

1. INTRODUCTION
The function of rapid eye movement (REM) sleep, a state identified in adult animals and humans by the simultaneous presence of a desynchronized cortical electroencephalogram (EEG), atonia and periodic bursts of rapid eye movements, remains one of the great biological mysteries of the twentieth century. This is despite the considerable amount that has been learned about the brainstem mechanisms underlying REM sleep since its discovery 30 years ago. In most behavioral systems, the situation is reversed. Thus, we know the biological functions of eating, sex, respiration, etc., and struggle to learn the precise neuronal mechanisms which generate these behaviors. In contrast, although the regional changes in neuronal firing that occur during REM sleep are well documented, little is known about the function of this state. Yet REM sleep occupies between 15 and 20% of the existence of most placental mammals199. Animals spend more time in REM sleep than they spend in other activities essential for survival. What is the selective advantage conveyed by this state?
2. UNIT ACTIVITY STUDIES
From the time of its discovery, attention has been focused on the dramatic neuronal activity changes occurring during REM sleep. Consideration of these changes should provide an important clue to the function of this state. Many studies have been directed at the identification of cell groups responsible for generating sleep states and state specific phenomena. In this review we will instead emphasize the likely functional consequences of the state related changes in neuronal activity. This approach allows a fresh perspective on the large body of information that has accumulated on central nervous system (CNS) activity during sleep states.
Most cell groups examined show substantial changes in their activity during the sleep cycle, and marked differences in their activity between REM and non-REM sleep. The general pattern can be summarized by saying that most cell groups behave similarly in REM sleep and in active waking104,168. In most cell groups, the active waking and REM sleep states are associated with high discharge rates, while quiet waking and non-REM sleep states are associated with minimal activity. However, in certain hypothalamic and basal forebrain regions, waking and REM sleep are associated with minimal discharge rates, and cells achieve their maximal discharge rates in non-REM sleep83,175.
While forebrain cell groups may contribute to the generation of REM sleep, it is now well established that they are not required for the generation of this state151. Is there a forebrain cell group which shows a pattern of activity in REM sleep that is distinct from that shown in non-REM sleep and active waking? If so, these cells might provide a clue to the functional role of REM sleep. Table I lists the neuronal discharge patterns across the sleep cycle reported in a number of representative studies. Most cell groups show reliable differences in their activity between REM and non-REM sleep. Thus, a number of neocortical and diencephalic regions show a relatively high ratio of REM sleep to non-REM sleep discharge rates. However, while many forebrain cells achieve high discharge rates in REM sleep, most discharge at similar rates in active waking, i.e. the high firing rates in REM sleep are not unique.
TABLE I.
Change in discharge rates in REM sleep
| Region | REM/non-REM | REM/active W | Ref. |
|---|---|---|---|
|
| |||
| Cortex | |||
| Parietal | 3.1 | 1.0 | 35 |
| Frontal | 0.8 | 0.9 | 35 |
| Motor (PTN) | 1.9 | 2.4 | 40 |
| Visual | 1.7 | 0.5 | 87 |
| Thalamus | |||
| VB | 2.0 | 1.5 | 15 |
| LGN | 4.5 | 2.2 | 96 |
| Hypothalamus | |||
| VH | 0.8 | 1.4 | 76 |
| BF | 1.4 | 1.0 | 175 |
| POA | 1.5 | 1.0 | 56 |
| Cerebellum | |||
| Simple | 1.9 | 0.6 | 95 |
| Complex | 0.8 | 0.8 | 95 |
| Brainstem | |||
| MRF | 2.0 | 2.0 | 74 |
| MPRF | 3.8 | 3.0 | 70 |
| MPRF | 6.4 | 0.8–3.2 | 185 |
| MPRF | 3.2 | 0.4–24.2 | 154 |
| PLCα | 12.5 | 3.0–∞ | 142 |
| Raphe | |||
| DRN | 0.2 | 0.1 | 103 |
| RM | 0.1 | 0.04 | 51 |
| Dorsolateral pons | |||
| LC | 0.3 | 0.1 | 69 |
| PBL | 0.07 | 0.05 | 69 |
| PBL | 0.06 | 0.02 | 140 |
| ⩾3 ⩽0.3 | |||
PTN, pyramidal tract neurons; VB, ventrobasal; LGN, lateral geniculate; VH, ventral hypothalamus; BF, basal forebrain; POA, preoptic area; MRF, midbrain reticular formation; MPRF, medial pontine reticular formation; PLCα, peri-locus coeruleus α; DRN, dorsal raphe nucleus; RM, raphe medianus; LC, locus coeruleus; PBL, parabrachialis lateralis.
As in the forebrain, most brainstem cell groups show dramatic modulation of discharge rate across the sleep cycle. The cells of the medial reticular formation, many of which fire slowly during quiet waking and during quiet REM sleep (the REM sleep intervals between bursts of rapid eye movements) show particularly high REM sleep to non-REM sleep discharge rate ratios101,154. However, they also show particularly high ratios of active waking to quiet waking discharge rates. In fact, the maximal discharge rates in active waking and REM sleep are positively and significantly correlated159.
One noteworthy exception to the general pattern of sleep related modulation of unit discharge rate is the regularity of discharge of the mesencephalic dopamine containing neurons. These cells have been found to show virtually no change in discharge rate across the sleep cycle110,167. Thus, this evidence suggests that dopamine containing cells and their receptors may not receive major trophic benefits from sleep or have an essential role in sleep generation.
Several brainstem cell groups do show a unique pattern of REM sleep related activity. Two of these cell groups are tonically active in REM sleep (‘REM-on’ cells), and are inactive in both non-REM sleep and waking. Two other cell groups are almost completely inactive in REM sleep and during the transition to REM sleep (‘REM-off’ cells), but are tonically active at all other times.
REM-on cells have been identified in the pons141,143,150 and in the medulla25,86,119,159. Their discharge has been shown to be highly selective for REM sleep, even in the freely moving animal. In contrast to medial reticular formation cells which are active in relation to movement in waking156,158, even vigorous waking movements are not associated with discharge in these REM-on cells. These cells show virtually continuous activity during REM sleep. In the pons of the cat, they are localized to a region between 1.8 and 5.0 mm lateral to the midline, lateral to the cells active in both waking and REM sleep (Fig. 1). In the medulla, REM-on cells are localized to the medial medullary reticular formation, intermixed with cells active in relation to waking movement and constituting less than 10% of the cell population in this region.
Fig. 1.
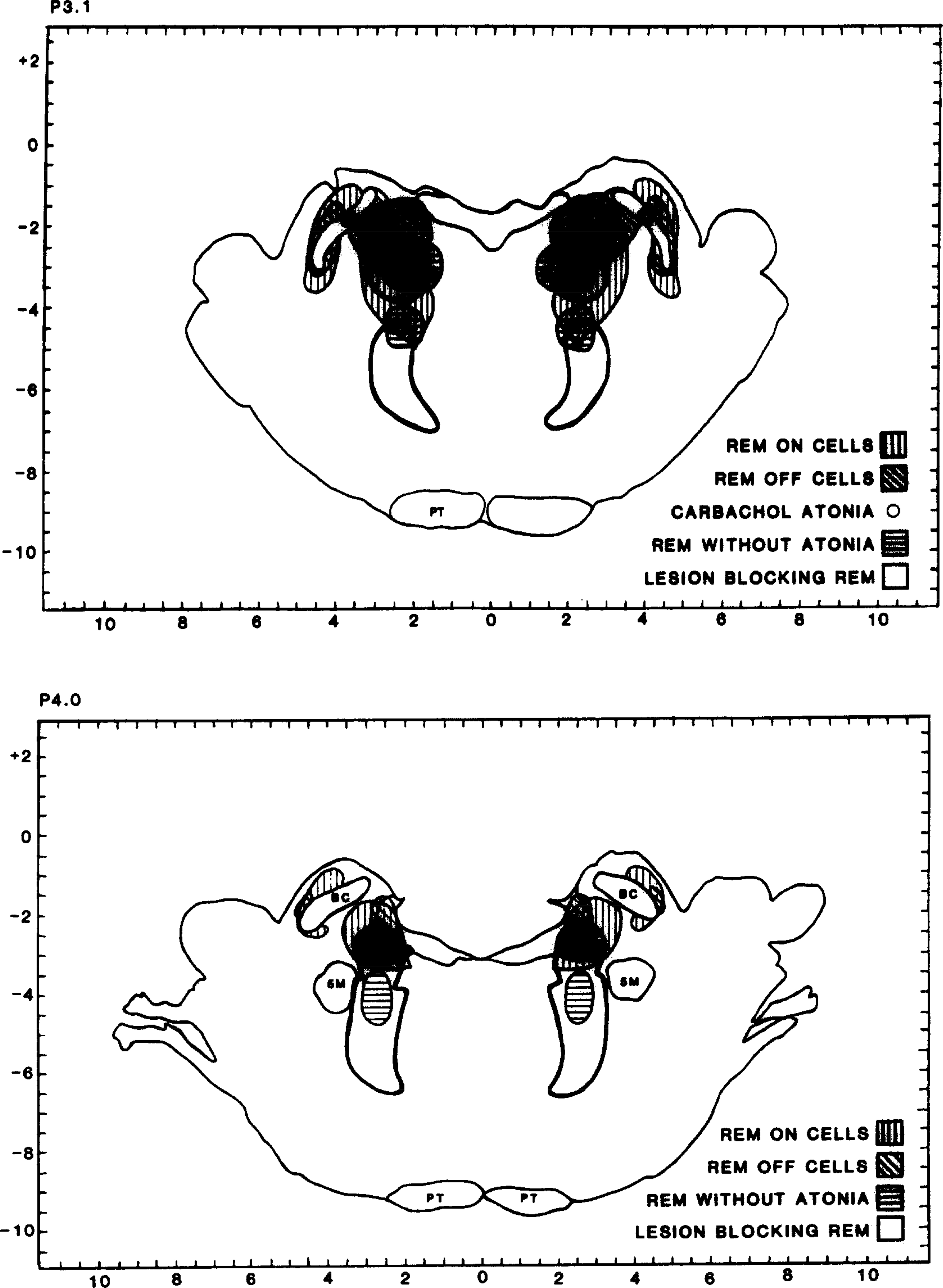
Location of cells selectively active in REM sleep (REM on cells), noradrenergic cells selectively inactive in REM sleep (REM off cells), bilaterally symmetrical lesions producing syndrome of REM sleep without atonia, and bilaterally symmetrical lesion suppressing REM sleep. See ref. 151 for separate maps of each cell type and lesion distribution.
Two major groups of REM-off cells have been investigated. One is in the dorsolateral pons in the locus coeruleus (LC) complex. The second is in the system of midline raphe nuclei. There is now over-whelming evidence that the pontine REM-off cell group is noradrenergic10,59,69,130 and the midline raphe REM-0ff cell group is serotonergic51,77,103. Recent work has also identified a population of REM sleep-off cells in the hypothalamus55,123. REM sleep-off cell groups are tonically active during virtually all waking behaviors and are almost completely inactive during REM sleep. A typical REM-off cell will never pause for more than a second or two in waking. These cells maintain a reduced level of activity in non-REM sleep, but are almost completely silent in REM sleep. Most cells will have periods of 30 s or more without a single discharge in each REM sleep period, and many are completely silent throughout REM sleep periods.
Cells which are selectively active or inactive in REM sleep may be hypothesized to be essential to the generation of REM sleep. Alternate hypotheses are that these cells mediate specialized functions occurring only during REM sleep or that REM sleep serves some ‘recuperative’ function for these cells or for the cells they project to. Of course, these hypotheses are not mutually exclusive. Is there any evidence that either the REM sleep-on or REM sleep-off cell groups are critical to REM sleep control?
3. BRAIN REGIONS CRITICAL FOR REM SLEEP
A series of lesion and transection studies has delimited the brainstem regions required for REM sleep. Structures rostral to the mid-pons are not required for REM sleep, since transection of the neuraxis at the mid-pontine level produces a preparation which has all the local signs of REM sleep caudal to the transection80. Transection at the pontomedullary junction produces a preparation which does not have REM sleep signs in the medulla157 but does show REM sleep signs in the forebrain155. These studies point to the caudal pons as the region critical for REM sleep. Lesion studies have shown that destruction of the pons between 2 and 5 mm lateral to the midline and 3 and 4 mm posterior to stereotaxic zero in the cat permanently suppresses REM sleep145. Lesions in more medial regions of the pons are without effect on REM sleep. The area whose destruction blocks REM sleep is virtually identical to the pontine area where REM-on cells are localized (Fig. 1). These cells project caudally to the second group of REM-on cells in the medial medulla142. This second cell group projects to the spinal cord and has been hypothesized to be involved in the motoneuron hyperpolarization that produces loss of muscle tone in REM sleep25,86,119,159. It would be most parsimonious to conclude that these cells have a critical role in the generation of REM sleep and particularly in controlling the motor phenomena of this state.
In contrast to REM-on cells, the REM-off cell populations do not appear to be essential for the REM sleep state. The midbrain decerebrate preparation, in which the transection is behind the dorsal raphe nucleus, has REM sleep in normal amounts caudal to the cut and no REM sleep signs in rostral structures80. Electrolytic lesion of the raphe nuclei does not block REM sleep160. Total serotonin (5-HT) depletion with p-chlorophenylalanine (PCPA)33,34,180 does not prevent REM sleep. While 5-HT is not necessary for the appearance of REM sleep, it does appear to have a vital role in the regulation of the ponto-geniculo-occipital (PGO) spike activity of REM sleep. PGO spikes, which are normally confined to the REM sleep state and the non-REM–REM transition state, are released into all states after 5-HT depletion or lesion of 5-HT containing neurons22,32,33,160.
The norepinephrine (NE) containing cells of the LC complex also are not required for REM sleep generation. Jones et al.79 have shown that electrolytic destruction of the LC with consequent depletion of NE does not prevent REM sleep. In fact, the critical lesion for blocking REM sleep is immediately ventral and caudal to the main noradrenergic REM sleep-off cell cluster (Fig. 1). Chemical destruction of NE cells by intraventricular injection of 6-hydroxydopamine does not prevent REM sleep63,78. Electrolytic lesions of the LC region release PGO waves into waking79. However, pharmacological depletion of NE does not release PGO waves and also does not block REM sleep58,170. Thus, neither the 5-HT nor the NE containing REM sleep-off neurons are essential for REM sleep, although they may contribute to the regulation of this state in the intact animal.
4. HYPOTHESIS
Since LC cells play no essential role in generating the REM sleep state, what is the significance of their complete cessation of activity in this state and their unique proximity to the cell groups required for REM sleep generation? While the experimental evidence does not support the concept that LC cells are required for REM sleep generation, the cessation of discharge occurring in LC cells in REM sleep may have significance for the functional role of REM sleep. We hypothesize that a central function of REM sleep is to reduce activity in the NE containing cells of the LC complex. This reduction in activity is required to maintain the sensitivity of NE receptors, with consequent benefits for all behaviors utilizing these receptors.
5. SIGNIFICANCE OF CESSATION OF LOCUS COERULEUS DISCHARGE IN REM SLEEP
Since NE does not appear to be essential for REM sleep, one may, of course, conclude that the cessation of LC discharge in REM sleep is an epiphenomenon with no functional significance. However, evidence from a number of lines of inquiry suggests that this cessation will have important consequences. Studies of the activity of LC cells in behaving animals have shown that they are almost continuously active. Very few cells in the adjacent areas of the medial pontine and medullary reticular formation have this property. Instead, most show extended periods of silence interspersed with periods of activity during waking 152,153,156. Pauses in LC cell discharge during waking lasting more than one or two seconds are rare 10,69. Thus, cells that receive projections from the LC will have a continuous supply of NE. This situation changes radically in REM sleep, since virtually all of the NE containing cells of the LC become silent. This silence begins during the transitional sleep prior to REM sleep onset and continues throughout REM sleep.
In addition to the property of relatively constant tonic firing throughout all behavioral states except REM sleep, the LC NE system is distinctive because its postsynaptic effects are typically slow to develop and prolonged in duration. For example, in recordings from single lateral geniculate neurons, several seconds of iontophoresis were required before cell responsiveness was altered by NE, and a similar delay occurred after electrical stimulation of the LC 136,139. Delays have also been reported for NE effects on cerebellar Purkinje cells71 and hippocampal pyramidal cells149. In contrast, γ-aminobutyric acid (GABA) and excitatory amino acids have near-immediate effects under the same conditions. In addition to the slow onset, the action of NE persists for seconds or minutes after the cessation of iontophoretic application or termination of LC stimulation113,114,136,137–139,149. In some cases, the effects of NE may be even more prolonged72,120,164,165. In contrast, the effects of GABA and excitatory amino acids are terminated within milliseconds.
The persistence of NE effects combined with the continuous activity of the LC cell population in waking and non-REM sleep, means that NE postsynaptic effects are continuously present throughout waking and non-REM sleep. However, during REM sleep, noradrenergic cells of the LC are silent or have greatly reduced discharge. Therefore during REM sleep and only during REM sleep, are postsynaptic cells free of noradrenergic influence. It can be said that NE neurons and receptors exist in two fundamentally different conditions. In waking and non-REM sleep, the NE cell population is tonically active and receptors are continuously exposed to NE, although the exact synaptic level of the transmitter is dependent upon the specific behaviors and stimuli occurring10. In REM sleep and during the transition to REM sleep there is a virtually complete cessation of activity in NE cells, and NE receptors are almost totally without exposure to NE.
The sensitivity of postsynaptic receptors for NE is regulated by the level of transmitter to which they are exposed. Reductions in NE levels result in ‘upregulation’ expressed as an increase in the number of receptors or in the sensitivity of the cyclic AMP response to NE, while increased levels of NE or its agonists cause the opposite changes. Indeed ‘the normal sensitivity of postsynaptic cells to [NE], corresponding to a ‘moderate’ noradrenergic action of release can be regarded as an intermediate state between hyper- and hyposensitive states’148. Major changes in receptor sensitivity can occur in relatively short time periods. Iontophoresis of NE on to medial reticular formation neurons for as little as one minute can cause a desensitization of response and a change in the pattern of discharge lasting over a minute73. Depletion of NE by reserpine treatment produces hypersensitivity of the cyclic AMP response to NE in 5 h148. Conversely. 5 h of treatment with dexamphetamine, a NE releasing agent, produces a reduction in NE sensitivity148. Withdrawal of the α2-aragonist clonidine, after chronic administration, produced a normalization of firing rate, presumably due to sensitization/upregulation of α2-receptors, within 70h39. As little as 3 h of cold restraint stress produced significant decreases in α2-receptors in a number of brain regions121. Changes in receptor sensitivity, as assessed by changes in the cyclic AMP response, occur well before the number of receptors changes89. Long duration drug treatments will cause marked changes in the number of postsynaptic receptors. Sporn et al.163 showed an upregulation of β-adrenergic receptors after 7–9 days of NE depletion with 6-hydroxy-dopamine. Menkes et al.108 demonstrated upregulation of α1-adrenoceptors in the thalamus 2–7 days after 6-hydroxy-dopamine treatment. These changes were accompanied by increases in receptor density, affinity and physiological responsiveness.
The dynamic nature of the control of NE receptors in the undrugged, undisturbed animal can be seen across the day. Wirz-Justice et al.195,196 have described a marked 24 h rhythm in binding of the α1-adrenoceptors. Nighttime levels are more than 10% higher than daytime levels. The most rapid increase occurs during the day portion of the light cycle, a time when rats are asleep. The circadian rhythm of the receptor population can be thought of as reflecting regulatory processes acting within the 24 h period, or may reflect processes with a longer time course which are synchronized to the 24 h cycle. The later possibility is suggested by the recent finding of Velly et al.184 that LC stimulation lasting as little as 4 h caused changes in α1- and α2-receptors. However, these changes were not detectable in most brain regions until 4 weeks after stimulation. Repeated transient perturbation of α1- and α2-receptors can, over a period of time, produce changes in their regulation174. Thus one should not rule out the possibility that REM sleep effects may be both cumulative and delayed.
The molecular mechanisms underlying the modulation of brain adrenoceptor sensitivity in response to NE availability have not yet been elucidated. However, in recent years, considerable progress has been made on this problem using peripheral adrenoceptor-bearing systems, such as avian erythrocytes and cultured smooth muscle cells. The presence of all of the required component enzymes and substrates in the CNS, suggests that similar regulatory mechanisms may account for changes in brain neuronal adrenoceptor sensitivity.
As a general rule, the regulation of adrenoceptor sensitivity occurs in a negative fashion. Thus, in the continued presence of agonist, specific molecular mechanisms cause the receptor-mediated physiological response to diminish. On the other hand, potentiation of activity can only result from restoration of the state prior to agonist exposure; no specific mechanisms provide for enhancement of receptor efficacy. Depending upon the duration of continued agonist activation, one of two distinct negative regulatory processes may occur. The first process (‘desensitization’) results within seconds to minutes and involves covalent modification of the receptor. Upon exposure to agonist for longer periods of time, the second form of modulation (‘downregulation’) comes into play which involves the actual loss of receptor protein.
Elegant studies by Lefkowitz and Caron92 have characterized the precise molecular events involved in adrenoceptor desensitization. In the case of the β-adrenoceptor, the most important desensitization mechanism appears to involve receptor phosphorylation catalyzed by a novel (and ubiquitous) protein kinase, referred to as the β-adrenergic receptor kinase (β-ARK). β-Adrenergic receptors are coupled to adenylate cyclase via the stimulatory GTP binding protein Ns. Upon exposure to agonist, β-ARK is translocated from the cytoplasm to the plasma membrane where it phosphorylates the agonist-occupied β-adrenoceptor. Phosphorylation leads to uncoupling of the β-adrenoceptor from Ns and the catalytic moiety of adenylate cyclase, resulting in a temporary loss of activity. Re-sensitization occurs when the receptor is dephosphorylated by a cytoplasmic phosphatase enzyme so that it can again bind Ns and adenylate cyclase. In addition to β-ARK, cAMP-dependent protein kinase (protein kinase A) can also phosphorylate the β-adrenergic receptor, thus providing an alternate pathway for acute desensitization.
Recent experiments have indicated that α1-aradrenoceptors also become acutely desensitized as a result of phosphorylation, However, this appears to occur via protein kinase C. Activation of α1-aradrenergic receptors results in phosphatidylinositol hydrolysis. Diacylglycerol produced by this reaction activates protein kinase C which phosphorylates the receptor.
Upon exposure to agonist for longer periods of time (~24 h), receptor ‘downregulation’ comes into play. Receptors become degraded and must be resynthesized for recovery of normal sensitivity to occur. This process probably requires the synthesis of new receptor protein, but the precise molecular details are not yet clearly understood.
In the context of sleep cycle related changes in adrenoceptor sensitivity, one can envisage a dynamic state where during waking and slow wave sleep varying degrees of desensitization and downregulation of central adrenoceptors are always present. Only during REM, when adrenoceptors are completely free of agonist for sufficiently long periods of time, can maximal receptor sensitivity be re-established.
We hypothesize that the silence of NE cells in REM sleep interrupts the desensitization/downregulation of NE receptors set in motion by prior waking periods. This hypothesis is a modification of Hartmann’s60,61 proposal, that a central function of REM sleep was the synthesis of NE stores depleted by prior waking behavior. This idea was based on pharmacological data. In fact, Hartmann was prescient in leaving open the possibility that changes in receptor sensitivity or in other aspects of catecholamine functioning could be behind the relationships he observed; ‘receptor mechanisms or other aspects of the physical structure of the synapse might be changed in order to render it again more sensitive to catecholamines....’61. Stern and Morgane171 proposed a similar role for REM sleep. Subsequent work has shown that the activity of tyrosine hydroxylase, the rate limiting enzyme in the NE biosynthetic pathway, is tightly regulated, so that NE synthesis increases with the enhanced firing of these neurons and decreases with reduced activity97. Thus, intracellular NE levels are held within a narrow range even with wide variations in the activity of NE neurons, and the maintenance of adequate NE levels is unlikely to require a long period of LC inactivity. However, much of the same pharmacological evidence that supported Hartmann’s, and Stern and Morgane’s hypotheses would apply to the present receptor regulation model.
The synaptic mechanisms producing the cessation of LC discharge during REM sleep are unknown. Active inhibition would appear to be involved, inasmuch as LC neurons fire spontaneously even in isolated brain slices where extrinsic excitatory inputs are absent8,193. This firing is apparently due to endogenous pacemaker mechanisms dependent upon the complex interplay between several membrane ionic currents including a novel low threshold inward current4 and a calcium-dependent potassium current. The receptors mediating this inhibition would be expected to show changes opposite to those seen in postsynaptic α1- and β-receptors during REM sleep.
6. MANIPULATIONS OF LOCUS COERULEUS AND NOREPINEPHRINE.
We hypothesize that physiological NE release or NE release induced by pharmacological agents sets in motion two processes having opposite effects on REM sleep duration. In the first process, NE release or its functional enhancement by pharmacological agents that prevent its reuptake or degradation, or the direct activation of NE receptors by agonists, is hypothesized to suppress or ‘substitute’ for REM sleep by increasing the activity of negative feedback circuits monitoring the efficiency of NE receptor action. In the second process, the release or potentiation of NE action is hypothesized to downregulate/desensitive NE receptors, this downregulation producing increased REM sleep ‘pressure’. Thus the actual level of REM sleep would be a function of the balance between these processes. As is discussed in Section 5., while the evidence that increased levels of NE lead to NE receptor downregulation is over-whelming, depending upon the manipulation involved, the time course is variable. Some studies report detectable effects after only a few minutes, while others find significant effects only after two weeks or more. The time course is likely to be a function of species, drug dosage and half-life, receptor type, and brain region. Therefore, while we can predict that longer term exposure to NE agonists and potentiators will produce greater downregulation and resulting REM sleep pressure and that short term administration will be more likely to reduce REM sleep with limited rebound, the precise balance between these processes cannot be predicted based on current knowledge.
Amphetamine promotes central noradrenergic neurotransmission primarily by releasing NE from nerve terminals, but also by blocking NE reuptake and by stimulating postsynaptic NE receptors directly. In REM sleep deprived rats, a single dose of D-amphetamine (3 mg/kg, i.p.) completely abolished the REM sleep rebound occurring in control animals during the 12 h period following REM sleep deprivation129. In addition the total REM sleep time during the 12 h period following REM sleep deprivation was markedly reduced (and this loss of REM sleep was not regained during a subsequent 48 h period), as if the enhanced stimulation of CNS noradrenergic receptors produced by amphetamine was able to ‘substitute’ for REM sleep. Decreased REM sleep, with some rebound, has been seen after d-amphetamine administration in humans53,131. Reduced REM sleep rebound after amphetamine suppression does not seem to be a non-specific effect of its stimulant properties inasmuch as caffeine, a stimulant that does not act primarily via central noradrenergic systems, does not produce this effect. REM sleep deprived rats treated with caffeine showed a suppression of REM sleep which was later followed by REM sleep rebound127. The total REM sleep time was unchanged and therefore caffeine, unlike amphetamine, does not appear to ‘substitute’ for REM sleep.
A similar pattern of effects is seen with certain tricyclic antidepressants, monoamine oxidase inhibitors, and electroconvulsive shock (ECT), which decrease REM sleep duration with little or no rebound171. (A recent study has provided direct evidence that ECT can induce a change in the discharge pattern of LC neurons to a mode favoring NE release198.) However, long-term maintenance of these treatments produces adrenoreceptor downregulation, with receptor levels maintained at a stable reduced level as long as treatment continues133,148. Withdrawal is accompanied by a compensatory upregulation to normal levels. During this withdrawal/upregulation period, REM sleep is elevated, as the hypothesis would predict171. In an analogous manner, withdrawal of amphetamine after chronic administration produces a marked REM sleep rebound in rats36 and humans44,181.
While many drugs depress REM sleep, some undoubtedly by non-specific disruption of sleep and others by affecting noradrenergic systems, very few treatments have been reported to increase REM sleep. However, if autonomic side effects are minimized by the use of low parenteral doses or by intraventricular administration, α-methylparatyrosine (α-MPT) and reserpine both produce elevations of REM sleep171. α-MPT inhibits catecholamine synthesis and reserpine depletes biogenic amines by interfering with vesicular storage. Thus, the increase in REM sleep can be seen as an attempt to upregulate NE receptors in response to this depletion. The proximity of the REM sleep-off cells to REM sleep generation mechanisms would facilitate feedback regulation of REM sleep duration in response to the downregulation/desensitization of NE receptors by pharmacological agents. The LC complex has major reciprocal connections with adjacent pontine regions where the REM sleep-on cells are located24,100,144, although a recent study indicates a relatively restricted distribution of direct afferents11. Destruction of the dorsal noradrenergic bundle is one of the few manipulations known to increase REM sleep21,124,166. This can be seen as a consequence of the interruption of a long loop feedback pathway suppressing REM sleep. Similarly, 6-hydroxydopamine and DSP4 lesions of the LC also produce increased REM sleep63,115 if non-specific effects are minimized62,171. Selective blockade of α1-receptors with prazosin produces a prompt and enduring increase in REM sleep66,67, suggesting that α1-adrenoceptors on postsynaptic neurons may, at least in part, mediate the feedback signal. Conversely, the α1-agonist methoxamine decreases REM sleep66. The non-specific α-adrenoceptor blockers phentolamine and phenoxybenzamine (which block both α1- and α2-adrenoceptors) eliminate REM sleep rebound as do treatments that enhance central noradrenergic transmission (i.e. they appear to ‘substitute’ for REM sleep)98,128. These drugs increase whole brain 3-methoxy4-hydroxyphenylethyleneglycol sulfate128, indicating that NE turnover and release is enhanced. At relatively high doses the α2-adrenoceptor stimulating drug clonidine decreases REM sleep65,125, possibly as a result of its agonist activity at α1- or postsynaptic α2-adrenoceptors94. β-Adrenoceptor blocking drugs have variable effects on REM sleep65. Although some drugs such as prenalterol increase REM sleep, other β-blockers decrease REM sleep84,85, and others have no effect. These inconsistencies could be due to non-specific effects of these drugs, some of which are highly membrane active.
Many of the behavioral changes produced by manipulation of the NE system are similar to those induced by REM sleep deprivation. REM sleep deprivation and depletion of NE with reserpine both increase sex drive and aggressive behaviors46,117. Destruction of NE neurons with 6-hydroxydopamine or depletion of catecholamines with α-MPT impair performance on avoidance tasks, decrease the stimulant effects of amphetamine and increase aggressive behaviors. The same effects are produced by REM sleep deprivation26,118,171,176. The similarity of the effects of REM sleep deprivation with these pharmacological effects can be seen as reflecting a common neurochemical cause, a hypofunctional NE system.
7. REM SLEEP AND RAPHE CELLS
Serotonin containing cells of the raphe nucleus are the other major group of neurons that have a selective cessation of discharge in REM sleep. Therefore, one might speculate that REM sleep would have a receptor regulating function for this system as well. However, behavioral evidence indicates that REM sleep is less closely tied to the functioning of 5-HT receptors than it is to NE receptors. Drugs which decrease the synaptic availability of serotonin, such as LSD, PCPA, and methysergide, do not produce a compensatory increase in REM sleep time as does the catecholamine synthesis inhibitor α-MPT171. Deficits in avoidance performance resulting from REM sleep deprivation can be reversed by drugs which enhance the synaptic availability of NE, such as l-DOPA, and amphetamine64,171. The combination of pargyline and PCPA, which elevates levels of NE while simultaneously decreasing 5-HT levels also effectively reverses performance decrements produced by REM sleep deprivation171. Therefore, the available evidence does not support a critical receptor regulating role for REM sleep with regard to control of 5-HT systems. Cessation of 5-HT neuron discharge during REM sleep might serve other functions. For example, the pauses might be more closely related to the raphe role of PGO spike inhibition. Nevertheless, direct measurements of sensitivity, using receptor binding techniques and/or biochemical and physiological measures of receptor activity will be required before a 5-HT receptor regulating role can be completely ruled out.
8. OTHER TRANSMITTER SYSTEMS
By emphasizing the role of REM sleep in the regulation of NE receptor function, we certainly do not mean to exclude a regulatory role for REM sleep in the maintenance of other receptors. It is unlikely that the NE receptor system could change without effects on other systems. Dopamine responsiveness has been reported to be affected by REM sleep deprivation23,42,179. Cholinergic mechanisms are intimately involved with the triggering and maintenance of REM sleep151 and in its pathology13. Changes in receptor sensitivity could occur in these systems during REM sleep. NE systems may either be involved in the mediation of these effects or they may represent independent consequences of REM sleep, occurring in parallel with NE receptor regulation. Further studies will be necessary to discriminate between these possibilities.
9. NARCOLEPSY
Narcolepsy is a disease characterized by excessive sleepiness and by the intrusion of various aspects of REM sleep into waking states19,57. Thus many narcoleptics experience cataplexy, an abrupt loss of muscle tone in waking similar to that normally seen only in REM sleep. Other symptoms include sleep paralysis, an inability to move which can occur upon awakening or falling asleep, and hypnagogic hallucinations, which are vivid, dream-like auditory or visual images appearing at sleep onset. Narcoleptics also have REM sleep at sleep onset188, while normals have non-REM sleep at sleep onset. Many of these signs of narcolepsy can be seen as indications of increased REM sleep ‘pressure’. Narcoleptics have marked abnormalities in their catecholamine and acetylcholine systems. Catecholamine turnover appears to be impaired as is indicated by low levels of catecholamine metabolites in the cerebrospinal fluid, and higher concentrations of dopamine and NE within the brain13,43,105. α1-Receptor binding in the amygdala is increased in narcoleptic animals, consistent with an impaired release of NE. Furthermore, blockade of α1-receptors with prazosin dramatically exacerbates cataplexy109. In fact, prazosin and the anticholinesterase physostigmine are the only drugs that consistently increase cataplexy. In the context of the present hypothesis, these findings can be interpreted as indicating an impairment in the NE mediated feedback signal regulating REM sleep. Impaired NE functioning leads to increased REM sleep pressure, just as NE receptor downregulation in the normal animal leads to increased REM sleep pressure.
10. SLEEP DEPRIVATION STUDIES
If REM sleep discharge cessation serves to upregulate or increase the sensitivity of the NE receptor system, then REM sleep deprivation might be predicted to cause a downregulation/desensitization of these receptors. However, REM sleep deprivation may not be a fully satisfactory way of testing the implications of the present hypothesis. If the cessation of NE neuron discharge is an important function of REM sleep, it is important to bear in mind that REM sleep-off cells cease discharging prior to the onset of REM sleep as defined by the usual polygraphic criteria69,103. REM sleep-off cells slow in non-REM sleep and actually turn off during the transitional or deep slow wave sleep state, and then continue off during REM sleep. The transitional state may last 30 s to a minute or more. Most REM sleep deprivation techniques involve arousing the animal shortly after REM sleep has begun. Thus, the initial moments of REM sleep combined with the preceding transitional sleep would produce a substantial period of silence in REM sleep-off cells. Since it has long been known that REM sleep deprivation is accompanied by a progressive increase in the number of REM sleep onsets, it is likely that REM sleep-off cells would show a relatively small change in ‘off time’ during REM sleep deprivation. Furthermore it is possible that average discharge rate in early non-REM sleep or even in waking states might change during deprivation. These effects would tend to minimize the physiological effects of REM sleep deprivation. Because of this, it is likely that the behavioral and physiological differences between REM sleep deprived and stress control animals might actually decrease during extended periods of REM sleep deprivation. A more direct test of the present hypothesis would be to deprive animals of the ‘REM sleep-off state’, as defined by LC unit recording.
One slow wave sign that is correlated with the ‘REM sleep-off state is the PGO spike, which is the defining characteristic of transitional sleep and which continues to occur in REM sleep. Dement33, showed that PGO spike deprivation, presumably correlated with ‘REM sleep-off deprivation’ was much more difficult to maintain and produced far more severe behavioral and REM sleep rebound effects than standard REM sleep deprivation.
However, even with PGO deprivation it would be essential to know if REM sleep-off cells reduced their discharge rate during non-REM sleep states to achieve the same upregulation/reversal of downregulation effect which would normally occur in REM sleep. Therefore, the activity of REM sleep-off cells should be recorded during REM sleep deprivation, and during total sleep deprivation to determine if lost REM sleep is compensated for by reduction of activity in NE cells during non-REM sleep states. If such compensation does not occur in any given experimental condition, then PGO deprivation is a valid test of our hypothesis. If a compensatory reduction of LC activity or NE release does occur during sleep deprivation conditions, it may be reasonable to view the REM sleep state as representing an efficient means for achieving a receptor sensitization which can occur in other states when sleep processes are disrupted. Thus, the ‘normal’ function of REM sleep would not be blocked by REM sleep deprivation, only shifted into other behavioral states.
Despite the possibility that slowing of LC discharge in transitional or non-REM sleep states reduces the effect of REM sleep deprivation, the REM sleep deprivation literature is generally consistent with the present hypothesis. Mogilnicka et al.111, in a study aimed at explaining the antidepressant effects of REM sleep deprivation189, reported that 72 h of REM sleep deprivation caused a significant decrease in cortical β-adrenergic receptor binding. This is precisely the result that would be predicted if the hypothesized upregulating function of REM sleep were lost. Radulovacki and Micovic126 found a non-significant decrease in β-adrenergic receptor density after a more extended 7 day REM sleep deprivation procedure. They speculate that the effects of REM sleep loss may be masked by the stress produced in both experimental and control groups during extended periods of deprivation. But, Abel et al.1, using a 72 h REM sleep deprivation procedure, found no significant change in β-receptor binding, They restricted their analysis to frontal cortex. Abel et al., conclude that Mogilnicka et al.’s111 results may have been caused by stress and non-REM sleep loss. A more recent study by Mogilnicka et al.112, utilized two different REM sleep deprivation techniques and three non-deprivation conditions to control for stress and non-REM sleep disruption. This study reaffirmed their previous findings of significant decreases in β-adrenoceptor sites in neocortex after REM sleep deprivation. Troncone et al.178 used NE stimulated cyclic AMP accumulation as an index of NE receptor status. They found a dramatic decrease in cyclic AMP accumulation (to 51–59% of control values), in slices from rat cortex after 96 h of REM sleep deprivation, indicating a downregulation of NE receptors. Thus, while further work is needed, the preponderance of recent evidence is consistent with the hypothesis that REM sleep deprivation causes NE receptor downregulation.
The reported downregulation/desensitization of β-receptors or decrease in sensitivity of the NE activated cyclic AMP system, would be consistent with the antidepressant effect of REM sleep deprivation191, since treatment with antidepressant drugs or with electroshock, which has antidepressant effects, are associated with β-downregulation122,133. Both REM sleep deprivation and treatment with antidepressant drugs are linked to an increase of NE turnover, which can be seen as a compensation for this downregulation18,126,147,172,197. Although one can view antidepressant drug treatments as acting directly on the presynaptic terminals of NE neurons or on β-receptors themselves, to produce downregulation, it is also possible that some portion of their effect is mediated by decreasing the duration of the ‘REM sleep-off’ periods, which in turn causes downregulation of β-receptors. Both antidepressant drug therapy and electroshock have been shown to produce a marked decrease in REM sleep duration28,189. Indeed the effectiveness of antidepressant drugs is directly proportional to their REM sleep depressant effects189,190. These treatments presumably act by blocking REM sleep rather than by fulfilling REM sleep ‘need’. This can be seen in the increased ‘REM sleep pressure’, manifested by a large rebound in REM sleep time, after both REM sleep deprivation and withdrawal of antidepressant medication. Only after antidepressant drugs have been administered for several weeks is an antidepressant and β-downregulation effect seen133. Throughout this period REM sleep is depressed. Similarly direct REM sleep deprivation requires 3 weeks to relieve depression189,191.
Many of the behavioral effects of amphetamine are thought to be mediated by a release of NE. If REM sleep deprivation produced a downregulation or desensitization of the NE receptor system, it would be predicted that, to the extent that behavioral responses to amphetamine are mediated by NE receptors, these responses would be reduced in REM sleep deprived animals. Indeed this is just what was seen in a behavioral avoidance task. Amphetamine greatly increased responding in stress control animals but not in REM sleep deprived animals. REM sleep deprivation also decreased the effect of amphetamine on general activity levels171. There is even some evidence that REM sleep deprivation may protect against the lethal effects of high doses of amphetamine45, a very surprising result, given that REM sleep deprivation itself can be lethal41.
REM sleep deprivation has been reported to impair performance in a number of learning tasks. If NE downregulation/desensitization contributed to these effects, it should be possible to restore function by potentiating the synaptic action of NE. Stern and Morgane171 reported just such an effect. Imipramine, which blocks NE reuptake, and pargyline, which blocks its degradation, were effective in reversing the deficit in performance caused in both active and passive avoidance tasks by REM sleep deprivation, but were without effect on the performance of control groups. Serotonin synthesis inhibition with PCPA did not interfere with this effect. l-DOPA, a catecholamine precursor, also improved performance in REM sleep deprived subjects, but actually impaired it in controls64.
Stress is known to produce increased activity in noradrenergic cells of the LC2,3,7,9,54,75. Indeed stress and alerting are the best correlates of increased LC unit discharge in waking2,3,75. The present hypothesis would predict that stress and REM sleep deprivation would be synergistic in their effects, since stress increases the activity of NE LC cells in waking, while REM sleep deprivation would block the ‘locus coeruleus-off’ state associated with REM sleep. REM sleep deprivation itself is normally confounded by some degree of waking stress38,90,107,182,189. We hypothesize that increased REM sleep, and/or reductions in LC activity during waking, occur during recovery from a sustained period of stress. The magnitude of REM sleep rebound or reduction in waking LC activity should be proportional to the extent of increased NE release during the stress and the degree of REM sleep deprivation resulting from the stress. Studies combining LC unit and sleep state recording can address this question.
A critical study directly examining changes in regional brain responsiveness after REM sleep deprivation was performed by Satinoff et al.146. In this study, cats were deprived of REM sleep while evoked potentials in the trigeminal system were monitored. It was found that trigeminal evoked potentials diminished by 33% after 24 h of REM sleep deprivation and were abolished by more extended deprivation. All potentials returned to baseline values after recovery sleep. Morilak and Jacobs116 have shown, in the intact rat, that NE released from LC neurons facilitates the trigeminal masseteric reflex. Removal of the NE input decreased the masseter muscle response to sensory nerve stimulation. Thus, the hypothesized downregulation/desensitization of NE receptors caused by REM sleep deprivation can explain the reduction in evoked potentials seen in the Satinoff et al.146 studies.
11. PREDICTIONS
A more direct test of the present hypothesis could be achieved by determining the sensitivity of adrenoceptors to iontophoresed NE before and after sustained periods of waking, and also before the offset of activity in NE cells and after onset following a single REM sleep period. These comparisons are preferable to deprivation studies, since they are not confounded by stress or by the brain’s attempts to compensate for deprivation. We would predict a decrease in response of β- and/or α1-NE receptors after sustained waking periods and an increase in response after REM sleep periods. Since it is now clear that there are regional differences in the sensitivity of NE receptors and of different receptor types to reductions in NE availability133 such measurements should be repeated in several brain areas, looking separately at the activity of pre- and postsynaptic receptor sites. Techniques for iontophoresis during waking and normal sleep cycles now are in regular use26 and could easily be applied to LC cells and their target cell populations. If α2-autoreceptors showed decreased responsiveness during sustained waking periods it might have the effect of compensating for any decline in sensitivity of postsynaptic receptors by decreasing feedback inhibition of NE release, while increased α2-responsiveness would slow α1- and β-downregulation. There is some evidence from in vitro studies for the direct reciprocal control of β- and α2-receptors93.
It is clear that the absolute magnitude of the effects produced by a single REM sleep period must be extremely small. We are not aware of any evidence that the loss of a single REM sleep period has ever been reported to produce a clear behavioral deficit. Evolution would long ago have eliminated any mechanism so sensitive to transient sleep loss. We hypothesize that the effects of REM sleep loss, and the correlated loss of LC-off periods, are small, but are additive over periods of time. Therefore, even if the deprivation of a single REM sleep period did not produce an easily measurable change in receptor sensitivity, the cumulative effect of the loss of ‘locus coeruleus-off’ activity should be detectable.
It is possible that the biochemical events set in motion by the offset of activity in REM sleep do not produce any immediate effect on receptor sensitivity, but instead produce effects only after some delay. If so, only extended deprivation of the ‘locus coeruleus-off’ state would be expected to produce reliable changes in receptor sensitivity. (As discussed above, while this LC-off state is more or less identical with REM sleep in the undisturbed animal, LC-off periods might well become dissociated from REM sleep under deprivation conditions.) The duration of REM sleep, the time course of behavioral deficits reported after REM sleep deprivation, and the time course of receptor changes need to be considered in evaluating the probable time course of relations between REM sleep and the hypothesized changes in receptor sensitivity. REM sleep has a duration which ranges between 2 and 30 min199. Most studies reporting significant behavioral deficits after REM sleep deprivation have utilized deprivation periods of approximately 2 weeks. Changes in receptor number have been reported in various studies to occur in as little as 2–3 min, or to require as long as 2 weeks. Changes in receptor sensitivity need to be assessed in the same situations in which behavioral and electrophysiological measures are made in order to clarify the relationships between these variables and test the present hypothesis.
12. PHYLOGENETIC EVIDENCE
Only one mammal has been found to lack REM sleep. This is the monotreme echidna. It appears to have a LC complex grossly similar to that seen in other mammals6. The present hypothesis suggests the question ‘does the echidna have an ‘off state’ for its LC neurons?’ We hypothesize that either NE LC neurons are not tonically active in waking in the echidna as they are in other mammalian species, making periods of upregulation/sensitization unnecessary, or that the LC neurons of the echidna are silent in non-REM sleep states.
It is in birds that the anlage of the mammalian LC first appears, albeit in rudimentary form177. In birds, REM sleep is clearly present, but occupies a much smaller percentage of sleep time than it does in mammals17,173,183,192. Teleosts and reptiles have not been found to have REM sleep states48,49,68,106. In the context of the present hypothesis it is interesting to note that, in contrast to the situation in mammals and birds, fish, reptiles and amphibia do not have widely ramifying catecholaminergic neurons in the rhombencephalon and medulla oblongata. In these species the forebrain is supplied with catecholamines by hypothalamic and upper brainstem neurons. The total number of tegmental catecholaminergic neurons in fish and amphibia is less than 10% that of birds and mammals177. Accordingly, mechanisms generating silent periods in the NE neurons in these animals would be expected to have different physiological and behavioral correlates than those generating similar periods in NE neurons localized to the pons of mammals.
We hypothesize that the evolution of REM sleep was linked to the need for continuous periods of high alertness. Such alertness became useful with the evolution of homeothermy, which enabled extended periods of muscle activity. NE produced by pontine LC neurons enabled long duration periods of high alertness. In turn, the responsiveness of receptors to sustained periods of LC activity in waking, was maintained by sustained silent periods coupled to REM sleep.
13. FUNCTIONAL CONSEQUENCES OF REM SLEEP INDUCED MODULATION OF ADRENOCEPTOR SENSITIVITY
A number of hypotheses of REM sleep function have been advanced. Among these are drive discharge33, binocular coordination16, periodic arousal from sleep162, memory consolidation47,91,161,169, protein synthesis102, forgetting excess memory traces31, programming of genetically determined behaviors82, reversal of a depression of excitability occurring in non-REM sleep186, information processing194, and brain development134. Since NE interacts with neurons throughout the brain, the consequences of any REM sleep generated effect on the functioning of NE systems would be seen in all of the above. Thus, the present hypothesis is not incompatible with any of these prior ideas, but suggests a more specific neural mechanism for REM sleep induced changes in behavior. For example, the high levels of REM sleep early in ontogeny, which have been cited as evidence for a REM sleep role in brain development134, can be seen more specifically as a necessary condition for the proliferation of NE receptors in the developing animal.
A considerable amount of evidence now supports the hypothesis that a principal function of NE in the CNS is to facilitate the excitability of target neurons to specific high priority signals12. This has been demonstrated in sensory relay nuclei such as the lateral geniculate nucleus and cochlear nucleus; in motor nuclei such as the facial or trigeminal motor nucleus; and in the neocortex, cerebellum and hippocampus135. Facilitation of target cell excitability can occur by several mechanisms, depending on the receptor type mediating the response. Activation of α1-adrenoceptors are typically associated with slow excitation during which responses to simultaneous excitatory inputs to the target cell are enhanced. The response to β-adrenoceptor activation is more complex, consisting of a decrease in spontaneous discharge in the face of a relative or absolute increase in the response to specific inputs. This has been described as an enhancement of the ‘signal-to-noise’ ratio for the neuron and the net effect of these changes would be to facilitate the throughput of specific signals and improve signal processing50,135. The corollary of this is that a reduction in NE availability decreases the accuracy and efficiency of signal processing in the brain, eventually leading to an upregulation/sensitization of NE receptors. Reductions in LC activity during waking (to reverse receptor downregulation caused by prior activity) would leave the organism vulnerable, by diminishing the ability to perceive and rapidly respond to threats. This problem is ameliorated by linking the LC-off state to sleep, a time when the animal seeks out a protected niche at an optimal time of day. Thus. we hypothesize that the evolution of a linkage between the ‘locus coeruleus-off’ state and sleep produces a selective advantage, particularly for animals that are not vulnerable to predation during sleep. Indeed, comparative studies have found that predators and other animals with safe sleeping sites have higher levels of REM sleep, while prey animals which must frequently and accurately monitor the environment during sleep have much lower levels of total REM sleep, and the REM sleep periods which do occur are brief5. We hypothesize that LC-off periods may be distributed throughout the day in short epochs in these animals, to compensate for their inability to have extended REM sleep periods.
While enhanced LC activity biases brain systems to cope with immediate environmental threats, depressed LC activity may allow internal programs and vegetative behaviors to predominate12. Indeed it has long been known that REM sleep, the state in which LC activity is minimal, is associated with a suppression of sympathetic tone132. This can be seen in the reduction of renal nerve and cervical sympathetic trunk activity14,52,132 and in the extreme miosis and relapse of the nictitating membrane which are classic signs of REM sleep132,187. Thus the autonomic nervous system and the LC, which has been hypothesized to represent a ‘central analogue of the sympathetic ganglia with the brain as its ‘end organ’ ‘7, operate in a largely parasympathetic or ‘internal’ mode during REM sleep. The nature and function of the central ‘internal programs’ that might be triggered during this state remain unknown; however, it has been speculated that REM sleep is a time when neural circuits mediating fixed motor patterns involved in innate aspects of behavior are strengthened and integrated with prior behavior81.
A cellular model of learning in the hippocampus, long term potentiation (LTP), has provided a specific example of how changes in the sensitivity of noradrenergic systems may affect performance and memory acquisition. LTP is a persistent increase in stimulation induced synchronous cell firing (field response) produced by a brief period of high frequency stimulation of the afferent pathway. LTP induced in dentate granule cells by perforant path stimulation is reduced by NE depletion20. Conversely NE can induce a persistent increase in the dentate gyrus population response that is similar to LTP120,165. NE appears to act via a β-adrenoreceptor linked to adenylate cyclase. β-Adrenoceptor modulation of LTP has also been demonstrated in the CA3 region of the hippocampus72. These studies demonstrate that NE pocampus plays a critical role in a form of long term plasticity that is possibly related to learning. Thus, during REM sleep, when LC neurons are silent, this form of hippocampal plasticity is restricted, perhaps implying a mechanism for the selective ‘forgetting’ of dreams. Downregulation of NE receptors by REM sleep deprivation would likewise modulate the effectivenss of LTP and related learning phenomena.
Diminution of NE inhibition in the limbic system may help explain the reduction in seizure thresholds and prolongation of seizures caused by REM sleep deprivation28. Treatments which decrease CNS noradrenergic activity potentiate seizures, while treatments that increase noradrenergic activity decrease seizures27,29,30,37,88,99. If REM sleep deprivation leads to decreased NE receptor sensitivity, this would have the same net functional effect as a decrease in NE levels. Therefore the effects of REM sleep deprivation on seizure threshold could be mediated, at least in part, via changes in NE receptor sensitivity.
The hypothesis that REM sleep functions to maintain NE receptor sensitivity explains recent findings of decreased receptor sensitivity after REM sleep deprivation. It makes further predictions that can be readily tested with presently available techniques. Furthermore, it provides a mechanism that allows explanation and integration of a number of experimental observations. Assessment of receptor sensitivity as a function of state, and changes in receptor sensitivity as a function of the duration of the intervening state would provide useful data on the function, as well as on the mechanisms generating REM sleep. The phylogenetic study of LC activity has just begun and will provide important information on the possible role of this system in the evolution of REM sleep.
14. SUMMARY
We hypothesize that REM sleep serves to upregulate and/or prevent downregulation of brain norepinephrine (NE) receptors. This hypothesis is based on the following observations: (1) NE neurons of the locus coeruleus (LC) are tonically active in waking and non-REM sleep, but the entire population of LC NE neurons is inactive during REM sleep. (2) Continuous presence of NE or adrenoceptor agonists downregulates NE receptors, while a reduction in NE availability upregulates these receptors. (3) The effects of REM sleep deprivation are similar to those of NE receptor downregulation. Recent biochemical studies of NE receptor sensitivity provide strong experimental support for this hypothesis. The functional consequence of enhanced NE receptor ‘tone’ brought about by REM sleep would be improved signal processing in diverse brain systems, thus endowing the organism with a selective advantage. This hypothesis makes a number of specific predictions which can be tested with currently available techniques, and suggests new ways of understanding the evolution and postnatal development of REM sleep.
ACKNOWLEDGEMENTS
Supported by the Medical Research Service of the Veterans Administration, USPHS Grant NS14610 and the National Institutes of Health. We thank Drs. E. Mignot, E. Hartman and M. Radulovacki for very helpful comments on an earlier version of this manuscript.
Footnotes
Presented in preliminary form at the Association of Professional Sleep Societies, Columbus, OH, June 17, 1986.
REFERENCES
- 1.Abel MS, Villegas F, Abreu J, Gimino F, Steiner S, Beer B and Meyerson LR, The effect of rapid eye movement sleep deprivation on cortical beta-adrenergic receptors, Brain Res. Bull, 11 (1983) 729–734. [DOI] [PubMed] [Google Scholar]
- 2.Abercrombie ED and Jacobs BL, Single-unit response of noradrenergic neurons in the locus coeruleus of freely moving cats. I. Acutely presented stressful and non-stressful stimuli, J. Neurosci, 7 (1987) 2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abercrombie ED and Jacobs BL, Single-unit response of noradrenergic neurons in the locus coeruleus of freely moving cats. II. Adaptation to chronically presented stressful stimuli, J. Neurosci, 7 (1987) 2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aghajanian GK and Wang YY, Common alpha 2-and opiate effector mechanisms in the locus coeruleus: intracellular studies in brain slices, Neuropharmacology, 26 (1987) 793–799. [DOI] [PubMed] [Google Scholar]
- 5.Allison T and Cicchetti DV, Sleep in mammals: ecological and constitutional correlates, Science, 194 (1976) 732–734. [DOI] [PubMed] [Google Scholar]
- 6.Allison T, Van Twyver H and Goff WR, Electrophysiological studies of the echidna, Tachyglossus aculeatus. I. Waking and sleep, Arch. Ital. Biol, 110 (1972) 145–184. [PubMed] [Google Scholar]
- 7.Amaral DG and Sinnamon HM, The locus coeruleus: neurobiology of a central noradrenergic nucleus, Progr. Neurobiol, 9 (1977) 147–196. [DOI] [PubMed] [Google Scholar]
- 8.Andrade R and Aghajanian GK, Locus coeruleus activity in vitro: intrinsic regulation by a calcium-dependent potassium conductance but not alpha 2-adrenoceptors, J. Neurosci, 4 (1984) 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anisman H, Kokkinidis L and Sklar LS, Neurochemical consequences of stress: contributions and adaptive processes. In Burchfield S (Ed.), Psychological and Physiological Interaction in Response to Stress, Hemisphere, New York, 1984, pp. 67–98. [Google Scholar]
- 10.Aston-Jones G and Bloom FE, Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle, J. Neurosci, 8 (1981) 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aston-Jones G, Ennis M, Pieribone VA, Nickell WT and Shipley MT, The brain nucleus locus coeruleus: restricted afferent control of a broad efferent network, Science, 234 (1986) 734–737. [DOI] [PubMed] [Google Scholar]
- 12.Aston-Jones G, Foote SL and Bloom FE, Anatomy and physiology of locus coeruleus neurons: functional implications. In Ziegler M and Lake R (Eds.), Norepinephrine, Williams and Wilkins, Baltimore, 1984, pp. 92–116. [Google Scholar]
- 13.Baker TL and Dement WC, Canine narcolepsy-cataplexy syndrome: evidence for an inherited monoaminergic-cholinergic imbalance. In McGinty DJ, Drucker-Colin R, Morrison A, and Parmeggiani PL (Eds.), Brain Mechanisms of Sleep, Raven, New York, 1985, pp. 199–234. [Google Scholar]
- 14.Baust W, Boehmke J and Blossfeld U, Somato-sympathetic reflexes during natural sleep and wakefulness in unrestrained cats, Exp. Brain Res, 12 (1971) 361–369. [DOI] [PubMed] [Google Scholar]
- 15.Benoit O, Thalamic unit activity during the transition from synchronized to desynchronized sleep: a comparative study in cats and macaque monkeys, Rev. Electroencephalogr. Neurophysiol. Clin, 3 (1973) 39–45. [DOI] [PubMed] [Google Scholar]
- 16.Berger RJ, The sleep and dream cycle. In Kales A (Ed.), Sleep Physiology and Pathology, Lippincott, Philadelphia, 1969, pp. 17–32. [Google Scholar]
- 17.Berger RJ and Walker JM, Sleep in the burrowing owl (Speotyto cunicularia hypugaea), Behav. Biol, 7 (1972) 183–194. [DOI] [PubMed] [Google Scholar]
- 18.Bergstrom DA and Kellar KJ, Adrenergic and serotonergic receptor binding in rat brain after chronic desmethylimipramine treatment, J. Pharmacol. Exp. Ther, 209 (1979) 256–261. [PubMed] [Google Scholar]
- 19.Billiard M, Narcolepsy. Clinical features and aetiology. Ann. Clin. Res, 17 (1985) 220–226. [PubMed] [Google Scholar]
- 20.Bliss TVP, Goddard GV and Riives M, Reduction of long-term potentiation in the dentate gyrus to the rat following selective depletion of monoamines. J. Physiol. (Lond.), 334 (1983) 475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blondaux C, Buda M, Petitjean F and Pujol JF, Hypersomnie par lesion isthmique chez le chat. I. Etude du metabolisme des monoamines cerebrales, Brain Res, 88 (1975) 425–437. [DOI] [PubMed] [Google Scholar]
- 22.Brooks DC and Gershon MD, An analysis of the effect of reserpine upon ponto-geniculo-occipital wave activity in the cat, Neuropharmacology, 11 (1972) 499–510. [DOI] [PubMed] [Google Scholar]
- 23.Carlini EA, REM sleep deprivation and dopamine in the CNS, Rev. Pure Appl. Pharmacol. Sci, 4 (1983) 1–25. [PubMed] [Google Scholar]
- 24.Cedarbaum JM and Aghajanian GK, Afferent projections to the rat locus coeruleus as determined by retrograde tracing technique, J. Comp. Neurol, 178 (1978) 1–16. [DOI] [PubMed] [Google Scholar]
- 25.Chase MH, Enomoto S, Murakami T, Nakamura Y and Taira M, Intracellular potential of medullary reticular neurons during sleep and wakefulness, Exp. Neurol, 71 (1981) 226–233. [DOI] [PubMed] [Google Scholar]
- 26.Chase MH, Morales FR, Soja PJ, Baranyi A and Navarrete DR, A pharmacological investigation of the phasic postsynaptic inhibition of lumbar motoneurons during the rapid eye movement episodes of active sleep, Sleep Res, 14 (1985) 4. [Google Scholar]
- 27.Chauvel P and Trottier S, Role of noradrenergic ascending system in extinction of epileptic phenomena. Adv. Neurol, 44 (1986) 475–487. [PubMed] [Google Scholar]
- 28.Cohen HB and Dement WC, Prolonged tonic convulsions in REM deprived mice, Brain Res, 22 (1970) 421–422. [DOI] [PubMed] [Google Scholar]
- 29.Corcoran ME, Fibinger HC, McCaughran JA and Wada JA, Potentiation of amygdaloid kindling and metrazol-induced seizures by 6-hydroxydopamine in rats, Exp. Neurol, 45 (1974) 118–133. [DOI] [PubMed] [Google Scholar]
- 30.Corcoran ME and Mason STC, Role of forebrain catecholamines in amygdaloid kindling, Brain Res, 190 (1980) 473–484. [DOI] [PubMed] [Google Scholar]
- 31.Crick F and Mitchison G, The function of dream sleep, Nature (Lond.), 304 (1983) 111–114. [DOI] [PubMed] [Google Scholar]
- 32.Delorme F, Froment JL and Jouvet M, Suppression du sommeil par la parachlorometamphetamine et la parachlorophenylalanine, C.R. Soc. Biol. (Paris), 160 (1966) 2347–2350. [PubMed] [Google Scholar]
- 33.Dement WC, The biological role of REM sleep (circa 1968). In Kales A (Ed.), Sleep Physiology and Pathology, Lippincott, Philadelphia, 1969, pp. 245–265. [Google Scholar]
- 34.Dement WC, Mitler MM and Henriksen SJ, Sleep changes during chronic administration of parachlorophenylalanine, Rev. Can. Biol, 31 (1972) 239–246. [PubMed] [Google Scholar]
- 35.Desiraju T, Discharge properties of neurons of the parietal association cortex during states of sleep and wakefulness in the monkey, Brain Res, 47 (1972) 69–75. [DOI] [PubMed] [Google Scholar]
- 36.Drucker-Colin R and Benitez J, REM sleep rebound during withdrawal from chronic amphetamine administration is blocked by chloramphenicol, Neurosci. Lett, 6 (1977) 267–271. [DOI] [PubMed] [Google Scholar]
- 37.Elazar Z and Hobson JA, Neuronal excitability control in health and disease: A neurophysiological comparison of REM sleep and epilepsy, Prog. Neurobiol, 25 (1985) 141–188. [DOI] [PubMed] [Google Scholar]
- 38.Elomaa E, The cuff pedestal: an alternative to flower-pots?, Physiol, Behave, 23 (1979) 669–672. [DOI] [PubMed] [Google Scholar]
- 39.Engberg G, Elam M and Svensson TH, Clonidine withdrawal: activation of brain noradrenergic neurons with specifically reduced alpha2-receptor sensitivity, Life sci, 30 (1982) 235–243. [DOI] [PubMed] [Google Scholar]
- 40.Evarts EV, Temporal patterns of discharge of pyramidal tract neurons during sleep and waking in the monkey, J. Neurophysiol, 27 (1964) 152–171. [DOI] [PubMed] [Google Scholar]
- 41.Everson CA, Kushida CA, Bergmann BM, Gilliland MA, Pilcher JJ, Fang VS and Rechtschaffen A, Recovery from chronic sleep deprivation in the rat, Sleep Res., 16 (1987) 521. [Google Scholar]
- 42.Farber J, Miller JD, Crawford KA and McMillen BA, Dopamine metabolism and receptor sensitivity in rat brain after REM sleep deprivation, Pharmacol. Biochem. Behav, 18 (1983) 509–513. [DOI] [PubMed] [Google Scholar]
- 43.Faull KF, ZellerDeAmicis L.c., Radde L, Bowersox SS, Baker T,L, Kilduff TS and Dement WC, Biogenic amine concentrations in the brains of normal and narcoleptic canines: current status, Sleep, 9 (1986) 107–110. [DOI] [PubMed] [Google Scholar]
- 44.Feinberg I, Hibi S, Braun M, Cavness C, Westermam G and Small A, Sleep amphetamine effects in MBDS and normal subjects, Arch. Gen, Psychiat, 31 (1974) 723–731. [DOI] [PubMed] [Google Scholar]
- 45.Ferguson J and Dement W, The effect of REM sleep deprivation on the lethality of dextroamphetamine sulfate in grouped rats, Psychophysiology, 4 (1967) 380. [Google Scholar]
- 46.Ferguson J and Dement W, The behavioral effects of amphetamine on REM deprived rats, J. Psychiat. Res, 7 (1969) 111–118. [DOI] [PubMed] [Google Scholar]
- 47.Fishbein W, Kastaniotis C and Chattman D, Paradoxical sleep: prolonged augmentation following learning, Brain Res, 79 (1974) 61–75. [DOI] [PubMed] [Google Scholar]
- 48.Flanigan WF Jr., Knight CP, Hartse KM and Rechtschaffen A, Sleep and wakefulness in chelonian reptiles. I. The box turtle, Terrapene carolina, Arch. Ital. Biol, 112 (1974) 227–252. [PubMed] [Google Scholar]
- 49.Flanigan WF Jr., Wilcox RH and Rechtschaffen A, The EEG and behavioral continuum of the crocodilian, Caiman sclerops, Electroencephologr. Clin. Neurophysiol, 34 (1973) 521–538. [DOI] [PubMed] [Google Scholar]
- 50.Foote SL, Bloom FE and Aston-Jones G, Nucleus locus ceruleus: new evidence of anatomical and physiological specificity, Physiol. Rev, 63 (1983) 844–914. [DOI] [PubMed] [Google Scholar]
- 51.Fornal C, Auerbach S and Jacobs BL, Activity of serotonin-containing neurons in nucleus raphe magnus in freely moving cats, Exp. Neurol, 88 (1985) 590–608. [DOI] [PubMed] [Google Scholar]
- 52.Futuro-Neto HA and Coote JH, Changes in sympathetic activity to heart and blood vessels during desynchronized sleep, Brain Res, 252 (1982) 259–268. [DOI] [PubMed] [Google Scholar]
- 53.Gillin JC, Van Kammen DP, Murphy DC, Graves J and Wyatt R, Differential effects of d- and l-amphetamine on the sleep of depressed psychiatric patients on and off lithum carbonate treatment. In Chase MH, Stern WC and Walter PL (Ed.,), Sleep Research, BIS/BRI, Los Angeles, 1974, p. 50. [Google Scholar]
- 54.Glavin GB, Stress and brain noradrenaline: a review, Neurosci. Biobehav, 9 (1985) 233–243. [DOI] [PubMed] [Google Scholar]
- 55.Glotzbach SF, Cornett CM and Heller HC, Activity of suprachiasmatic and hypothalamic neurons during sleep and wakefulness in the rat, Brain Res, 419 (1987) 279–286. [DOI] [PubMed] [Google Scholar]
- 56.Glotzbach SF and Heller CH, Changes in the thermal characteristics of hypothalamic neurons during sleep and wakefulness, Brain Res, 309 (1984) 17–26. [DOI] [PubMed] [Google Scholar]
- 57.Guilleminault C, Cataplexy. In Guilleminault C, Dement WC and Passouant P (Eds.), Narcolepsy, Spectrum, New York, 1976, pp. 125–143. [Google Scholar]
- 58.Haefely W, Wet physiology of REM sleep generation, Behav. Brain sci, 9 (1986) 407. [Google Scholar]
- 59.Harper RM and Sieck GC, Discharge correlations between neurons in the nucleus parabrachialis medialis during sleep-waking states, Brain Res, 199 (1980) 343–358. [DOI] [PubMed] [Google Scholar]
- 60.Hartmann E, Functions of Sleep, Yale University Press, New Haven, 1973. [Google Scholar]
- 61.Hartmann E, The D-state and norepinephrine-dependent systems, Int. Psychiat. Clin, 7 (1970) 308–328. [PubMed] [Google Scholar]
- 62.Hartmann E, Role and regulation of sleep. In Jovanovic UJ (Ed.), The Nature of S1eep, Thieme, Stuttgart, 1973, pp. 238–252. [Google Scholar]
- 63.Hartmann E, Chung R, Draskoczy PR and Schildkraut. J.J., Effects of 6-hydroxydopamine on sleep in the rat, Nature (Lond.), 233 (1971) 425–427. [DOI] [PubMed] [Google Scholar]
- 64.Hartmann E and Stern WC, Desynchronized sleep deprivation: learning deficit and its reversal by increased catecholamines, Physiol Behav, 8 (1972) 585–587. [DOI] [PubMed] [Google Scholar]
- 65.Hilakivi I, The role of beta- and alpha-adrenoreceptors in the regulation of the stages of the sleep-waking cycle in the cat, Brain Res, 277 (1983) 109–118. [DOI] [PubMed] [Google Scholar]
- 66.Hilakivi I and Leppavuori A, Effects of methoxamine, an alpha-1 adrenoceptor agonist, and prazosin, an alpha-1 antagonist, on the stages of the sleep-waking cycle in the rat, Acta Physiol. Scand, 120 (1984) 363–372. [DOI] [PubMed] [Google Scholar]
- 67.Hilakivi I, Leppavuori A and Putkonen PTS, Prazosin increases paradoxical sleep, Eur. J. Pharmacol, 65 (1980) 417–420. [DOI] [PubMed] [Google Scholar]
- 68.Hobson JA, Goin OB and Goin CJ, Electrographic correlates of behavior in tree frogs, Nature (Lond.), 220 (1968) 386–387. [DOI] [PubMed] [Google Scholar]
- 69.Hobson JA, McCarley RW and Nelson JP, Location and spike-train characteristics of cells in anterodorsal pons having selective decreases in firing rat during desynchronized sleep, J. Neurophysiol, 50 (1983) 770–783. [DOI] [PubMed] [Google Scholar]
- 70.Hobson JA, McCarley RW, Pivik T and Freedman R, Selective firing by cat pontine brain stem neurons in desynchronized sleep, J. Neurophysiol, 37 (1974) 497–511. [DOI] [PubMed] [Google Scholar]
- 71.Hoffer BJ, Siggins GR and Bloom FE, Studies on norepinephrine-containing afferents to Purkinje cells of rat cerebellum. II. Sensitivity of Purkinje cells to norepinephrine and related substances administered by microiontophoresis, Brain Res, 25 (1971) 523–534. [DOI] [PubMed] [Google Scholar]
- 72.Hopkins WF and Johnston D, Frequency-dependent noradrenergic modulation of long-term potentiation in the hippocampus, Science, 226 (1984) 350–352. [DOI] [PubMed] [Google Scholar]
- 73.Hricovini M and Pavlasek J, Desensitization of reticular neurons after iontophoretically applied noradrenalin in rats, Endocrin. Exp, 19 (1985) 291–296. [PubMed] [Google Scholar]
- 74.Huttenlocher PR, Evoked and spontaneous activity in single units of medial brain stem during natural sleep and waking, J. Neurophysiol, 24 (1961) 451–468. [Google Scholar]
- 75.Jacobs BL, Single unit activity of locus coeruleus neurons in behaving animals, Prog. Neurobiol, 27 (1986) 183–194. [DOI] [PubMed] [Google Scholar]
- 76.Jacobs BL, Harper RM and McGinty DJ, Neuronal coding of motivational level during sleep, Physiol. Behav, 5 (1970) 1139–1143. [DOI] [PubMed] [Google Scholar]
- 77.Jacobs BL, Heym J and Trulson ME, Behavioral and physiological correlates of brain serotoninergic unit activity, J. Physiol. (Paris), 77 (1981) 431–436. [PubMed] [Google Scholar]
- 78.Jacobs BL and Jones BE, The role of central monoamine and acetylcholine systems in sleep-wakefulness states: mediation or modulation?. In Butcher LL (Ed.). Cholinergic-Monoaminergic Interactions of the Brain, Academic, New York, 1978, pp. 271–290. [Google Scholar]
- 79.Jones BE, Harper ST and Halaris AE, Effects of locus coeruleus lesions upon cerebral monoamine content, sleep wakefulness states and the response to amphetamine in the cat, Brain Res, 124 (1977) 473–496. [DOI] [PubMed] [Google Scholar]
- 80.Jouvet M, Recherches sur les structures nerveuses et les mechanismes responsables des differentes phases du sommeil physiologique, Arch. Ital. Biol, 100 (1962) 125–206. [PubMed] [Google Scholar]
- 81.Jouvet M, Does a genetic programming of the brain occur during paradoxical sleep?. In Buser PA and Rougeul-Buser A (Eds.), Cerebral Correlates of Conscious Experience, INSERM Symposium, Elsevier/North-Holland, Amsterdam, 1978, pp. 245–261. [Google Scholar]
- 82.Jouvet M, Paradoxical sleep and the nature-nurture controversy, Prog. Brain Res, 53 (1980) 331–346. [DOI] [PubMed] [Google Scholar]
- 83.Kaitin KI, Prepotic area unit activity during sleep and wakefulness in the cat, Exp. Neurol, 83 (1984) 347–357. [DOI] [PubMed] [Google Scholar]
- 84.Kales A, Cadieux R, Soldatos CR and Tan T, successful treatment of narcolepsy with propranolol, Arch. Neurol, 36 (1979) 650–651. [DOI] [PubMed] [Google Scholar]
- 85.Kales A, Soldatos CR, Cadieux R, Bixler EO, Tan T and Scharf MB, Propranolol in the treatment of narcolepsy, Ann. Int. Med, 91 (1979) 742–743. [DOI] [PubMed] [Google Scholar]
- 86.Kanamori N, Sakai K and Jouvet M, Neuronal activity specific to paradoxical sleep in the ventromedial medullary reticular formation of unrestrained cats, Brain Res, 189 (1980) 251–255. [DOI] [PubMed] [Google Scholar]
- 87.Kasamatsu T and Adey WR, Excitability changes in various types of visual cortical units in freely behaving cats. Physiol. Behav, 13 (1974) 101–112. [DOI] [PubMed] [Google Scholar]
- 88.Kilian M and Frey HH, Central monoamines and convulsive thresholds in mice and rats, Neuropharmacology, 12 (1973) 681–692. [DOI] [PubMed] [Google Scholar]
- 89.Koe KB and Vinick FJ, Adaptive changes in central nervous system receptor systems, Annu. Rep. Med. Chem, 19 (1984) 41–50. [Google Scholar]
- 90.Kovalzon VM and Tsibulsky VL, REM sleep deprivation without stress in rats, Sleep 1978, Fourth European Congress on Sleep Research, 1978, pp. 411–414. [Google Scholar]
- 91.Leconte P, Hennevin E and Bloch V, Duration of paradoxical sleep necessary for the acquisition of conditioned avoidance in the rat, Physiol. Behav, 13 (1974) 675–681. [DOI] [PubMed] [Google Scholar]
- 92.Lefkowitz RJ and Caron MG, Molecular and regulatory properties of adrenergic receptors, Rec. Prog. Horm. Res, 43 (1987) 469–497. [DOI] [PubMed] [Google Scholar]
- 93.Maggi A, U’Prichard DC and Enna SJ, Beta-adrenergic regulation of alpha2-adrenergic receptors in the central nervous system, Science, 207 (1980) 645–647. [DOI] [PubMed] [Google Scholar]
- 94.Makela JP and Hilakivi IT, Evidence for the involvement of alpha-2 adrenoceptors in the sedation but not REM sleep inhibition by clonidine in the rat, Med. Biol, 64 (1986) 355–360. [PubMed] [Google Scholar]
- 95.Mano NI, Changes of simple and complex spike activity of cerebellar Purkinje cells with sleep and waking, Science, 170 (1970) 1325–1327. [DOI] [PubMed] [Google Scholar]
- 96.Marks GA, Farber J and Roffwarg HP, Phasic influences during REM sleep upon dorsal lateral geniculate nucleus unit activity in the rat, Brain Res, 222 (1981) 388–394. [DOI] [PubMed] [Google Scholar]
- 97.Masserano JM and Weiner N, Tyrosine hydroxylase regulation in the central nervous system, Mol, Cell Biochem, 53/54 (1983) 129–152. [DOI] [PubMed] [Google Scholar]
- 98.Matsumoto J and Watanabe S, Paradoxical sleep: effects of adrenergic blocking agents, Proc. Jpn. Acad, 43 (1967) 680–683. [Google Scholar]
- 99.Maynert EW, Marczynski TJ and Browning RA, The role of neurotransmitters in the epilepsies. In Friedlander WJ (Ed.), Adv. Neurol, Raven, New York, 1975, pp. 79–147. [PubMed] [Google Scholar]
- 100.McBride RL and Sutin J, Projections of the locus coeruleus and adjacent pontine tegmentum in the cat, J. comp. Neurol, 165 (1976) 265–284. [DOI] [PubMed] [Google Scholar]
- 101.McCarley RW and Hobson JA, Single neuron activity in cat gigantocellular tegmental field: Selectivity of discharge in desynchronized sleep, Science, 174 (1971) 1250–1252. [DOI] [PubMed] [Google Scholar]
- 102.McGinty DJ and Drucker-Colin RR, Sleep mechanisms: biology and control of REM sleep. Int. Rev. Neurobiol, 23 (1982) 391–436. [DOI] [PubMed] [Google Scholar]
- 103.McGinty DJ and Harper RM, Dorsal raphe neurons: depression of firing during sleep in cats, Brain Res, 101 (1976) 569–575. [DOI] [PubMed] [Google Scholar]
- 104.McGinty DJ and Siegel JM, Sleep states. In Satinoff E and Teitelbaum P (Eds.), Handbook of Behavioral Neurobiology: Motivation, Plenum, New York, 1983, pp. 105–181. [Google Scholar]
- 105.Mefford IN, Baker. TL, Boehme R, Foutz AS, Ciaranello RD, Barchas JD and Dement WC, Narcolepsy: biogenic amino deficits in an animal model, Science, 220 (1983) 629–632. [DOI] [PubMed] [Google Scholar]
- 106.Meglasson MD and Huggins SE, Sleep in a crocodilian, Caiman sclerops, Comp. Biochem. Physiol, 63 (1979) 561–567. [Google Scholar]
- 107.Mendelson W, Guthrie RD, Guynn R, Harris RL and Wyatt RJ, Rapid eye movement (REM) sleep deprivation, stress and intermediary metabolism, J. Neurochem, 22 (1974) 1157–1159. [DOI] [PubMed] [Google Scholar]
- 108.Menkes DB, Gallager DW, Reinhard JF Jr. and Aghajanian GK, Alpha 1-adrenoceptor denervation supersensitivity in brain: physiological and receptor binding studies, Brain Res, 272 (1983) 1–12. [DOI] [PubMed] [Google Scholar]
- 109.Mignot E, Guilleminault C, Bowersox S, Rappaport A and Dement WC, Effect of alpha-1 adrenoceptors blockade with prazosin in canine narcolepsy, Brain Res, in press. [DOI] [PubMed] [Google Scholar]
- 110.Miller JD, Farbar J, Gatz P, Roffwarg H and German DC, Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and waking in the rat, Brain Res, 273 (1983) 133–141. [DOI] [PubMed] [Google Scholar]
- 111.Mogilnicka E, Arbilla S, Depoortere H and Langer SZ, Rapid-eye-movement sleep deprivation decreases the density of 3H-dihydroalprenolol and 3H-imipramine binding sites in the rat cerebral cortex, Eur. J. Pharmacol, 65 (1980) 289–292. [DOI] [PubMed] [Google Scholar]
- 112.Mogilnicka E, Przewlocka B, van Luijtelaar ELJM, Klimek V and Coenen AML, Effects of REM sleep deprivation on central alpha 1- and beta-adrenoceptors in rat brain, Pharmacol. Biochem. Behav, 25 (1986) 329–332. [DOI] [PubMed] [Google Scholar]
- 113.Moises HC, Hoffer BJ and Woodward DJ, GABA facilitation by noradrenaline shows supersensitivity in cerebellum after 6-hydroxydopamine, Naunyn-Schmiedeberg’s Arch. Pharmacol, 315 (1980) 37–46. [DOI] [PubMed] [Google Scholar]
- 114.Moises HC and Woodward DJ, Potentiation of GABA inhibitory action in cerebellum by locus coeruleus stimulation, Brain Res, 182 (1980) 327–344. [DOI] [PubMed] [Google Scholar]
- 115.Monti JM, Pellejero T, Jantos H, D’Angelo L, Barbeito L and Abo V, Sleep-wake cycle and sensitivity to drugs acting on adrenergic receptors after DSP-4, a selective noradrenergic neurotoxin, Sleep Res, 16 (1987) 114. [DOI] [PubMed] [Google Scholar]
- 116.Morilak DA and Jacobs BL, Noradrenergic modulation of sensorimotor processes in intact rats: the masseteric reflex as a model system, J. Neurosci, 5 (1985) 1300–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morpugo C and Theobald W, Behavioral reaction to amphetamine in reserpinized rats, Int. J. Neuropharmacol, 5 (1966) 375–377. [DOI] [PubMed] [Google Scholar]
- 118.Nakamura K and Thoenen H, Increased irritability: a permanent behavior change induced in the rat by intraventricular administration of 6-hydroxydopamine, Psychopharmacology, 24 (1972) 359–372. [DOI] [PubMed] [Google Scholar]
- 119.Netick A, Orem J and Dement W, Neuronal activity specific to REM sleep and its relationship to breathing, Brain Res, 120 (1977) 197–207. [DOI] [PubMed] [Google Scholar]
- 120.Neuman RS and Harley CW, Long-lasting potentiation of the dentate gyrus population spike by norepinephrine, Brain Res, 273 (1983) 162–165. [DOI] [PubMed] [Google Scholar]
- 121.Nukina I, Glavin GB and LaBella FS, Acute cold-restraint stress affects alpha 2-adrenoceptors in specific brain regions of the rat, Brain Res, 401 (1987) 30–33. [DOI] [PubMed] [Google Scholar]
- 122.Pandey GN, Heinze WJ, Brown BD and Davis JM, Electroconvulsive shock treatment decreases beta-adrenergic receptors sensitivity in rat brain, Nature (Lond.), 280 (1979) 148. [DOI] [PubMed] [Google Scholar]
- 123.Parmeggiani PL, Cevolani D, Azzaroni A and Ferrari G, Thermosensitivity of anterior hypothalamic-preoptic neurons during the waking-sleeping cycle: a study in brain functional states, Brain Res, 415 (1987) 79–89. [DOI] [PubMed] [Google Scholar]
- 124.Petitjean F, Sakai K, Blondaux C and Jouvet M, Hypersomnie par lesion isthmique chez le chat. II. Etude neurophysiologique et pharmacologique, Brain Res, 88 (1975) 439–453. [DOI] [PubMed] [Google Scholar]
- 125.Putkonen PTS and Bergstrom L, Clonidine alleviates cataplectic symptoms in narcolepsy. In Koella WP (Ed.), Sleep, Karger, Basel, 1980, pp. 414–416. [Google Scholar]
- 126.Radulovacki M and Micovic N, Effects of REM sleep deprivation and desipramine on beta-adrenergic binding sites in rat brain, Brain Res, 235 (1982) 393–396. [DOI] [PubMed] [Google Scholar]
- 127.Radulovacki M, Walovitch R and Yanik G, Caffeine produces REM sleep rebounds in rats, Brain Res, 201 (1980) 497–500. [DOI] [PubMed] [Google Scholar]
- 128.Radulovacki M, Wojcik WJ, Fornal C and Miletich R, Elimination of REM sleep rebound in rats by alpha-adrenoreceptor blockers, phentolamine and phenoxybenzamine, Pharmacology Biochem. Behav, 13 (1980) 51–55. [DOI] [PubMed] [Google Scholar]
- 129.Radulovacki M and Zak R, Amphetamine abolishes REM sleep rebound in rats: effect of a single injection, Brain Res, 217 (1981) 420–424. [DOI] [PubMed] [Google Scholar]
- 130.Rasmussen K, Morilak DA and Jacobs BL, Single unit activity of locus coeruleus neurons in the freely moving cat. I. During naturalistic behaviors and in response to simple and complex stimuli, Brain Res, 371 (1986) 324–334. [DOI] [PubMed] [Google Scholar]
- 131.Rechtschaffen A and Maron L, The effect of amphetamine on the sleep cycle, Electroencephalogr. Clint Neurophysiol, 16 (1964) 438–445. [DOI] [PubMed] [Google Scholar]
- 132.Reiner PB, Correlation analysis of central noradrenergic neuronal activity and sympathetic tone in behaving cats, Brain Res, 378 (1986) 86–96. [DOI] [PubMed] [Google Scholar]
- 133.Reisine T, Adaptive changes in catecholamine receptors in the central nervous system, Neuroscience, 6 (1981) 1471–1502. [DOI] [PubMed] [Google Scholar]
- 134.Roffwarg HP, Muzio JN and Dement WC, Ontogenetic development of the human sleep-dream cycle, Science, 152 (1966) 604–619. [DOI] [PubMed] [Google Scholar]
- 135.Rogawski MA, Norepinephrine. In Rogawski MA and Barker JL (Eds.), Neurotransmitter Actions in the Vertebrate Nervous System, Plenum, New York, 1985, pp. 241–284. [Google Scholar]
- 136.Rogawski MA and Aghajanian GK, Activation of lateral geniculate neurons by norepinephrine: mediation by an alpha-adrenergic receptor, Brain Res, 182 (1980) 345–349. [DOI] [PubMed] [Google Scholar]
- 137.Rogawski MA and Aghajanian GK, Norepinephrine and serotonin: opposite effects on the activity of lateral geniculate neurons evoked by optic pathway stimulation, Exp. Neurol, 69 (1980) 678–694. [DOI] [PubMed] [Google Scholar]
- 138.Rogawski MA and Aghajanian GK, Modulation of lateral geniculate neurone excitability by noradrenalin microiontophoresis or locus coeruleus stimulation, Nature (Lond.), 287 (1980) 731–734. [DOI] [PubMed] [Google Scholar]
- 139.Rogawski MA and Aghajanian GK, Activation of lateral geniculate neurons by locus coeruleus or dorsal noradrenergic bundle stimulation: selective blockade by the alpha1-adrenoceptor antagonist prazosin, Brain Res, 250 (1982) 31–39. [DOI] [PubMed] [Google Scholar]
- 140.Saito H, Sakai K and Jouvet M, Discharge patterns of the nucleus parabrachialis lateralis neurons of the cat during sleep and waking, Brain Res, 134 (1977) 59–72. [DOI] [PubMed] [Google Scholar]
- 141.Sakai K, Some anatomical and physiological properties of pontomesencephalic tegmental neurons with special reference to the PGO waves and postural atonia during paradoxical sleep in the cat. In Hobson JA and Brazier MA (Eds.), The Reticular Formation Revisited, Raven, New York, 1980, pp. 427–447. [Google Scholar]
- 142.Sakai K, Anatomical and physiological basis of paradoxical sleep. In McGinty DJ, Drucker-Colin R, Morrison AR and Parmeggiani PL (Eds.), Brain Mechanisms of Sleep, Raven, New York, 1985, pp. 111–138. [Google Scholar]
- 143.Sakai K, Sastre JP, Kanamori N and Jouvet M, State-specific neurons in the ponto-medullary reticular formation with special reference to the postural atonia during paradoxical sleep in the cat. In Pompeiano O and Ajmone-Marsan C (Eds.), Brain Mechanisms and Perceptual Awareness, Raven, New York, 1981, pp. 405–429. [Google Scholar]
- 144.Sakai K, Sastre JP, Salvert D, Touret M, Tohyama M and Jouvet M, Tegmentoreticular projections with special reference to the muscular atonia during paradoxical sleep in the cat: an HRP study, Brain Res, 176 (1979) 233–254. [DOI] [PubMed] [Google Scholar]
- 145.Sastre JP, Sakai K and Jouvet. M, Persistance du sommeil paradoxal chez le chat apres destruction de l’aire gigantocellulaire du tegmentum pontique par l’acide kainique, C.R. Acad. Sci, 289D (1979) 959–964. [PubMed] [Google Scholar]
- 146.Satinoff. E, Drucker-Colin RR and Hernandez-Peon R, Paleocortical excitability and sensory filtering during REM sleep deprivation, Physiol. Behav, 7 (1971) 103–106. [DOI] [PubMed] [Google Scholar]
- 147.Schildkraut JJ and Hartmann E, Turnover and metabolism of norepinephrine in rat brain after 72 hours on a D-deprivation island, Psychopharmacology. 27 (1972) 17–27. [DOI] [PubMed] [Google Scholar]
- 148.Schwartz JC, Costentin J, Martres MP, Protais P and Baudry M, Modulation of receptor mechanisms in the CNS: hyper- and hyposensitivity to catecholamines, Neuropharmacology, 17 (1978) 665–685. [DOI] [PubMed] [Google Scholar]
- 149.Segal M and Bloom. FE, The action of norepinephrine in the rat hippocampus. I. Iontophoretic studies, Brain Res, 72 (1974) 79–97. [DOI] [PubMed] [Google Scholar]
- 150.Shiromani PJ, Armstrong. DM, Bruce G, Hersh LB, Groves PJ and Gillin C, Relation of pontine choline acetyltransferase immunoreactive neurons with cells which increase discharge during REM sleep, Brain Res. Bull, 18 (1987) 447–455. [DOI] [PubMed] [Google Scholar]
- 151.Siegel JM, Brainstem mechanisms generating REM sleep. In Kryger MK (Ed.), Principals and Practice of Sleep Medicine, Saunders, New York, in press. [Google Scholar]
- 152.Siegel JM and McGinty DJ, Brainstem neurons without spontaneous unit discharge, Science, 193 (1976) 240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Siegel JM and McGinty DJ, Pontine reticular formation neurons: relationship of discharge to motor activity, Science, 196 (1977) 678–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Siegel JM, McGinty DJ and Breedlove SM, Sleep and waking activity of pontine gigantocellular field neurons, Exp. Neural, 56 (1977) 553–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Siegel JM, Nienhuis R and Tomaszewski KS, REM sleep signs rostral to chronic transections at the pontomedullary junction, Neurosci. Lett, 45 (1984) 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Siegel JM and Tomaszewski KS, Behavioral organization of reticular formation: studies in the unrestrained cat. I. Cells related to axial, limb, eye, and other movements, J. Neurophysiol, 50 (1983) 696–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Siegel JM, Tomaszewski KS and Nienhuis R, Behavioral states in the chronic medullary and mid-pontine cat, Electroencephalogr. Clin. Neurophysiol, 63 (1986) 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Siegel JM, Tomaszewski KS and Wheeler RL, Behavioral organization of reticular formation: studies in the unrestrained eat. II. Cells related to facial movements, J. Neurophysiol, 50 (1983) 717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Siegel JM, Wheeler RL and McGinty. DJ, Activity of medullary reticular formation neurons in the unrestrained cat during waking and sleep, Brain Res, 179 (1979) 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Simon RP, Gershon MD and Brooks DC, The role of the raphe nuclei in the regulation of ponto-geniculo-occipital wave activity, Brain Res, 58 (1973) 313–330. [DOI] [PubMed] [Google Scholar]
- 161.Smith C, Young J and Young W, Prolonged increases in paradoxical sleep during and after avoidance-task acquisition, Sleep, 3 (1980) 67–81 [PubMed] [Google Scholar]
- 162.Snyder F, Toward an evolutionary theory of dreaming, Am. J. Psychiat, 123(1966) 121–136. [DOI] [PubMed] [Google Scholar]
- 163.Sporn JR, Wolfe BB, Harden TK and Molinoff PB, Supersensitivity in rat cerebral cortex: pre- and postsynaptic effects of 6-hydroxydopamine at noradrenergic synapses, Mol. Pharmacol, 13 (1977) 1170–1180. [PubMed] [Google Scholar]
- 164.Stanton PK and Sarvey. JM, Blockade of norepinephrine-induced long-lasting potentintion in the hippocampal dentate gyrus by an inhibitor of protein synthesis, Brain Res, 361 (1985) 276–283. [DOI] [PubMed] [Google Scholar]
- 165.Stanton PK and Sarvey JM, Depletion of norepinephrine, but not serotonin, reduces long-term potentiation in the dentate gyrus of rat hippocampal slices, J. Neurosci, 5 (1985) 2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stein D, Jouvet M and Pujol JF, Effects of alpha-methyl-p-tyrosine upon cerebral amine metabolism and sleep states in cat, Brain Res, 72 (1974) 360–365. [DOI] [PubMed] [Google Scholar]
- 167.Steinfels GF, Heym J, Strecker RE and Jacobs BL, Response of dopaminergic neurons in cat to auditory stimuli presented across the sleep-waking cycle. Brain Res, 277 (1983) 150–154. [DOI] [PubMed] [Google Scholar]
- 168.Steriade M and Hobson JA, Neuronal activity during the sleep-waking cycle, Prog. Neurobiol, 6 (1976) 155–376. [PubMed] [Google Scholar]
- 169.Stern. WC, Acquisition impairments following rapid eye movement sleep deprivation in rats, Physiol. Behav, 7 (1971) 345–352. [DOI] [PubMed] [Google Scholar]
- 170.Stern WC and Morgane PJ, Effects of alpha-methyltyrosine on REM sleep and brain amine levels in the cat, Biol. Psychiat, 6 (1973) 301–306. [PubMed] [Google Scholar]
- 171.Stern WC and Morgane PJ, Theoretical view of REM sleep function: maintenance of catecholamine systems in the central nervous system, Behav. Biol, 11 (1974) 1–32. [DOI] [PubMed] [Google Scholar]
- 172.Sugrue MF, Changes in rat brain monoamine turnover following chronic antidepressant administration, Life Sci, 26 (1980) 423–429. [DOI] [PubMed] [Google Scholar]
- 173.Susic VT and Kovacevic. RM, Sleep patterns in the owl, Strix aluco, Physiol. Behav, 11 (1973) 313–317. [DOI] [PubMed] [Google Scholar]
- 174.Swan AC, Grant SJ, Hattox. SE and Maas JW, Adrenoreceptor regulation in rat brain: chronic effects of alpha- or alpha 2-receptor blockers, Eur. J. Pharmacol, 73 (1981) 301–305. [Google Scholar]
- 175.Szymusiak R and McGinty DJ, Sleep-related neuronal discharge in the basal forebrain of cats, Brain Res, 370 (1986) 82–92. [DOI] [PubMed] [Google Scholar]
- 176.Thoa. NB, Eichelman B and Ng LKY, Shock-induced aggression: effects of 6-hydroxydopamine and other pharmacological agents, Brain Res, 43 (1972) 467–475. [DOI] [PubMed] [Google Scholar]
- 177.Tohyama M, Comparative anatomy of cerebellar catecholamine innervation from teleosts to mammals, J. Hirnforsch, 17 (1976) 43–60. [PubMed] [Google Scholar]
- 178.Troncone LRP, Braz S, Benedito MAC and Tufik S, REM sleep deprivation induces a decrease in norepinephrine-stimulated 3H-cyclic AMP accumulation in slices from rat brain, Pharmacol. Biochem. Behav, 25 (1986) 223–225. [DOI] [PubMed] [Google Scholar]
- 179.Tufik S, Lindsey CJ and Carlini EA, Does REM sleep deprivation induce a supersensitivity of dopaminergic receptors in the rat brain?, Pharmacology, 16 (1978) 98–105. [DOI] [PubMed] [Google Scholar]
- 180.Ursin R, Differential effect of para-chlorophenylalanine on the two slow wave sleep stages in the cat. Acta Physiol. Scand, 86 (1972) 278–285. [DOI] [PubMed] [Google Scholar]
- 181.Valerde C, Pastrana LS, Ruiz JA, Solis H, Jurado JL, Sordo CM, Fernandez-Guardiola A and Maisterrena JA, Neuroendocrine and electroencephalographic sleep changes due to acute amphetamine ingestion in human beings, Neuroendocrinology, 22 (1976) 57–71. [DOI] [PubMed] [Google Scholar]
- 182.Van Hulzen ZJM and Coenen AML, Effects of paradoxical sleep deprivation on two-way avoidance acquisition. Physiol. Behav, 29 (1982) 581–587. [DOI] [PubMed] [Google Scholar]
- 183.Van Twyver H and Allison T, A polygraphic and behavioral study of sleep in the pigeon (Columbia livia), Exp. Neurol, 35 (1972) 138–153. [DOI] [PubMed] [Google Scholar]
- 184.Velly J, Cardo B and Velly L, Delayed up-regulation of alpha-adrenoceptor populations in particular regions of the nucleus locus coeruleus, Neuroscience, 18 (1986) 321–328. [DOI] [PubMed] [Google Scholar]
- 185.Vertes RP, Brainstem gigantocellular neurons: patterns of activity during behavior and sleep in the freely moving rat, J. Neurophysiol, 42 (1979) 214–228. [DOI] [PubMed] [Google Scholar]
- 186.Vertes RP, A life-sustaining function for REM sleep: a theory, Neurosci. Biobehav. Rev, 10 (1986) 371–376. [DOI] [PubMed] [Google Scholar]
- 187.Villablanca J, Behavioral and polygraphic study of ‘sleep’ and ‘wakefulness’ in chronic decerebrate cats, Electroencephalogr. Clin. Neurophysiol, 21 (1966) 562–577. [DOI] [PubMed] [Google Scholar]
- 188.Vogel G, Studies in the psychophysiology of dreams. III. The dream of narcolepsy, Arch. Gen. Psychiat, 3 (1960) 421. [DOI] [PubMed] [Google Scholar]
- 189.Vogel GW, A review of REM sleep deprivation, Arch. Gen. Psychiat, 32 (1975) 749–761. [DOI] [PubMed] [Google Scholar]
- 190.Vogel GW, Minter K and Woolwine B, Effects of chronically administered antidepressant drugs on animal behavior, Physiol. Behav, 36 (1986) 659–666. [DOI] [PubMed] [Google Scholar]
- 191.Vogel GW, Thurmond A, Gibbons P, Sloan K, Boyd M and Walker M, REM sleep reduction effects on depression syndromes, Arch. Gen. Psychiat, 32 (1975) 765–777. [DOI] [PubMed] [Google Scholar]
- 192.Walker JM and Berger RJ, Sleep in the domestic pigeon (Columbia livia), Behav. Biol, 7 (1972) 195–203. [DOI] [PubMed] [Google Scholar]
- 193.Williams JT, Egan TM and North RA, Enkephalin opens potassium channels on mammalian central neurons, Nature (Lond.), 299 (1982) 1982. [DOI] [PubMed] [Google Scholar]
- 194.Winson J, Brain and Psyche, Anchor Press/Doubleday, New York, 1985. [Google Scholar]
- 195.Wirz-Justice A, Kafka MS, Naber D and Wehr TA, Circadian rhythms in rat brain alpha- and beta-adrenergic receptors are modified by chronic imipramine, Life sci, 27 (1980) 341–347. [DOI] [PubMed] [Google Scholar]
- 196.Wirz-Justice A, Tobler I, Kafka MS, Naber D, Marangos PJ, Borbely AA and Wehr TA, Sleep deprivation: effects on circadian rhythms of tat brain neurotransmitter receptors, Psychiat. Res, 5 (1981) 67–76 [DOI] [PubMed] [Google Scholar]
- 197.Wojcik CJ and Radulovacki M, Selective increase in brain dopamine metabolism during REM sleep rebound in the rat, Physiol. Behav, 27 (1981) 305–312. [DOI] [PubMed] [Google Scholar]
- 198.Yamaguchi S and Rogawski MA, Spike firing of single locus coeruleus neurons during experimental epileptic seizures in the rat, Soc. Neurosci Abstr, 13 (1987) 912. [Google Scholar]
- 199.Zepelin H and Rechtschaffen A, Mammalian sleep, longevity and energy metabolism, Brain Behav. Evol, 10 (1974) 425–470. [DOI] [PubMed] [Google Scholar]