

Abstract
Background and Objective
Abrocitinib is a Janus kinase 1-selective inhibitor for the treatment of moderate-to-severe atopic dermatitis. Abrocitinib is eliminated primarily by metabolism involving cytochrome P450 (CYP) enzymes. Abrocitinib pharmacologic activity is attributable to the unbound concentrations of the parent molecule and 2 active metabolites, which are substrates of organic anion transporter 3 (OAT3). The sum of potency-adjusted unbound exposures of abrocitinib and its 2 active metabolites is termed the abrocitinib active moiety. We evaluated effects of CYP inhibition, CYP induction, and OAT3 inhibition on the pharmacokinetics of abrocitinib, its metabolites, and active moiety.
Methods
Three fixed-sequence, open-label, phase I studies in healthy adult volunteers examined the drug–drug interactions (DDIs) of oral abrocitinib with fluvoxamine and fluconazole, rifampin, and probenecid.
Results
Co-administration of abrocitinib with fluvoxamine or fluconazole increased the area under the plasma concentration–time curve from time 0 to infinity (AUCinf) of the unbound active moiety of abrocitinib by 91% and 155%, respectively. Co-administration with rifampin decreased the unbound active moiety AUCinf by 56%. The OAT3 inhibitor probenecid increased the AUCinf of the unbound active moiety by 66%.
Conclusions
It is important to consider the effects of DDIs on the abrocitinib active moiety when making dosing recommendations. Co-administration of strong CYP2C19/2C9 inhibitors or CYP inducers impacted exposure to the abrocitinib active moiety. A dose reduction by half is recommended if abrocitinib is co-administered with strong CYP2C19 inhibitors, whereas co-administration with strong CYP2C19/2C9 inducers is not recommended. No dose adjustment is required when abrocitinib is administered with OAT3 inhibitors.
Clinical Trials Registration IDs
Supplementary Information
The online version contains supplementary material available at 10.1007/s13318-021-00745-6.
Key Points
| Assessing drug–drug interactions (DDIs) between abrocitinib and fluconazole provided an estimation of a worst-case scenario in the metabolism of abrocitinib involving fluconazole-driven inhibition of CYP2C19, CYP2C9, and CYP3A. The DDI between abrocitinib and probenecid provided an estimation of the effect on the OAT3-mediated clearance of the active metabolites. |
| The abrocitinib parent drug is a victim of DDIs with strong CYP2C19 inducers or inhibitors. However, the active moiety is less affected by DDIs. The OAT3 inhibitor, probenecid, increased the area under the plasma concentration–time curve from time 0 to infinity of the unbound active moiety by 66%. |
| A dose reduction by half is recommended if abrocitinib is co-administered with strong CYP2C19 inhibitors. Co-administration with strong CYP2C19/2C9 inducers is not recommended. No dose adjustment is required when abrocitinib is administered with OAT3 inhibitors. |
Introduction
Abrocitinib is a Janus kinase (JAK) 1-selective inhibitor [1] for the treatment of moderate-to-severe atopic dermatitis (AD) [2–4]. A small percentage (1.0–4.4%) of the abrocitinib parent drug is excreted unchanged in the urine [5], indicating that the majority of the parent drug undergoes hepatic metabolism. In vitro [6] and in vivo studies (data on file) have demonstrated that abrocitinib is a substrate of cytochrome P450 (CYP) 2C19 (contributing to 53% of abrocitinib metabolism) and to a lesser extent CYP2C9 (30%), CYP3A4 (11%), and CYP2B6 (6%).
After a single oral dose of abrocitinib, the parent drug is the most prevalent circulating species (26%), along with 3 more polar monohydroxylated metabolites: M1 (3-hydroxypropyl, 11%), M2 (2-hydroxypropyl, 12%), and M4 (pyrrolidinone pyrimidine, 14%) (data on file). Of the 3 metabolites, M1 and M2 have a JAK1 inhibitory profile similar to that of abrocitinib, whereas M4 is pharmacologically inactive. Therefore, the overall pharmacologic activity of abrocitinib is attributable to the unbound exposures of abrocitinib (~ 60%), M1 (~ 10%), and M2 (~ 30%) in the systemic circulation. The sum of unbound exposures of abrocitinib, M1, and M2, each in molar units and adjusted for relative potencies, is termed the active moiety of abrocitinib.
Results of urine recovery studies in vivo and transporter profiling studies in vitro of the M1, M2, and M4 metabolites suggest that each is primarily eliminated in the urine and is in part a substrate for the renal organic anion transporter 3 (OAT3) (data on file). With the known metabolism and excretion profile of abrocitinib, drug–drug interaction (DDI) studies evaluating the effect of CYP inhibition, CYP induction, and OAT3 inhibition on the active moiety of abrocitinib are warranted.
The objective of this analysis was to evaluate the effects of CYP inhibition with fluvoxamine and fluconazole, CYP induction with rifampin, and OAT3 inhibition with probenecid on the pharmacokinetics of abrocitinib, its metabolites, and active moiety. Fluvoxamine is a selective serotonin reuptake inhibitor widely used for the treatment of obsessive-compulsive disorder [7]. It is a strong inhibitor of CYP2C19 and a moderate inhibitor of CYP3A [8]. Fluconazole is a synthetic triazole antifungal agent [9], which is a strong inhibitor of CYP2C19 and a moderate inhibitor of CYP2C9 and CYP3A [8]. Assessing DDIs between abrocitinib and fluconazole provided the estimation of a worst-case scenario in the near-complete inhibition of the metabolism of abrocitinib by CYP2C19, CYP2C9, and CYP3A. Note that a strong inhibitor of CYP2C9 is currently not available, based on US Food and Drug Administration guidance [8]. Rifampin is an antibacterial drug [10] used in this study as a strong inducer of CYP2C19 and CYP3A and a moderate inducer of CYP2C9 and CYP2B6 [8]. Probenecid is widely used as a uricosuric agent [11] and is employed as a strong OAT3 inhibitor in clinical drug interaction studies [8].
Methods
Study Designs and Participants
Three open-label, fixed-sequence, phase I studies in healthy volunteers have examined the DDI effect of (1) multiple doses of fluvoxamine [50 mg once daily (QD)] or fluconazole (200 mg QD following a 400-mg loading dose; NCT03634345), (2) multiple doses of rifampin (600 mg QD; NCT03637790), and (3) multiple doses of probenecid [1000 mg twice daily (BID); NCT03937258] on the pharmacokinetics of abrocitinib and its active moiety. In addition, multiple-dose (steady-state) pharmacokinetics of abrocitinib and its active moiety was assessed in the probenecid study. Abrocitinib 100 mg was administered as a single dose in the fluvoxamine and fluconazole study, and as a 200-mg single dose in the rifampin and probenecid studies; both doses are within the linear range of dose proportionality and are clinically relevant. The treatment schedules in each study are shown in Fig 1. On pharmacokinetics sampling days, abrocitinib was administered after overnight fasted conditions. No food was allowed for at least 4 h following dosing. Subjects were not permitted to lie down during the first 4 h after abrocitinib dosing to standardize the conditions. This research was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments. The final protocol, any amendments, and informed consent documentation were reviewed and approved by the Independent Ethics Committee at the investigational centers participating in the study. Written informed consent was obtained from all participants prior to enrollment.
Fig. 1.

Study treatment schedule. Abrocitinib was administered 3 h after fluvoxamine, 1 h after fluconazole, 1 h after rifampin, and 2 h after probenecid; BID twice daily
Main inclusion criteria were: female or male subjects aged 18–55 years; body mass index of 17.5–30.5 kg/m2 and total body weight > 50 kg; and healthy subjects, which was defined as no clinically relevant abnormalities identified by a detailed medical history, full physical examination, including blood pressure and pulse measurement, 12-lead electrocardiogram, or clinical laboratory tests. Main exclusion criteria were: evidence or history of clinically significant dermatologic condition or visible rash present during physical examination; any history of chronic infections, any history of recurrent infections, any history of latent infections, or any acute infection within 2 weeks of screening; positive urine drug test; use of tobacco or nicotine-containing products; use of prescription or non-prescription drugs (except for acetaminophen/paracetamol at doses of ≤ 1 g/day) and dietary supplements within 7–14 days or 5 half-lives (whichever is longer) prior to first dose. Hormone replacement therapy was required to be discontinued at least 28 days prior to the first dose of investigational product.
Dietary and Activity Restrictions
The daily caloric intake per subject did not exceed 3200 kcal, and the total daily nutritional composition was approximately 55% carbohydrate, 30% fat, and 15% protein. Subjects were not allowed to eat or drink grapefruit or grapefruit-related citrus fruits from 7 days before the first dose of investigational product until collection of the final blood sample. Subjects were required to abstain from alcohol and caffeine-containing products for 24 h before the study and continue to abstain until collection of the final blood sample. Subjects were required to abstain from strenuous exercise for at least 48 h before each blood collection.
Blood Sample Collection
Blood samples were collected for plasma pharmacokinetics and CYP2C19 and CYP2C9 genotyping analyses. In the fluvoxamine study, serial blood samples (3 mL) were collected for 48 h following dosing in Periods 1 and 2. In the fluconazole study, serial blood samples (3 mL) were collected for 48 h after the dose in Period 1 and 72 h after dose in Period 2. In the rifampin study, serial blood samples (6 mL) were collected for 24 h following receipt of dose in Periods 1 and 2. In the probenecid study, serial blood samples (10 mL) were collected for 48 h after the dose on Period 1 Day 1, Period 2 Day 4, and Period 3 Day 2. Details of plasma preparation and sampling time points are described in the supplementary material. Pharmacokinetics sampling schedules were determined based on the pharmacokinetics profile of abrocitinib and anticipated DDI effect. Blood samples were collected into tubes containing dipotassium ethylenediaminetetraacetic acid. Plasma samples for pharmacokinetics analyses were stored at – 80 °C to – 20 °C until analysis.
Pharmacokinetics Sample Analysis
Plasma samples were assayed for abrocitinib and 3 metabolites (M1, M2, and M4) at Syneos Health (Princeton, NJ, USA) using 3 separate validated, sensitive, and specific high-performance liquid chromatography-tandem mass spectrometric (HPLC-MS/MS) bioanalytical methods [12]. Acquity UPLC (Waters, Milford, MA, USA) and API 5000 (Sciex, Concord, Ontario, Canada) were used for abrocitinib and its stable labeled internal standard, PF-06651703. Agilent 1100 Series LC pumps (Agilent Technologies, Wilmington, DE, USA) and API 5000 (Sciex) were used for M1, M2, and M4 and their respective internal standards, PF-07222472, PF-07222473, and PF-07222475.
Abrocitinib and its stable labeled internal standard PF-06651703 were extracted from human plasma using a liquid–liquid extraction procedure [12]. The samples were analyzed by HPLC-MS/MS using positive electrospray ionization mode. The calibration range of the method was 1–2000 ng/mL, and the quality control (QC) concentrations were 3.00 ng/mL, 60 ng/mL, 1000 ng/mL, 1600 ng/mL, and 5000 ng/mL. The concentration of 5000 ng/mL was validated per bioanalytic method validation guidelines, where a dilution QC is used at a concentration of at least 2 times the upper limit of quantitation to cover for any over-the-range sample concentration measurement; this dilution QC was only used in the probenecid study. The inter-day assay accuracy (%) ranged from − 3.33 to 1.00%, and the intra-day precision was ≤ 5.00% in the fluvoxamine and fluconazole study; inter-day assay accuracy ranged from − 4.00 to 0.00%, and the intra-day precision was ≤ 2.79% in the rifampin study; and inter-day assay accuracy ranged from − 1.33 to 0.00%, and the intra-day precision was ≤ 5.01% in the probenecid study.
M1, M2, and M4 and their respective internal standards, PF-07222472, PF-07222473, and PF-07222475, were extracted from human plasma using a protein precipitation extraction procedure [12]. The samples were analyzed by HPLC-MS/MS using positive electrospray ionization mode for M1 and M4 and using a positive atmospheric pressure chemical ionization mode for M2. The calibration range of the method for M1 and M4 was 1–1000 ng/mL, and the QC concentrations were 3.00 ng/mL, 50 ng/mL, and 750 ng/mL. The inter-day assay accuracy ranged from − 0.933 to 6.40% and − 3.47 to 4.00% in the fluvoxamine and fluconazole study; 3.80 to 6.27% and 0.600 to 1.60% in the rifampin study; and − 5.87 to 7.20% and − 3.20 to 3.40% in the probenecid study for M1 and M4, respectively. The intra-day assay precision was ≤ 13.3% and ≤ 9.60% in the fluconazole and fluvoxamine study, ≤ 10.6% and ≤ 8.51% in the rifampin study, and ≤ 7.97% and ≤ 5.49% in the probenecid study for M1 and M4, respectively. The calibration range of the method for M2 was 5–5000 ng/mL, and the QC concentrations were 15.0 ng/mL, 75 ng/mL, 250 ng/mL, and 3750 ng/mL. The inter-day assay accuracy range and intra-day assay precision, respectively, were − 4.27 to − 2.53% and ≤ 5.01% in the fluvoxamine and fluconazole study; inter-day assay accuracy range and intra-day assay precision, respectively, were − 1.33 to 0.00% and ≤ 3.77% in the rifampin study; and inter-day assay accuracy range and intra-day assay precision, respectively, were − 2.93 to 1.33% and ≤ 6.07% in the probenecid study.
Statistical Methods and Pharmacokinetic Parameters
A sample size of 12 subjects was enrolled in each study (or per cohort in the fluvoxamine and fluconazole study) to estimate the DDI effects on the pharmacokinetics of abrocitinib and its active moiety.
Pharmacokinetics parameters of abrocitinib and its metabolites [e.g., area under the concentration–time curve from 0 to infinity (AUCinf), area under the concentration–time curve from time 0 to the time of last quantifiable concentration (AUClast), and the maximum observed plasma concentration (Cmax)] were derived from the concentration–time profiles using standard non-compartmental methods. Pharmacokinetic parameter values were calculated using Pfizer’s internally validated electronic non-compartmental analysis software (eNCA, v.2.2.4). The statistical software used was SAS (SAS Institute, Cary, NC, USA). Samples below the lower limit of quantitation (1 ng/mL for abrocitinib, M1, and M4, and 5 ng/mL for M2) were set to 0 ng/mL for the pharmacokinetics analysis. Actual pharmacokinetics sampling times were used in the derivation of the pharmacokinetics parameters. The pharmacokinetics parameters were summarized descriptively by treatment. No subgroup analyses by CYP2C19 or CYP2C9 genotype status were performed.
Natural log-transformed parameters of abrocitinib and its metabolites were analyzed by treatment using a mixed effects model with treatment as a fixed effect and subject as a random effect. Estimates of the adjusted mean differences (Test – Reference) and corresponding 90% confidence intervals (CIs) were obtained from the model. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (Test/Reference) and 90% CI for the ratios.
The safety population was defined as all subjects that received at least 1 dose of study medication. Safety data are summarized descriptively.
Active Moiety Calculation
Unbound AUCinf, AUClast, and Cmax for the active moiety were calculated for each subject as the sum of the respective unbound pharmacokinetics parameters for abrocitinib and its active metabolites, M1 and M2, each in molar units and adjusted for relative potencies for the interferon alpha (IFN-α) receptor.[12] Taking AUCinf as an example, Eq. (1) was used [13]:
| 1 |
in which AUCinf,u,AM is the unbound AUCinf of the active moiety; AUCinf,u,P, AUCinf,u,M1, and AUCinf,u,M2 are the unbound AUCinf of abrocitinib, M1, and M2, respectively; and IC50,u,P/IC50,u,M1 and IC50,u,P/IC50,u,M2 are the relative potencies of abrocitinib to metabolites M1 and M2, respectively, for the IFN-α receptor.
In vitro IFN-α potency (unbound IFN-α IC50,u) and plasma protein binding [fraction unbound in plasma (fu)] for abrocitinib, M1, and M2 were 59 nM, 165 nM, and 50.6 nM, and 0.36, 0.63, and 0.71, respectively (data on file). These in vitro values were generated using methods that have been previously described [14]. IFN-α inhibition, which signals through a JAK1-tyrosine kinase 2 dimer, was chosen as representative of JAK1-dependent pharmacology of abrocitinib and its metabolites.
Results
Subject Demographics and Metabolizer Status
Twelve subjects were enrolled in each study cohort; all completed the studies. Most participants were male (Table 1). Genotyping data on metabolizer status were available for CYP2C19 and CYP2C9 (Table S1).
Table 1.
Subject demographics
| Characteristic | Fluvoxamine cohort (n = 12) | Fluconazole cohort (n = 12) | Rifampin study (n = 12) | Probenecid study (n = 12) |
|---|---|---|---|---|
| Sex, n (%) | ||||
| Male | 10 (83.3) | 12 (100.0) | 10 (83.3) | 7 (58.3) |
| Female | 2 (16.7) | 0 | 2 (16.7) | 5 (41.7) |
| Age, years | ||||
| Median (range) | 34.5 (22.0, 49.0) | 29.0 (23.0, 49.0) | 36.5 (24.0, 54.0) | 34.5 (27.0, 56.0) |
| Mean (standard deviation) | 34.3 (10.4) | 32.4 (9.4) | 37.6 (9.68) | 38.3 (9.3) |
| Race, n (%) | ||||
| White | 10 (83.3) | 10 (83.3) | 8 (66.7) | 6 (50.0) |
| Black or African American | 1 (8.3) | 1 (8.3) | 2 (16.7) | 6 (50.0) |
| Asian | 0 | 0 | 2 (16.7) | 0 |
| Native Hawaiian or other Pacific Islander | 0 | 1 (8.3) | 0 | 0 |
| Other | 1 (8.3) | 0 | 0 | 0 |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 3 (25.0) | 1 (8.3) | 0 | 5 (41.7) |
| Not Hispanic or Latino | 9 (75.0) | 11 (91.7) | 12 (100.0) | 7 (58.3) |
| Body mass index, kg/m2 | ||||
| Mean (standard deviation) | 24.4 (3.2) | 25.3 (2.5) | 23.9 (1.9) | 27.2 (2.7) |
Abrocitinib Pharmacokinetics with Fluvoxamine and Fluconazole
The median plasma concentration–time curves for the abrocitinib parent drug after a single dose of abrocitinib in the presence and absence of fluvoxamine and fluconazole are shown in Fig. 2. CYP inhibition with fluvoxamine or fluconazole increased the parent drug Cmax; the increase was more pronounced with fluconazole. Pharmacokinetic parameters [Cmax, AUCinf, time to Cmax (Tmax), terminal plasma half-life (t½), apparent oral clearance (CL/F), apparent volume of distribution following oral administration (Vz/F)] for the abrocitinib parent drug and active moiety are listed in Tables S2 and S3. Abrocitinib was absorbed rapidly, with a median Tmax of ~ 0.525–1 h under fasting conditions. Pharmacokinetic parameters (Cmax, AUCinf, AUClast, t½) for the abrocitinib metabolites M1, M2, and M4 are listed in Tables S6 and S7.
Fig. 2.
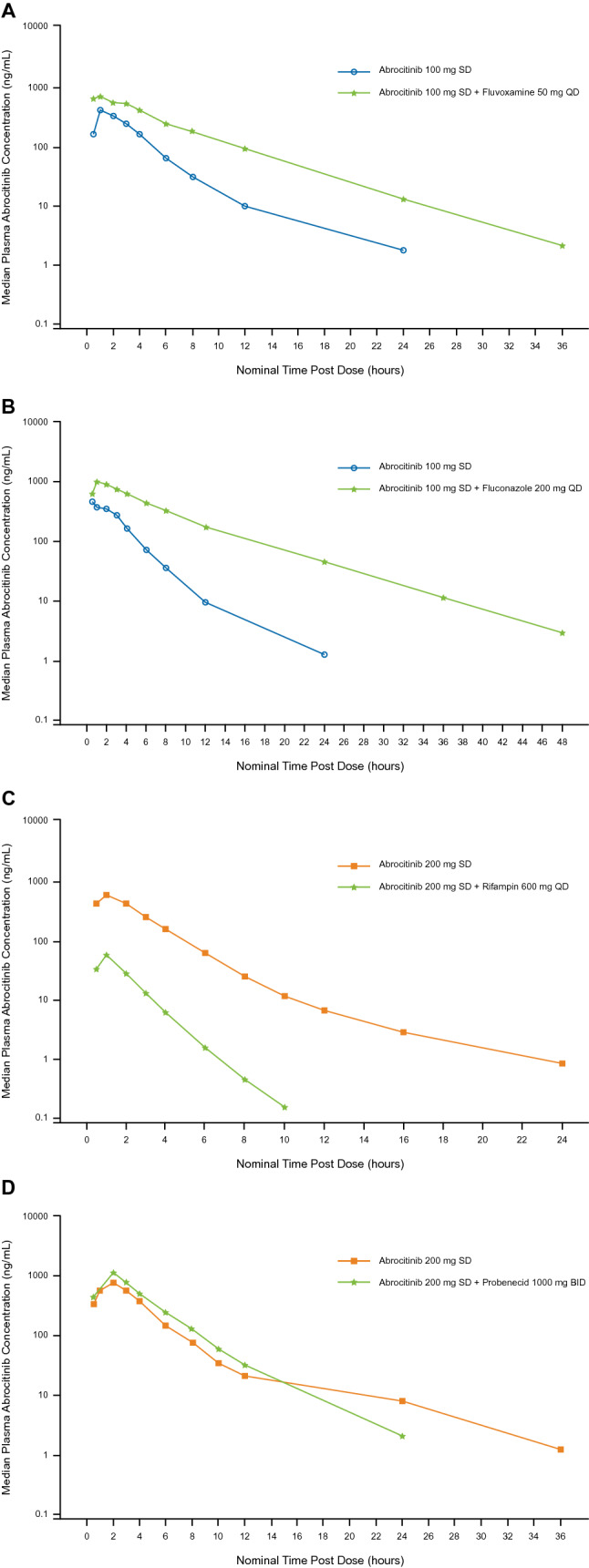
Median plasma concentration–time curves (semilog scale) for the abrocitinib parent drug in the presence and absence of fluvoxamine (a), fluconazole (b), rifampin (c), and probenecid (d); BID twice daily, QD once daily, SD single dose
Ratios (abrocitinib co-administration with fluvoxamine or fluconazole vs. abrocitinib alone) of adjusted geometric means of Cmax and AUCinf for the abrocitinib parent drug and its active moiety are shown in Table 2. Fluvoxamine increased abrocitinib Cmax by 1.8-fold and AUCinf by 2.8-fold, and increased unbound active moiety Cmax by 1.3-fold and AUCinf by 1.9-fold. Fluconazole increased abrocitinib Cmax by 1.9-fold and AUCinf by 4.8-fold, and increased unbound active moiety Cmax by 1.2-fold and AUCinf by 2.5-fold.
Table 2.
Ratio of adjusted geometric means (90% CI) of Cmax and AUCinf for abrocitinib parent drug and its active moiety in the presence of fluvoxamine, fluconazole, rifampin, and probenecid
| Abrocitinib | Active moiety | |||
|---|---|---|---|---|
| Cmax | AUCinf | Cmax | AUCinf | |
| Effect of fluvoxamine | 184.44% (133.27, 255.24) | 275.22% (238.77, 317.24) | 133.08% (99.58, 177.86) | 191.24% (173.81, 210.43) |
| Effect of fluconazole | 192.10% (154.15, 239.39) | 482.86% (383.94, 606) | 123.46% (107.58, 141.70) | 254.86% (241.75, 268.67) |
| Effect of rifampin | 20.86% (14.31, 30.41) | 12.45% (9.33, 16.60) | 68.91% (50.28, 94.46) | 43.86% (40.94, 46.98) |
| Effect of probenecid | 121.38% (92.93, 158.52) | 127.60% (114.97, 141.61) | 130.13% (104.10, 162.65) | 165.54% (152.00, 180.29) |
AUCinf area under the concentration–time curve from time 0 to infinity, CI confidence interval, Cmax maximum observed plasma concentration
Metabolite (M1, M2, and M4)-to-parent drug ratios for unadjusted geometric means of Cmax and AUCinf are shown in Tables S10 and S11. Fluvoxamine and fluconazole decreased exposure ratios of metabolites relative to the parent drug, showing a reduced metabolism due to these CYP inhibitors.
Abrocitinib Pharmacokinetics with Rifampin
Median plasma concentration–time curves for the abrocitinib parent drug and metabolites after a single dose of abrocitinib in the presence and absence of rifampin are shown in Fig. 2; pharmacokinetics parameters are listed in Table S4 for the parent drug and active moiety, and Table S8 for the metabolites. Rifampin decreased abrocitinib Cmax by 79.1% and AUCinf by 87.6%, and decreased unbound active moiety Cmax by 31.1% and AUCinf by 56.1% (Table 2). As expected, CYP induction with rifampin increased the metabolite-to-parent drug ratios (Tables S10 and S11).
Abrocitinib Pharmacokinetics with Probenecid
The median plasma concentration–time curves for the abrocitinib parent drug and metabolites after a single dose of abrocitinib in the presence and absence of probenecid are shown in Figs. 2 and 3, respectively; pharmacokinetics parameters are listed in Table S5 for the parent drug and Table S9 for the metabolites. OAT3 inhibition with probenecid increased the exposures of each metabolite (Table S9) to a higher extent than the parent drug (Table S5). Probenecid increased abrocitinib Cmax by 1.2-fold and AUCinf by 1.3-fold, and increased unbound active moiety Cmax by 1.3-fold and AUCinf by 1.7-fold (Table 2).
Fig. 3.
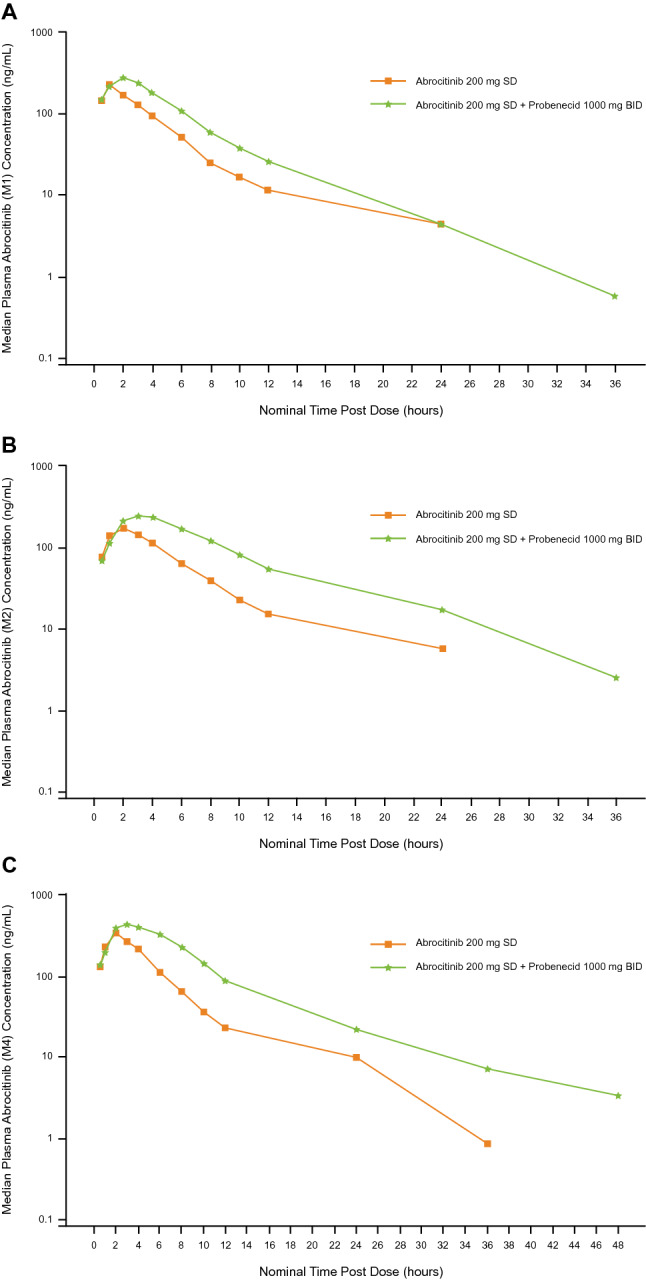
Median plasma concentration–time curves (semilog scale) for abrocitinib metabolites M1 (a), M2 (b), and M4 (c) in the presence and absence of probenecid; BID twice daily, SD single dose
Ratios (abrocitinib co-administration with probenecid vs. abrocitinib alone) of adjusted geometric means of Cmax and AUCinf for abrocitinib metabolites (M1, M2, and M4) are shown in Table 3. Probenecid increased Cmax of M1 by 1.4-fold, M2 by 1.3-fold, and M4 by 1.4-fold. Probenecid increased AUCinf of M1 by 1.8-fold, M2 by 2.2-fold, and M4 by 2.2-fold. Probenecid increased metabolite-to-parent ratios for AUCinf, but not Cmax, which is consistent with the fact that the elimination process of the metabolites was impacted by probenecid co-administration.
Table 3.
Ratio of adjusted geometric means (90% CI) of Cmax and AUCinf for abrocitinib metabolites (M1, M2, M4) in the presence of probenecid
| M1 | M2 | M4 | ||||
|---|---|---|---|---|---|---|
| Cmax | AUCinf | Cmax | AUCinf | Cmax | AUCinf | |
| Effect of probenecid | 136.69% (116.33, 160.61) | 177.17% (164.48, 190.84) | 134.60% (115.08, 157.44) | 224.85% (207.95, 243.12) | 144.81% (117.47, 178.51) | 216.74% (190.82, 246.18) |
AUCinf area under the concentration–time curve from time 0 to infinity, CI confidence interval, Cmax maximum observed plasma concentration, M metabolite
Abrocitinib Pharmacokinetics with Single vs. Multiple Dosing
Median plasma concentration–time curves for the abrocitinib parent drug and the metabolites, M1, M2, and M4, after single and multiple dosing are presented in Fig. S1, and the pharmacokinetics parameters are summarized in Table S12. Accumulation was observed for abrocitinib, M4, and the active moiety after multiple doses, but not for M1 and M2 (Table S12). After 4 days of 200-mg QD dosing, the accumulation ratio (Rac) for abrocitinib was 1.54, the steady-state accumulation ratio (Rss) was 1.47, and the observed accumulation ratio for Cmax (Rac,Cmax) was 1.56. The accumulation ratios for the active moiety were 1.31 for Rac, 1.16 for Rss, and 1.37 for Rac,Cmax. Ratios of metabolite (M1, M2, and M4)-to-parent drug for unadjusted geometric means of Cmax and AUCtau are shown in Table S13.
As shown in Fig. 4, the mean (SD) plasma concentrations for abrocitinib before dosing on Day 2, Day 3, and Day 4, and at 24 h after dose on Day 4 were 9.16 (7.96), 15.01 (21.38), 14.95 (15.88), and 11.34 (14.76) ng/mL, respectively. The concentrations on Days 3, 4, and 5 were relatively constant, and therefore, these results indicate that steady-state plasma concentrations of abrocitinib are achieved by Day 3 of dosing, i.e., within 48 h after once-daily administration.
Fig. 4.
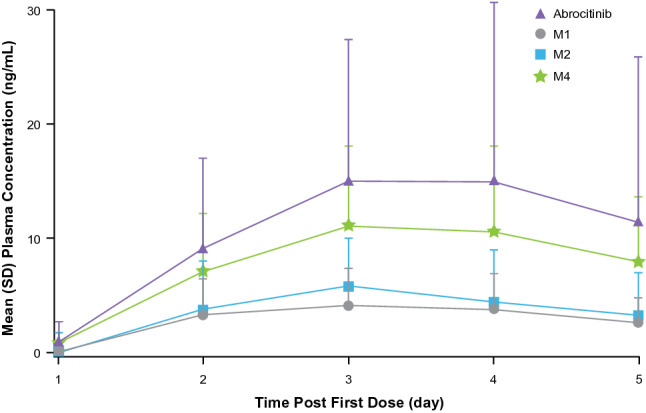
Mean (SD) plasma concentrations of abrocitinib and metabolites prior to dosing on Days 1–4, and at 24 h following dosing on Day 4 following 4 days of abrocitinib 200 mg once-daily dosing to assess the steady state of abrocitinib M1, M2, and M4
Safety
No serious or severe adverse events (AEs) were reported. There were no AE-related dose reductions or temporary discontinuations due to AEs. The incidence of treatment-emergent adverse events (TEAEs) and treatment-related adverse events (TRAEs) are shown in Table 4. TRAEs reported in 2 or more subjects treated with abrocitinib in the fluvoxamine study included headache in 2 subjects treated with abrocitinib alone and nausea in 2 subjects treated with abrocitinib plus fluvoxamine; in the fluconazole study, these were headache in 4 subjects and fatigue in 2 subjects treated with abrocitinib plus fluconazole; none occurred in the rifampin study; and in the probenecid study, these were nausea in 3 subjects treated with abrocitinib alone and nausea in 2 subjects and headache in 2 subjects treated with abrocitinib plus probenecid. More treatment-related AEs were observed after co-administration of a single dose of abrocitinib with fluconazole, the strongest CYP inhibitor. Increased toxicity is a risk after multiple doses of abrocitinib when co-administered with CYP inhibitors. Increased plasma exposure resulted in more TRAEs, although limited conclusions can be drawn from a single-dose study.
Table 4.
Treatment-emergent and treatment-related adverse events
| Abrocitinib 100 mg single dose (n = 12) | Abrocitinib 100 mg single dose + fluvoxamine 50 mg QD (n = 12) | Abrocitinib 100 mg single dose (n = 12) | Abrocitinib 100 mg single dose + fluconazole 200 mg QD (n = 12) | Abrocitinib 100 mg single dose (n = 12) | Rifampin 600 mg QD (n = 12) | Abrocitinib 200 mg single dose + Rifampin 600 mg QD (n = 12) | Abrocitinib 100 mg single dose (n = 12) | Probenecid 1000 mg BID (n = 12) | Abrocitinib 200 mg single dose + Probenecid 1000 mg BID (n = 12) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Fluvoxamine study | Fluconazole study | Rifampin study | Probenecid study | |||||||
| Subjects with TEAEs, n (%) | 3 (25.0) | 6 (50.0) | 2 (16.7) | 7 (58.3) | 2 (16.7) | 10 (83.3) | 4 (33.3) | 6 (50.0) | 2 (16.7) | 5 (41.7) |
| Subjects with TRAEs, n (%) | 3 (25.0) | 3 (25.0) | 2 (16.7) | 6 (50.0) | 2 (16.7) | 10 (83.3) | 4 (33.3) | 2 (16.7) | 2 (16.7) | 4 (33.3) |
BID twice daily, QD once daily, TEAEs treatment-emergent adverse events, TRAEs treatment-related adverse events
Discussion
Co-administration of abrocitinib with the CYP inhibitors fluvoxamine (a strong CYP2C19 and moderate CYP3A inhibitor) or fluconazole (a strong CYP2C19 inhibitor, moderate CYP2C9 and CYP3A inhibitor) increased active moiety total exposure (AUCinf) by 1.9-fold and 2.5-fold, respectively, and co-administration with the strong CYP2C19/2C9/3A4 inducer rifampin decreased AUCinf by 56%. The OAT3 inhibitor probenecid increased the AUCinf of the active moiety by 1.7-fold. The effect of probenecid was higher on the AUCinf of M1, M2, and M4 (approximately 1.8-fold, 2.2-fold, and 2.2-fold increases, respectively) than that of the abrocitinib parent drug (an approximately 1.3-fold increase), consistent with in vitro results indicating that metabolites M1, M2, and M4 are substrates of the OAT3 transporter, and that the abrocitinib parent drug is not a substrate. These changes to the total active moiety were not as pronounced as those for the abrocitinib parent drug.
Evaluation of the abrocitinib victim DDI risk is important for both the parent drug and its active metabolites, while consideration of the total active moiety is crucial when assessing DDIs of abrocitinib to determine dose adjustment. The proposed recommended dose of abrocitinib for treatment of patients with moderate-to-severe AD aged ≥ 12 years is 100 mg or 200 mg QD. The efficacy and safety of abrocitinib 100 mg and 200 mg QD is supported by the results of exposure–response analysis of the Eczema Area and Severity Index efficacy end point and analysis of platelets for safety [15, 16].
This report presents the effects on the pharmacokinetics of abrocitinib in the presence of CYP inhibitors or inducers. The effects of abrocitinib on CYP enzymes and transporters have been evaluated in both in vitro and in vivo studies. These studies showed that there was a lack of clinically relevant inhibition or induction effect of abrocitinib on CYP3A [17], and abrocitinib did not affect OAT3 transporter activity (Vourvahis et al, in prep.). Hence, no clinically relevant changes in plasma exposures are expected for drugs that are substrates for the evaluated CYP enzymes when co-administered with abrocitinib.
Based on the results of the DDI studies summarized here, the recommended dose adjustments for abrocitinib due to concomitant medicines are listed in Table 5. Co-administration of abrocitinib with strong inducers of CYP2C19 and CYP2C9 (e.g., rifampin) is not recommended, because it could reduce the effectiveness of abrocitinib. Increasing the abrocitinib dose in patients who are taking concomitant strong inducer(s) of CYP2C19/CYP2C9 enzymes has not been evaluated, and can be explored in future studies. An increase in active moiety exposure up to 70% will not result in clinically meaningful changes in the efficacy or safety, based on results from the phase III abrocitinib development program (data on file). The abrocitinib dose should be reduced to half the original dose (from 100 mg to 50 mg QD or from 200 mg to 100 mg QD) in patients receiving drugs that are strong CYP2C19 inhibitors (e.g., fluconazole and fluvoxamine). The OAT3 inhibitor probenecid increased the AUCinf of the unbound active moiety by 66%, an effect that was not considered clinically relevant. Thus, no abrocitinib dose adjustment is necessary in patients taking OAT3 inhibitors such as probenecid.
Table 5.
Recommended doses for abrocitinib due to drug–drug interactions
| Dosing recommendation | Perpetrator drugs | |
|---|---|---|
| Strong CYP2C19 inhibitors | The recommended dose of abrocitinib should be reduced by half to 100 mg or 50 mg once daily | Fluconazole, fluvoxamine, fluoxetine, ticlopidine [8, 22] |
| Strong CYP2C9/CYP2C19 inducers | Not recommended | Rifampin, apalutamide [8, 22] |
| OAT3 inhibitor | No adjustment | Probenecid, teriflunomide [8] |
Evaluating the impact of intrinsic and extrinsic factors on the exposure of a drug in terms of total active moiety has been explored previously [18–21]. In the present DDI evaluations, we focused on changes in the active moiety as opposed to changes in the parent drug alone, because the pharmacologic activity of abrocitinib is attributable to the unbound exposures of abrocitinib, M1, and M2 in the systemic circulation. At steady state with 200 mg QD dosing, the contribution of unbound parent drug, adjusted for relative potencies, to the total active moiety is only ~ 60%, with the other ~ 30% from M2 and ~ 10% from M1 (Table S12). In addition, the inhibition of CYP2C19 and CYP2C9 enzymes resulted in the reduced formation of M1, M2, and M4 metabolites, whereas induction of the enzymes led to enhanced formation of the metabolites. As a result, the overall impact of DDIs on the active moiety is less than that on the parent drug alone.
The steady-state pharmacokinetics profiles of abrocitinib, M1, M2, and M4 suggest formation-rate-limited pharmacokinetics of the 3 metabolites (Table S12). The elimination phases of the metabolites are in parallel with that for the parent drug at steady-state, with comparable values for Tmax between the metabolites and the parent drug, as are the values of t½.
Overall, AEs were classified as mild to moderate in severity. The most common TEAEs that occurred in subjects treated with abrocitinib across studies were headache and nausea. There were no deaths, severe AEs, serious AEs, or AE-related discontinuations in the studies.
The studies were conducted in healthy volunteers in accordance with regulatory DDI guidance. A limitation is that the data analyzed in this report were largely generated from White male healthy volunteers, with only 14 non-White (29%) participants and only 9 females (19%) out of 48 participants enrolled in the studies. Population pharmacokinetic analyses have been performed for abrocitinib and its metabolites using the data collected across the clinical development program, which included approximately 40% female participants and 35% non-White participants (data on file). Based on the population analyses, no sex-related differences were identified in the pharmacokinetics of abrocitinib and its metabolites. Asian subjects demonstrated a 43% increase in Cmax and a 51% increase in AUC at steady-state compared to White and Black subjects of the same weight (data on file). An assessment of DDIs in a patient population with AD through population pharmacokinetics analysis is not possible, because concomitant use of inhibitors and inducers of CYP2C19 and CYP2C9 was not permitted in phase II and III trials. Studies were carried out with strong inhibitors of CYP2C19 and moderate inhibitors of CYP3A4 and CYP2C9. Based on the study results and separate physiological-based pharmacokinetic modeling and simulation analysis using Simcyp® (data on file), it is predicted that interactions with moderate and weak CYP2C9 inhibitors would not require dose adjustment. CYP2C19 and CYP2C9 genotyping data were collected, consistent with other abrocitinib phase I studies. The results were pooled to evaluate the overall impact of genetic polymorphisms on the exposures of abrocitinib and its active moiety and will be reported in a separate analysis.
Conclusion
In summary, these studies demonstrate that exposures of abrocitinib total active moiety are influenced by the co-administration of medicinal products that strongly inhibit or induce enzymes CYP2C19 or CYP2C9. The abrocitinib dose should be reduced to half of the original dose (from 100 mg to 50 mg QD or from 200 mg to 100 mg QD) in patients receiving drugs that are strong CYP2C19 inhibitors (e.g., fluconazole and fluvoxamine). The co-administration of abrocitinib with strong inducers of CYP2C19 and CYP2C9 (e.g., rifampin) is not recommended as it could potentially reduce the effectiveness of abrocitinib. In addition, inhibition of the OAT3 transporter does not have a clinically relevant effect on abrocitinib active moiety exposure; therefore, no abrocitinib dose adjustment is required in patients taking OAT3 inhibitors such as probenecid. It is important to evaluate the abrocitinib victim DDI risk for both the parent drug and its active metabolites, and it is crucial to consider the total active moiety when assessing DDIs with abrocitinib for determining dose adjustment.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
Editorial/medical writing support under the guidance of authors was provided by Renee Gordon, PhD, CMPP, and Mariana Ovnic, PhD, at ApotheCom, Philadelphia, PA, USA, and was funded by Pfizer Inc., New York, NY, USA, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163:461–464).
Declarations
Funding
These trials were sponsored by Pfizer Inc.
Conflicts of interest
All authors are employees and stockholders of Pfizer Inc.
Availability of data and material
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Code availability
Not applicable.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by XW, MO, AW, JAW, and ST. Bioanalytical data preparation and interpretation were performed by VL and ST. VL performed all statistical analyses of pharmacokinetics data. AW and ST contributed to study safety. The first draft of the manuscript was written by XW; all authors critically reviewed the drafts and approved the final manuscript.
Ethics approval
This research was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments. The final protocol, any amendments, and informed consent documentation were reviewed and approved by the Independent Ethics Committee at the investigational center participating in the study.
Consent to participate
Written informed consent was obtained from all participants prior to enrollment.
Consent for publication
Not applicable.
References
- 1.Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfo namide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61:1130–1152. doi: 10.1021/acs.jmedchem.7b01598. [DOI] [PubMed] [Google Scholar]
- 2.Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396:255–266. doi: 10.1016/s0140-6736(20)30732-7. [DOI] [PubMed] [Google Scholar]
- 3.Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:863–873. doi: 10.1001/jamadermatol.2020.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Efficacy and safety of oral janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 2019;155:1371–1379. doi: 10.1001/jamadermatol.2019.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peeva E, Hodge MR, Kieras E, Vazquez ML, Goteti K, Tarabar SG, et al. Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: a phase 1, randomized, placebo-controlled, dose-escalation study. Br J Clin Pharmacol. 2018;84:1776–1788. doi: 10.1111/bcp.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dowty M, Yang X, Lin J, Bauman J, Doran A, Goosen T, et al. P190—The effect of CYP2C9 and CYP2C19 genotype on the pharmacokinetics of PF 04965842, A JAK1 inhibitor in clinical development. Drug Metab Pharmacokinet. 2020;35:S80. doi: 10.1016/j.dmpk.2020.04.191. [DOI] [Google Scholar]
- 7.Luvox (fluvoxamine maleate) tablets, for oral use. Prescribing information. ANI Pharmaceuticals, Inc. 2019.
- 8.US Food and Drug Administration. Drug development and drug interactions: table of substrates, inhibitors and inducers. 2020. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed 30 Nov 2021.
- 9.Diflucan (fluconazole tablets) (fluconazole for oral suspension). Prescribing information. Pfizer Inc. 2020.
- 10.Rifadin (rifampin capsules USP) (rifampin for injection USP). Prescribing information. Sanofi Aventis Inc. 2020.
- 11.Probenecid (probenecid tablet, film coated). Prescribing information. Mylan Pharmaceuticals Inc. 2006.
- 12.Wang EQ, Le V, O'Gorman M, Tripathy S, Dowty ME, Wang L, et al. Effects of hepatic impairment on the pharmacokinetics of abrocitinib and its metabolites. J Clin Pharmacol. 2021;61:1311–1323. doi: 10.1002/jcph.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leclercq L, Cuyckens F, Mannens GS, de Vries R, Timmerman P, Evans DC. Which human metabolites have we MIST? Retrospective analysis, practical aspects, and perspectives for metabolite identification and quantification in pharmaceutical development. Chem Res Toxicol. 2009;22:280–293. doi: 10.1021/tx800432c. [DOI] [PubMed] [Google Scholar]
- 14.Dowty ME, Lin TH, Jesson MI, Hegen M, Martin DA, Katkade V, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. 2019;7:e00537. doi: 10.1002/prp2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojciechowski J, Malhotra BK, Wang X, Fostvedt L, Valdez H, Nicholas T. Population pharmacokinetic-pharmacodynamic modeling of platelet time courses after administration of abrocitinib. In: Presented at: 11th American Conference on Pharmacometrics; November 9–13, 2020; Virtual.
- 16.Fostvedt L, Wojciechowski J, Malhotra BK, Wang X, Valdez H, Rojo R, et al. Zero-inflated beta regression exposure-response modeling of the eczema area and severity index for patients taking abrocitinib. In: Presented at: 11th American Conference on Pharmacometrics; November 9–13, 2020; Virtual.
- 17.Wang X, Huh Y, Curto M, Chan G, Dowty ME, O’Gorman M, et al. Assessment of cytochrome P450 3A inhibition and induction of abrocitinib: midazolam drug drug interaction (DDI) study and oral contraceptive DDI study. J Am Acad Dermatol. 2020;83:S(AB151). doi: 10.1016/j.jaad.2020.06.691. [DOI] [Google Scholar]
- 18.Brynne N, Dalen P, Alvan G, Bertilsson L, Gabrielsson J. Influence of CYP2D6 polymorphism on the pharmacokinetics and pharmacodynamic of tolterodine. Clin Pharmacol Ther. 1998;63:529–539. doi: 10.1016/S0009-9236(98)90104-7. [DOI] [PubMed] [Google Scholar]
- 19.Adams VR, Leggas M. Sunitinib malate for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors. Clin Ther. 2007;29:1338–1353. doi: 10.1016/j.clinthera.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 20.Namour F, Diderichsen PM, Cox E, Vayssiere B, Van der Aa A, Tasset C, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet. 2015;54:859–874. doi: 10.1007/s40262-015-0240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scordo MG, Spina E, Facciolà G, Avenoso A, Johansson I, Dahl ML. Cytochrome P450 2D6 genotype and steady state plasma levels of risperidone and 9-hydroxyrisperidone. Psychopharmacology. 1999;147:300–305. doi: 10.1007/s002130051171. [DOI] [PubMed] [Google Scholar]
- 22.W Drug Interactions Solutions, School of Pharmacy. DIDB—The Drug Interaction Database. Seattle, WA: University of Washington. https://www.druginteractionsolutions.org/solutions/drug-interaction-database/. Accessed 30 Nov 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.