

Summary
The development and advent of mutagenesis tools for solventogenic clostridial species in recent years has allowed for the increased refinement of industrially relevant strains. In this study we have utilised CLEAVE™, a CRISPR/Cas genome editing system developed by Green Biologics Ltd., to engineer a strain of Clostridium saccharoperbutylacetonicum N1‐4(HMT) with potentially useful solvents titres and energy metabolism. As one of two enzymes responsible for the conversion of glyceraldehyde‐3‐phosphate (GAP) to 3‐phosphoglyceric acid in glycolysis, it was hypothesised that deletion of gapN would increase ATP and NADH production that could in turn improve solvent production. Herein, whole genome sequencing has been used to evaluate CLEAVE™ and the successful knockout of gapN, demonstrating a clean knockout with no other detectable variations from the wild type sequence. Elevated solvent levels were detected during the first 24 h of batch fermentation, indicating an earlier shift to solventogenesis. A 2.4‐fold increase in ATP concentration was observed, and quantitation of NAD(P)H derivatives revealed a more reducing cytoplasm for the gapN strain. These findings expand our understanding of clostridium carbon metabolism and report a new approach to optimising biofuel production.
This article employs a combination of genetic, genomic and microbiological techniques to demonstrate that manipulation of glyceraldehyde‐3‐phosphate activity in a solventogenic clostridium species can enhance butanol and acetone titres. These findings expand our understanding of clostridium carbon metabolism and report a new approach to optimising biofuel production.
Introduction
ABE fermentation using solventogenic clostridial species has been used for the industrial production of acetone, butanol and ethanol for over a century (for a review see Green, 2011). In the century that has passed since the discovery of the original Weizmann strain, species such as Clostridium beijerinckii (Jones and Keis, 1995) and Clostridium saccharoperbutylacetonicum (Hongo et al., 1969) have surpassed the original Weizmann strain as the preferred choice for ABE fermentation when grown on carbohydrate material. Unlike the well‐characterised strain Clostridium acetobutylicum, C. saccharoperbutylacetonicum N1‐4(HMT) is a relative newcomer to the world of industrial ABE production. First discovered in 1969 (Hongo et al., 1969) the full genome sequence of the strain was not published until 2014 (Poehlein et al., 2014). Over the years, fermentation experiments have been performed using a wide variety of feedstocks that have confirmed this strain as a butanol hyperproducer (Tashiro et al., 2007; Al‐Shorgani et al., 2012; Noguchi et al., 2013). Early work demonstrated that unlike C. acetobutylicum strains, loss of solvent production in C. saccharoperbutylacetonicum N1‐4(HMT) did not result from loss of the sol genes but more so from the dysregulation of the sol operon and upstream genes encoding other pathway enzymes (Kostan et al., 2010). Further to this, the mega plasmid found in C. saccharoperbutylacetonicum N1‐4(HMT) has also been linked to ester production of butyl acetate and butyl butyrate (Gu et al., 2019). To better understand butanol production in C. saccharoperbutylacetonicum N1‐4(HMT), the key biosynthetic genes (either endogenous or exogenous) including the sol operon (bld‐ctfA‐ ctfB‐adc), adhE1, adhE1D485G, thl, thlA1V5A, thlAV5A and the expression cassette EC (thl‐hbd‐crt‐bcd) were overexpressed in the strain (Wang et al., 2017b). In summary, overexpression of the sol operon resulted in a 400% increase in the production of ethanol with the highest increase in butanol (13.7%) seen in the strain with the over expression of the EC cassette. In an attempt to better understand and elevate the process of carbon catabolite repression (CCR), the sucrose metabolic pathway was shut down via inactivation of the gene scrO, resulting in a decrease in sucrose consumption by 28.9% and a decrease in ABE production by 44.1% using sucrose as the main carbon source. Additionally, deletion of the scrR gene alleviated CCR in the glucose/sucrose mixed fermentation, and overexpression of the endogenous sucrose pathway resulted in increased ABE production (Zhang et al., 2018).
Despite significant understanding of the Clostridium genus and its genome it is notoriously difficult to engineer. As a result, tools available for genetic engineering of Clostridium have lagged behind those for Gram‐negative species such as E. coli. Moreover, the development of tools for creation of robust strain development in C. saccharoperbutylacetonicum N1‐4(HMT) has lagged behind that of other Clostridium strains such as C. acetobutylicum and C. beijerinckii, where the ClosTron system developed by Heap et al. (2007) has been widely used (Underwood et al., 2009; Cartman et al., 2010; Cooksley et al., 2010, 2012; Antunes et al., 2011; Wietzke and Bahl, 2012; Xu et al., 2015). However, CRISPR/Cas technology, a powerful counter‐selection method, has recently been used to screen against homologous recombination events in Clostridium (Bruder et al., 2016; Huang et al., 2016; Nagaraju et al., 2016; Wang, et al., 2017; Wasels et al., 2017). Moreover, CLEAVE™ technology developed by Green Biologics has previously been shown to successfully generate strains carrying desired SNPs as well as those with deletions and insertions (Atmadjaja et al., 2019), although detailed whole genome sequencing (WGS) analyses have not previously been performed on the engineered strains to screen for unwanted mutations. Hence, this method was chosen for chromosomal deletions in the current study and CLEAVE™ was assessed using WGS.
The enzyme of interest in the current study is GapN, a cytosolic non‐phosphorylating NADP‐dependent glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; EC.1.2.1.9) that catalyses the irreversible oxidation of glyceraldehyde‐3‐phosphate (G3P) to 3‐phospholglycerate (Fig. 1; Iddar et al., 2002). However, three classes of GAPDH exist that are known to be involved in the central carbon metabolism pathway, with the other two being; (i) a NAD+‐dependent glycolytic enzyme (EC.1.2.1.12, GapA in C. saccharoperbutylacetonicum) found in the cytoplasm of all organisms that plays a key role in the Embden‐Meyerhoff pathway (Fothergill‐Gilmore and Michels, 1993); and (ii) a NADP+‐dependent GAPDH found in photosynthetic organisms (EC. 1.2.1.13, absent in C. saccharoperbutylacetonicum), that is a key component of the reductive pentose‐phosphate cycle (Brinkmann et al., 1989). Originally discovered in photosynthetic eukaryotes (Mateos and Serrano, 1992), GapN was subsequently identified in Streptococcus mutans and S. salivarius (Boyd et al., 1995). Recombinant GapN from Streptococcus mutans has been expressed in Corynebacterium glutamicum as a route for NADPH generation to facilitate l‐lysine production (Takeno et al., 2010), and recombinant GapN from C. acetobutylicum has been expressed in E. coli and has been shown to have an absolute specificity for NADPH (Iddar et al., 2002). GapN activity is likely to diminish the production of NADH via GAPDH (Fig. 1), decreasing this valuable source of reducing power that is obligately required solvent production via ABE fermentation. Furthermore, GapN activity also diminishes the production of ATP by phosphoglycerate kinase (PGK; Fig. 1), which is likely to influence cell growth and potentially impact upon ABE yield. Hence, it was hypothesised that deletion of gapN would enhance ABE yield, presumably via increasing the production of NADH and ATP. The study herein describes the deletion of gapN from C. saccharoperbutylacetonicum using CLEAVETM, assessment of this mutagenesis tool using WGS, and assessment of the impact of gapN deletion upon growth, acid/solvent titre, redox poise and ATP yield.
Fig. 1.
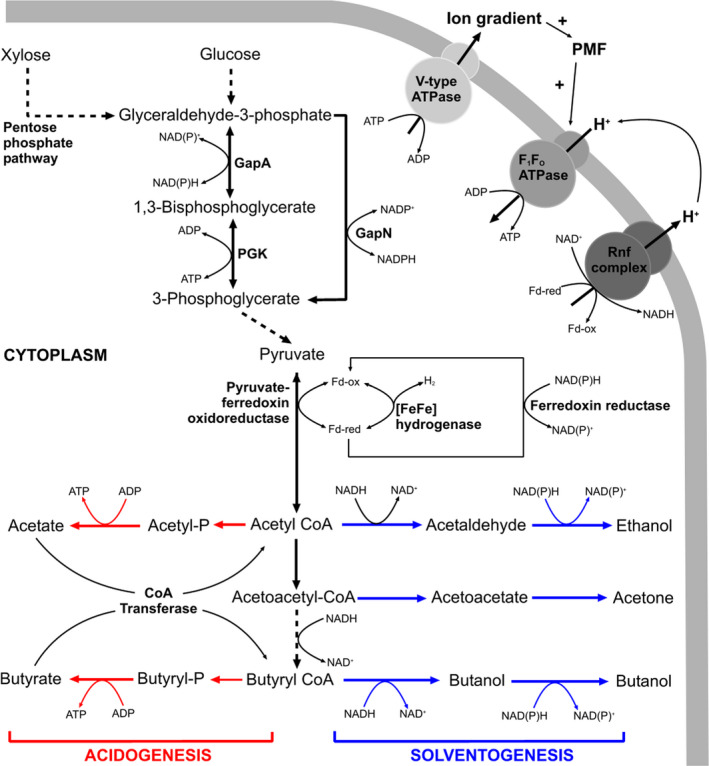
GapN is the monophosphorylating NADP‐dependant GAPDH found in C. saccharoperbutylacetonicum N1‐4(HMT). Along with GapA, GapN is responsible for the conversion of glyceraldehyde‐3‐phosphate to 3‐phosphoglycerate. With the GapN reaction producing NADPH rather than NADH and also circumventing ATP generation by PGK, it is hypothesised that deletion of gapN will result in an increase in the concentration of ATP and elevate the NADH:NAD+ ratio. Pathways for acidogenesis, solventogenesis and generation of PMF are shown to highlight their involvement in consumption/production of NADH and ATP.
Results
Deletion of gapN in C. saccharoperbutylacetonicum N1‐4(HMT) using CLEAVE™
Deletion of gapN was carried out using the proprietary CRISPR/Cas technology CLEAVE™ developed by Green Biologics Limited (Atmadjaja et al., 2019). After successful construction of the pMTL82154‐gapN‐HR vector (Fig. S1 and Table S1) and transformation into E. coli, this construct was isolated and was used to transform C. saccharoperbutylacetonicum N1‐4(HMT). Successful transformants were subjected to several rounds of sub‐culturing intended to promote a double recombination event between pMTL82154‐gapN‐HR and the chromosome, yielding a clean deletion of the entire gapN gene.
Following sub‐culturing, the cells were transformed with the killing vector pMTL83251_Ldr_DR_Sp_DR (Fig. S2) that is capable of producing crRNA that targets the PAM protospacer site derived from the middle of the gapN gene. The crRNA encoded by the killing vector was designed to recognize the PAM/protospacer within cells that have been unable to undergo the double recombination event and still contain the wild type gapN locus, resulting in killing of only these wild type cells. Following successful transformation of the killing vector, colonies were screened using the HR_F1 and HR_R2 primers that anneal upstream and downstream of gapN and were used in the construction of the HR vector (Fig. S1A and Table S1). Colonies that lacked gapN resulted in a PCR product of 1256 bp compared to the wild type at ~ 3000 bp containing the gapN gene. Successful colonies were sent for Sanger sequencing to confirm successful deletion of gapN.
WGS of ∆gapN and wild type C. saccharoperbutylacetonicum N1‐4(HMT)
To determine whether the genome editing via CLEAVE™ introduced any unwanted mutations, WGS was carried out on both wild type and ∆gapN C. saccharoperbutylacetonicum N1‐4(HMT). Genome libraries were prepared using the Nextera XT v2 protocol and sequenced using an Illumina MiSeq benchtop sequencer according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Raw reads have been uploaded to the European Nucleotide Archive and are available under accession numbers ERS6580404 (wild type C. saccharoperbutylacetonicum N1‐4(HMT); ‘C1’) and ERS6580405 (∆gapN C. saccharoperbutylacetonicum N1‐4(HMT); ‘C2’). The resulting short paired‐end reads were assessed using FastQC before and after trimming of the reads using Trimmomatic (Bolger et al., 2014). Trimmomatic removes adaptor sequences inserted during library preparation, as well as low quality reads and bases from each dataset. The final trimmed reads from the wild type and the ∆gapN strain were mapped to the reference C. saccharoperbutylacetonicum N1‐4(HMT) genome obtained from Genbank (CP004121.1). The mapping was carried out using BWA and Samtools (Li, 2013), and then Qualimap (García‐Alcalde et al., 2012) was used to assess the quality of genome mapping of both strains (Table 1). Qualimap revealed that the coverage depth varied, with 68.6 ± 33.2 seen for the ∆gapN strain compared to 18.8 ± 11.3 for the wild type strain. Although coverage varied, 97.12% of the total 596 655 reads for the wild type strain were mapped and 95.21% of the total 2 093 890 reads from the ∆gapN data were successfully mapped to the reference genome.
Table 1.
Qualimap results of ∆gapN and wild type C. saccharoperbutylacetonicum N1‐4(HMT) genome sequences following read mapping to the C. saccharoperbutylacetonicum N1‐4(HMT) reference genome from Genbank (CP004121.1).
| Characteristic | ∆gapN | Wild type |
|---|---|---|
| Reference size (bp) | 6 530 257 | 6 530 257 |
| Number of reads | 2093 890 | 596 655 |
| Mapped reads | 1 993 694/95.21% | 579 484/97.12% |
| Supplementary alignments | 1509/0.07% | 247/0.04% |
| Unmapped reads | 100 196/4.79% | 17 171/2.88% |
| Read min/max/mean length (bp) | 30/251/224.9 | 30/251/212.68 |
| Clipped reads | 21 456/1.02% | 4726/0.79% |
| Mapping quality | 59.11 | 59.30 |
| Mean coverage | 68.59 ± 33.2 | 18.8 ± 11.3 |
The mapped Illumina reads for the ∆gapN strain were analysed for variations compared to the reference genome using Snippy (https://github.com/tseemann/snippy). All 17 variations that were identified in the ∆gapN WGS (Table 2) were manually compared to our wild type WGS using the Integrated Genome Viewer (Robinson et al., 2011), and were found to be present in both our wild type parent strain and the ∆gapN strain. This indicates that these variations are not due to the CLEAVETM genome editing process and merely reflect minor differences in our genome sequences compared to the reference strain (CP004121.1).
Table 2.
Nucleotide variations between assembled ∆gapN strain and reference strain from Genbank (CP004121.1).
| Position | Gene | Product | Type | Strand | Reference | ∆gapN |
|---|---|---|---|---|---|---|
| 41500 | – | – | del | GTTTTTG | GTTTTG | |
| 297173 | mdtN_1 | Multidrug resistance protein MdtN | ins | + | GAAGTAAA | GAAGTAAAAGTAAA |
| 807822 | – | – | snp | G | T | |
| 2136980 | ybdL | Methionine aminotransferase | del | + | TAG | TG |
| 2136989 | ybdL | Methionine aminotransferase | complex | + | AAAGA | AG |
| 2137002 | ybdL | Methionine aminotransferase | del | + | ATTTTTTG | ATTTTTG |
| 2169690 | 01968 | hypothetical protein | snp | – | G | A |
| 2170090 | 01968 | hypothetical protein | snp | – | T | C |
| 2170579 | 01968 | hypothetical protein | snp | – | C | T |
| 2170673 | 01968 | hypothetical protein | del | – | CGCCTTGACGACCTTGAGAG | CG |
| 2171011 | 01968 | hypothetical protein | snp | – | C | T |
| 3257186 | rocR_1 | Arginine utilisation regulatory protein RocR | snp | + | T | G |
| 3506222 | 03225 | Nucleotidase | snp | – | T | C |
| 3651781 | rsgI_2 | Antisigma‐I factor RsgI | ins | – | GG | GATGGAGTTG |
| 4705891 | – | – | snp | G | A | |
| 6036488 | fdtB_2 | dTDP‐3‐amino‐3,6‐dideoxy‐alpha‐d‐galactopyranose transaminase | snp | C | T | |
| 6036553 | fdtB_2 | dTDP‐3‐amino‐3,6‐dideoxy‐alpha‐d‐galactopyranose transaminase | snp | G | A |
del, deletion; ins; insertion; snp; single nucleotide polymorphism.
Comparison was performed using Snippy. The ∆gapN deletion is not shown.
Solvent quantitation reveals an earlier entry to solventogenesis for the ∆gapN strain
The ∆gapN and wild type C. saccharoperbutylacetonicum N1‐4(HMT) strains were cultured via batch fermentation in YETM medium containing 40 g l−1 glucose and supplemented with 0.1 M MES for pH control. Both strains reached similar cell densities, with maximal OD600 values of 7.2 ± 0.2 for the wild type and 7.4 ± 0.3 for the ∆gapN strain (Fig. 2A). Between 9 h and 21 h in the fermentation, the media pH for ∆gapN strain was less acidic throughout compared to the wild type C. saccharoperbutylacetonicum N1‐4(HMT) (Fig. 2B). The redox potential of the growth media for both strains behaved in a predictable manner, both dropping to ~ −300 mV vs. NHE in the first few hours (Fig. 2C), and the rate of glucose consumption was greater between 10 h and 20 h the ∆gapN strain (Fig. 2D).
Fig. 2.
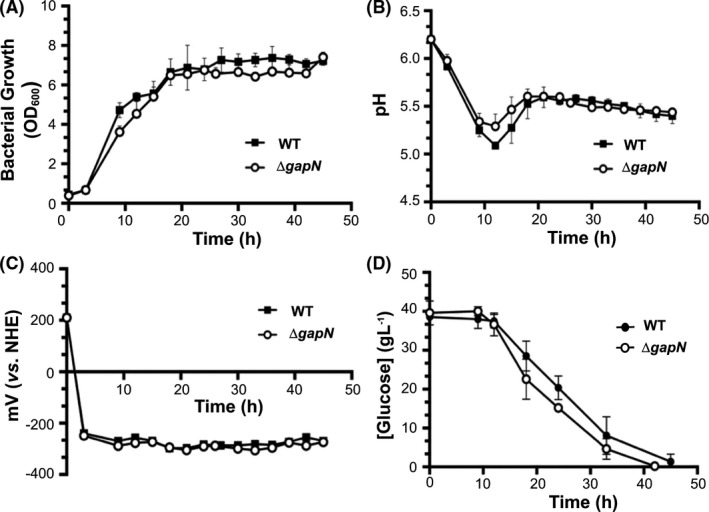
Fermentation measurements for ∆gapN (o) and wild type (•) C. saccharoperbutylacetonicum N1‐4(HMT). Cultures were grown on 40 g l−1 glucose in YETM media in bespoke bioreactors (Monaghan et al., 2021); 0.1 M MES was supplemented as a pH buffering agent and the following parameters were monitored: (A) OD600; (B) pH; (C) redox potential of media (mV vs. NHE); (D) Glucose concentrations in growth media. Data points are mean values of a total of six repeats including three biological repeats. Error bars represent the standard deviation of mean.
The products of acidogenesis and solventogenesis were then analysed for the ∆gapN and wild type strains for the growth experiments described in Fig. 2. The ∆gapN strain exhibited reduced concentrations of acids throughout the fermentation (Fig. 3A and B). In addition, depletion of acid concentrations in the ∆gapN strain also occurred at a faster rate in the ∆gapN strain compared to the wild type (Fig. 3C and D). The largest observed discrepancies between the strains for acetic acid and butyric acid measurements were observed at 15 h: 0.057 g l−1 of butyric acid and 0.073 g l−1 of acetic acid were measured for the ∆gapN strain, compared to 0.170 g l−1 of butyric acid and 0.29 g l−1 of acetic acid for the wild type strain. The concentrations of acetone and butanol produced by the ∆gapN strain were higher during the first 30 h of the fermentation, and the largest observed discrepancies between the strains for acetone and butanol measurements occurred at 15 h: the concentration of acetone produced by the ∆gapN strain was 3.2 g l−1 compared with only 1.1 g l−1 in the wild type strain; [butanol] produced by the ∆gapN strain was 6.87 g l−1 compared to 3.5 g l−1 in the wild type strain. Peak concentrations of butanol in the ∆gapN, however, were similar to those of the wild type cultures at ~ 13 g l−1.
Fig. 3.
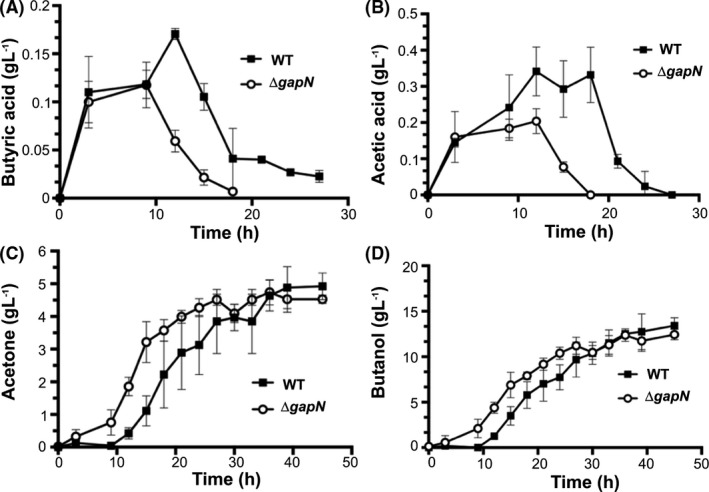
Acid and solvent profiles for ∆gapN (o) and wild type (•) C. saccharoperbutylacetonicum N1‐4(HMT) grown on 40 g l−1 glucose in YETM media in bespoke bioreactors (Monaghan et al., 2021); 0.1 M MES was supplemented as a pH buffering agent and concentrations of the following metabolites were monitored: (A) butyric acid; (B) acetic acid; (C) acetone; (D) butanol. Data points are mean values of a total of six repeats including three biological repeats. Error bars represent the standard deviation of mean.
Since the maximal concentrations of butanol and acetone were similar for ∆gapN and wild type strains, it was hypothesised that solvent toxicity was the limiting factor for these measurements. To test this hypothesis, a butanol toxicity test was carried out that measured growth rates in the presence of varying concentrations of solvents. These data indicate that deletion of gapN does not affect the ability of C. saccharoperbutylacetonicum to tolerate solvent toxicity (Fig. S3).
Deletion of gapN results in a reducing cytoplasm with a higher ATP pool
Herein, it was hypothesised that loss of gapN may increase the NADH:NAD+ ratio, which could enhance the reducing power required for ABE fermentation (Fig. 1). Hence, it was of interest to quantify the NAD(P)H derivatives during fermentation. Due to the labile nature of NAD(P)H derivatives, cells were grown in serum bottles and assayed at 4 and 24 h to provide concentrations during early exponential phase (acidogenesis) as well as in late exponential/stationary phase (solventogenesis). Deletion of gapN did not appear to affect the concentrations of NADH and NADPH although a marked reduction in the concentrations of NAD+ and NADP+ during both acidogensis and solventogenesis was observed (Table S2), which is indicative of a more reducing environment in the ∆gapN strain compared to the wild type (Fig. 4).
Fig. 4.
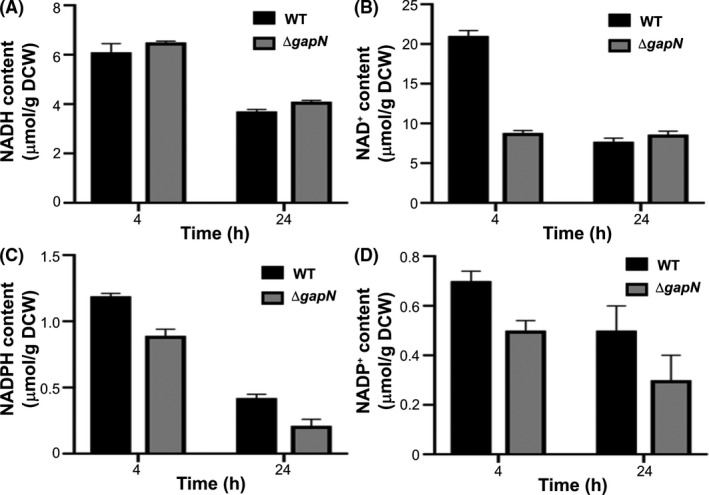
Quantitation of NAD(P)H derivatives in wild type and ∆gapN strains during acidogensis (4 h) and solventogenesis (24 h). Levels of NADH (A), NAD+ (B), NADPH (C) and NADP+ (D) were measured in C. saccharoperbutylacetonicum cells after 4 h and 24 h of growth. Cells were grown in YETM 40 g l−1 glucose in 50 ml serum bottles. Data points are mean values of a total of six repeats including three biological repeats. Error bars represent the standard deviation of mean.
Deletion of gapN was also hypothesised to elevate ATP levels, so the concentrations of ATP were also monitored during fermentation in bespoke bioreactors (Monaghan et al., 2021) and were found to be higher in the ∆gapN strain compared to wild type for the majority of fermentation period (Fig. 5). The greatest difference in ATP concentrations between the ∆gapN and wild type strains was observed at 15 h, where 11.2 µmol g−1 cell dry mass was recorded for the ∆gapN strain compared to 4.7 µmol g−1 cell dry mass for the wild type.
Fig. 5.
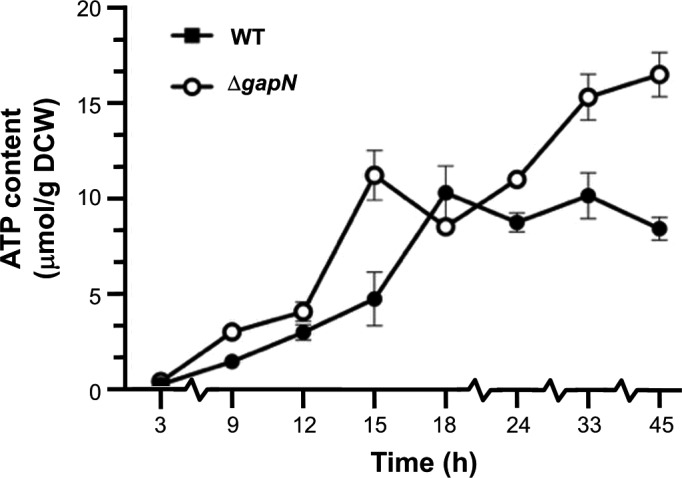
ATP measurements for ∆gapN (o) and wild type (•) strains grown in 40 g l−1 glucose in YETM media in bespoke bioreactors (Monaghan et al., 2021); 0.1 M MES was supplemented as a pH buffering agent. Data points are mean values of a total of six repeats including three biological repeats. Error bars represent the standard deviation of mean.
Discussion
The gapN gene was successfully deleted in C. saccharoperbutylacetonicum and no unwanted mutations were detected in the ∆gapN strain compared to the isogenic wild type parent strain, as confirmed by WGS. The ∆gapN strain grew at a similar rate to the wild type, with a slightly more rapid consumption of glucose during exponential phase (Fig. 2). However, the most striking difference in the ∆gapN strain was that acetic acid and butyric acid were converted to acetone and butanol much earlier in the fermentation (Fig. 3), reflecting an earlier shift into solventogenesis and higher solvent titres in exponential phase compared to the wild type. Ultimately, the final titre of butanol was the same as the wild type strain, probably reflecting butanol toxicity as the limiting factor. As there was no difference in butanol tolerance between the wild type and mutant strains (Fig. S3), it would be interesting to engineer butanol tolerance into ∆gapN and wild type strains to investigate the limits of butanol production during the early solventogenic shift.
In solventogenic Clostridia, acid production facilitates the generation of the majority of cellular ATP as a result of kinase activity of the enzymes involved in acetic and butyric acid production (Grupe and Gottschalk, 1992; Dürre and Hollergschwandner, 2004; Fig. 1). The associated ATP generation in acidogenesis is important as it enables the cells to produce adequate concentrations of ATP for vegetative growth, as well as to establish an ion gradient via V‐type ATPases that will enhance the electrical component of the proton motive force (PMF) that drives NADH production via the RnF complex with concomitant oxidation of reduced ferredoxin (Poehlein et al., 2017). This ion gradient enables ADP + Pi recycling when the cells shift into solventogensis via the use of F1FO ATPases (Jones and Woods, 1986), as illustrated in Fig. 1. The ∆gapN strain was shown to have elevated concentrations of ATP (compared to the wild type strain) for a sustained period during batch fermentation (Fig. 5). Between 15 and 24 h there is an observed drop in the measured ATP concentration of the ∆gapN strain over the wild type. This observed decrease in ATP corresponds to the time points with the lowest acid titres and the most rapid rate of solvent production in the ∆gapN strain (Fig. 3). These data suggest that the deletion of ∆gapN increases the ATP concentration such that the ∆gapN strain is able to diminish acid production and allow for an earlier shift into solventogenesis.
As well as an increase in ATP production, there is an increase in the ratio of NADH:NAD+ in the ∆gapN strain. ATP production, ∆pH generated by acid production and reducing conditions within the cell play a key role in the shift to solventogenesis (Wang et al., 2012; Wietzke and Bahl, 2012; Zhang et al., 2014; Liu et al., 2018). Previous work on C. acetobutylicum reported that overexpression of both 6‐phosphofructokinase (PfkA) and pyruvate kinase (PykA) resulted in an increase in intracellular ATP and NADH, which was also accompanied by elevated butanol and ethanol production (Ventura et al., 2013). It has been shown that in cultures with lower ATP levels acidogenesis is a major route for energy generation, whereas cultures with elevated ATP levels produce more solvents (Meyer and Papoutsakis, 1989). Enhanced butanol production has also been observed as a result of blocking NAD(P)H consumption in Clostridium beijerinckii NCIMB 8052 where insertional inactivation of a NADH‐quinone oxidoreductase (nuoG) resulted in increased NAD(P)H and ATP as well as elevated butanol production (Liu et al., 2016). Overall, it can be seen that increases in both ATP and NAD(P)H in solventogenic Clostridium aids in maximising the solvent producing potential of the bacteria.
Previous metabolic flux analysis in in C. acetobutylicum (Yoo et al., 2015) has revealed that GapN is poorly expressed during solventogenesis, with 0.56 mRNA molecules per cell in comparison to 66 mRNA molecules per cell of GAPDH. This low expression results in only 3500 molecules of GapN protein per cell compared to 190 000 molecules of GAPDH. This study estimated that GapN would be responsible for only 5% of flux through the glyceraldehyde‐3‐phosphate oxidation pathway, so it was initially surprising that in the current study deletion of gapN in C. saccharoperbutyacetonicum elicited such significant changes in solvent production, NADH:NAD+ ratio and ATP yield. However, there are significant metabolic differences between C. acetobutylicum and C. saccharoperbutyacetonicum, including the absence of the Rnf complex in the former, that could account for variations between species.
In conclusion, we have engineered a C. saccharoperbutylaconicum strain that switches earlier to solventogenesis and have provided new insights into the role of GapN in controlling redox poise and ATP yield.
Experimental procedures
Bacterial strains, plasmids and culture conditions
The clostridium strain used in this study was Clostridium saccharoperbutylacetonicum N1‐4(HMT) and plasmids used were based upon pMTL82154 or pMTL83251 (Heap et al., 2007). Liquid cultures were recovered from 15% glycerol stocks and grown at 32°C in reinforced clostridium media (RCM) (Sigma) that had been autoclaved in sealed serum bottles. E. coli strains were grown aerobically at 37°C in Luria‐Bertani media supplemented with the appropriate antibiotic when required.
Chromosomal mutagenesis
The gapN gene was deleted in C. saccharoperbutylacetonicum N1‐4(HMT) using CLEAVE as previously described (Atmadjaja et al., 2019), and all oligonucleotides are listed in Table S1. Firstly, a homologous recombination vector was generated with a deletion cassette that contains homology arms that are able to replace all of or part of the intended site of mutation. To construct the gapN deletion cassette, colony PCR of C. saccharoperbutylacetonicum N1‐4(HMT) was carried out (Fig. S1A) to amplify 2 × 1 Kbp regions upstream and downstream of gapN, containing complementary sequences such that it was possible to generate a seamless in‐frame deletion cassette fragment that lacked the gapN gene. Following successful PCR reactions, fragment isolation and Sanger sequencing to confirm the correct sequence of the deletion cassette, it was successfully blunt‐end cloned into the StuI restriction site of pMTL82154 (Fig. S1B). The new pMTL82154_gapN_HR vector was first transformed into E. coli. The HR vector pMTL82154 is a lineage of the previously described pMTL80000 shuttle vector system (Heap et al., 2007).
The killing vector, pMTL83251_Ldr_DR_Sp_DR, is also derived from the pMTL8000 shuttle vector system (Heap et al., 2007). The parent pMTL83251 vector contains a Gram‐positive replicon pCB102, ermB antibiotic marker, ColE1 + tra Gram‐negative replicon and a multiple cloning site (MCS). Pre‐engineered into the MCS of pMTL83251 is the native leader sequence (Ldr), a 181 bp sequence that is found downstream of the Cas2 machinery in C. saccharoperbutylacetonicum N1‐4(HMT) (Atmadjaja et al., 2019). With the leader sequence pre‐engineered into pMTL83251, it allows for the construction of CRISPR/Cas clusters on the plasmid that target the cleavage of specific chromosomal loci. The CRISPR/Cas targeting system seen in C. saccharoperbutylacetonicum N1‐4(HMT) is comprised of a target‐specific spacer flanked by direct repeats (DR_Sp_DR) that is downstream of the Cas2 sequence. For the successful deletion of gapN, a DR_Sp_DR cluster was designed (Fig. S2A), synthesised, and successfully cloned into pMTL83125 (Fig. S2B). The 35 bp spacer sequence that was used in the DR_Sp_DR cluster is present within the native gapN gene.
Whole genome sequencing
WGS was carried out using the Illumina MiSeq system. Genome libraries for both the wild type and ∆gapN strains were prepared using the Nextera® XT DNA Library Prep Kit, which is optimised for small genomes. Once the libraries were prepared, they were sequenced using an Illumina MiSeq benchtop sequencer. Following Illumina® MiSeq, the read quality was assessed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). To improve read quality, reads were trimmed using Trimmomatic (Bolger et al., 2014). Trimmomatic improves read quality of the Illumina reads by removing sequence adapters, trimming bases at the beginning and end of the reads if they fall below the stipulated threshold, followed by eliminating any of the reads that fall below the minimum read length (default setting of 36 bp). Following this, reads were mapped using BWA (Li, 2013) to the reference genome for C. saccharoperbutylacetonicum N1‐4(HMT) obtained from Genbank (CP004121.1). The quality of the genome maps were assessed using Qualimap (García‐Alcalde et al., 2012). Functional annotation was performed using PROKKA (Seemann, 2014) and Roary (Page et al., 2015) was used for analysis of gene content and the pangenome.
Fermentation conditions
ABE fermentation was carried out as recently described using a bespoke anoxic bioreactor system (Monaghan et al., 2021). Briefly, 1 l Pyrex Quickfit culture vessels were used (SciLabware, Hartlepool, United Kingdom) with 500 ml culture volumes at 32°C and media consisted of yeast extract tryptone media (YETM) (40 g l−1 glucose, 2.5 g l−1 yeast extract, 2.5 g l−1 tryptone, 0.5 g l−1 ammonium sulphate and 0.025 g l−1 iron sulphate) at pH 6.2, supplemented with 0.1 M 2‐(N‐morpholino)ethanesulfonic acid (MES) free acid (Merck) for pH control. MES buffer was chosen as it has a pKa of 6 and has previously been shown to be the very effective at the pH range observed during ABE fermentation. Anaerobic conditions in the fermentation were generated by sparging filtered (0.2 µm pore size) oxygen‐free nitrogen through the fermentation media for 20 min pre‐inoculation and then 5 min post‐inoculation. Seed cultures were established by growing recovered RCM grown cells, in 80 ml YETM in serum bottles overnight to an OD600 of ~ 4.0. The final inoculation was 10% (v/v). Fermentations were carried out in triplicate. Throughout the fermentations OD600, pH and redox poise (Inlab Redox Micro electrode from Mettler Toledo, Leicester, United Kingdom) of the fermentation media was measured. The redox electrode was calibrated against quinhydrone (87 mV at pH 7.0, 264 mV at pH 4.0, Normal Hydrogen Electrode (NHE) correction for a Ag/AgCl electrode in 3 M KCl = +210 mV). Following sampling, supernatant and cells were separated by centrifugation at 8000 g for 10 min. Supernatant and cell pellet were separated and frozen at −80°C for later use.
Acid and solvent quantitation
Acids and solvents from culture supernatants were quantitated using Gas Chromatography Mass Spectrometry (GCMS) using an Agilent 6890N instrument as previously described (Monaghan et al., 2021). The GC was equipped with a Phenomenex 7HG‐6013‐11 Zebron column. Helium (> 99.999%) was used as the carrier gas, with a constant flow rate of 1 ml min−1. A 0.2 µl water sample was injected with a 100:1 split. Injection temperature was set to 150°C, the GCMS transfer line temperature was set to 280°C, ion source to 230°C, and quadrapole to 150°C. After injection, column temperature was held at 30°C for 5 min, after which this increased at a linear gradient to 150°C at the 20 min mark. Compounds were identified by comparing retention times of each of the compounds with retention times of reference compounds.
Quantitation of NADH and NADPH
NADH, NADPH and their oxidised derivatives were extracted essentially as previously described (Beri et al., 2016). Briefly, 2 ml of bacterial culture was added to 1 ml of 1 M HCl for NAD(P)+ extraction and 1 ml of 1 M KCL for NAD(P)H extraction. Cells were incubated at 55°C for 10 min and then the pH was adjusted to pH 6.5 for acidic samples and pH 7.5 for basic samples using 1 M HCl and 1 M KOH dropwise with continual vortexing. Samples were centrifuged for 15 min at 4400 g, with the supernatant being saved for subsequent analysis. To determine the dry cell weight (DCW), cells were harvested were centrifuged at 12 000 rpm for 5 min, washed twice with distilled water, and dried to a constant weight at 80°C.
Concentrations of NADH and NADPH were measured using previously described methods (Nisselbaum and Green, 1969; Bernofsky and Swan, 1973; Baker, 2016). In brief, the reaction mixture for the NADH assay consisted of 100 µl 1 M tricine‐NaOH (pH 8), 100 µl 40 mM EDTA, 100 µl 0.1 M NaCl, 100 µl 4.2 mM MTT, 100 µl 16.6 mM PES, 100 µl 100% EtOH. The reaction mixture for NADPH assay consisted of 100 µl 1 M tricine‐NaOH (pH 8), 100 µl 40 mM EDTA, 100 µl 0.1 M NaCl, 100 µl 4.2 mM MTT, 100 µl 16.6 mM PES, 100 µl 10 mM Glucose‐6‐phosphate; 500 µl of each assay mixture was added to 100 µl of extracted sample, topped up to 900 µl with 0.1 mM NaCl and incubated at 37°C for 5 min. To generate standard curves, similar reactions were set up where the extracted sample was replaced with nucleotide solutions of known concentration; 100 µl of alcohol dehydrogenase (Sigma Aldrich, Burlington, MA, USA) and 100 µl glucose‐6‐phosphate dehydrogenase (Sigma Aldrich) were then added (10 U of each) per reaction. The reaction was incubated at 37°C for 1 h in the dark. 500 µl of 5 M NaCl was then added to stop the reaction and precipitate the MTT. Samples were centrifuged at 10 000 g and 4°C for 5 min. Supernatants were decanted, MTT pellets were resuspended in 1 ml of ethanol and absorbance was measured at 570 nm.
ATP Luminescence assay
ATP was quantified in cells growing in YETM medium using a luminescence assay according to the manufacturer’s instructions (Abcam ab113849, Cambridge, United Kingdom). To determine the DCW, cells were harvested were centrifuged at 12 000 rpm for 5 min, washed twice with distilled water, and dried to a constant weight at 80°C.
Sugar quantitation
Sugars were quantitated essentially as previously described (Monaghan et al., 2021). Briefly, culture supernatants were homogenised and centrifuged at 13 400 g for 5 min; 200 µl of the sample was then added to 600 µl of HPLC grade water, achieving a ×4 dilution and a total volume of 800 µl. Glucose concentrations were measured using cation exchange chromatography at 60°C using a Phenomenex Rezex ROA H+ column at 1 ml min−1 5 mM sulphuric acid using an Agilent 1100 series refractive index detector to monitor glucose elution. Concentrations of samples were determined by comparison to a standard curve for glucose with integrated peak areas used for the determination of glucose concentration.
Butanol toxicity
To test butanol toxicity, 30 ml serum bottles were filled with RCM, inoculated with 15% C. saccharoperbutylacetonicum glycerol stocks, and were grown overnight at 32°C. The overnights were then used to inoculate (10% v/v) YETM media containing 40 g l−1 glucose in serum bottles under anaerobic conditions. The cells were left to grown for 4 h at 32°C until an OD600 of ~ 1 was reached, when they were challenged with various concentrations of butanol ranging from 0.5% to 5% (v/v). Subsequent growth rates were then monitored to assess the impact of butanol toxicity.
Conflict of interest
The authors declare no conflicts of interest.
Author contribution
TIM contributed to experimental design, performed the experiments and wrote the paper; JAB developed the sugar analysis methodology; PK, ETD and ERJ discussed the experiments and results; IBG supervised the genome sequencing/assembly/annotation work; GKR contributed to experimental design and edited the paper; MS was responsible for overseeing experimental design and wrote the paper.
Supporting information
Fig. S1. Generation of a homologous recombination vector for gapN deletion. A) PCR approach to generate the deletion cassette: (i) Amplification of 1 kb fragments upstream and downstream of the gapN gene with 48 bp of complementary sequences; (ii) Two 1 kb PCR products with complementary ends. (iii) Product of overlap‐extension PCR, ready to be blunt‐end ligated into pMTL82154. B) Vector map of ‘pMTL82154_gapN_HR’ that contains the homologous recombination (HR) fragment (i.e. the deletion cassette) cloned into the StuI site of pMTL82154 (verified via StuI restriction digests and sequencing). pMTL82154_gapN_HR contains a pBP1 Gram‐positive replicon, catP antibiotic maker, ColE1 +tra Gram‐negative replicon and a catP reporter gene.
Fig S2. Generation of a killing vector for elimination of transformants that do not contain the gapN deletion. A) Overview of the killing vector targeting cassette for endogenous CRISPR‐Cas for genome editing. The native leader sequence (Ldr) is a 181 bp sequence found downstream of the Cas2 machinery in C. saccharoperbutylacetonicum N1‐4(HMT) (Atmadjaja et al., 2019). The CRISPR/Cas targeting system is comprised of a target‐specific spacer (i.e. gapN spacer) flanked by direct repeats (DR_Sp_DR) that is downstream of the Cas2 sequence. B) Vector map of ‘pMTL83251_Ldr_HR_Sp_HR’ that contains the targeting cassette from panel A. Successful cloning was confirmed via colony PCR and sequencing.
Fig. S3. Butanol toxicity test of wild type (black bars) and ∆gapN (grey bars) strains of C. saccharoperbutylacetonicum N1‐4(HMT). Cells were grown to an OD600 of 1 and were then challenged with varying [butanol]. Doubling times were calculated for the 48 h of growth that followed solvent addition.
Table S1. Oligonucleotides used in this study.
Table S2. Ratio of nucleotide cofactors in wild type (WT) and ∆gapN C. saccharoperbutylacetonicum N1‐4(HMT).
Acknowledgements
The authors thank Kevin Howland (University of Kent) for guidance on the HPLC/GCMS measurements and Amanda Nicolle/Christopher Hills for support with the CLEAVETM mutagenesis.
Microbial Biotechnology (2022) 15(5), 1574–1585
Funding Information
This work was funded by a BBSRC iCASE studentship (BB/M016048/1 to MS) and a BBSRC NIBB proof‐of‐concept award (HD‐RD0300H to MS).
References
- Al‐Shorgani, N.K.N. , Ali, E. , Kalil, M.S. , and Yusoff, W.M.W. (2012) Bioconversion of butyric acid to butanol by Clostridium saccharoperbutylacetonicum N1–4 (ATCC 13564) in a limited nutrient medium. BioEnergy Res 5: 287–293. [Google Scholar]
- Antunes, A. , Martin‐Verstraete, I. , and Dupuy, B. (2011) CcpA‐mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79: 882–899. [DOI] [PubMed] [Google Scholar]
- Atmadjaja, A.N. , Holby, V. , Harding, A.J. , Krabben, P. , Smith, H.K. , and Jenkinson, E.R. (2019) CRISPR‐Cas, a highly effective tool for genome editing in Clostridium saccharoperbutylacetonicum N1–4(HMT). FEMS Microbiol Lett 366: fnz059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, J. (2016) Modified chromogenic assay for determination of the ratio of free intracellular NAD+/NADH in Streptococcus mutans . Bio‐protocols 6: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beri, D. , Olson, D.G. , Holwerda, E.K. , and Lynd, L.R. (2016) Nicotinamide cofactor ratios in engineered strains of Clostridium thermocellum and Thermoanaerobacterium saccharolyticum . FEMS Microbiol Lett 363: fnw091. [DOI] [PubMed] [Google Scholar]
- Bernofsky, C. , and Swan, M. (1973) An improved cycling assay for nicotinamide adenine dinucleotide. Anal Biochem 53: 452–458. [DOI] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. , and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, D.A. , Cvitkovitch, D.G. , and Hamilton, I.R. (1995) Sequence, expression, and function of the gene for the nonphosphorylating, NADP‐dependent glyceraldehyde‐3‐phosphate dehydrogenase of Streptococcus mutans . J Bacteriol 177: 2622–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann, H. , Cerff, R. , Salomon, M. , and Soll, J. (1989) Cloning and sequence analysis of cDNAs encoding the cytosolic precursors of subunits GapA and GapB of chloroplast glyceraldehyde‐3‐phosphate dehydrogenase from pea and spinach. Plant Mol Biol 13: 81–94. [DOI] [PubMed] [Google Scholar]
- Bruder, M.R. , Pyne, M.E. , Moo‐Young, M. , Chung, D.A. , and Chou, C.P. (2016) Extending CRISPR‐Cas9 technology from genome editing to transcriptional engineering in the genus Clostridium. Appl Env Microbiol 82: 6109–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartman, S.T. , Heap, J.T. , Kuehne, S.A. , Cockayne, A. , and Minton, N.P. (2010) The emergence of ‘hypervirulence’ in Clostridium difficile . Int J Med Microbiol 300: 387–395. [DOI] [PubMed] [Google Scholar]
- Cooksley, C.M. , Davis, I.J. , Winzer, K. , Chan, W.C. , Peck, M.W. , and Minton, N.P. (2010) Regulation of neurotoxin production and sporulation by a putative agrBD signaling system in proteolytic Clostridium botulinum . Appl Environ Microbiol 76: 4448–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooksley, C.M. , Zhang, Y. , Wang, H. , Redl, S. , Winzer, K. , and Minton, N.P. (2012) Targeted mutagenesis of the Clostridium acetobutylicum acetone–butanol–ethanol fermentation pathway. Metab Eng 14: 630–641. [DOI] [PubMed] [Google Scholar]
- Dürre, P. , and Hollergschwandner, C. (2004) Initiation of endospore formation in Clostridium acetobutylicum . Anaerobe 10: 69–74. [DOI] [PubMed] [Google Scholar]
- Fothergill‐Gilmore, L.A. , and Michels, P.A.M. (1993) Evolution of glycolysis. Prog Biophys Mol Biol 59: 105–235. [DOI] [PubMed] [Google Scholar]
- García‐Alcalde, F. , Okonechnikov, K. , Carbonell, J. , Cruz, L.M. , Götz, S. , Tarazona, S. , et al. (2012) Qualimap: evaluating next‐generation sequencing alignment data. Bioinformatics 28: 2678–2679. [DOI] [PubMed] [Google Scholar]
- Green, E.M. (2011) Fermentative production of butanol ‐ the industrial perspective. Curr Opin Biotechnol 22: 337–343. [DOI] [PubMed] [Google Scholar]
- Grupe, H. , and Gottschalk, G. (1992) Physiological events in Clostridium acetobutylicum during the shift from acidogenesis to solventogenesis in continuous culture and presentation of a model for shift induction. Appl Environ Microbiol 58: 3896–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, Y. , Feng, J. , Zhang, Z.‐T. , Wang, S. , Guo, L. , Wang, Y. , and Wang, Y.I. (2019) Curing the endogenous megaplasmid in Clostridium saccharoperbutylacetonicum N1–4 (HMT) using CRISPR‐Cas9 and preliminary investigation of the role of the plasmid for the strain metabolism. Fuel 236: 1559–1566. [Google Scholar]
- Heap, J.T. , Pennington, O.J. , Cartman, S.T. , Carter, G.P. , and Minton, N.P. (2007) The ClosTron: a universal gene knock‐out system for the genus Clostridium. J Microbiol Methods 70: 452–464. [DOI] [PubMed] [Google Scholar]
- Hongo, M. , Murata, A. , and Ogata, S. (1969) Bacteriophages of Clostridium saccharoperbutylacetonicum . Agric Biol Chem 33: 331–342. [Google Scholar]
- Huang, H. , Chai, C. , Li, N. , Rowe, P. , Minton, N.P. , Yang, S. , et al. (2016) CRISPR/Cas9‐based efficient genome editing in Clostridium ljungdahlii, an autotrophic gas‐fermenting bacterium. ACS Synth Biol 5: 1355–1361. [DOI] [PubMed] [Google Scholar]
- Iddar, A. , Valverde, F. , Serrano, A. , and Soukri, A. (2002) Expression, purification, and characterization of recombinant nonphosphorylating NADP‐dependent glyceraldehyde‐3‐phosphate dehydrogenase from Clostridium acetobutylicum . Protein Expr Purif 25: 519–526. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. , and Keis, S. (1995) Origins and relationships of industrial solvent‐producing clostridial strains. FEMS Microbiol Rev 17: 223–232. [Google Scholar]
- Jones, D.T. , and Woods, D.R. (1986) Acetone‐butanol fermentation revisited. Microbiol Rev 50: 484–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostan, J. , Sjöblom, B. , Maixner, F. , Mlynek, G. , Furtmüller, P.G. , Obinger, C. , et al. (2010) Structural and functional characterisation of the chlorite dismutase from the nitrite‐oxidizing bacterium “Candidatus Nitrospira defluvii”: Identification of a catalytically important amino acid residue. J Struct Biol 172: 331–342. [DOI] [PubMed] [Google Scholar]
- Li, H. (2013) Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA‐MEM: arXiv:1303.3997.
- Liu, D. , Yang, Z. , Wang, P. , Niu, H. , Zhuang, W. , Chen, Y. , et al. (2018) Towards acetone‐uncoupled biofuels production in solventogenic Clostridium through reducing power conservation. Metab Eng 47: 102–112. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Guo, T. , Wang, D. , Shen, X. , Liu, D. , Niu, H. , et al. (2016) Enhanced butanol production by increasing NADH and ATP levels in Clostridium beijerinckii NCIMB 8052 by insertional inactivation of Cbei_4110. Appl Microbiol Biotechnol 100: 4985–4996. [DOI] [PubMed] [Google Scholar]
- Mateos, M.I. , and Serrano, A. (1992) Occurrence of phosphorylating and non‐phosphorylating NADP+‐dependent glyceraldehyde‐3‐phosphate dehydrogenases in photosynthetic organisms. Plant Sci 84: 163–170. [Google Scholar]
- Meyer, C.L. , and Papoutsakis, E.T. (1989) Increased levels of ATP and NADH are associated with increased solvent production in continuous cultures of Clostridium acetobutylicum . Appl Microbiol Biotechnol 30: 450–459. [Google Scholar]
- Monaghan, T.I. , Baker, J.A. , Robinson, G.K. , and Shepherd, M. (2021) Parallel bioreactor system for accessible and reproducible anaerobic culture. Access Microbiol 3: 000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju, S. , Davies, N.K. , Walker, D.J.F. , Köpke, M. , and Simpson, S.D. (2016) Genome editing of Clostridium autoethanogenum using CRISPR/Cas9. Biotechnol Biofuels 9: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisselbaum, J.S. , and Green, S. (1969) A simple ultramicro method for determination of pyridine nucleotides in tissues. Anal Biochem 27: 212–217. [DOI] [PubMed] [Google Scholar]
- Noguchi, T. , Tashiro, Y. , Yoshida, T. , Zheng, J. , Sakai, K. , and Sonomoto, K. (2013) Efficient butanol production without carbon catabolite repression from mixed sugars with Clostridium saccharoperbutylacetonicum N1–4. J Biosci Bioeng 116: 716–721. [DOI] [PubMed] [Google Scholar]
- Page, A.J. , Cummins, C.A. , Hunt, M. , Wong, V.K. , Reuter, S. , Holden, M.T.G. , et al. (2015) Roary: rapid large‐scale prokaryote pan genome analysis. Bioinformatics 31: 3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poehlein, A. , Krabben, P. , Dürre, P. , and Daniel, R. (2014) Complete genome sequence of the solvent producer Clostridium saccharoperbutylacetonicum strain DSM 14923. Genome Announc 2: e01056–e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poehlein, A. , Solano, J.D.M. , Flitsch, S.K. , Krabben, P. , Winzer, K. , Reid, S.J. , et al. (2017) Microbial solvent formation revisited by comparative genome analysis. Biotechnol Biofuels 10: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. , and Mesirov, J.P. (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. [DOI] [PubMed] [Google Scholar]
- Takeno, S. , Murata, R. , Kobayashi, R. , Mitsuhashi, S. , and Ikeda, M. (2010) Engineering of Corynebacterium glutamicum with an NADPH‐generating glycolytic pathway for L‐lysine production. Appl Environ Microbiol 76: 7154–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro, Y. , Shinto, H. , Hayashi, M. , Baba, S. , Kobayashi, G. , and Sonomoto, K. (2007) Novel high‐efficient butanol production from butyrate by non‐growing Clostridium saccharoperbutylacetonicum N1–4 (ATCC 13564) with methyl viologen. J Biosci Bioeng 104: 238–240. [DOI] [PubMed] [Google Scholar]
- Underwood, S. , Guan, S. , Vijayasubhash, V. , Baines, S. D. , Graham, L. , Lewis, R. J. , et al. (2009) Characterization of the sporulation initiation pathway of Clostridium difficile and its role in toxin production. J Bacteriol 191: 7296–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura, J.‐R.‐ S. , Hu, H. , and Jahng, D. (2013) Enhanced butanol production in Clostridium acetobutylicum ATCC 824 by double overexpression of 6‐phosphofructokinase and pyruvate kinase genes. Appl Microbiol Biotechnol 97: 7505–7516. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Dong, S. , Wang, P. , Tao, Y. , and Wang, Y. (2017) Genome editing in Clostridium saccharoperbutylacetonicum N1–4 with the CRISPR‐Cas9 system. Appl Environ Microbiol 83: e00233–e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Dong, S. , and Wang, Y. (2017) Enhancement of solvent production by overexpressing key genes of the acetone‐butanol‐ethanol fermentation pathway in Clostridium saccharoperbutylacetonicum N1–4. Bioresour Technol 245: 426–433. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Zhu, Y. , Zhang, Y. , and Li, Y. (2012) Controlling the oxidoreduction potential of the culture of Clostridium acetobutylicum leads to an earlier initiation of solventogenesis, thus increasing solvent productivity. Appl Microbiol Biotechnol 93: 1021–1030. [DOI] [PubMed] [Google Scholar]
- Wasels, F. , Jean‐Marie, J. , Collas, F. , López‐Contreras, A.M. , and Lopes Ferreira, N. (2017) A two‐plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum . J Microbiol Methods 140: 5–11. [DOI] [PubMed] [Google Scholar]
- Wietzke, M. , and Bahl, H. (2012) The redox‐sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum . App Microbiol Biotechnol 96: 749–761. [DOI] [PubMed] [Google Scholar]
- Xu, M. , Zhao, J. , Yu, L. , Tang, I.‐C. , Xue, C. , and Yang, S.‐T. (2015) Engineering Clostridium acetobutylicum with a histidine kinase knockout for enhanced n‐butanol tolerance and production. Appl Microbiol Biotechnol 99: 1011–1022. [DOI] [PubMed] [Google Scholar]
- Yoo, M. , Bestel‐Corre, G. , Croux, C. , Riviere, A. , Meynial‐Salles, I. , and Soucaille, P. (2015) A Quantitative system‐scale characterization of the metabolism of Clostridium acetobutylicum . MBio 6: e01808–e1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Wang, P. , Wang, X. , Feng, J. , Sandhu, H.S. , and Wang, Y. (2018) Enhancement of sucrose metabolism in Clostridium saccharoperbutylacetonicum N1–4 through metabolic engineering for improved acetone–butanol–ethanol (ABE) fermentation. Bioresour Technol 270: 430–438. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Nie, X. , Ravcheev, D.A. , Rodionov, D.A. , Sheng, J. , Gu, Y. , et al. (2014) Redox‐responsive repressor Rex modulates alcohol production and oxidative stress tolerance in Clostridium acetobutylicum . J Bacteriol 196: 3949–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Generation of a homologous recombination vector for gapN deletion. A) PCR approach to generate the deletion cassette: (i) Amplification of 1 kb fragments upstream and downstream of the gapN gene with 48 bp of complementary sequences; (ii) Two 1 kb PCR products with complementary ends. (iii) Product of overlap‐extension PCR, ready to be blunt‐end ligated into pMTL82154. B) Vector map of ‘pMTL82154_gapN_HR’ that contains the homologous recombination (HR) fragment (i.e. the deletion cassette) cloned into the StuI site of pMTL82154 (verified via StuI restriction digests and sequencing). pMTL82154_gapN_HR contains a pBP1 Gram‐positive replicon, catP antibiotic maker, ColE1 +tra Gram‐negative replicon and a catP reporter gene.
Fig S2. Generation of a killing vector for elimination of transformants that do not contain the gapN deletion. A) Overview of the killing vector targeting cassette for endogenous CRISPR‐Cas for genome editing. The native leader sequence (Ldr) is a 181 bp sequence found downstream of the Cas2 machinery in C. saccharoperbutylacetonicum N1‐4(HMT) (Atmadjaja et al., 2019). The CRISPR/Cas targeting system is comprised of a target‐specific spacer (i.e. gapN spacer) flanked by direct repeats (DR_Sp_DR) that is downstream of the Cas2 sequence. B) Vector map of ‘pMTL83251_Ldr_HR_Sp_HR’ that contains the targeting cassette from panel A. Successful cloning was confirmed via colony PCR and sequencing.
Fig. S3. Butanol toxicity test of wild type (black bars) and ∆gapN (grey bars) strains of C. saccharoperbutylacetonicum N1‐4(HMT). Cells were grown to an OD600 of 1 and were then challenged with varying [butanol]. Doubling times were calculated for the 48 h of growth that followed solvent addition.
Table S1. Oligonucleotides used in this study.
Table S2. Ratio of nucleotide cofactors in wild type (WT) and ∆gapN C. saccharoperbutylacetonicum N1‐4(HMT).