

Abstract
A variety of aminated bipyridines and bipyridine sultams are prepared by intramolecular radical [1,5]-ipso and [1,6]-ortho substitutions, using a sulfonamide as a linker to connect the pyridyl radical to the pyridine under attack. For the cases studied, different regiochemistries are observed depending on the initial position of the sulfonamide linker.
Results show that intramolecular radical [1,5]-ipso and [1,6]-ortho substitutions offer a method for the preparation of aminobipyridines or bipyridine sultams.
Introduction
Intramolecular aryl and heteroaryl radical addition to arenes represents a valuable and straightforward pathway to access biaryls and heterocyclic aryl derivatives. However, although this radical process has been widely studied in these areas,1a–h the preparation of an important class of bisheteroaryls, namely bipyridines, has received little attention.
Bipyridines are important scaffolds in many natural alkaloids and synthetic derivatives with a wide range of pharmacological activities (Fig. 1).2a–g In addition, they are widely used as ligands in organometallic chemistry and are important cores in novel polymeric electro-transporting materials and organic solar cells, amongst others.3a–d Moreover, although considerable attention has been devoted to the synthesis, study and characterization of dibenzosultams,4a–j no references have been found for the corresponding bipyridine derivatives.
Fig. 1. Significant bioactive bipyridines compounds.

As part of a research program devoted to studying the bis(hetero)arylation process, via a radical pathway, in the absence of transition metals, we have previously described a new preparation of the bisheterobiaryl sultam 1, which contains a β-carboline core. The behavior of the starting pyridine sulfonamide 2 was studied under different conditions and only the bisheteroaryl sultam 1 was detected under all conditions.5
In order to study this cyclization in other related and simpler systems, and to shed some light on this finding, herein we report our studies of radical intramolecular bisheteroarylations in the presence of tris(trimethylsilyl)silane and azobisisobutyronitrile (TTMSS/AIBN), using sulfonamide 3, which is easy to prepare, as a linker to connect the pyridyl radical to the pyridine under attack, to give bipyridines 4 and/or bipyridine sultams 5, depending on the initial substitution pattern (Scheme 1).
Scheme 1. Background and objectives of this work.

Results and discussion
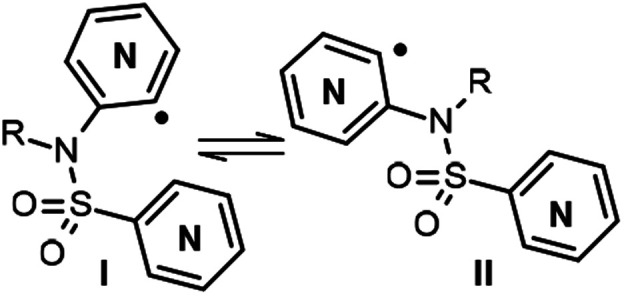
Our work started with the preparation of sulfonamides 3, as starting material for the radical cyclization process. According to Motherwell,1d,6a,b Togo7 and others,8 and taking into consideration our previous work,9 it has been shown that the presence of the free NH can negatively affect the radical arylation reaction by facilitating reduction of the radical generated. In addition, the corresponding group in an N-alkylsulfonamide increases the stiffness of the sulfonamide bond with respect to a free NH, thus meaning that the cis conformation (IversusII, Fig. 2) turns out to be the most suitable for radical cyclization as the radical and the heteroaryl are closest to each other in space. In contrast, although a methyl group is most commonly used for this purpose, it is difficult to remove subsequently, therefore in addition to carrying out our experiments with a methyl group as substituent, we also used MOM (methoxymethyl), which is suitable for protecting sulfonamides but not amines.10 Initial experiments were conducted with pyridine 2-sulfonyl chloride (6b)11 (X = CH, Y = N, Scheme 2) and 2-bromo-3-aminopyridine (7a; Z = N, W = CH, Scheme 2), in refluxing Et3N/CH2Cl2, although only a 34% yield of the desired compound 8c was obtained (Method A).
Fig. 2. cis and trans conformations for the sulfonamides radicals.
Scheme 2. Synthesis of 8a–c.

Even worse results were obtained with the same starting products when using NaH as the base in refluxing MeCN or DMF (Method B).
The best results for the formation of sulfonamides 8a–c, which contain a 2- or 3-aminopyridine core and a sulfonic acid residue formed from the chlorides of 3-12 or 2-sulfonylpyridine 6, were obtained by addition of 2 equiv. of pyridine sulfonyl chloride to a solution of 1 equiv. of amine in pyridine and stirring the mixture at 80 °C for 5 h (Method C).13
However, when the same method was applied for the formation of sulfonamide 8d, using pyridine 2-sulfonyl chloride 6b and the 2-aminopyridine 7b as starting products, the desired product was not obtained and the main product was the double sulfonylation derivative 9 (Scheme 3). Product 9 is likely to be obtained as a consequence of the acidity of the NH of the sulfonamide in product 8d. Thus, once 8d has formed, and under the basic conditions of the reaction, the NH group deprotonates to give the conjugated base of acid III with additional resonance forms IV and V, in which the negative charge is delocalized onto the sulfone group and the endocyclic nitrogen, respectively (Scheme 4).
Scheme 3. Reaction of pyridine 2-sulfonyl chloride (6b) in the presence of 3-bromo-2-aminopyridine (7b).

Scheme 4. Proposed in situ deprotonation of pyridine sulfonamide 8d to give the double sulfonylation product 9.

As this conjugated base is acylated again, the only detectable product is 9 (Scheme 3). In view of this finding, and in order to avoid formation of the double sulfonylation product 9, the reaction between pyridine 2-sulfonyl chloride (6b) and the corresponding N-methyl-3-bromo-2-aminopyridines 10a,b (Scheme 5), prepared by methods previously described in the literature,14a,b was considered as an alternative for the synthesis of pyridines 3d and 3e. In addition 3ba was obtained from the methylated aminopyridine 10a. Again, the sulfonylation reaction was carried out with 10a and 10b using Method C, for all cases, giving the corresponding sulfonamides 3ba, 3d and 3e in good yields (Scheme 6). N-Alkylation of sulfonamides 8a–c was carried out by addition of 1.1 equiv. of sodium hydride to a solution of 1 equiv. of sulfonamides in DMF and subsequent treatment with 1.1 equiv. of methyl iodide or methoxymethyl chloride. Scheme 6 shows our results in this process for N-alkyl sulfonamides 3aa, 3ab, 3bb, 3ca and 3cb, as well as the result for 3ba, 3d and 3e.
Scheme 5. Methylation of aminopyridines 7b and 7c.

Scheme 6. Results in the preparation of N-alkyl sulfonamides 3.

At this point, we turned our attention to the intramolecular radical bisheteroarylation process, using 3 as starting material and under our optimized conditions, in the presence of tris(trimethylsilyl)silane and azobisisobutyronitrile (TTMSS/AIBN) under thermal conditions. Previous studies by Motherwell1d and Chatgilialoglu15 suggest that different reaction pathways are possible, depending on the structure of the starting pyridine 3, with the main ones being shown in Scheme 7. Thus, 3ca and 3cb (X = CH, Y = N and Z = N, A, W = CH) can undergo a non-radical SNAr process to give the cyclic derivative 11 due to the nucleophilicity of the pyridine nitrogen and the favourable position of bromide, which can act as a good leaving group, on the other pyridine nitrogen. However, in accordance with previous reports, products 11 were never detected. The other pathways involve the initial formation of heteroaryl radical VI, which would evolve by [1,5]-ipso-addition to give the spirocyclic radical VII and then bipyridines 4, and/or by [1,6]-ortho-addition, in different positions, depending on the starting compound 3, to give radical VIII and, subsequently, 5.
Scheme 7. Some alternative reaction pathways for the bromo sulfonamides 3.

For the starting material 3 with the sulfonamide group in position C3 (X = N, Y = CH; compounds 3aa-3bb), in all cases the initial radical VI undergoes a [1,6]-ortho-attack to yield compounds 5, whereas the alternative [1,5]-ipso-attack was only detected in the case of sulfonamide 3aa, with product 4aa being obtained as a by-product of the reaction (<20%; Scheme 8). The experimental evidence points to a trend in the reactivity depending on whether the Br substituent is positioned on either C2′ (3aa and 3ab) or C3′ (3ba and 3bb) of the attacking pyridine.16
Scheme 8. Results in the preparation of compound 5aa-bb.

Thus, those substrates with a Br in the C2′ position of the pyridine (3aa and 3ab) yield the C4-substituted derivatives as the main products (5aa and 5ab, respectively, Scheme 8) due to the repulsion generated by the proximity of the radical to the non-binding pairs of both nitrogens in the adduct intermediate. In contrast, for those with a Br in position C3′of the pyridine (3ba and 3bb), the main product results from the attack at C2, which yields products 5ba and 5bb, respectively.
Conversely, for compounds 3 with the sulfonamide group placed in position C2 (Y = N, X = CH; compounds 3ca-b, 3d and 3e) and a Br substituent in positions C2′ and C3′ (Scheme 9), the initial radical VI undergoes [1,5]-ipso-attack to yield the spirocyclic radical VII and, subsequently, compounds 4. For the alternative [1,6]-ortho-attack, we were only able to detect traces of compounds 5 (if at all). The experimental data indicate that, irrespective of the position of the pyridine nitrogen in N1′, the reaction evolves towards the formation of product 4 with an intramolecular hydrogen bond in all cases, with the primary or secondary amine acting as H-donor and the pyridine N1 as H-acceptor.
Scheme 9. Results in the preparation of compound 4ca-b, 4d and 4e.

In order to check other referable cases, we tried to prepare sulfonamides 3 from the commercially available products 4-bromo-3-aminopyridine 7c and 3-bromo-4-aminopyridine 7d, but our attempts were unsuccessful. Notably, for compound 3cb, which contains a MOM group in the starting material, a concomitant deprotection of MOM group was observed during work-up, thus giving easy access to the unsubstituted amine.17 These results are summarized in Scheme 9.
The identity of these compounds was elucidated by NMR experiments and the structure of one of them (compound 5ba) unequivocally confirmed by X-ray crystallographic analysis (Fig. 3 and ESI†).
Fig. 3. ORTEP diagram for compound 5ba. Thermal ellipsoids are drawn at the 50% probability level.

Conclusions
In summary, our results show that intramolecular radical [1,5]-ipso and [1,6]-ortho substitutions offer a method for the preparation of aminobipyridines or bipyridine sultams when a sulfonamide linker connects the attacking pyridyl radical to the pyridine under attack. For derivatives with a sulfonamide group at the C3-position of the starting pyridine, the [1,6]-ortho substitution product was identified in all cases. For substrates with the sulfonamide group at the C2-position, mainly aminobipyridines were obtained, being the leading event the formation of an intramolecular hydrogen bond between the primary or secondary amine as H-donor, and the pyridine N1 as H-acceptor. These sulfonamides with a MOM group in the starting material underwent deprotection of the MOM group during work-up, thus allowing easy access to the unsubstituted amine.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from FEDER and the Comunidad de Madrid (CAM, project B2017/BMD-3688 MULTI-TARGET&VIEW-CM FEDER FUNDS), the Ministerio de Economia, Industria y Competitividad (project CTQ2017-85203-P), Instituto de Salud Carlos III (FEDER funds, ISCIII RETIC REDINREN RD16/0009/0015 FEDER FUNDS) and the Universidad de Alcalá (CCG2017/EXP-021, CCG2018/EXP-008 and CCG2018/EXP-051). J. R. also thanks the Universidad de Alcalá for a predoctoral grant.
Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C NMR spectra for new compounds and X-ray crystallographic analysis for 5ba (PDF). CCDC 1967009. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02026e
Notes and references
- (a) Zhou Y. Deng S. Mai S. Song Q. Org. Lett. 2018;20:6161. doi: 10.1021/acs.orglett.8b02629. [DOI] [PubMed] [Google Scholar]; (b) Chatgilialoglu C. Ferreri C. Landais Y. Timokhin V. I. Chem. Rev. 2018;118:6516. doi: 10.1021/acs.chemrev.8b00109. [DOI] [PubMed] [Google Scholar]; (c) Murakami K. Yamada S. Kaneda T. Itami K. Chem. Rev. 2017;117:9302. doi: 10.1021/acs.chemrev.7b00021. [DOI] [PubMed] [Google Scholar]; (d) Ujjainwalla F. da Mata M. L. E. N. Pennell A. M. K. Escolano C. Motherwell W. B. Vazquez S. Tetrahedron. 2015;71:6701. doi: 10.1016/j.tet.2015.07.048. [DOI] [Google Scholar]; (e) Kawamoto T. Sato A. Ryu I. Org. Lett. 2014;16:2111. doi: 10.1021/ol500614q. [DOI] [PubMed] [Google Scholar]; (f) Bonin H. Sauthier M. Felpin F.-X. Adv. Synth. Catal. 2014;356:645. doi: 10.1002/adsc.201300865. [DOI] [Google Scholar]; (g) Dewanji A. Murarka S. Curran D. P. Studer A. Org. Lett. 2013;15:6102. doi: 10.1021/ol402995e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Vaillard S. and Studer A.Radical arylations. Ed. C. Chatgilialoglu and A. Studer; Encyclopedia of Radicals in Chemistry, Biology and Materials 2012, vol. 2, p. 1059 [Google Scholar]
- (a) Werner F.-M. Covenas R. Curr. Pharm. Des. 2019;25:396. doi: 10.2174/1381612825666190319121505. [DOI] [PubMed] [Google Scholar]; (b) Chen D. Zhao Q. Liu W. J. Ind. Microbiol. Biotechnol. 2019;46:459. doi: 10.1007/s10295-018-2092-7. [DOI] [PubMed] [Google Scholar]; (c) Chen M. Zhang Y. Du Y. Zhao Q. Zhang Q. Wu J. Liu W. Org. Biomol. Chem. 2017;15:5472. doi: 10.1039/C7OB01284E. [DOI] [PubMed] [Google Scholar]; (d) Garcia-Barrantes P. M. Harp J. R. Lindsley C. W. Tetrahedron Lett. 2016;57:2194. doi: 10.1016/j.tetlet.2016.04.019. [DOI] [Google Scholar]; (e) Zhu Y. Picard M.-E. Zhang Q. Barma J. Despres X. M. Mei X. Zhang L. Duvignaud J.-B. Couture M. Zhu W. Shi R. Zhang C. Chem. Sci. 2016;7:4868. doi: 10.1039/c6sc00771f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fu P. Liu P. Li X. Wang Y. Wang S. Hong K. Zhu W. Org. Lett. 2011;13:5948. doi: 10.1021/ol202245s. [DOI] [PubMed] [Google Scholar]; (g) Croom K. F. Siddiqui M. A. A. Drugs. 2009;69:1513. doi: 10.2165/00003495-200969110-00008. [DOI] [PubMed] [Google Scholar]
- (a) Samiee S. Taghvaeian S. Appl. Organomet. Chem. 2019;33:1. doi: 10.1002/aoc.4626. [DOI] [Google Scholar]; (b) Long X. Gao Y. Tian H. Dou C. Yan D. Geng Y. Liu J. Wang L. Chem. Commun. 2017;53:1649. doi: 10.1039/C6CC09684K. [DOI] [PubMed] [Google Scholar]; (c) Zhang Z. Ding Z. Long X. Dou C. Liu J. Wang L. J. Mater. Chem. C. 2017;5:6812. doi: 10.1039/C7TC01996C. [DOI] [Google Scholar]; (d) Shabunina O. V. Kapustina D. Y. Krinochkin A. P. Kim G. A. Kopchuk D. S. Zyryanov G. V. Li F. Chupakhin O. N. Mendeleev Commun. 2017;27:602. doi: 10.1016/j.mencom.2017.11.021. [DOI] [Google Scholar]
- (a) Feng S. Li S. Li J. Wei J. Org. Chem. Front. 2019;6:517. doi: 10.1039/C8QO01311J. [DOI] [Google Scholar]; (b) Ma Y.-N. Guo C.-Y. Zhao Q. Zhang J. Chen X. Green Chem. 2018;20:2953. doi: 10.1039/C8GC01057A. [DOI] [Google Scholar]; (c) Nacsa E. D. Lambert T. H. Org. Chem. Front. 2018;5:64. doi: 10.1039/C7QO00731K. [DOI] [Google Scholar]; (d) Chattopadhyay S. K. Synth. Commun. 2018;48:3033. doi: 10.1080/00397911.2018.1527355. [DOI] [Google Scholar]; (e) Maheshwar Rao B. Yadav J. S. Sridhar B. Subba Reddy B. V. Org. Biomol. Chem. 2018;16:5163. doi: 10.1039/C8OB00918J. [DOI] [PubMed] [Google Scholar]; (f) Debnath S. Mondal S. Eur. J. Org. Chem. 2018:933. doi: 10.1002/ejoc.201701491. [DOI] [Google Scholar]; (g) Virk T. S. Ilawe N. V. Zhang G. Yu C. P. Wong B. M. Chan J. M. W. ACS Omega. 2016;1:1336. doi: 10.1021/acsomega.6b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Guerra W. D. Rossi R. A. Pierini A. P. Barolo S. M. J. Org. Chem. 2016;81:4965. doi: 10.1021/acs.joc.6b00330. [DOI] [PubMed] [Google Scholar]; (i) Han Y.-Y. Wang H. Yu S. Org. Chem. Front. 2016;3:953. doi: 10.1039/C6QO00116E. [DOI] [Google Scholar]; (j) Majumdar K. C. Mondal S. Chem. Rev. 2011;111:7749. doi: 10.1021/cr1003776. [DOI] [PubMed] [Google Scholar]
- Recio J. Perez-Redondo A. Alvarez-Builla J. Burgos C. Org. Chem. Front. 2019;6:3300. doi: 10.1039/C9QO00944B. [DOI] [Google Scholar]
- (a) da Mata M. L. E. N. Motherwell W. B. Ujjainwalla F. Tetrahedron Lett. 1997;38:141. doi: 10.1016/S0040-4039(96)02237-X. [DOI] [Google Scholar]; (b) da Mata M. L. E. N. Motherwell W. B. Ujjainwalla F. Tetrahedron Lett. 1997;38:137. doi: 10.1016/S0040-4039(96)02236-8. [DOI] [Google Scholar]
- Ryokawa A. Togo H. Tetrahedron. 2001;57:5915. doi: 10.1016/S0040-4020(01)00560-9. [DOI] [Google Scholar]
- Yamasaki R. Tanatani A. Azumaya I. Saito S. Yamaguchi K. Kagechika H. Org. Lett. 2003;5:1265. doi: 10.1021/ol034344g. [DOI] [PubMed] [Google Scholar]
- (a) Camacho-Artacho M. Abet V. Frutos L. M. Gago F. Alvarez-Builla J. Burgos C. J. Org. Chem. 2011;76:1452. doi: 10.1021/jo102122h. [DOI] [PubMed] [Google Scholar]; (b) Sanchez A. Nunez A. Alvarez-Builla J. Burgos C. Tetrahedron. 2004;60:11843. doi: 10.1016/j.tet.2004.09.099. [DOI] [Google Scholar]
- Beltran A. Alvarez E. Diaz-Requejo M. M. Perez P. J. Adv. Synth. Catal. 2015;357:2821. doi: 10.1002/adsc.201500588. [DOI] [Google Scholar]
- Dupont-Passelaigue E., Le Roy I. and Pignier C., PCT. Int. Appl., WO 2012069503 A1, 2012
- Birch M., Sibley G. E. M., Law D. and Oliver J. D., PCT. Int. Appl., WO 2009144473 A1, 2009
- Caldwell W. T. Tyson F. T. Lauer L. J. Am. Chem. Soc. 1944;66:1479. doi: 10.1021/ja01237a018. [DOI] [Google Scholar]
- (a) Blanchard S. Rodriguez I. Kuehm-Caubere C. Renard P. Pfeiffer B. Guillaumet G. Caubere P. Tetrahedron. 2002;58:3513. doi: 10.1016/S0040-4020(02)00310-1. [DOI] [Google Scholar]; (b) Abbiati G. Beccalli E. M. Broggini G. Paladino G. Rossi E. Synthesis. 2005:2881. [Google Scholar]
- Chatgilialoglu C. Ferreri C. Landais Y. Timokhin V. I. Chem. Rev. 2018;118:6516. doi: 10.1021/acs.chemrev.8b00109. [DOI] [PubMed] [Google Scholar]
- Filace F. Sanchez-Murcia P. A. Sucunza D. Perez-Redondo A. Alvarez-Builla J. Gago F. Burgos C. Eur. J. Org. Chem. 2016:1891. doi: 10.1002/ejoc.201600170. [DOI] [Google Scholar]
- Kaczmarek L. Bull. Pol. Acad. Sci., Chem. 1985;33:401. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.