

Summary
The probiotic strain Escherichia coli Nissle 1917 (EcN) is a common bacterial chassis in synthetic biology developments for therapeutic applications given its long track record of safe administration in humans. Chromosomal integration of the genes of interest (GOIs) in the engineered bacterium offers significant advantages in genetic stability and to control gene dose, but common methodologies relying on the transformation of EcN are inefficient. In this work, we implement in EcN the use of bacterial conjugation in combination with markerless genome engineering to efficiently insert multiple GOIs at different loci of EcN chromosome, leaving no antibiotic resistance genes, vector sequences or scars in the modified bacterium. The resolution of cointegrants that leads to markerless insertion of the GOIs requires expression of I‐SceI endonuclease and its efficiency is enhanced by λ Red proteins. We show the potential of this strategy by integrating different genes encoding fluorescent and bioluminescent reporters (i.e. GFP, mKate2, luxCDABE) both individually and sequentially. We also demonstrate its application for gene deletions in EcN (ΔflhDC) and to replace the endogenous regulation of chromosomal locus (i.e. flhDC) by heterologous regulatory elements (e.g. tetR‐Ptet) in order to have an ectopic control of gene expression in EcN with an external inducer to alter bacterial behaviour (e.g. flagellar motility). Whole‐genome sequencing confirmed the introduction of the designed modifications without off‐target alterations in the genome. This straightforward approach accelerates the generation of multiple modifications in EcN chromosome for the generation of living bacterial therapeutics.
In this work, we implement in the probiotic strain Escherichia coli Nissle 1917 (EcN) the use of bacterial conjugation in combination with markerless genome engineering to efficiently insert multiple GOIs at different loci of EcN chromosome, leaving no antibiotic resistance genes, vector sequences, or scars in the modified bacterium. This straightforward approach accelerates the generation of multiple modifications in EcN chromosome for the generation of living bacterial therapeutics.
![]()
Introduction
The probiotic bacterium Escherichia coli Nissle 1917 (EcN) was isolated from the faeces of a healthy German soldier during a severe Shigella epidemic in World War I, being originally reported as having antagonistic activity against pathogenic enterobacteria (Sonnenborn and Schulze, 2009; Sonnenborn, 2016). Since then, the ability of EcN to colonize and outcompete other bacteria in the gastrointestinal (GI) tract has been extensively demonstrated (Altenhoefer et al., 2004; Hancock and Dahl, 2010; Rund et al., 2013), which is partially due to the production of two microcins (Patzer et al., 2003) and the expression of different iron acquisition systems (Deriu et al., 2013). EcN is nowadays commercialized under the trade name Mutaflor® (Sonnenborn, 2016) and has been administered in adults and infants as probiotic therapy to treat enteric infections and various inflammatory disorders in the gut (Henker et al., 2007, 2008; Schultz, 2008; Losurdo et al., 2015; Kotłowski, 2016). Given its long and safe record of human administration, EcN has become one of the ‘chassis’ of choice in synthetic biology applications for engineering therapeutic bacteria against infections, metabolic diseases, GI inflammatory disorders and cancer (Piñero‐Lambea et al., 2015a; Ozdemir et al., 2018; Charbonneau et al., 2020; Kelly and Liang, 2020; Yu et al., 2020).
Plasmids are the most common vectors in synthetic biology given its simple manipulation and high modularity (Martinez‐Garcia et al., 2015). In EcN different plasmids have been successfully used in vivo for expression of gene(s) of interest (GOI) in the GI, including some common E. coli vectors (Rao et al., 2005; Loessner et al., 2009), derivatives of cryptic plasmids found in EcN (Ou et al., 2016; Kan et al., 2021) and vectors ensuring killing of bacteria losing the recombinant plasmid (Fedorec et al., 2019). Nonetheless, gene integration in the chromosome of bacteria offers significant advantages over the use of plasmids, increasing genetic stability without the need of toxin–antitoxin systems (Gerdes and Thisted, 1990; Fedorec et al., 2019), antibiotic resistance genes (which cannot be deployed in natural or industrial environments) (Kroll et al., 2010; Theriot et al., 2014) or other selection markers (e.g. metabolic genes complementing auxotrophies) (Kang et al., 2018). In addition, chromosomal integration improves control of gene dose (copy number), expression levels, and reduces the risk of horizontal gene transfer (HGT) to other bacteria in the microbiome and the environment (Aminov, 2011; Brito, 2021). Hence, chromosomal integration of GOIs minimizes many common concerns associated with live bacterial biotherapeutic products (Wright and Stan, 2013; Wegmann et al., 2017; Rouanet et al., 2020).
Insertions and deletions of GOIs in the chromosome of EcN have been done using the well‐characterized one‐step integration of linear PCR products using the bacteriophage λ Red recombineering system (Datsenko and Wanner, 2000; Sawitzke et al., 2007). EcN strains modified with this technology have been administered in both preclinical mouse models and clinical assays (Duan and March, 2010; Isabella et al., 2018; Kurtz et al., 2019; Charbonneau et al., 2020; Leventhal et al., 2020). In λ Red recombineering, the GOI is amplified by PCR and fused to an antibiotic resistance (AbR) gene and flanking DNA homology regions (HRs) for recombination with the chromosomal site of choice. The AbR gene is flanked by short FRT‐sequences recognized by the flipase (FLP), a site‐specific recombinase enabling subsequent deletion (curing) of the AbR gene, but leaving the recombined FRT‐sequences ("scars" of ca. 80 bp) in the chromosome (Datsenko and Wanner, 2000; Sawitzke et al., 2007). When multiple GOIs are inserted and/or deleted at different locations of the chromosome, FLP‐mediated recombination between distant scars may elicit unwanted chromosomal deletion and rearrangements. In addition, given the low transformation efficiency of EcN compared to common laboratory E. coli strains, performing multiple insertions/deletions become a time‐consuming and labour‐intensive process requiring the preparation of electrocompetent cells for every modification step. Hence, the insertion of multiple GOIs in the chromosome of EcN for synthetic biology applications continues to be challenging.
In this work, we report the implementation in EcN of the markerless and scarless genome engineering approach (Posfai et al., 1999; Feher et al., 2008) in combination with bacterial conjugation (Cabezón et al., 2015) to efficiently transfer GOIs on designed mobilizable suicide vectors carrying I‐SceI recognition sites that are integrated at selected loci in EcN chromosome by homologous recombination. Vector sequences in the cointegrants are deleted from EcN chromosome after repair of double‐strand breaks (DSBs) generated in vivo by expression of I‐SceI meganuclease, which has no cleavage sites in the bacterial genome except the integrated vector. We also show that DSBs in EcN chromosome are repaired more efficiently in the presence of λ Red proteins. This process leaves no AbR genes, vector sequences or DNA scars in the chromosome. Multiple rounds of deletion and/or integration of GOIs can be performed in EcN following this methodology. We demonstrate the potential of this approach by integrating at different sites of EcN chromosome genes encoding fluorescent protein reporters (GFP, mKate2), a bioluminescence operon (luxCDABE) and the tetracycline promoter region (tetR‐Ptet) to control the expression of chromosomal flhDC regulators of EcN flagella. The accuracy of the genetic manipulations was confirmed by whole‐genome sequencing.
Results
Exploring the conjugation efficiency of oriT‐RP4 plasmids in EcN
We first evaluated the efficiency of conjugation as an alternative method to transformation for introducing foreign DNA into EcN. Although bacterial conjugation has been widely used to transfer DNA from a donor strain to diverse bacteria (de Lorenzo et al., 1990; Martinez‐Garcia and Lorenzo, 2012; Cabezón et al., 2015), EcN was reported to be a poor recipient of foreign DNA with conjugative plasmids from multiple incompatibility (Inc) groups (Sonnenborn and Schulze, 2009). This former study did not use donor E. coli K‐12 strains with the genes encoding the conjugative machinery of the broad‐host‐range IncPα plasmid RP4 integrated in the chromosome (Pansegrau et al., 1994; Grahn et al., 2000). We decided to evaluate conjugation to EcN using an E. coli K‐12 Mu‐free donor strain, named MFDpir, which encodes the RP4 conjugation machinery and was engineered to avoid the undesirable transfer of parts of the donor genome via Mu bacteriophage mobilization or high‐frequency recombination (hfr) origin of transfer of the integrated RP4 (oriT chrRP4) (Ferrières et al., 2010). E. coli MFDpir has a mutation in the oriT chrRP4 (Δnic), is also deleted of the Mu bacteriophage (ΔMu) and is recombination‐deficient (ΔrecA). In addition, it contains a chromosomal dapA::pir fusion, which encodes the π‐protein supporting the replication of plasmids with R6K‐ori, and makes this strain an auxotroph for diaminopimelic acid (DAPA), facilitating its elimination in media lacking DAPA. To test the conjugation frequency to EcN, we used two mobilizable plasmids with RP4 oriT and a kanamycin resistance (KmR) gene, pSEVA237R and pSEVA241, containing pBBR1 and pRO1600/ColE1 replication origins respectively (Fig. 1A). Conjugation assays were performed by spotting a small sample (~20 μl) of overnight liquid cultures of both strains (in 1:1 proportion by OD600) on LB‐agar plates containing DAPA (for the correct growth of the auxotrophic donor; Experimental procedures). After 3.5 h incubation at 37 °C, transconjugants were selected on LB+Km agar plates lacking DAPA to ensure donor removal. The conjugation frequency was measured as the number of transconjugants (T) per recipient (R) EcN cell in four independent assays, showing a mean of 3.87 × 10−2 and 2.67 × 10−2 for pSEVA237R and pSEVA241 respectively (Fig. 1B). These data indicated that a significant proportion of EcN bacteria were taking these conjugative plasmids from MFDpir donor under the conditions tested.
Fig. 1.
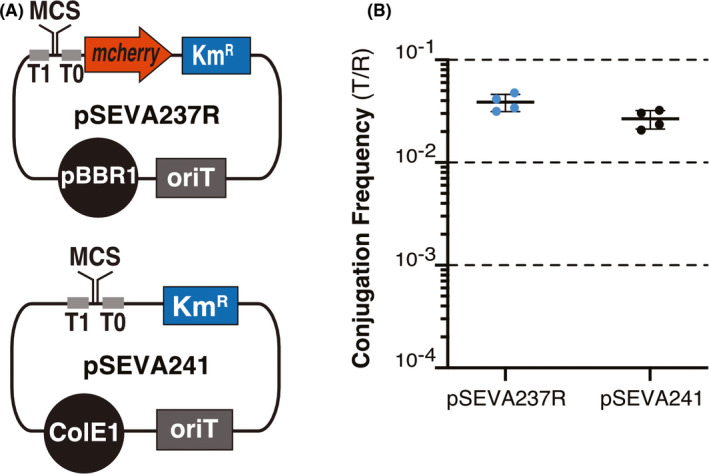
Conjugation frequency to EcN of replicative plasmids with oriTRP4 transferred from E. coli MFDpir donor.
A. Replicative mobilizable plasmids pSEVA237R and pSEVA241 (Martinez‐Garcia et al., 2015), both carrying the origin of transfer oriT of RP4. Other genetic elements of these plasmids shown are as follows: the kanamycin resistance gene (KmR), the replication origins (ori‐pBBR1 or ori‐ColE1), mCherry, multiple cloning site (MCS) and transcriptional terminators (T1, T0).
B. Conjugation frequency of plasmids pSEVA237R and pSEVA241 indicated as the ratio of EcN transconjugants (T) versus EcN recipients (R). Horizontal lines indicate the mean of four independent experiments (individual dots, n = 4). Vertical bars indicate standard deviation.
Chromosomal integration upon conjugation in EcN
The high conjugation frequency observed in EcN with MFDpir donor prompted us to explore chromosomal integration after conjugation of mobilizable suicide plasmids carrying homology regions (HRs) with the chromosome of EcN (Fig. 2A). We had previously constructed a suicide plasmid vector carrying I‐SceI recognition sites, called pGE, for integration of different GOIs in the chromosome of E. coli K‐12 MG1655 by homologous recombination (Piñero‐Lambea et al., 2015b; Ruano‐Gallego and Álvarez, 2015) using the markerless gene replacement strategy (Posfai et al., 1999; Feher et al., 2008). To test this strategy in combination with conjugation in EcN, we constructed a mobilizable pGE‐derivative having the oriT of RP4, named pGEC (Table S1). As its parental pGE, the mobilizable pGEC vector has a π‐dependent R6K‐ori, a KmR cassette and a multiple cloning site (MCS) flanked with I‐SceI recognition sites for cloning of the GOI and homology regions (HRs) of the targeted chromosomal locus (Fig. 2B and Fig. S1). To investigate the frequency of cointegrants in EcN after conjugation, we constructed pGEC derivatives, called pGECfluEcN_ gfp and pGECfimEcN_ gfp (Table S1; Figs S2 and S3). These suicide plasmids carry a fusion between the Ptac promoter (Brunner and Bujard, 1987) and a synthetic gfp gene (Corcoran and Cameron, 2010), having flanking HRs of ~500 bp for recombination in two different loci of EcN chromosome, the flu gene encoding Ag43 adhesin (van der Woude and Henderson, 2008) and fimAICDFGH operon encoding type 1 fimbriae (Connell et al., 1996) respectively. Schemes of the targeted loci of EcN are shown in Figs S2 and S3. We chose integration in these adhesin genes because their deletion was not expected to affect the growth or viability of EcN, similar to the situation in E. coli K‐12 MG1655 (Piñero‐Lambea et al., 2015a).
Fig. 2.
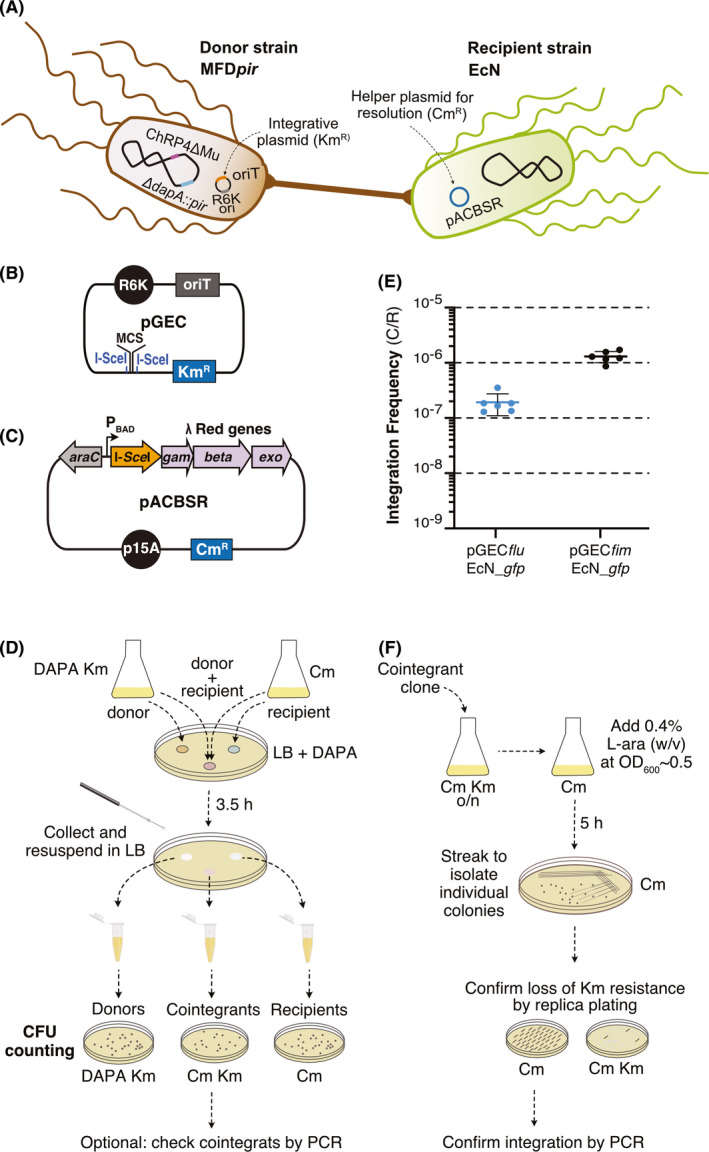
Markerless integration of suicide pGEC‐derivatives in EcN chromosome.
A. Scheme of both donor (MFDpir with pGEC‐derivative) and recipient (EcN with pACBSR) bacteria used for conjugation. The donor strain has integrated in its chromosome the genes encoding the RP4 conjugative machinery (ChRP4) and the π‐protein for R6K‐ori replication (ΔdapA::pir). The MFDpir strain (Ferrières et al., 2010) is also recombination deficient (ΔrecA), Mu‐free (ΔMu) and auxotroph for diaminopimelic acid (DAPA; ΔdapA).
B. Scheme of suicide plasmid pGEC indicating R6K‐ori, oriT, KmR gene and multiple cloning site (MCS) with flanking I‐SceI sites.
C. Scheme of plasmid pACBSR (Herring et al., 2003) for resolution of cointegrants, indicating the p15A‐ori, the regulatory region araC‐PBAD responding to L‐ara inducer, the genes encoding I‐SceI enzyme, λ Red functions (gam, beta, exo) and the CmR gene.
D. Scheme of the procedure for conjugative transfer of pGEC‐derivatives and isolation of EcN cointegrants.
E. Integration frequency of the pGECfluEcN_gfp and pGECfimEcN_gfp indicated as the ratio of cointegrants (C) versus recipients (R). Horizontal lines indicate the mean of six independent experiments (individual dots, n = 6). Vertical bars indicate standard deviation.
F. Scheme of the procedure for resolution of cointegrants and isolation of EcN with the markerless chromosomal insertion.
MFDpir bacteria carrying pGECfluEcN_gfp or pGECfimEcN_gfp, were used as donors in conjugation assays with EcN recipients, carrying the chloramphenicol resistant (CmR) helper plasmid pACBSR (Table S1, Fig. 2C) (Herring and Glasner, 2003). This helper plasmid is later used for the resolution of the cointegrants (see below) by inducing the expression of I‐SceI endonuclease and λ Red proteins from the PBAD promoter with L‐arabinose (L‐ara). Upon conjugation, cointegrants were selected on LB‐agar plates containing Km and Cm (without DAPA) to eliminate both donor and recipient bacteria (Fig. 2D, see Experimental procedures for details). Integration frequency in EcN was calculated as the ratio of cointegrants versus recipient cells in six independent assays, with mean values of 1.4 × 10−7 and 1.3 × 10−6 for pGECfluEcN_gfp and pGECfimEcN_gfp respectively (Fig. 2E). As expected, integration frequencies were much lower than conjugation frequencies (~4 orders of magnitude), due to the double molecular event of conjugation and integration of the transferred plasmid. Nonetheless, cointegrants were obtained in sufficient numbers in both loci in every conjugation assay, from 35 and 105 colonies when flu was used for integration, and between 235 and 510 colonies when the target was the fim operon. Plasmids could be integrated by either HR1 or HR2, giving rise to two alternative cointegrants (Figs S2A and S3A). PCR analysis with primer pairs that hybridize at the DNA insert (e.g. gfp) and at the chromosome, external to the HRs used for recombination, confirmed that the corresponding pGEC derivative was integrated in either flu (Fig. S2B and C) or fim (Fig. S3B and C) locus of EcN chromosome. Of note, PCR products from cointegrants often result in DNA bands compatible with integration in both HR1 and HR2 (see Fig. S3B and C). However, this is not a reflection of a double recombination event, rather a consequence of the duplication of both HR1 and HR2 in the chromosome of the cointegrant that enables in vitro assembly of extended ssDNA products hybridizing through the HR.
Resolution of cointegrants in EcN
Next, we evaluated the resolution of cointegrants in EcN. To this end, one cointegrant clone from each integration site (flu and fim) was grown in liquid LB+Cm cultures and expression of I‐SceI endonuclease and λ Red proteins from pACBSR was induced with 0.4% (w/v) L‐ara for 5 h. Then, individual colonies were isolated by streaking on LB+Cm agar plates (Fig. 2F). During resolution, DSBs generated by I‐SceI digestion of the integrated pGEC are repaired by homologous recombination assisted by the λ Red proteins and leads to the markerless integration of the GOI or the reversion to the wild‐type allele with equal probabilities (Fig. S1) (Posfai et al., 1999; Feher et al., 2008). Forty colonies grown on LB+Cm plates after L‐ara induction were randomly picked and checked for loss of KmR resistance by replica plating on LB+Cm plates with and without Km, which indicated that all 40 colonies from the resolution in the flu site were Km‐sensitive (Fig. S4A). Similarly, all 40 colonies randomly picked from the resolution in the fim site were Km‐sensitive (Fig. S5A). This indicates a very effective resolution of cointegrants in EcN after expression of I‐SceI and λ Red proteins. These Km‐sensitive colonies may have resolved to the wild‐type allele or to the insertion mutant. Colony PCR of 10 Km‐sensitive colonies from each resolution event, with primers pairs hybridizing in the EcN chromosome outside the HRs, showed that four colonies of flu (Fig. S4B–D) and three colonies of fim (Fig. S5B–D) were insertion mutants whereas the rest had reverted to the wild‐type allele. Flow cytometry analysis of EcN bacteria with Ptac‐gfp fusions inserted in fim (EcNΔfim_gfp) and flu (EcNΔflu_gfp) confirmed expression of GFP in both strains (Fig. S6).
λ Red proteins increase the resolution frequency of cointegrants
The λ Red system consists of three genes, namely, gam, bet and exo (Caldwell and Bell, 2019). The λ Gam inhibits nuclease activities, preserving linear double‐stranded DNA (dsDNA). The λ Exo is a 5'‐3'exonuclease that requires a dsDNA end to bind and remains bound to one strand while degrading the other in a 5'‐3' direction leading to dsDNA with a 3' ssDNA overhang, which is the substrate for the λ Beta protein binding. To determine whether λ Red proteins are involved in the recombination process leading to the resolution of cointegrants in the EcN chromosome, we constructed a new helper plasmid, named pACBS, encoding I‐SceI but lacking all λ Red genes (Table S1). EcN strains carrying pACBS, or its parental pACBSR, were tested for integration and resolution of cointegrants. As expected, similar integration frequencies of pGECfimEcN_gfp were obtained in the recipient EcN strains carrying pACBS or pACBSR (Fig. S7), likely because λ Red proteins are not induced nor needed in this initial recombination step that generates the cointegrants. Cointegrants carrying pACBS or pACBSR were then tested for resolution after induction with L‐ara for 5 h (Experimental Procedures). The resolution was calculated in five independent assays as the fraction of CFU sensitive to Km (i.e. losing integrated vector) compared to the total CFU of the induced culture. We found that the average percentage for resolution of cointegrants was ~ 31% in the absence of λ Red proteins (pACBS) compared to ~ 99% with pACBSR (Fig. 3A). These data indicate that λ Red proteins are not essential for the resolution of cointegrants but they enhance the efficiency of the process by ~threefold.
Fig. 3.
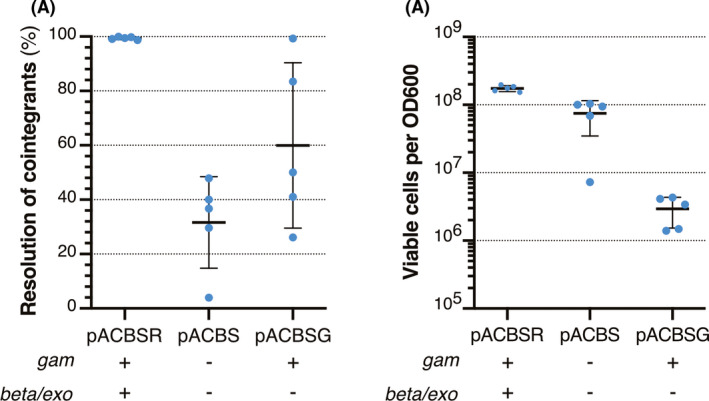
Role of λ Red proteins in the efficient resolution of cointegrants.
A. Percentage of cointegrant resolution (EcN‐pGECfimEcN_gfp) after 5 h induction with L‐ara of bacteria carrying helper plasmids pACBSR, pACBS or pACBSG, as indicated. These helper plasmids express I‐SceI, whereas the presence of gam and beta/exo genes is indicated under each plasmid (+).
B. Viable bacteria after 5 h induction with L‐ara of EcN‐pGECfimEcN_gfp cointegrants carrying pACBSR, pACBS or pACBSG, as indicated. Viability was calculated as CFU per unit of OD600 of the cultures.
A and B. Horizontal lines indicate the mean of five independent experiments (individual dots, n = 5) and vertical bars indicate standard deviation.
Since the λ Gam protein inhibits nuclease activities in the bacterium and protects linear DNA (Sawitzke et al., 2007), we wondered whether the λ Red proteins could only participate in the protection of linear DSBs generated after I‐SceI digestion or could have additional roles in the recombination step leading to resolution. For this purpose, we constructed a new helper plasmid encoding I‐SceI and only Gam, named pACBSG (Table S1). EcN bacteria carrying pACBSG were tested for cointegrants resolution after initial pGECfimEcN_gfp integration, as above, which revealed a lower average percentage of resolution of ~60%, compared to ~99% of pACBSR, and a much higher variability between independent experiments (Fig. 3A). This variability was associated to a decrease in CFU of the cultures of this strain after I‐SceI expression. We compared bacterial viability after L‐ara induction from pACBSR, pACBS and pACBSG (Fig. 3B), revealing that the number of viable EcN bacteria moderately decreases ~twofold in the absence of λ Red proteins (pACBS) but is dramatically reduced about two orders of magnitude in the strain carrying pACBSG, expressing only the Gam protein. Hence, both higher resolution frequencies and bacterial viabilities are found in EcN carrying pACBSR helper plasmid, indicating that the expression of all three λ Red proteins have an overall positive effect on the resolution of cointegrants after I‐SceI digestion.
Chromosomal integration of genes encoding fluorescent and bioluminescence protein reporters in EcN
Since fluorescent proteins (FPs) and bioluminescent reporters are extremely useful to tag live biotherapeutic bacterial strains during in vivo preclinical assays (Cronin et al., 2012; Gahan, 2012; Tiwari and Tiwari, 2020), in addition to GFP‐expressing EcN, we constructed EcN strains expressing a monomeric far‐red FP (mKATE2) (Shcherbo et al., 2009; Chudakov et al., 2010) and the bioluminescence luxCDABE operon from Photorhabdus luminescens (Winson et al., 1998; Gahan, 2012). We followed the conjugation, integration and resolution strategy described previously with Ptac‐gfp fusions.
Ptac‐mKATE2 gene fusion was inserted in the EcN chromosome replacing the fim operon using pGECfimEcN_mKATE2 (Fig. 4A), similar to the integration of pGECfluEcN_gfp replacing flu (Fig. 4B). Expression of FPs GFP and mKATE2 was analysed in EcNΔflu_gfp and EcNΔfim_mKATE2 strains. Bright green or red fluorescence was clearly visible under a blue light transilluminator in bacterial pellets obtained after centrifugation of overnight cultures from these strains grown in LB with 0.1 mM IPTG, but not in similar cultures from the parental EcN (Fig. 4C). The expression of FPS in the modified EcN strains was confirmed by flow cytometry showing a high‐level expression for both GFP (Fig. 4D) and mKATE2 (Fig. 4E). A ~threefold increase in the mean fluorescence intensity (MFI) values of these EcN strains was observed in the presence of IPTG, but a strong expression of these FPs is also seen in the absence of the inducer due to the leaky expression of Ptac promoter. These data indicate the expression of the chromosomal lacI gene in EcN is not sufficient to effectively repress leaky expression of the strong Ptac promoter (de Boer and Comstock, 1983). We also showed that fluorescent EcN bacteria with the chromosomal expression of GFP and mKATE2 can be visualized by fluorescence microscopy (Fig. 4F and G).
Fig. 4.
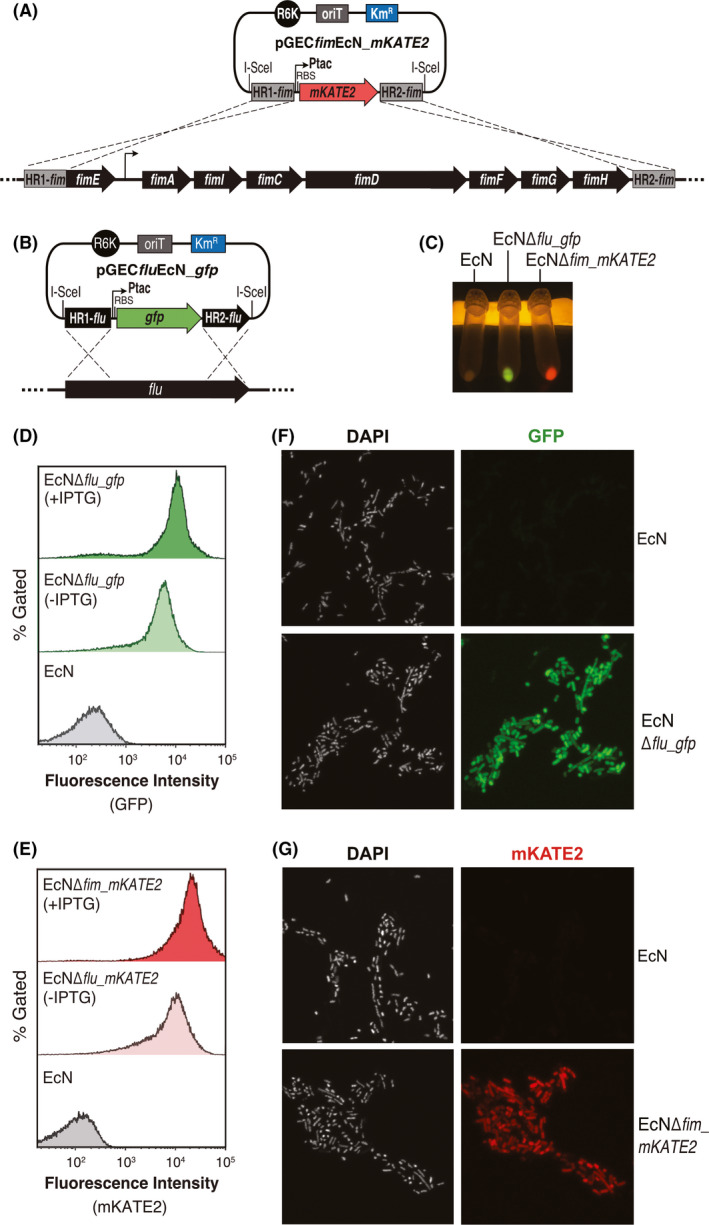
Chromosomal integration of fluorescent gene reporters in EcN.
A and B. Chromosomal integration strategy for Ptac_mKATE2 and Ptac_gfp in the EcN fim or flu locus respectively.
C. Bacterial pellets observed with a blue light transilluminator (Safe Imager 2.0, ThermoFisher) after centrifugation of liquid o/n cultures of the indicated strains (EcN, EcNΔflu_gfp and EcNΔfim_mKATE2) grown in LB cultures with 0.1 mM IPTG.
D and E. Flow cytometry analysis (Cytoflex, Beckman Coulter) to detect GFP (D) and mKATE2 (E) fluorescence of parental EcN and modified EcNΔflu_gfp or EcNΔfim_mKATE2 strains, grown in LB in the presence of inducer (+IPTG) or not (‐IPTG), as indicated.
F and G. Fluorescence microscopy images of parental EcN and modified EcNΔflu_gfp (F) or EcNΔfim_mKATE2 (G) bacteria to visualize GFP (green) or mKATE2 (red) expression. Bacterial DNA stained with DAPI is also shown. Microscopy images were obtained in a Leica TCS SP8 multispectral confocal microscope.
Lastly, a genetic fusion between a constitutive P2 promoter and luxCDABE (Piñero‐Lambea et al., 2015a) was integrated in EcN using pGECmatEcN_lux (Fig. 5A; Table S1), replacing the ecpA gene of EcN, which encodes the major fimbrial subunit MatB of E. coli common pili (ecp/mat) (Rendon et al., 2007). The resulting strain, EcNΔmat_lux, was checked for light emission showing a clear bioluminescence of individual colonies compared to the parental EcN (Fig. 5B).
Fig. 5.
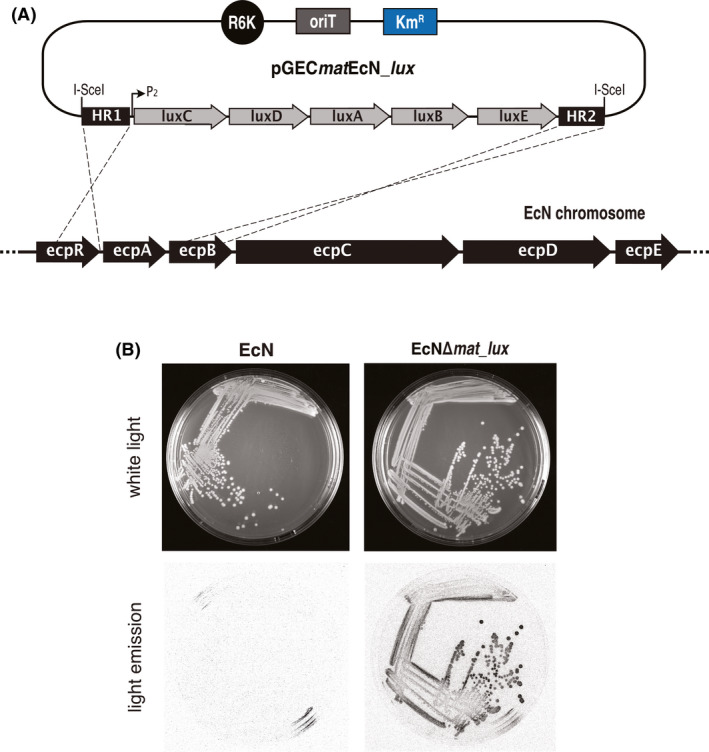
Chromosomal integration of bioluminescent gene reporter operon in EcN.
A. Scheme showing the strategy for chromosomal integration of luxCDABE operon from Photorhabdus luminescens with suicide conjugative plasmid pGECmatEcN_lux in the EcN ecpA gene.
B. Bioluminescence from EcNΔmat‐lux bacterial colonies streaked on LB agar plates. White light and light emission images from the plates were acquired showing a clear bioluminescence of individual colonies of EcNΔmat_lux compared to the parental EcN strain. Bacterial light emission from LB plates was captured using a Chemidoc XRS system (Bio‐Rad).
Replacement of the endogenous promoter of flhDC to control flagellar expression in EcN
We wanted to show the applicability of the conjugation and markerless gene replacement strategy to engineer a heterologous control over a biological function in EcN. Flagellar motility and chemotaxis allow bacteria to reach environments with optimal nutrients for growth and run away from harmful chemicals (Sourjik, 2004; Erhardt and Namba, 2010). In EcN, flagella have been reported to act also as a major adhesin binding intestinal mucins (Troge et al., 2012). Flagella are complex nanomachines encoded by more than 50 genes, and its synthesis is highly regulated in a hierarchical transcriptional network (Soutourina and Bertin, 2003; Anderson and Smith, 2010). In E. coli and other enterobacteria, the flhDC operon encodes the master transcriptional regulator FlhDC that activates, either directly or indirectly, the expression of all structural and regulatory components of the flaglellar apparatus (Wang et al., 2006; Fitzgerald and Bonocora, 2014). Expression of flhDC operon is controlled by multiple environmental and metabolic signals that are integrated in the promoter and 5' untranslated region (UTR) of this operon (Fig. 6A), which is the target for several regulatory proteins and small regulatory RNAs (sRNAs) (Soutourina et al., 1999; Sperandio and Torres, 2002; Soutourina and Bertin, 2003; De Lay and Gottesman, 2012). We decided to replace the native promoter and 5'UTR of the flhDC operon in EcN chromosome with the tetracycline repressor and promoter region (tetR‐Ptet), which can be induced by anhydrotetracycline (aTc) (Bertram and Hillen, 2008). To this end, we constructed pGEC_tetR‐Ptet‐flhDC' (Table S1, Fig. 6A), carrying the upstream chromosomal region to the promoter and UTR of flhDC, followed by tetR‐Ptet‐flhD and 5' of flhC (flhC'). This plasmid was integrated in the chromosome of EcN resulting in EcN_Ptet‐flhDC strain. In addition, we followed the same strategy to generate an flhDC deletion strain (EcNΔflhDC) using pGECΔflhDC plasmid (Table S1). The secretion of the main flagellar subunit (FliC; ~51 kDa) was analysed in EcN_Ptet‐flhDC strain along with the parental EcN and the EcNΔflhDC strains, as controls. Proteins from culture supernatants of liquid LB cultures of these strains, either uninduced or induced with aTc, were precipitated with trichloroacetic (TCA) acid and analysed by SDS‐PAGE and Coomassie staining (Fig. 6B). The results demonstrated the expression of flagellin (FliC) in the EcN_Ptet‐flhDC strain in the presence of aTc (lane 3), with higher levels than those found in wild‐type EcN (lane 1). FliC protein was not detected in the culture supernatants of the deletion mutant EcNΔflhDC and of the EcN_Ptet‐flhDC strain in the absence of aTc (lanes 2 and 4 respectively). Next, we conducted bacterial motility assays by growing these strains in LB soft‐agar plates (Experimental procedures). Motility of EcN_Ptet‐flhDC was found to be higher than that of wild‐type EcN in media containing aTc (Fig. 6C). However, in the absence of aTc the motility of EcN_Ptet‐flhDC bacteria was similar to EcN wild‐type strain, and not to ΔflhDC strain (Fig. 6C), indicating a leaky expression of flhDC from tetR‐Ptet promoter under these conditions, which was not noticeable in the FliC levels in liquid cultures (Fig. 6B).
Fig. 6.
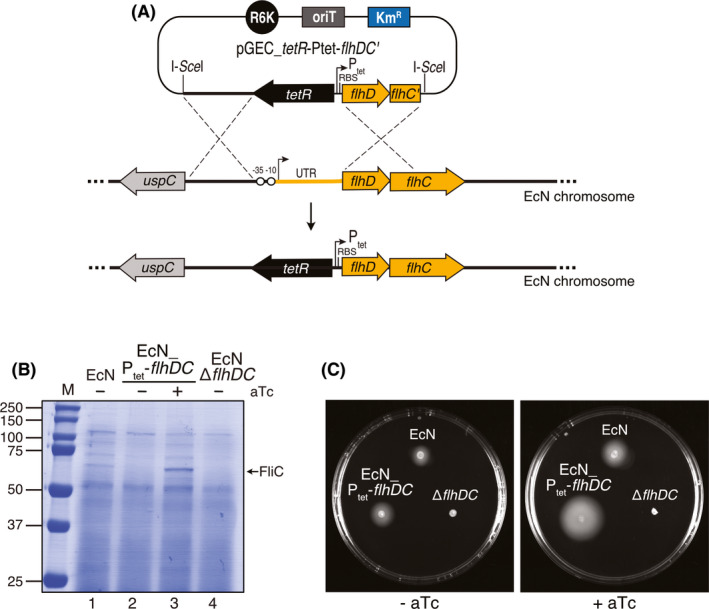
Replacement of the endogenous promoter of flhDC for heterologous control of flagellar expression in EcN.
A. Scheme of the strategy for chromosomal integration using plasmid pGEC_TetR_Ptet‐flhDC' for replacement of the endogenous native promoter and UTR of flhDC by the inducible tetR‐Ptet.
B. Coomassie staining of 10% SDS‐PAGE loaded with TCA‐precipitated protein samples from culture supernatants of the indicated strains (EcN, EcN_Ptet‐flhDC, EcNΔflhDC) grown in LB induced (+) or not (−) with anhydrotetracycline (aTc). The protein band of flagellin (FliC) is labelled with an arrow on the right. Mass of molecular markers (M) are on the left (in kDa).
C. Flagellar motility assay of EcN strains as in B, as indicated, grown in LB‐soft agar plates in the absence (−aTc) or presence (+aTc) of inducer.
Sequential chromosomal integrations in EcN
An advantage of markerless gene replacement is that sequential integrations can be made with the same approach, enabling the construction of bacterial strains with multiple insertions in different loci of their genomes. To demonstrate multiple markerless insertions in EcN, we constructed an EcN strain with chromosomal integrations expressing mKATE2, Lux and aTc‐inducible FlhDC by sequential conjugation, integration and resolution of the corresponding pGEC‐derivatives. The acceptor EcN strains maintain pACBSR helper plasmid throughout the process, and this plasmid is only cured at the end of all integrations (by growth in the absence of Cm) to make the modified bacteria free of any antibiotic resistance gene. The resulting strain, named EcNΔfim_mKATE2 Δmat_lux Ptet‐flhDC (Table S1), showed red fluorescence, bioluminescence and enhanced motility in aTc like the strains with individual integrations (Fig. 7).
Fig. 7.
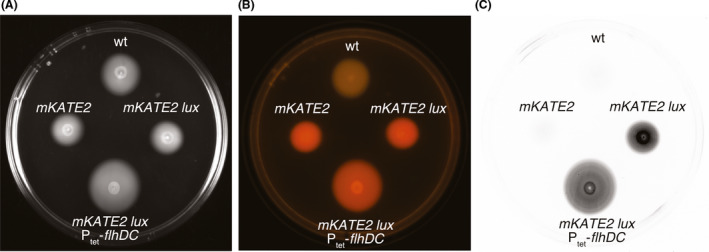
Sequential chromosomal integrations in EcN. Images of a motility assay plate in LB‐soft agar with aTc inoculated with EcN wild type (wt) and modified EcN strains carrying sequential insertions of mKATE2, lux (luxCDABE) and tetR‐Ptet‐flhDC, as indicated. The same plate was imaged under different conditions to observe the expression of fluorescence and bioluminescence gene reporters and the swimming behaviour of the bacteria.
A. Image under while light (ChemiDoc XRS, Bio‐Rad).
B. Image under blue light transilluminator (Safe Imager 2.0, ThermoFisher).
C. Bacterial light emission (ChemiDoc XRS, Bio‐Rad).
Lastly, we performed whole‐genome sequencing of the parental wild‐type EcN and the final strain EcNΔfim_mKATE2 Δmat_lux Ptet‐flhDC to confirm the expected deletions and integrations and rule out the existence of other mutations in the EcN genome that potentially could have been introduced during conjugation and/or recombination steps. Next‐generation sequencing (NGS) reads from both strains were compared with the reference EcN genome CP007799.1 of 5 441 200 bp (Experimental procedures). This analysis revealed that the genome of the modified strain shared the same genome sequence of the parental EcN strain, except for the designed gene replacements. In both strains, we found variations with respect to the reference EcN genome CP007799.1, mostly single nucleotide polymorphisms (SNPs) and small insertions and/or deletions (indels) scattered in the genome (Data S1). These variations were shared by the modified strain, except two deletions of 1 bp in the promoter region of the fim operon (positions 4982169 and 4982544), which are deleted by the Δfim_mKATE2 insertion of this strain. No exclusive variants were detected in the genome of the modified strain, except the designed deletions and insertions. Comparison of NGS data with the expected sequences of the DNA insertions did not reveal any variation of the inserts, except for a SNP in the starting codon of luxC gene. Sanger sequencing of the plasmid preparation of pGECmatEcN_lux used for construction of the modified EcN strain showed the existence of the same SNP (Fig. S8), indicating that this mutation was generated during plasmid propagation in E. coli BW25141 and not through the conjugation and insertion process (see Discussion). In conclusion, analysis of whole‐genome sequencing data demonstrates that the reported conjugation and markerless insertion methodology is accurate and does not introduce off‐target mutations in EcN genome nor in the DNA insertions.
Discussion
Escherichia coli Nissle 1917 is one of the preferred probiotic chassis for engineering living therapeutics given its extensive record of safe administration in humans (Behnsen et al., 2013; Ozdemir et al., 2018; Charbonneau et al., 2020; Kelly et al., 2020). Nonetheless, the lower efficiency of transformation of EcN compared to conventional E. coli K‐12 strains has made difficult its genetic manipulation without the use of replicative plasmids or leaving no scars or antibiotic resistance genes in the chromosome (Ou et al., 2016). In this work, we have reported a simple and efficient methodology to transfer suicide plasmids to EcN by bacterial conjugation in order to obtain the stable integration of multiple GOIs at targeted loci of choice using a markerless and scarless gene replacement strategy (Posfai et al., 1999; Feher et al., 2008). In our study, we used gene reporters encoding fluorescent and bioluminescent proteins that are extremely useful to visualize bacteria in microscopy or using in vivo imaging systems. The same approach can be used to make gene deletions in EcN (e.g. ΔflhDC). We have also demonstrated that this methodology can be applied to precisely replace the endogenous regulatory promoter region of a GOI (e.g. flhDC) for an exogenous regulatory element (e.g. tetR‐Ptet) in order to have an ectopic control of gene expression in EcN (e.g. flagella expression) with an external inducer (e.g. aTc). Bacterial conjugation and markerless genome engineering had been used to generate multiple gene deletions in Pseudomonas putida (Martínez‐García and Lorenzo, 2011), but it has never been adapted for its use in EcN or other probiotic bacteria, maybe because of earlier reports indicating that bacterial conjugation of different Inc groups is not efficient in EcN (Sonnenborn and Schulze, 2009). However, we have found that the integrated RP4 system in the E. coli MFDpir strain (Ferrières et al., 2010) used as donor in this study has very high efficiency in transferring exogenous plasmid DNA into EcN. This enables integration frequencies of suicide plasmids sufficiently high to readily obtain multiple cointegrant clones in EcN by simply mixing small samples (~ 20 μl) of unconcentrated cultures of the donor (MFDpir) and recipient (EcN) strains. Besides high conjugation frequencies, E. coli MFDpir strain has other important characteristics that make it a suitable donor, like its deficiency for recombination (ΔrecA), its mutated transfer chromosomal oriT chrRP4 (Δnic) that avoids transfer of chromosomal genes, and its lack of λ and Mu bacteriophages (Ferrières et al., 2010). Another important feature of E. coli MFDpir strain is the dapA::pir genetic fusion in its chromosome encoding the π‐protein for replication of plasmids with R6K‐ori, like pGEC. This fusion also makes E. coli MFDpir an auxotroph for DAPA, which allows its simple elimination from conjugation mixtures in media lacking this essential component of the peptidoglycan (Egan and Errington, 2020). In EcN, deletion of the dapA gene has been used for biocontainment and to prevent bacterial replication in vivo (Isabella et al., 2018). If an EcN dapA mutant would be used as receptor in conjugations with E. coli MFDpir donor, all media should contain DAPA and cointegrants can be still selected by their double antibiotic resistance (KmR due insertion of pGEC and CmR due to the pACBSR helper plasmid). The Km resistance gene is later lost with the resolution of the cointegrants. The helper plasmid can be maintained in the strain while making sequential genetic insertions/deletions and is cured in the final EcN strain carrying all designed modifications by 2–3 serial passages in liquid media lacking Cm followed by streaking on LB plates lacking Cm. Colonies that grow after these passages are tested for sensitivity to Cm by replica plating, thus generating modified EcN strain‐free of any antibiotic resistance gene. Although we have not experienced any difficulties in curing pACBSR in EcN strains, alternative helper plasmids expressing I‐SceI and having a thermosensitive origin of replication have been reported (de Moraes and Teplitski, 2015), which could accelerate this process.
One interesting observation of our work is that the λ Red proteins co‐expressed with I‐SceI from the helper plasmid pACBSR (Herring et al., 2003) are not essential for the resolution of the initial EcN cointegrants, although they enhance the efficiency of this process by at least threefold. RecA, the native protein for homologous recombination in E. coli (Bell and Kowalczykowski, 2016), is likely the major actor in the recombination events taking place for resolution of cointegrants after I‐SceI digestion. However, the improved efficiency of resolution elicited by λ Red suggests that these proteins assist RecA‐dependent homologous recombination to repair the DSBs. In this sense, it has been described that the presence of RecA improves λ Red recombineering (Wang et al., 2006), which also suggests that both systems can cooperate in vivo. Interestingly, we observed a high instability of cointegrants when only the λ Red Gam protein is expressed, with a major decrease in bacterial viability (ca. 100‐fold) after I‐SceI expression. These data indicate that cointegrants of EcN bacteria expressing only Gam have an impaired ability to repair DSBs, causing bacterial cell death and reducing viability. This result is consistent with a Gam inhibition of the RecBCD nuclease and helicase complex (Wilkinson et al., 2016), which is reported to be involved in DSBs repair by recruiting RecA in E. coli (Churchill and Anderson, 1999; Dillingham and Kowalczykowski, 2008).
Importantly, the enhancement of cointegrants resolution elicited by the λ Red proteins is not associated with the generation of SNPs or indels in the genome, as we have demonstrated by whole‐genome sequencing of the parental and modified EcN strains. λ Red recombineering has been associated with offtarget mutations in the genome (Sharan et al., 2009; Pines et al., 2015). However, most mutations can be traced down to mismatches and errors in the oligonucleotides and PCR products used. Also, the expression of the flipase to excise AbR genes with flanking FRT‐sites may lead to unexpected recombination events between distant FRT sites (Datsenko and Wanner, 2000). None of these elements concurred in our system, which avoids the use of PCR products in the recombination step and does not require flipase nor FRT‐sites to remove AbR genes from the EcN genome. However, mutations during propagation of multicopy suicide plasmids may still occur and become inadvertently inserted in the genome of the modified strain, as it happened in our case in pGECmatEcN_lux with a SNP mutation in the starting codon of luxC. This mutation detected after whole‐genome sequencing does not abolish the bioluminescence in our strain but reduces its activity compared to that obtained by integration of the lux construct in E. coli K‐12 (Piñero‐Lambea et al., 2015a). The metabolic burden due to the overexpression of Lux proteins from the suicide plasmid in E. coli BW25141 may have been the cause for the selection of this mutation. In this sense, it is important to note that the expression of gene constructs, including P2‐lux, is very stable upon integration in the E. coli chromosome (Piñero‐Lambea et al., 2015a), but mutations are more frequent when cloned in multicopy plasmids. It is also interesting to highlight that, apart from the designed modifications, genome‐sequencing analysis did not detect any exogenous DNA in the genome of the modified EcN strain. This indicates that bacterial conjugation with the donor strain MFDpir did not transfer parts of its genome to the modified EcN strain, which is in agreement with the engineered properties of MDFpir strain (ΔMu‐bacteriophage, ΔrecA and Δnic oriT chrRP4) to avoid chromosomal DNA transfer from the donor to recipient strains (Ferrières et al., 2010).
Another important aspect of the reported system is that multiple integration sites in EcN chromosome can be selected by the final user to harbour multiple GOIs. Besides obvious considerations for the selection of the integration sites, like avoiding essential genes, it is important to conveniently select the homologous regions (HR1 and HR2) cloned in the suicide pGEC vector. Both non‐essential genes and intergenic regions can be targets of insertions. Distant HRs can be selected to produce a gene replacement (deletion) along with the insertion, as we have shown, but this is not a requirement of the system, as HRs can be selected without any intervening DNA, avoiding deletion of any bp in the chromosome along with the insertion. It is also important that the selected HRs are unique in EcN chromosome, lacking repetitive sequences or any other genetic duplication. For instance for the integration sites used in this work, three different flu genes and four fimAICDFGH operons exist on EcN chromosome (GeneBank CP007799.1). We have selected the gene flu located in the positions 3399340‐3402462 from CP007799.1 and the fimAICDFGH operon located between 4981488 and 4989160 in CP007799.1. The 500 bp DNA regions upstream and downstream used as HRs from the fim operon were unique. However, in the case of flu, repetitions of the DNA regions upstream and downstream were detected in other sites of the EcN genome, which led us to the selection of HRs located within the 5' and 3' ends of flu ORF. With this type of simple analysis, unique HRs were selected and gene insertions were found always at the targeted site and not in other loci of EcN genome. Also importantly, we found that all KmR and CmR EcN colonies growing in LB media (lacking DAPA) after conjugation are bona‐fide cointegrants at the expected site, making unnecessary the PCR test of the cointegrants prior to their resolution. In our constructs, we have used a standard length of the HRs of ca. 500 bp for insertions in EcN. However, it is likely that this length could be reduced if necessary to avoid any repetitive element in the genome. Our previous experience with suicide integrative vectors in E. coli K‐12 (Piñero‐Lambea et al., 2015a; Ruano‐Gallego et al., 2015) and enteropathogenic E. coli (Cepeda‐Molero et al., 2017), indicates that the length of the HRs can be reduced to ca. 300 bp without an apparent decline in the insertion frequency.
In conclusion, the straightforward genetic approach described in this work can accelerate the generation of multiple markerless insertions/deletions in EcN chromosome needed for the generation of living bacterial therapeutics like, for instance the expression of large nanomachines for protein delivery (Ruano‐Gallego et al., 2015) or complex genetic regulatory circuits responding to environmental cues (Riglar et al., 2017). Given the broad host range of RP4 conjugation (Cabezón et al., 2015; Silbert and Lorenzo, 2021), this approach could be also applicable for different commensal and probiotic E. coli strains (Wassenaar, 2016) and adapted to other Gram‐negative bacteria of interest as biotherapeutic chassis (Chua et al., 2017; Wegmann et al., 2017; Taketani et al., 2020).
Experimental procedures
Bacterial strains and growth conditions
Escherichia coli strains used in the work are detailed in Table S1. Details of the generation of the recombinant EcN strains are described in the Supporting Information. EcN strains were grown at 37 °C in Lysogeny broth (LB) agar plates (1.5% w/v) or in liquid LB medium unless otherwise indicated. Antibiotics or inductors were added at the following concentrations: chloramphenicol (Cm) at 30 μg ml−1; kanamycin (Km) at 50 μg ml−1; anhydrotetracycline (aTc) at 200 ng ml−1; and, isopropyl‐ß‐D‐thiogalactopyranoside (IPTG) at 0.1 mM; L‐arabinose (L‐ara) at 0.4% (w/v). When indicated, diaminopimelic acid (DAPA) was added at 0.3 mM to LB media for complementation of dapA mutants.
Plasmids, primers and cloning procedures
Plasmids used in this study are listed in Table S1. Oligonucleotides (MilliporeSigma, Merck, Darmstadt, Germany) used as primers are listed in Table S2. Cloning procedures were performed following standard protocols of DNA digestion with restriction enzymes and ligation (Sambrook and Russel, 2001). Details of DNA constructs are described in the Supporting Information. All DNA constructs were sequenced by the chain‐termination Sanger method (Macrogen Inc., Seoul, South Korea).
Conjugation assay
Escherichia coli MFDpir strain (RP4, π‐expressing, Mu‐deficient, ΔdapA) (Ferrières et al., 2010) was transformed with the pGEC‐derivative (KmR, π‐dependent R6K‐ori and oriT RP4) previously constructed and replicated in the π‐expressing E. coli strain BW25141 (Datsenko and Wanner, 2000). MFDpir carrying the pGEC‐derivative served as donor and EcN as recipient strain during conjugation. Both strains were grown overnight at 37 °C in LB with agitation 250 rpm, in the presence of DAPA and Km for the donor strain. In case of conjugation with integrative plasmids, the recipient EcN strain was previously transformed with pACBSR (CmR; I‐SceI endonuclease and λ Red genes) (Herring et al., 2003) for the resolution of the cointegrants. Drops between 18 and 25 μl of overnight cultures corresponding to an Optical Density at 600 nm (OD600) of 0.1 were used of both donor and recipient strain. At first, the drop containing the donor strain was deposited on a LB‐agar medium containing DAPA for the correct growth of the auxotrophic donor strain. Once the drop of the donor strain dried, a second drop of the recipient strain was added on top. Drops of each strain were also deposited on the plate at a different site to check the correct growth of donor and recipient strains. After 3.5 h of incubation at 37 °C (plate face up), bacteria on the conjugation spot were harvested with a sterile loop and resuspended in 1 ml of LB. Serial dilutions were plated on LB‐Km (without DAPA) for selection of transconjugants and colony‐forming units (CFU) counting. In the case of integrative plasmids, bacteria were plated on LB+Km+Cm for the correct selection of cointegrants with pACBSR. Drops with only the donor or the recipient bacteria were also harvested and counted by serial dilutions on plates with appropriated media for donor (LB+Km+DAPA) or recipient (LB+Cm). The conjugation frequency was calculated as the ratio of EcN transconjugants versus EcN recipients.
Resolution of cointegrants
A colony of a cointegrant containing plasmid pACBSR (CmR) expressing the I‐SceI endonuclease and the λ Red protein under the control of PBAD promoter (inducible with L‐ara) was overnight grown in a flask with 10 ml of liquid LB with Cm and Km for the selection of pACBSR plasmid and gene of interest integration respectively. Next day, this culture was used to inoculate 5 ml LB with Cm at OD600 0.01 in agitation (250 rpm). When the culture reached an OD600 ~ 0.5, L‐ara 0.4% (w/v) was added with agitation to induce the expression of λ Red genes and I‐SceI endonuclease for the corresponding cleavage in the I‐SceI recognition sites flanking the integrated gene of interest. After 5 h of incubation, a sample of this culture was streaked on LB‐Cm plates using an inoculating loop and incubated overnight to isolate individual colonies, which were replicated in LB‐Cm and LB‐Cm‐Km to identify Km‐sensitive colonies. Using specific primers for each locus, see Supporting Experimental procedures, the resolution to the wild type or to the insertion mutant could be confirmed by PCR.
The percentage of cointegrants resolution was calculated from the indicated cultures, grown in LB‐Cm and induced 5 h with L‐ara, by plating serial dilutions in LB‐agar plates containing Cm (total CFU) or Cm and Km (cointegrants CFU). Cointegrants resolution was calculated as the percentage of Km‐sensitive colonies with respect to the total Cm‐resistant colonies.
Flow cytometry analysis
EcN bacteria corresponding to an OD600 of 1.0 were harvested for overnight LB cultures by centrifugation (4000 g, 3 min). When indicated, cultures were induced with IPTG 0.1 mM. Bacteria were washed twice with 1 ml of PBS and resuspended in 1 ml of PBS for analysis in a flow cytometer Cytoflex S (Beckman Coulter Inc., Brea, CA, USA). GFP was measured with a blue laser in the detection channel FITC (525/40) and mKATE2 with a yellow laser in the channel ECD (610/20).
Immunofluorescence microscopy
Pellets of 1 ml of EcN grown to OD600 ~ 0.5 were washed with 1 ml of cold PBS and resuspended in 100 μl of 3% (w/v) paraformaldehyde (PFA) and incubated 10 min at room temperature (RT) for bacteria fixation. PBS was added to dilute the PFA to 1%. Seventy microlitres of 0.01% (w/v) poly‐L‐lysine was added to the round coverslip and stayed for 10 min. Poly‐L‐Lysine drop was discarded and coverslip was dried for 5–10 min at RT. Then, 5 μl of fixed bacteria was added to the coverslip. After 10 min of incubation, the excess liquid was taken out and coverslip was washed three times with 50 μl of PBS. Finally, coverslips were mounted on glass slides with 2.5 μl of Prolong (Invitrogen) containing 0.5 μg l−1 DAPI solution (previously diluted from the stock). Samples were examined by confocal microscopy (Leica TCS SP8 multispectral confocal system, Leica Microsystems GmbH, Wetzlar, Germany).
Supernatant proteins preparation and SDS‐PAGE
EcN bacteria were grown in 10 ml of LB up to an OD600 ~ 2. Cells were harvested by centrifugation (4000 g, 10 min at 4 °C). One millilitre of the supernatant, after centrifugation at the maximal velocity (20 000 g, 10 min at 4 °C), was incubated for 60 min at 4 °C in the presence of trichloroacetic acid (TCA 20% w/v; Merck) for precipitation. After centrifugation (20 000 g, 15 min at 4°C), TCA‐precipitated protein pellets were rinsed with cold acetone (−20 °C), air‐dried and resuspended in 25 μl of SDS‐PAGE sample buffer. Proteins separated by SDS‐PAGE were stained with Coomassie Blue R‐250 (Biorad, Hercules, CA, USA).
Motility assays
Amounts of bacteria corresponding to an OD600 of 0.02 were taken from overnight static cultures (volumes from 7 to 10 μl) and introduced at the middle of soft LB‐agar medium plates (agar 0.3% w/v). Plates were incubated for ~ 8 h.
Genome sequencing and analysis
Genomic DNA samples from parental EcN wild type and modified EcNΔfim_mKATE2 Δmat_lux Ptet‐flhDC strains were isolated using the Gnome DNA kit (MP Biomedicals, Santa Ana, CA, USA). Next‐generation sequencing (NGS) was performed by whole‐genome libraries prepared following the Illumina TruSeq DNA PCR Free kit (Illumina, San Diego, CA, USA). Libraries from both DNA samples (numbered 118 and 185 respectively) were sequenced on an Illumina NovaSeq600 as 150‐bp paired‐ends reads (Macrogen Inc). Illumina genomic paired‐ends FASTQ files for both samples were provided by Macrogen Inc. Analysis of these sequencing files and their comparison with the reference genome CP007799.1 of E coli Nissle 1917, as well as with fasta sequence files of the inserts, was performed by the BioinfoGP service of CNB‐CSIC (https://bioinfogp.cnb.csic). Bioinformatic tools and procedures used in the analysis of genomic sequencing data to identify variations in the genome of the modified strain and in the inserted DNA are described in detail in the Supporting Information. Results of this analysis are shown in the Data S1 (.xlsx file, Microsoft). Whole‐genome sequencing data have been deposited in the National Center for Biotechnology Information under Sequence Read Archive (SRA) accession SRP341149 (BioProject PRJNA770875, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA770875).
Statistics
The mean and standard deviation of experimental values were calculated using prism 8.0 (GraphPad software Inc., San Diego, CA, USA). Data from independent experiments are shown as individual dots.
Author contributions
L.A.F. conceived this study and secured funding. E.M.S. and L.A.F. designed the experiments, analysed the results and interpreted the data. E.M.S. performed the experiments and prepared the draft manuscript and Figures. L.A.F. wrote the final manuscript and edited the final Figures. E.M.S. and L.A.F. revised and approved the final manuscript.
Conflict of interest
The authors declare that they have no competing interests.
Supporting information
Table S1. E. coli strains and plasmids used in this study.
Table S2. Oligonucleotides used as primers in this work.
Fig. S1. Site‐specific markerless integration in EcN chromosome. Scheme showing the integration of a gene of interest (GOI) under the control of a promoter (P) in the EcN chromosome using a suicide conjugative plasmid pGEC derivative containing the origin of transfer (oriT), the replication origin R6K, the KmR gene, and the GOI flanked by two homologous regions (HR1 and HR2) corresponding to regions located upstream and downstream of the target gene. The cassette formed by the GOI and the HRs is flanked by two I‐SceI restriction sites. The first recombination event (Recombination I) leads to the cointegrants obtaining which are further resolved by the expression of the I‐SceI endonuclease and the λ Red protein from the helper plasmid pACBSR. Double strand breaks generated by the I‐SceI endonuclease are repaired a second homologous recombination event (Recombination II) assisted by the λ Red protein that may produce either the wild type allele (i) or the insertion mutant (ii).
Fig. S2. PCR analysis of flu_Ptac‐gfp cointegrants. A. Scheme showing that cointegrants can be generated after homologous recombination using HR1‐flu (i) or HR2‐flu (ii). B‐C. Colony PCR of five cointegrants using primer oligonucleotides 1 and 2 to check homologous recombination with HR1‐flu (B) or with oligonucleotides 3 and 4 to check homologous recombination with HR2‐flu (C). PCR of EcN wt colony was used as negative control in both panels B and C.
Fig. S3. PCR analysis of fim_Ptac‐gfp cointegrants. A. Scheme showing that cointegrants can be generated after homologous recombination using HR1‐fim (i) or HR2‐fim (ii). B‐C. Colony PCR of five cointegrants using primer oligonucleotides 5 and 2 to check homologous recombination with HR1‐fim (B) or with oligonucleotides 3 and 6 to check homologous recombination with HR2‐fim (C). PCR of EcN wt colony was used as negative control in both panels B and C.
Fig. S4. PCR analysis of the resolution of flu_Ptac‐gfp cointegrants. A. Replica plating of 40 colonies randomly picked after resolution of EcN flu_Ptac‐gfp cointegrants using LB Cm plates, with and without Km, to check the loss of Km resistance. B. Scheme of the cointegrants resolved to the Ptac‐gfp insertion. C‐D. Colony PCR of 10 colonies sensitive to Km with oligonucleotides 1 and 2 to check the insertion Ptac‐gfp using the upstream region of flu locus (C) or with oligonucleotides 3 and 4 to check the insertion using the downstream region of flu locus (D). PCR of EcN wt colony was used as negative control in both panels C and D.
Fig. S5. PCR analysis of the resolution of fim_Ptac‐gfp cointegrants. A. Replica plating of 40 colonies randomly picked after resolution of EcN fim_Ptac‐gfp cointegrants using LB Cm plates, with and without Km, to check the loss of Km resistance. B. Scheme of the cointegrants resolved to the Ptac‐gfp insertion. C‐D. Colony PCR of 10 colonies sensitive to Km with oligonucleotides 5 and 2 to check the insertion Ptac‐gfp using the upstream region of fim locus (C) or with oligonucleotides 3 and 6 to check the insertion using the downstream region of fim locus (D). PCR of EcN wt colony was used as negative control in both panels C and D.
Fig. S6. Expression of GFP in EcNΔflu_Ptac‐gfp and EcNΔfim_Ptac‐gfp cointegrants. Flow cytometry analysis of EcN wild type bacteria (EcN) and derivative strains EcNΔflu_gfp and EcNΔfim_gfp carrying insertion of Ptac‐gfp replacing flu or fim locus. Bacteria were harvested from cultures of the corresponding strain in LB with IPTG 0.1 mM.
Fig. S7. Integration frequency of pGECfimEcN_gfp in EcN carrying pACBSR or pACBS. Frequencies are calculated as the ratio of cointegrants (C) vs. recipients (R). Horizontal lines indicate means of four independent assays (n=4). Vertical bars indicated standard deviation. Plasmid pACBS lacks the λRed genes found in pACBSR.
Fig. S8. DNA Sanger sequencing of the plasmid preparation of pGEC_matEcN‐lux used for integration of lux operon in EcN. The mutated nucleotide (G to A) in the start codon of luxC gene is labelled with an arrow.
Data S1.
Acknowledgements
We specially thank Dr. Rafael Torres‐Pérez and Dr. Juan Carlos Oliveros (BioinfoGP, CNB‐CSIC) for the bioinformatic analysis of whole‐genome sequencing data. We also thank the excellent technical support of CNB‐CSIC core scientific facilities ‘Advanced Light Microscopy’ and ‘Flow Cytometry’.
Microbial Biotechnology (2022) 15(5), 1374–1391
Funding information
This work was supported by research Grants BIO2017‐89081‐R (Agencia Española de Investigación AEI/MICIU/FEDER, EU), and FET Open 965018‐ BIOCELLPHE of the European Union’s Horizon 2020 Future and Emerging Technologies research and innovation programme.
References
- Altenhoefer, A. , Oswald, S. , Sonnenborn, U. , Enders, C. , Schulze, J. , Hacker, J. , and Oelschlaeger, T.A. (2004) The probiotic Escherichia coli strain Nissle 1917 interferes with invasion of human intestinal epithelial cells by different enteroinvasive bacterial pathogens. FEMS Immunol Med Microbiol 40: 223–229. [DOI] [PubMed] [Google Scholar]
- Aminov, R.I. (2011) Horizontal gene exchange in environmental microbiota. Front Microbiol 2: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, J.K. , Smith, T.G. , and Hoover, T.R. (2010) Sense and sensibility: flagellum‐mediated gene regulation. Trends Microbiol 18: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnsen, J. , Deriu, E. , Sassone‐Corsi, M. , and Raffatellu, M. (2013) Probiotics: properties, examples, and specific applications. Cold Spring Harb Perspect Med 3: a010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, J.C. , and Kowalczykowski, S.C. (2016) RecA: regulation and mechanism of a molecular search engine. Trends Biochem Sci 41: 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram, R. , and Hillen, W. (2008) The application of Tet repressor in prokaryotic gene regulation and expression. Microb Biotechnol 1: 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, H.A. , Comstock, L.J. , and Vasser, M. (1983) The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci USA 80: 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito, I.L. (2021) Examining horizontal gene transfer in microbial communities. Nat Rev Microbiol 19: 442–453. [DOI] [PubMed] [Google Scholar]
- Brunner, M. , and Bujard, H. (1987) Promoter recognition and promoter strength in the Escherichia coli system. EMBO J 6: 3139–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezón, E. , Ripoll‐Rozada, J. , Peña, A. , de la Cruz, F. , and Arechaga, I. (2014) Towards an integrated model of bacterial conjugation. FEMS Microbiol Rev 39: 81–95. 10.1111/1574-6976.12085. [DOI] [PubMed] [Google Scholar]
- Caldwell, B.J. , and Bell, C.E. (2019) Structure and mechanism of the Red recombination system of bacteriophage λ. Prog Biophys Mol Biol 147: 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda‐Molero, M. , Berger, C.N. , Walsham, A.D.S. , Ellis, S.J. , Wemyss‐Holden, S. , Schüller, S. , et al. (2017) Attaching and effacing (A/E) lesion formation by enteropathogenic E. coli on human intestinal mucosa is dependent on non‐LEE effectors. PLoS Pathog 13: e1006706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau, M.R. , Isabella, V.M. , Li, N. , and Kurtz, C.B. (2020) Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat Commun 11: 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, K.J. , Kwok, W.C. , Aggarwal, N. , Sun, T. , and Chang, M.W. (2017) Designer probiotics for the prevention and treatment of human diseases. Curr Opin Chem Biol 40: 8–16. [DOI] [PubMed] [Google Scholar]
- Chudakov, D.M. , Matz, M.V. , Lukyanov, S. , and Lukyanov, K.A. (2010) Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90: 1103–1163. [DOI] [PubMed] [Google Scholar]
- Churchill, J.J. , Anderson, D.G. , and Kowalczykowski, S.C. (1999) The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of chi, resulting in constitutive recombination activation. Genes Dev 13: 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell, I. , Agace, W. , Klemm, P. , Schembri, M. , Marild, S. , and Svanborg, C. (1996) Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc Natl Acad Sci USA 93: 9827–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran, C.P. , Cameron, A.D. , and Dorman, C.J. (2010) H‐NS silences gfp, the green fluorescent protein gene: gfpTCD is a genetically remastered gfp gene with reduced susceptibility to H‐NS‐mediated transcription silencing and with enhanced translation. J Bacteriol 192: 4790–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin, M. , Akin, A.R. , Collins, S.A. , Meganck, J. , Kim, J.‐B. , Baban, C.K. , et al. (2012) High resolution in vivo bioluminescent imaging for the study of bacterial tumour targeting. PLoS One 7: e30940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K.A. , and Wanner, B.L. (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay, N. , and Gottesman, S. (2012) A complex network of small non‐coding RNAs regulate motility in Escherichia coli . Mol Microbiol 86: 524–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriu, E. , Liu, J.Z. , Pezeshki, M. , Edwards, R.A. , Ochoa, R.J. , Contreras, H. , et al. (2013) Probiotic bacteria reduce salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 14: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham, M.S. , and Kowalczykowski, S.C. (2008) RecBCD enzyme and the repair of double‐stranded DNA breaks. Microbiol Mol Biol Rev 72: 642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, F. , and March, J.C. (2010) Engineered bacterial communication prevents Vibrio cholerae virulence in an infant mouse model. Proc Natl Acad Sci USA 107: 11260–11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, A.J.F. , Errington, J. , and Vollmer, W. (2020) Regulation of peptidoglycan synthesis and remodelling. Nat Rev Microbiol 18: 446–460. [DOI] [PubMed] [Google Scholar]
- Erhardt, M. , Namba, K. , and Hughes, K.T. (2010) Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harbor Perspect Biol 2: a000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorec, A.J.H. , Ozdemir, T. , Doshi, A. , Ho, Y.‐K. , Rosa, L. , Rutter, J. , et al. (2019) Two new plasmid post‐segregational killing mechanisms for the implementation of synthetic gene networks in Escherichia coli . iScience 14: 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher, T. , Karcagi, I. , Gyorfy, Z. , Umenhoffer, K. , Csorgo, B. , and Posfai, G. (2008) Scarless engineering of the Escherichia coli genome. Methods Mol Biol 416: 251–259. 10.1007/978-1-59745-321-9_16. [DOI] [PubMed] [Google Scholar]
- Ferrières, L. , Hémery, G. , Nham, T. , Guerout, A.M. , Mazel, D. , Beloin, C. , and Ghigo, J.M. (2010) Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad‐host‐range RP4 conjugative machinery. J Bacteriol 192: 6418–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, D.M. , Bonocora, R.P. , and Wade, J.T. (2014) Comprehensive mapping of the Escherichia coli flagellar regulatory network. PLoS Genet 10: e1004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahan, C.G. (2012) The bacterial lux reporter system: applications in bacterial localisation studies. Curr Gene Ther 12: 12–19. [DOI] [PubMed] [Google Scholar]
- Gerdes, K. , Thisted, T. , and Martinussen, J. (1990) Mechanism of post‐segregational killing by the hoklsok system of plasmid R1: sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid‐free cells. Mol Microbiol 4: 1807–1818. [DOI] [PubMed] [Google Scholar]
- Grahn, A.M. , Haase, J. , Bamford, D.H. , and Lanka, E. (2000) Components of the RP4 conjugative transfer apparatus form an envelope structure bridging inner and outer membranes of donor cells: implications for related macromolecule transport systems. J Bacteriol 182: 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, V. , Dahl, M. , and Klemm, P. (2010) Probiotic Escherichia coli strain Nissle 1917 outcompetes intestinal pathogens during biofilm formation. J Med Microbiol 59: 392–399. [DOI] [PubMed] [Google Scholar]
- Henker, J. , Laass, M. , Blokhin, B.M. , Bolbot, Y.K. , Maydannik, V.G. , Elze, M. , et al. (2007) The probiotic Escherichia coli strain Nissle 1917 (EcN) stops acute diarrhoea in infants and toddlers. Eur J Pediatr 166: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henker, J. , Muller, S. , Laass, M.W. , Schreiner, A. , and Schulze, J. (2008) Probiotic Escherichia coli Nissle 1917 (EcN) for successful remission maintenance of ulcerative colitis in children and adolescents: an open‐label pilot study. Z Gastroenterol 46: 874–875. [DOI] [PubMed] [Google Scholar]
- Herring, C.D. , Glasner, J.D. , and Blattner, F.R. (2003) Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli . Gene 311: 153–163. [DOI] [PubMed] [Google Scholar]
- Isabella, V.M. , Ha, B.N. , Castillo, M.J. , Lubkowicz, D.J. , Rowe, S.E. , Millet, Y.A. , et al. (2018) Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat Biotechnol 36: 857–864. [DOI] [PubMed] [Google Scholar]
- Kan, A. , Gelfat, I. , Emani, S. , Praveschotinunt, P. , and Joshi, N.S. (2021) Plasmid vectors for in vivo selection‐free use with the probiotic E. coli Nissle 1917. ACS Synth Biol 10: 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, C.W. , Lim, H.G. , Yang, J. , Noh, M.H. , Seo, S.W. , and Jung, G.Y. (2018) Synthetic auxotrophs for stable and tunable maintenance of plasmid copy number. Metab Eng 48: 121–128. [DOI] [PubMed] [Google Scholar]
- Kelly, V.W. , Liang, B.K. , and Sirk, S.J. (2020) Living therapeutics: the next frontier of precision medicine. ACS Synth Biol 9: 3184–3201. [DOI] [PubMed] [Google Scholar]
- Kotłowski, R. (2016) Use of Escherichia coli Nissle 1917 producing recombinant colicins for treatment of IBD patients. Med Hypotheses 93: 8–10. [DOI] [PubMed] [Google Scholar]
- Kroll, J. , Klinter, S. , Schneider, C. , Voß, I. , and Steinbüchel, A. (2010) Plasmid addiction systems: perspectives and applications in biotechnology. Microb Biotechnol 3: 634–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, C.B. , Millet, Y.A. , Puurunen, M.K. , Perreault, M. , Charbonneau, M.R. , Isabella, V.M. , et al. (2019) An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose‐dependent exposure in healthy humans. Sci Transl Med 11: eaau7975. [DOI] [PubMed] [Google Scholar]
- Leventhal, D.S. , Sokolovska, A. , Li, N. , Plescia, C. , Kolodziej, S.A. , Gallant, C.W. , et al. (2020) Immunotherapy with engineered bacteria by targeting the STING pathway for anti‐tumor immunity. Nat Commun 11: 2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner, H. , Leschner, S. , Endmann, A. , Westphal, K. , Wolf, K. , Kochruebe, K. , et al. (2009) Drug‐inducible remote control of gene expression by probiotic Escherichia coli Nissle 1917 in intestine, tumor and gall bladder of mice. Microbes Infect 11: 1097–1105. [DOI] [PubMed] [Google Scholar]
- de Lorenzo, V. , Herrero, M. , Jakubzik, U. , and Timmis, K.N. (1990) Mini‐Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram‐negative eubacteria. J Bacteriol 172: 6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losurdo, G. , Iannone, A. , Contaldo, A. , Ierardi, E. , Di Leo, A. , and Principi, M. (2015) Escherichia coli Nissle 1917 in ulcerative colitis treatment: systematic review and meta‐analysis. J Gastrointestin Liver Dis 24: 499–505. [DOI] [PubMed] [Google Scholar]
- Martinez‐Garcia, E. , Aparicio, T. , Goni‐Moreno, A. , Fraile, S. , and de Lorenzo, V. (2015) SEVA 2.0: an update of the Standard European Vector Architecture for de‐/re‐construction of bacterial functionalities. Nucleic Acids Res 43: D1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García, E. , and de Lorenzo, V. (2011) Engineering multiple genomic deletions in Gram‐negative bacteria: analysis of the multi‐resistant antibiotic profile of Pseudomonas putida KT2440. Environ Microbiol 13: 2702–2716. [DOI] [PubMed] [Google Scholar]
- Martinez‐Garcia, E. , and de Lorenzo, V. (2012) Transposon‐based and plasmid‐based genetic tools for editing genomes of gram‐negative bacteria. Methods Mol Biol 813: 267–283. [DOI] [PubMed] [Google Scholar]
- de Moraes, M.H. , and Teplitski, M. (2015) Fast and efficient three‐step target‐specific curing of a virulence plasmid in Salmonella enterica. AMB Express 5: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, B. , Yang, Y. , Tham, W.L. , Chen, L. , Guo, J. , and Zhu, G. (2016) Genetic engineering of probiotic Escherichia coli Nissle 1917 for clinical application. Appl Microbiol Biotechnol 100: 8693–8699. [DOI] [PubMed] [Google Scholar]
- Ozdemir, T. , Fedorec, A.J.H. , Danino, T. , and Barnes, C.P. (2018) Synthetic biology and engineered live biotherapeutics: toward increasing system complexity. Cell Syst 7: 5–16. [DOI] [PubMed] [Google Scholar]
- Pansegrau, W. , Lanka, E. , Barth, P.T. , Figurski, D.H. , Guiney, D.G. , Haas, D. , et al. (1994) Complete nucleotide sequence of Birmingham IncP alpha plasmids. Compilation and comparative analysis. J Mol Biol 239: 623–663. [DOI] [PubMed] [Google Scholar]
- Patzer, S.I. , Baquero, M.R. , Bravo, D. , Moreno, F. , and Hantke, K. (2003) The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IroN. Microbiology 149: 2557–2570. [DOI] [PubMed] [Google Scholar]
- Piñero‐Lambea, C. , Bodelón, G. , Fernández‐Periáñez, R. , Cuesta, A.M. , Álvarez‐Vallina, L. , and Fernández, L.A. (2015) Programming controlled adhesion of E. coli to target surfaces, cells, and tumors with synthetic adhesins. ACS Synth Biol 4: 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero‐Lambea, C. , Ruano‐Gallego, D. , and Fernández, L.Á. (2015) Engineered bacteria as therapeutic agents. Curr Opin Biotechnol 35: 94–102. [DOI] [PubMed] [Google Scholar]
- Pines, G. , Freed, E.F. , Winkler, J.D. , and Gill, R.T. (2015) Bacterial recombineering: genome engineering via phage‐based homologous recombination. ACS Synth Biol 4: 1176–1185. [DOI] [PubMed] [Google Scholar]
- Posfai, G. , Kolisnychenko, V. , Bereczki, Z. , and Blattner, F.R. (1999) Markerless gene replacement in Escherichia coli stimulated by a double‐strand break in the chromosome. Nucleic Acids Res 27: 4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. , Hu, S. , McHugh, L. , Lueders, K. , Henry, K. , Zhao, Q. , et al. (2005) Toward a live microbial microbicide for HIV: commensal bacteria secreting an HIV fusion inhibitor peptide. Proc Natl Acad Sci USA 102: 11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendon, M.A. , Saldana, Z. , Erdem, A.L. , Monteiro‐Neto, V. , Vazquez, A. , Kaper, J.B. , et al. (2007) Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc Natl Acad Sci USA 104: 10637–10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riglar, D.T. , Giessen, T.W. , Baym, M. , Kerns, S.J. , Niederhuber, M.J. , Bronson, R.T. , et al. (2017) Engineered bacteria can function in the mammalian gut long‐term as live diagnostics of inflammation. Nat Biotechnol 35: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouanet, A. , Bolca, S. , Bru, A. , Claes, I. , Cvejic, H. , Girgis, H. , et al. (2020) Live biotherapeutic products, a road map for safety assessment. Front Med 7: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano‐Gallego, D. , Álvarez, B. , and Fernández, L.Á. (2015) Engineering the controlled assembly of filamentous injectisomes in E. coli K‐12 for protein translocation into mammalian cells. ACS Synth Biol 4: 1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rund, S.A. , Rohde, H. , Sonnenborn, U. , and Oelschlaeger, T.A. (2013) Antagonistic effects of probiotic Escherichia coli Nissle 1917 on EHEC strains of serotype O104:H4 and O157:H7. Int J Med Microbiol 303: 1–8. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , and Russel, D.W. (2001) Molecular Cloning. A Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sawitzke, J.A. , Thomason, L.C. , Costantino, N. , Bubunenko, M. , Datta, S. , and Court, D.L. (2007) Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. In Advanced Bacterial Genetics: Use of Transposons and Phage for Genomic Engineering. Hughes, K.T. , and Maloy, S.R. (eds.), Amsterdam, Netherlands: Elsevier, pp. 171–199. URL https://pubmed.ncbi.nlm.nih.gov/17352923/ [DOI] [PubMed] [Google Scholar]
- Schultz, M. (2008) Clinical use of E. coli Nissle 1917 in inflammatory bowel disease. Inflamm Bowel Dis 14: 1012–1018. [DOI] [PubMed] [Google Scholar]
- Sharan, S.K. , Thomason, L.C. , Kuznetsov, S.G. , and Court, D.L. (2009) Recombineering: a homologous recombination‐based method of genetic engineering. Nat Protoc 4: 206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbo, D. , Murphy, C.S. , Ermakova, G.V. , Solovieva, E.A. , Chepurnykh, T.V. , Shcheglov, A.S. , et al. (2009) Far‐red fluorescent tags for protein imaging in living tissues. Biochem J 418: 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbert, J. , Lorenzo, V. , and Aparicio, T. (2021) Refactoring the conjugation machinery of promiscuous plasmid RP4 into a device for conversion of gram‐negative isolates to Hfr strains. ACS Synth Biol 10: 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenborn, U. (2016) Escherichia coli strain Nissle 1917‐from bench to bedside and back: history of a special Escherichia coli strain with probiotic properties. FEMS Microbiol Lett 363: 1–6. 10.1093/femsle/fnw212. [DOI] [PubMed] [Google Scholar]
- Sonnenborn, U. , and Schulze, J. (2009) The non‐pathogenic Escherichia coli strain Nissle 1917 – features of a versatile probiotic. Microbial Ecol Health Dis 21: 122–158. [Google Scholar]
- Sourjik, V. (2004) Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol 12: 569–576. [DOI] [PubMed] [Google Scholar]
- Soutourina, O.A. , and Bertin, P.N. (2003) Regulation cascade of flagellar expression in Gram‐negative bacteria. FEMS Microbiol Rev 27: 505–523. [DOI] [PubMed] [Google Scholar]
- Soutourina, O. , Kolb, A. , Krin, E. , Laurent‐Winter, C. , Rimsky, S. , Danchin, A. , and Bertin, P. (1999) Multiple control of flagellum biosynthesis in Escherichia coli: role of H‐NS protein and the cyclic AMP‐catabolite activator protein complex in transcription of the flhDC master operon. J Bacteriol 181: 7500–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio, V. , Torres, A.G. , and Kaper, J.B. (2002) Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two‐component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli . Mol Microbiol 43: 809–821. [DOI] [PubMed] [Google Scholar]
- Taketani, M. , Zhang, J. , Zhang, S. , Triassi, A.J. , Huang, Y.J. , Griffith, L.G. , and Voigt, C.A. (2020) Genetic circuit design automation for the gut resident species Bacteroides thetaiotaomicron. Nat Biotechnol 38: 962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot, C.M. , Koenigsknecht, M.J. , Carlson, P.E. , Hatton, G.E. , Nelson, A.M. , Li, B.O. , et al. (2014) Antibiotic‐induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5: 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari, D.K. , Tiwari, M. , and Jin, T. (2020) Near‐infrared fluorescent protein and bioluminescence‐based probes for high‐resolution in vivo optical imaging. Mater Adv 1: 967–987. URL https://pubs.rsc.org/en/content/articlelanding/2020/ma/d0ma00273a [Google Scholar]
- Troge, A. , Scheppach, W. , Schroeder, B.O. , Rund, S.A. , Heuner, K. , Wehkamp, J. , et al. (2012) More than a marine propeller – the flagellum of the probiotic Escherichia coli strain Nissle 1917 is the major adhesin mediating binding to human mucus. Int J Med Microbiol 302: 304–314. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Sarov, M. , Rientjes, J. , Fu, J. , Hollak, H. , Kranz, H. , et al. (2006) An improved recombineering approach by adding RecA to lambda Red recombination. Mol Biotechnol 32: 43–53. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Fleming, R.T. , Westbrook, E.M. , Matsumura, P. , and McKay, D.B. (2006) Structure of the Escherichia coli FlhDC complex, a prokaryotic heteromeric regulator of transcription. J Mol Biol 355: 798–808. [DOI] [PubMed] [Google Scholar]
- Wassenaar, T.M. (2016) Insights from 100 years of research with probiotic E. coli . Eur J Microbiol Immunol 6: 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann, U. , Carvalho, A.L. , Stocks, M. , and Carding, S.R. (2017) Use of genetically modified bacteria for drug delivery in humans: revisiting the safety aspect. Sci Rep 7: 2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, M. , Troman, L. , Wan Nur Ismah, W.A.K. , Chaban, Y. , Avison, M.B. , Dillingham, M.S. , and Wigley, D.B. (2016) Structural basis for the inhibition of RecBCD by Gam and its synergistic antibacterial effect with quinolones. eLife 5: e22963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winson, M.K. , Swift, S. , Hill, P.J. , Sims, C.M. , Griesmayr, G. , Bycroft, B.W. , et al. (1998) Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini‐Tn5 constructs. FEMS Microbiol Lett 163: 193–202. [DOI] [PubMed] [Google Scholar]
- van der Woude, M.W. , and Henderson, I.R. (2008) Regulation and function of Ag43 (flu). Annu Rev Microbiol 62: 153–169. [DOI] [PubMed] [Google Scholar]
- Wright, O. , Stan, G.B. , and Ellis, T. (2013) Building‐in biosafety for synthetic biology. Microbiology 159: 1221–1235. [DOI] [PubMed] [Google Scholar]
- Yu, X. , Lin, C. , Yu, J. , Qi, Q. , and Wang, Q. (2020) Bioengineered Escherichia coli Nissle 1917 for tumour‐targeting therapy. Microb Biotechnol 13: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. E. coli strains and plasmids used in this study.
Table S2. Oligonucleotides used as primers in this work.
Fig. S1. Site‐specific markerless integration in EcN chromosome. Scheme showing the integration of a gene of interest (GOI) under the control of a promoter (P) in the EcN chromosome using a suicide conjugative plasmid pGEC derivative containing the origin of transfer (oriT), the replication origin R6K, the KmR gene, and the GOI flanked by two homologous regions (HR1 and HR2) corresponding to regions located upstream and downstream of the target gene. The cassette formed by the GOI and the HRs is flanked by two I‐SceI restriction sites. The first recombination event (Recombination I) leads to the cointegrants obtaining which are further resolved by the expression of the I‐SceI endonuclease and the λ Red protein from the helper plasmid pACBSR. Double strand breaks generated by the I‐SceI endonuclease are repaired a second homologous recombination event (Recombination II) assisted by the λ Red protein that may produce either the wild type allele (i) or the insertion mutant (ii).
Fig. S2. PCR analysis of flu_Ptac‐gfp cointegrants. A. Scheme showing that cointegrants can be generated after homologous recombination using HR1‐flu (i) or HR2‐flu (ii). B‐C. Colony PCR of five cointegrants using primer oligonucleotides 1 and 2 to check homologous recombination with HR1‐flu (B) or with oligonucleotides 3 and 4 to check homologous recombination with HR2‐flu (C). PCR of EcN wt colony was used as negative control in both panels B and C.
Fig. S3. PCR analysis of fim_Ptac‐gfp cointegrants. A. Scheme showing that cointegrants can be generated after homologous recombination using HR1‐fim (i) or HR2‐fim (ii). B‐C. Colony PCR of five cointegrants using primer oligonucleotides 5 and 2 to check homologous recombination with HR1‐fim (B) or with oligonucleotides 3 and 6 to check homologous recombination with HR2‐fim (C). PCR of EcN wt colony was used as negative control in both panels B and C.
Fig. S4. PCR analysis of the resolution of flu_Ptac‐gfp cointegrants. A. Replica plating of 40 colonies randomly picked after resolution of EcN flu_Ptac‐gfp cointegrants using LB Cm plates, with and without Km, to check the loss of Km resistance. B. Scheme of the cointegrants resolved to the Ptac‐gfp insertion. C‐D. Colony PCR of 10 colonies sensitive to Km with oligonucleotides 1 and 2 to check the insertion Ptac‐gfp using the upstream region of flu locus (C) or with oligonucleotides 3 and 4 to check the insertion using the downstream region of flu locus (D). PCR of EcN wt colony was used as negative control in both panels C and D.
Fig. S5. PCR analysis of the resolution of fim_Ptac‐gfp cointegrants. A. Replica plating of 40 colonies randomly picked after resolution of EcN fim_Ptac‐gfp cointegrants using LB Cm plates, with and without Km, to check the loss of Km resistance. B. Scheme of the cointegrants resolved to the Ptac‐gfp insertion. C‐D. Colony PCR of 10 colonies sensitive to Km with oligonucleotides 5 and 2 to check the insertion Ptac‐gfp using the upstream region of fim locus (C) or with oligonucleotides 3 and 6 to check the insertion using the downstream region of fim locus (D). PCR of EcN wt colony was used as negative control in both panels C and D.
Fig. S6. Expression of GFP in EcNΔflu_Ptac‐gfp and EcNΔfim_Ptac‐gfp cointegrants. Flow cytometry analysis of EcN wild type bacteria (EcN) and derivative strains EcNΔflu_gfp and EcNΔfim_gfp carrying insertion of Ptac‐gfp replacing flu or fim locus. Bacteria were harvested from cultures of the corresponding strain in LB with IPTG 0.1 mM.
Fig. S7. Integration frequency of pGECfimEcN_gfp in EcN carrying pACBSR or pACBS. Frequencies are calculated as the ratio of cointegrants (C) vs. recipients (R). Horizontal lines indicate means of four independent assays (n=4). Vertical bars indicated standard deviation. Plasmid pACBS lacks the λRed genes found in pACBSR.
Fig. S8. DNA Sanger sequencing of the plasmid preparation of pGEC_matEcN‐lux used for integration of lux operon in EcN. The mutated nucleotide (G to A) in the start codon of luxC gene is labelled with an arrow.
Data S1.