

Abstract
A major challenge of targeted cancer therapy is the selection for drug-resistant mutations in tumor cells leading to loss of treatment effectiveness. p97/VCP is central regulator of protein homeostasis and a promising anticancer target because of its vital role in cell growth and survival. One ATP-competitive p97 inhibitor, CB-5083, has entered clinical trials. Selective pressure on HCT116 cells dosed with CB-5083 identified 5 different resistant mutants. Identification of p97 inhibitors with different mechanisms of action would offer the potential to overcome this class of resistance mutations. Our results demonstrate that two CB-5083 resistant p97 mutants, N660K and T688A, were also resistant to several other ATP-competitive p97 inhibitors, whereas inhibition by two allosteric p97 inhibitors NMS-873 and UPCDC-30245 were unaffected by these mutations. We also established a CB-5083 resistant cell line that harbors a new p97 double mutation (D649A/T688A). While CB-5083, NMS-873, and UPCDC-30245 all effectively inhibited proliferation of the parental HCT116 cell line, NMS-873 and UPCDC-30245 were 30-fold more potent in inhibiting the CB-5083 resistant D649A/T688A double mutant than CB-5083. Our results suggest that allosteric p97 inhibitors are promising alternatives when resistance to ATP-competitive p97 inhibitors arises during anticancer treatment.
Keywords: p97 VCP, resistance, small molecule inhibitor, ATPase, biological activity, cancer
Graphical Abstract

We established a new CB-5083 resistant HCT116 cell line and demonstrate that NMS-873 and UPCDC-30245 exhibited a 30-fold higher potency in inhibiting its proliferation than CB-5083. Our studies indicated that allosteric p97 inhibitors are promising alternatives to overcome resistance to ATP-competitive p97 inhibitors.
Introduction
p97/VCP is an abundant AAA-ATPase and is essential for life. Amongst its multiple cellular functions, its role in mediating degradation of ubiquitinated proteins via the endoplasmic reticulum associated degradation (ERAD) pathway has been well-studied and is considered to be relevant to cancer cell growth and survival [1]. Rapid growth of cancer cells makes them more dependent on the ubiquitin-proteasome system (UPS) to alleviate proteotoxic stress caused by an imbalance of protein synthesis and degradation. Increased expression of p97/VCP is associated with metastasis and poor prognosis in multiple cancer types [2]. Recently, another essential role of p97 in the clearance of damaged lysosomes by autophagy has been identified. Specifically, upon damage to lysosomes, p97 will translocate to lysosomes and cooperate with cofactors such as UBXD1 and PLAA to promote autophagosome formation [3]. Overall, p97VCP targets proteins to two major degradation systems, the proteasome and autophagy machinery. Inhibition of p97/VCP ATPase activity can block two major protein degradation pathways, and therefore can potentially be effective for cells that are resistant to proteasome inhibitors.
p97 is composed of two ATPase domains, D1 and D2, and assembles into a hexameric structure resembling 2 rings, one formed by the D1 domains and the other by the D2 domains. D2 is responsible for most of the ATPase activity, and D1 is required for the formation of stable hexamers. p97 also contains the N domain, which coordinates the multiple functions of p97 by binding to distinct cofactors [4]. ATP-binding and hydrolysis in the D2 domain triggers significant conformational changes, which are transmitted via long flexible linkers from the D2 domain to the D1 domain and further to the N domain [5].
Since p97 plays an important role in cancer progression, several groups have sought to discover p97 inhibitors that could be eventually be developed for cancer treatment. DBeQ is a reversible, selective ATP-competitive inhibitor of p97, which can inhibit cancer cell proliferation by impairing both ubiquitin-dependent and autophagic protein clearance pathways [6]. Modifications of DBeQ led to the discovery of the more potent p97 inhibitors ML240 and ML241 [7]. From these molecules Cleave Biosciences subsequently developed CB-5083, the most advanced p97 inhibitor [1]. CB-5083 induces apoptosis of cancer cells due to the retention of endoplasmic reticulum-associated degradation (ERAD) substrates [8], which has been shown to be a novel approach to cancer treatment. CB-5083 entered two phase I clinical trials in 2015; however, the trials were halted when toxicities were identified that arose due to off-target effects of the compound [9]. Recently, a crystal structure with CB-5083 bound to D1-D2 p97 (lacking N domain) was solved and revealed the molecular basis for CB-5083’s selective inhibition of the D2 domain of p97 [10].
As observed with other molecularly targeted oncogene therapies, drug resistance is likely to become an obstacle to effective, durable p97 inhibitor therapy in cancer patients. Several CB-5083 resistant cancer cell lines have been established and p97 mutants have been identified in the laboratory [1, 10–11] (Fig. 1). Since CB-5083 has shown promise as a potential therapy for cancers, it is crucial to identify p97 inhibitors with different mechanisms of action, thereby offering patients additional treatment options upon appearance of drug resistance. To overcome the resistance that developed to clinically use ATP-competitive kinase inhibitors that results from the selection of drug-resistant mutant forms of the kinase target, two approaches have been described. The first one is the development of new ATP-competitive inhibitors that have different binding modes; another strategy is to find non-ATP competitive inhibitors that can regulate kinase activity allosterically [12]. Allosteric inhibitors have been developed for multiple kinases including mTor30, Mek31, Akt32, IkB kinase33, Chk1, and BCR-ABL1 [13]. Based on these successes, we sought to identify if allosteric p97 inhibitors could be effective against CB-5083 resistant p97 mutant proteins and cancer cell lines.
Figure 1.
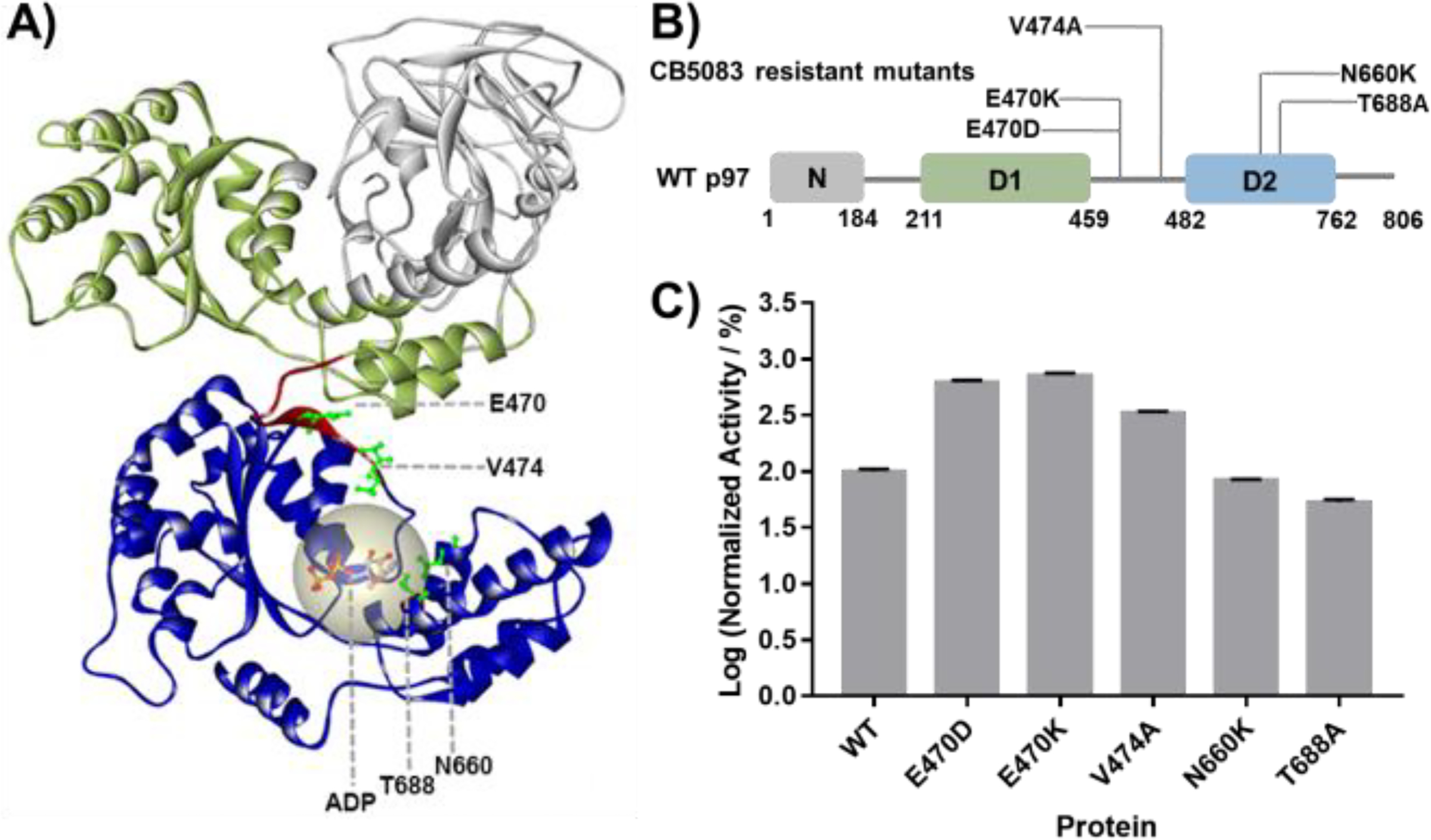
Structures and ATPase activity of WT p97 and p97 mutants. A) 3D structure of a p97 monomer displaying sites of CB-5083 resistant mutations (PDB: 5FTJ). B) CB-5083 resistant p97 mutants generated in this study. C) ATPase activity of p97 and p97 mutants, normalized to WT. Data are shown as Mean ± SEM taken from triplicate experiments.
Two different allosteric p97 inhibitors have been reported in independent high throughput screening campaigns and structure-activity relationship optimization. Genentech/Nerviano published the discovery and optimization of NMS-873 [14] and a multi-institutional consortium identified UPCDC-30245 [15]. The binding site of NMS-873 was mapped to a pocket defined by residues from D1 and D2 domains of adjacent protomers. By binding to p97, NMS-873 changes the binding affinity of nucleotide with p97 and inhibited ATPase activity. UPCDC-30245 is a structurally distinct allosteric p97 inhibitor and was showed by single particle cryo-EM to occupy a site at the D1 to D2 interface of a p97 monomer [9]. UPCDC-30245 prevents propagation of the conformational changes necessary for p97 function [9].
Based upon the different properties and binding sites for these two allosteric inhibitors, NMS-873 and UPCDC-30245 were chosen to evaluate whether allosteric p97 inhibitors could be viable alternatives or follow-ons to CB-5083 in cancer therapy when resistance to CB-5083 treatment occurs.
Results and Discussion
1. The D1-D2 linker region and D2 domain of p97 ATPase are mutational hot spots for CB-5083 resistance.
CB-5083 resistant cell lines were selected following treatment of the colon cancer cell line HCT116 and the ovarian cancer cell line OVSAHO. p97 mutations V474A, N660K, and T688A were identified in CB-5083 resistant HCT116 colon cancer cells [1], whereas E470K and E470D mutations were identified in the CB-5083 resistant OVSAHO ovarian cancer cell line[16]. These mutations, considered to be the cause of drug resistance, are located in the p97 D2 ATPase domain and in the linker that connects the D1 and D2 domains (Fig. 1A). CB-5083 has been characterized as a D2-selective p97 inhibitor; therefore, the identification of CB-5083 resistant mutations around the ATP binding site in D2 is not surprising. The presence of mutations in the linker region was somewhat unexpected and attests to the important of this region in the interdomain transmission of energy from ATP hydrolysis and may help explain the specificity. To elucidate the mechanism of drug-resistance conferred by these mutants and to evaluate if different p97 inhibitors that can overcome CB-5083 resistance, we purified recombinant p97 proteins harboring these mutations (Fig. 1 B) and characterized their enzymatic activity (Fig. 1 C). Our results showed that all the linker mutants have dramatically increased ATP hydrolysis activity (3.3 to 7.2 folds of WT activity), while the D2 domain mutants decreased p97 ATPase activity slightly in the case of N660K (83% of WT activity) or significantly in the case of T688A (53% of WT activity).
2. Most of CB-5083 resistant mutants are globally resistant to ATP competitive inhibitors (DBeQ analogs).
To understand how CB-5083 resistant mutants affect other ATP competitive p97 inhibitors, we tested the sensitivity of these mutants to the CB-5083 related compounds, DBeQ, ML240 and ML241 (Fig. 2 A). Interestingly, our results (Fig. 2 B–F) showed that all the CB-5083 resistant mutants are resistant to all the DBeQ analogs tested, with resistance ranging from 2- to 580-fold.
Figure 2.
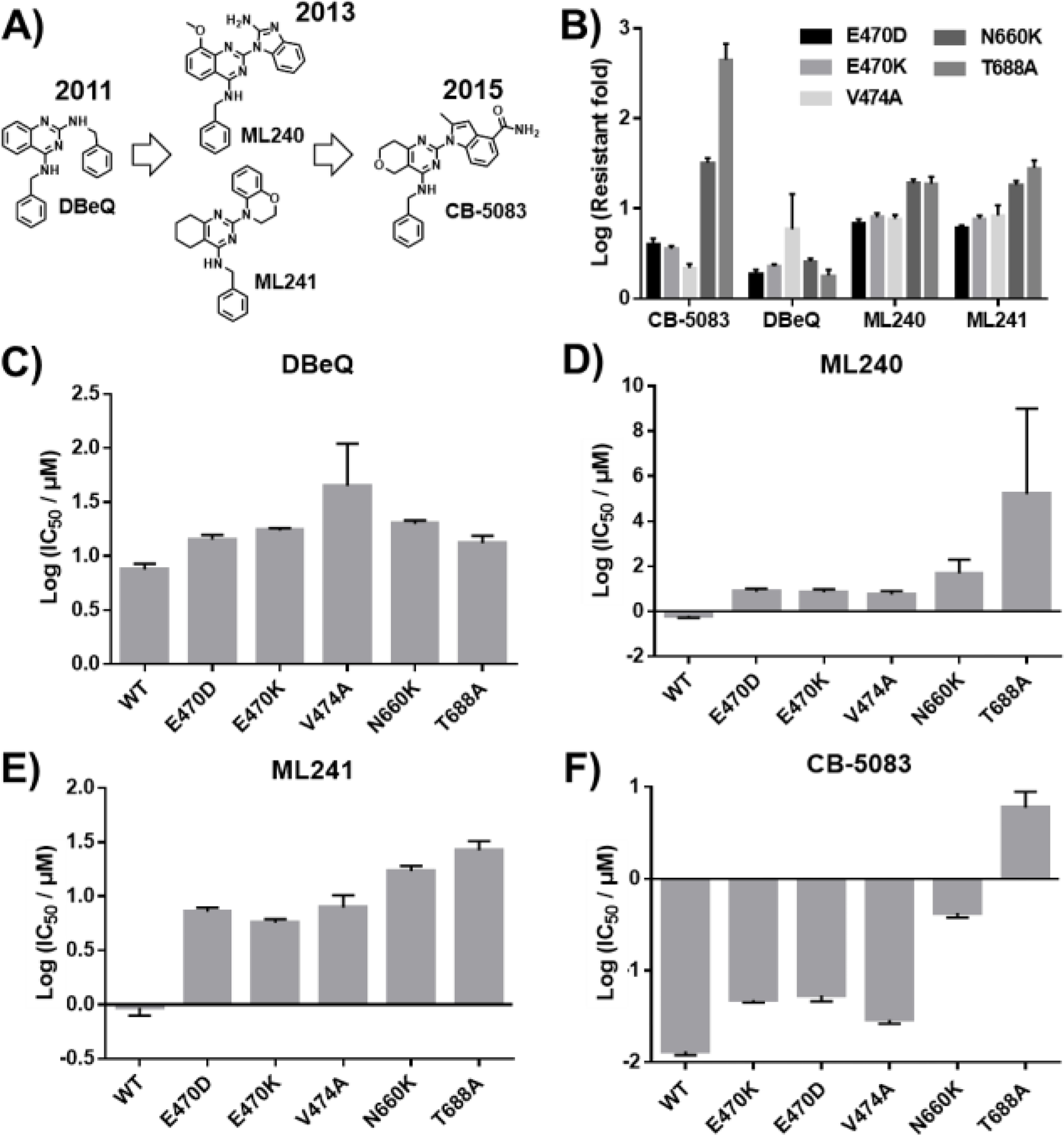
Structures of ATP competitive p97 inhibitors and inhibition of different p97 mutants. A) Structures of CB-5083 and related structures DBeQ, ML240 and ML241. B) Relative resistance to inhibition by ATP competitive p97 inhibitors for each p97 mutant. IC50 for mutant divided by IC50 for WT p97. C-F) IC50 values of each p97 inhibitor tested against WT p97 and each mutant. CB-5083 resistant mutants are globally resistant to ATP competitive inhibitors. Data are shown as Mean ± SEM taken from six replicates.
Among those mutants, N660K and T688A are the most resistant to inhibition by CB-5083, ML240, and ML241. Because the D2 domain is responsible for the majority of the p97 ATPase activity, it was not surprising that N660K and T688A, which are located close to the ATP binding site in the D2 domain, were the most resistant mutants (Fig. 1 A). DBeQ inhibits both D1 and D2 domains and the fold resistance is only 2–3 fold. These results suggest that residues N660 and T688 are located where the 2-quinazoline substituents bind to p97, and that the more flexible benzylamine substituent in DBeQ is less affected by these mutations. This conclusion is supported by the recent X-ray structure of CB-5083 bound to p97 (6MCK).
Drug-resistant mutations have been used to characterize the site of inhibitor binding in EGFR and other anticancer targets [17]. For p97, the effects of the N660K and T688A mutations on binding of the ATP-competitive p97 inhibitors have not been examined. Thermal shift assays (TSA) and microscale thermophoresis (MST) are widely used methods to investigate protein-ligand interactions [18]. Here we applied TSA to investigate whether these mutations weaken the direct interaction between CB-5083 and p97 (Fig. 3 and S5). Consistent with previously reported data, p97 showed two melting transitions in our assay [19]. Following the addition of CB-5083, the melting temperature (Tm) of wild type p97 increased dose-dependently. In the presence of 10 μM CB-5083, Tm1 of WT p97 increased 2.4 °C while Tm1 of N660K and T688A increased only 0.6 °C and 0.4 °C respectively. Those results suggest that each mutation decreased the direct interaction of CB-5083 with p97. Moreover, by performing competitive MST experiments, we observed that these two mutations impaired CB-5083’s ability to compete with ATP binding to p97 (Fig. S1). In the presence of 1 μM CB-5083, the Kd of ATP-γ-S binding to WT p97 was 2-fold higher than that without CB-5083, demonstrating that CB-5083 competed with ATP-γ-S for binding to WT p97. However, the binding affinity of ATP-γ-S with N660K and T688A exhibited a similar Kd in the absence or presence of CB-5083.
Figure 3.

Thermal shift assay (TSA) of WT p97 and p97 mutants in the presence of CB-5083. A) The first derivative of the fluorescence for 1 μM WT p97 and p97 mutants was measured with respect to temperature. B and C) The addition of CB-5083 resulted in a dramatic shift in the melting temperature of WT p97, but not of N660K and T688A mutants. Every point in the dose response curve represents Mean ± SEM taken from triplicate samples.
3. N660K and T688A mutant p97 are sensitive to allosteric p97 inhibitors.
Allosteric inhibitors of protein kinases can be distinguished from ATP-competitive inhibitors by binding at different sites. They are unaffected by resistance mutations in the ATP-binding-site, and when used in combination with ATP-competitive inhibitors the emergence of resistance in vitro has been suppressed [12]. A similar paradigm could be applied to p97. NMS-873 and UPCDC-30245 represent two different type of allosteric p97 inhibitors.
To compare the mechanisms of action of CB-5083, NMS-873, and UPCDC-30245, we performed steady-state kinetic experiments at ten concentrations of ATP and 2 or 3 different concentrations of p97 inhibitors. We confirmed that CB-5083 is an ATP-competitive inhibitor (Fig. 5 C) as suggested by Anderson and coauthors [1]. We also confirmed that NMS-873 is a noncompetitive inhibitor (Fig. 5 A) as suggested by Magnaghi and coauthors [20]. UPCDC-30245 exhibited noncompetitive inhibition similar to that of NMS-873 (Fig. 5 B). Compared to CB-5083, the different inhibition mechanism of NMS-873 and UPCDC-30245 suggests that they may retain their inhibitory potency toward N660K and T688A. ATPase assays (Fig. 4) with these mutants demonstrate that, as hypothesized, the potency of UPCDC-30245 and NMS-873 in inhibiting N660K is similar to WT, while CB-5083 was 30-fold less potent. For the T688A p97 mutant, however, the results were somewhat different. UPCDC-30245 showed 3-fold resistance and NMS-873 showed 30-fold resistance, while the IC50 for CB-5083 increased by more than 500-fold in ATPase assays. According to the cryo-EM structure of p97 reported by Banerjee et al [9], the binding site of UPCDC-30245 is close to the linker region between the D1 and D2 domains. Based on cross-linking studies, Magnaghi et al [20] concluded that NMS-873 also binds between the D1-D2 linker of p97. Both binding sites of UPCDC-30245 (29.17 Å from V493 to T688) and NMS-873 (34.26 Å from N616 to T688) are far from the mutation site T688 (Fig. 6 A and Fig. S4). To examine whether these two mutations affect the binding affinity of NMS-873 and UPCDC-30245, TSA was performed. As shown in Fig. 6 B, NMS-873 had a similar effect on increasing Tm1 of WT, N660K, and T688A, indicating N660K and T688A may not affect the binding of NMS-873. UPCDC-30245, unlike CB-5083 and NMS-873, did not stabilize WT or either mutant p97 protein (Fig. 6 C), indicating a distinct binding mode for UPCDC-30245.
Figure 5.

Chemical structures of three p97 inhibitors and analysis of the mechanism by which three inhibitors inhibited p97 ATPase (A) NMS-873, (B) UPCDC-30245, and (C) CB-5083. Every point in the dose response curve represents Mean ± SEM taken from triplicates.
Figure 4.

N660K and T688A are more sensitive to NMS-873 and UPCDC-30245 than CB-5083. The IC50 of 3 p97 inhibitors in inhibiting N660K (A) and T688A (B). Resistant fold of p97 inhibitors against mutants comparing to WT p97 (C). Data are represented as Mean ± SEM taken from triplicate experiments
Figure 6.
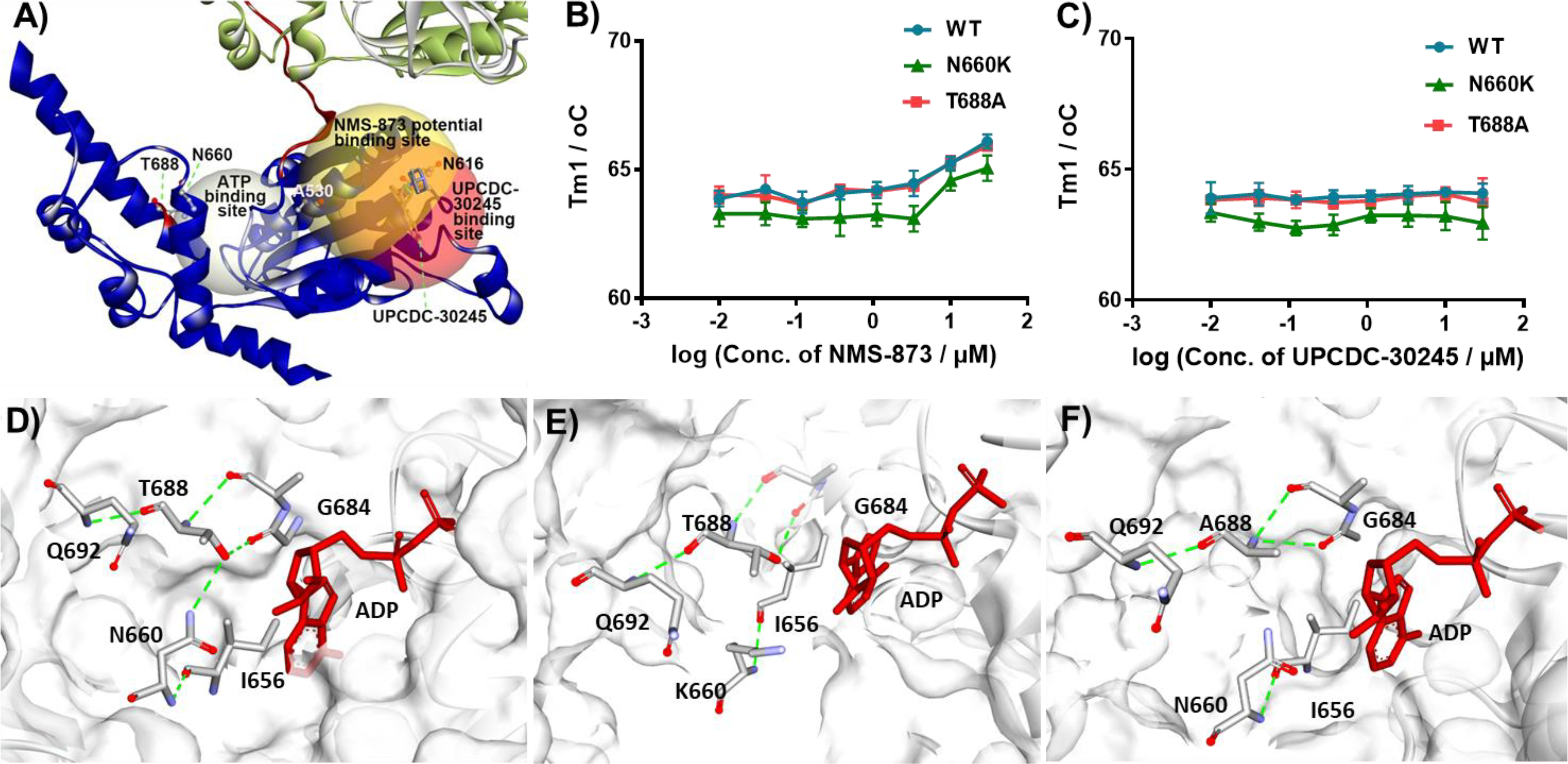
Structural models of CB-5083 resistant p97 mutants. A) D2 domain structure of WT p97 (PDB: 5FTJ); B) Tm of WT and mutant p97 with increasing concentration of NMS-873; C) Tm of WT and mutant p97 with increasing concentration of UPCDC-30245; D) interactions of N660, T688 and G684 in D2 of WT p97; computationally built models of mutant p97 proteins N660K (E) and T688A (F). Every point in the dose response curve represents Mean ± SEM taken from triplicate samples.
As we can see from Fig. 3 A, the second melting transition of WT p97 is almost undetectable in the N660K and T688A mutants. Considering the impaired ATPase activity of T688A (Fig. 1 C), the mutation at T688 is likely having a functional impact on the conformation or dynamics of p97 that may contribute to the resistance against UPCDC-30245 and NMS-873. To understand how these mutations might affect the structure of p97, we used Discovery Studio 4.0 to build a protein structure model of mutants based on the structure of WT p97 (PDB: 5FTJ). In wild-type p97, the hydroxy group of T688 sits between G684 and N660, making hydrogen bonds with both the carbonyl of G684 and the amide side-chain nitrogen of N660. These hydrogen bonds link the α-helix containing N660 to the α-helix containing T688 and may contribute additional rigidity to the D2 domain (Fig. 6 D). The hydrogen bond between T688 and N660 is eliminated when asparagine 660 is replaced by lysine, and the absence of this interaction may lead to the separation of the two α-helices and “loosening” of the D2 domain (Fig. 6 E). Similarly, the connection is also lost when T688 was mutated to A688. Furthermore, the backbone amide nitrogen atom now makes a hydrogen bond with the carbonyl of G684, rather than the hydroxy group of T688. This new hydrogen bond turns the angle of α-helix containing T688A and further modifies the conformation of the D2 domain (Fig. 6 F). Potentially the conformational changes caused by drug-resistant mutations might alter the energy generation and passage from the D2 domain to the D1 domain. Past experiments have shown that when the function of the D2 domain was impaired, the D1 domain was able to contribute more to total ATPase activity [25]. This is consistent with previous results demonstrating that when D1 domain is inactive the kcat of the D2 domain increases and vice versa [25]. This data may explain why T688A is more sensitive to DBeQ than N660K since DBeQ inhibits both the D1 and D2 domains. If the mechanism of action of the allosteric inhibitors is to alter or block conformational changes and the passage of energy then these factors, rather than simply decreased binding affinity, may explain the resistance of T688A to the allosteric p97 inhibitors NMS-873 and UPCDC-30245.
Although the T688A mutant of p97 showed resistance to NMS-873 and UPCDC-30245 at different levels, compounds in this class are still potential alternatives to CB-5083 due to their lower IC50 against T688A (Fig 4 B). However, such compounds are able to overcome the resistance to CB-5083 at cellular level remains need to be examined.
4. Antiproliferative effect of p97 inhibitors in cancer cell lines.
We next explored whether NMS-873 and UPCDC-30245 could be effective alternatives when CB-5083 resistance arises. First, we tested to ability of NMS-873 and UPCDC-30245 to inhibit cancer cell proliferation (Fig. 7 A). All three p97 inhibitors inhibited proliferation of all seven cancer lines tested. Compared to CB-5083, the two allosteric p97 inhibitors were less potent. In biochemical assays, the IC50 of CB-5083 toward WT p97 ATPase activity was 10 nM, while the IC50 of NMS-873 and UPCDC-30245 were 26 nM and 300 nM, respectively. However, UPCDC-30245 showed similar activity to that of NMS-873 at inhibiting cell proliferation in most of the cell lines we tested, and appeared more potent in HT29 cells, despite being 12-fold less potent than NMS-873 in the ATPase activity assay. One potential explanation for this discrepancy would be if the physical properties of UPCDC-30245 allowed it to more readily enter cells, via active transport or passive diffusion, or if it might be preferentially retained inside cells. To examine this possibility, we measured the intracellular concentrations of NMS-873 and UPCDC-30245 following published methods [21] (Fig. 7 B and 7 C, table S5, standard curves for compounds are provided in Fig. S7). Both compounds dose-dependently entered into A549 cells and 10 μM UPCDC-30245 caused cellular toxicity and reduced its intracellular concentration. For UPCDC-30245, the steady-state intracellular concentration appeared to have been reached after 3 hours of treatment. Meanwhile, the intracellular concentration of NMS-873 increased between the 3 and 6 hour time points. At 5 μM, the intracellular concentration of UPCDC-30245 was 12-fold higher that of NMS-873, potentially explaining the inconsistency between p97 ATPase inhibition potencies and antiproliferation potencies of UPCDC-30245 and NMS-873. Our results indicated that both compounds appear to be highly cell penetrant, accumulating to millimolar concentrations in cells.
Figure 7.
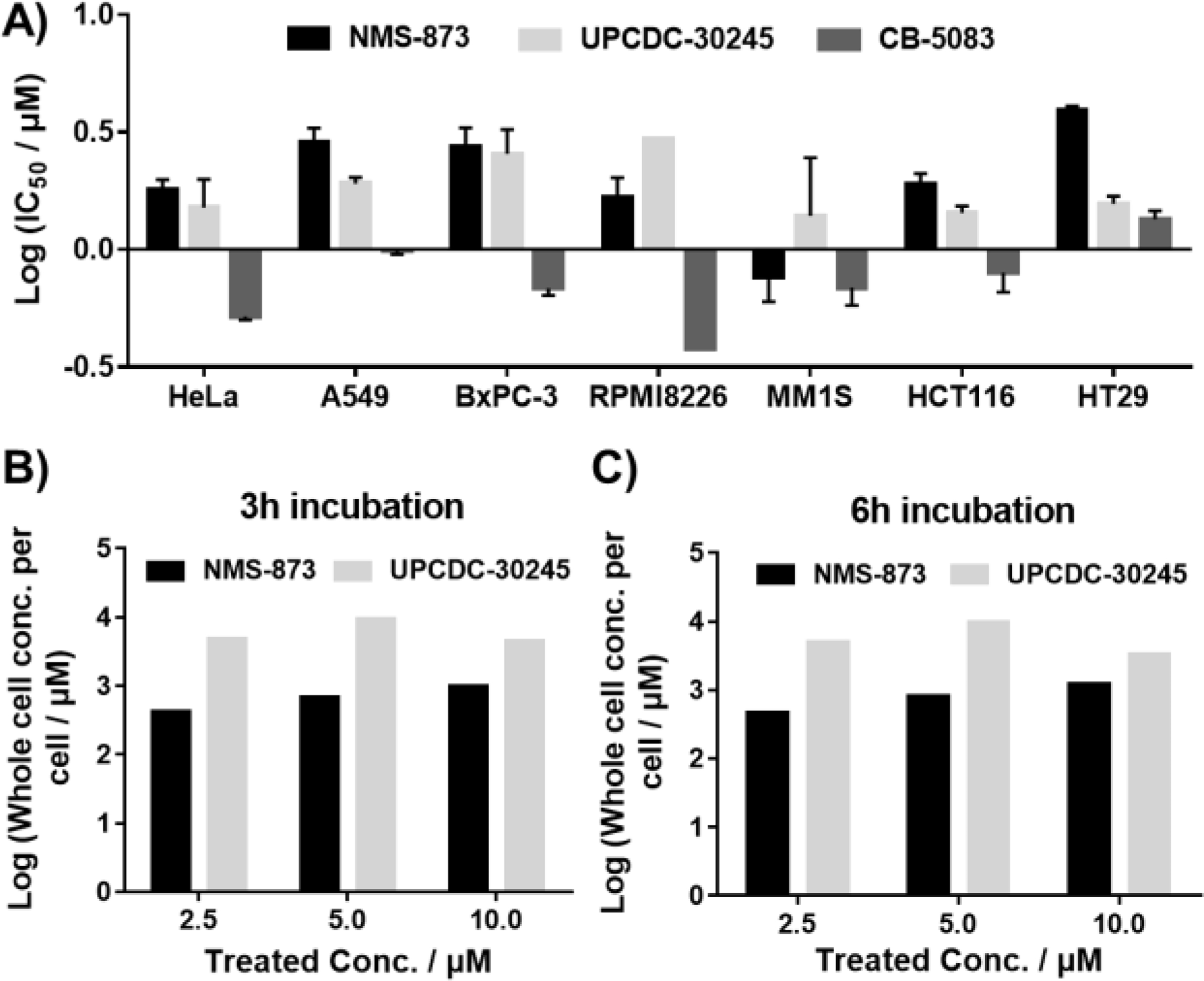
Antiproliferation activity of p97 inhibitors against multiple cancer cell lines (A) and cellular concentrations of NMS-873 and UPCDC-30245 in A549 cells after 3h (B) and 6h (C) treatment. Data are shown as Mean ± SEM taken from triplicate experiments.
5. Cellular effect of allosteric p97 inhibitors in CB-5083 resistant HCT116 colon cancer cell line.
NMS-873 and UPCDC-30245 demonstrated good potency (IC50s of 0.1 – 2 uM, Fig 4) in ATPase assays of the N660K and T688A, CB-5083 resistant mutants and low micromolar IC50s in cancer cell proliferation assays. To test whether they also were effective in inhibiting the proliferation of CB-5083 resistant cancer cells, we established a CB-5083 resistant HCT-116 cell line (CB-R-HCT116) according to published methods [22]. We cloned and sequenced p97 cDNA from this line and identified a double mutant of p97, namely D649A/T688A (Fig. S2). We purified recombinant D649A/T688A and conducted steady-state kinetic analysis. Surprisingly, the specific activity of D649A/T688A was almost undetectable using our typical ATPase assay conditions. By increasing the concentration of D649A/T688A 4-fold and extending the reaction time 4-fold, we were able to obtain a good signal-to-noise ratio. Under these conditions, the IC50 for CB-5083 toward the double mutant D649A/T688A in ATPase assays was about 400-fold greater than for WT p97, while IC50s for UPCDC-30245 and NMS-873 only increased 2-fold and 5-fold respectively (Fig. S3). In cell proliferation experiments, CB-R-HCT116 was over 30-fold less sensitive to CB-5083 than the parental HCT116 cell line. Proliferation of CB-R-HCT116 cells was only slightly inhibited by the presence of 10.5 μM CB-5083 (Fig. 8 A). In contrast, both allosteric p97 inhibitors inhibited the growth of CB-R-HCT116 and parental HCT116 cells with similar IC50s (Fig. 8 B and C). Changes in biomarkers of the UPR response and p97 inhibition were examined by Western blot. After treatment of HCT116 cells with 2.5 μM of CB-5083 for 6 hours, accumulation of K48 ubiquitination, ATF4 and CHOP were observed along with a decrease in p62 protein. These results are consistent with published data [1, 6]. After 6 hours of treatment of CB-R-HCT116 cells with 10 μM CB-5083, no effects on these biomarkers of p97 related activity were observed, consistent with CB-5083’s inability to impair growth of these cells harboring the D649A/T688A double mutant of p97 (Fig. 8 D). After 6 hours of treatment with NMS-873 or UPCDC-30245, both parental HCT116 and CB-R-HCT116 cells displayed similar changes in levels of these biomarkers (Fig. 8 E and F), indicating that their activity toward p97 was unaffected by the mutations. NMS-873 induced changes in the biomarkers are stronger than those observed following treatment with UPCDC-30245 at concentrations where similar effects on viability are measured. This difference suggests that perhaps the effects of UPCDC-30245 on viability might not entirely be mediated through p97 and the UPR. Taken together, our results demonstrate that allosteric p97 inhibitors, such as NMS-873 and UPCDC-30245, could be effective alternatives when resistance occurs in response to CB-5083 cancer therapy.
Figure 8.

Cellular effects of three p97 inhibitors. Antiproliferative effects on parental and CB-5083 resistant HCT116 cell lines for (A) CB-5083, (B) NMS-873, (C) UPCDC-30245. Every point in the dose response curve represents Mean ± SEM taken from four replicates. Western blot analysis following treatment with (D) CB-5083, (E) NMS-873, (F) UPCDC-30245 in parental and CB-5083 resistant HCT116 cell lines. CB-5083 resistant HCT116 (CB-R-HCT116) is unaffected by CB-5083, while it is slightly more sensitive to NMS-873 but similarly sensitive to UPCDC-30245. All western blot experiments were repeated twice and the second blots are shown in Fig. S6.
Conclusions
Targeted therapy for human cancers has significant advantages over conventional therapies in prolonging patient survival while minimizing toxicity [17b]. Kinase inhibitors represent a major advance in this regard. Although kinase inhibitors have been extremely effective in specific patient populations, the accumulating clinical experience suggests that most patients will develop resistance, often caused by mutations in the targeted kinase [23]. The AAA ATPase protein p97 plays an important role in regulating protein homeostasis. And since proteasome inhibitors, which impair protein degradation via blocking proteasome function, have been approved by FDA as anticancer therapies, targeting this pathway via inhibition of p97 is a promising new targeted approach. However, as observed in the clinic with protein kinases, studies with ATP-competitive p97 inhibitors have shown that resistance rapidly arises from mutations in the p97 protein, and thus strategies need to be developed to overcome this resistance.
The hot spots for CB-5083 resistance occur in the D1-D2 linker region and near the ATP binding site in the D2 ATPase domain of p97 [1],[16]. Currently reported ATP-competitive p97 inhibitors, CB-5083, ML240 and ML241, are analogs of DBeQ. CB-5083, ML240 and ML241 are considered D2 domain selective p97 inhibitors and were predicted to share a similar binding mode [7]. By measuring ATPase activity of purified CB-5083 resistant p97 mutants, we confirmed that these three inhibitors all lose inhibitory potency against the mutants. DBeQ inhibits ATPase activity of both D1 and D2 domains and is less affected by the CB-5083 resistant mutations than the other, more selective analogs. The CB-5083 resistant p97 mutants, N660K and T688A, are located near the adenosine binding site for ADP (Figure 6). As CB-5083 binds competitively with ATP to the D2 domain of p97, it is perhaps not surprising that these resistance mutants have arisen in the D2 domain; however, these mutations have a greater effect on the binding of CB-5083 than they do on the ATPase activity. We have demonstrated here that the resistance of CB-5083 to N660K and T688A is due to reduced binding affinity of these mutant proteins for the inhibitor, reminiscent of the drug-resistance mechanisms of ATP-competitive kinase inhibitors. Our results suggest that CB-5083 resistant p97 mutants will likely be resistant to most or all other p97 inhibitors that share the same ATP-competitive binding mode of inhibition.
In contrast, allosteric inhibitors have been successfully developed to overcome drug resistance of clinically approved ATP-competitive kinase inhibitors [12], [13]. To examine whether allosteric p97 inhibitors could be an alternative approach to overcome CB-5083 resistance, we studied two different types of allosteric p97 inhibitors, NMS-873 and UPCDC-30245. Steady-state kinetic experiments demonstrated that these compounds exhibited a different mode of inhibition than CB-5083 and could thus potentially overcome CB-5083 resistance. Our molecular modeling studies with N660K and T688A suggest that conformation changes caused by the loss of a hydrogen-bonding network in the D2 domain may contribute to the slight resistance that we observe toward NMS-873 and UPCDC-30245. Nonetheless, both allosteric p97 inhibitors were still potent in inhibiting the ATPase activities of N660K and T688A.
CB-5083, NMS-873, and UPCDC-30245 were all able to inhibit the proliferation of multiple cancer cell lines, with IC50s ranging from 0.5 to 4 μM. We also found that UPCDC-30245 accumulated to higher concentrations in cells than the other two compounds. For solid tumors, efficacy of anticancer drugs can be limited by the ability of the blood supply to penetrate the extravascular space and to deliver drug to the cancer cells in sufficient concentrations to cause lethal toxicity [24]. The high cell penetration of UPCDC-30245 may be a beneficial property in a candidate for treatment of solid tumors.
To investigate whether CB-5083 resistant cells remain sensitive to allosteric p97 inhibitors, we generated a CB-5083 resistant HCT116 cell line, CB-R-HCT116, that is 127-fold less sensitive to CB-5083 than parental HCT116 cells. cDNA sequencing identified a p97 gene with two missense mutations, D649A/T688A. ATPase activity assays on the purified p97 double mutant showed that it was ~400-fold resistant to CB-5083. Resistance to cell-killing by CB-5083 correlated with a lack of response in p97 biomarkers. Due to the important role of p97 in ERAD, inhibition of p97 causes ER stress and leads to the accumulation of ubiquitinated proteins. Parental HCT116 cells treated with CB-5083 exhibit increased levels of ER stress biomarkers (ATF4 and CHOP), as well as accumulation of proteins ubiquitinated on lysine48 (K48). However, there was no activation of ER stress biomarkers when CB-R-HCT116 was treated with CB-5083. These results indicated that the resistance of CB-R-HCT116 to killing by CB-5083 was caused by the presence of the p97 mutations and the consequent absence of ER stress. As discussed above, both NMS-873 and UPCDC-30245 retain potency toward previously known CB-5083 resistant p97 mutants. Here we confirmed that these two allosteric p97 inhibitors also retain more activity toward our newly-identified double mutant p97 D649A/T688A than does CB-5083, and that they exhibited the same potency in inhibiting the proliferation of CB-R-HCT116 cells as parental HCT116 cells. The findings here support allosteric p97 inhibitors as promising alternatives to overcome resistance to ATP-competitive p97 inhibitors, reminiscent of the drug-resistant mechanisms well chronicled for kinase inhibitors in anticancer therapy.
Experimental Section
Generation of CB-5083 resistant cell line.
A CB-5083 resistant HCT116 cell line was derived using the method described by McDermott et al [25] with certain modifications. Briefly, 105 cells were plated into a 10 cm plate and cultured in the presence of 1.25 μM or 2.5 μM of CB-5083 (Active Biochem) until drug resistant clones were visible. Drug resistant clones were transferred into a new plate for passaging, and subsequently used to study drug resistance and identify the p97 mutations.
Intracellular compound concentration determination.
The intracellular concentration of NMS-873 (Xcess Biosciences Inc.) and UPCDC-30245 (Millipore Sigma or University of Pittsburgh Chemical Diversity Center) was measured according to Mateus et al[21] with modification. Generally, 20,000 A549 cells per well in 90 μL of DMEM with 2.5% FBS and 1% Penicillin/Streptomycin were plated in a 96 well plate. After 24 hours, 10 μL of 10x compound stock was added into each well (final DMSO concentration is 1%) and incubated for 3h or 6h. Medium was removed, cells were washed twice with ice-cold DPBS, solubilized in 50 μL acetonitrile and shaken at 800 rpm for 60 min at room temperature. After centrifugation of the 96 well plate at 4000 rpm for 15 min, supernatant was transferred into a fresh plate and mixed with internal reference Verapamil at 1:1 ratio. The mixtures were injected into LC/MS/MS to measure compound concentration. A Shimadzu LC-20AD HPLC system was coupled to an AB Sciex API5000 mass spectrometer. A Hypersil Gold column (50 × 2.1 mm, 5 μm, Thermo Fisher) was used to separate NMS-873 treated samples and Atlantis T3 column (50 × 4.6 mm, 5 μm, Waters) was used to separate UPCDC-30245 treated samples, and the appropriate positive MS/MS transition was used to detect the analyze response. The detailed HPLC conditions are supplied in Table S3 and S4. Compound concentration in cells was determined by counting the number of cells and assuming 1670 μm3 /cell volume. Standard curves results for compounds were provided in Figure S7, and the preparation method for the standard curves also described in the supplemental file.
Thermal Shift assay.
Protein Thermal Shift assays were performed on a QuantStudio 5 Real-Time PCR System with Protein Thermal Shift™ Dye Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. WT and mutant p97 were diluted to 1 μM with 1x ATPase assay buffer (50 mM Tris pH 7.4, 20 mM MgCl2, 1 mM EDTA, 0.5 mM TCEP, 0.01% Triton X-100). p97 inhibitors were titrated against 1 μM of p97 in three-fold steps from 30 μM to 40 nM. Assays were performed in MicroAmp™ EnduraPlate™ Optical 384-Well Blue Reaction Plates with Barcode (Thermo, Cat# 4483320) in four replicates. Data were analyzed using Protein Thermal Shift™ Software.
The experimental details of protein purification, ATPase activity, cell viability, RNA extraction, molecular cloning and sequencing, western blot, MST assay, and protein structure modelling are available in the supplementary information.
Supplementary Material
Acknowledgments
We thank the University of Kansas Specialized Chemistry Center for providing DeBQ, ML240, ML241 and University of Pittsburgh Chemical Diversity Center for providing UPCDC-30245. We thank Jingying Xu for helping on maintaining the CB-5083 resistant line, Xiaoyi Zhang and Lin Gui for assistance with preparing samples to determine intracellular compound concentration, and Paul Sternberg for editorial suggestions. We thank Quintara Discovery for LC-MS/MS analysis. We thank Neal Green for helpful suggestion. The project was supported in part with funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E, through the NExT Chemical Biology Consortium.
Footnotes
Supporting information for this article can be found under: https://doi.org/10.1002/cmdc.201900722.
References:
- [1].Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M, Cancer Cell 2015, 28(5), 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Yamamoto S, Tomita Y, Hoshida Y, Iizuka N, Monden M, Yamamoto S, Iuchi K, Aozasa K, Annals of surgical oncology 2004, 11(7), 697–704; [DOI] [PubMed] [Google Scholar]; b) Yamamoto S, Tomita Y, Hoshida Y, Takiguchi S, Fujiwara Y, Yasuda T, Yano M, Nakamori S, Sakon M, Monden M, Aozasa K, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2003, 21(13), 2537–2544; [DOI] [PubMed] [Google Scholar]; c) Yamamoto S, Tomita Y, Nakamori S, Hoshida Y, Iizuka N, Okami J, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H, Aozasa K, Monden M, Oncology 2004, 66(6), 468–475; [DOI] [PubMed] [Google Scholar]; d) Yamamoto S, Tomita Y, Nakamori S, Hoshida Y, Nagano H, Dono K, Umeshita K, Sakon M, Monden M, Aozasa K, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2003, 21(3), 447–452. [DOI] [PubMed] [Google Scholar]
- [3].Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, Lux V, Ehrmann M, Weihl CC, Meyer H, EMBO J 2017, 36(2), 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang Q, Song C, Yang X, Li C-CH, Journal of Biological Chemistry 2003, 278(35), 32784–32793. [DOI] [PubMed] [Google Scholar]
- [5].Tang WK, Xia D, Frontiers in molecular biosciences 2016, 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chou T-F, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Proceedings of the National Academy of Sciences 2011, 108(12), 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ, ChemMedChem 2013, 8(2), 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chou T-F, Deshaies RJ, Autophagy 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, Science 2016, 351(6275), 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tang WK, Odzorig T, Jin W, Xia D, Molecular pharmacology 2019, 95(3), 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Her NG, Toth JI, Ma CT, Wei Y, Motamedchaboki K, Sergienko E, Petroski MD, Cell Chem Biol 2016, 23(4), 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS, Nature 2010, 463(7280), 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Converso A, Hartingh T, Garbaccio RM, Tasber E, Rickert K, Fraley ME, Yan Y, Kreatsoulas C, Stirdivant S, Drakas B, Bioorganic & medicinal chemistry letters 2009, 19(4), 1240–1244. [DOI] [PubMed] [Google Scholar]
- [14].Magnaghi P, et al. and Isacchi A, Nat. Chem. Biol. 2013, 9(9), 548–556. [DOI] [PubMed] [Google Scholar]
- [15].a) Alverez C, Arkin MR, Bulfer SL, Colombo R, Kovaliov M, LaPorte MG, Lim C, Liang M, Moore WJ, Neitz RJ, Yan Y, Yue Z, Huryn DM, Wipf P, ACS medicinal chemistry letters 2015, 6(12), 1225–1230; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) LaPorte MG, Burnett JC, Colombo R, Bulfer SL, Alverez C, Chou T-F, Neitz RJ, Green N, Moore WJ, Yue Z, ACS medicinal chemistry letters 2018, 9(11), 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bastola P, Wang F, Schaich MA, Gan T, Freudenthal BD, Chou TF, Chien J, Cell death discovery 2017, 3, 17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ, Proceedings of the National Academy of Sciences of the United States of America 2008, 105(6), 2070–2075; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ellis LM, Hicklin DJ, Clinical cancer research : an official journal of the American Association for Cancer Research 2009, 15(24), 7471–7478. [DOI] [PubMed] [Google Scholar]
- [18].Grøftehauge MK, Hajizadeh NR, Swann MJ, Pohl E, Acta Crystallographica Section D: Biological Crystallography 2015, 71(1), 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chou T-F, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, Schoenen FJ, Lin HJ, Deshaies RJ, Arkin MR, Journal of molecular biology 2014, 426(15), 2886–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A, Donati D, O’Brien T, Jackson PK, Isacchi A, Nat Chem Biol 2013, 9(9), 548–556. [DOI] [PubMed] [Google Scholar]
- [21].Mateus A, Matsson P, Artursson P, Molecular pharmaceutics 2013, 10(6), 2467–2478. [DOI] [PubMed] [Google Scholar]
- [22].McDermott M, Eustace AJ, Busschots S, Breen L, Crown J, Clynes M, O’Donovan N, Stordal B, Frontiers in oncology 2014, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, Edeen PT, Proceedings of the National Academy of Sciences 2005, 102(31), 11011–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ, Clinical cancer research 2002, 8(3), 878–884. [PubMed] [Google Scholar]
- [25].McDermott M, Eustace A, Busschots S, Breen L, Clynes M, O’Donovan N, Stordal B, Frontiers in oncology 2014, 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.