

Abstract
Intraneuronal accumulation of aggregated α-synuclein is a pathological hallmark of Parkinson’s disease. Therefore, mechanisms capable of promoting α-synuclein deposition bear important pathogenetic implications. Mutations of the glucocerebrosidase 1 (GBA) gene represent a prevalent Parkinson’s disease risk factor. They are associated with loss of activity of a key enzyme involved in lipid metabolism, glucocerebrosidase, supporting a mechanistic relationship between abnormal α-synuclein–lipid interactions and the development of Parkinson pathology.
In this study, the lipid membrane composition of fibroblasts isolated from control subjects, patients with idiopathic Parkinson’s disease and Parkinson's disease patients carrying the L444P GBA mutation (PD-GBA) was assayed using shotgun lipidomics.
The lipid profile of PD-GBA fibroblasts differed significantly from that of control and idiopathic Parkinson’s disease cells. It was characterized by an overall increase in sphingolipid levels. It also featured a significant increase in the proportion of ceramide, sphingomyelin and hexosylceramide molecules with shorter chain length and a decrease in the percentage of longer-chain sphingolipids. The extent of this shift was correlated to the degree of reduction of fibroblast glucocerebrosidase activity. Lipid extracts from control and PD-GBA fibroblasts were added to recombinant α-synuclein solutions. The kinetics of α-synuclein aggregation were significantly accelerated after addition of PD-GBA extracts as compared to control samples. Amyloid fibrils collected at the end of these incubations contained lipids, indicating α-synuclein–lipid co-assembly. Lipids extracted from α-synuclein fibrils were also analysed by shotgun lipidomics. Data revealed that the lipid content of these fibrils was significantly enriched by shorter-chain sphingolipids. In a final set of experiments, control and PD-GBA fibroblasts were incubated in the presence of the small molecule chaperone ambroxol. This treatment restored glucocerebrosidase activity and sphingolipid levels and composition of PD-GBA cells. It also reversed the pro-aggregation effect that lipid extracts from PD-GBA fibroblasts had on α-synuclein.
Taken together, the findings of this study indicate that the L444P GBA mutation and consequent enzymatic loss are associated with a distinctly altered membrane lipid profile that provides a biological fingerprint of this mutation in Parkinson fibroblasts. This altered lipid profile could also be an indicator of increased risk for α-synuclein aggregate pathology.
Keywords: fibroblasts, GBA, α-synuclein, lipidomics, Parkinson’s disease
Galvagnion et al. report that fibroblast lipid extracts from patients with Parkinson’s disease carrying the L444P GBA mutation feature an increased ratio of short chain: long chain sphingolipids. This lipid profile promotes faster α-synuclein aggregation. Ambroxol treatment reverses both sphingolipid and protein aggregation changes.
Introduction
Homozygous mutations of the glucocerebrosidase 1 (GBA) gene have long been associated with autosomal recessive Gaucher’s disease. The same GBA mutations, such as L444P and N370S, also represent the commonest known genetic risk factor for Parkinson’s disease.1,2 Parkinson's disease risk is significantly higher in heterozygous mutation carriers, with variable odds ratios that depend on the specific GBA mutation and racial characteristics of the population tested.3 Disease penetrance also varies with age and has been estimated to range from 10% to 30% in mutation carriers aged 50–80 years old.4 Clinical features are indistinguishable between Parkinson's disease patients carrying GBA mutations (PD-GBA) and patients affected by idiopathic Parkinson’s disease, as underscored by investigations showing that a significant proportion of patients with idiopathic Parkinson’s disease (∼5–10%), once specifically tested, were in fact carriers of GBA mutations.1,5-7 Genotype–phenotype correlation studies have confirmed the similarities in clinical and pathological manifestations between PD-GBA and idiopathic Parkinson’s disease. They have also revealed that PD-GBA is characterized by a slightly earlier disease onset and higher prevalence of cognitive impairment and other non-motor symptoms.5,8,9 Different GBA mutations may be associated with different clinical phenotypes, with more severe parkinsonian features and a more aggressive disease course affecting PD-GBA patients carrying specific ‘severe’ variants, such as the p.L444P mutation.10,11
GBA encodes the lysosomal enzyme glucocerebrosidase (GCase), and GBA mutations cause a decrease in GCase activity and GCase-catalysed hydrolysis of glucosylceramide (GluCer) to glucose and ceramide (Cer). Loss of GCase activity has been reported in the brain and blood of PD-GBA patients as well as in patient-derived cells, such as skin fibroblasts and dopaminergic neurons generated from induced pluripotent stem cells (iPSCs).12–18 Decreased GCase activity would be expected to result in an accumulation of GCase substrates as seen, for example, in iPSCs from PD-GBA patients containing higher levels of GluCer and glucosylsphingosine (GluSph).13,15 It is noteworthy, however, that due to the close interrelationship between pathways of lipid metabolism and the central role played by Cer in sphingolipid homeostasis, lower GCase activity is likely to have broader consequences on cell lipid composition and, in particular, sphingolipid synthesis, maintenance and breakdown (Fig. 1).19 To date, only limited information is available from lipidomic analyses of cell or tissue samples from PD-GBA patients.12,13,15 More detailed and comprehensive studies are, therefore, warranted for the identification of PD-GBA lipid signatures. These studies could also shed light upon the role that specific changes in lipid profile may play in increasing the risk for parkinsonian pathology.
Figure 1.

Simplified sphingolipid metabolic pathway.
Mechanisms contributing to Parkinson’s disease pathogenesis in carriers of GBA mutations are not fully understood. Of likely relevance, however, is evidence indicating a reciprocal relationship between GCase activity and intracellular levels and toxic properties of α-synuclein (α-syn).20,21 α-Syn is a key player in the pathogenesis of Parkinson’s disease. It is a major component of Lewy bodies and Lewy neurites, the intraneuronal inclusions pathognomonic of idiopathic Parkinson’s disease.22 Furthermore, single-point and multiplication mutations of the SNCA (α-syn) gene are causally associated with familial forms of parkinsonism.23–25 The tendency of α-syn to assemble into oligomeric and fibrillar aggregates is thought to be a gain of toxic function involved in inclusion formation and other neurodegenerative effects. Interestingly, loss of GCase activity, as induced by GBA mutations, causes intracellular accumulation of monomeric as well as aggregated α-syn.13,15,20,26–28 Increased α-syn levels are in turn capable of reducing GCase activity, giving rise to a reciprocal self-amplifying cycle of protein accumulation and enzyme inhibition.12,20,21,29 Changes in lipid composition caused by loss of GCase activity may also contribute to this chain of toxic events triggered by GBA mutations and involving α-syn. This possibility is supported by findings showing that α-syn-lipid interactions modulate protein aggregation, can promote α-syn assembly and accelerate the rate of α-syn amyloid formation.30–36
In this study, we first performed a comprehensive lipidomic analysis of membrane preparations from human fibroblasts and compared data in cells from control individuals versus cells from patients with idiopathic Parkinson’s disease without GBA mutations and PD-GBA patients. Results revealed that loss of GCase activity in PD-GBA fibroblasts was associated with a specific lipid profile. Subsequent experiments assessed the potential link between PD-GBA-related lipid changes and α-syn aggregation. Findings provided evidence of such a relationship, showing accelerated formation of amyloid fibrils after addition of lipids extracted from PD-GBA fibroblasts to α-syn solutions. In a final set of experiments, the relationship between decreased GCase activity, altered lipid profile and enhanced lipid-induced α-syn aggregation was further evaluated in fibroblast cultures treated with ambroxol, a GCase pharmacological chaperone. Treatment with ambroxol was found to reverse all cellular abnormalities induced by the mutant enzyme in PD-GBA fibroblasts.
Materials and methods
Fibroblasts
Collection and use of human tissue were done in agreement with the principles of the Declaration of Helsinki. Fibroblasts were obtained from skin biopsies that were performed at the IRCCS Mondino Foundation under a research protocol previously approved by the institutional Ethic Committee. Informed consent was obtained from all subjects who underwent the procedure. Fibroblast cultures were grown in RPMI-1640 Medium (Sigma Aldrich) with 10% serum (Sigma Aldrich), 2 mM L-glutamine (Sigma Aldrich), 100 μg/ml streptomycin and 100 units/ml penicillin. Analyses were carried out at low culture passages, and disease and control cultures were matched for passage number. In some experiments, fibroblast cultures were treated with ambroxol. Ambroxol hydrochloride (Sigma Aldrich) was dissolved in dimethylsulphoxide (DMSO; 20 mM stock concentration) and diluted in cell culture medium. Cells were treated with 60 μM (final concentration) ambroxol or DMSO for 15 days and, during this treatment time, culture medium was changed every third day. Fresh medium also contained ambroxol or DMSO.
GCase activity assay
Fibroblasts were trypsinized and resuspended in lysis buffer (0.1 M sodium citrate, 0.1% Triton X-100, 6.7 mM sodium taurocholate). The resulting cell suspensions were lysed by sonication at 4°C, and lysates were incubated on ice for 20 min before being centrifuged at 15 000g for 15 min. The supernatants were collected into new tubes and their protein content determined using bicinchoninic acid assay. Lysates were then diluted to a protein concentration of 0.1 μg/μl with assay buffer (0.1 M sodium citrate, 0.1% Triton X-100, 6.5 mM sodium taurocholate, 2.5 mM 4-methylumbelliferyl β-D-glucopyranoside) and incubated in non-binding plates (Corning 3881) while shaking (300 rpm) at 37°C. Fluorescence was measured using a FLUOstar Omega plate reader (BMG) with excitation/emission filters of 355–20/460 nm. The fluorescence of 25 μM 4-methylumbelliferone was measured under the same conditions and used to convert fluorescence units into μM of 4-methylumbelliferone. GCase activity was calculated using the following equation:
where is the change in fluorescence intensity per hour (fluorescence unit/h), standard is the fluorescence (fluorescence unit/μM) of 4-methylumbelliferone and [protein] is the total protein concentration in the sample (0.1 mg/ml).
Western blot analysis
Fibroblasts were trypsinized and resuspended in lysis buffer with protease inhibitors. Cell lysates were incubated on ice with shaking and then centrifuged at 16 000g for 15 min. The supernatants were transferred to fresh tubes, and lysates containing 30 µg of protein were electrophoresed on a NuPageTM 4%–12% Bis–Tris Protein gel (Thermo Fisher Scientific). Proteins were transferred to a PVDF membrane (Amersham), blocked in 5% bovine serum albumin and treated with primary and secondary antibodies. Antibody binding was detected using an ECL chemiluminescence kit. The following antibodies were used: glucocerebrosidase (ab55080 Abcam, dilution 1:1000) and β-actin (ab8227, Abcam, dilution 1:7500).
Lipid extraction for mass spectrometry lipidomics
Mass spectrometry-based lipid analysis was performed by Lipotype GmbH as described.37 Lipids were extracted using a two-step chloroform/methanol procedure.38 Samples were spiked with internal lipid standard mixture containing: cardiolipin (CL) 16:1/15:0/15:0/15:0, Cer 18:1; 2/17:0, diacylglycerol (DAG) 17:0/17:0, hexosylceramide (HexCer) 18:1; 2/12:0, lyso-phosphatidate (LPA) 17:0, lyso-phosphatidylcholine (LPC) 12:0, lyso-phosphatidylethanolamine (LPE) 17:1, lyso-phosphatidylglycerol (LPG) 17:1, lyso-phosphatidylinositol (LPI) 17:1, lyso-phosphatidylserine (LPS) 17:1, phosphatidate (PA) 17:0/17:0, phosphatidylcholine (PC) 17:0/17:0, phosphatidylethanolamine (PE) 17:0/17:0, phosphatidylglycerol (PG) 17:0/17:0, phosphatidylinositol (PI) 16:0/16:0, phosphatidylserine (PS) 17:0/17:0, cholesterol ester (CE) 20:0, sphingomyelin (SM) 18:1; 2/12:0; 0, triacylglycerol (TAG) 17:0/17:0/17:0 and cholesterol (Chol) D6. After extraction, the organic phase was transferred to an infusion plate and dried in a speed vacuum concentrator. First-step dry extract was resuspended in 7.5 mM ammonium acetate in chloroform/methanol/propanol (1:2:4, v:v:v) and second-step dry extract in 33% ethanol solution of methylamine in chloroform/methanol (0.003:5:1; v:v:v). All liquid handling steps were performed using Hamilton Robotics STARlet robotic platform with the Anti Droplet Control feature for organic solvents pipetting.
Mass spectrometry data acquisition
Samples were analysed by direct infusion on a QExactive mass spectrometer (Thermo Scientific) equipped with a TriVersa NanoMate ion source (Advion Biosciences). Samples were analysed in both positive and negative ion modes with a resolution of Rm/z = 200 = 280 000 for mass spectrometry (MS) and Rm/z = 200 = 17 500 for tandem mass spectrometry (MSMS) experiments, in a single acquisition. MSMS was triggered by an inclusion list encompassing corresponding MS mass ranges scanned in 1 Da increments.39 Both MS and MSMS data were combined to monitor CE, DAG and TAG ions as ammonium adducts; PC, PC O–, as acetate adducts; and CL, PA, PE, PE O–, PG, PI and PS as deprotonated anions. MS only was used to monitor LPA, LPE, LPE O–, LPI and LPS as deprotonated anions; Cer, HexCer, SM, LPC and LPC O– as acetate adducts and cholesterol as ammonium adduct of an acetylated derivative.40 Of note, separate measurements of GluCer and GluSph were not carried out as part of this study. Instead, measurements of HexCer included combined assessments of both GluCer and galactosylceramide.
Data analysis and post-processing
Data were analysed with in-house developed lipid identification software based on LipidXplorer.41,42 Data post-processing and normalization were performed using an in-house developed data management system. Only lipid identifications with a signal-to-noise ratio >5 and a signal intensity 5-fold higher than in corresponding blank samples were considered for further data analysis.
Aggregation kinetics measurements
Recombinant wild-type α-syn was produced as previously described.33 For experiments assessing the effect of lipid extracts in untreated fibroblast cultures, lipids were extracted from 106 cells resuspended in 200 μl PBS using 1 ml chloroform:methanol mixture (10:1, v:v). The organic phase was then evaporated and the lipids incubated in the presence of 50 μM α-syn and 50 μM thioflavin-T (ThT) in MES buffer (10 mM MES, pH 5.0, 0.01% sodium azide) at 37°C in high binding plates (Corning 3601) under quiescent conditions. For experiments assessing the effect of lipid extracts in ambroxol-treated fibroblasts, lipids were extracted from 300 000 cells resuspended in 200 μl PBS using 1 ml chloroform:methanol mixture (10:1, v:v). The organic phase was then evaporated and the lipids incubated in the presence of 50 μM α-syn and 50 μM ThT in acetate buffer (10 mM sodium acetate, pH 5.0, 0.01% sodium azide) at 37°C in high binding plates (Corning 3601) under quiescent conditions. The fluorescence intensity was measured on a FLUOstar Omega plate reader (BMG) with excitation/emission filters of 448–10/482–10 nm.
Statistics
Data are shown as individual values and mean ± SEM (standard error of the mean). Unpaired t-test (two-tailed P-value) was used for comparisons of means between two groups. Statistical analysis was performed using GraphPad Prism v8, GraphPad Software, CA, USA. Correlations were analysed with Pearson correlation analysis. Statistical significance was set at P < 0.05.
Data availability
Data are available from the corresponding author upon reasonable request.
Results
GBA mutation and GCase activity in human fibroblasts
Fibroblasts were generated from skin biopsies of four control subjects with no history of neurological disorders (two males and two females), four idiopathic Parkinson’s disease patients (two males and two females) and five PD-GBA patients carrying a heterozygous L444P GBA mutation (three males and two females). Average age of onset of clinical manifestations was 57 ± 3.5 and 47 ± 4.3 years in the idiopathic Parkinson’s disease and PD-GBA groups, respectively, consistent with an earlier disease onset of GBA-associated Parkinson’s disease.1 GCase protein levels, as assessed by western blot analysis of lysed cells, were unchanged between the control and the two patient groups (Fig. 2A and B). GCase enzyme activity was also similar in fibroblasts from control subjects and idiopathic Parkinson’s disease patients, but was significantly decreased by ∼25% in fibroblasts from PD-GBA patients (Fig. 2C).
Figure 2.

GCase protein levels and activity in fibroblasts. Measurements were made in fibroblasts from control subjects and idiopathic Parkinson’s disease (iPD) and PD-GBA patients. (A) Western blots showing immunoreactivity for human GCase and β-actin (one replicate per cell line). (B) Semi-quantitative analysis of band intensities of the blots shown in A. (C) Measurements of GCase activity in fibroblasts from the control (n = 4), idiopathic Parkinson’s disease (n = 4) and PD-GBA (n = 5) groups (three replicates per cell line). Data are shown as individual values and mean ± SEM. Unpaired t-test was performed to compare data in control subjects versus values in the idiopathic Parkinson’s disease or PD-GBA groups. Control versus PD-GBA: P = 0.0258; F(4,3) = 3.734. *P < 0.05.
Lipidome of human fibroblasts
Mass spectrometry after direct infusion (‘shotgun lipidomics’) was used for the first time to perform a comprehensive quantitative and qualitative analysis of the lipid composition of fibroblasts from control subjects and Parkinson’s disease patients with and without a GBA mutation. The lipidome of control cells mainly consisted of phospholipids (74.5% of total lipids), cholesterol (19%), sphingolipid (4.2%) and glycerides (1.6%; Fig. 3A–D). A detailed quantification of phospholipid species is reported in Supplementary Fig. 1. The 4.2% of total sphingolipid was composed of 3.6%, 0.4% and 0.2% SM, Cer and HexCer, respectively (Fig. 3E–G). Fibroblast lipid composition did not differ significantly between control subjects and idiopathic Parkinson’s disease patients (Fig. 3 and Supplementary Fig. 1). Specific changes were instead observed in the lipidome of cells from PD-GBA patients. In these cells, the most prominent variation concerned the levels of total sphingolipid that were significantly increased by ∼25% (Fig. 3D). The per cent of specific sphingolipid species, namely SM, Cer and HexCer, also tended to be higher in PD-GBA fibroblasts, although these changes did not reach statistical significance (Fig. 3E–G). Levels of cholesteroland glycerides were similar to control values, whereas a small but significant 3% decrease in the percentage of total phospholipids was detected in samples from PD-GBA patients (Fig. 3A).
Figure 3.

Lipid levels in control, idiopathic Parkinson’s disease and PD-GBA fibroblasts. Levels of (A) phospholipids, (B) cholesterol, (C) diacyl glycerides and (D) total and (E–G) specific (E, SM; F, Cer; and G HexCer) sphingolipids were assayed in fibroblast lipid extracts. Measurements were made in extracts from control subjects (n = 4) and idiopathic Parkinson’s disease (iPD; n = 4) and PD-GBA (n = 5) patients (one replicate per cell line). Data are expressed as percentage of the total lipid content. Data are shown as individual values and mean ± SEM. Unpaired t-test was performed to compare data in control subjects versus values in the idiopathic Parkinson’s disease or PD-GBA groups. Control versus PD-GBA: phospholipids [P = 0.0130; F(4,3) = 7.330] and sphingolipids [P = 0.0485; F(4,3) = 4.766]. *P < 0.05.
Chemical properties of sphingolipids in human fibroblasts
Our lipidomic analysis of human fibroblasts also assessed levels of specific sphingolipid molecules and, in particular, identified and quantified SM, Cer and HexCer molecules with different hydrocarbon chain length and degree of unsaturation. Data showed that most sphingolipid (80%–90% of SM, Cer and HexCer) had either 34 (C34) or 42 (C42) hydrocarbons (Fig. 4A–C). SM with 34 hydrocarbons and a single double bond (SM 34:1; 18:1/16:0) and SM with 42 hydrocarbons and two double bonds (SM 42:2; 18:1/24:1) were the two main SM species detected in our fibroblast preparations; they accounted for approximately 55% and 15%, respectively, of the total SM levels (Fig. 4D). Other abundant sphingolipid molecules were Cer 34:1 (18:1/16:0) and Cer 42:1 (18:1/24:0), which accounted for 40% and 25% of all Cer (Fig. 4E), and HexCer 34:1 (18:1/16:0) and HexCer 42:1 (18:1/24:0), representing 20% and 35% of the total HexCer (Fig. 4F).
Figure 4.

Levels of sphingolipid molecules with different acyl chain length. Measurements of SM, Cer and HexCer molecules were made in fibroblast lipid extracts from control subjects (n = 4) and idiopathic Parkinson’s disease (PD; n = 4) and PD-GBA (n = 5) patients (one replicate per cell line). (A–C) Levels of SM, Cer and HexCer with different hydrocarbon chain lengths (C32 to C44) are shown as per cent of the respective SM, Cer and HexCer total content. Data are shown as individual values and mean ± SEM. Multiple t-test was performed to compare means between two groups, control versus idiopathic Parkinson’s disease, control versus PD-GBA and idiopathic Parkinson’s disease versus PD-GBA. Control versus PD-GBA: SM C32 (P = 0.0269), SM C34 (P = 0.0113), SM C42 (P = 0.0154), Cer C36 (P = 0.0188), Cer C44 (P = 0.0197), HexCer C34 (P = 0.0285). Idiopathic Parkinson’s disease versus PD-GBA: SM C34 (P = 0.0124), SM C42 (P = 0.0144), Cer C34 (P < 0.001). Control versus idiopathic Parkinson’s disease: Cer C36 (P < 0.001). (D) Levels of the two main SM molecules, SM 34:1 and SM 42:2, are shown as percentage of the total SM content. Control versus PD-GBA: SM 34:1 (P = 0.0034), SM 42:2 (P = 0.0024). Idiopathic Parkinson’s disease versus PD-GBA: SM 34:1 (P = 0.0258), SM 42:2 (P = 0.0282). (E) Levels of the two main Cer molecules, Cer 34:1 and Cer 42:1, are shown as percentage of the total Cer content. Control versus PD-GBA: Cer 42:1 (P = 0.0477). Idiopathic Parkinson’s disease versus PD-GBA: Cer 34:1 (P < 0.001), Cer 42:1 (P = 0.0329). (F) Levels of the two main HexCer molecules, HexCer 34:1 and HexCer 42:1, are shown as percentage of the total HexCer content. Control versus PD-GBA: HexCer 34:1 (P = 0.029). *P < 0.05, **P < 0.01, ***P < 0.001.
Potential changes in sphingolipid with shorter and longer hydrocarbon chains were then compared between control cells and fibroblasts from idiopathic Parkinson’s disease and PD-GBA patients. Levels of C34 and C42 sphingolipids were not significantly different in control versus idiopathic Parkinson’s disease-derived preparations (Fig. 4A–C). In contrast, when measurements were compared between PD-GBA versus control or idiopathic Parkinson’s disease fibroblasts, data revealed an increase in short-chain (C34) SM, Cer and HexCer and a consistent reduction of long-chain (C42) SM, Cer and HexCer (Fig. 4A–C). A comparison of levels of specific sphingolipid molecules confirmed these differences. SM 34:1, Cer 34:1 and HexCer 34:1 were all increased by 15–35% in mutation-carrying versus control and/or idiopathic Parkinson’s disease fibroblasts (Fig. 4D–F). In the same PD-GBA cells, measurements of SM 42:2, Cer 42:1 and HexCer 42:1 showed a decrease that ranged between 25% and 50% (Fig. 4D–F).
Fibroblast sphingolipid composition and GCase activity
Taken together, results obtained from our lipidomic analysis indicated that loss of GCase activity in PD-GBA fibroblasts was accompanied not only by an overall increase in sphingolipid levels but also higher levels of short-chain and lower levels of long-chain sphingolipid. In these cells, the ratios C34:C42 SM, C34:C42 Cer and C34:C42 HexCer were indeed increased by 50%, 95% and 70%, respectively (Fig. 5A–C). To assess whether changes in acyl chain length were specific for sphingolipid molecules, levels of phospholipids with different carbon numbers were also measured and compared in control subjects and idiopathic Parkinson’s disease and PD-GBA patients. Data showed small differences in the per cent of specific phospholipid molecules among the three groups but no trend towards an increase in short-chain and decrease in long-chain phospholipids in fibroblast extracts from PD-GBA patients (Supplementary Fig. 2). To evaluate further the relationship between sphingolipid composition and GCase activity, C34:C42 SM, Cer and HexCer ratio values were calculated for each control, idiopathic Parkinson’s disease and PD-GBA cell preparation and then plotted against the corresponding values of enzyme activity. This analysis revealed a significant inverse correlation. Higher enzyme activity was associated with a lower ratio and vice versa, supporting the conclusion that changes in GCase activity alter the content and relative proportion of short- and long-chain sphingolipid (Fig. 5D–F). The increase in C34:C42 sphingolipid ratio, as detected in our study, is therefore likely to be a specific consequence of reduced GCase activity and a feature of altered lipid metabolism caused by the L444P GBA mutation.
Figure 5.

Correlation between the ratio short- over long-chain sphingolipids and GCase activity. (A–C) The ratios C34:C42 (percentage C34 over percentage C42) SM, C34:C42 Cer and C34:C42 HexCer were calculated from lipid measurements made in fibroblasts from control subjects (n = 4) and idiopathic Parkinson’s disease (iPD; n = 4) and PD-GBA (n = 5) patients (one replicate per cell line). Data are shown as individual values and mean ± SEM. Unpaired t-test was performed to compare data in control subjects versus values in the idiopathic Parkinson’s disease or PD-GBA groups. Control versus PD-GBA: SM C34:C42 [P = 0.0147; F(4,3) = 1.504], Cer C34:C42 [P = 0.0221, F(3,4) = 4.173]. Idiopathic Parkinson’s disease versus PD-GBA: SM C34:C42 [P = 0.0149; F(4,3) = 2.791], Cer C34:C42 [P = 0.0352, F(4,3) = 2.246]. (D–F) C34:C42 SM, Cer and HexCer ratio values for each control (n = 4, black), idiopathic Parkinson’s disease (n = 4, orange) and PD-GBA (n = 5, blue–grey) fibroblast preparation were plotted against the corresponding values of GCase activity. Pearson correlation analysis was performed to assess the strength of the association between: C34:C42 SM ratio and GCase activity (P = 0.0053; r = −0.7226), C34:C42 Cer ratio and GCase activity (P = 0.1546; r = −0.4186) and C34:C42 HexCer ratio and GCase activity (P = 0.0020; r = −0.7729). *P < 0.05.
Relationship between fibroblast lipid composition and α-synuclein aggregation
Specific lipid–α-syn interactions modulate α-syn aggregation.30–36 The next set of experiments was therefore designed to test the hypothesis that differences in lipid composition observed in fibroblasts from PD-GBA patients may affect α-syn in vitro fibrillation. For these experiments, lipids were obtained from control (LIPIDcontrol) and PD-GBA (LIPIDPD-GBA) fibroblasts using the same extraction procedure that was employed for our lipidomic analysis. The kinetics of α-syn aggregation were monitored over a 90-min incubation time following the shift in fluorescence caused by the binding of ThT to amyloid structures. Addition of fibroblast-derived lipids, which were resuspended as vesicles, triggered α-syn aggregation as indicated by an increase in ThT fluorescence. Interestingly, kinetics curves of the ThT signal generated from PD-GBA samples were distinctly shifted to the left, with fluorescence rising and reaching its plateau at earlier time points (Fig. 6A). When reaction half-times (t½) were plotted and compared between incubations of α-syn with LIPIDcontrol versus LIPIDPD-GBA, a significantly lower t½ was observed under the latter condition, consistent with a pro-aggregation effect of LIPIDPD-GBA (Fig. 6B).
Figure 6.
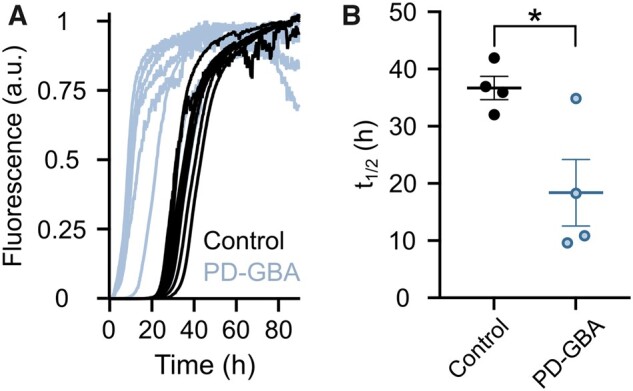
Kinetics of α-syn aggregation in the presence of fibroblast lipid extracts. (A) Lipid extracts from control (n = 4; black) and PD-GBA (n = 4; blue–grey) fibroblast preparations were added to solutions of α-syn and ThT. ThT fluorescence was measured as an indicator of amyloid fibril formation. Kinetics curves are shown from two separate incubations of each subject/patient extract. (Β) Half-times of the reaction of α-syn fibril formation after addition of lipid extracts from control subjects (n = 4) and PD-GBA patients (n = 4). Each circle represents the average of duplicate measurements. Data are shown as individual values and mean ± SEM. Unpaired t-test was performed to compare data. P = 0.0248; F(3,3) = 8.102. *P < 0.05.
Amyloid fibrils that were formed after incubations of α-syn with LIPIDcontrol or LIPIDPD-GBA were collected (at the time when ThT signals reached their plateau) and subjected to lipid extraction. Solvent mixtures were then analysed using MS to determine if specific lipid molecules were co-assembled with α-syn fibrils.43,44 Results showed that the composition of fibril-derived lipid mixtures consisted mainly of PC (∼75% of total lipids), DAG (10%) and sphingolipid (12%); the 12% of total sphingolipid was comprised of 7.1%, 1.4% and 3.4% SM, Cer and HexCer, respectively (Fig. 7A). Lipid composition of α-syn fibrils did not differ significantly between samples collected after incubations with LIPIDcontrol or LIPIDPD-GBA (Fig. 7A). Fibril lipid mixtures were also analysed for the presence of short- and long-chain sphingolipid. Similar to results obtained from the lipidomic analysis of whole fibroblasts, most sphingolipid extracted from α-syn fibrils had 34 or 42 hydrocarbons (Fig. 7B–D). The relative proportion of short- (C34) and long- (C42) chain molecules was different, however, between whole fibroblast- versus fibril-derived samples (cf. data in Fig. 4 and Fig. 7B–D). In particular, marked changes were observed in the per cent of short- and long-chain SM and HexCer. C34 SM and C34 HexCer represented approximately 60% and 20% of the total SM and HexCer levels in extracts of whole fibroblasts (Fig. 4A and C); they instead accounted for >90% of SM and HexCer that were extracted from amyloid fibrils after incubations of α-syn with either LIPIDcontrol or LIPIDPD-GBA (Fig. 7B and D). On the other hand, levels of C42 SM and C42 HexCer, which accounted for a significant percentage of total SM and HexCer in whole fibroblasts, were barely detected in lipid mixtures from α-syn fibrils. Taken together, these data indicate an enrichment of C34 SM and HexCer in fibril-derived lipid extracts and are consistent with a high propensity of these short-chain molecules to interact with and be incorporated into amyloid fibrils during the process of α-syn aggregation.
Figure 7.

Lipid composition of α-syn fibrils. Amyloid fibrils were collected at the end of incubations of α-syn in the presence of fibroblast lipid extracts from control subjects (n = 4) and PD-GBA patients (n = 4; one replicate per cell line). These fibrils were subjected to lipid extraction, and fibril lipid content was measured by mass spectrometry. (A) Data show the percentage of lipid species, phosphatidylcholine (PC), diacyl glyceride (DAG), HexCer, SM and Cer, detected in extracts from α-syn fibrils. (B–D) Levels of SM, Cer and HexCer with different hydrocarbon chain lengths (C32 to C44) are shown as percentage of the respective SM, Cer and HexCer total content. Data are shown as individual values and mean ± SEM.
Effects of ambroxol on fibroblast sphingolipid composition and lipid-induced α-synuclein aggregation
Ambroxol is a small molecule chaperone capable of restoring enzyme activity of mutant GCase both in vitro and in vivo.45–47 Consistent with these earlier results, addition of ambroxol to our fibroblast cultures caused a marked enhancement of GCase protein levels and enzyme activity in both control cells and fibroblasts from PD-GBA patients (Fig. 8A–C). The western blot of proteins extracted from vehicle- and ambroxol-treated fibroblasts showed slightly lower GCase bands in the latter samples (Fig. 8A). This protein shift caused by ambroxol can be seen in western blot images from earlier reports and may be due to conformational protein changes after binding of ambroxol to GCase.16,46,48 GCase activity was markedly lower in vehicle-treated PD-GBA versus control cells. This reduction was effectively reversed by addition of ambroxol, because activity levels in ambroxol-treated PD-GBA fibroblasts became comparable to levels in vehicle-treated controls (Fig. 8C). The next set of analyses was aimed at determining whether this restoration of GCase activity also reversed the effects of the mutant GCase protein on fibroblast sphingolipid composition. Lipid extracts were obtained from ambroxol-treated fibroblasts and processed for shotgun lipidomics; sample processing and analytical procedures were the same as the ones used for untreated cells. As already shown in Figs 3 and 5, in the absence of ambroxol, total sphingolipid levels and the ratios C34:C42 SM, C34:C42 Cer and C34:C42 HexCer were all significantly increased in PD-GBA samples. Quite in contrast, when analyses were carried out in control and PD-GBA fibroblasts treated with ambroxol, data revealed that sphingolipid levels as well as short-chain:long-chain sphingolipid ratios did not differ between these two groups of cells (Fig. 8D). Final experiments were then carried out using lipid extracts from ambroxol-treated fibroblasts that were added to a solution containing recombinant human α-syn and ThT. Lipid-induced increase in ThT fluorescence indicated the formation of amyloid structures and generated kinetics curves of α-syn fibrillation that were similar upon addition of samples from control cells or PD-GBA fibroblasts (Fig. 8F). Thus, treatment with ambroxol not only restored GCase activity and sphingolipid composition of PD-GBA fibroblasts but also reversed the pro-aggregation effect of LIPIDPD-GBA (cf. Fig. 6A with Fig. 8F), further supporting a relationship between GBA mutation-induced sphingolipid changes and enhanced α-syn assembly.
Figure 8.

Effects of ambroxol treatment. (A–C) GCase protein levels and activity were assayed in fibroblasts treated with vehicle (DMSO, A−) or ambroxol (A+). Cells were obtained from control subjects (Ctrl) and PD-GBA patients. (A) Western blots showing immunoreactivity for human GCase and β-actin. (B) Semi-quantitative analysis of band intensities of the blots shown in A. Data are shown as individual values and mean ± SEM. Unpaired t-test was performed to compare data in Ctrl A− versus Ctrl A+ [P = 0.0056; F(3,3) = 13.16] and PD-GBA A− versus PD-GBA A+ [P = 0.0101; F(4,4) = 9.401]. *P < 0.05; **P < 0.01. (C) Measurements of GCase activity in fibroblasts from the control (n = 4) and PD-GBA (n = 5) groups. Data are shown as individual values and mean ± SEM. Unpaired t-test was performed to compare data in Ctrl A− versus Ctrl A+ [P = 0.0038; F(3,3) = 2.351], PD-GBA A− versus PD-GBA A+ [P = 0.0011; F(4,4) = 2.044] and Ctrl A− versus PD-GBA A– [P = 0.0144; F(3,4) = 3.879]. *P < 0.05; **P < 0.01. (D–G) Levels of sphingolipid molecules were assayed in lipid extracts from fibroblast cultures treated with ambroxol (A+) (one replicate per cell line). Cells were obtained from control subjects (ctrl, n = 4) and PD-GBA patients (n = 5). (D) Levels of sphingolipids (SL) are expressed as percentage of the total lipid content. (E–G) The ratios C34:C42 SM (E), Cer (F) and HexCer (G) were calculated from measurements of these sphingolipid molecules in fibroblast lipid extracts. Data are shown as individual values and mean ± SEM. (H) Lipid extracts were prepared from control (n = 4; black) and PD-GBA (n = 4; blue–grey) fibroblast cultures treated with ambroxol. They were added to solutions of α-syn and ThT, and ThT fluorescence was measured as an indicator of amyloid fibril formation. Kinetics curves are shown from three separate incubations of each subject/patient extract.
Discussion
Results of this study indicate the potential value of patient-derived fibroblasts as an ex vivo system for the identification and monitoring of changes in lipid metabolism caused by GBA mutations. A unique lipid profile was detected by shotgun lipidomics of cultures from carriers of the severe L444P GBA mutation; this profile distinguished PD-GBA fibroblasts from cells that were obtained from control subjects as well as cells from idiopathic Parkinson’s disease patients without GBA mutations. Detailed analyses revealed not only that levels of sphingolipid were increased but also that the ratio of short- over long-chain sphingolipid was distinctly altered in fibroblasts from mutation carriers, thus providing a new biological fingerprint of the mutation in these Parkinson’s disease patients. An additional important finding of this study was that membrane lipids isolated from PD-GBA cultures were significantly more effective than lipids from control cells in triggering in vitro fibrillation of recombinant human α-syn. This finding suggests a mechanism of likely pathophysiological relevance linking changes in cell lipid composition caused by GBA mutations to α-syn aggregate pathology. Evidence in support of this mechanism was obtained from experiments in which control fibroblasts and fibroblasts from PD-GBA patients were treated with the pharmacological GCase chaperone ambroxol. This treatment reversed the α-syn pro-aggregation effect of membrane lipids isolated from PD-GBA cultures. Different pharmacological properties of ambroxol may contribute to this effect. Nonetheless, our results strongly support a mechanistic action involving restoration of GCase activity and sphingolipid composition in ambroxol-treated PD-GBA cells.
A correlation was found between the unique lipid profile of PD-GBA fibroblasts and the activity of GCase that was significantly decreased in L444P mutation carriers as compared to control subjects and idiopathic Parkinson’s disease patients. This latter finding, i.e. lack of GCase changes in Parkinson’s disease patients without a GBA mutation, is consistent with data of earlier studies using control- and patient-derived fibroblasts.16,17,46,48 It is apparently at odds, however, with the results of previous investigations in which GCase activity was measured and found to be lowered in brain tissue specimens from idiopathic Parkinson’s disease as compared to control subjects.12,49,50 In contrast to neurons, fibroblasts do not express α-syn. Thus, a plausible explanation for the different results in fibroblasts and brain tissue may be that, in the latter, loss of GCase activity specifically arises from a pathological accumulation of α-syn.21 Consistent with this interpretation, a region-specific reduction of GCase activity has been reported in the brain of patients with sporadic Parkinson’s disease; this effect was correlated with α-syn burden and early development of α-syn pathology.51
Reduced GCase activity in PD-GBA fibroblasts significantly affected sphingolipid content and metabolism, resulting in higher levels of total sphingolipid, enhanced amounts of short-chain sphingolipid molecules and decreased content of long-chain sphingolipid. Earlier studies have reported accumulation of sphingolipid and, in particular, GluCer and GluSph as a consequence of reduced GCase activity in cell lines (i.e. iPSC-derived neurons) from Gaucher’s disease and PD-GBA patients.13,15 Our current data not only extend these observations but reveal for the first time that changes in acyl chain length of sphingolipid significantly contribute to the altered lipid profile of Parkinson’s disease patients with GBA mutations. Interestingly, the hydrocarbon chain length of Cer, HexCer and SM molecules were all affected by a loss of GCase activity, underscoring the interrelated nature of sphingolipid metabolism and suggesting that an initial GCase impairment ultimately alters the function of other enzymes involved in this metabolism. Indeed, as indicated by the results of previous investigations, a shift from long- to short-chain sphingolipid may result from changes in the expression/activity of enzymes such as α-sphingomyelinase and ceramide synthases (CerS).52–55 For example, accumulation of short-chain C18 GluCer was observed in different brain regions of a Gaucher’s disease mouse model (4 L/PS/NA) homozygous for a mutant GCase (V394L [4L]) and expressing a prosaposin hypomorphic (PS-NA) transgene; this accumulation appeared to be correlated with the regional distribution of CerS1 and CerS2.56 It is also noteworthy that a significant shift in Cer acyl chain composition towards shorter chain length was measured in the anterior cingulate cortex of brains from idiopathic Parkinson’s disease patients and attributed to an upregulation of CerS1 expression.57
Sphingolipids are bioactive molecules playing both structural and signalling roles in a range of cellular processes including cell growth, differentiation and migration, autophagy, inflammation, response to trophic factors and apoptosis.58–60 Thus, by altering cellular sphingolipid composition, GBA mutations could have broad functional and pathological consequences, most of all within the brain where SM, HexCer and Cer molecules are particularly abundant and tightly regulated.61,62 Structural and signalling functions of sphingolipid are also significantly affected by their saturation and length of acyl chain. In particular, accumulation of short-chain at the expenses of long-chain sphingolipid could alter the structural order of membrane lipid bilayers, modify membrane curvature and disrupt membrane fluidity.63–65 A switch from long- to short-chain molecules could also interfere with lipid–protein interactions at the plasma membrane level and affect signalling responses involved, for example, in lipid and protein trafficking and degradation and in cell death pathways.66–71
Implications of the results of this study on mechanisms of α-syn pathology are particularly relevant not only due to the important role played by α-syn in Parkinson’s disease pathogenesis but also in view of evidence linking GBA mutations to α-syn alterations. Findings of earlier investigations strongly support a relationship between GCase deficiency, increased levels of sphingolipid and accumulation and aggregation of α-syn in cells treated with a GCase inhibitor, iPSC-derived dopaminergic neurons and brain tissue from Gaucher’s disease and PD-GBA patients and brain tissue from Gaucher’s disease mouse models and GBA-mutant mice.13,15,20,28,72–78 In another earlier in vitro study, accumulation of specific sphingolipid molecules was found to promote the conversion of high molecular weight physiological α-syn conformers into toxic oligomers; these assembly-state intermediates were then capable of seeding the aggregation of monomeric α-syn and promoted the formation of ThT-binding α-syn amyloid structures.79 An important role of lipid–α-syn interactions in pathogenetic processes triggered by GBA mutations is also suggested by earlier biochemical and biophysical studies showing that the chemical properties of lipids significantly affect the binding of α-syn to membranes and the kinetics of membrane-induced α-syn aggregation.77,80 α-Syn binds preferentially to membranes in a fluid state and with high curvature.34,81 Therefore, changes in membrane lipid composition that, as discussed above, could alter these biophysical features could also interfere with α-syn function and promote its toxic potential.
Synthetic membrane preparations have been effectively used to gain insight into the properties of membranes with different lipid compositions. In an earlier study, formation of α-syn amyloid fibrils was compared in the presence of synthetic membranes composed of lipids with the same head group, i.e. phosphatidylserine, but with hydrocarbon chains of different length. The reported results showed that α-syn aggregation was significantly enhanced in the presence of lipids with shorter hydrocarbon chains.34 Our present findings are in line with these earlier observations. Fibril formation was indeed accelerated when α-syn was incubated with membrane lipids from PD-GBA cells that contained higher levels of Cer, HexCer and SM molecules with shorter hydrocarbon chains. Short-chain SM and HexCer species were also found to be enriched in lipid extracts from α-syn fibrils collected at the end of lipid/α-syn incubations. This latter new finding suggests that short chain sphingolipid have a high propensity to co-assemble with α-syn fibrils and may act as preferred reactants during lipid-induced α-syn amyloid formation.
Extrapolation of results from ex vivo models (patient-derived fibroblasts) and in vitro experiments (incubations of α-syn with membrane lipids) to pathological processes always requires caution. Further reasons for a prudent interpretation of our present findings include the fact that data obtained in fibroblast cultures may or may not fully reflect biological and pathological processes (including lipid changes and lipid-α-syn interactions) involving neuronal cells. Nevertheless, taken together, the results of our study support the concept that changes in lipid chemical properties induced by GBA mutations are of likely pathophysiological relevance. In particular, an altered lipid profile induced by loss of GCase activity may itself represent a risk factor for the development of α-syn aggregate pathology in carriers of GBA mutations. Follow-up work is warranted to further elucidate specific lipid abnormalities (e.g. changes in individual sphingolipid molecules, their chain length and saturation) that are directly responsible for promoting α-syn aggregation. Findings of this study and future investigations into the effects of GBA mutations on fibroblast lipid content also bear potential clinical implications. Sphingolipid changes could conceivably be more pronounced in fibroblasts from PD-GBA patients as compared to aged-matched carriers without clinical manifestations. They may therefore be considered as a biomarker of disease risk and disease conversion. Clinical trials are currently assessing safety and effectiveness of drugs capable of reversing a loss of GCase activity in PD-GBA patients.82 In particular, data of a recent report indicated safety, tolerability, CSF penetration and target engagement (e.g. enhanced CSF GCase protein levels) of ambroxol in Parkinson’s disease patients with and without GBA mutations.82 A therapeutic use of ambroxol is supported by our present findings showing its ability to rectify cellular abnormalities induced by GBA mutations. Our data also suggest that, in future clinical trials, measurements of fibroblast lipid profile could be used as an indicator of therapeutic response and may help elucidate the relationship between GCase activity, membrane lipid composition, α-syn aggregation and Parkinson’s disease progression.
Supplementary Material
Acknowledgements
We thank Drs Roberta Zangaglia and Micol Avenali (IRCCS Mondino Foundation) for their assistance with the enrolment and skin biopsy procedures and Ms Laura Demmer for her technical assistance with fibroblast cultures and lipid extract preparations.
Funding
This work was supported by a Marie Skłodowska-Curie Actions-Individual Fellowship (H2020-MSCA, 706551) (C.G.), and by grants from the Lundbeck Foundation (R314-2018–3493) (C.G., F.R.M.), the Carlsberg Foundation (CF19-0382) (C.G., F.R.M.), the EU Innovative Medicines Initiative (IMI 821522, PD-MitoQUANT) (D.D.M.), the EU Joint Programme-Neurodegenerative Disease Research (JPND 01ED2005B, GBA-PaCTS) (D.D.M., A.H.V.S. and F.B.), and The Michael J. Fox Foundation for Parkinson’s Research and the Aligning Science Across Parkinson’s (ASAP-000420) (D.D.M., A.H.V.S. and F.B.).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- α-syn
α-synuclein
- Cer
ceramide
- GCase
glucocerebrosidase
- GluCer
glucosylceramide
- HexCer
hexosylceramide
- PD-GBA
Parkinson's disease patients carrying the L444P GBA mutation
- SM
sphingomyelin
- ThT
thioflavin T
References
- 1. Schapira AHV. Glucocerebrosidase and Parkinson disease: Recent advances. Mol Cell Neurosci. 2015;66(Pt A):37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ryan E, Seehra G, Sharma P, Sidransky E. GBA1-associated Parkinsonism: New insights and therapeutic opportunities. Curr Opin Neurol. 2019;32(4):589–596. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y, Shu L, Sun Q, et al. . Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: A meta-analysis. Front Mol Neurosci. 2018;11:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anheim M, Elbaz A, Lesage S, et al. ; French Parkinson Disease Genetic Group. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78(6):417–420. [DOI] [PubMed] [Google Scholar]
- 5. Neumann J, Bras J, Deas E, et al. . Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132(Pt 7):1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sidransky E, Nalls MA, Aasly JO, et al. . Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alcalay RN, Caccappolo E, Mejia-Santana H, et al. . Cognitive performance of GBA mutation carriers with early-onset PD. Neurology. 2012;78(18):1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brockmann K, Srulijes K, Hauser AK, et al. . GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77(3):276–280. [DOI] [PubMed] [Google Scholar]
- 9. Winder-Rhodes SE, Evans JR, Ban M, et al. . Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain. 2013;136(Pt 2):392–399. [DOI] [PubMed] [Google Scholar]
- 10. Gan-Or Z, Amshalom I, Kilarski LL, et al. . Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. 2015;84(9):880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cilia R, Tunesi S, Marotta G, et al. . Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol. 2016;80(5):662–673. [DOI] [PubMed] [Google Scholar]
- 12. Gegg ME, Burke D, Heales SJR, et al. . Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol. 2012;72(3):455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schöndorf DC, Aureli M, McAllister FE, et al. . iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014;5:4028. [DOI] [PubMed] [Google Scholar]
- 14. Alcalay RN, Levy OA, Waters CH, et al. . Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138(9):2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aflaki E, Borger DK, Moaven N, et al. . A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J Neurosci. 2016;36(28):7441–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanchez-Martinez A, Beavan M, Gegg ME, Chau K-Y, Whitworth AJ, Schapira AHV. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep. 2016;6(1):31380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collins LM, Drouin-Ouellet J, Kuan W-L, Cox T, Barker RA. Dermal fibroblasts from patients with Parkinson’s disease have normal GCase activity and autophagy compared to patients with PD and GBA mutations. F1000Research. 2017;6:1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moors TE, Paciotti S, Ingrassia A, et al. . Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurobiol. 2019;56(2):1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Futerman AH, Platt FM. The metabolism of glucocerebrosides—From 1965 to the present. Mol Genet Metab. 2017;120(1–2):22–26. [DOI] [PubMed] [Google Scholar]
- 20. Mazzulli JR, Xu YH, Sun Y, et al. . Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schapira AHV, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet Lond Engl. 2014;384(9942):545–555. [DOI] [PubMed] [Google Scholar]
- 22. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- 23. Polymeropoulos MH, Lavedan C, Leroy E, et al. . Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. [DOI] [PubMed] [Google Scholar]
- 24. Nussbaum RL. Genetics of synucleinopathies. Cold Spring Harb Perspect Med. 2018;8(6):a024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol Dis. 2020;137:104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang SY, Beavan M, Chau KY, Taanman JW, Schapira AH. A human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Rep. 2017;8(3):728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang S, Gegg M, Chau D, Schapira A. Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol Dis. 2020;134:104620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gündner AL, Duran-Pacheco G, Zimmermann S, et al. . Path mediation analysis reveals GBA impacts Lewy body disease status by increasing α-synuclein levels. Neurobiol Dis. 2019;121:205–213. [DOI] [PubMed] [Google Scholar]
- 29. Yap TL, Velayati A, Sidransky E, Lee JC. Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol Genet Metab. 2013;108(1):56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem. 2003;278(19):16873–16877. [DOI] [PubMed] [Google Scholar]
- 31. Martinez Z, Zhu M, Han S, Fink AL. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry. 2007;46(7):1868–1877. [DOI] [PubMed] [Google Scholar]
- 32. Franceschi GD, Frare E, Pivato M, et al. . Structural and morphological characterization of aggregated species of α-synuclein induced by docosahexaenoic acid. J Biol Chem. 2011;286(25):22262–22274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Galvagnion C, Buell AK, Meisl G, et al. . Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. 2015;11(3):229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galvagnion C, Brown JW, Ouberai MM, et al. . Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc Natl Acad Sci U S A. 2016;113(26):7065–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grey M, Dunning CJ, Gaspar R, et al. . Acceleration of alpha-synuclein aggregation by exosomes. J Biol Chem. 2015;290(5):2969–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gaspar R, Pallbo J, Weininger U, Linse S, Sparr E. Ganglioside lipids accelerate α-synuclein amyloid formation. Biochim Biophys Acta BBA—Proteins Proteomics. 2018;1866(10):1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sampaio JL, Gerl MJ, Klose C, et al. . Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A. 2011;108(5):1903–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ejsing CS, Sampaio JL, Surendranath V, et al. . Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci U S A. 2009;106(7):2136–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Surma MA, Herzog R, Vasilj A, et al. . An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. Eur J Lipid Sci Technol. 2015;117(10):1540–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liebisch G, Binder M, Schifferer R, Langmann T, Schulz B, Schmitz G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim Biophys Acta. 2006;1761(1):121–128. [DOI] [PubMed] [Google Scholar]
- 41. Herzog R, Schwudke D, Schuhmann K, et al. . A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011;12(1):R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herzog R, Schuhmann K, Schwudke D, et al. . LipidXplorer: A software for consensual cross-platform lipidomics. PLoS One. 2012;7(1):e29851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hellstrand E, Nowacka A, Topgaard D, Linse S, Sparr E. Membrane lipid co-aggregation with alpha-synuclein fibrils. PLoS One. 2013;8(10):e77235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Galvagnion C, Topgaard D, Makasewicz K, et al. . Lipid dynamics and phase transition within α-synuclein amyloid fibrils. J Phys Chem Lett. 2019;10(24):7872–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bendikov-Bar I, Maor G, Filocamo M, Horowitz M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol Dis. 2013;50(2):141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McNeill A, Magalhaes J, Shen C, et al. . Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137(5):1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol. 2016;80(5):766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis. 2015;82:235–242. [DOI] [PubMed] [Google Scholar]
- 49. Rocha EM, Smith GA, Park E, et al. . Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol. 2015;2(4):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huebecker M, Moloney EB, van der Spoel AC, et al. . Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener. 2019;14(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murphy KE, Halliday GM. Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy. 2014;10(7):1350–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Imgrund S, Hartmann D, Farwanah H, et al. . Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem. 2009;284(48):33549–33560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ben-David O, Pewzner-Jung Y, Brenner O, et al. . Encephalopathy caused by ablation of very long acyl chain ceramide synthesis may be largely due to reduced galactosylceramide levels. J Biol Chem. 2011;286(34):30022–30033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mosbech M-B, Olsen ASB, Neess D, et al. . Reduced ceramide synthase 2 activity causes progressive myoclonic epilepsy. Ann Clin Transl Neurol. 2014;1(2):88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van Smeden J, Janssens M, Boiten WA, et al. . Intercellular skin barrier lipid composition and organization in Netherton syndrome patients. J Invest Dermatol. 2014;134(5):1238–1245. [DOI] [PubMed] [Google Scholar]
- 56. Jones EE, Zhang W, Zhao X, et al. . Tissue localization of glycosphingolipid accumulation in a Gaucher Disease mouse brain by LC-ESI-MS/MS and high-resolution MALDI imaging mass spectrometry. SLAS Discov Adv Life Sci R D. 2017;22(10):1218–1228. [DOI] [PubMed] [Google Scholar]
- 57. Abbott SK, Li H, Muñoz SS, et al. . Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov Disord. 2014;29(4):518–526. [DOI] [PubMed] [Google Scholar]
- 58. Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327(5961):46–50. [DOI] [PubMed] [Google Scholar]
- 59. Lippincott-Schwartz J, Phair RD. Lipids and cholesterol as regulators of traffic in the endomembrane system. Annu Rev Biophys. 2010;39:559–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Brien JS, Sampson EL. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J Lipid Res. 1965;6(4):537–544. [PubMed] [Google Scholar]
- 62. Olsen ASB, Færgeman NJ. Sphingolipids: Membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017;7(5):170069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niemelä PS, Hyvönen MT, Vattulainen I. Influence of chain length and unsaturation on sphingomyelin bilayers. Biophys J. 2006;90(3):851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ben-David O, Futerman AH. The role of the ceramide acyl chain length in neurodegeneration: Involvement of ceramide synthases. Neuromolecular Med. 2010;12(4):341–350. [DOI] [PubMed] [Google Scholar]
- 65. Mencarelli C, Martinez-Martinez P. Ceramide function in the brain: When a slight tilt is enough. Cell Mol Life Sci. 2013;70(2):181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sillence DJ, Puri V, Marks DL, et al. . Glucosylceramide modulates membrane traffic along the endocytic pathway. J Lipid Res. 2002;43(11):1837–1845. [DOI] [PubMed] [Google Scholar]
- 67. Kroesen B-J, Jacobs S, Pettus BJ, et al. . BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome. J Biol Chem. 2003;278(17):14723–14731. [DOI] [PubMed] [Google Scholar]
- 68. Koivusalo M, Jansen M, Somerharju P, Ikonen E. Endocytic trafficking of sphingomyelin depends on its acyl chain length. Mol Biol Cell. 2007;18(12):5113–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sassa T, Suto S, Okayasu Y, Kihara A. A shift in sphingolipid composition from C24 to C16 increases susceptibility to apoptosis in HeLa cells. Biochim Biophys Acta. 2012;1821(7):1031–1037. [DOI] [PubMed] [Google Scholar]
- 70. Ali M, Fritsch J, Zigdon H, Pewzner-Jung Y, Schütze S, Futerman AH. Altering the sphingolipid acyl chain composition prevents LPS/GLN-mediated hepatic failure in mice by disrupting TNFR1 internalization. Cell Death Dis. 2013;4:e929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Backman APE, Halin J, Nurmi H, Möuts A, Kjellberg MA, Mattjus P. Glucosylceramide acyl chain length is sensed by the glycolipid transfer protein. PLoS One. 2018;13(12):e0209230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sun Y, Quinn B, Witte DP, Grabowski GA. Gaucher disease mouse models: Point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J Lipid Res. 2005;46(10):2102–2113. [DOI] [PubMed] [Google Scholar]
- 73. Sardi SP, Clarke J, Kinnecom C, et al. . CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A. 2011;108(29):12101–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xu YH, Sun Y, Ran H, Quinn B, Witte D, Grabowski GA. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab. 2011;102(4):436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cleeter MW, Chau KY, Gluck C, et al. . Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int. 2013;62(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fernandes HJR, Hartfield EM, Christian HC, et al. . ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 2016;6(3):342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Muñoz SS, Petersen D, Marlet FR, Kücükköse E, Galvagnion C. The interplay between GCase, α-synuclein and lipids in human models of Parkinson’s disease. Biophys Chem. 2020;273:106534. [DOI] [PubMed] [Google Scholar]
- 78. Migdalska-Richards A, Wegrzynowicz M, Rusconi R, et al. . The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain J Neurol. 2017;140(10):2706–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zunke F, Moise AC, Belur NR, et al. . Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97(1):92–107.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Galvagnion C. The role of lipids interacting with alpha-synuclein in the pathogenesis of Parkinson’s disease. J Parkinsons Dis. 2017;7(3):433–450. [DOI] [PubMed] [Google Scholar]
- 81. Middleton ER, Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys J. 2010;99(7):2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mullin S, Smith L, Lee K, et al. . Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol. 2020;77(4):427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request.