

Abstract
Objectives.
Current treatments are effective only in 30% of lupus nephritis patients emphasizing the need for novel therapeutic strategies. To develop mechanistic hypotheses and explore novel biomarkers, we analyzed the longitudinal urinary proteomic profiles in patients with lupus nephritis undergoing treatment.
Methods.
We quantified 1,000 urinary proteins in 30 patients with lupus nephritis at the time of the diagnostic renal biopsy and after 3, 6, and 12 months. The proteins and molecular pathways detected in the urine proteome were then analyzed with respect to baseline clinical features and longitudinal trajectories. The intrarenal expression of candidate biomarkers was evaluated using single cell transcriptomics of renal biopsies from lupus nephritis patients.
Results.
Our analysis revealed multiple biological pathways including chemotaxis, neutrophil activation, platelet degranulation, and extracellular matrix organization that could be noninvasively quantified and monitored in the urine. We identified 237 urinary biomarkers associated with lupus nephritis as compared to controls without SLE. IL-16, CD163, and TGF-β mirrored intrarenal nephritis activity. Response to treatment was paralleled by a reduction of urinary IL-16, a CD4 ligand with proinflammatory and chemotactic properties. Single cell RNA sequencing independently demonstrated that IL16 is the second most expressed cytokine by most infiltrating immune cells in lupus nephritis kidneys. IL-16 producing cells were found at key sites of kidney injury.
Conclusion.
Urine proteomics may profoundly change the diagnosis and management of lupus nephritis by noninvasively monitor active intrarenal biological pathways. These findings implicate IL-16 in lupus nephritis pathogenesis designating it as a potentially treatable target and biomarker.
Introduction
Lupus nephritis (LN) is a severe manifestation of systemic lupus erythematosus (SLE) that frequently leads to end-stage kidney disease despite treatment 1. Diagnosis and treatment of LN rely on histopathological features of kidney biopsies in patients with proteinuria. Kidney biopsies have an indispensable role in that they can distinguish active nephritis from chronic damage, both of which manifest with proteinuria. However, kidney biopsies have limitations. Most notably, histology does not capture patient-specific active biological pathways. Further, the histological class frequently changes on repeat kidney biopsies, suggesting that the histological classification may artificially divide patients based on one point in time 2,3. Procedure-related complications may occur 4, and up to 35% of kidney biopsies may fail to obtain an adequate sample 5. Access to kidney biopsies may delay diagnosis and treatment, and can be limited by antithrombotic and anticoagulation treatments, severe thrombocytopenia, and in resource poor settings. Finally, because the presence of proteinuria implies that underlying kidney damage has already happened, kidney biopsies are a lagging indicator. Thus, there is a pressing need for a noninvasive biomarker to probe in “real-time” the active molecular pathological processes in the kidney and to monitor them over time in response to treatment.
Several available biomarkers correlate with histological features, but none are currently used in clinical practice 6,7. These lack the sensitivity and specificity to detect active renal inflammation, predict flares, and reliably inform prognosis, and do not add actionable information in addition proteinuria or renal function 6,7. Unbiased proteomic screenings carry a high potential for discovery, but these have been limited to the evaluation of proteins or peptides sufficiently abundant to be detectable by mass spectrometry 8,9. More sensitive aptamer-based arrays identified candidate urinary biomarkers associated with proteinuria, but their ability to predict nephritis activity and clinical outcomes is still to be determined 10.
Management of LN could be greatly enhanced by a resource which can identify candidate biomarkers that predict histological features and clinical outcomes, as well as infer the renally active biologically pathways. Here, we used a glass slide based protein microarray to screen and quantify 1000 proteins covering a wide range of biological processes in longitudinal urine samples of patients with LN (starting at the time of biopsy) to develop mechanistic hypotheses and explore novel biomarkers. This array allowed the unbiased, precise and sensitive quantification of the concentration of each of the 1000 proteins as validated in previous studies11–13. We found that protein expression patterns define distinct molecular pathways that are differentially expressed among LN patients. We also discovered that IL-16, a proinflammatory chemokine, is tightly associated with LN activity and may have role in LN pathogenesis thus nominating IL-16 as a potentially treatable target.
Methods
Patients and sample collection.
This study enrolled SLE patients with urine protein/creatinine ratio greater than 0.5 undergoing clinically indicated renal biopsy. Only patients with a pathology report confirming LN were included in the study. Renal biopsies were scored by one renal pathologist at each site of the two sites according to the International Society of Nephrology/Renal Pathology Society (ISN/RPS) guidelines and NIH activity and chronicity indices14. Clinical information, including serologies, were collected at the most recent visit before the biopsy. Response status at week 52 was defined as follows. Complete: pr/cr ≤ 0.5, normal serum creatinine (sCr) or <25% increase from baseline if abnormal, and prednisone ≤ 10mg daily; partial: pr/cr > 0.5 but ≤ 50% of the baseline value and identical sCr and prednisone rules as complete response; no response: pr/cr > 50% of baseline value or new abnormal elevation of sCr or ≥ 25% from baseline or prednisone ≥ 10mg daily. Urine samples from healthy volunteers (all females, median age 42 years [32–54], 3 identifying as Caucasian and 4 as African American) were included. Urine specimens were acquired on the day of the biopsy (before the procedure) at 2 clinical sites in the United (Johns Hopkins University, JHU, and New York University, NYU). For the validation cohort (n=101), urine samples were collected on the day of (73%) or within 3 weeks (27%) of kidney biopsy. Serological features and complement levels were determined at the clinical visit preceding the biopsy. Proteinuria was measured on or near the day of the biopsy.
Study approval.
Human study protocols were approved by the institutional review boards at JHU and NYU, and written informed consent was received from all participants.
For healthy controls, IRB approval was obtained from the Oklahoma Medical Research Foundation. After informed consent, controls were recruited through the Oklahoma Rheumatic Disease Research Cores Center, cohort matching for gender, race, ethnicity and age. Subjects were screened by a questionnaire and tested negative for ANA, dsDNA, Chromatin, RiboP, Ro, La, Sm, SmRNP, RNP, Centromere B, Scl-70, and Jo-1 antibodies. Samples were processed, stored, and shipped using the AMP RA/SLE protocols to align with the patient samples.
Urine Quantibody assay
The Kiloplex Quantibody protein array platform (Raybiotech) was used for screening urine samples as previously described12. Validation was performed using an immunoquantitative (PCR-Based) IL-16 ELISA (Raybiotech) to match and improve the sensitivity and dynamic range provided by the kiloplex array. These were summarized in the Supplementary Methods.
Renal tissue single cell RNA sequencing
Renal tissue was collected, stored and processed as previously described 15. Briefly, research biopsy cores were collected from consented subjects as an additional biopsy pass or tissue from routine clinical passes. Only biopsies with confirmed LN were included. Kidney tissue was frozen on site and shipped to a central processing location where it was thawed and disaggregated. Individual cells were retrieved and sorted by flow cytometry. For each sample, 10% of the sample was allocated to sort CD10+CD45− epithelial cells as single cells, and the remaining 90% of the sample was used to sort CD45+ leukocytes as single cells. For each single cell, the whole gene expression profile was sequenced using the CEL-Seq2 method.
Statistical analyses.
The differential protein abundance was calculated using a moderated t statistic. To achieve normal distribution, the protein abundances were log-transformed after adding 10% (arbitrary constant empirically shown not to significantly alter distributions) of the lowest measured abundance to remove zeros. With 30 LN and 7 HD samples, using a two-sided .05-level test adjusting for 1000 comparisons (Bonferroni), there was 80% power to detect a difference in mean peptide magnitude of 1.2 standard deviations (i.e., an effect size of 1.2). Concentrations of all urinary proteins for all urine samples were available without missing data. Clustering was performed using the Ward’s minimum variance method. ROC curves and areas under the curve (AUCs) were calculated using the function roc within the pROC R package. The impact of confounders on the association between the ISN Activity Index and the urinary abundance of a biomarker was tested using one confounder at the time (given limited sample size) using a linear regression model as follows: activity ~ biomarker_abundance + confounder. The models were fitted using the lm function within the stats R package. See Supplementary Methods for pathway enrichment analysis. Pearson correlation coefficients were used throughout the manuscript. All analyses were performed in R.
Prevalence of cytokine positive cells.
Analysis of cytokine-positive cells was based on a compendium of 237 cytokines obtained from Gene Ontology 16 and manually extended using the Cytokine Registry (https://www.immport.org/resources/cytokineRegistry), the iTalk database 17, and the International Union of Basic and Clinical Pharmacology (IUPHAR) and British Pharmacological Society (BPS) database. For each cytokine, we calculated the prevalence of the cells with at least one transcript over the total number of cells.
Immunohistochemistry.
Results
Urine proteomics identifies biologically relevant active pathways in LN
Urine samples from 30 subjects with active LN were collected near or at the time of a renal biopsy. Clinical and demographic characteristics are summarized in Supplementary Table 1. Compared to healthy donors, there were 237 proteins significantly elevated in the urine of patients with LN (FDR < 10%) as displayed in Figure 1A. This list included both novel and previously described urinary biomarkers (Supplementary File 1). Pathway enrichment analysis of the proteins the proteins significantly elevated in LN identified 12 enriched non-overlapping pathways, including relevant biological processes such as chemotaxis, neutrophil activation, platelet degranulation, and extracellular matrix organization (Figure S1). Hierarchical clustering using enriched pathways segregated LN patients into 2 groups, with 80% of those who later achieved a complete renal response being in the same group with overall less inflammatory pathways (OR 12.6, p=0.03) (Figure 1B). Baseline parameters such as proteinuria, creatinine, histologic activity or chronicity scores, or class were present in similar frequencies in both clusters suggesting that urine proteomics may provide unique informative features (Figure 1B).
Figure 1. Identification of pathogenic pathways by urine proteomics.
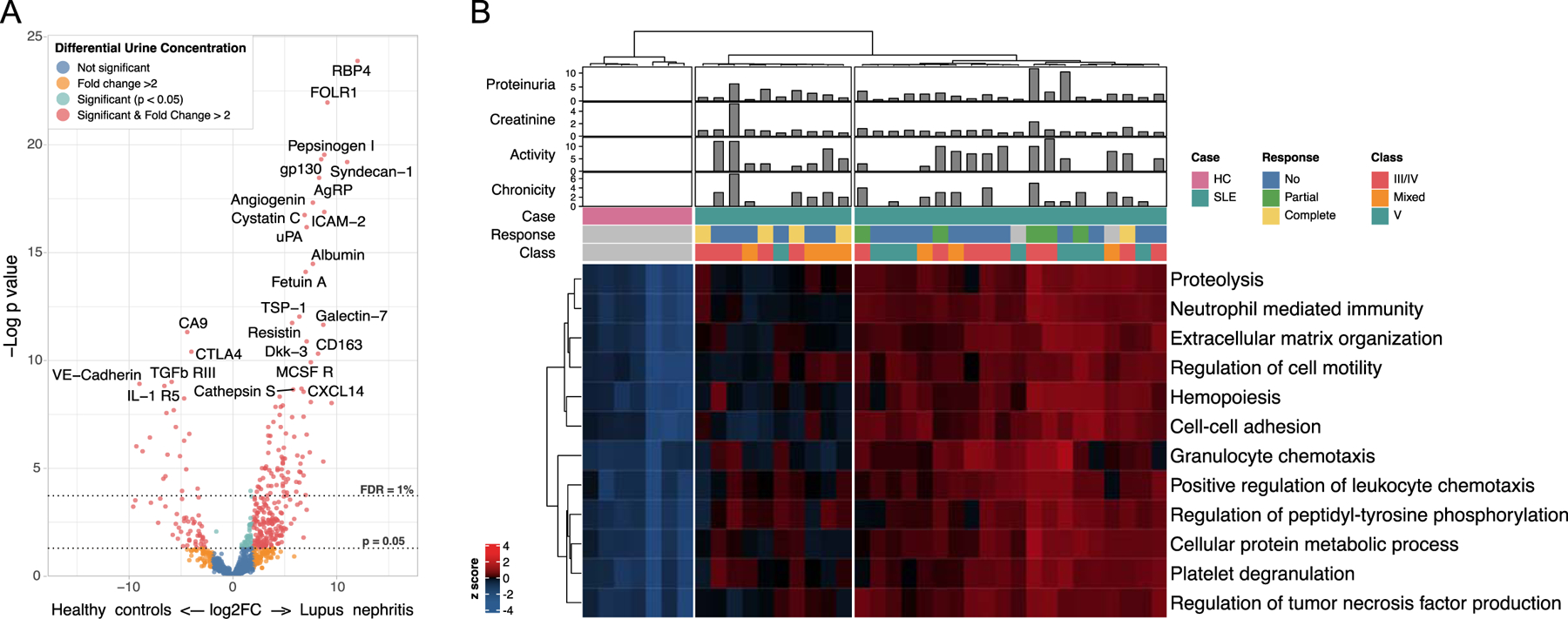
(A) Volcano plot illustrating the differential abundance of 1000 urinary proteins in lupus nephritis (LN, n=30) and healthy controls (HC, n=7). There were 237 proteins significantly more abundant in LN (>2 fold, false discovery rate (FDR) <10%, moderated t test). Thresholds for an FDR of 1% and nominal p=0.05 are shown by dotted lines. (B) Heatmap of the abundance of the 12 non-overlapping pathways enriched in LN urine samples by pathway enrichment analysis (GO Biological Process). Twenty of the thirty patients displayed a LN cluster with higher abundance of all pathways, whereas the patients in the other cluster exhibited an intermediate abundance as compared to healthy controls. Clustering was otherwise not explained by other clinical variables such as proteinuria, renal function, nephritis activity, chronic damage, or class. Values were scaled by rows. Clustering was performed using the Ward’s minimum variance method.
Identification of urinary biomarkers of renal histology.
We sought to identify urinary proteins that could identify renal histology. LN can be classified in two broad categories based on the presence of a glomerular endocapillary immune infiltrate or “proliferation”. Proliferative LN (ISN class III or IV) is a more aggressive phenotype associated with glomerular endocapillary hypercellularity, abundant immune cell infiltration and higher risk of permanent renal damage. Compared to pure membranous LN (n=9), patients with proliferative LN (n=14) showed higher concentration of several urine cytokines and molecules involved in immune activation and chemotaxis (Figure 2A-B). IL-16 was the most significantly enriched urinary protein in proliferative LN (Figure 2A). Pathway enrichment analysis revealed that the pattern of chemokines matched the chemokine released in response to interferon-gamma (IFN-γ), IL-1β, and TNF (Figure 2B).
Figure 2. Proteomic profile of proliferative lupus nephritis.
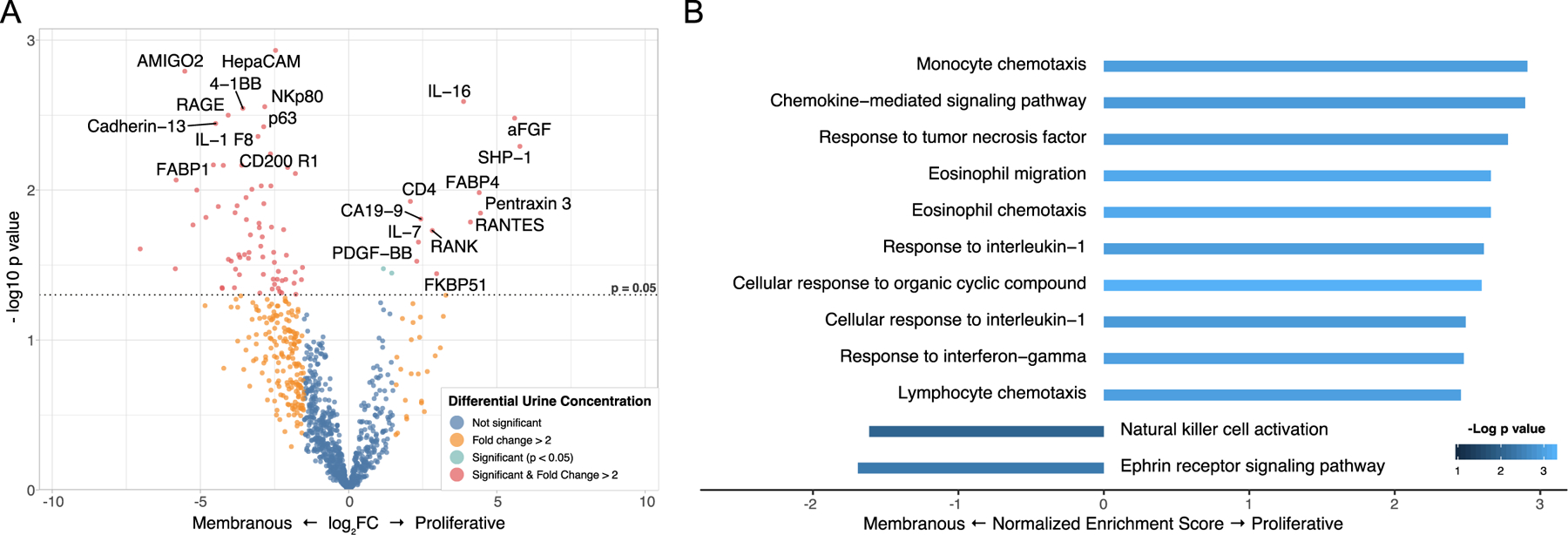
(A) Volcano plot illustrating the differential abundance of 1000 urinary proteins in proliferative LN (n=14) and pure membranous LN (n=9). (B) Pathway enrichment analysis (GO Biological Process) of the urinary proteomic profile revealed that chemotaxis was the process most enriched in proliferative LN. In particular, these were chemokines secreted in response to TNF, IL-1, and IFN-γ. The enrichment FDR (GSEA rank permutation) was <5% for all pathways except for “Natural killer activation” (16%).
Many of the urinary proteins that were differentially abundant when comparing proliferative and membranous were not significantly more abundant when comparing all LN patients to healthy controls. In fact, although most of the proteins enriched in proliferative LN were generally more abundant in LN vs healthy controls, these were not among the most abundant (> 2 SD) (Figure S2 A-B). This is because the first comparison (LN vs healthy) is aimed to identify proteins that are generally more abundant in all LN patients, regardless of class. Not surprisingly, the most abundant protein in all LN was RBP4, a general marker of tubular impairment 18. These findings indicate that contrasting well defined subgroups allowed to identify relevant biomarkers that could have been missed by analyzing all LN patients together. Different pathogenic processes may underlie each histological subgroup and thus these biomarkers may provide insight into the relative active pathways.
Urinary IL-16 reflects histological activity.
The degree of histological activity is often used to inform clinical decisions, so we sought to identify noninvasive urinary biomarkers that reflect histological activity. We studied the correlation of the urinary abundances of all 1000 biomarkers in urine samples collected at the time of biopsy with the histological NIH activity index. We found that IL-16 was the urinary protein most strongly positively correlated with the NIH activity index (Pearson’s r 0.73, p = 1.2*10−5, FDR <10%, n=28) followed by CD163, and TGF-β (FDR <10%) (Figure 3A-D). We validated the significant concurrent correlation between urinary IL-16 abundance and NIH activity index in an independent cohort of 101 patients (r =0.59, p = 9.3*10−11, Figure S3 and Supplementary Table 2) and with a PCR-based ELISA (Figure S4). Notably, IL-16 was the only one not associated with proteinuria (Figure 3H), suggesting the potential to provide actionable information in addition to classic biomarkers such as proteinuria. In multivariate models, IL-16, CD163, and TGF-β retained their association with histological activity after adjustment for multiple confounders, including proteinuria (Supplementary Table 3). The pathways associated with histological activity are displayed in Figure S4.
Figure 3. Urinary biomarkers of histological nephritis activity.
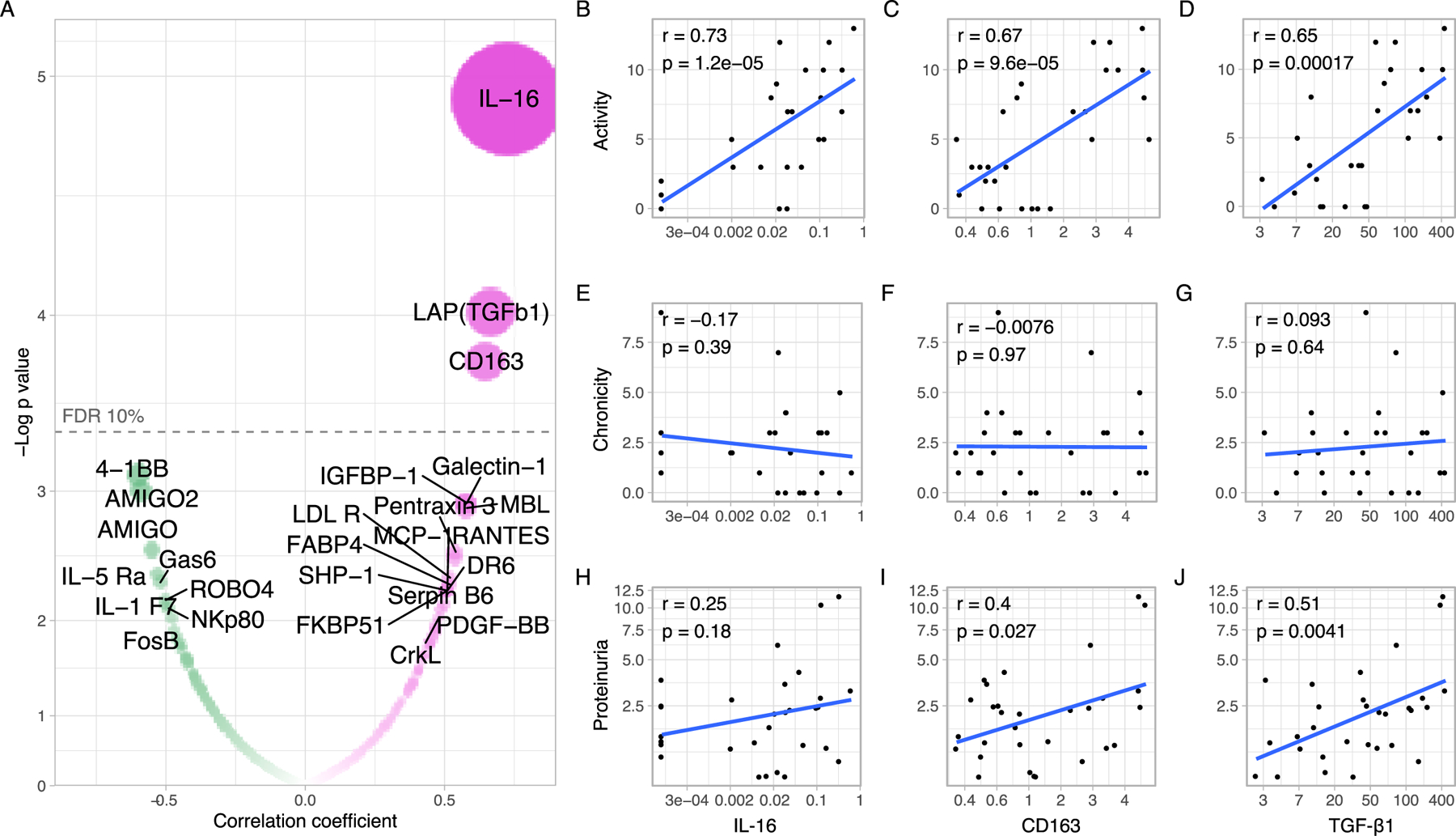
(A) Pearson’s correlations of the urinary abundance of 1000 proteins and the histological NIH Activity Index in near or same day renal biopsies. Each dot represents a protein within the array. The dashed line marks the significance threshold after correcting for multiple comparisons (FDR 10%). The area of the dot is proportional to the absolute of the correlation coefficient. Three proteins showed an FDR <10%. The FDR of IL-16 was 1.2%. Scatter plots displaying the Pearson’s r and p values of correlations of the urinary abundance of IL-16, CD163, and TGF-β1 with the NIH Activity Score (B-D), NIH Chronicity Score (E-G), and proteinuria (H-J).
In addition to having the strongest correlation with histological activity, IL-16 was the urinary protein most associated with proliferative LN (Figure 2A). The receiver operating characteristic curve revealed that IL-16 was a promising urinary biomarker to identify patients with proliferative LN with an area under the curve (AUC) of 0.85 (p = 0.016) and 0.89 (p = 0.037) in association with CD163 and TGF-β (Figure S5).
Urinary biomarkers correlating with activity decrease with clinical response in longitudinal samples.
A goal of immunosuppression in LN is to eradicate pathological renal inflammation to ultimately prevent irreversible renal damage and preserve function. The NIH activity index captures many renal inflammatory features and, as a consequence, it improves with treatment in patients achieving renal remission 2,19. However, it is impractical to monitor in clinical practice as it requires frequent repeat renal biopsies. Thus, we hypothesized that the 3 urinary biomarkers associated with histological activity would decline over time in patients who are responding to treatment and might serve as noninvasive biomarkers of response. The urinary concentration of all 3 candidate biomarkers declined in complete and partial responders but not in non-responders (Figure 4A-C). The average decline was most striking in IL-16 with a decrease in partial and complete responders by week 12. CD163 concentration improved by week 12 in complete responders but not in partial responders. TGF-β showed a more modest decline.
Figure 4. Biomarkers associated with nephritis activity decrease in responders.
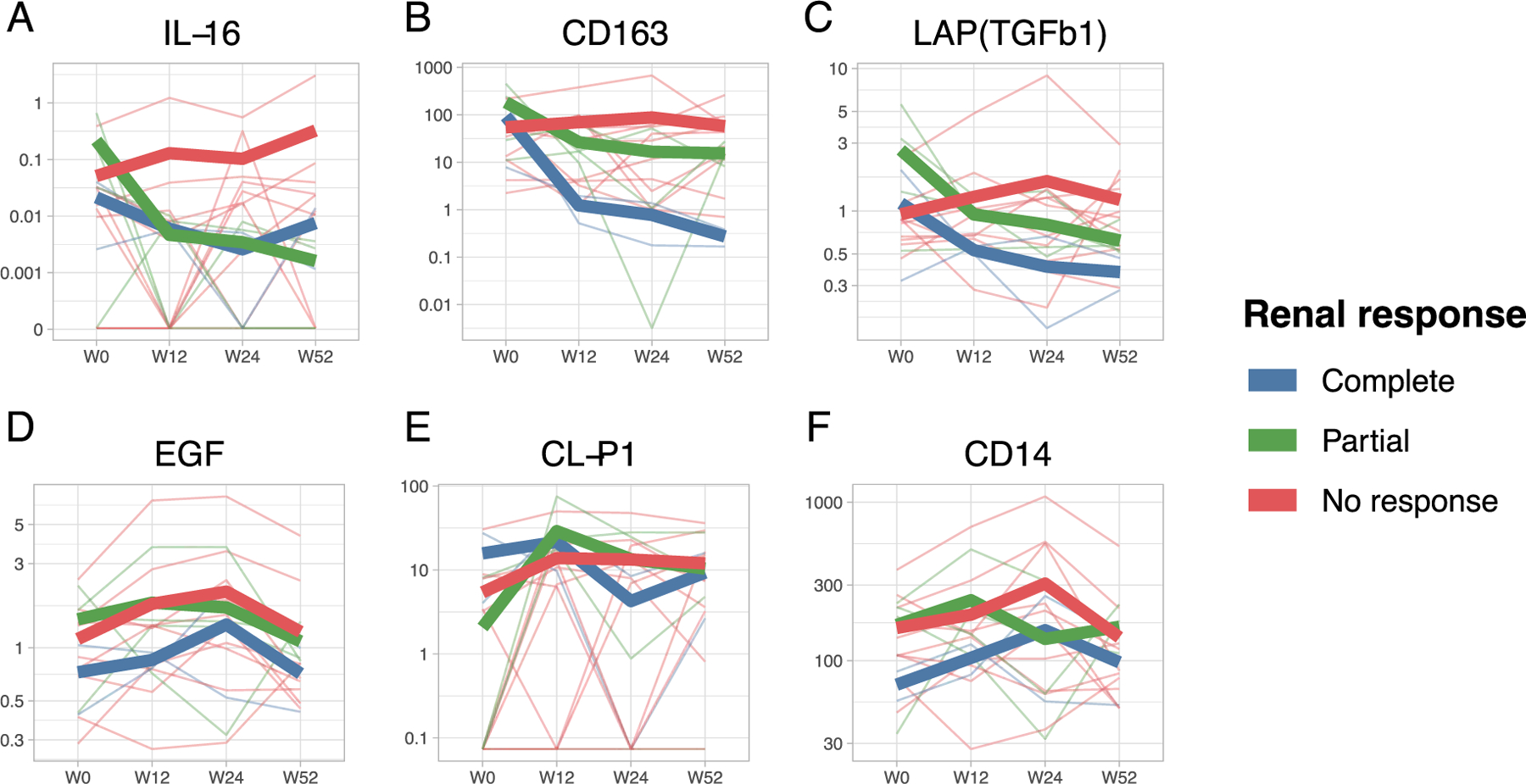
Urinary concentration of all biomarkers was measured at time of biopsy (or “week 0”, W0) and after 12, 24, and 52 weeks. Thin lines depict the trajectories of each patient categorized based on the response status determined at week 52. Thick lines represent the average for each group. The urinary concentration of the 3 biomarkers significantly correlated with histological activity declined in complete and partial responders but not in non-responders (A-C). In contrast, 3 biomarkers that did not correlate with histological activity (Pearson’s r range −0.0018 – 0.0015, p=ns) did not show a decline over time.
Since response status is defined by reduction of proteinuria, we wanted to ensure that the observed biomarker trajectories were not simply a reflection of a decline in all urinary protein in responders. The trajectories of 3 urinary proteins that were selected among those that did not correlate with histological activity demonstrated that there was no non-specific decline (Figure 4D-F). These findings indicate that IL-16, CD163, and TGF-β trajectories represent a specific decrease in the production and excretion of these molecules and, as they correlated with activity at baseline, likely reflect a corresponding improvement of intrarenal LN activity supporting their value as biomarkers.
IL16 is one the most expressed cytokines in kidney infiltrating immune cells in LN.
To determine whether the urinary concentration of the 3 candidate biomarkers reflects an active intrarenal process rather than passive filtration through a damaged glomerular membrane, we evaluated the intrarenal relative gene expression using single cell RNA sequencing of LN renal biopsies. IL16 was abundantly expressed by most immune infiltrating cells, CD163 by a subset of myeloid cells, and TGFB1 mostly by NK cells (Figure 5A-D).
Figure 5. High expression of IL16 in lupus nephritis kidney.
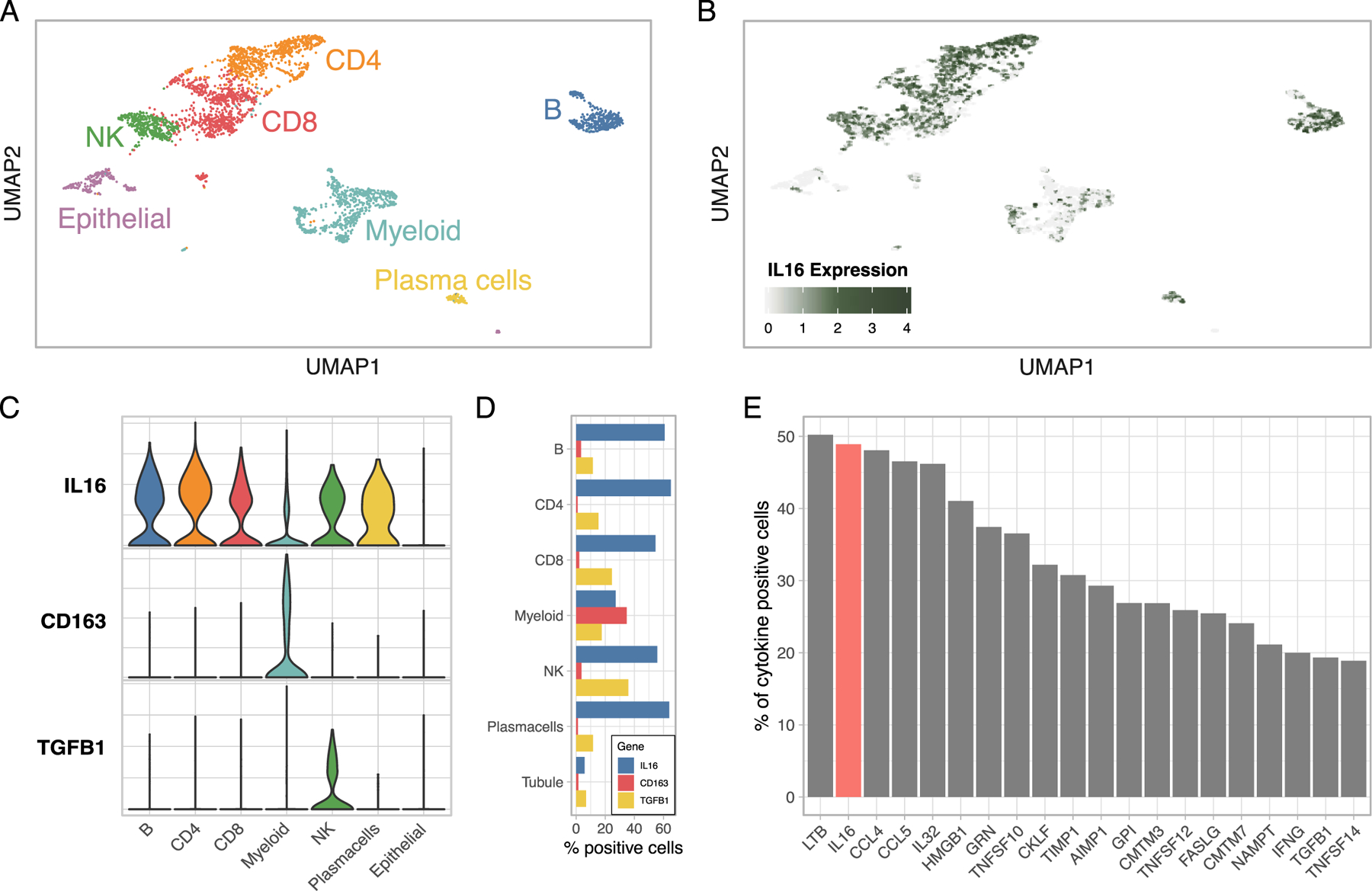
(A) UMAP plot of scRNA-seq of renal biopsies (3131 cell) by lineage. (B) Feature plot displaying IL16 expression at single cell level. Violin (C) and bar (D) plots summarizing the expression of the genes coding for the urinary proteins associated with nephritis activity. IL16 was abundantly expressed by most kidney infiltrating immune cells, CD163 mostly by macrophages, and TGFB1 by NK cells. (E) Prevalence (%) of cytokine positive cells out of a compendium of 237 cytokines ranked decreasingly (top 20 are shown). IL16 (in red) was the second most expressed cytokine in LN kidneys.
In LN, most of IL-16 expression was in immune infiltrating cells, especially the lymphoid lineage (Figure 5C-D). In renal allograft rejection, single cell RNA-seq showed that IL16 is expressed by endothelial, epithelial, and immune cells, but immune cells were the main source (Figure S6A)20. Conversely, in the healthy kidney, single nuclear RNA sequencing and ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) revealed substantial IL16 expression by podocytes, fibroblasts, endothelial, mesangial, and proximal tubular cells (Figure S5B-C)21,22. These findings suggest that while immune cells are likely the major intrarenal source of IL-16 in LN, IL-16 secretion by endothelial and tubular cells may precede immune infiltration. Speculating, this initial event can then be amplified by infiltrating immune cells as seen in LN and allograft rejection.
Finally, we explored whether IL16 was disproportionally more expressed as compared to other cytokines in LN. Out of a compendium of 237 cytokines, IL16 was the second most commonly expressed cytokine (49% of all infiltrating immune cells) (Figure 5E). These findings independently nominate IL-16 as a major cytokine involved in LN.
Tissue expression of IL-16 correlate with LN activity and urinary IL-16 abundance.
To establish the location of IL-16 secreting cells in renal tissue, we performed immunohistochemical staining of human IL-16 in 7 LN kidney biopsies with matching urine IL-16 collected at or near the time of biopsy. We observed abundant interstitial and glomerular IL-16 expression in proliferative LN (Figure 6 and S7A-C), with the exception of one case (Figure S7D) in which the activity index was uncharacteristically low (2) and IL-16 was not detectable in the urine. In contrast, there was very scant IL-16 positivity in membranous LN (Figure 6 and S7E-G) and marginal in a class I LN biopsy used as negative control (Figure S7H). These findings matched the urinary IL-16 profile. Furthermore, there was a qualitative correlation between the number of IL-16 positive cells and urinary IL-16 abundance as well as the NIH activity index (Figure 6). This was particularly evident for glomerular IL-16 positive cells. These findings indicate that IL-16 is intrarenally produced in proliferative LN and urinary IL-16 reflects the abundance of intrarenal IL-16 positive cells and LN activity.
Figure 6. IL-16 positive cells are abundant in proliferative LN and qualitatively correlate with urinary IL-16 and LN activity.
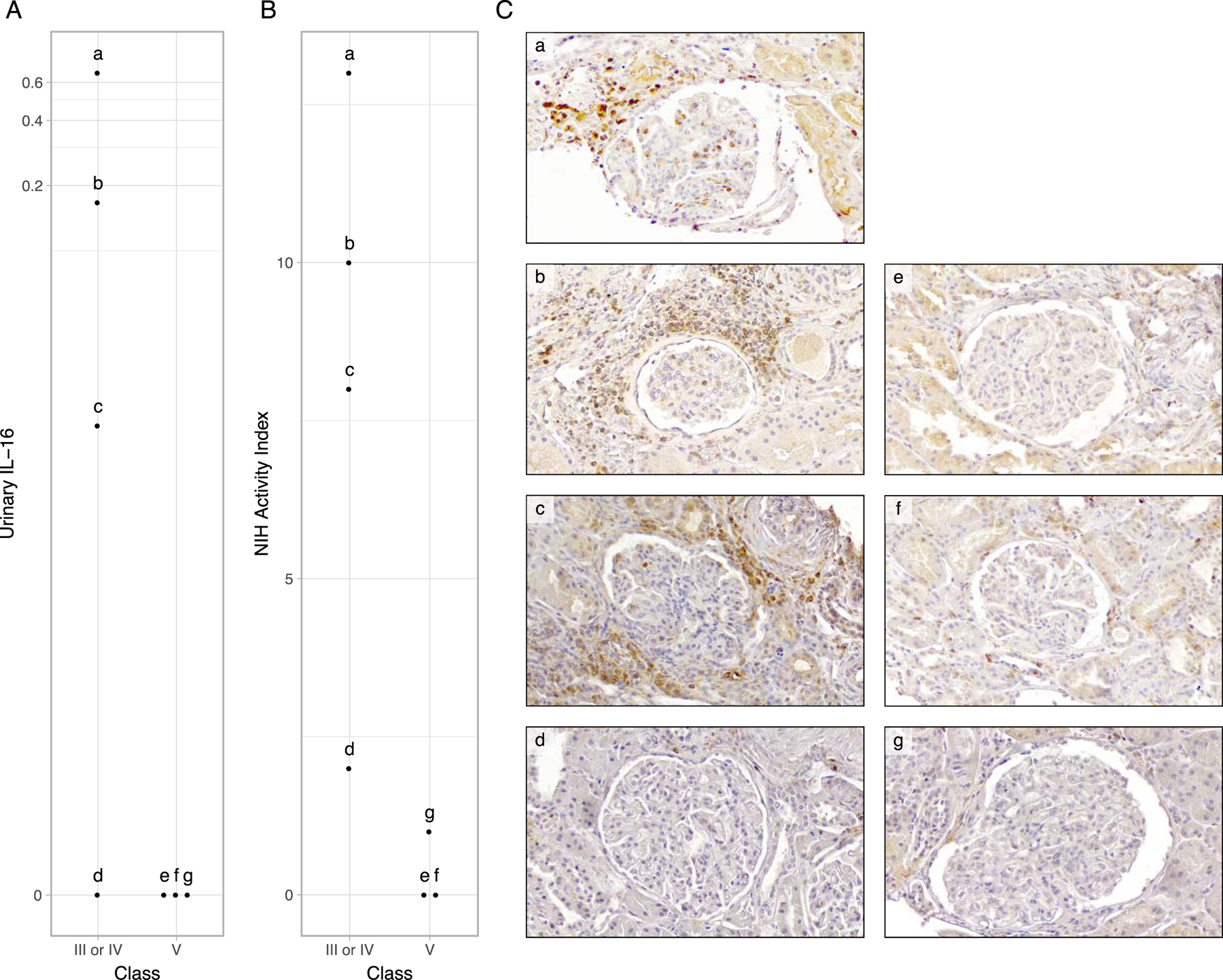
Immunohistochemical (IHC) staining of human IL-16 were performed in 7 LN kidney biopsies with matching urine IL-16 collected at or near the time of biopsy. The corresponding urinary abundance of IL-16 (A) and NIH Activity Index (B) of the patients whose biopsy depicted in C are plotted according to ISN class. Lower case letters (“a” to “g”) in A, B, and C identify information from the same patients. (C) IHC of IL-16 in 4 proliferative LN biopsy sections (a-d) and 3 pure membranous LN (e-g). Abundance of IL-16+ cells was noted in proliferative LN (C, a-d) with qualitatively more prominent intraglomerular IL-16 positivity in patients with higher urinary IL-16 and NIH Activity Index. Images are magnified 33.6x. Lower magnification images with larger representation of the interstitium are displayed in Supplementary Figure 6.
Discussion
Leveraging urine proteomics in patients with LN and healthy controls, the results of this study confirmed that the pathological processes in LN can be noninvasively captured and monitored over time. We found: (1) 237 urinary proteins associated with LN that represented at least 12 distinct molecular pathways; (2) a strong chemokine signature characterizing the urine of patients with proliferative LN; and (3) several candidate biomarkers to detect active nephritis that can be monitored over time to assess response to treatment. Overall IL-16 emerged as the most robust correlate of histological activity implying a role in LN pathogenesis and thus subsequent translation to clinical application both as a biomarker and treatable target.
Proteomic analysis revealed that the intrarenal activation of several pathogenic mechanisms contributing to LN can be quantified in the urine. These biological processes were previously implicated in LN including neutrophil immunity 23,24, platelet degranulation 25, extracellular matrix organization 26, and chemotaxis 27. Patients did not cluster based on the abundance of a single or a group of signatures. Rather, we observed two clusters characterized by high and intermediate abundance of all signatures, respectively. This is consistent with previous findings from an agnostic approach to urine proteomics in LN that showed that patients stratify on a gradient 27. Importantly, 80% of complete responders clustered in the intermediate abundance group. The predictive value of this approach needs to be validated in a larger cohort given the small number of responders.
In this study, urinary abundance of proteomic signatures was independent from proteinuria, suggesting that these signatures specifically reflect active biological processes rather than a non-specific increase or decrease of all urine proteins. In particular, pathway enrichment analysis revealed a strong chemokine signature in proliferative LN suggesting active recruiting of immune cells in the kidney in these patients. This is biologically consistent with the abundant immune cell infiltration and more aggressive phenotype observed in class III and IV LN, further supporting the ability of urine proteomics to infer intrarenal biological processes.
Ideal biomarkers in LN should noninvasively infer nephritis activity, longitudinally track response to treatment, and ideally capture the intrarenal biology. Based on feasibility, the current management of LN hinges on monitoring proteinuria to establish renal activity rather than frequent biopsies. However, proteinuria is a poor marker of nephritis activity. Six-month repeat biopsies after induction therapy revealed that about half of the patients in complete clinical remission (proteinuria <0.5 g/24h and no increase in sCr) had persistent histologically active proliferative nephritis 28. Conversely, > 50% of patients who achieved complete histological remission had persistent proteinuria >0.5 g/24h. Moreover, patients in clinical remission 3 years after induction treatment may show persistent nephritis activity on per protocol biopsies which is associated with flares of nephritis as immunosuppression is tapered 2. Using an unbiased approach, we discovered a previously unrecognized biomarker of intrarenal activity, IL-16, in addition to two previously recognized LN biomarkers, CD163 29 and TGF-β 30. IL-16 showed the strongest and most significant association with the renal activity index of any marker measured, and urinary abundance of IL-16 decreased over time in patients who ultimately responded to treatment after 1 year. IL-16, CD163, and TGF-beta were selected based on their correlation with histological activity; thus, it is conceivable that their decreasing urinary abundance mirrored an improvement of intrarenal histological activity. In fact, urinary proteins that did not correlate with activity did not decrease over time in responders.
Renal single cell RNA sequencing revealed that IL16, CD163, and TGFB1 are actively expressed by immune infiltrating cells in LN kidney biopsies, suggesting that their detection in the urine reflects intrarenal immune activity. Because their expression was in distinct immune cell types, their urinary abundance could identify the activity of distinct immune processes. We discovered that IL16 was the second most expressed cytokine in LN kidneys (49% of all infiltrating immune cells). This striking concordant result was independent of the urine proteomics dataset, thus demonstrating the relevance of IL-16 in LN in an orthogonal approach. Furthermore, we demonstrated prominent intraglomerular and interstitial renal production of IL-16 in proliferative LN by immunohistochemistry. Although we did not evaluate circulating cells or serum, IL-16 urinary abundance correlated with intrarenal IL-16 positive cells implicating that urinary IL-16 is the direct consequence of intrarenal IL-16 secretion. Because urinary IL-16, intrarenal IL-16 positive cells, and histological activity are positively co-correlated and IL16 is one the most expressed cytokines in LN, our findings suggest that IL-16 may be implicated in LN pathogenesis and this process can be non-invasively measured in urine.
IL-16 is a proinflammatory chemokine secreted by immune cells and non-immune cells (endothelial, epithelial cells, fibroblasts, and neurons) in response to several stimuli such as complement activation, antigen stimulation, interferon, hypoxia, and cell injury31–34. Because the release of bioactive IL-16 depends on caspase 3 activation 33, apoptosis and pro-apoptotic stimuli including sublethal doses of granzymes may also lead to its release. IL-16 can also be released upon cleavage by proteinase 3 35 suggesting that urinary IL-16 may indicate neutrophil degranulation. IL-16 is the natural ligand for CD4 and CD9, and is a strong chemoattractant for CD4+ T cells (especially Th1 cells) as well as CD8 T, NK, B cells monocytes, neutrophils, dendritic cells, and mast cells 31. IL-16 can activate CD4 T cells independently of T-cell receptor (TCR) activation 36 and may lead to the release of proinflammatory cytokines such as TNF, IL-1β, IL-6, IL-15, and IL-1231. IL16 polymorphisms were associated with increased risk of SLE (OR 3.3–10.4) suggesting a potential causal role37. Plasma IL-16 levels were associated with SLE severity including renal involvement 38. Finally, IL-16 was mechanistically linked to lung disease in the pristane model of SLE 39. The role of IL-16 in LN is yet to be fully understood, but it has been implicated in several other immune mediated diseases such as multiple sclerosis, scleroderma, rheumatoid arthritis, and allograft rejection31,40,41. Further studies are needed to address the efficacy of IL-16 blockade in LN.
Our study demonstrated the power of integrating urinary proteomic screening platforms with matching clinical and pathological information and with tissue single cell transcriptomics 42. In fact, in addition to a newly discovered biomarker, our approach detected CD163 and TGF-β that are proven biomarkers in LN. Similar to our findings, soluble CD163 was shown to correlate with LN nephritis activity and improve with treatment29. CD163 is a scavenger receptor expressed on phagocytic monocytes, especially in M2c polarized macrophages that infiltrate tissues during the healing phase of inflammation and are implicated in fibrosis resolution 43. Notably, M2c macrophage are inducible by TGF-β44. CD163+ cells are a dominant macrophage subtype in LN 44, thus again supporting the capability of urinary proteomic to infer intrarenal biology. CD163+ cells have been detected in proliferative glomerular lesions and in tubulointerstitial inflammation 45 and they constitute ~80% of the urinary cells in LN 46. Similarly consistent with our results, urinary TGF-β correlated with nephritis activity and response in previous studies 30,47,48, but sensitive immunoassays (such as the one used here) are required to reliably detect urinary TGF-β48. TGF-β regulates inflammation and progression of renal fibrosis. Notably, TGF-β increased IL-16 release in synovial fibroblasts suggesting a possible similar interplay between these two cytokines in LN 49. Here, we have shown that NK cells are the major immune cell type expressing TGFB1 in LN, whether NK or tubular cells 50 are responsible for urinary TGF-β in LN is to be determined.
We acknowledge the limitations of our study. Since we did not analyze serum or plasma, we could not establish with definitive certainty whether the concentration of specific proteins in the urine was the consequence of extrarenal leakage from the circulation through a damaged glomerular basement membrane or of intrarenal production. For example, plasma IL-16 levels were associated with disease severity including renal involvement in a group of SLE patients38, but whether the source IL-16 was intra or extrarenal was not established. We have unequivocally demonstrated that there is high intrarenal production of IL-16 in LN indicating that urinary IL-16 derives, at least in part, from active intrarenal secretion. Importantly, the association between urinary IL-16 and proliferative LN activity was independent of proteinuria suggesting that a change in urinary IL-16 abundance is an independent process rather than nonspecific leakage from plasma. Future studies will be needed to address the power of urinary IL-16 to discriminate “active” from “non active” proliferative LN. In addition, as there was a limited number of complete responders, we could not study biomarkers to predict future response with statistically robust confidence nor confidently test if the longitudinal trajectories were statistically significant. Ongoing studies as part of the AMP RA/SLE consortium will allow us to address these questions.
In summary, this study linked IL-16 release with lupus nephritis activity suggesting a possible role as a biomarker and in LN pathogenesis thus nominating IL-16 as a potentially treatable target. Further, our study demonstrated the feasibility to detect novel and biologically relevant biomarkers in LN using a urine proteomic platform in a well characterized longitudinal cohort. Further ongoing studies are required to confirm the clinical applicability of these findings, this unprecedented dataset may further discovery by allowing investigators to research and validate new biomarkers, test new hypotheses, and complement mechanistic studies in LN.
Supplementary Material
Acknowledgments:
We thank the participating AMP network clinical sites and participants. We thank Felipe Andrade, Erika Darrah, and René Ferretti for their critical review of the interpretation of the results. This work was supported by the AMP Rheumatoid Arthritis and Lupus Network. AMP is a public-private partnership (AbbVie Inc., Arthritis Foundation, Bristol-Myers Squibb Co., GlaxoSmithKline plc, Lupus Foundation of America, Lupus Research Alliance, Janssen Pharmaceutica, Merck Sharp & Dohme Corp., National Institute of Allergy and Infectious Diseases, National Institute of Arthritis and Musculoskeletal and Skin Diseases, Pfizer Inc., Rheumatology Research Foundation, Sanofi, and Takeda Pharmaceuticals International Inc.) created to develop new ways of identifying and validating promising biological targets for diagnostics and drug development. See Supplemental Acknowledgments for details on network authors. Part of this work was presented at the American College of Rheumatology Convergence 2020.
Funding:
NIH grants UH2-AR067676, UH2-AR067677, UH2-AR067679, UH2-AR067681, UH2-AR067685, UH2- AR067688, UH2-AR067689, UH2-AR067690, UH2-AR067691, UH2-AR067694, UM2- AR067678, and AR074096. The Oklahoma Rheumatic Disease Research Cores Center is funded by NIH P30AR073750. The Hopkins Lupus Cohort is funded by NIH AR 69572. Dr Fava is supported by the Jerome L. Greene Foundation and the Cupid Foundation.
Footnotes
None of the authors have a conflict of interest with regard to this work.
Financial disclosure: JB is a consultant for Bristol-Myers Squibb and received research grant support from Exagen; HMB is a consultant for Exagen and in the speakers bureau for Ra Pharmaceuticals; CM has consulted for Equillium; PI was a consultant for GSK; MP is a consultant for AbbVie, Aleon, Amgen, Annenberg Center for Health Sciences, AstraZeneca, Blackrock, Bristol-Myers Squibb, Decision Resources, Exagen, Glenmark, GSK, INOVA, IQVIA, Janssen, Lilly, Merck EMD Serono, Novartis, Sanofi Japan, Thermo Fisher, and UCB, and received research grant support from AstraZeneca, Exagen, GSK, and Lilly. BD is a consultant for Ashai Kasei, Nextcure, Biogen, ISD, Glycoera, Cyxone, Asylia, and Abbvie; has equity in Cyteir; and participates in the DSMB for BMS. PI was a consultant for GSK and Momenta/Jansen. NH consults and has equity in Danger Bio, and has equity in BioNtech. SR consults for Gilead, Pfizer, Rheos Medicines, and Janssen and is founder for Mestag.
Data and materials availability: The data reported in this publication, including the clinical and serological data of the study participants, are deposited in the ImmPort repository (accession code SDY997). The raw single-cell RNA-seq data are also deposited in dbGAP (accession code phs001457.v1.p1).
References:
- 1.Fava A & Petri M Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun 96, 0–1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Rosa M et al. A prospective observational cohort study highlights kidney biopsy findings of lupus nephritis patients in remission who flare following withdrawal of maintenance therapy. Kidney Int 94, 788–794 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Greloni G et al. Value of repeat biopsy in lupus nephritis flares. Lupus Sci. Med 1, 1–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luciano RL & Moeckel GW Update on the Native Kidney Biopsy: Core Curriculum 2019. Am. J. Kidney Dis 73, 404–415 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Geldenhuys L et al. Percutaneous native renal biopsy adequacy: A successful interdepartmental quality improvement activity. Can. J. Kidney Heal. Dis 2, 1–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birmingham DJ et al. Biomarkers of lupus nephritis histology and flare: deciphering the relevant amidst the noise. Nephrol. Dial. Transplant 32, i71–i79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soliman S & Mohan C Lupus nephritis biomarkers. Clin. Immunol 185, 10–20 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Pejchinovski M et al. Urine peptidomic biomarkers for diagnosis of patients with systematic lupus erythematosus. Lupus 27, 6–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson A What is damaging the kidney in lupus nephritis? Nat. Rev. Rheumatol 12, 143–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanley S et al. Comprehensive aptamer-based screening identifies a spectrum of urinary biomarkers of lupus nephritis across ethnicities. Nat. Commun 11, 2197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang T, Duran V, Vanarsa K & Mohan C Targeted urine proteomics in lupus nephritis – a meta-analysis. Expert Rev. Proteomics 17, 767–776 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Vanarsa K et al. Quantitative planar array screen of 1000 proteins uncovers novel urinary protein biomarkers of lupus nephritis. Ann. Rheum. Dis 79, 1349–1361 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platonov ME et al. KISS1 tumor suppressor restricts angiogenesis of breast cancer brain metastases and sensitizes them to oncolytic virotherapy in vitro. Cancer Lett 417, 75–88 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Bajema IM et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int 93, 789–796 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Arazi A et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol 20, 902–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y et al. iTALK: an R Package to Characterize and Illustrate Intercellular Communication. bioRxiv 507871 (2019). doi: 10.1101/507871 [DOI]
- 18.Norden AGW, Lapsley M & Unwin RJ Urine retinol-binding protein 4. A functional biomarker of the proximal renal tubule. Advances in Clinical Chemistry 63, (Elsevier Inc., 2014). [DOI] [PubMed] [Google Scholar]
- 19.Alvarado AS et al. The value of repeat kidney biopsy in quiescent Argentinian lupus nephritis patients. Lupus 23, 840–847 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Wu H et al. Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J. Am. Soc. Nephrol 29, 2069–2080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H et al. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 23, 869–881.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muto Y, Wilson PC, Wu H, Waikar SS & Humphreys B Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. bioRxiv 2020.06.14.151167 (2020). doi: 10.1101/2020.06.14.151167 [DOI] [PMC free article] [PubMed]
- 23.Banchereau R et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165, 551–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villanueva E et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J. Immunol 187, 538–552 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linge P, Fortin PR, Lood C, Bengtsson AA & Boilard E The non-haemostatic role of platelets in systemic lupus erythematosus. Nat. Rev. Rheumatol 14, 195–213 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Wei R et al. Alterations in urinary collagen peptides in lupus nephritis subjects correlate with renal dysfunction and renal histopathology. Nephrol. Dial. Transplant 32, 1468–1477 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Fava A et al. Integration of urine proteomics and renal single-cell genomics identifies an interferon-gamma response gradient in lupus nephritis. JCI Insight in press, (2020). [DOI] [PMC free article] [PubMed]
- 28.Malvar A et al. Histologic versus clinical remission in proliferative lupus nephritis. Nephrol. Dial. Transplant 32, 1338–1344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mejia-Vilet JM et al. Urinary Soluble CD163: a Novel Noninvasive Biomarker of Activity for Lupus Nephritis. J. Am. Soc. Nephrol ASN.2019121285 (2020). doi: 10.1681/ASN.2019121285 [DOI] [PMC free article] [PubMed]
- 30.Torabinejad S et al. Urinary monocyte chemotactic protein-1 and transforming growth factor-β in systemic lupus erythematosus. Indian J. Nephrol 22, 5–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glass WG, Sarisky RT & Del Vecchio AM Not-so-sweet sixteen: The role of IL-16 in infectious and immune-mediated inflammatory diseases. J. Interf. Cytokine Res 26, 511–520 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Roth S et al. Secondary necrotic neutrophils release interleukin-16C and macrophage migration inhibitory factor from stores in the cytosol. Cell Death Discov 1, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y et al. Processing and activation of pro-interleukin-16 by caspase-3. J. Biol. Chem 273, 1144–1149 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Cruikshank W & Little F Interleukin-16: The ins and outs of regulating T-cell activation. Crit. Rev. Immunol 28, 467–483 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Kerstein-Staehle A et al. OP0054 New role for proteinase 3 in IL-16 bioactivity control in granulomatosis with polyangiitis. Ann. Rheum. Dis 80, 28 LP–29 (2021). [Google Scholar]
- 36.Lun WH et al. Loss of virus-specific CD4+ T cells with increases in viral loads in the chronic phase after vaccine-based partial control of primary simian immunodeficiency virus replication in macaques. J. Gen. Virol 85, 1955–1963 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Xue H et al. The IL-16 gene polymorphisms and the risk of the systemic lupus erythematosus. Clin. Chim. Acta 403, 223–225 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Lard LR, Roep BO, Verburgh CA, Zwinderman AH & Huizinga TWJ Elevated IL-16 levels in patients with systemic lupus erythematosus are associated with disease severity but not with genetic susceptibility to lupus. Lupus 11, 181–185 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Mariani LH et al. Interstitial fibrosis scored on whole-slide digital imaging of kidney biopsies is a predictor of outcome in proteinuric glomerulopathies. Nephrol. Dial. Transplant 33, 310–318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Oliveira JGG, Xavier PDP, Sampaio SM, Tavares IS & Mendes AA The synthesis by fine-needle aspiration biopsy cultures of IL-7, IL-16 and IL-18 is significantly associated with acute rejection in kidney transplants. Nephron 92, 622–628 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Kawabata K et al. IL-16 expression is increased in the skin and sera of patients with systemic sclerosis. Rheumatol. (United Kingdom) 59, 519–523 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Fava A, Raychaudhuri S & Rao DA The Power of Systems Biology. Rheum. Dis. Clin. North Am 47, 335–350 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Lu J et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int 84, 745–755 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Olmes G et al. CD163+ M2c-like macrophages predominate in renal biopsies from patients with lupus nephritis. Arthritis Res. Ther 18, 1–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li YR et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med 21, 1018–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arazi A et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol 20, 902–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avihingsanon Y et al. Measurement of urinary chemokine and growth factor messenger RNAs: A noninvasive monitoring in lupus nephritis. Kidney Int 69, 747–753 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Tsakas S & Goumenos DS Accurate measurement and clinical significance of urinary transforming growth factor-beta. Am. J. Nephrol 26, 186–193 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Warstat K et al. Transforming growth factor β1 and laminin-111 cooperate in the induction of interleukin-16 expression in synovial fibroblasts from patients with rheumatoid arthritis. Ann. Rheum. Dis 69, 270–275 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Lee JS, Lim JY & Kim J Mechanical stretch induces angiotensinogen expression through PARP1 activation in kidney proximal tubular cells. Vitr. Cell. Dev. Biol. - Anim 51, 72–78 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.