

Abstract
Although water-borne viruses have important implications for the health of humans and other animals, little is known about the impact of human land use on viral diversity and evolution in water systems such as rivers. We used metatranscriptomic sequencing to compare the diversity and abundance of viruses at sampling sites along a single river in New Zealand that differed in human land-use impacts, ranging from pristine to urban. From this, we identified 504 putative virus species, of which 97 per cent were novel. Many of the novel viruses were highly divergent and likely included a new subfamily within the Parvoviridae. We identified at least sixty-three virus species that may infect vertebrates—most likely fish and water birds—from the Astroviridae, Birnaviridae, Parvoviridae, and Picornaviridae. No putative human viruses were detected. Importantly, we observed differences in the composition of viral communities at sites impacted by human land use (farming and urban) compared to native forest sites (pristine). At the viral species level, the urban sites had higher diversity (327 virus species) than the farming (n = 150) and pristine sites (n = 119), and more viruses were shared between the urban and farming sites (n = 76) than between the pristine and farming or urban sites (n = 24). The two farming sites had a lower viral abundance across all host types, while the pristine sites had a higher abundance of viruses associated with animals, plants, and fungi. We also identified viruses linked to agriculture and human impact at the river sampling sites in farming and urban areas that were not present at the native forest sites. Although based on a small sample size, our study suggests that human land use can impact viral communities in rivers, such that further work is needed to reduce the impact of intensive farming and urbanisation on water systems.
Keywords: metagenomics, metatranscriptomics, viral ecology, Parvoviridae, freshwater
1. Introduction
As viruses likely infect all life forms, and often at high abundance, they can be considered an integral part of global ecosystems (Zhang, Shi, and Holmes 2018; French and Holmes 2020; Sommers et al. 2021). Until recently, however, there has been a strong bias towards studying viruses in the context of individual disease-causing pathogens, particularly in humans, domestic animals, and plants (Zhang, Shi, and Holmes 2018). Although understandable, such a bias limits our understanding of their ecology and evolution, how viral abundance and diversity might be shaped by anthropogenic activities and their role at the ecosystem scale (French and Holmes 2020; Sommers et al. 2021). Clearly, a better understanding of these processes will enable virus evolution and disease emergence to be placed in their true ecological context. As most viruses do not cause disease in their hosts (Roossinck 2015), characterising non-pathogenic viruses will greatly expand our understanding of the composition of the global virosphere.
Metagenomic sequencing enables the entire virome of a sample to be characterised in an unbiased manner, giving studies of RNA virus diversity and evolution a new perspective (Zhang, Shi, and Holmes 2018; Wolf et al. 2020; Nayfach et al. 2021). In particular, metagenomics enables the comparison of viral abundance and diversity between groups (animal populations, environments, etc.) that was previously not possible on large scales. To date, however, most metagenomic studies of viromes have focused on describing viral diversity without placing it in an appropriate ecological context (Zhang, Shi, and Holmes 2018; French and Holmes 2020; Sommers et al. 2021).
Rivers collect water from the land they flow through. As such, their microbial community necessarily reflects the ecological properties of this adjacent land (Van Rossum et al. 2015). Run-off from farmland, urban areas, and sewage discharge directly introduce human and livestock-infecting microbes into rivers, sometimes causing water-borne disease (Ferguson et al. 2003; Alegbeleye and Sant’Ana 2020). For bacteria, it is well understood that human activity on land impacts the environment within rivers, in turn affecting bacterial abundance and diversity (Van Rossum et al. 2015; Chen et al. 2018; Phiri et al. 2020; Qiu et al. 2020). However, even though some water-borne viruses have important implications for human health, such as enteroviruses (Amvrosieva et al. 2001), hepatitis E virus (Sedyaningsih-Mamahit et al. 2002; Martolia et al. 2009), and norovirus (Jack, Bell, and Hewitt 2013; Sekwadi et al. 2018), we know little about how human land use impacts viral abundance and diversity in rivers. A study of an agricultural river basin in Ontario, Canada, found that higher levels of human viruses and coliphages were associated with greater upstream human land development (Jones et al. 2017), and Adriaenssens et al. (2021) found geographically distinct virus communities associated with different wastewater treatment plants in the Conwy river catchment and coastal zone in the United Kingdom. Similarly, land use in Singapore was the main driver of the viral community structure in reservoirs used for potable water supplies and recreational activities (Gu et al. 2018). However, because such comparisons often involved different catchments that are likely to have contrasting viral communities, it may not be possible to isolate the effect of human activity on viral ecology. To date, there has been no study directly comparing the viral ecology of river water flowing through different land-use types within the same river catchment.
New Zealand freshwater communities have been isolated since New Zealand split from Gondwanaland approximately 80 million years ago (Mortimer et al. 2019). Freshwater communities within New Zealand are also generally isolated from each other, with little opportunity for non-migratory species to colonise new water catchments (Burridge and Waters 2020). This is reflected in the evolution of freshwater plant, vertebrate, and invertebrate species. For example, there are high levels of endemism within New Zealand's non-migratory galaxiid fish, with many species found only in one water catchment (Dunn et al. 2018; Burridge and Waters 2020). It might therefore be expected that the freshwater communities of New Zealand would similarly contain many highly divergent viruses and locally unique viruses that have co-evolved with their isolated hosts. In contrast, human activity has had a large impact on New Zealand freshwater communities, including run-off from intensive agriculture and urbanisation and the introduction of invasive species such as rainbow trout (Oncorhynchus mykiss). It is likely that these changes would also have affected the viral community in the rivers, introducing viruses associated with plants and animals grown for food, as well as viruses that infect humans.
Very little is known about the viral ecology of New Zealand rivers, with research generally limited to targeted testing for known pathogens. Two river sites—the Waikato River in the North Island and the Oreti River in the South Island—that supply drinking water to urban populations have been screened for enteric viruses, with positive results in 97 per cent of samples (Williamson et al. 2011). In the Manawatū region, the Manawatū and Pohangina rivers and Turitea creek have been screened for plant viruses, with three tombusviruses detected (Mukherjee 2011; Mukherjee et al. 2012). New variants of tobacco mosaic virus and tomato mosaic virus were also identified (Mukherjee 2011), and human polyomaviruses have been found in the Matai river in Nelson (Kirs et al. 2011). Sclerotinia sclerotiorum hypovirulence–associated virus 1 (which infects a fungus often found in agricultural plants) was detected in a Christchurch river using metagenomic sequencing of DNA from sediment samples (Kraberger et al. 2013). To our knowledge, viral metatranscriptomics has not yet been performed on a river system in New Zealand.
The core aim of this study was to compare the viral (particularly RNA virus) abundance and diversity between sites with differing human land-use impacts in a New Zealand river catchment and from this determine how virome ecology and evolution are shaped by human activity. Accordingly, six sites on the Manawatū River, North Island, were selected based on their differing land-use types. Two sites were at the edge of the Ruahine forest park, containing water that has only flowed through the pristine native forest (pristine sites, denoted P1 and P2). Two sites contain water that has flowed through farmland (farmland sites, at least 25 km for F1 and 50 km for F2). The final two sites have flowed first through the pristine native forest, then farmland, and finally urbanised areas (urban sites)—Feilding and Palmerston North (named U1 and U2, respectively). Water samples were taken at these sites and subjected to total RNA sequencing (i.e. metatranscriptomics).
2. Methods
2.1. The Manawatū River
The Manawatū River is a 180-km river located in the North Island of New Zealand (Fig. 1). Importantly, it flows through three very different land-use types, allowing a direct comparison between them. The river begins in the Ruahine forest park that encompasses the Ruahine mountain range (Department of Conservation 2021b). The park is dominated by native vegetation, including podocarp forest at lower altitudes and sub-alpine shrubland and tussock grasslands at higher altitudes. Between the 1800s and the 1970s, there was considerable forest clearing and logging, but since 1976, the area has been protected as a forest park with no farming or logging (Department of Conservation 2021a). There is little to no human habitation or activity in these ranges, with the exception of recreational hikers, hunters, and rangers. A variety of endemic New Zealand animals inhabit these ranges, including parrots (kakariki Cyanoramphus novaezelandiae and kaka Nestor meridionalis), ducks (whio Hymenolaimus malacorhynchos), New Zealand long-tailed bats (Chalinolobus tuberculatus), and large carnivorous land snails (Powelliphanta marchanti). Introduced pest species are also found there, including red deer (Cervus elaphus), feral pigs (Sus scrofa), goats (Capra hircus), brush-tailed possums (Trichosurus vulpecula), stoats (Mustela erminea), and rainbow trout (O. mykiss) (Department of Conservation 2021b).
Figure 1.
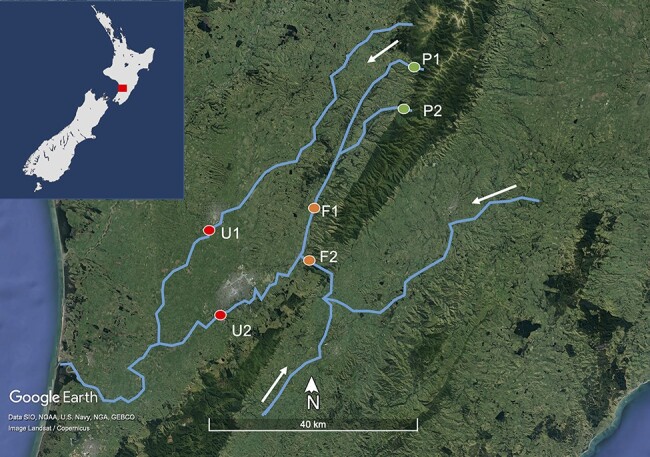
Map of the Manawatū River catchment in blue and the six sampling sites as coloured circles, with the inset showing the river catchment location on the North Island of New Zealand as a red square. P1 and P2 are ‘pristine’ sites having only flowed through native bush (seen as dark green areas on the satellite image). F1 and F2 are ‘farming’ sites flowing through intensive agricultural land (light green on the satellite image). U1 and U2 are ‘urban’ sites, flowing through two urban areas: Feilding and Palmerston North (grey on the satellite image). The water flows from the east and south towards the sea on the west coast, as indicated by the white arrows. Satellite image created using Google Earth.
After flowing through the Ruahine Forest Park, the river passes through intensively farmed areas, primarily consisting of sheep, beef, and dairy farming. This land use is known to impact the river, with the Manawatū region having among the highest nitrogen and phosphorus concentrations (nutrients associated with pastoral agriculture) in New Zealand (Roygard, McArthur, and Clark 2012). It then flows through two urban centres: Feilding (town, population 17,050) and Palmerston North (city, population 81,500), before flowing out into the Tasman Sea. Both these urban centres discharge treated wastewater into the Manawatū River, and in some years, this negatively impacts aquatic life through the discharge of nutrients and a corresponding increase in periphyton cover (Hamill 2012).
2.2. Sample collection
Of water samples, 2 l were collected at each of the six sites in the Manawatū River catchment (Fig. 1). For consistency, all samples were collected on the same day (13 July 2019). At each site, 1 l was collected from the water’s edge, and 1 l was collected 1.2 m from the bank in the main flow of the river using a sampling pole, with the aim of obtaining a representative sample of the river water. In the case of P2, which was less than 1.2 m wide, the second sample was taken by hand in the main flow of the river. These samples were combined to obtain 2 l of water per site and 12 l in total. Once collected, the water samples were kept at approximately 4°C using icepacks until processing.
At each site, a separate 250-ml sample was collected from the main flow of the river to measure additional variables [temperature, salinity, conductivity, pH, total dissolved solids (TDS), and turbidity]. These were measured using a multiparameter tester (Waterproof PCSTestr 35, Thermo Scientific) and a Turbidimetre (2100P Turbidimetre, Hach). The temperature was measured on-site, and the remaining variables were measured in the laboratory.
2.3. Sample processing and sequencing
Filtering of all samples was completed within 30 h of sample collection. Samples with a large amount of silt (farming and urban sites) were first filtered through a glass fibre filter of 47-mm diameter and 0.7-μm pore size (Microscience). All samples were then filtered through polyether sulphone (PES) membrane filters, of 47-mm diameter and 0.2-μm pore size (Microscience).
Samples were concentrated from 2 l to ∼100 µl in two steps using tangential ultra-filtration and ultra-centrifugation. The samples were first concentrated using ultra-filtration from 2 l down to 40 ml with the Vivaflow 200 (Sartorius). The water samples were pumped through the cross-flow cassette for 1–2 h until concentrated to ∼40 ml. The samples were then further concentrated using ultra-centrifugation from 40 to ∼2 ml with Vivaspin 20 ultrafiltration units (Sartorius). Each sample was placed into a separate ultrafiltration unit, which was spun in a centrifuge with a swing bucket at 3,000 g until concentrated to ∼2 ml. The time to achieve this concentration varied from 30 min to 1.5 h. The sample was then further concentrated to ∼100 µl (again using ultra-centrifugation) with Vivaspin 2 ultrafiltration units (Sartorius). The samples were spun at 3,000 g for 2–8 min until concentrated to ∼100 µl. All units had PES filters with a molecular weight cut-off of 10 kDa. Samples were stored at −80°C until nucleic acid extraction.
RNA and DNA were extracted from the concentrated water samples using the AllPrep® PowerViral® DNA/RNA Kit following the kit protocol. After RNA extraction, the DNA was removed by DNase digestion (Qiagen RNase-Free DNase I Set), then in the same column, the RNA was concentrated to 15 µl using the MN NucleoSpin RNA Clean-up XS (Macherey-Nagel), following the kit protocol. The same process was conducted on 2 × 200 µl of sterile water to create two blank control libraries. Complementary DNA libraries were prepared using the SMARTer® Universal Low Input RNA Kit for Sequencing (Takara Bio), without ribosomal RNA depletion. Libraries were sequenced on the Illumina Novaseq platform (150 bp, paired-end sequencing). The corresponding sequencing data have been deposited in the Sequence Read Archive (SRA) under accession numbers SRR17234948-53. The trimmed alignment FASTA files used to infer the phylogenetic trees are available at https://github.com/RKFrench/Viral-Diversity-NZ-River. The consensus sequences of all novel viruses have been submitted to GenBank and assigned accession numbers OM953841–OM954332.
2.4. Quality control, assembly, and virus identification
TruSeq3 adapters were trimmed using Trimmomatic (0.38) (Bolger, Lohse, and Usadel 2014). Bases below a quality score of five were trimmed with a sliding window approach (window size of four). Bases at the beginning and end of the reads were similarly excluded if below a quality of three. SMART adapters were trimmed using bbduk in BBtools (bbmap 37.98) (Bushnell 2018). Sequences below an average quality of ten were removed.
Sequence reads were assembled de novo using Trinity (2.5.1) (Grabherr et al. 2011), with a kmer size of thirty-two and a minimum contig length of 300. BLASTN (BLAST+ 2.9.0) and Diamond BLASTX (Diamond 0.9.32) were used to identify viruses by comparing the assembled contigs to the National Center for Biotechnology Information (NCBI) nucleotide (nt) database and the non-redundant (nr) protein database, downloaded in June 2020 (Camacho et al. 2009; Buchfink, Reuter, and Drost 2021). Contigs with hits to viruses were retained. To avoid false positives, sequence similarity cut-off e-values of 1E-5 and 1E-10 were used for the nt and nr databases, respectively. Virus abundances were estimated using RSEM (1.3.0), allowing us to determine the expected count according to the expectation–maximization algorithm for each contig (Li and Dewey 2011). This was expressed as the percentage of the total number of reads in each library. Eukaryotic and prokaryotic diversity was characterised using CCMetagen (v 1.2.4) and the NCBI nt database (Clausen, Aarestrup, and Lund 2018; Marcelino et al. 2020).
2.5. Evolutionary and ecological analysis
Using the nucleotide sequences identified as encoding viral replication proteins (i.e. as identified by BLAST), the getorf program from EMBOSS (6.6.0) was used to find and extract open reading frames and translate them into amino acid sequences using the standard genetic code with a minimum size of 100 amino acids (Rice, Longden, and Bleasby 2000). For each sequence identified, representative viruses from the viral family in question and the top ten blast hits from the nr database (determined by the e-value) were used to estimate phylogenetic trees. To improve phylogenetic inference, where possible larger polyprotein sequences containing the viral replication protein were utilised rather than simply the replication protein in isolation. Amino acid sequences were aligned using the E-INS-I algorithm in MAFFT (7.402) (Katoh and Standley 2013) and trimmed using Trimal (1.4.1) (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009) with a gap threshold of 0.9, and at least 20 per cent of the sequence conserved (Supplementary Table S1). Maximum likelihood phylogenetic trees for each virus family were then estimated using IQ-TREE (1.6.12) (Nguyen et al. 2015), with the best fit substitution model determined by the program and employing 1,000 bootstrap replications to assess node robustness. Any sequences with >95 per cent amino acid similarity to each other or known species were assumed to represent the same virus species, with only one representative of each then included in the phylogenetic analysis. All novel viruses identified were given names that include the word ‘flumine’ (Latin for ‘of the river’) to convey where the virus was found.
Host relationships and patterns of virus diversity were described by characterising the broad host range of each viral family and creating stacked bar graphs of virus abundance per family for each host range using ggplot2 (Wickham 2016). Families were characterised as infecting either (1) animals, (2) plants, fungi, or protists, (3) bacteria, or (4) unknown, as identified in previous studies. APE (5.4) and ggtree (2.4.1) were used to visualise the phylogenetic trees and produce figures (Paradis and Schliep 2019; Yu 2020). Alpha diversity (i.e. the diversity within each sample) was analysed using viral family richness (number of viral families), the Shannon index, and the Shannon effective number of species (ENS). The Shannon index reflects the number of taxa and the evenness of the taxa abundances. The Shannon ENS is the effective number of taxa present in the community if the abundances were equal (Hill 1973). Beta diversity (i.e. diversity across land-use types) was analysed using a principal co-ordinate analysis with a Bray–Curtis dissimilarity matrix, presented as an ordination plot. Alpha and Beta diversity analyses were conducted using Phyloseq (v1.34.0) in R (v 4.0.5) (McMurdie and Holmes 2013; R Core Team 2021). Other graphs were generated using ggplot2 (Wickham 2016) and venneuler (Wilkinson 2011).
2.6. Identifying possible reagent contamination
Any virus found in the blank negative control libraries (i.e. a sterile water and reagent mix) was assumed to have resulted from contamination likely associated with laboratory reagents. This included several unclassified viral species as well as those from the Autographiviridae, Dicistroviridae, Tombusviridae, Leviviridae, Myoviridae, Podoviridae, Solemoviridae, Marnaviridae, and Virgaviridae. Accordingly, these viruses were removed from the river sample libraries and excluded from all analyses. Additionally, any viruses that fell into the same clades as those found in blank libraries were conservatively assumed to be contaminants (Porter et al. 2021) and similarly removed. These included six circo-like viruses in the Circoviridae (single-strand DNA viruses) and nineteen tombus-like viruses from the Tombusviridae (single-strand, positive-sense RNA viruses).
3. Results
We characterised the RNA viromes from six Manawatū River water samples using total RNA sequencing.
3.1. Water measurements
Our water measurements indicated that the two pristine sites had a different abiotic environment from the farming and urban sites, while the farming and urban sites were similar to each other (Fig. 2). Specifically, the pristine sites had lower salinity, pH, TDS, turbidity, conductivity, and temperature than the farming and urban sites. The farming and urban sites also had a larger variation between sites, with the exception of pH and temperature.
Figure 2.
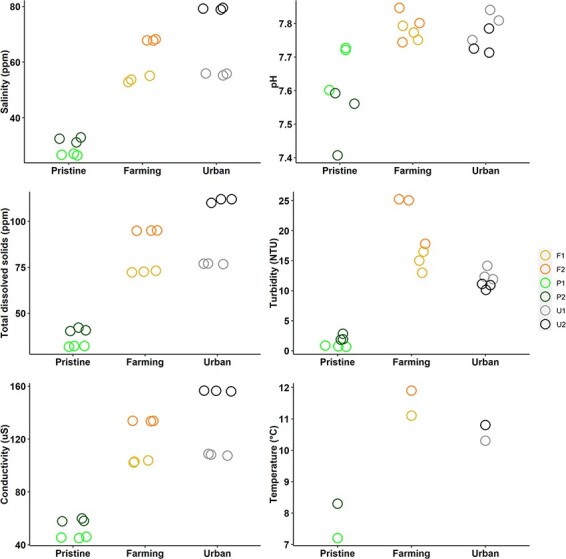
Measurements of salinity, pH, TDS, turbidity, conductivity, and temperature at the six different sites on the Manawatū River, grouped into the different site types (pristine, farming, and urban). For all measurements except temperature, there are three values per sampling site that have been jittered to reduce overplotting.
3.2. Virus identification
The six sequencing libraries generated had an average of 90 million reads per library, of which an average of 0.7 per cent was derived from viruses. This is within the usual range found in faecal samples, cloacal swabs, and invertebrate tissue, but higher than commonly observed in vertebrates (Zhang, Shi, and Holmes 2018; Campbell et al. 2020; Le Lay et al. 2020; Mahar et al. 2020; Wille et al. 2020, 2021) in studies using similar metagenomic techniques. However, it was lower than observed in urban streams in Ecuador (Guerrero-Latorre et al. 2018). P2 and U2 had the highest number of total reads and the highest number of viral reads (Fig. 3). Notably, the two pristine sites had the highest percentage of viral reads, at 1.31 and 1.35 per cent. F1, F2, and U1 all had lower total reads, total viral reads, and percentage viral reads. The analysis of eukaryotic and prokaryotic diversity showed that all samples primarily consisted of bacteria (accounting for 71–86 per cent of assembled contigs) followed by eukaryotes (5–19 per cent).
Figure 3.
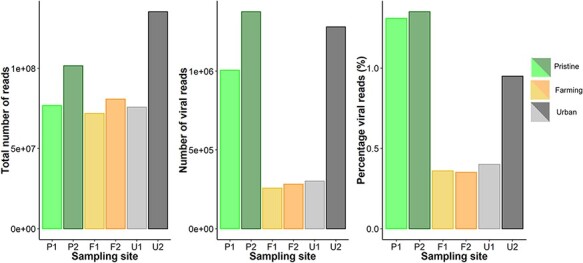
Read counts and the percentage of viral reads (per cent) for libraries from six sites on the Manawatū River.
In total, we identified 504 putative virus species from twenty-seven viral families, of which 491 (97 per cent) were novel using a cut-off of 95 per cent amino acid similarity, primarily in replication-associated proteins (although these await formal verification by the International Committee on Taxonomy of Viruses). These included multiple members of the Nodaviridae (n = 74 novel viruses), Tombusviridae (n = 64), and Dicistroviridae (n = 61). If a more conservative cut-off of <90 per cent amino acid sequence similarity is used to define a novel virus species, then the samples analysed here contain 470 novel viruses. We also detected previously described viruses (i.e. with > 95 per cent amino acid similarity to viruses detected previously), including S. sclerotiorum hypovirulence–associated DNA virus 1 (Genomoviridae), white clover mosaic virus (Alphaflexiviridae), Rhopalosiphum padi virus (Dicistroviridae), carnation Italian ringspot virus (Tombusviridae), pepper mild mottle virus (Virgaviridae), Norway luteo-like virus 4 (Solemoviridae). Importantly, we identified at least sixty-three virus species from the Astroviridae, Birnaviridae, Parvoviridae, and Picornaviridae that may infect vertebrates. No likely human viruses were detected. Below we describe, in more detail, those families with high virus diversity in our study and those containing viruses that may infect vertebrates.
3.3. Families with a high diversity of novel virus species
3.3.1. Tombusviridae
We identified a high diversity and abundance of novel Tombusviridae (Supplementary Figure 24), a family of single-strand positive-sense RNA viruses that infect plants (Sit and Lommel 2015). Of the viruses identified, eighteen fell into the subfamily Procedovirinae, found in all land-use types. Of these, one virus in U2 clustered within the genus Betacarmovirus and was most closely related to cardamine chlorotic fleck virus (Skotnicki et al. 1993), although with only 45 per cent amino acid similarity. Another, found in U2 and F2, belonged to the genus Gammacarmovirus and is most closely related to melon necrotic spot virus with 71–73 per cent amino acid similarity (Riviere and Rochon 1990). We also found carnation Italian ringspot virus (genus Tombusvirus) at both farming sites, with 98–99 per cent amino acid similarity. The Procedovirinae also contain a clade of closely related novel tombus-like viruses that appear basal to any currently described genera and were found at the pristine sites. Another clade was found in the urban and farming sites and appears to fall within the subfamily Regressovirinae.
3.3.2. Dicistroviridae
We similarly found a high diversity of viruses belonging to the Dicistroviridae (Supplementary Figure 4), a family of single-strand positive-sense RNA viruses commonly associated with arthropods (Valles et al. 2017). Thirty-nine viruses were part of a highly divergent clade that fell basal to the three currently recognised genera, to which it exhibited less than 50 per cent amino acid similarity. There were also fifteen newly identified viruses that fell into the genus Cripavirus found across all the land-use types. In addition, we detected two previously described cripaviruses: R. padi virus (97–100 per cent amino acid similarity) and cricket paralysis virus (96–97 per cent), both only at the urban site U2.
3.3.3. Nodaviridae
We identified thirteen novel viruses from the genus Alphanodavirus of the Nodaviridae (single-strand positive-sense RNA viruses) (Supplementary Figure 14). In addition, we identified one known virus—black beetle virus (Alphanodavirus; 95–98 per cent amino acid similarity)—in all farming and urban sites, but not at the pristine sites. Although we did not find any viruses belonging to the genus Betanodavirus, the only other genus of Nodaviridae, we did identify sixty-one other novel viruses from a divergent clade that fell outside of the Alphanodavirus and Betanodavirus genera. Many of these were most closely related to Barns Ness serrated wrack noda-like virus 2 isolated from marine algae (Waldron, Stone, and Obbard 2018), although with less than 50 per cent amino acid similarity.
3.4. Vertebrate-infecting families
3.4.1. Astroviridae
The Astroviridae are a family of single-stranded positive-sense RNA viruses that infect mammals and birds (Lukashov and Goudsmit 2002). We identified twenty-eight novel astroviruses, including three new species within the Bastrovirus clade found at the farming and urban sites (Fig. 4). These three viruses were most closely related to a bastrovirus detected in sewage in Brazil (Dos Anjos, Nagata, and de Melo 2017), although with only 57–67 per cent amino acid similarity. Twenty-four novel viruses fell into a divergent clade outside of the genus Avastrovirus and the Bastrovirus clade: these were most closely related to ‘Astroviridae sp.’ viruses found in metagenomic studies of grassland soil in California, USA (Starr et al. 2019). The viruses found in our study that fell into this clade (denoted flumine astroviruses 1–24) all had less than 62 per cent amino acid similarity with the soil viruses. Notably, they were found across all our river sites, with a higher diversity at the farming and urban sites (n = 11 and 21 species, respectively) than the pristine sites (n = 4). However, flumine astrovirus 3 found at a pristine site had the highest abundance, representing 0.04 per cent of the total reads in the library.
Figure 4.
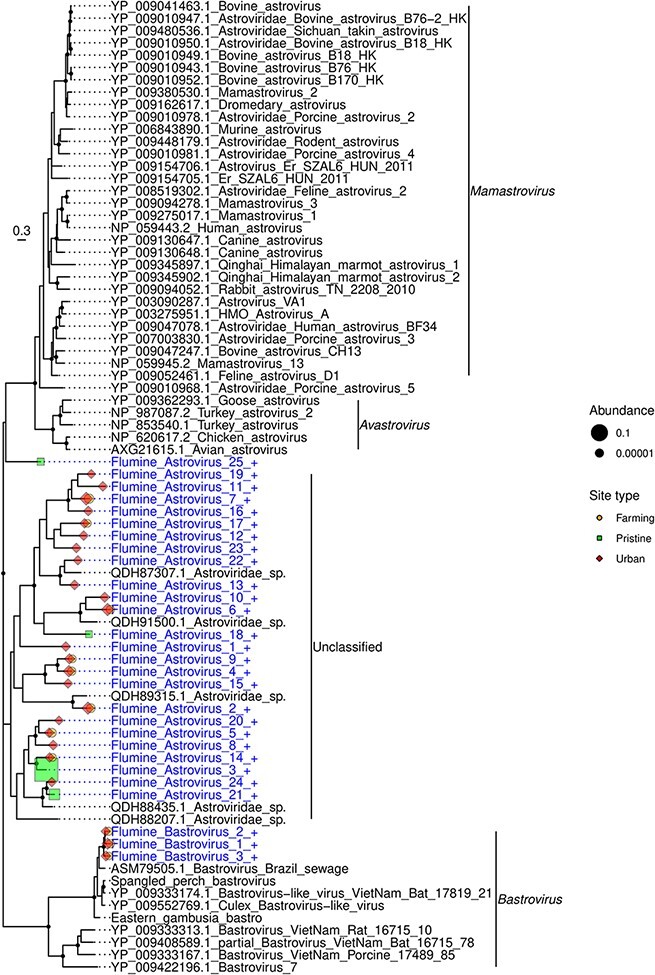
Phylogeny of the Astroviridae based on the non-structural polyprotein sequence (alignment length of 733 amino acids). Viruses obtained in this study are shown in blue and have a ‘+’ after their name. Related viruses are shown in black. Virus abundance is expressed as the percentage of the total number of reads and represented by the size of each coloured symbol. The colour of each symbol refers to the site type. Black circles on nodes show bootstrap support values greater than 90 per cent. Branches are scaled according to the number of amino acid substitutions per site, shown in the scale bar. The tree is midpoint rooted.
3.4.2. Birnaviridae
The Birnaviridae are a family of double-stranded RNA viruses that infect fish, birds, and insects. We identified one virus from this family at a pristine site (P1), which was most closely related (although with only 30 per cent amino acid similarity) to blotched snakehead virus (Da Costa et al. 2003) and Lates calcarifer birnavirus (Chen et al. 2019), both of which are associated with fish (Supplementary Figure 2).
3.4.3. Parvoviridae
The Parvoviridae are a family of small double-stranded DNA viruses that infect both vertebrates and invertebrates. We identified twenty-one parvoviruses: all were present at the Feilding urban site (U1), with one (flumine parvovirus 17) found at both P1 and U1. Most (n = 18) of these viruses fell into a distinct clade, separate from other previously described subfamilies and genera (Fig. 5). This clade had less than 18 per cent amino acid similarity with other parvoviruses, but up to 81 per cent similarity (average of 41 per cent) with each other. A new subfamily Hamaparvovirinae was created in 2020 that exhibits less than 20 per cent amino acid sequence identity to other parvoviruses (Pénzes et al. 2020). This is comparable to the level of sequence similarity observed in the novel clade identified here, indicating that this may also represent a new subfamily that we have tentatively called the Flumenparvovirinae. The remaining three viruses fell into the subfamily Densovirinae. These viruses most likely infect invertebrates as they were most closely related to Planococcus citri densovirus (Thao et al. 2001) (76 per cent amino acid identity) and Blattella germanica densovirus 1 (Kapelinskaya et al. 2011) (55–61 per cent) that infect mealy bugs and cockroaches, respectively.
Figure 5.
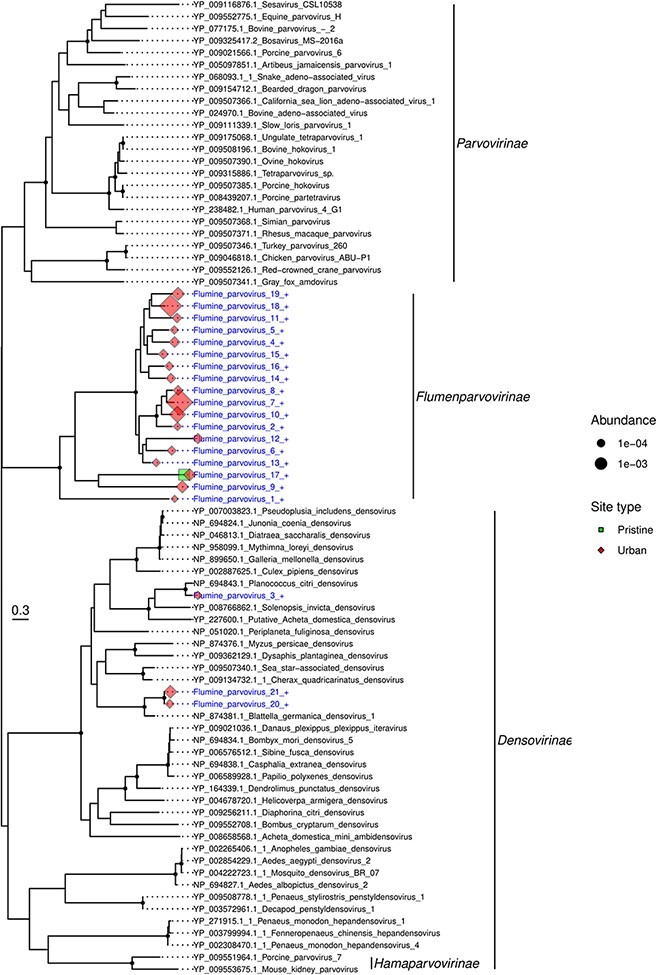
Phylogeny of the Parvoviridae based on the non-structural protein sequence (alignment length of 452 amino acids). Viruses obtained in this study are shown in blue and have a ‘+’ after their name. Related viruses are in black. Virus abundance is expressed as the percentage of the total number of reads and represented by the size of each coloured symbol. The colour of each symbol refers to the site type. Black circles on nodes show bootstrap support greater than 90 per cent. Branches are scaled according to the number of amino acid substitutions per site, shown in the scale bar. The tree is midpoint rooted.
3.4.4. Picornaviridae
The Picornaviridae are a large family of single-stranded RNA viruses associated with both vertebrates and invertebrates. We identified fifteen novel picornaviruses, most (n = 11) from the urban sites, although they were also identified at both farming and pristine sites (Supplementary Figure 17). The pristine sites had the lowest diversity of picornaviruses (n = 2), but the highest abundance. Eight of the picornaviruses fell into a clade with fur seal picorna-like virus (Krumbholz et al. 2017) and ampivirus A1 associated with the smooth newt Lissotriton vulgaris (Reuter et al. 2015). This clade was most closely related to a cluster of genera Tremovirus, Harkavirus, and Hepatovirus found in birds and mammals.
3.4.5. Genomoviridae
The Genomoviridae are a family of single-stranded DNA viruses. Although they are commonly associated with fungi, they have also been identified in mammals and birds (Varsani and Krupovic 2021). With the exception of one virus species (flumine genomovirus 1), members of the Genomoviridae were only found at the urban sites (Supplementary Figure 5). Nine of the fourteen viruses were most closely related to viruses found in sewages or faeces. Interestingly, a number of these viruses are closely related to viruses previously documented in New Zealand. Flumine genomovirus 11 fell into a clade of five sewage–derived gemycircularviruses, exhibiting 85 per cent amino acid similarity to its closest match—sewage-associated gemycircularvirus 7a sampled from a sewage oxidation pond in Christchurch, New Zealand (Kraberger et al. 2015). Similarly, flumine genomoviruses 8 and 9 share 85 and 86 per cent amino acid similarity, respectively, with sewage-associated gemycircularvirus 3 also isolated from a New Zealand oxidation pond (Kraberger et al. 2015), while flumine genomoviruses 12, 4, and 6 share 87, 76, and 82 per cent amino acid similarity, respectively, with faeces-associated gemycircularvirus 21 isolated from llama faeces in New Zealand (Steel et al. 2016). Finally, the single Genomoviridae virus found in the pristine sites (flumine genomovirus 1) was most closely related (81 per cent amino acid similarity) to a virus isolated from minnow tissue in the USA.
3.5. Host relationships and patterns of virus diversity
We next characterised each viral family according to their usual assigned host (as identified in previous studies) and used this to visualise patterns of virus abundance and diversity (Fig. 6). This revealed that the two farming sites had a lower viral abundance across all host types, while the two pristine sites had a higher abundance of viruses with animals, plants/fungi (such as the Astroviridae and Tombusviridae), and unknown hosts. In turn, U1 had a very high abundance of prokaryote-infecting viruses but a lower abundance of all other virus types (Fig. 6). Virus abundance was the highest for those viral families where the host was unknown, accounting for 2.35 per cent of reads. Plant-infecting viruses were the next-most abundant (1 per cent), followed by animal-infecting viruses (0.56 per cent). At the family level, we found a high diversity of plant- and animal-infecting viruses (eighteen and sixteen families, respectively). There was a surprisingly low abundance and diversity of known prokaryote-infecting viruses: these were the least abundant (0.096 per cent) and the least diverse (six families) group. All sites had a proportionally high abundance of Nodaviridae, Tombusviridae, and ‘unclassified Picornavirales’ compared to other viral families.
Figure 6.
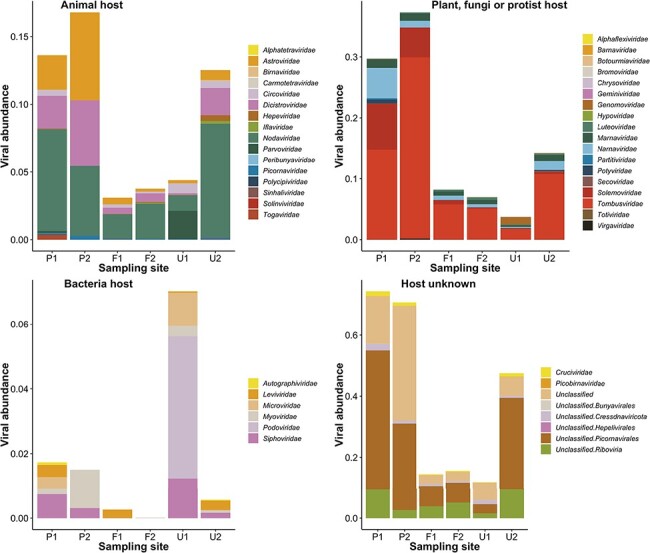
Virus abundance (as a percentage of the total number of reads) of viral families from each site on the Manawatū River. Virus families are divided into each panel depending on their usual host—animals, plants/fungi, prokaryotes, and unknown. Unclassified viruses did not have a classification according to the current NCBI taxonomy.
3.6. Alpha and beta virus diversity
Sites P1 and U2 had the highest viral family richness, manifesting as thirty-eight and forty virus families, respectively, while P2 had the lowest at sixteen (Table 1). U1 had higher Shannon and Shannon ENS values than any other site, but only the third highest viral family richness. The two pristine sites had very different levels of diversity, with P1 having much higher viral family richness (with more than twice the number of viral families), Shannon, and Shannon ENS values than P2. The two farming sites had a similar viral family richness, Shannon, and Shannon ENS to each other, and the two urban sites were also similar with respect to viral family richness (with U1 having 20 per cent fewer virus families than U2).
Table 1.
Viral family richness, Shannon, and Shannon ENS values for each site on the Manawatū River. These were calculated at a family level and include unclassified virus groups such as ‘unclassified Riboviria’.
| Richness | Shannon | Shannon ENS | |
|---|---|---|---|
| P1 | 38 | 2.13 | 8.39 |
| P2 | 16 | 1.88 | 6.54 |
| F1 | 28 | 2.21 | 9.07 |
| F2 | 24 | 2.11 | 8.28 |
| U1 | 32 | 2.52 | 12.45 |
| U2 | 40 | 2.00 | 7.39 |
At the level of virus family, we used a principal co-ordinate analysis to examine the differences between viral communities, examining ‘intra-type’ (i.e. comparing sites with the same land-use type) and ‘inter-type’ (comparing sites across different land-use types) differences (Fig. 7). Accordingly, the pristine and farming sites had high intra-type similarity and low inter-type similarity, such that their viral communities were more similar within a land-use type than to viral communities from different land-use types (Fig. 7). The two pristine sites displayed the most similar viral communities. In contrast, the two urban sites differed markedly from each other and all other sites and, hence, had a very different viral community, both from each other and to that at any other site (i.e. both intra- and inter-type). The urban site U2 (Palmerston North) was closer to F1 and F2 than to U1 and, hence, had a viral community that was more similar to the farming sites than to the other urban site. At the viral species level, the urban sites had a higher diversity (n = 327 species) than the farming (n = 150) and pristine sites (n = 119). There were many more viruses shared between the urban and farming sites (n = 76) than between the pristine and farming or urban sites (n = 24). Finally, only eight of 504 species were found in all land-use types, indicative of a relatively high level of local differentiation.
Figure 7.
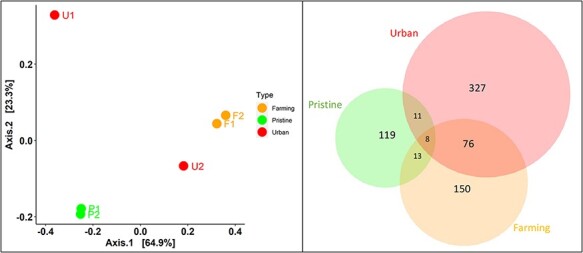
Viral community similarities and differences at the taxonomic family level (left) and species level (right). Left—A PCoA applying the Bray–Curtis dissimilarity matrix for viral abundance and virus family diversity, showing the relative similarity/differences in viral community between sites with differing land-use types—pristine (green), farming (orange), and urban (red). Points closer to one another are more similar in virome structures than those further away. Right—a Venn diagram showing the number of virus species shared between the three land-use types (with an amino acid similarity >95 per cent).
4. Discussion
Using a metagenomic approach, we identified a high diversity of novel and highly divergent viruses in a single New Zealand river system and revealed differences of virome composition in the river between sampling sites associated with different land-use types. In total, we observed 504 putative virus species, of which 491 (97 per cent) were potentially novel and included a new subfamily within the Parvoviridae. Notably, there were considerable differences in the viral community structure between the land-use types. In particular, the two pristine sites had a higher abundance of viruses that infect animals, plants, and fungi, while at the viral species level, the urban sites had the highest virome diversity. In addition, there were many more viruses shared between the urban and farming sites (n = 76) than between the pristine and farming or urban sites (n = 24).
The abiotic environment within the Manawatū River system differed considerably between sampling sites that had different land-use types, with the pristine sites having lower measurements of all variables. TDS, salinity, conductivity, and turbidity represent different ways of measuring the presence of dissolved solids and ions in the water, and generally, the lower they are then the higher the water quality (Davies-Colley 2013). Increases in TDS can adversely affect plants and animals in freshwater environments due to changes in osmotic conditions, making this an important indicator of the health of the freshwater ecosystem (Chapman, Bailey, and Canaria 2000). These measures are all elevated in effluents, and their presence in freshwater can therefore indicate contamination from fertilisers, urban run-off, and animal and human waste (Chapman, Bailey, and Canaria 2000; de Sousa et al. 2014). They can also naturally be elevated due to changes in climate and differing geology, which impact the amount of dissolved substance from the weathering of rocks (Davies-Colley 2013). However, as the sites in this study were all in the same river catchment and within 50 km of each other, the climate and geology are unlikely to be markedly different. Water temperature was also lower in the pristine sites than in the farming and urban sites, likely due to the pristine sites being at higher altitudes, closer to the Ruahine Ranges (a source of snowmelt), and also the thicker vegetation cover blocking solar radiation. The pristine sites had a pH closer to neutral, which may be related to differences in dissolved solids from the surrounding soils (Baldisserotto 2011). All these indicators show that the riverine environment was very different between the pristine and farming/urban sites, but not substantially different between the farming and urban sites.
A key observation of our study was the high diversity of viruses in the Manawatū River system, with an average of thirty virus families detected per site. Indeed, we found a very high diversity of novel viruses, and only a small number (n = 13) described previously, again indicating that only a tiny fraction of the virosphere has been described to date. Of particular note, we identified a novel clade of parvoviruses that was phylogenetically distinct from previously described subfamilies and genera, and which may represent a new subfamily within the Parvoviridae. Little is known about the viruses that characterise freshwater habitats, although our study suggests that they may have highly diverse viromes. Indeed, we show that isolated New Zealand freshwater environments can harbour many novel and highly divergent viruses. This may in part reflect New Zealand’s distinctive ecological history, including high levels of endemic species (Walker, Monks, and Innes 2021). The novel viruses present in this water catchment may therefore reflect the ‘undisturbed’ diversity of the river, rather than the result of human land use. A high diversity of bacteria have similarly been found in New Zealand freshwater (Phiri et al. 2021). Indeed, it is notable that the pristine sites had the highest abundance of viruses that infect animals, plants, and fungi and had little overlap (in terms of species shared) with the farming and urban sites (Figs 6 and 7). This may reflect a greater abundance of native flora and fauna in these pristine sections of the river that are surrounded by native forests. Interestingly, trends in virus abundance and species richness differed in the pristine and the farming or urban sites, with the urban and farming sites having a higher diversity of viruses at the species level (Fig. 7), but the pristine sites displaying a higher abundance and percentage of viral reads (Fig. 3). Hence, the viruses naturally present in the water catchment may be an important part of river communities, compatible with the idea that viruses should be viewed as integral parts of natural ecosystems (Suttle 2016; French and Holmes 2020). It also suggests that, while increasing the overall diversity, the viruses introduced into the river by human land use are not present at high abundance. However, it is also possible that changes in the abiotic (Fig. 2) and biotic environment within the river at the urban and farming sites have reduced the abundance of those viruses naturally present, resulting in a viral community with high diversity but low abundance.
The two pristine sites had very different levels of diversity, with P1 harbouring more than twice the number of viral families than P2, despite P2 having a higher number of viral reads. This may be because the P2 stream was smaller in volume with a smaller catchment, with less opportunity for a high diversity of terrestrial viruses to enter the river from the surrounding land. However, despite these differences, the beta analysis (Fig. 7) indicated that the two samples were very similar to each other. This likely reflects the fact that the families in P1 that are not shared between the two sites are at lower abundances (such as the bacteriophage families; Fig. 6) than the families that both sites have in common. Indeed, the Bray–Curtis similarity matrix [used to create the Principal Coordinates Analysis (PCoA) plots] is sensitive to differences in abundance between species, with abundant taxa weighted more than rare taxa (Ricotta and Podani 2017).
Virus families that infect fungi, plants, and algae were found in particularly high abundance, with the highest being the Tombusviridae associated with plants. This presumably reflects a high abundance of plant and algae matter in the water, including both aquatic plants living in the river and terrestrial plants from the surrounding land. However, there was a very low abundance and diversity of viruses associated with prokaryotes, although this is likely because we filtered out bacteria prior to RNA extraction (using PES membrane filters of 0.2-μm pore size) which have in turn removed many bacteriophages. In contrast, an earlier study of urban streams in Ecuador using a similar filtering system found that prokaryote infecting viruses had the highest abundance (Guerrero-Latorre et al. 2018). Interestingly, these Ecuadorian streams are known to be contaminated with untreated sewage, which may increase the bacterial load (and therefore bacteriophage abundance). However, because our study is based on total RNA sequencing, it is expected that there would be ascertainment bias towards RNA viruses and against those with DNA genomes, which may also have impacted the detection of bacteriophages. Finally, it is also possible that the high abundance of divergent viruses belonging to families in which the host is unknown (Fig. 6) are in fact bacteriophages.
Metagenomics frequently identifies highly divergent viruses in environmental samples, and in most such studies, the host is unknown. We observed a similar pattern. The novel viruses identified often clustered into clades that fell in basal positions on family-level phylogenies and were most closely related to viral species found in other metagenomic studies (Zhang, Shi, and Holmes 2018; Starr et al. 2019). As a case in point, many of the Tombusviridae-like viruses were highly divergent and fell into the ‘tombus-like’ virus clade. This is similar to a previous study that identified 199 tombus-like viruses in seawater from the Yangshan Deep-Water Harbour, Shanghai, China (Wolf et al. 2020). We also found many divergent viruses from the Astroviridae that were most closely related to viruses found using metagenomics from grassland soil in California, USA (Starr et al. 2019). As astroviruses are routinely associated with vertebrates, it is surprising to find these viruses in soil and water. Hence, it is likely that these were shed from a vertebrate host, although this clearly needs to be studied in greater detail.
As the sampling sites were all from the same river catchment, in some cases the water from one site would flow to other sampling sites downstream. For example, water from P1 and P2 would flow through F1 and U2. However, there did not appear to be a trend in the number of reads and the number of viral hits when comparing upstream and downstream sites (Fig. 3). In contrast, a study of a river catchment in the United Kingdom found increasing viral richness moving downstream, related to inputs of wastewater from treatment plants (Adriaenssens et al. 2021). Notably, for the pristine and farming sites, there was considerably more overlap in the viral community at sites of the same land-use type: P1 and P2 were very similar to each other, as were F1 and F2 (Fig. 7). The exception to this was the urban site U2 (Palmerston North) that was more similar to the two farming sites than to U1 (Feilding). Indeed, U1 was on a different tributary of the Manawatū River than the other sites (Fig. 1), so the viral ecology in that part of the river may be different. Feilding (population 17,050) is also a much smaller urban centre than Palmerston North (population 81,500). In addition, while the sample sites from both urban sites were downstream of the urban area at the edge of the town/city, Feilding releases wastewater far downstream in a rural area, whereas Palmerston North releases wastewater upstream of our sampling site. This wastewater may have affected the viral ecology of the river downstream, as wastewater inputs commonly deposit viral genetic material into the river environment (Adriaenssens et al. 2021).
Some novel viruses were found only in the sites adjacent to human land use (i.e. farming and urban sites), including all the bastroviruses (Astroviridae), all of which were most closely related to a bastrovirus found in sewage (Dos Anjos, Nagata, and de Melo 2017). The pattern of presence/absence in our study and the link to sewage suggest that the presence of these viruses is likely related to human land use. Similarly, the presence of viruses from the Genomoviridae appears to be related to urban land use: nine of the fourteen viruses were most closely related to viruses found in sewage or faeces. Despite the generally large differences in viral community between the two urban sites, they both contained viruses from the Genomoviridae: thirteen viral species were identified in U1 and four in U2 (including two viruses found in both sites), suggesting a link between human land use and the presence of these viruses. Notably, however, we did not detect any human viruses. This contrasts with a PCR study that detected enteric viruses (adenovirus, norovirus, enterovirus, rotavirus, and hepatitis E virus) in two other rivers in New Zealand (Williamson et al. 2011), which may reflect the greater sensitivity of PCR assays specifically designed to detect these viruses. In general, more viruses were found in common between urban and farming sites. For example, eleven Astroviridae-like viruses were found in both urban and farming sites, but no Astroviridae-like viruses found in either of the pristine sites were identified in urban or farming sites. Hence, within viral families at the species level, the farming and urban sites had a more similar viral community to each other than to the pristine sites, which may be a result of human land use adjacent to the river.
The only viruses we identified that were previously described were all found in the urban and farming sites, again indicative of an anthropogenic influence, including agriculture and introduced species. For example, we identified the black beetle virus in all farming and urban sites. The host (Heteronychus arator) is a major invasive pasture pest species that was introduced from Africa in the 1930s (Wilson et al. 2016), and the virus was first identified in 1975 in New Zealand (Longworth and Carey 1976). Similarly, at the Palmerston North urban site (U2), we detected R. padi virus that infects the bird cherry-oat aphid, a pest of cereal crops that was first recorded in New Zealand in 1921 (Bulman et al. 2005). Surprisingly, we found Norway luteo-like virus 4 with 96–100 per cent amino acid identity in one farming and one urban site (F1 and U2), previously associated with the castor bean tick Ixodes ricinus (Pettersson et al. 2017). As this species of tick has not been described in New Zealand, this result suggests that this virus has a wider host range than is currently known. Similarly, we identified pepper mild mottle virus in U2. This virus has previously been proposed as a water quality indicator and an indicator of the presence of human faeces in freshwater, as it is the most abundant RNA virus in human faeces but is rarely found in animal faecal matter (Rosario et al. 2009; Kitajima, Sassi, and Torrey 2018). Our ability to detect these viruses suggests that this technique could be used for ongoing monitoring, including detecting the presence of pest species in the surrounding area and human-related viruses indicating contamination of faecal matter in the river. That none of these viruses were found in the pristine sites suggests these viruses are being introduced into the river from agricultural and urban run-off, and possibly also discharge of treated wastewater into the river. Indeed, at pristine sites, we would generally expect viruses associated with New Zealand native plants and animals, most of which are yet to be described.
This study represents the first characterisation of the virome of a New Zealand river. We observed a high abundance and diversity of viruses, including many that are both novel and highly divergent. Within the same river catchment, we identified viruses linked to agriculture and human presence (including possible links to sewage) in the farming and urban sites that were not present in the pristine sites. More broadly, this work provides the foundation for more detailed research on the impacts of human land use on river viromes, which will require larger sample sizes across multiple river systems. As the size of our study is small, with all samples collected at the same time of year and in a large river system, it is clear that the trends observed need to be validated with larger surveys. In addition to widening the scope to include more river catchments and sampling sites, repeated sampling across different seasons and different river levels would also be beneficial to determine whether the observed differences between sites persist across different environmental conditions. Our results indicate that human land use has likely impacted the viral community in the river, such that further work is needed to reduce the impact of intensive farming and urbanisation on the land and rivers.
Supplementary Material
Acknowledgements
We thank Wendy Kay and Nigel French for their assistance in the field and Anthony Pita for assistance with sample processing. We thank Ci-Xiu Li, Wei-Shan Chang, Jackie Mahar, and Sabrina Sadiq for their bioinformatics advice. We thank the Molecular Epidemiology and Public Health Laboratory at Massey University and the Behaviour, Ecology and Evolution Lab at the University of Sydney for use of their laboratory space. We thank two anonymous reviewers for their helpful comments. This research utilised the high-performance computing service, Artemis, provided by the Sydney Informatics Hub, Core Research Facility, University of Sydney.
Contributor Information
Rebecca French, Sydney Institute for Infectious Diseases, Westmead Institute for Medical Research, 176 Hawkesbury Road, Westmead NSW 2145, Australia; School of Life and Environmental Sciences, The University of Sydney, Sydney, NSW 2006, Australia; Sydney Medical School, The University of Sydney, Sydney, NSW 2006 Australia.
Justine Charon, Sydney Institute for Infectious Diseases, Westmead Institute for Medical Research, 176 Hawkesbury Road, Westmead NSW 2145, Australia; School of Life and Environmental Sciences, The University of Sydney, Sydney, NSW 2006, Australia; Sydney Medical School, The University of Sydney, Sydney, NSW 2006 Australia.
Callum Le Lay, Sydney Institute for Infectious Diseases, Westmead Institute for Medical Research, 176 Hawkesbury Road, Westmead NSW 2145, Australia; School of Life and Environmental Sciences, The University of Sydney, Sydney, NSW 2006, Australia; Sydney Medical School, The University of Sydney, Sydney, NSW 2006 Australia.
Chris Muller, Wildbase, School of Veterinary Sciences, Massey University, Palmerston North 4442, New Zealand; Wildlife and Ecology Group, School of Agriculture and Environment, Massey University, Palmerston North 4442, New Zealand.
Edward C Holmes, Sydney Institute for Infectious Diseases, Westmead Institute for Medical Research, 176 Hawkesbury Road, Westmead NSW 2145, Australia; School of Life and Environmental Sciences, The University of Sydney, Sydney, NSW 2006, Australia; Sydney Medical School, The University of Sydney, Sydney, NSW 2006 Australia.
Data availability
Sequence data have been deposited in the SRA under accession numbers SRR17234948-53. The trimmed alignment FASTA files used to infer the phylogenetic trees are available at https://github.com/RKFrench/Viral-Diversity-NZ-River. The consensus sequences of all novel viruses are available on NCBI/GenBank with accession numbers OM953841–OM954332.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
ARC Australian Laureate Fellowship held by ECH (FL170100022).
Conflict of interest:
None declared.
References
- Adriaenssens E. et al. (2021) ‘Tracing the Fate of Wastewater Viruses Reveals Catchment-scale Virome Diversity and Connectivity’, Water Research, 203: 117568. [DOI] [PubMed] [Google Scholar]
- Alegbeleye O. O., and Sant’Ana A. S. (2020) ‘Manure-borne Pathogens as an Important Source of Water Contamination: An Update on the Dynamics of Pathogen Survival/Transport as Well as Practical Risk Mitigation Strategies’, International Journal of Hygiene and Environmental Health, 227: 113524. [DOI] [PubMed] [Google Scholar]
- Amvrosieva T. et al. (2001) ‘Viral Water Contamination as the Cause of Aseptic Meningitis Outbreak in Belarus’, Central European Journal of Public Health, 9: 154–7. [PubMed] [Google Scholar]
- Baldisserotto B. (2011) ‘Water pH and Hardness Affect Growth of Freshwater Teleosts’, Brazilian Journal of Animal Science, 40: 138–44. [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics, 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Reuter K., and Drost H.-G. (2021) ‘Sensitive Protein Alignments at Tree-of-life Scale Using DIAMOND’, Nature Methods, 18: 366–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulman S. et al. (2005) ‘Rhopalosiphum Aphids in New Zealand. I. RAPD Markers Reveal Limited Variability in Lineages of Rhopalosiphum padi’, New Zealand Journal of Zoology, 32: 29–36. [Google Scholar]
- Burridge C. P., and Waters J. M. (2020) ‘Does Migration Promote or Inhibit Diversification? A Case Study Involving the Dominant Radiation of Temperate Southern Hemisphere Freshwater Fishes’, Evolution, 74: 1954–65. [DOI] [PubMed] [Google Scholar]
- Bushnell B. (2018) BBMap Short-read Aligner, and Other Bioinformatics Tools. Berkeley, CA: University of California. [Google Scholar]
- Camacho C. et al. (2009) ‘BLAST+: Architecture and Applications’, BMC Bioinformatics, 10: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S. J. et al. (2020) ‘Red Fox Viromes in Urban and Rural Landscapes’, Virus Evolution, 6: veaa065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P. M., Bailey H., and Canaria E. (2000) ‘Toxicity of Total Dissolved Solids Associated with Two Mine Effluents to Chironomid Larvae and Early Life Stages of Rainbow Trout’, Environmental Toxicology and Chemistry, 19: 210–4. [Google Scholar]
- Chen J. et al. (2019) ‘Detection and Characterization of a Novel Marine Birnavirus Isolated from Asian Seabass in Singapore’, Virology Journal, 16: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. et al. (2018) ‘Aquatic Bacterial Communities Associated with Land Use and Environmental Factors in Agricultural Landscapes Using a Metabarcoding Approach’, Frontiers in Microbiology, 9: 2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen P. T., Aarestrup F. M., and Lund O. (2018) ‘Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA’, BMC Bioinformatics, 19: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Costa B. et al. (2003) ‘Blotched Snakehead Virus Is a New Aquatic Birnavirus That Is Slightly More Related to Avibirnavirus than to Aquabirnavirus’, Journal of Virology, 77: 719–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies-Colley R. J. (2013) ‘River Water Quality in New Zealand: An Introduction and Overview’, in John Dymond (ed.) Ecosystem Services in New Zealand: Conditions and Trends, pp. 432–47. Lincoln: Manaaki Whenua Press. [Google Scholar]
- de Sousa D. N. R. et al. (2014) ‘Electrical Conductivity and Emerging Contaminant as Markers of Surface Freshwater Contamination by Wastewater’, Science of the Total Environment, 484: 19–26. [DOI] [PubMed] [Google Scholar]
- Department of Conservation . (2021a), History and Culture <https://www.doc.govt.nz/parks-and-recreation/places-to-go/manawatu-whanganui/places/ruahine-forest-park/history-and-culture/> accessed 1 Jun 2021.
- ———. (2021b), Nature and Conservation <https://www.doc.govt.nz/parks-and-recreation/places-to-go/manawatu-whanganui/places/ruahine-forest-park/nature-and-conservation/> accessed 1 Jun 2021.
- Dos Anjos K., Nagata T., and de Melo F. L. (2017) ‘Complete Genome Sequence of a Novel Bastrovirus Isolated from Raw Sewage’, Genome Announcements, 5: e01010-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn N. R. et al. (2018) Conservation Status of New Zealand Freshwater Fishes, 2017. Wellington, New Zealand: Publishing Team, Department of Conservation. [Google Scholar]
- Ferguson C. et al. (2003) ‘Fate and Transport of Surface Water Pathogens in Watersheds’, Critical Reviews in Environmental Science and Technology, 33: 299–361. [Google Scholar]
- French R. K., and Holmes E. C. (2020) ‘An Ecosystems Perspective on Virus Evolution and Emergence’, Trends in Microbiology, 28: 165–75. [DOI] [PubMed] [Google Scholar]
- Grabherr M. G. et al. (2011) ‘Full-length Transcriptome Assembly from RNA-Seq Data without a Reference Genome’, Nature Biotechnology, 29: 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X. et al. (2018) ‘Geospatial Distribution of Viromes in Tropical Freshwater Ecosystems’, Water Research, 137: 220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Latorre L. et al. (2018) ‘Quito’s Virome: Metagenomic Analysis of Viral Diversity in Urban Streams of Ecuador’s Capital City’, Science of the Total Environment, 645: 1334–43. [DOI] [PubMed] [Google Scholar]
- Hamill K. (2012) Effects of Palmerston North City’s Wastewater Treatment Plant Discharge on Water Quality and Aquatic Life in the Manawatu River. Palmerston North, New Zealand: Palmerston North City Council and Horizons Regional Council. [Google Scholar]
- Hill M. O. (1973) ‘Diversity and Evenness: A Unifying Notation and Its Consequences’, Ecology, 54: 427–32. [Google Scholar]
- Jack S., Bell D., and Hewitt J. (2013) ‘Norovirus Contamination of a Drinking Water Supply at a Hotel Resort’, New Zealand Medical Journal, 126: 98–107. [PubMed] [Google Scholar]
- Jones T. H. et al. (2017) ‘Waterborne Viruses and F-specific Coliphages in Mixed-use Watersheds: Microbial Associations, Host Specificities, and Affinities with Environmental/Land Use Factors’, Applied and Environmental Microbiology, 83: e02763-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapelinskaya T. V. et al. (2011) ‘Expression Strategy of Densonucleosis Virus from the German Cockroach, Blattella germanica’, Journal of Virology, 85: 11855–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirs M. et al. (2011) ‘Source Tracking Faecal Contamination in an Urbanised and a Rural Waterway in the Nelson-Tasman Region, New Zealand’, New Zealand Journal of Marine and Freshwater Research, 45: 43–58. [Google Scholar]
- Kitajima M., Sassi H. P., and Torrey J. R. (2018) ‘Pepper Mild Mottle Virus as a Water Quality Indicator’, NPJ Clean Water, 1: 1–9. [Google Scholar]
- Kraberger S. et al. (2013) ‘Discovery of Sclerotinia sclerotiorum Hypovirulence-associated Virus-1 in Urban River Sediments of Heathcote and Styx Rivers in Christchurch City, New Zealand’, Genome Announcements, 1: e00559-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2015) ‘Characterisation of a Diverse Range of Circular Replication-associated Protein Encoding DNA Viruses Recovered from a Sewage Treatment Oxidation Pond’, Infection, Genetics and Evolution, 31: 73–86. [DOI] [PubMed] [Google Scholar]
- Krumbholz A. et al. (2017) ‘Genome Sequence of a Novel Picorna-like RNA Virus from Feces of the Antarctic Fur Seal (Arctocephalus gazella)’, Genome Announcements, 5: e01001–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay C. et al. (2020) ‘Unmapped RNA Virus Diversity in Termites and Their Symbionts’, Viruses, 12: 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., and Dewey C. N. (2011) ‘RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome’, BMC Bioinformatics, 12: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth J., and Carey G. (1976) ‘A Small RNA Virus with a Divided Genome from Heteronychus arator (F.)[Coleoptera: Scarabaeidae]’, Journal of General Virology, 33: 31–40. [DOI] [PubMed] [Google Scholar]
- Lukashov V. V., and Goudsmit J. (2002) ‘Evolutionary Relationships among Astroviridae.’, Journal of General Virology, 83: 1397–405. [DOI] [PubMed] [Google Scholar]
- Mahar J. E. et al. (2020) ‘Comparative Analysis of RNA Virome Composition in Rabbits and Associated Ectoparasites’, Journal of Virology, 94: e02119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelino V. R. et al. (2020) ‘CCMetagen: Comprehensive and Accurate Identification of Eukaryotes and Prokaryotes in Metagenomic Data’, Genome Biology, 21: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martolia H. C. S. et al. (2009) ‘An Outbreak of Hepatitis E Tracked to a Spring in the Foothills of the Himalayas, India, 2005’, Indian Journal of Gastroenterology, 28: 99–101. [DOI] [PubMed] [Google Scholar]
- McMurdie P. J., and Holmes S. (2013) ‘Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data’, PLoS One, 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer N. et al. (2019) ‘Late Cretaceous Oceanic Plate Reorganization and the Breakup of Zealandia and Gondwana’, Gondwana Research, 65: 31–42. [Google Scholar]
- Mukherjee S. S. (2011) ‘Identification and Characterization of Tobamo and Tombusviruses Isolated from New Zealand Waters’, PhD thesis, State University of New York College of Environmental Science and Forestry. [Google Scholar]
- ——— et al. (2012) ‘New Tombusviruses Isolated from Surface Waters in New Zealand’, Australasian Plant Pathology, 41: 79–84. [Google Scholar]
- Nayfach S. et al. (2021) ‘Metagenomic Compendium of 189,680 DNA Viruses from the Human Gut Microbiome’, Nature Microbiology, 6: 960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E., and Schliep K. (2019) ‘Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R’, Bioinformatics, 35: 526–8. [DOI] [PubMed] [Google Scholar]
- Pénzes J. J. et al. (2020) ‘Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association’, Archives of Virology, 165: 2133–46. [DOI] [PubMed] [Google Scholar]
- Pettersson J. H.-O. et al. (2017) ‘Characterizing the Virome of Ixodes ricinus Ticks from Northern Europe’, Scientific Reports, 7: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiri B. J. et al. (2020) ‘Does Land Use Affect Pathogen Presence in New Zealand Drinking Water Supplies?’ Water Research, 185: 116229. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2021) ‘Microbial Diversity in Water and Animal Faeces: A Metagenomic Analysis to Assess Public Health Risk’, New Zealand Journal of Zoology, 48: 188–201. [Google Scholar]
- Porter A. F. et al. (2021) ‘Metagenomic Identification of Viral Sequences in Laboratory Reagents’, Viruses, 13: 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H. et al. (2020) ‘Metagenomic Analysis Revealed that the Terrestrial Pollutants Override the Effects of Seasonal Variation on Microbiome in River Sediments’, Bulletin of Environmental Contamination and Toxicology, 105: 892–8. [DOI] [PubMed] [Google Scholar]
- R Core Team . (2021) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Reuter G. et al. (2015) ‘A Highly Divergent Picornavirus in an Amphibian, the Smooth Newt (Lissotriton vulgaris)’, Journal of General Virology, 96: 2607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., and Bleasby A. (2000) ‘EMBOSS: The European Molecular Biology Open Software Suite’, Trends in Genetics, 16: 276–7. [DOI] [PubMed] [Google Scholar]
- Ricotta C., and Podani J. (2017) ‘On Some Properties of the Bray-Curtis Dissimilarity and Their Ecological Meaning’, Ecological Complexity, 31: 201–5. [Google Scholar]
- Riviere C., and Rochon D. (1990) ‘Nucleotide Sequence and Genomic Organization of Melon Necrotic Spot Virus’, Journal of General Virology, 71: 1887–96. [DOI] [PubMed] [Google Scholar]
- Roossinck M. J. (2015) ‘Plants, Viruses and the Environment: Ecology and Mutualism’, Virology, 479: 271–7. [DOI] [PubMed] [Google Scholar]
- Rosario K. et al. (2009) ‘Pepper Mild Mottle Virus as an Indicator of Fecal Pollution’, Applied and Environmental Microbiology, 75: 7261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roygard J., McArthur K., and Clark M. (2012) ‘Diffuse Contributions Dominate over Point Sources of Soluble Nutrients in Two Sub-catchments of the Manawatu River, New Zealand’, New Zealand Journal of Marine and Freshwater Research, 46: 219–41. [Google Scholar]
- Sedyaningsih-Mamahit E. et al. (2002) ‘First Documented Outbreak of Hepatitis E Virus Transmission in Java, Indonesia’, Transactions of the Royal Society of Tropical Medicine and Hygiene, 96: 398–404. [DOI] [PubMed] [Google Scholar]
- Sekwadi P. et al. (2018) ‘Waterborne Outbreak of Gastroenteritis on the KwaZulu-natal Coast, South Africa, December 2016/January 2017’, Epidemiology and Infection, 146: 1318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit T. L., and Lommel S. A. (2015) ‘Tombusviridae’, eLS, 1–9. [Google Scholar]
- Skotnicki M. et al. (1993) ‘The Genomic Sequence of Cardamine Chlorotic Fleck Carmovirus’, Journal of General Virology, 74: 1933–7. [DOI] [PubMed] [Google Scholar]
- Sommers P. et al. (2021) ‘Integrating Viral Metagenomics into an Ecological Framework’, Annual Review of Virology, 8: 133–58. [DOI] [PubMed] [Google Scholar]
- Starr E. P. et al. (2019) ‘Metatranscriptomic Reconstruction Reveals RNA Viruses with the Potential to Shape Carbon Cycling in Soil’, Proceedings of the National Academy of Sciences USA, 116: 25900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel O. et al. (2016) ‘Circular Replication-associated Protein Encoding DNA Viruses Identified in the Faecal Matter of Various Animals in New Zealand’, Infection, Genetics and Evolution, 43: 151–64. [DOI] [PubMed] [Google Scholar]
- Suttle C. (2016) ‘Environmental Microbiology: Viral Diversity on the Global Stage’, Nature Microbiology, 1: 1–2. [DOI] [PubMed] [Google Scholar]
- Thao M. L. et al. (2001) ‘Genetic Characterization of a Putative Densovirus from the Mealybug Planococcus citri’, Current Microbiology, 43: 457–8. [DOI] [PubMed] [Google Scholar]
- Valles S. et al. (2017) ‘ICTV Virus Taxonomy Profile: Dicistroviridae’, Journal of General Virology, 98: 355–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rossum T. et al. (2015) ‘Year-long Metagenomic Study of River Microbiomes across Land Use and Water Quality’, Frontiers in Microbiology, 6: 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varsani A., and Krupovic M. (2021) ‘Family Genomoviridae: 2021 Taxonomy Update’, Archives of Virology, 166: 2911–26. [DOI] [PubMed] [Google Scholar]
- Waldron F. M., Stone G. N., and Obbard D. J. (2018) ‘Metagenomic Sequencing Suggests a Diversity of RNA Interference-like Responses to Viruses across Multicellular Eukaryotes’, PLoS Genetics, 14: e1007533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S., Monks A., and Innes J. G. (2021) ‘Life History Traits Explain Vulnerability of Endemic Forest Birds and Predict Recovery after Predator Suppression’, New Zealand Journal of Ecology, 45: 3447. [Google Scholar]
- Wickham H. (2016) Ggplot2: Elegant Graphics for Data Analysis. New York: Springer. [Google Scholar]
- Wilkinson L. (2011), Venneuler: Venn and Euler Diagrams. R package version 1.1-0.
- Wille M. et al. (2020) ‘Sustained RNA Virome Diversity in Antarctic Penguins and Their Ticks’, The ISME Journal, 14: 1768–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021) ‘RNA Virome Abundance and Diversity Is Associated with Host Age in a Bird Species’, Virology, 561: 98–106. [DOI] [PubMed] [Google Scholar]
- Williamson W. et al. (2011) ‘Enteric Viruses in New Zealand Drinking-water Sources’, Water Science and Technology, 63: 1744–51. [DOI] [PubMed] [Google Scholar]
- Wilson M. J. et al. (2016) ‘Developing a Strategy for Using Entomopathogenic Nematodes to Control the African Black Beetle (Heteronychus arator) in New Zealand Pastures and Investigating Temperature Constraints’, Biological Control, 93: 1–7. [Google Scholar]
- Wolf Y. I. et al. (2020) ‘Doubling of the Known Set of RNA Viruses by Metagenomic Analysis of an Aquatic Virome’, Nature Microbiology, 5: 1262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. (2020) ‘Using Ggtree to Visualize Data on Tree‐like Structures’, Current Protocols in Bioinformatics, 69: e96. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-Z., Shi M., and Holmes E. C. (2018) ‘Using Metagenomics to Characterize an Expanding Virosphere’, Cell, 172: 1168–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data have been deposited in the SRA under accession numbers SRR17234948-53. The trimmed alignment FASTA files used to infer the phylogenetic trees are available at https://github.com/RKFrench/Viral-Diversity-NZ-River. The consensus sequences of all novel viruses are available on NCBI/GenBank with accession numbers OM953841–OM954332.