol glucosides inside the cell.
In the presence of a sink, monolignol export does not require ATP hydrolysis
One critical implication of a sink-driven diffusion model is that monolignol export, in the presence of a strong sink of oxidative enzymes such as laccases or peroxidases, should be considered an energetically favorable process. In planta, the facilitated diffusion of monolignols could occur through membrane channels, analogous to aquaporins, although no such channels have been yet identified. An energetically favorable export process explains our present lack of well-characterized ATP-dependent monolignol transporters or gene products, and the observation that vanadate treatment does not decrease lignification (Figure 3).
If ABC transporters were solely responsible for monolignol transport it would be expected, however, that inhibition would attenuate the lignin signal to some measurable degree, which it did not. Although lignification was normal following vanadate treatment, there was evidence to show that the vanadate inhibition of ABC transporters in the Arabidopsis roots was effective. First, vanadate treatment phenocopied the mdr4-1 (abcb4) mutant (Lewis et al., 2007), indicating auxin-related ABC transporters were inhibited. Second, vanadate treatment led to slowed growth and increased root hairs, as reported by Lin et al. (2015). Although vanadate-treated roots were still alive and continued to grow and differentiate xylem, the toxicity of vanadate was apparent. Finally, there was increased autofluorescence in the cytoplasm of endodermal, cortical, and epidermal cells in vanadate-treated roots compared to control roots (Figure 3C). It is possible that P-type transporters in the tonoplast and/or plasma membrane in these cells were inhibited, leading to autofluorescence in the cytoplasm, despite the normal lignification.
Sink-driven diffusion could operate in parallel with other export mechanisms
While the liposomes and laccase mutants show that sink-driven diffusion is a larger factor in monolignol export than previously known, these data do not rule out the possibility that, in some lignifying cell types, other transport mechanisms including active transporters or channels, may operate in parallel. In this way, monolignol diffusion can be thought of as analogous to the diffusion of plant auxins, such as indole acetic acid (IAA), which is also a small aromatic molecule that diffuses across the plasma membrane. In the case of IAA, the cell wall-localized protonated form crosses the bilayer, while the anionic form remains trapped inside the cell (Naramoto, 2017). In the case of monolignols, the monolignol crosses the bilayer, is oxidized by laccases and/or peroxidases, and is trapped in the cell wall. As with auxin transport, a suite of membrane proteins could fine-tune the bulk flow. ABC transporters and proton-dependent monolignol glucoside transport activities (Alejandro et al., 2012; Tsuyama et al., 2013) are likely examples of complementary and perhaps fine-tuning mechanisms that could accompany diffusion. The low peripheral phenolic signal in the endodermis following vanadate treatment hints at potential fine-tuning, e.g. vanadate could have inhibited an ABCG29-based phenolic transport in the endodermal cells (Alejandro et al., 2012). Still, the contrast between the low lignin in the cell wall accompanied by intracellular accumulation of phenolics in laccase mutants, compared with the control levels of lignin signal in the xylem of vanadate-treated roots point to sink-driven diffusion as the major component of monolignol export in this system.
Liposomes conservatively model the energetic barrier of plant membranes
Plant membranes vary a great deal in composition between cell types, species, and environmental conditions (Furt et al., 2011). In all cases, they can readily transport lignin monomers, either during development or when induced by wounding or infection. The lipid composition of our liposomes was chosen for its stability and low permeability (Edwards et al., 1997). The in silico modeling of monolignol diffusion by Vermaas et al. (2019) suggests that biological membranes containing sterols reduce the permeability of membranes to monolignols. The permeability coefficient for sterol-rich Zea mays membranes indicates that this membrane represents a more significant barrier than bilayers of homogeneous phospholipids. Not only does our experimental system include sterol, it also includes only saturated fatty acids rather than the mix of saturated and unsaturated fatty acids used by Vermaas et al. (2019). We focused our experiments on CA, which makes up a large proportion of lignin in all biomass, as our model monolignol compound. The permeability coefficients between CA and sinapyl alcohol were determined to be identical while, due to the lack of methoxy groups, the permeability coefficient of p-coumaroyl alcohol was even lower (Vermaas et al., 2019). Overall, our experimental system models the energetic barrier of the membrane conservatively, and it may underestimate the ability of monolignols to cross the bilayer.
Laccase-containing liposomes validate in silico models
If monolignols diffuse into bilayers as predicted by Vermaas et al. (2019), then they should also diffuse out, reaching some equilibrium of zero net flux unless they are trapped by polymer formation. In the case of the laccase-containing liposomes, it is evident that a significant net flux of monomer passed through the membrane, since the laccase was present only in the interior of the liposomes, and the total monomer level declined precipitously while dimers formed (Figure 4). Monitoring the declining levels of monolignols and increasing levels of dimers provided a read-out of the interaction of monolignols with laccases, as for technical reasons, our analysis did not distinguish between monolignols that were inside and outside of the liposome. The dimers initially formed were biased towards the β-5 dimer rather than the β-O-4 bonds that are more common in lignin. It is known, however, that the presence of xylans during polymer formation favors the formation of β-O-4 bonds (Li et al., 2015). Our model system did not contain any xylan and this may explain the discrepancy in bond abundance. Over later time points of the liposome experiments, dimers declined and the presence of polymer product was confirmed by gel permeation chromatography and TIRF microscopy. The super-resolution TIRF experiment clearly distinguished the small regular puncta of polymer within liposomes from the DHP product made by unencapsulated laccases free in solution. By the end, the overwhelming majority of monomer added to the solution had passed through the membrane and was trapped there by incorporation into the polymer (Figure 5). Laccase-containing liposomes provide empirical evidence supporting the predicted ability of monolignols to diffuse across lipid bilayers, and demonstrate the importance of monolignol polymerization in this process.
Conclusion
Using two-photon microscopy to peer into lignifying tissues to see how monolignol export is affected in mutants or with inhibitors provides insight into the important role of oxidative enzymes. Of course, in lignifying tissues, both laccases and peroxidases are predicted to contribute to the consumption of monolignols in the cell wall (Sterjiades et al., 1993; Blokhina et al., 2019; Hoffmann et al., 2020). Our work used laccases, as the laccase mutants in Arabidopsis have clear and well-described phenotypes (Berthet et al., 2011; Zhao et al., 2013), unlike the peroxidase mutants whose phenotypes are more subtle (reviewed by Shigeto and Tsutsumi, 2016). Laccases do not require co-factors such as hydrogen peroxide, so the liposome experiments contained just the minimal components needed for monolignol translocation across a membrane: monolignols, lipid bilayers, and oxidative enzymes, without confounding considerations such as diffusion and stability of the supplied peroxide over time.
Overall, a sink-driven diffusion model explains many previously reported features of monolignol export: It is flexible and promiscuous to different forms of monolignols, it does not rely on programmed cell death, and even nonlignifying tissues can export monolignols during defense or wounding responses.
Materials and methods
Plant growth
Arabidopsis plant lines were WT (Columbia-0 accession), lac4-2 lac17 double mutants, described in Berthet et al. (2011), were the kind gift from Richard Sibout and Lise Jouanin from the Institut Jean Pierre Bourgin, INRA, Versailles, France.
For all mutant lines and inhibitor treatments, Arabidopsis seeds were plated on half-strength MS 1% sucrose germination medium (GM) in a 0.2% agar solution, then subjected to 72 h of stratification in the dark at 4°C. Plates were wrapped in aluminum foil to maintain darkness, arranged vertically, and transferred to a 21°C growth chamber for 4 days. For inhibitor experiments, seedlings were manually transferred to vertical square petri plates containing GM medium with or without inhibitors as appropriate. PA (Aldrich P49805) prepared in water was added to a final concentration of 5 μM, sodium metavanadate (Sigma S-6383) solubilized in 1-M NaOH was added to a final concentration of 50 μM. The lac4 lac17 line and WT control seedlings were grown for 6 days of growth on GM plates in the dark. Light grown seedlings were grown under continuous (24 h) cool white fluorescent lighting at an intensity of 210 µmol m−2 s−1.
Soluble phenolic quantification
For phenolic analysis of roots, tissue was harvested from dark-grown seedlings grown on vertical plates, as these were the conditions used for imaging intracellular phenolics. About 100–200 mg of tissue was combined with 49.5% methanol:49.5%H2O:1% acetic acid to a final concentration of 150 mg·mL−1 and incubated at 45°C for 4 h to extract soluble phenolic compounds. The samples were centrifuged at 21K g for 15 min and the supernatant was transferred to glass vials. Samples were filter-sterilized into HPLC vials.
The phenolic profiles were determined using an Agilent 1200 HPLC system equipped with G4212A diode array detector (DAD) and 6530B quadrupole time-of-flight (QTOF) detectors (Agilent, Palo Alto, CA, USA). The HPLC conditions for the separation of the phenolic compounds were performed according to Pico et al. (2022). Briefly, the separation was accomplished using a Zorbax SB-C18 column (50 mm × 4.6 mm internal diameter) at 40°C with a flow rate of 0.8 mL·min−1 and a gradient of water with 2% formic acid (v/v) (solvent A) and acetonitrile with 0.1% formic acid (v/v) (solvent B). The injection volume was 5 μL, and the tray was maintained at 4°C to avoid degradation of phenolic compounds.
The detection of targeted compounds (Supplemental Figure S3) implied a double identification of phenolics with their corresponding standards by means of MS and DAD (520 nm and 280 nm). The MS mode conditions of the QTOF detector were as follow: ionization mode in electrospray ionisation (ESI) (+); drying gas (nitrogen) flow and temperature of 5 mL min−1 and 325°C, respectively; sheath gas (nitrogen) flow and temperature of 12 mL min−1 and 275°C, respectively; nebulizer pressure of 40 psi; capillary and nozzle voltages of 2,500 and 2,000 V, respectively; fragmentor, skimmer, and octapole voltages of + 200, 65, and 750 V, respectively.
To identify untargeted phenolic compounds (i.e. unknown peaks for which we do not have standard), the QTOF detector was also operated in product ion mode (i.e. tandem mass spectroscopy [MS/MS]). Following the same HPLC and QTOF conditions as for the MS mode, the collision gas (nitrogen) cell was activated and defined at 20 psi and 35 eV for the three main compounds that showed differences among the wild and mutant species. For the unknown peaks labeled in the Supplemental Figure S3 as # 5, # 6, and # 8, we used the parent ions with m/z 520.3315, m/z 564.3578, and m/z 558.2806, respectively, at their corresponding retention times.
Generation and analysis of liposomes
Laccase enzyme purified from T. versicolor (#38429) and BSA (#85040C) were obtained from Sigma-Aldrich. DHP was produced by adding 1 mM of CA to a 1-U mL−1 solution of the T. versicolor laccase and incubating at room temperature for 72 h.
Laccase or BSA was resuspended in 10-mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer pH 5.1. 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), cholesterol, and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) lipophilic membrane stain were dissolved in chloroform at a 54.9:45:0.1 mol:mol:mol ratio. DPPC and cholesterol were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Chloroform was evaporated by blowing the samples with a constant stream of nitrogen gas until the samples became thick. The samples were dried completely in a vacuum for 3 h to remove any residual chloroform. The dried lipid films were rehydrated in entrapment solution of choice (laccase enzyme solution, BSA solution, or 10-mM MES buffer) at 1-mL 50 µmol−1 of lipids for 3 h at 50°C. Rehydrated lipids were extruded through two stacked 0.1-µm filters in a 10-mL thermobarrel extruder (Extruder, Northern lipids, Vancouver, BC, Canada). The resulting lipid nanoparticles had a mean diameter of 100 ± 20 nm as determined by Phase Analysis Light Scattering methods (ZetaPALS, Brookhaven Instruments Corp., Holtsville, NY, USA). The extruded laccase-containing liposomes were separated from unencapsulated laccase by passing through a Sepharose column (Sigma-Aldrich) pre-equilibrated with 10-mM MES buffer to exchange the laccase-containing extra-liposomal buffer with 10-mM MES.
Control experiments were performed to verify the efficacy of the above column purification. Empty liposomes were prepared and mixed with laccase solution to test if unencapsulated laccases could stick to the liposome exterior. In short, dried lipid films were rehydrated in 10-mM MES buffer as described previously. Rehydrated lipids were extruded as above. The lipid nanoparticles were concentrated to 2× the desired lipid concentration by tangential flow filtration using an ultrafiltration hollow fiber column (molecular weight cut-off 500 kDa; GE Healthcare). The 2× liposome solution was mixed at equal volumes with a 2× laccase solution. The mixture was subsequently separated via size exclusion chromatography using a Sepharose column to remove unencapsulated laccases (Supplemental Figure S9A).
Quantification of liposome concentrations was performed using serial dilutions of all liposome preparations and subsequent analysis of DiI fluorescent intensity using a 515-nm excitation and 590-nm emission filter combination on a plate reader (BioTek Instruments, Inc.). Liposome concentrations for all treatments were normalized for consistency.
Liposome monomer uptake assays
CA was purchased from Sigma-Aldrich (#223735) and was resuspended in 100% methanol at a concentration of 1 M. CA was added to a final concentration of 1 mM and incubated at room temperature for up to 24 h. CA concentrations were measured as total CA in the system (both liposomes and media). All reaction were stopped at specific time points by addition of ethylenediaminetetraacetic acid (EDTA; 10-mM final concentration as a copper chelating agent) and boiling for 10 min. Methanol (100%) was added to the stopped reactions to a final concentration of 50% methanol and stored at −20°C. HPLC analysis was performed as described for soluble phenolics and the CA remaining in the reactions was quantified using a standard curve generated from serial dilutions of a CA standard.
To identify the CA oligomers that were formed in liposome experiments, we used an Agilent 1100 LC/MSD Trap XCT Plus instrument with a Waters Atlantis T3 column (3 µm, 2.1 × 50 mm) in negative electrospray mode. The reaction mixtures were eluted from the column over a gradient from 95% A (water and 0.2% formic acid) to B (acetonitrile and 0.2% formic acid) over 25 min. CA dimers were identified from characteristic MS/MS (Morreel et al., 2010).
The buffer surrounding liposomes was tested for laccase activity by filtering liposomes out of the liquid buffer using Amicon 100-kDa filter device (Millipore-Sigma). It was confirmed using sodium dodecyl sulphate–polyacrylamide gel electrophoresis that T. versicolor laccase at ∼60 kDa can readily pass through the 100-kDa filter device (Supplemental Figure S8A). Assays for CA uptake were performed as described above. A subsample was taken and processed as above, while the remainder was filtered before the addition of EDTA and topped up to the starting concentration of CA and incubated for an additional 24 h before being processed as above and analyzed by HPLC as above.
TIRF microscopy
A Zeiss Observer Z1 TIRF microscope with 405-nm and 561-nm excitation lasers was used to detect liposomes and their contents. Lipid membranes were prepared using DiI stain (DiIC18(3)) causing them to emit a signal when excited by the 561-nm laser. The poly-phenolic material forming within laccase-containing liposomes was excited by the 405-nm laser. As a control, DHP was made from a mixture of 1-mM CA and free laccase in solution (1-U mL−1 of T. versicolor laccase), and incubated at room temperature for 72 h. Liposomes were immobilized on poly-L-lysine-coated coverslips (Sigma P8920). Five images were taken with Hamamatsu Flash 4.0 camera and averaged to minimize signal from nonimmobilized and out-of-plane liposomes. Images were processed using ImageJ to analyze particles. Background was subtracted using a rolling ball background subtraction with a 25-pixel radius. Images had a threshold applied with the RenyiEntropy algorithm. The area and number of particles were calculated.
Cryo-TEM
Liposomes, diluted in 10-mM MES pH 5.0, were incubated with 0.3-mM and 1.0-mM CA for 24 h. The bulk of the buffer was removed by filtration through an Amicon 100-kDa filter device (Millipore-Sigma). Cryo-TEM was performed as described elsewhere (Kulkarni et al., 2018). Briefly, 3–6 μL of the liposome suspension was added to glow-discharged copper TEM grids (Ted Pella, Redding, CA, USA) and plunge-frozen with a FEI Vitrobot Mark IV (FEI, Hillsboro, OR, USA). Following vitrification, all grids were stored under liquid nitrogen until imaged. A FEI Tecnai LaB6 G2 was used to image the samples under low-dose conditions at a magnification of 55,000–62,000×. Images were captured using a bottom-mount FEI Eagle 4k CCD detector.
Gel permeation chromatography
A protocol for extracting and analyzing liposome samples by gel permeation chromatography was developed according to Lange et al. (2016). Liposomes containing T. versicolor laccase were added to MES buffer at pH 5.0 and incubated at 21°C. At the prescribed time interval (0–92 h), 50 µL of 0.5-M EDTA/0.75 mL of sample was added and the samples mixed. Samples were immediately heated to 90°C for 10 min. Methanol (100%) was added to a concentration of 50% to disrupt liposomes. Analysis of T0 controls suggests that this protocol is sufficient to fully inactivate laccase activity given the absence of coupling products in the T0 samples. This is evidence that during further processing, no coupling activity was observed. Samples were stored at −20°C prior to further processing. Samples were dried in a vacuum centrifuge at 60°C (∼4 h) until dry. Pellets were resuspended in 100 µL of MeOH (∼30-min incubation to solubilize EDTA precipitate). Following solubilization of the pellet, 200 µL of 95% dimethylformamide (DMF): 5% acetonitrile was added and samples were mixed by inverting (approximately 10 times), and incubated at room temperature for 10 min. To remove methanol, samples were dried under vacuum centrifugation at 30°C for 30 min. To settle out precipitated EDTA, samples were centrifuged for 20 min at 15,000g. Supernatants were transferred to new tubes, stored at −20°C for 48 h, and re-centrifuged for 20 min at 15,000g to pellet residual EDTA crystals. As the final stage of sample preparation, the supernatant was filter sterilized into HPLC vials. Samples were eluted from a 5-µm particle size DVB (divinylbenzene) 1,000 Å 5-µm 250 mm × 10 mm column (Jordi Labs) maintained at 40°C. Samples were eluted from the column with a mixture of HPLC grade 95% DMF and 5% acetonitrile. Signals were detected with a PDA-100 photodiode array detector.
Root xylem two-photon microscopy
Roots were prepared for imaging by placing them in the distilled water filled channel formed in between two parallel #1.5 coverslips on a microscope slide with a 2–5 mm gap in between. That assembly was topped with an additional coverslip, and the position of the coverslips adjusted such that a narrow ∼1 mm water filled channel containing the root remained. The perimeter of the assembly was sealed with wax.
Stacks of images were collected using an Olympus FV1000 MPE Multi-photon Laser Scanning Microscope with a Spectra-Physics Mai Tai DeepSee pulsed laser and an Olympus XLPLN 25X WMP water-dipping objective. Images of lignin and soluble phenolic autofluorescence were acquired using 730 nm excitation and emission was captured at 520–560nm. Images of mCherry-tagged laccase fluorescent proteins were acquired using two-photon excitation at 1,000-nm and 630-nm emission filter. For consistency laser intensity was kept constant at 10%, photomultiplier voltage was kept at 715 V, z thickness 0.96 μm, image dimensions of 2,048 × 640, zoom 1.4× and dwell time was set to 4 μs pixel−1. Auxin was visualized using a PromotorDR5:GFP reporter line (Benková et al., 2003), as described by Lewis et al. (2007). Roots were grown in the dark on vertical plates for 5 days then rotated 90° and transferred to 50 μM or control GM media vertical plates to stimulate a gravitropic response for 6 h. A two-photon laser wavelength of 910 nm was used to excite the GFP fluorophore and images were captured at 380–560 nm. Cross-sections were reconstructed from a z projection of the root tip.
For quantification of lignin in protoxylem vessels, stacks of six images that encompassed the youngest lignifying protoxylem vessel in the root were extracted and a maximum intensity z projection of that optical sub-sample was made. Lignin signal was measured in the sub-sample by drawing a roughly rectangular region of interest around the vessel and the average pixel intensity normalized by area recorded. A background region in the adjacent nonlignified young xylem was selected and used for background subtraction. Vacuolar fluorescence was measured in the same manner using an intracellular region of interest.
To detect if there was growth of new root tissue during the treatment with inhibitors, fluorescent beads were used as a reference point. Roots were dipped in a 1:10 diluted solution of fluorescent beads (MagSphere Orange Fluorescent Aminated PS Microspheres 6.8 μm AMOF007UM) immediately before transferring to inhibitor and control plates for 24 h. The beads were readily visible using the same microscope setting used for lignin autofluorescence. All images were processed and quantified using ImageJ/FIJI (Schneider et al., 2012).
To stain the plasma membrane, endomembranes and tonoplast seedlings were stained in 45-µM FM4–64 (Invitrogen) for 6 h in the dark. Images of FM 4–64 stained cells were acquired using two-photon excitation at 950-nm (Nuriya et al., 2016) and 630-nm emission filter. To stain the cytoplasm seedling were stained in 5 µg·mL−1 FDA for 1 h in the dark. Images of FDA-stained cells were acquired using two-photon excitation at 780 nm and 630 nm emission filter. As above, images of lignin and soluble phenolic autofluorescence were acquired using 730 nm excitation and emission was captured at 520–560 nm.
Statistical analysis
All statistical tests were performed in RStudio (RStudio Team, 2020). Differences in particle size and quantity in TIRF micrographs were analyzed with the nonparametric Dunn’s test. Differences in lignin and vacuole fluorescence intensity were analyzed using a one-way analysis of variance followed by Tukey’s test. Mander’s overlap co-efficient was calculated using the COLOC2 plugin in FIJI (Schindelin et al., 2012). A summary of statistical tests is included in Supplemental Table S1.
Accession numbers
Sequence data for genes referenced in this article can be found in the EMBL/GenBank databases using the following accession numbers: LAC4 (AT2G38080); LAC17 (AT5G60020). The lac4-2 lac17 double mutants were generated by Berthet et al. (2011) from lac4-2 (GK-720G02) and lac17 (SALK_016748).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Vacuolar localization of soluble autofluorescent signals.
Supplemental Figure S2. Quantified changes in soluble phenolics in lac4 lac17 roots compared to WT Col-0 determined by HPLC/QTOF.
Supplemental Figure S3 . Characterization of targeted and untargeted soluble phenolics by HPLC/QTOF-MS.
Supplemental Figure S4. No size difference was detected between WT Col-0 and lac4 lac17 roots.
Supplemental Figure S5. Effect of inhibitor treatments on the viability of Arabidopsis roots.
Supplemental Figure S6. Vanadate slows root growth but cell division and differentiation continue.
Supplemental Figure S7. Positive controls for effective ABC transporter inhibition by vanadate in gravitropic auxin responses and root hair development.
Supplemental Figure S8. Laccase activity remains within liposomes throughout the duration of monolignol uptake assays.
Supplemental Figure S9. Laccase activity is not carried over on the exterior of liposomes following purification.
Supplemental Table S1 . Summary of statistical tests performed in the figures and supplementary figures.
Supplementary Material
Acknowledgments
We thank the UBC Bioimaging Facility (RRID: SCR_021304) and the Bioimaging Facility staff Miki Fujita, Eun Kyoung Lee, and Kevin Hodgson for their training, technical support and ongoing maintenance of the optical and electron microscopes used in this study. We thank Laryssa Halat and Geoffery Wasteneys for training and use of the TIRF microscope.
Funding
This work was funded by Natural Sciences and Engineering Research Council (NSERC) Postgraduate Scholarship-Doctoral to M.L.P, and NSERC Discovery Grants to A.L.S., S.D.M., and S.D.C.
Conflict of interest statement. None declared.
Contributor Information
Mendel L Perkins, Department of Botany, University of British Columbia, Vancouver, BC, Canada.
Mathias Schuetz, Department of Botany, University of British Columbia, Vancouver, BC, Canada.
Faride Unda, Department of Wood Science, University of British Columbia, Vancouver, BC, Canada.
Kent T Chen, Department of Experimental Therapeutics, BC Cancer Research Centre, Vancouver, BC, Canada; Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada.
Marcel B Bally, Department of Experimental Therapeutics, BC Cancer Research Centre, Vancouver, BC, Canada; Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada.
Jayesh A Kulkarni, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC, Canada.
Yifan Yan, Wine Research Centre, University of British Columbia, Vancouver, BC, Canada.
Joana Pico, Wine Research Centre, University of British Columbia, Vancouver, BC, Canada.
Simone D Castellarin, Wine Research Centre, University of British Columbia, Vancouver, BC, Canada.
Shawn D Mansfield, Department of Wood Science, University of British Columbia, Vancouver, BC, Canada.
A Lacey Samuels, Department of Botany, University of British Columbia, Vancouver, BC, Canada.
M.S., M.L.P., and A.L.S. conceived and designed the experiments; M.L.P., M.S., and F.U. performed the HPLC; J.A.K. and M.P. performed the cryo-TEM; M.L.P. performed the two-photon microscopy; M.L.P. and S.M. performed the gel permeation chromatography; M.B. and T.J.C. contributed materials and generated liposomes; Y.Y., J.P., and S.D.C. performed mass spectrometric analyses (HPLC/QTOF); M.L.P. performed the statistical analysis; M.L.P. and L.S.. wrote the article; M.L.P., L.S., M.S.. and S.M. edited the article.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: A. Lacey Samuels (lsamuels@mail.ubc.ca).
References
- Alejandro S, Lee Y, Tohge T, Sudre D, Osorio S, Park J, Bovet L, Lee Y, Geldner N, Fernie AR, et al. (2012) AtABCG29 is a monolignol transporter involved in lignin biosynthesis. Curr Biol 22: 1207–1212 [DOI] [PubMed] [Google Scholar]
- Aoki D, Hanaya Y, Akita T, Matsushita Y, Yoshida M, Kuroda K, Yagami S, Takama R, Fukushima K (2016) Distribution of coniferin in freeze-fixed stem of Ginkgo biloba L. by cryo-TOF-SIMS/SEM. Sci Rep 6: 31525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balat M, Ayar G (2005) Biomass energy in the world, use of biomass and potential trends. Energy Sources 27: 931–940 [Google Scholar]
- Barros J, Serk H, Granlund I, Pesquet E (2015) The cell biology of lignification in higher plants. Ann Bot 115: 1053–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benková E, Michniewicz M, Sauer M, Teichmann T, Seifertová D, Jürgens G, Friml J (2003) Local, efflux-dependent auxin gradients as a common module for plant organ formation. Cell 115: 591–602 [DOI] [PubMed] [Google Scholar]
- Berthet S, Demont-Caulet N, Pollet B, Bidzinski P, Cézard L, Le Bris P, Borrega N, Hervé J, Blondet E, Balzergue S, et al. (2011) Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell 23: 1124–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokhina O, Laitinen T, Hatakeyama Y, Delhomme N, Paasela T, Zhao L, Street NR, Wada H, Kärkönen A, Fagerstedt K (2019) Ray parenchymal cells contribute to lignification of tracheids in developing xylem of Norway Spruce. Plant Physiol 181: 1552–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerjan W, Ralph J, Baucher M (2003) Lignin biosynthesis. Annu Rev Plant Biol 54: 519–546 [DOI] [PubMed] [Google Scholar]
- Bohn M, Heinz E, Lüthje S (2001) Lipid composition and fluidity of plasma membranes isolated from corn (Zea mays L.) roots. Arch Biochem Biophys 387: 35–40 [DOI] [PubMed] [Google Scholar]
- Boija E, Johansson G (2006) Interactions between model membranes and lignin-related compounds studied by immobilized liposome chromatography. Biochim Biophys Acta - Biomembr 1758: 620–626 [DOI] [PubMed] [Google Scholar]
- Boija E, Lundquist A, Edwards K, Johansson G (2007) Evaluation of bilayer disks as plant cell membrane models in partition studies. Anal Biochem 364: 145–152 [DOI] [PubMed] [Google Scholar]
- Chou E, Schuetz M, Hoffmann N, Watanabe Y, Sibout R, Samuels AL (2018) Distribution, mobility, and anchoring of lignin-related oxidative enzymes in Arabidopsis secondary cell walls. J Exp Bot 69: 1849–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dima O, Morreel K, Vanholme B, Kim H, Ralph J, Boerjan W (2015) Small glycosylated lignin oligomers are stored in Arabidopsis leaf vacuoles. Plant Cell 27: 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RA, Barros J (2019) Lignin biosynthesis: old roads revisited and new roads explored. Open Biol 9: 190215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi UN, Singh P, Pandey VP, Kumar A (2011) Structure–function relationship among bacterial, fungal and plant laccases. J Mol Catal B 68: 117–128 [Google Scholar]
- Edwards K, Johnsson M, Karlsson G, Silvander M (1997) Preparations of small unilamellar liposomes. Biophys J 73: 258–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg K (1959) Biosynthesis and constitution of lignin. Nature 183: 1152–1155 [DOI] [PubMed] [Google Scholar]
- Furt F, Simon-Plas F, Mongrand S (2011) The Plant Plasma Membrane. Springer Berlin Heidelberg, Berlin, Heidelberg [Google Scholar]
- Gou M, Ran X, Martin DW, Liu C (2018) The scaffold proteins of lignin biosynthetic cytochrome P450 enzymes. Nat Plants 4: 299–310 [DOI] [PubMed] [Google Scholar]
- Hemm MR, Rider SD, Ogas J, Murry DJ, Chapple C (2004) Light induces phenylpropanoid metabolism in Arabidopsis roots. Plant J 38: 765–778 [DOI] [PubMed] [Google Scholar]
- Hiraide H, Tobimatsu Y, Yoshinaga A, Lam PY, Kobayashi M, Matsushita Y, Fukushima K, Takabe K (2021) Localised laccase activity modulates distribution of lignin polymers in gymnosperm compression wood. New Phytol 230: 2186–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann N, Benske A, Betz H, Schuetz M, Samuels AL, Lacey Samuels A, Samuels AL (2020) Laccases and peroxidases co-localize in lignified secondary cell walls throughout stem development. Plant Physiol 184: 806–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni JA, Darjuan MM, Mercer JE, Chen S, Meel R, van der Thewalt JL, Tam YYC, Cullis PR (2018) On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 12: 4787–4795 [DOI] [PubMed] [Google Scholar]
- Lange H, Rulli F, Crestini C (2016) Gel permeation chromatography in determining molecular weights of lignins: critical aspects revisited for improved utility in the development of novel materials. ACS Sustain Chem Eng 4: 5167–5180 [Google Scholar]
- Lee Y, Rubio MC, Alassimone J, Geldner N (2013) A mechanism for localized lignin deposition in the endodermis. Cell 153: 402–412 [DOI] [PubMed] [Google Scholar]
- LeRoy J, Huss B, Creach A, Hawkins S, Neutelings G (2016) Glycosylation is a major regulator of phenylpropanoid availability and biological activity in plants. Front Plant Sci 7: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DR, Miller ND, Splitt BL, Wu G, Spalding EP (2007) Separating the roles of acropetal and basipetal auxin transport on gravitropism with mutations in two Arabidopsis multidrug resistance-like ABC transporter genes. Plant Cell 19: 1838–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Koda K, Yoshinaga A, Takabe K, Shimomura M, Hirai Y, Tamai Y, Uraki Y (2015) Dehydrogenative polymerization of coniferyl alcohol in artificial polysaccharides matrices: effects of xylan on the polymerization. J Agric Food Chem 63: 4613–4620 [DOI] [PubMed] [Google Scholar]
- Lin CY, Huang LY, Chi WC, Huang TL, Kakimoto T, Tsai CR, Huang HJ (2015) Pathways involved in vanadate-induced root hair formation in Arabidopsis. Physiol Plant 153: 137–148 [DOI] [PubMed] [Google Scholar]
- Miao Y-C, Liu C-J (2010) ATP-binding cassette-like transporters are involved in the transport of lignin precursors across plasma and vacuolar membranes. Proc Natl Acad Sci USA 107: 22728–22733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morreel K, Dima O, Kim H, Lu F, Niculaes C, Vanholme R, Dauwe R, Goeminne G, Inzé D, Messens E, et al. (2010) Mass spectrometry-based sequencing of lignin oligomers. Plant Physiol 153: 1464–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottiar Y, Vanholme R, Boerjan W, Ralph J, Mansfield SD (2016) Designer lignins: harnessing the plasticity of lignification. Curr Opin Biotechnol 37: 190–200 [DOI] [PubMed] [Google Scholar]
- Naramoto S (2017) Polar transport in plants mediated by membrane transporters: focus on mechanisms of polar auxin transport. Curr Opin Plant Biol 40: 8–14 [DOI] [PubMed] [Google Scholar]
- Novaes E, Kirst M, Chiang V, Winter-Sederoff H, Sederoff R (2010) Lignin and biomass: a negative correlation for wood formation and lignin content in trees. Plant Physiol 154: 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriya M, Fukushima S, Momotake A, Shinotsuka T, Yasui M, Arai T (2016) Multimodal two-photon imaging using a second harmonic generation-specific dye. Nat Commun 7: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren M, Morsomme P (2019) The plasma membrane H+ -ATPase, a simple polypeptide with a long history. Yeast 36: 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins M, Smith RA, Samuels L (2019) The transport of monomers during lignification in plants: anything goes but how? Curr Opin Biotechnol 56: 69–74 [DOI] [PubMed] [Google Scholar]
- Perkins ML, Schuetz M, Unda F, Smith RA, Sibout R, Hoffmann NJ, Wong DCJ, Castellarin SD, Mansfield SD, Samuels L (2020) Dwarfism of high‐monolignol Arabidopsis plants is rescued by ectopic LACCASE overexpression. Plant Direct 4: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico J, Yifan Y, Gerbrandt EM, Castellarin SD (2022) Determination of free and bound phenolics in northern highbush blueberries by a validated HPLC/QTOF methodology. J Food Compos Anal (in press) [Google Scholar]
- Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH, et al. (2004) Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem Rev 3: 29–60 [Google Scholar]
- Rea PA (2007) Plant ATP-binding cassette transporters. Annu Rev Plant Biol 58: 347–375 [DOI] [PubMed] [Google Scholar]
- Reszczyńska E, Hanaka A (2020) Lipids composition in plant membranes. Cell Biochem Biophys 78: 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalk M, Cabello-Hurtado F, Pierrel M-A, Atanossova R, Saindrenan P, Werck-Reichhart D (1998) Piperonylic acid, a selective, mechanism-based inactivator of the trans-cinnamate 4-hydroxylase: a new tool to control the flux of metabolites in the phenylpropanoid pathway. Plant Physiol 118: 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuring D, Scholler M, Kleine-Vehn J, Lofke C (2015) Vacuolar staining methods in plant cells. In Estevez JM, ed, Plant Cell Expansion: Methods and Protocols, Methods in Molecular Biology. Springer Science+Business Media, New York, pp 83–92 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) HISTORICAL commentary NIH Image to ImageJ : 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz M, Benske A, Smith RA, Watanabe Y, Tobimatsu Y, Ralph J, Demura T, Ellis B, Samuels AL (2014) Laccases direct lignification in the discrete secondary cell wall domains of protoxylem. Plant Physiol 166: 798–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeto J, Tsutsumi Y (2016) Diverse functions and reactions of class III peroxidases. New Phytol 209: 1395–1402 [DOI] [PubMed] [Google Scholar]
- Sibout R, Höfte H (2012) Plant cell biology: the ABC of monolignol transport. Curr Biol 22: 533–535 [DOI] [PubMed] [Google Scholar]
- Smith RA, Schuetz M, Roach M, Mansfield SD, Ellis B, Samuels L (2013) Neighboring parenchyma cells contribute to Arabidopsis xylem lignification, while lignification of interfascicular fibers is cell autonomous. Plant Cell 25: 3988–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speeckaert N, Adamou NM, Hassane HA, Baldacci-Cresp F, Mol A, Goeminne G, Boerjan W, Duez P, Hawkins S, Neutelings G, et al. (2020) Characterization of the UDP-glycosyltransferase UGT72 family in poplar and identification of genes involved in the glycosylation of monolignols. Int J Mol Sci 21: 5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterjiades R, Dean JFD, Gamble G, Himmelsbach DS, Eriksson K-E (1993) Extracellular laccases and peroxidases from sycamore maple (Acer pseudoplatanus) cell-suspension cultures. Planta 190: 75–87 [Google Scholar]
- Tobimatsu Y, Schuetz M (2019) Lignin polymerization: how do plants manage the chemistry so well? Curr Opin Biotechnol 56: 75–81 [DOI] [PubMed] [Google Scholar]
- Tsuyama T, Kawai R, Shitan N, Matoh T, Sugiyama J, Yoshinaga A, Takabe K, Fujita M, Yazaki K (2013) Proton-dependent coniferin transport, a common major transport event in differentiating xylem tissue of woody plants. Plant Physiol 162: 918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyama T, Matsushita Y, Fukushima K, Takabe K, Yazaki K, Kamei I (2019) Proton gradient-dependent transport of p-glucocoumaryl alcohol in differentiating xylem of woody plants. Sci Rep 9: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väisänen E, Takahashi J, Obudulu O, Bygdell J, Karhunen P, Blokhina O, Laitinen T, Teeri TH, Wingsle G, Fagerstedt KV, et al. (2020) Hunting monolignol transporters: membrane proteomics and biochemical transport assays with membrane vesicles of Norway spruce. J Exp Bot 71: 6379–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väisänen EE, Smeds AI, Fagerstedt KV, Teeri TH, Willför SM, Kärkönen A (2015) Coniferyl alcohol hinders the growth of tobacco BY-2 cells and Nicotiana benthamiana seedlings. Planta 242: 747–760 [DOI] [PubMed] [Google Scholar]
- Vermaas JV, Dixon RA, Chen F, Mansfield SD, Boerjan W, Ralph J, Crowley MF, Beckham GT (2019) Passive membrane transport of lignin-related compounds. Proc Natl Acad Sci USA 116: 23117–23123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warinowski T, Koutaniemi S, Kärkönen A, Sundberg I, Toikka M, Simola LK, Kilpeläinen I, Teeri TH (2016) Peroxidases bound to the growing lignin polymer produce natural like extracellular lignin in a cell culture of Norway spruce . Front Plant Sci 7: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennberg CL, van der Spoel D, Hub JS (2012) Large influence of cholesterol on solute partitioning into lipid membranes. J Am Chem Soc 134: 5351–5361 [DOI] [PubMed] [Google Scholar]
- Zhao Q, Nakashima J, Chen F, Yin Y, Fu C, Yun J, Shao H, Wang X, Wang Z-Y, Dixon RA (2013) LACCASE is necessary and nonredundant with PEROXIDASE for lignin polymerization during vascular development in Arabidopsis. Plant Cell 25: 3976–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.