

Summary
The COVID‐19 pandemic goes into its third year and the world population is longing for an end to the pandemic. Computer simulations of the future development of the pandemic have wide error margins and predictions on the evolution of new viral variants of SARS‐CoV‐2 are uncertain. It is thus tempting to look into the development of historical viral respiratory pandemics for insight into the dynamic of pandemics. The Spanish flu pandemic of 1918 caused by the influenza virus H1N1 can here serve as a potential model case. Epidemiological observations on the shift of influenza mortality from very young and old subjects to high mortality in young adults delimitate the pandemic phase of the Spanish flu from 1918 to 1920. The identification and sequencing of the Spanish flu agent allowed following the H1N1 influenza virus after the acute pandemic phase. During the 1920s H1N1 influenza virus epidemics with substantial mortality were still observed. As late as 1951, H1N1 strains of high virulence evolved but remained geographically limited. Until 1957, the H1N1 virus evolved by accumulation of mutations (‘antigenic drift’) and some intratypic reassortment. H1N1 viruses were then replaced by the pandemic H2N2 influenza virus from 1957, which was in 1968 replaced by the pandemic H3N2 influenza virus; both viruses were descendants from the Spanish flu agent but showed the exchange of entire gene segments (‘antigenic shift’). In 1977, H1N1 reappeared from an unknown source but caused only mild disease. However, H1N1 achieved again circulation in the human population and is now together with the H3N2 influenza virus an agent of seasonal influenza winter epidemics.
Introduction
Two years after the onset of the COVID‐19 pandemic, people are asking when it will end. Some scientists see in the infection wave with the omicron variant signs for the beginning of the end of the pandemic, while others call for caution with respect to the emergence of new variants. Such variants could come from developing countries where large parts of the population are still unvaccinated and could thus create new variants with novel and potentially disturbing phenotypes. Current predictions of the future trajectory of the COVID‐19 pandemic rely on general concepts of viral evolution and population immunity development assisted by computer simulations. Basic assumptions for novel variants of SARS‐CoV‐2 used in mathematical simulations have wide error margins, hence one might be tempted instead to look into the historical past how respiratory pandemics evolved and ended to obtain a framework for possible future developments of the COVID‐19 pandemic. There are four respiratory pandemics that can serve as potential models: the Russian flu from 1889, the Spanish flu from 1918, the Asian flu from 1957 and the Hong Kong flu from 1968.
Recently, we looked into the Russian flu from 1889 for indications about its dynamics and ending (Brüssow, 2021, 2021). This choice was further motivated by circumstantial virological evidence as well as epidemiological and clinical evidence (Brüssow and Brüssow, 2021) that it might have been a possible prior coronavirus pandemic. Based on the interpretation of clinical symptoms noted in contemporary reports the Russian flu might have shown recurrences up to 9 years after the onset of the pandemic. However, the lack of virological data and the scarcity of clinical and epidemiological data makes it difficult to link the subsequent infection waves occurring until 1900 with the initial 1889 pandemic. Therefore, it might be instructive to analyse the time development and the ending of the Spanish flu pandemic which was caused by an H1N1 influenza A virus. While this represents a respiratory pandemic with a distinct viral etiology from COVID‐19, this approach has the advantage that the H1N1 virus can be used as a tracer for the development of the H1N1 virus after the acute phase of the Spanish flu pandemic. The Asian flu from 1957 and the Hong Kong flu from 1968 were caused by H2N2 and H3N2 influenza A viruses, both are descendants from the H1N1 virus from 1918 resulting from antigenic shifts (reassortment with avian influenza virus genes), but they caused much less mortality. According to estimates, the Spanish flu caused 675 000, the Asian flu 86 000 and the Hong Kong flu 56 300 excess deaths in the United States respectively. Since CDC calculated 1 million excess deaths by COVID‐19 in the United States (Excess Deaths Associated with COVID‐19 (cdc.gov)), only the Spanish flu pandemic can serve as relevant reference for COVID‐19. However, one should always keep in mind that an influenza virus pandemic might behave differently than a coronavirus pandemic even if both were described as pandemics of the century or the largest pandemics in the medical and scientific literature. In addition, the mortality of the Spanish flu might not be a direct consequence of the viral infection, but a sequel of subsequent bacterial superinfection. Recut lung tissue sections from 1918 to 1919 influenza case material revealed, in virtually all cases, compelling histological evidence of severe acute bacterial pneumonia. A review of pathological and bacteriological findings showed that 92% of autopsy lung cultures from Spanish flu victims were positive for bacterial pneumonia pathogens, mostly pneumococci, or pneumococci mixed with streptococci, less with staphylococci (Morens et al., 2008). Since bacterial proteases cleave the influenza virus hemagglutinin, they are likely to contribute to the development of influenza pneumonia (Tashiro et al., 1987). These caveats should be kept in mind when using the Spanish flu as a paradigm for the future development of COVID‐19.
The outset
To start with the name ‘Spanish flu’ – it does not mean that the epidemic started in Spain. Spain was neutral during the First World War and newspaper reports were not controlled by military censorship. Therefore, Spanish newspapers were only the first to report on the new disease. The geographical region where the epidemic started could not be clearly defined since a US military camp in Kansas as well as military camps in Western France were later claimed to be at the origin of the pandemic (Oxford, 2000). The origin could not be clearly defined since frequent and massive troop transports between the United States and Europe and within Europe clearly helped to spread the disease. Since the Spanish flu set in with relative mild ‘herald waves’, a differentiation from preceding respiratory infections is difficult. Epidemiologists associated the Kansas outbreak with the disease in pigs, but greater outbreaks of an influenza‐like disease in pigs were only observed after an October 1918 Swine Show in Iowa (Zimmer and Burke, 2009). Infections in pigs were caused by the Classical H1N1 North American Swine influenza virus, which was serologically closely related to the Spanish flu influenza virus (Shope, 1936). The ultimate origin of the Spanish flu virus is an avian influenza virus, however, no close relative of the Spanish flu virus was found within available influenza virus isolates from birds, which could mean that it came from an unknown bird species or that it evolved for some time in a mammalian species as also suggested by signature mammalian sequences in the Spanish flu virus. It is unknown whether the precursor of the pandemic virus replicated in pigs or humans, but a direct transfer from birds seems unlikely.
In a 300‐page report on the pandemic written in 1921 (Vaughan, 1921), the United States origin of the pandemic virus seemed to be followed by a strikingly rapid worldwide spread of the pandemic. The report displays in a comprehensive way the geographical spread of the pandemic. Early cases were seen in March 1918 in the United States and France (the author mentioned also China and Japan). In May 1918, the virus had spread across Europe. In June, cases probably imported from Europe were seen in South America and India, the latter probably also by troop transports. In August 1918, Europe was in the grip of the infection, as was the United States, Central and South America, India, China, Japan, Indonesia and the Philippines. In September 1918, the infection reached Africa and in October 1918 Australia. In November/December 1918, large parts of the world suffered from influenza infections (Fig. 1). Contemporary medical researchers observed a strikingly synchronized course of the pandemic: when overlaying all‐cause mortality data for major cities on both sides of the Atlantic such as New York, London, Paris and Berlin (Fig. 2) there were only small temporal shifts for the major peak all‐cause mortality in October/November 1918. They noted a mild herald wave in spring 1918 followed by a major mortality peak in fall/winter 1918 and then a recrudescent wave extending into 1920. However, the details of the dynamics of the pandemic showed marked differences in different geographical areas which will be summarized in the following sections.
Fig. 1.
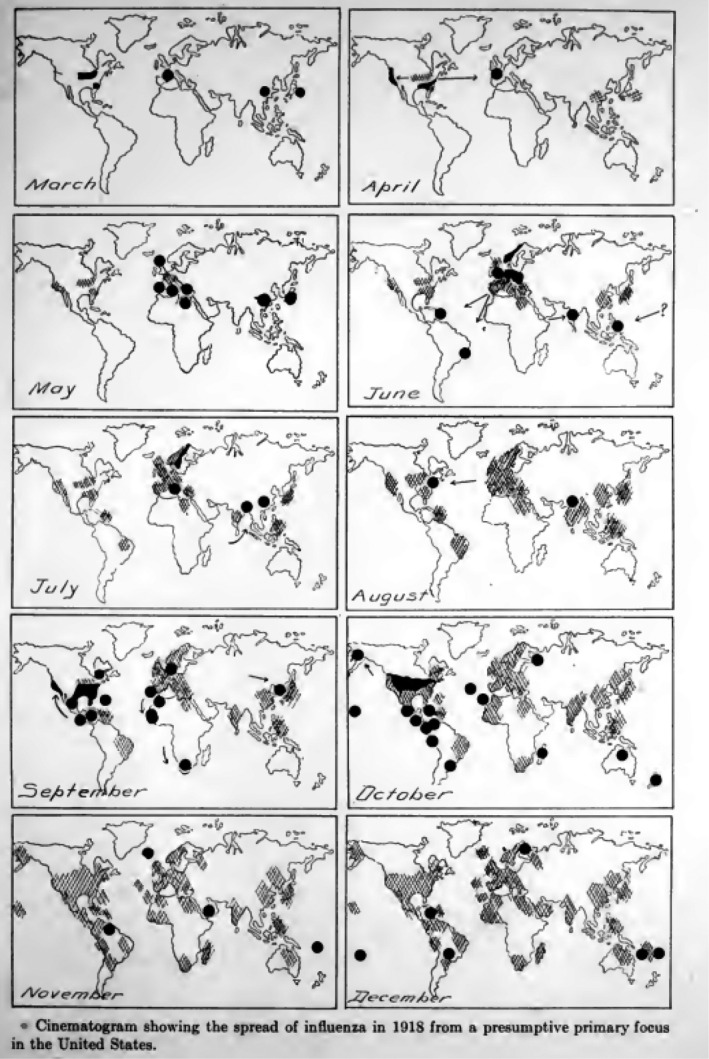
The monthly spread of the Spanish flu pandemic from March to December 1918. Source: Vaughan (1921).
Fig. 2.
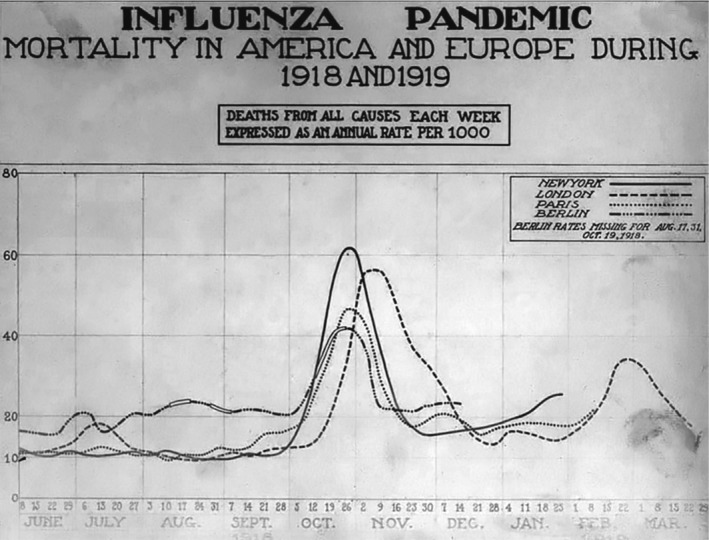
All‐cause mortality in New York, London, Paris and Berlin between June 1918 and March 1919. Source: Wikipedia Public Domain; Image: courtesy of the National Museum of Health and Medicine.
The Spanish flu pandemic in the Americas
North America
The start of the pandemic was not clearly defined. Contemporary reports from the United States mentioned ‘herald waves’ but they are difficult to distinguish from ‘seasonal’ peaks of influenza and pneumonia deaths that were the winter rule in the United States in the decade preceding the Spanish flu. Physicians from Chicago reported for example for December 1915/January 1916 an infection wave in large US cities with frequent fatal bronchopneumonia that physicians had not seen since 1889/1890 (‘Russian flu’) pandemic (Capps and Moody, 1916). However, from the contemporary US mortality charts, a clearly distinct situation only became evident in the fall of 1918 with a 10‐fold mortality rate increase. Modern re‐analysis of the mortality statistics shed more light on the timing of the pandemic (Fig. 3).
Fig. 3.
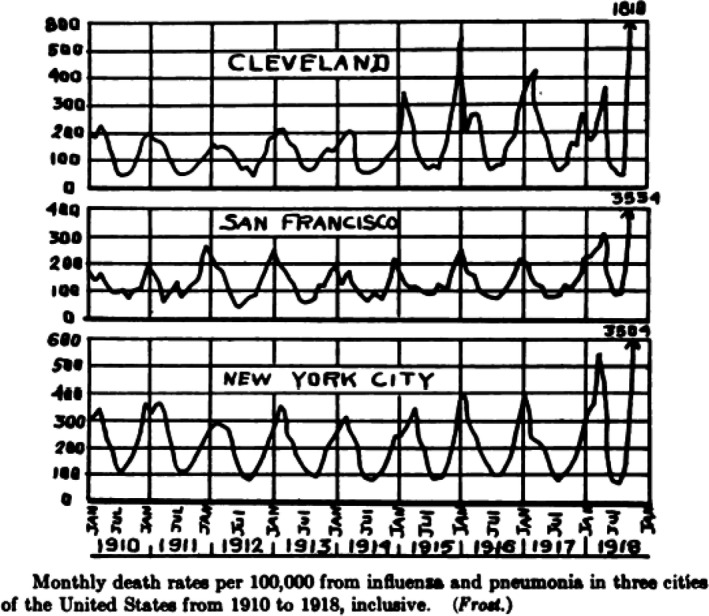
Monthly death rate per 100 000 for three US cities between 1910 and 1918. The fall 1918 peak is not to scale and exceeds that of the preceding years by more than a factor of 10 for New York. Source: Vaughan (1921).
A key observation that allowed the differentiation of pandemic versus epidemic influenza mortality was published in 1998 by researchers from the CDC (Simonsen et al., 1998). In all influenza pandemics of the 20th century, people younger than 65 years represented an important part of the influenza‐associated fatalities. This was particularly apparent for the Spanish flu pandemic from 1918 to 1919 where < 65 years old people represented 99% of the excess mortality. This pandemic pattern was described as a ‘W‐shaped’ mortality curve with peaks in infants, young adults and seniors while the seasonal influenza epidemic is characterized by a ‘U‐shaped’ mortality curve with mortality maxima in infants and seniors and a broad minimum in age groups between these extremes. The CDC researchers observed that the percentage of excess mortality in people younger than 65 years decreased in the years following the Spanish flu pandemic. They observed another increase of excess mortality in < 65 years old subjects during the 1957–1958 H2N2 influenza A pandemic and the 1968–1969 H3N2 influenza pandemic, albeit with lower proportions (35% and 55% of excess mortality in < 65 years old respectively). In the decade following these two later influenza pandemics, the excess mortality in younger adults dropped to less than 5%. Since this age profile differs so clearly from the mortality statistics of both the Russian flu from 1889/1890 (where mostly seniors, but few young adults and young children died) (Brüssow and Brüssow, 2021; Brüssow, 2021, 2021) and seasonal influenza (where young children and particularly seniors die while young adults are mostly spared), this age‐related mortality analysis became a standard approach for defining the pandemic phase of the Spanish flu outbreak.
When US researchers applied this criterion to all‐cause mortality data from New York City (NYC) they observed that the smaller January 1916 and January 1917 excess mortality peaks were mostly contributed by persons > 65 years. Between January and March 1918 they noted a double peak in all‐cause mortality (including pneumonia and influenza). The smaller January peak was due to excess mortality in the > 65 years age group, while the bigger March peak was mostly contributed by 15–24 years old people. Between September 1918 and April 1919 large excess mortality with split fall and winter peaks were observed. People older than 65 years were not affected, while mortality in 15–44 years old subjects constituted the majority of the excess deaths (with some contribution of child mortality to the fall 1918 peak). In NYC, another moderately large excess mortality peak was observed between January and March 1920. Young adults were still the major contributors to excess mortality, but less exclusively than during the 1918/1919 peak suggesting a transition from a pandemic into an epidemic phase (Olson et al., 2005). In 1921, no excess mortality was observed in NYC and between 1922 and 1924 only small excess winter mortality peaks were noted. When using this ‘signature’ age shift criterion, the pandemic phase of the Spanish flu in NYC extended from March 1918 to March 1920. During these four waves 42 000 New Yorkers died from the pandemic corresponding to 0.7% of the NYC population. The first wave was mild, while the second wave (fall 1918) was severe causing a massive mortality increase. The third and fourth waves were associated with more moderate mortality increases. People older than 40 years were largely spared from lethal infections. The growth rate of the epidemic expressed as the effective reproductive number Re did not differ between the four waves. It started with high R values at the beginning of each wave and then R decreased with time (Yang et al., 2014). W‐shaped mortality by age was also found when analysing death certificates from Kentucky during the Spanish flu pandemic. Peak mortality was found in children < 1 year, in young adults with a median age of 25 years and in persons older than 80 years; no difference was seen between males and females. The distinct mortality in the different age groups could not be explained by exposure to prior epidemics excluding immune memory as an explanation for this age profile of mortality (Viboud et al., 2013). Data from Arizona also showed four waves at the same time periods as observed in New York. Native Americans were more affected and income differences explained part of the mortality differences (Dahal et al., 2018). Even remote regions of North America such as the Canadian Newfoundland Island showed a pandemic pattern synchronized with observations from New York with a herald wave, a two‐peak major mortality wave and a recrudescence wave. Overall, the pandemic claimed the life of 1% of the islanders (Sattenspiel, 2011).
Latin America
Mortality data were analysed for a large (Mexico City) and a small city (Toluca) in Mexico. Three successive waves of increased mortality were observed in April–May 1918, September–December 1918 and January–April 1920. Mortality was high across all age groups. The mortality rate was twofold higher in Toluca than in Mexico City and the 1920 mortality was limited to older subjects (Chowell et al., 2010). A study from Columbia revealed neither a ‘herald’ wave in spring 1918 nor a recrudescence wave in winter 1920; excess mortality formed a single peak between October 1918 and January 1919 with a W‐shaped age profile. As in Mexico but unlike the United States, seniors were not spared from excess mortality (Chowell et al., 2012). The study of mortality records from three regions in Peru revealed a mild herald wave in July–September 1918, followed by a second wave in November 1918 to February 1919. From January to March 1920, Lima experienced a recrudescence wave that claimed more lives in Lima than during the rest of the pandemic. Ica (southern coast) experienced a later recrudescence wave in July to October 1920, while Iquitos (northeast) was spared in 1920. The highest risk of death shifted from 25 to 44 years age group during the beginning to the 15–24 years age group in the later phase of the pandemic (Chowell et al., 2011). Analysis of death certificates between 1913 and 1921 in a town of tropical Brazil revealed a single Spanish flu mortality peak in November/December 1918 claiming the life of 0.3% of the townspeople (Alonso et al., 2011). The Spanish flu spread heterogeneously in multiple waves across Chile. Until February 1919, the epidemic was clinically mild and mortality peaked only in August 1919 with excess mortality observed until February 1920. A recrudescence wave was seen in the second half of 1921 in the most densely populated area, the capital Santiago (Chowell et al., 2014).
The Spanish flu in Europe
An overall similar pandemic development as in the United States was seen in Europe, although with substantial local variation. For example, influenza deaths in Madrid during 1916 showed peak mortality in persons older than 65 years, particularly women. This situation changed in Madrid with the May–June 1918 influenza mortality peak which occurred in young adults. This pattern was again observed during the October–November 1918 wave in Madrid. In 1920, a mixed‐age profile of influenza mortality was noted in Madrid with three peaks, namely in young children, young adults and persons older than 65 years. In 1921, influenza mortality in Madrid showed the pre‐pandemic pattern of seasonal influenza, with the highest mortality in the young and the old. These data define for Spain a pandemic phase of influenza during 1918–1919, a transition period in 1920 and a return to seasonal influenza in 1921 (Erkoreka, 2010). Likewise, the highest death rates from influenza were also seen in Paris among young adults during the years 1918 and 1919, unlike the situation in 1917 (Erkoreka, 2010). In neighboring Portugal weakened by participation in World War I, hunger, food shortage, poverty and social conflicts, only one major excess mortality peak occurred between September 1918 and February 1919, causing 178 deaths for 10 000 inhabitants after migrant farmers had introduced the first cases from Spain in May 1918. All provinces were affected even remote islands such as the Acores (Nunes et al., 2018). Northern Europe was also severely affected by the Spanish flu. A quarter of the population from Copenhagen sought medical attention for influenza disease; 5% during the summer 1918 wave, 12% during the fall 1918 wave and 7% during the first months of 1919. About 2% of the population was hospitalized; the temporal development of the hospitalizations was proportional to the observed cases. With respect to mortality, the summer wave was sevenfold less lethal than the fall wave which displayed a 2.3% case‐fatality rate. The death occurred mostly in young adults while older subjects were spared. In the neighboring Swedish city of Gothenburg the influenza epidemic developed the same dynamics and lethality as in Copenhagen while in Stockholm few lethal infections were observed. Oslo in Norway showed most cases during the summer 1918 wave (Andreasen et al., 2008). Finland in contrast displayed between April 1918 and April 1919 four distinct mortality peaks followed by a recrudescence of influenza deaths in February 1920 (Ansart et al., 2009). This variability of the wave dynamics of the Spanish flu pandemic observed in Scandinavia was also observed when excess mortality was compared across European countries. French scientists calculated from excess all‐cause mortality data about 2 million influenza victims in Europe. Mortality increases were highest in Italy, Bulgaria, Portugal and Spain, which all showed a single mortality peak in November 1918. Germany and Switzerland showed a complex wave pattern with herald waves in spring and summer 1918, a major late fall 1918 peak and a recrudescence peak in March 1920. These researchers calculated a fourfold lower total mortality in North America than in Europe (0.5 vs. 2.6 million deaths), which they attributed to massive troop movements across Europe favoring the pandemic spread and an infection encountering a frail population, weakened by a long war (Ansart et al., 2009).
The United Kingdom also experienced infection dynamics that mirrored the herald, major peak and recrudescence wave pattern when analysed by mortality data. Areas with larger populations experienced an early pandemic onset. Urban death rates were higher than rural rates, but overall no clear correlation with population size was seen, nor was a correlation with residential crowding or pre‐pandemic childhood mortality found. The researchers suspected that the build‐up of immunity might explain the wave structure better than epidemiological factors (Chowell et al., 2008). Anecdotal evidence from contemporary medical reports noted that subjects infected in the summer 1918 wave escaped clinical infection in the autumn 1918 wave, suggesting cross‐immunity between the viruses circulating in successive waves (Anonymous, 1919). Epidemiologists analysing influenza morbidity and mortality in US military camps confirmed this observation. Among troops exposed to influenza in the spring, 7% became sick with influenza in the fall. In contrast, among the troops not exposed to influenza in the spring, 49% became sick in the fall. Based on repeated illness data, the first wave provided 35%–94% protection against clinical illness during the second wave and 56%–89% protection against death. In the British Grand Fleet population comprising 90 000 sailors, exposure to influenza during the first wave provided 72% protection against influenza in the fall. In the British civilian population, the first wave conferred only a 35% protection against second wave infection, while first and second wave influenza exposure did not protect this population against the third wave in winter 1918/1919 (Barry et al., 2008). Other researchers had proposed that viral evolution (which modifies transmissibility or immunological escape), environmental change (weather) and behavioural change of people in response to the pandemic caused this peculiar wave structure of the pandemic. Mathematicians studied the effect of school closure, ambient temperature changes and behavioural changes in computer simulations. Only when behaviour changes (e.g. social distancing) were included, was the observed wave structure reproduced by the model (He et al., 2013) suggesting behavioural changes as a major explanation for the wave structure of the Spanish flu pandemic in United Kindom.
The Spanish flu elsewhere
The Spanish flu claimed between 8 and 14 million victims in British India. With that number about 1 of every 25 Indians died during the epidemic and Indians might represent 1 out of 4 Spanish flu victims worldwide. The pandemic entered Bombay in September 1918 with British troop transports and the infection wave spread from the western coast eastward. The northern and central parts suffered the highest death burden. There was substantial temporal heterogeneity in the timing of the pandemic in the different Indian provinces, but cases were observed in each district only over 2–13 weeks. The majority of cases were observed between October and December 1918 without recurrence waves. Death rates were high for all age groups, but excess death was fivefold higher than the seasonal death rate in the 20–30 years age group. The pandemic influenza occurred in a period of low rainfall, while seasonal influenza was associated with periods of high rainfall. Epidemiological analysis showed that long‐distance railway transport was a motor of pandemic spread (Reyes et al., 2018). Singapore experienced a first infection wave in June–July 1918 that was relatively mild, displaying a high illness but a low mortality rate, while a second wave occurred in October and November 1918, leading to frequent pneumonia associated with a high mortality rate (excess mortality 11 per 1000). By the end of November 1918, the epidemic was over in Singapore, with no recurrence wave despite continuing influenza case reporting in Indonesia, New Zealand and Japan. Government’s advice that infected persons should stay home, that public places should be disinfected, that people should avoid crowded places, that schools were closed during the second peak and that indoor ventilation should be increased might have contributed to the end of the pandemic in Singapore (Lee et al., 2007). When analysing mortality data between 1917 and 1921 for Taiwan, two peaks of excess mortality were observed, one in November–December 1918 and another in January–February 1920. In 1918, young adults (30–39 years) experienced a high death rate while older people were spared. In 1920, young adults had a noticeably decreased deaths rate compared with 1918 which was interpreted as a sign of acquired immunity possibly suggesting that both waves were caused by the same virus strain (Hsieh, 2009). A reanalysis of vital statistics for 27 countries by United States and Australian epidemiologists confirmed the unequal impact of the Spanish flu pandemic. With 4.4 excess deaths per 100 people India leads the list, followed by countries from southern Europe (Portugal 2.6; Spain 1.5; Italy 1.4 per 100), central Europe was less affected (0.8 except Austria with 1.6), even less so the United States (0.4 deaths per 100). Overall, the population mortality varied over 30‐fold across countries. The epidemiologists observed that the per‐head income explained a large fraction of this variation in mortality with a marked increase in developing countries, while the association with geographical latitude was not significant. Mortality was overall concentrated in young adults, not in older individuals and was higher in males than in females, but substantial variation was also found for these rules (Murray et al., 2006).
Reconstructing the genome of the 1918 pandemic virus
It is unknown whether the different waves represent infections with different viral variants or the effect of other factors (bacterial co‐pathogens, immunity and behaviour). Epidemiology alone would have left knowledge around the beginning and ending of the pandemic in a limbo without the heroic effort to isolate the pathogen of the Spanish flu which allowed to follow the virus through time.
The detective work started with formalin‐fixed, paraffin‐embedded and stained tissue samples from US servicemen that were killed by the 1918 pandemic wave, preserved in the Armed Forces Institute of Pathology in Washington, D.C. The majority died of acute bacterial pneumonia, one of the most common complications of the Spanish flu. However, one victim was identified (case 1) who showed bacterial lobar pneumonia in the left lung and viral pneumonia in the right lung. The pathologists designed degenerate, consensus primers and obtain 200‐bp DNA fragments by RT‐PCR for several influenza virus genes from the right lung. Sequence analysis placed the fragments for the hemagglutinin (HA) gene (HA mediates receptor recognition of sialic acid residues on cell membranes) close to a swine H1 influenza virus isolated in Iowa in 1930 and a human H1 virus isolated in Puerto Rico in 1934, historically the earliest isolates of influenza viruses in the pioneer period of virology. The neuraminidase (NA) gene fragments (NA is a sialidase that allows the virus to leave infected cells and to spread in the body) again placed the 1918 isolate next to the same swine and human N1 virus isolates. The nucleocapsid gene was a close relative of an H1N1 human virus isolated in 1933 from the United Kingdom (Taubenberger et al., 1997). The close relationship of the 1918 virus with swine influenza virus isolates from the 1930s fits with serology data published in 1936: subjects older than 20 years in contrast to younger subjects showed frequent antibodies neutralizing the swine virus. A virology pioneer, R. Shope, concluded that the virus of swine influenza is the surviving prototype of the viral agent responsible for the great human pandemic of 1918 (Shope, 1936).
Subsequently, the complete coding sequence for the HA gene was obtained from two soldiers who died on September 20, 1918, in South Carolina and on September 30, 1918, in New York (their lung tissue was also conserved on ancient pathology slides) and from an Inuit woman who died in November 1918 in Brevig/Alaska. The woman was exhumed from a mass grave conserved in permafrost and RNA segments smaller than 120 nucleotides were extracted from the lung. Notably, all three viral HA sequences differed by just two nucleotides causing one amino acid change (Reid et al., 1999). Conserved tissue samples from influenza victims in London who died in November 1918 and February 1919 showed in partial HA sequences 2 and 3 nucleotide changes documenting a high degree of sequence conservation for the pandemic virus across continents and a lapse of 5 months (Reid et al., 2003).
The 1918 HA gene was at the root of both the swine and the human H1 clades and was distant from all known avian HA sequences. It differed less from the earliest swine influenza virus HA than from the earliest human influenza virus isolates from the early 1930s. This might not indicate a closer relationship to pig influenza virus, but less accumulation of mutations in pigs due to less immune selection (shorter life span of commercial pigs?). Since veterinary records noted an influenza epidemic in swine only in fall 1918, the researchers concluded that the 1918 virus entered from humans into the swine population. By phylogenetic analysis, the scientists estimated that the 1918 virus had entered the human population sometime between 1900 and 1915. Notably, derivatives of the HA gene from the 1918 virus were still detected in H1 human influenza viruses isolated throughout the 20th century. No close avian influenza virus precursor was found in birds, where HA is under low selective pressure (Reid et al., 1999). However, the later human H1 isolates showed however a gradual accumulation of glycosylation sites masking antigenic sites probably selected for immune escape. Subsequently, the US pathologists sequenced the complete NA gene from the Inuit woman from Brevig. The partial sequences from the two US servicemen showed no nucleotide differences, but the Brevig sample contained two distinct nucleotides at one position. Phylogenetic analysis placed the Brevig sequence again close to swine and human influenza virus isolates from the 1930s. Sequences related to the 1918 virus NA gene were found in human influenza viruses isolated through the 20th century. When not considering the closely related swine influenza viruses from the United States in the 1930s, the 1918 virus HA and NA showed closer relatedness to avian than to other mammalian influenza viruses, which suggested that the 1918 virus was ultimately transmitted to mammals or directly to humans from birds.
The researchers noted that both human and swine NA proteins accumulate phylogenetically informative amino acid replacements in a linear fashion over time. The extrapolation of the regression line indicated a common ancestor which circulated from 1910 to 1915 (Reid et al., 2000). The sequencing of the nonstructural NS gene from the Brevig sample once again placed the 1918 virus at the root of the swine and human H1N1 influenza virus isolates from the 1930s (Basler et al., 2001). The sequence of RNA segment 7 from the 1918 pandemic virus encoding the two matrix genes M1 and M2 also turned out to be very similar to the common ancestor of all subsequent human and classical swine matrix gene segments. Several sequence matches in the Brevig 1918 virus with mammalian viral matrix genes, which were not found in avian viral isolates, indicated to the researchers that the pandemic virus circulated in mammals or humans for several years before 1918 (Reid et al., 2002).
Finally, the US pathologists also sequenced segment 1 (encoding viral protein PB2), segment 2 (PB1) and segment 3 (PA) of the Brevig 1918 pandemic virus, which encode the viral heterotrimeric polymerase complex PA/PB1/PB2 known to be involved in viral replication and interaction with host factors. Their sequence analysis suggested that the polymerase protein sequences from the 1918 virus differed from avian consensus sequences at only a small number of amino acids in line with the hypothesis that they were derived from an avian source. Once the researchers had then the entire viral genome from the 1918 virus at hand, it became clear that the 1918 pandemic strain was not a reassortant virus such as the 1957 H2N2 and 1968 H3N2 pandemic influenza strains, but likely an in‐toto avian‐like virus that had adapted to humans. However, they observed an unexpectedly large number of synonymous nucleotide changes when the 1918 virus was compared with avian influenza viruses. This suggested a substantial distance to avian viruses and indicated that the ancestor of the 1918 virus had circulated in mammals or humans for several years (possibly as early as 1900) and had adapted before causing the pandemic in 1918. A US‐Chinese consortium tried to date the emergence of the 1918 pandemic virus by phylogenetic analysis. According to their analysis components of the 1918 H1N1 pandemic virus circulated in mammalian hosts (swine and humans) as early as 1911 and was not a recently introduced avian virus. According to their interpretation of the sequence data, seasonal and classic swine H1N1 viruses were not directly derived from Brevig 1918 pandemic strain, but their precursors co‐circulated during the pandemic (Smith et al., 2009). The co‐circulation of the Brevig1918 virus and seasonal H1N1 viruses might explain reports of influenza outbreaks of varying severity during the 1918 pandemic. As for the other gene segments of the 1918 virus, the closest relatives of the three polymerase genes were found in human and swine influenza virus isolates from the 1930s. Later human influenza virus isolates showed a gradually increased distance to the 1918 virus sequence suggesting about 0.4 amino acid changes per year for the PB1 protein (Taubenberger et al., 2005).
Further evolution of the 1918 pandemic virus
A consortium of US public health scientists, genomics researchers and the US pathologists who sequenced the 1918 virus developed phylogenetic trees for all eight genome segments of 71 influenza A/H1N1 viruses isolated from the 1930s (when the first direct isolations of influenza virus became technically possible) to 2006. Notably, all eight gene phylogenies (influenza viruses have a segmented negative single‐strand RNA genome consisting of 8 segments encoding 10 proteins) showed generally similar evolutionary patterns. At the root was the Brevig 1918 pandemic virus; its closest relatives were virus isolates from the 1930s. Interestingly, the trees also showed a strong temporal structure, comprising a main trunk lineage that links viruses from successive epidemics in the 1930s, 1940s, 1950s until 1957, followed by a gap. The tree resumes with isolates from 1977 and then with gradually greater divergence through the 1980s, 1990s and into the 21st century (Nelson et al., 2008). This phylogenetic tree finds an explanation in the complex natural history of influenza infections in the last century. Based on a wealth of research, which I will not retrace here, the following grand picture of influenza infections has emerged. The 1918 pandemic virus has survived, and its descendants still circulate today. In the words of the researchers who recovered the Spanish flu virus sequences, the 1918 Influenza is the Mother of All Pandemics (Taubenberger and Morens, 2006). Until 1957, the human influenza A /H1N1 virus has not acquired new gene segments from avian or other viral sources. Each new yearly strain successively showed an accumulation of mutations over time (‘antigenic drift’). In reality, the pattern is more complicated since there is some phylogenetic evidence of distinct intrasubtype reassortant events among viruses from various H1N1 sublineages, so that the overall evolutionary pattern is not truly linear but tightly networked (Zimmer and Burke, 2009). The H1N1 virus got extinct in 1957 when new influenza A H2N2 reassortant virus appeared which caused the Asian flu pandemic. This virus contained three new segments as a result of genetic reassortment (‘antigenic shift’) encoding the PB1, HA and NA proteins acquired from an avian source, but maintained the other five segments from the H1N1 strains of the 1918 Brevig lineage. H2N2 circulated in the human population until 1968 when it was itself replaced by new influenza A H3N2 virus which caused the Hong Kong flu pandemic from 1968 to 1969. The pandemic H3N2 virus strain is a descendant of the H2N2 strain (thus still maintaining 5 gene segments of the Spanish flu 1918 H1N1 virus lineage) by another round of gene segment replacements encoding PB1 and HA protein from a distinct avian influenza virus. Exclusively H3N2 influenza viruses circulating in the human population until 1977 when unexpectedly an H1N1 influenza virus resurfaced in China, the Soviet Union and then also in the United States and became known as Russian flu from 1977 (not to be mixed up with the Russian flu from 1890 which might not have been an influenza virus pandemic, Brüssow and Brüssow, 2021). The origin of this reemerging H1N1 virus is still unclear. Virologists showed in 1978 by oligonucleotide mapping that the 1977 H1N1 virus closely resembled H1N1 virus isolates from 1950 (Nakajima et al., 1978). This apparent evolutionary stasis of an influenza virus over a quarter‐century has startled the researchers and various explanations were proposed. Proposals that some influenza viruses survived in frozen ice of Siberian lakes to explain such an evolutionary stasis were rejected (Worobey, 2008). Accidental release of stored viral samples from a scientific or military laboratory was discussed, but release from a vaccine trial of insufficiently attenuated live virus or use as challenge virus in vaccination trials were considered as more likely explanations (Rozo and Gronvall, 2015). Whatever the origin, the 1950‐like H1N1 viral strain was successfully reintroduced into the human population, it caused the Russian flu epidemic from 1977 to 1979, and cocirculates since then with H3N2 as a cause of seasonal winter influenza epidemics until today. Viruses derived by antigenic drift (H1N1) and by antigenic shift (H3N2) from the 1918 Spanish flu virus are therefore still part of the annual influenza vaccines.
In 2009 another H1N1 virus caused a pandemic, which started in Mexico with severe pneumonia and high mortality (Chowell et al., 2009). CDC estimated that between 150 000 to 570 000 people died from this influenza A H1N1pdm09 (pdm stands for pandemic, 09 for 2009) virus during two waves in 2009. This virus contains genes from North American avian viruses (PB2 and PA); PB1 from human H3N2; HA, NP and NS genes from classical swine influenza virus and NA and M genes from Eurasian avian virus origin. Its origin is thus complex and it is excluded from this overview of human infections with more or less close descendants of the Spanish flu virus from 1918. Likewise excluded are avian influenza viruses H5N1, H7N7 and H9N2, which sporadically infect humans (Neumann et al., 2021).
Influenza mortality in the 20th century
The detailed virological investigation of the influenza virus genomes allowed scientists to trace the legacy of the Spanish flu virus after the pandemic phase. Good data are available for the United States from 1900 to 2004 (Doshi, 2008). All‐cause mortality in the United States showed a constant decrease from 1900 to the mid‐1950s, only interrupted by a sharp mortality increase during 1918–1919 by the Spanish flu pandemic. Yearly data for influenza mortality are available from 1930 onwards: high mortality was seen in the 1930s with up to 40 deaths per 100 000 people. In the first half of the 1940s influenza classified mortality decreased to 20 deaths per 100 000, and remained below 5 deaths per 100 000 since the early 1950s. Very similar data were reported for the United States in an analysis of 10 major infectious diseases. After the 1918/1919 peak for pneumonia and influenza mortality, several smaller peaks were observed between 1922 and 1936, followed by a general decline to low mortality values that do not differentiate the pandemics of 1957 and 1968 from background mortality (Armstrong et al., 1999). The influenza mortality decline followed the general mortality decline for infectious disease. This study noted that many non‐pandemic seasons were more deadly than subsequent pandemics. This observation questions the distinction of pandemic influenza from seasonal influenza with respect to causing more than usual excess mortality per influenza season (Doshi, 2008). In addition, some researchers claim that the 1957 and 1968 pandemic displayed the U‐shaped mortality age profile, not the W‐shaped age mortality observed during the Spanish flu pandemic (Luk et al., 2001). In the US dataset, each pandemic was less mortal than the preceding pandemic. Both seasonal and pandemic influenza mortality showed a secular decrease over time which could not be explained by vaccination, as vaccines did not become available until the 1940s and were not widely used until the late 1980s. Researchers suspected improvement of the living conditions for this secular trend, but one could also suspect a decrease in the virulence of the H1N1 derivatives from the Spanish flu virus. The exceptional lethality of the Spanish flu compared with subsequent influenza epidemics might also find an explanation that many if not most the fatal cases are believed to have occurred because of secondary complications caused by bacterial pathogens/infections (Taubenberger et al., 1997; Morens and Fauci, 2007). Bacterial superinfections were also found in fatal influenza infections during later influenza epidemics (Brundage, 2006), but did not cause this rampant mortality.
Major post‐Spanish flu influenza epidemics
Leading experts distinguished several major pre‐1957 influenza epidemics: one occurred in 1928–1929 with an H1N1 strain modified by an antigenic drift that was associated with 100 excess all‐cause deaths per 100 000 persons/year. The 1918 pandemic was in comparison associated with 600 excess deaths/100 000 subjects and year. The next notable influenza epidemic occurred in 1934–1936 with an H1N1 strain modified by additional antigenic drift. This epidemic was associated with half as many excess deaths (50/100 000 and year) than the 1928–1929 epidemic. Then, followed two influenza epidemics with H1N1 viruses that were intrasubtypic reassortants: 1947–1948 with an H1N1 A‐prime strain which was associated with 10 and another in 1951–1953 with 32 excess deaths per 100 000 people per year respectively (Collins and Lehmann, 1953; Morens et al., 2009).
Similar data as for the United States were reported for Germany. Data on pneumonia mortality were collected for the 1892–1970 time period. They showed a high plateau of 130 deaths per 100 000 people for the years before the Spanish flu despite a marked decrease in overall mortality. During the 1918/1919 pandemic, the mortality peaked at 690 deaths per 100 000 people when 25% of the population was infected. Subsequently, pneumonia mortality decreased continuously. A small mortality peak was seen in 1928–1929 while the Asian and Hong Kong flu pandemics could hardly be differentiated from the pneumonia mortality baseline despite the fact that 31% and 21% of the German population was infected during the 1957–1958 and 1968–1969 pandemic respectively. The 2009 pandemic was associated with even lower mortality of 0.4 deaths per 100 000 people (Buchholz et al., 2016).
Detailed data on influenza medical visits and respiratory mortality were also documented in Copenhagen/Denmark for the period 1904–1937. In 1918–1919, respiratory mortality was sevenfold higher than in the surrounding years while respiratory morbidity was only twofold increased indicating a high virulence of the Spanish flu strain. In the pre‐pandemic years 1904–1917, people older than 65 years displayed a 10‐fold higher risk of influenza‐related death per capita than the younger population; this pattern was again observed between 1925 and 1937. In contrast, seniors were – as in the United States – spared in 1918–1919 while young and old citizens experienced comparable excess mortality rates in the years following the Spanish flu. A series of influenza mortality peaks were seen in the 1920s and early 1930s that reached high mortality rates but were of shorter duration than the Spanish flu pandemic (Saglanmak et al., 2011).
Epidemiology of the influenza epidemics in the 1920s
Statisticians from the US Public Health Service reported during the 1920s six influenza epidemics, most showed only small excess mortality increases of short duration. Only the 1928–1929 epidemic caused 50 000 excess influenza‐pneumonia deaths, half as much as the recrudescence Spanish flu wave from February 1920 and one‐tenth of the major wave of the Spanish flu (Fig. 4). The excess mortality peak was first seen at the Pacific coast and traveled eastwards to reach New England 7 weeks later (Fig. 5). The other and smaller influenza epidemics from the 1920s did not follow this geographical spread, with one epidemic starting in Chicago. In 1928–1929, half as many cases of influenza were reported as during the Spanish flu pandemic, but the case fatality rate was smaller (0.6% vs. 1.6%) possibly explained by a lower association with pneumonia complications (2% vs. 6%). Those developing pneumonia showed however comparable mortality of 21% vs. 26% in 1928 and 1918 respectively. The highest mortality was seen in 10‐year‐old children and in 30–40‐year‐old adults, while no sparing of seniors seen during the Spanish flu was noted in the 1928 epidemic (Fig. 6) (Collins, 1930).
Fig. 4.
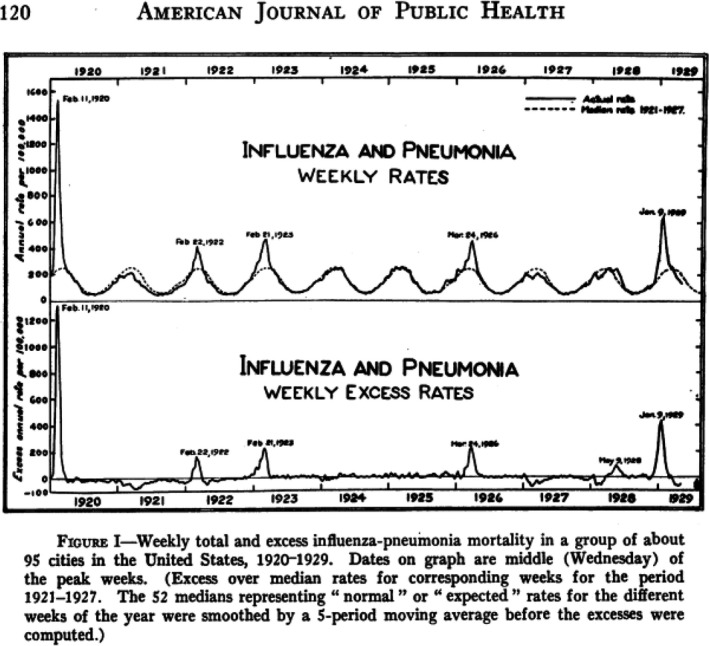
Weekly total (top) and excess rate (bottom) of influenza and pneumonia mortality for 95 US cities between 1920 and 1929. Source: Collins (1930).
Fig. 5.
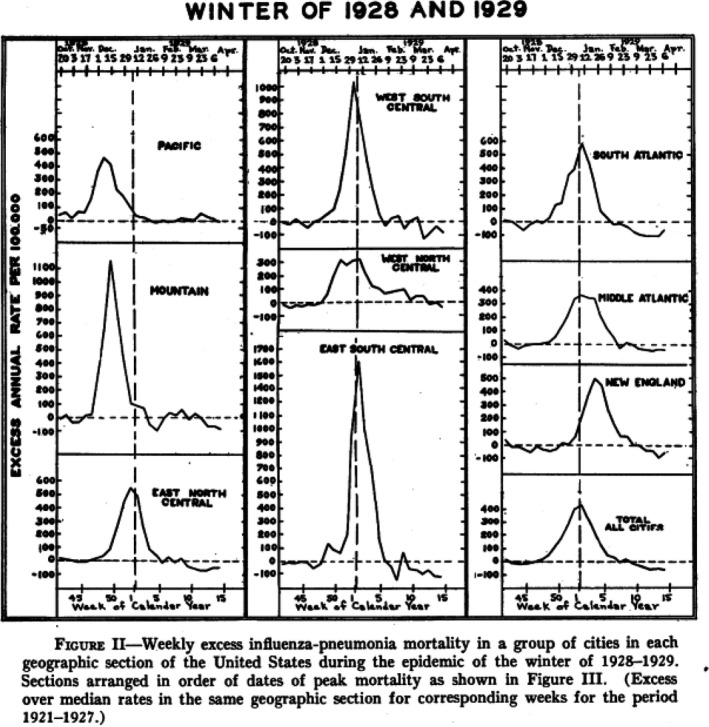
Weekly excess influenza and pneumonia mortality for selected geographical regions across the United States during the influenza winter epidemic 1928–1929. Source: Collins (1930).
Fig. 6.
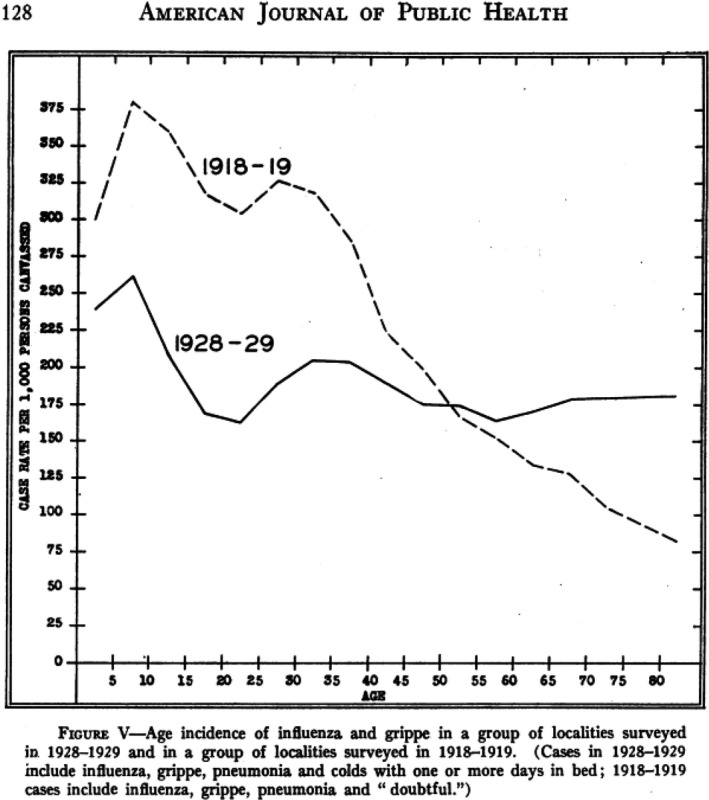
Age profile for influenza case incidence in US cities during the winter epidemic 1928 to 1929 compared with the age profile of influenza cases during the 1918–1919 pandemic. Source: Collins (1930).
Influenza epidemics between 1930 and 1957
British researchers analysed the antigenic drift in the NA gene from H1N1 viruses isolated between 1933 and 1957. Significant antigenic differences were detected among them; viruses isolated 1 year apart could be distinguished serologically (Luther et al., 1984).
Over the period 1933–1946, there were three major influenza A epidemics in England and when studying influenza in several districts in Yorkshire the researchers observed a significant negative correlation between influenza incidence in 1933 and 1936–1937, suggesting the persistence of immunity to serologically cross‐reacting viral strains for 4 years (Pickels et al., 1947).
US pioneers of vaccinology observed that a subcutaneous vaccine consisting of influenza A and B viruses which protected vaccinees from influenza disease during three successive seasons failed to protect in January and May 1948 when compared with vaccinees who received the same vaccine complemented by a newly emerged A prime influenza strain, highlighting the need for adding new strains to the virus cocktail (Salk and Suriano, 1949). In 2002, US researchers re‐investigated the epidemiological situation and detected important intrasubtypic viral differences which could explain the failure of the 1947 vaccine. Infections in 1947/1948 were caused by H1N1 virus strains, but cross‐immunity diminished since mutations had accumulated by antigenic drift within the same virus subtype. This antigenic drift necessitated frequent changes in the vaccine composition, but older vaccines were usually at least partially protective which was however not the case in 1947. Serological analysis showed a lack of neutralization of the 1947 strains by antisera to 1943 viruses and a lack of inhibition in lung replication of the 1947 virus in mice immunized with the 1943 virus vaccine. The 1947 viral HA differed in five H1 antigenic sites from the 1943 H1 protein while the N1 proteins from the 1943 and 1947 viruses were nearly identical. The 1947 virus spread widely but failed to cause notable excess mortality (Kilbourne et al., 2002).
In 1951, Liverpool experienced an influence epidemic that surpassed the mortality seen in that city during the Spanish flu pandemic. This H1N1 outbreak was observed across the whole of England with a death rate for influenza and pneumonia superior to that of other influenza epidemics of the 1950s, including that of the 1957 and 1968 pandemics. A similar marked influenza mortality was seen in 1951 for Canada, which represented the most severe influenza epidemic seen there between 1950 and 2000. Paradoxically, no such epidemic was seen in the United States (Viboud et al., 2006). This discrepancy might be explained by the co‐circulating of two antigenically distinct influenza A/H1N1 strains in the Northern Hemisphere during the 1951 epidemic, a virulent ‘Liverpool’ and a less virulent ‘Scandinavian’ strain which could be differentiated serologically.
Also in the tropical globally connected city‐state of Singapore, a substantial excess mortality peak was seen in 1951 that was greater than that of the 1968 pandemic and comparable to that of the 1957 pandemic. In addition, about 20 further, but much smaller excess mortality peaks were seen between 1950 and 2000. Only a few of these smaller excess mortality peaks were associated with more than 50% influenza virus detection in patients with respiratory symptoms. H1N1 epidemics were only associated with less than 6 excess deaths per 100 000 citizens (Lee et al., 2009). Poland also reported peaks of influenza cases in 1951 and 1954 during a 10 year survey period starting in 1947 (Przesmycki et al., 1959).
The reappearance of the H1N1 influenza virus in 1977
The emergence of the H2N2 influenza virus with the Asian flu pandemic led to the disappearance of the H1N1 influenza virus from the human population. After 20 years of non‐observation human H1N1 influenza virus was again isolated in 1977. Notably, the re‐emerging H1N1 strain from 1977 resembled relatively closely the ‘Scandinavian’ strain (Kendal et al., 1978). Earlier reports had shown that the NA of the re‐emerged 1977 H1N1 was serologically close to 1950 virus isolates (Luther et al., 1984). The resemblance of the 1977 re‐emerging H1N1 virus with the less virulent ‘Scandinavian’ H1N1 strain from 1951 concurs with epidemiological data from China where its spread was first reported. The virus was characterized by slow spread, unevenness of attack rates, and the occurrence of many mild cases and inapparent infections. The 7–20 years age group suffered the highest morbidity (Kung et al., 1978). In the winter, 1977/1978 people younger than 25 years in the Soviet Union also experienced a widespread epidemic of mild influenza with this re‐emerged H1N1 virus (Gregg et al., 1978). In February 1978, several US university campuses also reported H1N1 outbreaks with high attack rates in students younger than 24 years, but the disease was mild and no complications were reported (Layde et al., 1980; Wright et al., 1980).
Despite its low virulence the re‐emerged H1N1 strain could establish cocirculation with the previously circulating H3N2 influenza A virus (Glass et al., 1978). A survey of virus isolation conducted between 1978 and 1990 in India showed that some years were dominated by H1N1 isolates, others by H3N2 isolates while still others showed the presence of both viral types. Serological investigations demonstrated that the 1986 H1N1 strain differed from the 1978 H1N1 isolate (Rao and Banerjee, 1993). A more detailed genomic analysis of H1N1 and H3N2 strains was conducted with viral isolates from New York State and New Zealand obtained between 1990 and 2006. Peaks in genetic diversity of H1N1 typically coincided with weak diversity peaks of H3N2 viruses. H1N1 only dominated in seasons following unusually mild H3N2 epidemics. Since its emergence in 1977, H1N1 epidemics exhibited lower mortality rates than H3N2 epidemics. A key observation was the return of H1N1 strains with comparable gene segment ancestry after a genetic bottleneck that led to its disappearance. To explain this situation the researchers developed a source‐sink model. Southern China or tropical regions, characterized by more sustained viral transmission dynamics across the year represent a ‘source’ region which feed regularly viruses into the northern and southern hemisphere, the ‘sink’ where an epidemic develops in the next cold season. The researchers observed greater variation which persisted among several epidemic seasons for H1N1 than for H3N2 strains which may reflect weaker immune selection on H1N1 than on H3N2 viruses (Rambaut et al., 2008). The Influenza Genome Sequencing Project also identified a segment exchange between H1N1 and H3N2 viruses, resulting in an H1N2 serotype in which the HA segment was exchanged (Ghedin et al., 2005).
Lessons
It is of course difficult to draw conclusions on the future trajectory of the COVID‐19 pandemic from the Spanish flu since the two pandemics were caused by two different viruses, which make extrapolations risky. However, the Spanish flu and the 1890 Russian flu pandemic are the only respiratory pandemics of comparable impact as COVID‐19 to serve as historical parallels. The 1890 Russian flu pandemic has the disadvantage of an undefined viral etiology, which prevents a follow‐up of the Russian flu beyond the acute phase from 1890 to 1893. The reconstruction of the Spanish flu virus allowed a follow‐up of the infection from a pandemic into an epidemic and endemic phase, which is a major topic of scientific discussion for the future of SARS‐CoV‐2 and COVID‐19.
With caution, some conclusions can be tentatively drawn. Prior respiratory pandemics showed development in waves and the pandemic phase extended over a 2–3 years course. The waves were characterized by different clinical severity and the pandemic phases seem to end with an attenuation of the viral virulence. The causes for the wave structure of the Spanish flu pandemic are not clearly determined not the least because no viral reconstructions are available for the first and fourth waves. We thus ignore to what extent changes in viral virulence, transmission and immune escape in combination with an immunity build‐up in the population and behavioural responses and public health mitigation measures have shaped the wave pattern.
Even without efficient vaccines and drugs, the pandemic phase of the Russian and Spanish flu ended after a wave associated with milder symptoms. It is of course a matter of conjecture whether we are with the omicron wave of COVID‐19 in a comparable situation as the Spanish flu in 1920 and the Russian flu in 1892. The higher transmissibility and the lower virulence of omicron compared with the delta variant with a preferential replication in the upper over the lower respiratory tract are hopeful signs and correspond also to general ideas about the evolution of transspecies infections. There is thus hope that we might see in 2022 the end of the pandemic phase of COVID‐19. However, if one takes the example of the Spanish flu, the SARS‐CoV‐2 virus will not go extinct. The genomics of H1N1 viruses showed that the pandemic virus persisted in the human population for nearly four decades causing annual epidemics in the temperate zones. This is in fact also the prediction of many virologists that SARS‐CoV‐2 will continue to circulate, mutate and in the future become another respiratory virus causing common cold or influenza‐like symptoms in regular time intervals where the length of their spacing might depend on the persistence of the immune response.
We know that the Spanish flu pandemic virus evolved over four decades (1918–1957) by rather conventional genetic means, namely the accumulation of nucleotide changes and concomitant amino acid changes in the viral proteins (‘antigenic drift’). However, there is some warning in the historical experience. Some of the antigenic drift‐products of H1N1 caused substantial epidemics. For example, the influenza epidemic from 1928 to 1929 showed still mortality that amounted to a sixth of the mortality of the pandemic phase. Throughout the 1920s and 1930s influenza mortality remained elevated and even as late as 1951 viral variants appeared (‘Liverpool’ strain) that showed a greater virulence than the Spanish flu virus itself while missing its capacity to spread. The same observation was made with the Russian flu: 9 years after the start of the pandemic a recrudescence wave was seen that was associated with high mortality. This parallel is less clear since neither for the 1890 nor the 1900 waves it was directly proven that it was caused by the same virus and it could also have been caused by another respiratory virus, and not an influenza virus. Clinical symptoms of patients in 1900 resembled those of patients in 1890 and showed, in addition, some resemblance to COVID‐19 symptoms (Brüssow and Brüssow, 2021). If the 1890 pandemic was caused by a coronavirus, this would strengthen the argument that not only for an influenza pandemic but also for a coronavirus pandemic, one must count on substantial epidemic activities a decade or more after the end of the pandemic phase. It is therefore likely that with the end of the pandemic phase of COVID‐19, SARS‐CoV‐2 will keep public health institutions on alert for at least a decade with winter epidemics. The historical H1N1 experience with the vaccine failure of 1947 as well as the reduced neutralization of the omicron variant by vaccination‐induced antibodies suggest that we will need regular surveillance of circulating virus strains and updates of vaccines if coverage problems are revealed. However, the progressive trend of decreasing mortality of influence epidemics in the 100 years following the Spanish flu pandemic which applies also to the Asian, Hong Kong and the Swine flu pandemic with reassortant viruses created by antigenic shift instills hope that viruses cannot easily reacquire the virulence of the initial transspecies infection event. Vigilance is nevertheless needed since new avian influenza viruses and new bat coronaviruses continue to represent a threat and zoonotic infections are by far not a problem restricted to these two viral families.
Conflict of interets
None declared.
Acknowledgements
I thank Dr. Sophie Zuber for critical reading of the manuscript.
Microbial Biotechnology (2022) 15(5), 1301–1317
Funding Information
No funding information provided.
References
- Alonso, W.J. , Nascimento, F.C. , Acuña‐Soto, R. , Schuck‐Paim, C. , and Miller, M.A. (2011) The 1918 influenza pandemic in Florianopolis: a subtropical city in Brazil. Vaccine 29(Suppl. 2): B16–B20. [DOI] [PubMed] [Google Scholar]
- Andreasen, V. , Viboud, C. , and Simonsen, L. (2008) Epidemiologic characterization of the 1918 influenza pandemic summer wave in Copenhagen: implications for pandemic control strategies. J Infect Dis 197: 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous . (1919) The influenza pandemic. Lancet 1919: 386–387. [Google Scholar]
- Ansart, S. , Pelat, C. , Boelle, P.Y. , Carrat, F. , Flahault, A. , and Valleron, A.J. (2009) Mortality burden of the 1918–1919 influenza pandemic in Europe. Influenza Other Respir Viruses 3: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, G.L. , Conn, L.A. , and Pinner, R.W. (1999) Trends in infectious disease mortality in the United States during the 20th century. JAMA 281: 61–66. [DOI] [PubMed] [Google Scholar]
- Barry, J.M. , Viboud, C. , and Simonsen, L. (2008) Cross‐protection between successive waves of the 1918–1919 influenza pandemic: epidemiological evidence from US army camps and from Britain. J Infect Dis 198: 1427–1434. 10.1086/592454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler, C.F. , Reid, A.H. , Dybing, J.K. , Janczewski, T.A. , Fanning, T.G. , Zheng, H. , et al. (2001) Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc Natl Acad Sci USA 98: 2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage, J.F. (2006) Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness. Lancet Infect Dis 6: 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow, H. (2021) What we can learn from the dynamics of the 1889 'Russian flu' pandemic for the future trajectory of COVID‐19. Microb Biotechnol 14: 2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow, H. , and Brüssow, L. (2021) Clinical evidence that the pandemic from 1889 to 1891 commonly called the Russian flu might have been an earlier coronavirus pandemic. Microb Biotechnol 14: 1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz, U. , Buda, S. , Reuß, A. , Haas, W. , and Uphoff, H. (2016) Influenza pandemic deaths in Germany from 1918 to 2009. Estimates based on literature and own calculations. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 59: 523–536. [DOI] [PubMed] [Google Scholar]
- Capps, J.A. , and Moody, A.M. (1916) The recent epidemic of grip. JAMA 67: 1349–1350. [Google Scholar]
- Chowell, G. , Bertozzi, S.M. , Colchero, M.A. , Lopez‐Gatell, H. , Alpuche‐Aranda, C. , Hernandez, M. , and Miller, M.A. (2009) Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med 361: 674–679. [DOI] [PubMed] [Google Scholar]
- Chowell, G. , Bettencourt, L.M. , Johnson, N. , Alonso, W.J. , and Viboud, C. (2008) The 1918–1919 influenza pandemic in England and Wales: spatial patterns in transmissibility and mortality impact. Proc Biol Sci 275: 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell, G. , Simonsen, L. , Flores, J. , Miller, M.A. , and Viboud, C. (2014) Death patterns during the 1918 influenza pandemic in Chile. Emerg Infect Dis 20: 1803–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell, G. , Viboud, C. , Simonsen, L. , Miller, M.A. , and Acuna‐Soto, R. (2010) Mortality patterns associated with the 1918 influenza pandemic in Mexico: evidence for a spring herald wave and lack of preexisting immunity in older populations. J Infect Dis 202: 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell, G. , Viboud, C. , Simonsen, L. , Miller, M.A. , Hurtado, J. , Soto, G. , et al. (2011) The 1918–1920 influenza pandemic in Peru. Vaccine 29(Suppl. 2): B21–B26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell, G. , Viboud, C. , Simonsen, L. , Miller, M.A. , Acuna‐Soto, R. , Díaz, J.M. , and Martínez‐Martín, A.F. (2012) The 1918–19 influenza pandemic in Boyacá, Colombia. Emerg Infect Dis 18: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S.D. (1930) The influenza epidemic of 1928–1929 with comparative data for 1918–1919. Am J Public Health Nations Health 20: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S.D. , and Lehmann, J. (1953) Excess Deaths from Influenza and Pneumonia and from Important Chronic Diseases During Epidemic Periods, 1918–51. Washington, DC: Government Printing Office. [PubMed] [Google Scholar]
- Dahal, S. , Jenner, M. , Dinh, L. , Mizumoto, K. , Viboud, C. , and Chowell, G. (2018) Excess mortality patterns during 1918–1921 influenza pandemic in the state of Arizona, USA. Ann Epidemiol 28: 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi, P. (2008) Trends in recorded influenza mortality: United States, 1900–2004. Am J Public Health 98: 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkoreka, A. (2010) The Spanish influenza pandemic in occidental Europe (1918‐1920) and victim age. Influenza Other Respir Viruses 4:81‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin, E. , Sengamalay, N.A. , Shumway, M. , Zaborsky, J. , Feldblyum, T. , Subbu, V. , et al. (2005) Large‐scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 437: 1162–1166. [DOI] [PubMed] [Google Scholar]
- Glass, R.I. , Brann, E.A. , Slade, J.D. , Jones, W.E. , Scally, M.J. , Craven, R.B. , and Gregg, M.G. (1978) Community‐wide surveillance of influenza after outbreaks due to H3N2 (A/Victoria/75 and A/Texas/77) and H1N1 (A/USSR/77) influenza viruses, Mercer County, New Jersey, 1978. J Infect Dis 138: 703–706. [DOI] [PubMed] [Google Scholar]
- Gregg, M.B. , Hinman, A.R. , and Craven, R.B. (1978) The Russian flu. Its history and implications for this year's influenza season. JAMA 240: 2260–2263. [DOI] [PubMed] [Google Scholar]
- He, D. , Dushoff, J. , Day, T. , Ma, J. , and Earn, D.J. (2013) Inferring the causes of the three waves of the 1918 influenza pandemic in England and Wales. Proc Biol Sci 280: 20131345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, Y.H. (2009) Excess deaths and immunoprotection during 1918–1920 influenza pandemic, Taiwan. Emerg Infect Dis 15: 1617–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendal, A.P. , Noble, G.R. , Skehel, J.J. , and Dowdle, W.R. (1978) Antigenic similarity of influenza A (H1N1) viruses from epidemics in 1977‐‐1978 to ‘Scandinavian’ strains isolated in epidemics of 1950‐‐1951. Virology 89:632‐636. [DOI] [PubMed] [Google Scholar]
- Kilbourne, E.D. , Smith, C. , Brett, I. , Pokorny, B.A. , Johansson, B. , and Cox, N. (2002) The total influenza vaccine failure of 1947 revisited: major intrasubtypic antigenic change can explain failure of vaccine in a post‐World War II epidemic. Proc Natl Acad Sci USA 99: 10748–10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung, H.C. , Jen, K.F. , Yuan, W.C. , Tien, S.F. , and Chu, C.M. (1978) Influenza in China in 1977: recurrence of influenzavirus A subtype H1N1. Bull World Health Organ 56: 913–918. [PMC free article] [PubMed] [Google Scholar]
- Layde, P.M. , Engelberg, A.L. , Dobbs, H.I. , Curtis, A.C. , Craven, R.B. , Graitcer, P.L. , et al. (1980) Outbreak of influenza A/USSR/77 at Marquette University. J Infect Dis 142: 347–352. [DOI] [PubMed] [Google Scholar]
- Lee, V.J. , Chen, M.I. , Chan, S.P. , Wong, C.S. , Cutter, J. , Goh, K.T. , and Tambyah, P.A. (2007) Influenza pandemics in Singapore, a tropical, globally connected city. Emerg Infect Dis 13: 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, V.J. , Yap, J. , Ong, J.B. , Chan, K.P. , Lin, R.T. , Chan, S.P. , et al. (2009) Influenza excess mortality from 1950–2000 in tropical Singapore. PLoS One 4: e8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk, J. , Gross, P. , and Thompson, W.W. (2001) Observations on mortality during the 1918 influenza pandemic. Clin Infect Dis 33: 1375–1378. [DOI] [PubMed] [Google Scholar]
- Luther, P. , Bergmann, K.C. , and Oxford, J.S. (1984) An investigation of antigenic drift of neuraminidases of influenza A (H1N1) viruses. J Hyg (Lond) 92: 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens, D.M. , and Fauci, A.S. (2007) The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 195:1018‐1028. [DOI] [PubMed] [Google Scholar]
- Morens, D.M. , Taubenberger, J.K. , and Fauci, A.S. (2008) Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 198: 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens, D.M. , Taubenberger, J.K. , and Fauci, A.S. (2009) The persistent legacy of the 1918 influenza virus. N Engl J Med 361:225‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, C.J. , Lopez, A.D. , Chin, B. , Feehan, D. , and Hill, K.H. (2006) Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918–20 pandemic: a quantitative analysis. Lancet 368: 2211–2218. [DOI] [PubMed] [Google Scholar]
- Nakajima, K. , Desselberger, U. , and Palese, P. (1978) Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature 274: 334–339. [DOI] [PubMed] [Google Scholar]
- Nelson, M.I. , Viboud, C. , Simonsen, L. , Bennett, R.T. , Griesemer, S.B. , St George, K. , et al. (2008) Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog 4: e1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann, G. , Treanor, J.J. , and Kawaoka, Y. (2021) Orthomyxoviruses. In Fields Virology. Volume 1: Emerging Viruses. Howley, P.M. , and Knipe, D.M. (eds). Philadelpjia: Wolters Kluwer, pp. 649‐705. [Google Scholar]
- Nunes, B. , Silva, S. , Rodrigues, A. , Roquette, R. , Batista, I. , and Rebelo‐de‐Andrade, H. (2018) The 1918–1919 influenza pandemic in portugal: a regional analysis of death impact. Am J Epidemiol 187: 2541–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, D.R. , Simonsen, L. , Edelson, P.J. , and Morse, S.S. (2005) Epidemiological evidence of an early wave of the 1918 influenza pandemic in New York City. Proc Natl Acad Sci USA 102: 11059–11063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford, J.S. (2000) Influenza A pandemics of the 20th century with special reference to 1918: virology, pathology and epidemiology. Rev Med Virol 10: 119–133. [DOI] [PubMed] [Google Scholar]
- Pickels, W.N. , Burnet, F.M. , and Mcarthur, N. (1947) Epidemic respiratory infection in a rural population with special reference to the influenza A epidemics of 1933, 1936–7 and 1943–4. J Hyg (Lond) 45: 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przesmycki, F. , Dobrowska, H. , and Sawicki, I. (1959) Epidemiology of influenza in Poland, 1947/57. Bull World Health Organ 20: 303–323. [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Pybus, O.G. , Nelson, M.I. , Viboud, C. , Taubenberger, J.K. , and Holmes, E.C. (2008) The genomic and epidemiological dynamics of human influenza A virus. Nature 453: 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, B.L. , and Banerjee, K. (1993) Influenza surveillance in Pune, India, 1978–90. Bull World Health Organ 71: 177–181. [PMC free article] [PubMed] [Google Scholar]
- Reid, A.H. , Fanning, T.G. , Hultin, J.V. , and Taubenberger, J.K. (1999) Origin and evolution of the 1918 ‘Spanish’ influenza virus hemagglutinin gene. Proc Natl Acad Sci USA 96: 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, A.H. , Fanning, T.G. , Janczewski, T.A. , and Taubenberger, J.K. (2000) Characterization of the 1918 ‘Spanish’ influenza virus neuraminidase gene. Proc Natl Acad Sci USA 97: 6785–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, A.H. , Fanning, T.G. , Janczewski, T.A. , McCall, S. , and Taubenberger, J.K. (2002) Characterization of the 1918 ‘Spanish’ influenza virus matrix gene segment. J Virol 76: 10717–10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, A.H. , Janczewski, T.A. , Lourens, R.M. , Elliot, A.J. , Daniels, R.S. , Berry, C.L. , et al. (2003) 1918 influenza pandemic caused by highly conserved viruses with two receptor‐binding variants. Emerg Infect Dis 9: 1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes, O. , Lee, E.C. , Sah, P. , Viboud, C. , Chandra, S. , and Bansal, S. (2018) Spatiotemporal patterns and diffusion of the 1918 influenza pandemic in British India. Am J Epidemiol 187: 2550–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozo, M. , and Gronvall, G.K. (2015) The reemergent 1977 H1N1 strain and the gain‐of‐function debate. MBio 6:e01013‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglanmak, N. , Andreasen, V. , Simonsen, L. , Mølbak, K. , Miller, M.A. , and Viboud, C. (2011) Gradual changes in the age distribution of excess deaths in the years following the 1918 influenza pandemic in Copenhagen: using epidemiological evidence to detect antigenic drift. Vaccine 29(Suppl. 2): B42–B48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salk, J.E. , and Suriano, P.C. (1949) Importance of antigenic composition of influenza virus vaccine in protecting against the natural disease; observations during the winter of 1947–1948. Am J Public Health Nations Health 39: 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattenspiel, L. (2011) Regional patterns of mortality during the 1918 influenza pandemic in Newfoundland. Vaccine 29(Suppl. 2): B33–B37. [DOI] [PubMed] [Google Scholar]
- Shope, R.E. (1936) The incidence of neutralizing antibodies for swine influenza virus in the sera of human beings of different ages. J Exp Med 63: 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, L. , Clarke, M.J. , Schonberger, L.B. , Arden, N.H. , Cox, N.J. , and Fukuda, K.J. (1998) Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J Infect Dis 178: 53–60. [DOI] [PubMed] [Google Scholar]
- Smith, G.J. , Bahl, J. , Vijaykrishna, D. , Zhang, J. , Poon, L.L. , Chen, H. , et al. (2009) Dating the emergence of pandemic influenza viruses. Proc Natl Acad Sci USA 106: 11709–11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro, M. , Ciborowski, P. , Klenk, H.D. , Pulverer, G. , and Rott, R. (1987) Role of Staphylococcus protease in the development of influenza pneumonia. Nature 325: 536–537. [DOI] [PubMed] [Google Scholar]
- Taubenberger, J.K. , and Morens, D.M. (2006) 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 12: 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger, J.K. , Reid, A.H. , Krafft, A.E. , Bijwaard, K.E. , and Fanning, T.G. (1997) Initial genetic characterization of the 1918 ‘Spanish’ influenza virus. Science 275: 1793–1796. [DOI] [PubMed] [Google Scholar]
- Taubenberger, J.K. , Reid, A.H. , Lourens, R.M. , Wang, R. , Jin, G. , and Fanning, T.G. (2005) Characterization of the 1918 influenza virus polymerase genes. Nature 437: 889–893. [DOI] [PubMed] [Google Scholar]
- Vaughan, W.T. (1921) Influenza: an epidemiological study. Am J Hyg (Monograph 1) 1–260. [Google Scholar]
- Viboud, C. , Eisenstein, J. , Reid, A.H. , Janczewski, T.A. , Morens, D.M. , and Taubenberger, J.K. (2013) Age‐ and sex‐specific mortality associated with the 1918–1919 influenza pandemic in Kentucky. J Infect Dis 207: 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viboud, C. , Tam, T. , Fleming, D. , Miller, M.A. , and Simonsen, L. (2006) 1951 influenza epidemic, England and Wales, Canada, and the United States. Emerg Infect Dis 12: 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey, M. (2008) Phylogenetic evidence against evolutionary stasis and natural abiotic reservoirs of influenza A virus. J Virol 82: 3769–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, P.F. , Thompson, J. , and Karzon, D.T. (1980) Differing virulence of H1N1 and H3N2 influenza strains. Am J Epidemiol 112: 814–819. [DOI] [PubMed] [Google Scholar]
- Yang, W. , Petkova, E. , and Shaman, J. (2014) The 1918 influenza pandemic in New York City: age‐specific timing, mortality, and transmission dynamics. Influenza Other Respir Viruses 8: 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, S.M. , and Burke, D.S. (2009) Historical perspective‐‐Emergence of influenza A (H1N1) viruses. N Engl J Med 361:279‐285. [DOI] [PubMed] [Google Scholar]