

Abstract
Hydrogels demonstrate superior properties that favor wound healing and have been widely used in clinical settings for wound dressing applications. However, commercial hydrogel dressings often lack flexibility/adhesiveness, and do not conform well to the irregular skin surfaces of complex wounds and/or wounds near joints. As a result, the wound is likely to be exposed to potential bacterial invasion. Herein, we designed and developed a mechanically flexible and self-adhesive polyurethane-poly(acrylamide) (PU-PAAm) hydrogel for wound healing applications. The hydrogel can be cured from a novel waterborne emulsion within 90 s under UV irradiation. The PU component within the PU-PAAm hydrogel plays a “bridging” role that accelerates the formation of an interpenetrating polymer network (IPN), which consists of a physically crosslinked PU network trapped within a chemically crosslinked PAAm network. The unique IPN structure endowes the hydrogel with superior stretchability and ductility. The hydrogen bonding formation and electrostatic interaction between the hydrogel and skin ensure strong adhesion without causing irritation to skin upon dressing removal. Animal studies further confirmed the PU-PAAm hydrogel's remarkable skin regeneration capability. This work shows our new hydrogel holds a promising prospect for treatment of complicated or challenging wounds such as burns and chronic wounds.
A fast UV-curable, mechanical flexible and self-adhesive PU-PAAm hydrogel has been prepared for clinical wound management.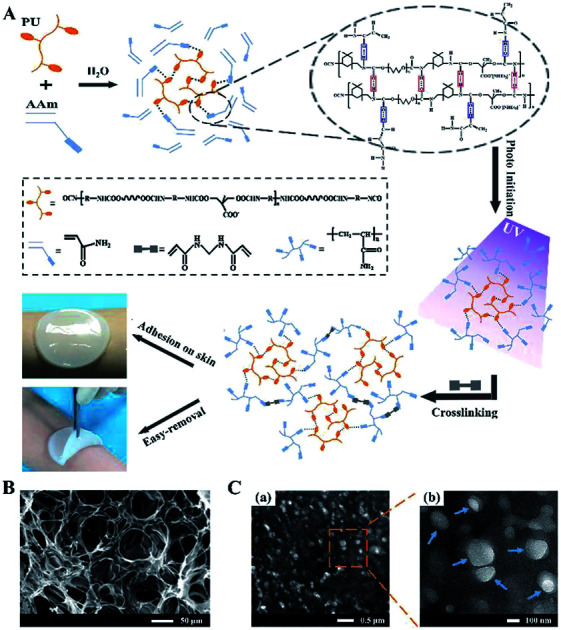
1. Introduction
Wound healing is a critical process for restoring the barrier function of skin. If damaged skin is not healed properly, a chronic wound may develop, which will not only compromise the patient's quality of life but also burden the social healthcare systems.1 It is reported that chronic wounds affect around 6.5 million patients in US alone,2 while out of 265 000 deaths occur every year due to burns, a majority are down to inappropriate wound treatment.3 As such, advanced wound care products are in great demand for more effective treatments at lower cost, given the longstanding challenges in wound managements (such as chronic wounds and burns), and the ever increasing cosmetic expectations from the wounded patients.
Traditional wound dressings made of textile fibers are often infiltrated by wound discharge and newly-formed soft tissues, and their difficulty in removal usually causes secondary skin damage.4–6 In recent years, hydrogels have attracted increasing attention for wound management/tissue generation applications. Their skin mimicking properties and high-water retention offer an environment with desirable moisture and temperature suitable for wound healing.7,8 Currently, hydrogels as wound dressings, have been extensively developed for treating skin injuries like burns, bruises, ulcers and so on. However, due to the lack of flexibility/adhesiveness, existing commercial hydrogels do not conform well to uneven, curved or folded skin surfaces in the case of complex wounds and/or wounds near joints (wrists, elbows, ankles, etc.). As a result, the wound is likely to be exposed to potential bacterial invasion, which will lead to hindered wound healing or even deteriorated wound conditions.9
To address this problem, much efforts have been made to endow hydrogels with self-adhesive property (i.e., hydrogel which can be applied directly onto wounded skin without need of additional adhesive) based on chemical bonding or physical interactions to extend their applications in wound dressing.10–12 Recently, bioinspired strategies based on catechol and its analogues have drawn much attention for developing such hydrogel.13,14 Unfortunately catechol can easily oxidize in air and lose its adhesiveness dramatically.15 Furthermore, time-consuming molding process and poor mechanical properties of such adhesive hydrogels often lead to high cost and impaired performance, which are the stumbling blocks for their practical applications.16 As such, developing a fast-curing hydrogel with desirable self-adhesive and mechanical properties as wound dressing is of a great interest.
PAAm is a wildly used polymer with excellent hydrophilicity, mechanical properties, and biocompatibility. PAAm can be synthesized by free radical polymerization of acrylamide (AAm) monomer with the aid of photo-initiators under UV-irradiation.17–19 Polyurethane (PU) possesses excellent mechanical properties, biocompatibility, anti-thrombosis properties, and has been widely used in biomedical applications.20–22 Waterborne PU contains a large number of functional groups (–COONH– and –COO−), which make it easily functionalized when interacting with other chemicals.23–25
In this work, we developed a fast UV-curing, mechanically flexible and self-adhesive PU-PAAm hydrogel as wound dressing. The presence of PU facilitated the crosslinking of an interpenetrating polymer network (IPN), in which a physically cross-linked PU network was trapped within a chemically crosslinked PAAm network. This in turn enhanced the IPN mechanical properties and accelerated its curing time (within 90 s) under UV irradiation. In addition, strong self-adhesion of the hydrogel has been achieved through hydrogen bonding and electrostatic interaction between the hydrogel and the skin. Furthermore, the excellent tissue adhesiveness and cell affinity of the hydrogel ensured its firm contact with the surrounding tissues and promotion of the skin regeneration.
2. Experimental section
2.1. Materials
Isophorone diisocyanate (IPDI), poly(tetramethylene ether glycol) (PTMG, Mw 2000), 2,2-dimethylol propionic acid (DMPA), N,N-dimethylformamide (DMF), were purchased from Shanghai Aladdin Co. Ltd., China. 2-Hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone (Irgacure 2959), acrylamide (AAm), N,N′-methylenebisacrylamide (MBA), and triethylamine (TEA) were purchased from Shandong Xiya Chemical Industry Co., Ltd., China. Dimethyl sulfoxide-d6 was obtained from Cambridge Isotope Laboratories, Lnc., USA.
2.2. Synthesis of PU emulsion
PU emulsion was prepared following procedure detailed in previous report.26 More specifically, PTMG-2000 (24.00 g, 0.012 mol) was first mixed with IPDI (13.34 g, 0.060 mol) and the mixture was stirred for 2 h under nitrogen atmosphere at 80 °C to obtain the precursor. After cooling to 65 °C, 2.4 g (0.018 mol) DMPA pre-dissolved in 8 ml DMF (serve as chain-extender) was added to the pre-polymer and stirred for 2 h. TEA (2.02 g, 0.02 mol) was then added into the mixture and stirred at room temperature for 10 min to neutralize the excessive carboxyl groups. Finally, PU emulsion (70% (w/w) water) was obtained by adding distilled water into the mixture followed by 1 h stirring. Fig. S1† provides further details on the associated chemical reaction process.
2.3. Preparation of PAAm hydrogels
PAAm hydrogel was prepared by the one-pot method.27 50% (w/w), 30% (w/w) and 10% (w/w) AAm solutions were obtained by dissolving appropriate amount of monomer in distilled water. MBA and Irgacure 2959 (0.03% (w/w) and 5% (w/w), respectively) were then introduced to the AAm solutions and stirred for 1 h at room temperature. The mixture was then transferred to glass molds (Φ × h = 70 mm × 3 mm) to form PAAm hydrogels with different solid content under UV irradiation (365 nm wavelength). The photoinitiated-reaction and crosslinking processes are described in Fig. S2.†
2.4. Preparation of PU-PAAm IPN hydrogels
PU-PAAm interpenetrating polymer network (IPN) hydrogels were prepared by the latex IPN method.28 First, AAm monomer was dissolved in deionized water to make AAm solutions with three different concentrations (50% (w/w), 30% (w/w), 10% (w/w) solid content). This was followed by adding PU emulsion synthesized in part 2.2. The mixture was stirred for 1 h at room temperature to obtain PU-AAm mixtures with 50% (w/w), 30% (w/w) and 10% (w/w) solid content, respectively. After that, MBA and Irgacure 2959 (0.03% (w/w) and 5% (w/w)) were added into the above mixtures followed by 0.5 h stirring. Finally, PU-PAAm hydrogels with IPN structure were obtained by ultraviolet (UV) irradiation.
PU-AAm emulsions with 10% (w/w), 30% (w/w) and 50% (w/w) solid contents were named as PA-10, PA-30 and PA-50, respectively; PAAm hydrogels with 50% (w/w), 30% (w/w) and 10% (w/w) solid contents, were named as P-50, P-30 and P-10, respectively; and PU-PAAm hydrogels with 50% (w/w), 30% (w/w) and 10% (w/w) solid contents were named as PP-50, PP-30 and PP-10, respectively.
2.5. Characterization
Lyophilized PAAm and PU-PAAm hydrogels were characterized by Fourier transform infrared spectrometer (FT-IR, Nicolet 6700, Thermo Scientific, USA) for their chemical composition. The thermal properties of PAAm and PU-PAAm hydrogels were analysed by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) at 10 °C min−1 scan rate from 25 to 600 °C under nitrogen atmosphere (DSC, STA449F3, NETZSCH, Germany). Lyophilized PAAm and PU-PAAm hydrogels before and after swelling experiment were observed using scanning electron microscopy (SEM, JSM-6500LV, JEOL, Japan). Transmission electron microscope (TEM, Tecnai G2 F20 S-TWIN, FEI, USA) was used to further analyse the microstructure of the hydrogels. The particle size distribution (PSD) in AAm solutions, PU and PU-AAm emulsions with different solid contents were characterized by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Instruments Ltd., UK). 1H-NMR (NMR, Bruker AV 400 MHz, Bruker, Switzerland) was used to analyze the spatial configuration of organic molecules within the polymers (dissolved in di-methyl-d6 sulfoxide (DMSO-d6)). The viscoelastic properties of PU-AAm emulsions, PAAm and PU-PAAm hydrogels were characterized by rotary rheometer (DHR-2, TA Instruments Ltd., USA). All measurements were performed using flat steel plate geometry (Φ = 30 mm) at 25 °C with a gap distance of 1000 μm. The viscosity of PU-AAm emulsions with different solid contents were measured using oscillatory time sweep mode for 5 min (1% strain, 1 Hz frequency). Also, oscillatory time sweep mode was used to determine the storage modulus (G′), viscous modulus (G′′) and loss factor (tan(δ)) of PAAm and PU-PAAm hydrogels. Frequency sweep tests were carried out over a frequency range from 0.1 to 100 Hz (strain 1%) to explore the stability of the internal structure of PAAm and PU-PAAm hydrogels.
2.6. Swelling experiments
PAAm and PU-PAAm hydrogels were molded into cylinders for testing their swelling properties using gravimetric methods.29 More specifically, samples in triplicate were submerged in PBS solution for a given time, then their weights were measured after removing the excessive surface water. Mass swelling ratio were calculated according to eqn (1).
 |
1 |
where, W1 is the weight of the initial hydrogel, and W2 is the weight of the swollen hydrogel.
2.7. Mechanical testing
Dumbbell shaped PAAm and PU-PAAm hydrogel samples (l × w × h = 75 mm × 4 mm × 2 mm) were tested using a universal testing machine (AGIC 50 KN, Shimadzu, Japan). The samples were stretched at a speed of 50 mm min−1 under a tensile load of 10 N for tensile tests. Cyclic tensile tests (3 cycles) were performed by stretching samples to 5 times of its initial length at a speed of 50 mm min−1, then unloaded to 0 N at the same speed. Silicone oil was applied to the surface of samples to prevent moisture evaporation, and all tests were repeated three times.
2.8. Adhesive property of PU-PAAm hydrogel
Lap-shear tensile stress measurements of PU-PAAm hydrogels were performed using universal test machine (AGIC 50 KN, Shimadzu, Japan). Glass slide and porcine skin (l × w = 75 mm × 25 mm) were chosen as adhesion substrates in the test. Square PU-PAAm hydrogels (l × w = 25 mm × 20 mm) were adhered between two substrates. The sandwiched structure was fixed to the clamps and sheared at a rate of 5 mm min−1. The maximum adhesive strength of PU-PAAm hydrogels was calculated according to force–displacement curves.
2.9. In vitro toxicity test
L929 fibroblasts (Cell Bank, Chinese Academy of Sciences, Shanghai, China) were used to evaluate the cell affinity of the hydrogels. Sterilized hydrogels were cocultured with L929 (5 × 103 cells per well) in 24-well plates, and the cells were allowed to adhere and grow for 1, 3 and 5 days. The morphologies of the cells adhered on hydrogels were observed using a fluorescence microscopy (TE 2000-U, Nikon Eclipse, Japan). The cell proliferation of L929 cultured on hydrogels were assessed by the MTT assay.30
2.10. In vivo animal study
Full skin wounds experiments
Animal experiment was conducted following the standards established by Animal Research Committee of the State Key Laboratory of Oral Diseases and West China School of Stomatology, Sichuan University (approval number: WCHSIRB-D-2018-003). 24 New Zealand white rabbits were used in animal experiments under general anesthesia. After shaving and sterilization by povidone-iodine (Zoletil, Vibarc, German), 15 mm × 15 mm full-thickness skin defects were created and covered by hydrogel. Penicillin was injected for infection prevention. Photos were taken every three days to record the change of the defect regions. More details of animal experiments can be found in Fig. S3.†
Histological observation
Immediately after the animal sacrifice, the skin defect samples were fixed in 4% neutral formalin buffer, decalcified with orthophosphoric acid, and embedded in paraffin (Technovit 7200 VCL, Exact Apparatebau, Norderstedt, Germany). The skin was sectioned, ground into 10 μm-thick slices using a SP1600 rotary diamond saw (Leica, Nussloch, Germany), and then stained with hematoxylin-eosin (H&E) and Masson' Trichrome staining solution. Histomorphometry was performed using a semi-automated digitizing image system, which consists of an Eclipse E600 stereomicroscope, a DXM1200 digital camera (Nikon, Tokyo, Japan), and NIS-Elements F2.20 image software.
2.11. Statistical analysis
Quantitative data were presented as the means ± standard deviation (SD). Statistical significance was determined using SPASS (v. 10.0) software. p < 0.05 was considered to be statistically significant.
3. Results and discussion
The formation mechanism of PU-PAAm hydrogel is illustrated in Fig. 1A. Upon physical mixing in the aqueous solution, the PU molecular chains were linked to the AAm chains through hydrogen bonding. The PU-AAm precursor can be cured within 90 s under irradiation of UV light to form a hydrogel membrane and self-adhere to the skin surface. SEM image (Fig. 1B) shows that the PU-PAAm hydrogel displays a 3D network structure with highly interconnected pores, and its IPN morphology was further confirmed by TEM images in Fig. 1C. Here, PU globules were uniformly dispersed in the continuous PAAm phase (Fig. 1C-a). The magnified image in Fig. 1C-b highlights the blurred boundaries between PU globules and the PAAm matrix, which correlate to the interpenetration of PU and PAAm molecular chains.28,31
Fig. 1. The formation mechanism and microstructures of PU-PAAm hydrogel. (A) Schematic illustration of the formation of PU-PAAm self-adhesive hydrogel and its potential application as sprayable wound dressing; (B) SEM image of PU-PAAm hydrogel; (C) TEM images of PU-PAAm hydrogel under different magnifications, arrows denote the PU globules.

3.1. Characterization of PU-PAAm hydrogel and its fast-curing mechanism
The non-covalent interaction, i.e., hydrogel bonding, between PU and AAm in the emulsion phase has been further confirmed by NMR, PSD analysis and the viscosity tests. As shown in the 1H NMR spectra in Fig. 2A-a, the peaks in the range of 5.57 to 5.60 and 6.15 to 6.22 ppm can be ascribed to H in CH2 groups (H1, H2), the peak around 6.08 ppm represents the CH– groups (H3), while the peaks at 7.07 and 7.51 ppm can be assigned to the H in –NH2 groups (H4, H5) of AAm.
Fig. 2. Characterization of the PU-AAm emulsion. (A) 1H NMR spectra (DMSO, 400 MHz, 25 °C) of AAm (a), PU (b) and PU-AAm emulsion (c); (B) ROESY spectrum of the PU-AAm emulsion; (C) particle-size distribution of PU-AAm emulsions (pure PU represents PU emulsion with 30% (w/w) solid content); (D) UV curing time of different hydrogels (n = 3); (E) FT-IR spectra of PU film, PAAm and PU-PAAm hydrogels.

In addition, the peak at 7.95 ppm (Hf, Hi) has been attributed to the –COONH– groups of PU, resulted from the reaction between –OH and –NCO groups (Fig. 2A-b). In comparison, the two peaks originally ascribed to –NH2 groups (7.51 and 7.07 ppm) for the PU-AAm emulsion mixture (i.e., the precursor of PU-PAAm hydrogel), have shifted to 7.62 and 7.11 ppm respectively, indicating the occurrence of non-covalent interaction between AAm monomer and PU chains upon mixing (Fig. 2A-c). The formation is further confirmed by 2D ROESY displayed in Fig. 2B, where Hf and Hi are close to H1 and H3 in 2D space. This can be associated to hydrogen bonds between CH2 and/or CH– groups of AAm and –COONH– groups of PU.
Fig. S4† shows that the particle size distribution of AAm solutions with varying solid contents is within the range of 0.5–3 nm, indicating the AAm concentration has no significant influence on the particle size of dispersoids. Fig. 2C shows that the mean particle size of pure PU (30% (w/w) solid content) is 37 nm, whereas that of the PU-AAm emulsion with the same solid content (PA-30) has increased to 54 nm. This could be due to the newly formed larger PU-AAm particles. The PSD of PU-AAm emulsions tends to increase with increasing solid content, indicating the increasing interaction between AAm and PU. The curing behavior of AAm solutions and PU-AAm emulsions under UV irradiation was evaluated by the inverted vial method, see Fig. S5.† Gelation for both AAm solutions and PU-AAm emulsions occurs upon exposure to UV light, and the curing time depends on the solid concentration, see Fig. 2D. PAAm hydrogel can be obtained by addition of AAm monomer and chemical crosslinking, and the process takes 350–420 s. Whereas when PU is added, the entangled PU chains can serve as physical cross-linking agents (i.e., “bridges”), leading to accelerated formation of PU-PAAm hydrogels (60–90 s). The recorded curing time is much faster than most existing photocurable PAAm based hydrogels (Table S1†). Fig. 2E shows FT-IR spectra of PU, PAAm and PAAm-PU hydrogels. It can be seen that the absorption peak of –NCO group in IPDI at 2263 cm−1 disappeared in PU, while the characteristic absorption peaks associated to urethane (–NHCOO–) groups of PU were found at 1542 cm−1 (C–N bending vibration), 1252 cm−1 (C–O stretching vibration), 1722 and 1665 cm−1 (C O stretching vibration) in the IR spectrum. Also, the characteristic peaks of PAAm emerged at 3348 cm−1 and 3201 cm−1, which can be assigned to N–H stretching vibration. The peaks at 1662, 1620, 1252 cm−1 can be associated with C O stretching, N–H bending, C–N stretching vibration of –CONH, respectively. For PU-PAAm hydrogels, the characteristic peaks corresponding to PU and PAAm were all present without any new adsorption, suggesting no formation of new substances in these hydrogels. Furthermore, the broadened and shifted C O band at 1660 cm−1 was due to the combined stretching vibrations of the C O groups within both PAAm (1662 cm−1) and PU (1722 cm−1). The intensity of this peak also decreased compared with that of PAAm, further confirming the presence of hydrogen bonding within the IPN structure.32,33
3.2. Rheological study of PU-PAAm hydrogel
Rheological tests have been carried out for the UV cured hydrogel samples. Fig. 3A shows that elastic modulus (G′) for both PAAm and PAAm-PU hydrogels increased with increasing solid content, suggesting a more compact cross-linked network with less water. Compared to PAAm, G′ of PAAm-PU with the same solid content decreased slightly, which may be due to the different network structure with PU chains being the flexible segments. Fig. 3B shows that the loss factor (tan(δ)) values for all cured hydrogel samples were below 1. The fact that the elastic modulus (G′) is always higher than the viscous modulus (G′′) suggests the materials' gel-like characteristic. G′ of PAAm-PU hydrogels as a function of curing time is plotted in Fig. 3C. It can be seen that G′ of all groups increased markedly over the curing period 45 s to 90 s, after that, G′ remained almost constant, indicating the formation of a stable network structure in PAAm-PU hydrogels under UV irradiation in 90 s. The maximum of G′ (up to 26.4 kPa) was seen for PP-50 after 90 s of UV irradiation, the value is higher than other existing wound dressing hydrogels.34,35 The network stability of PU-PAAm was further inspected by frequency sweep analysis. Fig. 3D shows the drastic decrease of G′ in PP-10 at 10 Hz (even lower than G′′), which indicates that damage occurred within the more fragile network structure. In contrast, G′ for PP-50 and PP-30 remained stable in the 0.1–100 Hz range, indicating that the hydrogel behaved like a viscoelastic solid suitable for wound dressing.
Fig. 3. Characterization of the PAAm and PU-PAAm hydrogels. (A) Elastic modulus–time curves of hydrogels with different solid contents; (B) tan(δ) of PAAm and PU-PAAm hydrogels in the time sweep mode; (C) elastic modulus (G′) of PU-PAAm hydrogels as a function of curing time (n = 3); (D) elastic modulus (G′) and viscous modulus (G′′) as a function of test frequency.

3.3. Mechanical properties of PU-PAAm hydrogel
Stretchability and notch-insensitivity of hydrogels are important characters for wound dressing applications. The incorporation of PU has remarkably enhanced the stretchability of PP-50 and PP-30. In particular, the elongation at break for PP-30 is as high as 2560% under 0.40 MPa tensile stress, far exceeding that of P-50 and P-30 (Fig. 4A). Such high stretchability can be attributed to the extension of the entangled and interpenetrated molecular chains under tensile stress.
Fig. 4. Mechanical properties of PAAm and PU-PAAm hydrogels. (A) Tensile stress–strain curves of different PAAm and PU-PAAm hydrogels; (B) loading–unloading cycle curves of P-30; (C) loading–unloading cycle curves of PP-30; (D) tensile experiment of knotted P-30 and PP-30; (E) notch sensitivity test of P-30 and PP-30.

SEM images (Fig. S6†) show that both PAAm and PU-PAAm hydrogels feature highly porous interconnected network structure, with the struts in PP-50 and PP-30 thicker than those in P-50 and P-30, further indicating a more robust network formation in the biphasic hydrogels. DSC thermal analysis (Fig. S7†) shows that the glass transition temperature (Tg) of PP-50 and PP-30 was lower than that of P-50 and P-30, which can be attributed to the bonding between PU and PAAm in the IPN structure.31 TEM images in Fig. S8† shows that interpenetrated PU particles in PP-30 were more evenly dispersed within the continuous PAAm phase as compared to PP-50, this may lead to better material integrity and hence better mechanical properties. The cyclic loading curves in Fig. 4B and C show that PP-30 had very little residual strain after several repeated cycles, indicating its potential fatigue resistance under cyclic loading. P-30 however, displays a bigger hysteresis loop (Fig. 4B), which may be due to the greater internal friction between the PAAm chains. While in PAAm the accumulated energy dissipated through irreversible chemical bond breakage (and subsequently the destruction of the network), in PU-PAAm hydrogel, reversible hydrogen bonds breaking and reformation were responsible for the energy dissipation.36 This has offered PP-30 greater stretchability and effectively protected the network structure from damage.
More specifically, Fig. 4D shows that PP-30 can be stretched to 15 times of its original length after being knotted, much better than P-30. Besides, centre-notched (Φ = 5 mm) PP-30 can be stretched to 10 times of its original length without fracture (Fig. 4E), while the notched P-30 failed at 5 times of its original length. These results further support that PU can effectively absorb and dissipate the elastic energy accumulated at the cracks tips initiated within the IPN structure.
3.4. Swelling and adhesive properties of PU-PAAm hydrogel
The mass swelling ratio of PAAm and PU-PAAm hydrogels was determined during 28 days of incubation in PBS at 37 °C. The curves of swelling ratio in Fig. 5A show that all hydrogels swell rapidly in the first 72 h, after which the swelling slows down and finally reaches saturation after 7 days. Due to the synergistic effect of the hydrophilic PAAm and PU, the swelling ratio of PU-PAAm is slightly higher than that of the PAAm hydrogel with the same solid content. The maximum mass swelling ratios of P-30, P-50, PP-30 and PP-50 are about 454%, 582%, 476% and 598%, respectively. In addition, upon swelling, PU-PAAm hydrogels still maintain their structural integrity with uniformed pore distribution, which can be seen from the SEM images in Fig. S9.† The above results suggest the potential of our hydrogel in absorbing exudate in wound dressing applications.
Fig. 5. Swelling performance and adhesive properties of PU-PAAm hydrogels. (A) Mass swelling curves of PAAm and PU-PAAm hydrogels; (B) adhesion force–displacement curve measured from glass substrate and porcine skin; (C) maximum adhesive strength of PP-50 and PP-30 adhered to glass slide and porcine skin (**p < 0.01); (D) PU-PAAm hydrogels adhered on (a) glass slide and (b) human skin with a hanging weight (100 g); (E) PU-PAAm adhesion experiments in air and under water; (F) conformability of PP-30 to human skin at different angles.

To investigate the self-adhesion property of our hydrogels, fresh porcine skins and glass slides were selected as representative adhesion substrates, and lap shear measurement was carried out using a universal test machine (UTM) under ambient conditions, see Fig. S10.† The force–displacement curves in Fig. 5B show that for both types of substrates, the displacement against the maximum stretching force was significantly larger for PP-30 than that of PP-50.
The stronger adhesion of PP-30 may be attributed to the more uniform distribution of smaller PU particles within the IPN (see Fig. S8†), that is, the greater specific surface area of PU. This would allow formation of more hydrogen bonds between –COONH– and –COO– groups. In Fig. 5C, the adhesion strength (lap shear stress) for PP-30 on glass slide and porcine skin exceeded 16 and 12 kPa, respectively, which was comparable to the performance of the reported PDA-PAAm hydrogel34 and typical commercial wound dressings (such as Evicel, Coseal).37 Furthermore, the strong adhesion between the hydrogel and the substrates were retained under various harsh conditions, such as under a 100 g hanging weight (Fig. 5D), under vigorous water flushing (Fig. S11†), or under repeated bending/twisting in water or in air (Fig. 5E). In addition, PP-30 can adhere well to human skin at any angle (Fig. 5F). The strong adhesion should be attributed to the existing –COONH– and –COO− groups in PU, which could form hydrogen bonding or interact electrostatically with the collagen in skin tissue,38,39 see mechanistic model illustrated in Fig. S12.†
3.5. In vitro cell behaviors & in vivo animal study
An ideal wound dressing should be mechanically robust while providing a moist/aseptic and breathable environment for effective extrudate absorption. The dressing should also be biocompatible, gentle to the skin and preferably be able to promote the healing process. Here, we carried out in vitro and in vivo experiments to investigate the biological properties of the hydrogels as wound dressing.
Live/dead staining images show that both PAAm and PU-PAAm hydrogel supported cell adhesion and spreading (in Fig. 6A). Compared with P-30 and P-50, PP-30 and PP-50 hydrogels showed better cell adhesiveness on day 5. As shown in Fig. 6B, L929 fibroblasts cultured on PP-30 and PP-50 exhibited the normal polygonal and elongated shape. What's more, the result of MTT assay indicates that the proliferation of L929 on PP-30 and PP-50 were faster than that on P-30 and P-50, suggesting that the hydrogel with PU has improved biocompatibility and cell affinity.
Fig. 6. In vitro cell behaviors on PAAm and PU-PAAm hydrogels. (A) Live/dead staining images of L929 fibroblasts cultured on P-30 (a), P-50 (b), PP-30 (c), PP-50 (d) on day 3 and day 5; and (B) confocal laser scanning microscopy (CLSM) micrographs of L929 fibroblasts cultured on P-30 (a), P-50 (b), PP-30 (c), PP-50 (d) on day 5; (C) MTT assay for the proliferation of L929 fibroblasts with different hydrogels for day 1, day 3 and day 5; ***p < 0.001, **p < 0.01, *p < 0.05.

The PU-PAAm hydrogels were further used to repair full-thickness skin defects in vivo (Fig. 7). 15 mm × 15 mm full-thickness skin defects were created by established protocol.40 PP-50 and PP-30 hydrogels were used as experimental groups, and the control group was set without any treatment. After 3, 6, 9, 13 and 17 days, the wound samples were harvested to detect the regeneration of skin. As shown in Fig. 7B, no sign of infection was observed in all groups. However, the wound repairing process shows distinct difference across different groups. Quantitative analysis (Fig. 7C) demonstrates that wounds covered by hydrogels showed better healing as compared to the control group, with PP-30 gave the best healing results.
Fig. 7. In vivo wound healing experiments. (A) Skin defects of a rabbit and application of PU-PAAm hydrogels; (B) visual inspection of the healings skin surface after surgery; (C) quantitative analysis of skin healing after surgery (***p < 0.001, **p < 0.01, *p < 0.05); (D) traces of wound-bed closure for each treatment; (E) defected skin tissue with HE and Masson staining after 3, 9, 17 days, respectively.

Histological analysis was then performed to evaluate the skin regeneration more specifically. During the process of wound recovery, damaged tissues were collected and visualized by hematoxylin-eosin (HE) staining. As shown in Fig. 7E and S13,† no noticeable infection or signs of excessive exudation were observed in all groups. On day 3 and day 6, there was a sign of exudation and blood crust on the control wound, while little effusion was observed from the surface of hydrogel group. On day 9, there was lack of epithelium on the control wound, whereas the epithelial layer well covered the wound in PP-50 and PP-30 groups. Especially in PP-30 group, the exposed defect was very narrow on day 9, and was almost healed on day 13. By the end of day 17, there was still a shallow wound in the control skin defect. In contrast, the wound regions covered with hydrogel were smoother and better recovered, and the PP-30 group was completely healed. The fibrosis of the healed tissue was also investigated by Masson's Trichrome staining, see Fig. 7E and S14.† On day 3, there was very large blood crust in all groups. However, little effusion was left on the surface of the hydrogel group in day 6, which was in accordance with the HE observation. On day 9, there was no regular fibrosis tissue in the control group, but the fibrosis beneath epithelial layer in PP hydrogel was well regenerated. Even on day 17, there was still lack of mature fibrosis in the control group. In the late healing phase (13–17 days), although there was no distinct difference in fiber quantity between PP-50 and PP-30, the fibers in PP-30 were better organized, suggesting better tissue regeneration.
There are three stages in wound healing process, which are inflammation, cellular proliferation and extracellular matrix regeneration.41 As a dynamic factor, inflammatory cells are reported to be the main cause for delayed healing and scar forming.42,43 As shown in Fig. 7, inflammatory infiltration was observed in all groups at the typical early stage (day 3). However, inflammatory cells infiltration for PP-50 and PP-30 markedly decreased in comparison with the control group. Notably, inflammation of wounds covered by PP-30 was less severe compared to other groups. Also, in the late phase (day 9), the fibrosis and granulation for PP-30 group was almost twice thicker than that of the control group. This may be because the PP-30 hydrogel has created an ideal microenvironment that minimized the formation of necrotic tissue and inflammatory cytokines, hence reducing the inflammatory reaction and increasing the vascularization.44,45 In contrast, dry crusted wounds with no treatment decreased the supply of blood and nutrients, resulting in a barrier to cell migration and slowdown of epithelialization.22 Besides, hydrogels attached to the wounds could remove redundant exudation and create a refreshed environment, hence benefiting the migration and proliferation of fibroblasts, and accelerating the recovery of skin defects.
4. Conclusion
In summary, a fast curing, self-adhesive and flexible PU-PAAm hydrogel with IPN structure has been successfully developed. Experimental results show that our hydrogel is extremely tough and stretchable, i.e., mechanically robust enough to protect and stabilize wound under various harsh conditions. The unique chemical composition of the hydrogel allows both strong self-adhesion to human skin, and easy removal. The highly porous structure and excellent swelling properties of the hydrogel can provide the wound with an ideal moist/aseptic and breathable environment, where excess extrudate can be effectively absorbed. Finally, the animal studies confirm our hydrogel is highly biocompatible and can promote skin tissue regeneration, hence is ideal for wound dressing applications.
Author contributions
Yi Hou and Nan Jiang contributed equally to this work. Yi Hou carried out all experimental work in relation to materials synthesis and characterization. Nan Jiang performed all biology-related work (cell and animal). All authors contributed to the analysis of data and result discussion as well as the writing of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
The authors would like to thank Shaolan Wang, Yong Luo, Pengchi Deng (Analytical & Testing Center, SCU) for their help with DSC-TG, TEM, NMR testing, respectively. This work was supported by the National Natural Science Foundation of China (No. 51673131, 51872190), National Key Research and Development Program of China (2016YFA0201703/2016YFA0201700) and Graduate Student’s Research and Innovation Fund of Sichuan University (No. 2018YJSY068). The authors would like to acknowledge the Engineering and Physical Sciences Research Council (EPSRC) for funding support (EP/P00394X/1).
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10666a
References
- Frykberg R. G. Banks J. Adv. Wound Care. 2015;4:560. doi: 10.1089/wound.2015.0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen C. K. Gordillo G. M. Roy S. Kirsner R. Lambert L. Hunt T. K. Gottrup F. Gurtner G. C. Longaker M. T. Wound Repair Regen. 2009;17:763. doi: 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi M. A. M. Zangabad P. S. Basri S. M. M. Zangabad K. S. Ghamarypour A. Aref A. R. Karimi M. Hamblin M. R. Adv. Drug Delivery Rev. 2018;123:33. doi: 10.1016/j.addr.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood A. Granick M. S. Tomaselli N. L. Adv. Wound Care. 2014;3:511. doi: 10.1089/wound.2012.0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhivya S. Padma V. V. Santhini E. BioMedicine. 2015;5:24. doi: 10.7603/s40681-015-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzun M. J. Journal of Textile Engineering & Fashion Technology. 2018;4:53. [Google Scholar]
- Winter G. D. Nature. 1962;193:293. doi: 10.1038/193293a0. [DOI] [PubMed] [Google Scholar]
- Dyson M. Young S. Pendle C. L. Webster D. F. Lang S. M. J. Invest. Dermatol. 1988;91:434. doi: 10.1111/1523-1747.ep12476467. [DOI] [PubMed] [Google Scholar]
- Bian S. Zheng Z. Liu Y. Ruan C. Pan H. Zhao X. J. Mater. Chem. B. 2019;7:6488. doi: 10.1039/C9TB01521C. [DOI] [PubMed] [Google Scholar]
- Yuk H. Zhang T. Lin S. et al. . Nat. Mater. 2016;15:190–196. doi: 10.1038/nmat4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. Y. O'Cearbhaill E. D. Sisk G. C. et al. . Nat. Commun. 2013;4:1702. doi: 10.1038/ncomms2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman A. H. van Hees I. A. Yang J. Kamperman M. Adv. Mater. 2018;30(19):1704640. doi: 10.1002/adma.201704640. [DOI] [PubMed] [Google Scholar]
- Cui J. Iturri J. Paez J. Shafiq Z. Serrano C. d’lschia M. del Campoet A. Macromol. Chem. Phys. 2014;215(24):2403–2413. doi: 10.1002/macp.201400366. [DOI] [Google Scholar]
- Han L. Yan L. Wang K. Fang L. Zhang H. Tang Y. Ding Y. Weng L. Xu J. Weng J. Liu Y. Ren F. NPG Asia Mater. 2017;9:e372. doi: 10.1038/am.2017.33. [DOI] [Google Scholar]
- Gan D. Xing W. Jiang L. Fang J. Zhao C. Ren F. Fang L. Wang K. Lu X. Nat. Commun. 2019;10:1487. doi: 10.1038/s41467-019-09351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annabi N. Rana D. Sani E. S. Portillo-Lara R. Gifford J. L. Fares M. M. Mithieux S. M. Weiss A. S. Biomaterials. 2017;139:229–243. doi: 10.1016/j.biomaterials.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X. Fang W. Xue J. Sun T. Dong L. Zha Z. Qian H. Song Y. Zhang M. Gong X. Lu Y. He T. ACS Nano. 2019;13:10074. doi: 10.1021/acsnano.9b02845. [DOI] [PubMed] [Google Scholar]
- Nguyen K. T. West J. L. Biomaterials. 2002;23:4307. doi: 10.1016/S0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- Baker B. A. Murff R. L. Milam V. T. Polymer. 2010;51:2207. doi: 10.1016/j.polymer.2010.02.022. [DOI] [Google Scholar]
- Deng Y. Huang M. Sun D. Hou Y. Li Y. Dong T. Wang X. Zhang L. Yang W. ACS Appl. Mater. Interfaces. 2018;10:37544. doi: 10.1021/acsami.8b15385. [DOI] [PubMed] [Google Scholar]
- Zdrahala R. J. Zdrahala I. J. J. Biomater. Appl. 1999;14:67. doi: 10.1177/088532829901400104. [DOI] [PubMed] [Google Scholar]
- Howard G. T. Int. Biodeterior. Biodegrad. 2002;49:245. doi: 10.1016/S0964-8305(02)00051-3. [DOI] [Google Scholar]
- Hou Y. Jiang N. Zhang L. Li Y. Meng Y. Han D. Chen C. Yang Y. Zhu S. ACS Appl. Mater. Interfaces. 2017;9:25808. doi: 10.1021/acsami.7b06673. [DOI] [PubMed] [Google Scholar]
- Strøbech C. Int. J. Adhes. Adhes. 1990;10:225. doi: 10.1016/0143-7496(90)90108-A. [DOI] [Google Scholar]
- Jia D. Pang Y. Liang X. J. Polym. Sci., Part B: Polym. Phys. 1994;32:817. doi: 10.1002/polb.1994.090320505. [DOI] [Google Scholar]
- Kim B. K. Lee J. C. J. Polym. Sci., Part A: Polym. Chem. 1996;34:1095. doi: 10.1002/(SICI)1099-0518(19960430)34:6<1095::AID-POLA19>3.0.CO;2-2. [DOI] [Google Scholar]
- Liu S. Li L. ACS Appl. Mater. Interfaces. 2016;8:29749. doi: 10.1021/acsami.6b11363. [DOI] [PubMed] [Google Scholar]
- Baek S. H. Kim B. K. Colloids Surf., A. 2003;220:191. doi: 10.1016/S0927-7757(03)00074-8. [DOI] [Google Scholar]
- Liu X. Zhang Q. Gao Z. Hou R. Gao G. ACS Appl. Mater. Interfaces. 2017;9:17645. doi: 10.1021/acsami.7b04832. [DOI] [PubMed] [Google Scholar]
- Li L. Zuo Y. Zou Q. Yang B. Lin L. Li J. Li Y. ACS Appl. Mater. Interfaces. 2015;7:22618. doi: 10.1021/acsami.5b07327. [DOI] [PubMed] [Google Scholar]
- Yang J. Winnik M. A. Ylitalo D. DeVoe R. J. Macromolecules. 1996;29:7047. doi: 10.1021/ma9601373. [DOI] [Google Scholar]
- Bao J. Xu G. Liu D. Ji X. Chin. J. Appl. Chem. 2008;25:587. [Google Scholar]
- Owens D. E. Jian Y. Fang J. E. Slaughter B. V. Chen Y. Pappas N. A. Macromolecules. 2007;40:7306. doi: 10.1021/ma071089x. [DOI] [Google Scholar]
- Wu F. Chen L. Wang Y. Fei B. J. Mater. Sci. 2019;54:12131. doi: 10.1007/s10853-019-03729-9. [DOI] [Google Scholar]
- Han L. Li P. Tang P. Wang X. Zhou T. Wang K. Ren F. Guo T. Lu X. Nanoscale. 2019;11:15846. doi: 10.1039/C9NR03095F. [DOI] [PubMed] [Google Scholar]
- Wang Z. Lu X. Sun S. Yu C. Xia H. J. Mater. Chem. B. 2019;7:4876. doi: 10.1039/C9TB00831D. [DOI] [PubMed] [Google Scholar]
- Zhao X. Wu H. Guo B. Dong R. Qiu Y. Ma P. X. Biomaterials. 2017;122:34. doi: 10.1016/j.biomaterials.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Liang S. Zhang Y. Wang H. Xu Z. Chen J. Bao R. Tan B. Cui Y. Fan G. Wang W. Wang W. Liu W. Adv. Mater. 2018;30:1704235. doi: 10.1002/adma.201704235. [DOI] [PubMed] [Google Scholar]
- Segura D. M., Nurse A. D., McCourt A., Phelps R. and Segura A., Handbook of Adhesives and Sealants, Elsevier Science Ltd, 2005, ch. 3 [Google Scholar]
- Quini J. G. Marinucci G. Mater. Res. 2012;15:434. doi: 10.1590/S1516-14392012005000042. [DOI] [Google Scholar]
- Qu J. Zhao X. Liang Y. Zhang T. Ma P. X. Guo B. Biomaterials. 2018;183:185. doi: 10.1016/j.biomaterials.2018.08.044. [DOI] [PubMed] [Google Scholar]
- Singer A. J. Clark R. A. N. Engl. J. Med. 1999;341:738. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Li Y. Tang L. Yu J. Dai X. Zhou W. Zhang W. Hu X. Xiao S. Ni W. Ma X. Wu Y. Yao M. Mu G. Wang G. Han W. Xia Z. Tang H. Zhao J. Trials. 2012;13:67. doi: 10.1186/1745-6215-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C. C. Miller C. L. Trunkey D. D. J. Trauma. 1979;19:641. doi: 10.1097/00005373-197909000-00001. [DOI] [PubMed] [Google Scholar]
- Church D. Elsayed S. Reid O. Winston B. Lindsay R. Clin. Microbiol. Rev. 2006;19:403. doi: 10.1128/CMR.19.2.403-434.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.