

Abstract
Objectives:
Defibrotide is a heterogenous mixture of polyanionic oligonucleotides currently approved for treatment of transplant-associated veno-occlusive disease. While defibrotide has a known role in limiting endothelial cell activation, some studies have also demonstrated anti-leukocyte properties. We recently revealed a role for neutrophil extracellular traps (NETs) in the thrombotic complications of antiphospholipid syndrome (APS). Here, we hypothesized that defibrotide might act to mitigate APS-relevant NET formation in vitro and in mouse models.
Methods:
We used in vitro assays and a mouse model to determine mechanisms by which defibrotide inhibits NET formation and venous thrombosis in APS.
Results:
At doses ranging from 1 to 10 μg ml−1, defibrotide significantly suppressed NET formation from control neutrophils stimulated with IgG isolated from APS patients. Defibrotide increased levels of intracellular cyclic AMP in neutrophils, and its suppressive effects on NET formation were mitigated by blocking adenosine A2A receptor or by inhibiting the cyclic AMP-dependent kinase, protein kinase A. Defibrotide at doses ranging from 15 to 150 mg/kg/day inhibited NET formation and venous thrombosis in a model of antiphospholipid antibody-accelerated thrombosis—an effect that was reduced in adenosine A2A receptor knockout mice.
Conclusion:
This study is the first to demonstrate mechanisms by which defibrotide counteracts neutrophil-mediated thrombo-inflammation inherent to APS.
INTRODUCTION
Antiphospholipid syndrome (APS) is a thrombo-inflammatory disease characterized by circulating antiphospholipid antibodies, classically anticardiolipin and anti-beta-2-glycoprotein I (anti-β2GPI). Meanwhile, additional relevant antibodies such as anti-phosphatidylserine/prothrombin can be detected by a functional screen called the lupus anticoagulant assay (1). APS is a leading acquired cause of both thrombotic events and pregnancy morbidity. Treatment of APS typically focuses on suppressing thrombosis with anticoagulation. However, anticoagulation does not fully protect against thrombotic events, conveys an increased risk of bleeding, and in many cases fails to restrain microvascular complications of APS such as diffuse alveolar hemorrhage, nephropathy, and livedoid vasculopathy.
Neutrophil extracellular traps (NETs) are web-like tangles of DNA, chromatin, and granule proteins released into the extracellular space by neutrophils in response to both infectious and sterile stimuli (2, 3). NETs have been revealed as pathogenic actors in numerous autoimmune and thrombo-inflammatory diseases ranging from lupus to sepsis to COVID-19. To this end, recent work has pointed to a multifaceted (and generally deleterious) intersection between NETs and the vasculature. The proteases and histones of NETs kill endothelial cells (4). NETs stimulate type I interferon production, which reduces the numbers and function of restorative endothelial progenitors (5). Furthermore, NET-derived DNA triggers coagulation, while histones activate platelets (6).
Our group and others have found a role for NETs in the thrombotic complications of APS. Neutrophils isolated from patients with APS have a diminished threshold for spontaneous NET formation, while neutrophils from healthy volunteers can be activated to release NETs by exposure to APS serum or purified antiphospholipid antibodies (7). In mouse models of antiphospholipid antibody-accelerated large-vein thrombosis, treatments that counteract NETs such as neutrophil depletion (8), administration of intravenous deoxyribonuclease (8), agonism of neutrophil adenosine A2A receptors (9), boosting neutrophil cyclic AMP (cAMP) levels (10), and interfering with adhesive interactions between neutrophils and the endothelium (11) are all protective.
Defibrotide is a mixture of polyanionic phosphodiester oligonucleotides isolated from porcine intestinal mucosa cells. Defibrotide is approved for the treatment of patients with veno-occlusive disease (VOD) following hematopoietic stem-cell transplantation (HSCT) that is complicated by hepatic, renal, or pulmonary dysfunction (12, 13). Defibrotide is considered a multi-target compound, and is best known for its ability to limit endothelial cell activation (14). At the same time, some older literature demonstrates anti-leukocyte and anti-neutrophil properties of defibrotide (15), with that work mostly completed prior to the first descriptions of NETs in 2004 (2). Almost 20 years ago, defibrotide was first suggested as a possible treatment for APS, especially the life-threatening microangiopathic variant known as catastrophic APS (CAPS) (16). This possibility has not though been investigated in trials, nor have possible mechanisms been explored in the laboratory. Here, we hypothesized that defibrotide might act to mitigate APS-relevant NET formation in vitro and in mouse models.
METHODS
Isolation of human IgG.
A Protein G Agarose Kit (Pierce) was used to isolate IgG from patient or healthy control sera. This was done by following the manufacturer’s instructions (Pierce) as we have reported previously (7, 9).
Human neutrophil isolation and NET formation assays.
Neutrophils were isolated from human blood as previously described by our group (7, 9). NET formation was monitored by an assay that quantifies nuclease-liberated myeloperoxidase (MPO) activity. Neutrophils were cultured in RPMI media (Gibco) supplemented with 0.5% heat-inactivated fetal bovine serum (Gibco) and 0.5% bovine serum albumin (Sigma) at 37°C. Neutrophils were seeded into 96-well plates at 1×105 neutrophils per well. Stimulation was for three hours with 100 nM phorbol 12-myristate 13-acetate (PMA, Sigma) or 10 μg ml−1 APS IgG (which was pooled from 5 primary APS patients). In some cases, cultures were also supplemented with different concentrations (1–40 μg ml−1) of defibrotide (Jazz Pharmaceuticals), 10 μM KT5720 (PKA inhibitor, Tocris), 10 μM 8-cyclopentyltheophylline (adenosine A1 receptor antagonist, Tocris), or 10 μM SCH442416 (adenosine A2A receptor antagonist, Tocris). After stimulation, the culture media was discarded, and the plate was gently emptied over a paper towel (to remove residual culture media containing soluble MPO). Discarded media was immediately replaced with RPMI media ± 10 units ml−1 Micrococcal nuclease (Thermo Fischer Scientific). The samples without nuclease were used to account for any NET-independent background signal. EDTA (10 mM) was used to stop the digestion of NETs after 10 minutes at 37°C. Supernatants were next transferred into a V-shaped 96-well plate, which was centrifuged at 350xg to remove debris. MPO activity was then measured in a fresh plate by adding an equal volume of 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (1 mg ml−1, Thermo Fischer Scientific). The reaction was stopped 10 minutes later by the addition of 1 mM sulfuric acid (50 μL). Finally, a Cytation 5 Cell Imaging Multi-Mode Reader was used to measure absorbance at 450 nm.
Qualitative immunofluorescence microscopy.
Neutrophils were seeded onto poly-L-lysine (Sigma) coated coverslips. After fixing with 4% paraformaldehyde for 15 minutes, blocking was with 1% bovine serum albumin overnight. Neutrophil elastase was labeled with an antibody from Abcam (21595, diluted 1:100). The primary antibody was detected with a FITC-conjugated secondary antibody (Southern Biotech 4052–02, diluted 1:250). Hoechst 33342 (Invitrogen) was used to stain DNA. A Cytation 5 Cell Imaging Multi-Mode Reader was used to capture images.
Measurement of intracellular cAMP.
Neutrophils were incubated for 30 minutes at room temperature with 1 μg ml−1 defibrotide or 1 μM CGS21680 (adenosine A2A receptor agonist, Tocris). Other neutrophils were incubated for 10 minutes with 100 μM forskolin (adenylyl cyclase activator, Tocris). cAMP levels were then measured using the Bridge-It cAMP Designer fluorescence assay kit (Mediomics, Catalog #122934) as instructed by the manufacturer and as we have done previously (10).
Animal housing and treatment.
Mice were fed standard chow and housed in a specific pathogen-free facility. The University of Michigan Institutional Animal Care and Use Committee approved all protocols. Male C57BL/6 mice were obtained from The Jackson Laboratory.
Adenosine A2A receptor knockout mice.
We introduced a conditional knockout of the adenosine A2A receptor in murine neutrophils (and other myeloid-lineage cells such as macrophages) using the Cre-loxP system. Mice with a ‘floxed’ adenosine A2A receptor gene (Adora2a+/fl) on the C57BL/6 genetic background were purchased from Jackson Laboratory (010687). Adora2a+/fl were bred to obtain homozygous Adora2afl/fl mice. The Adora2afl/fl mice were then crossed with hemizygote MRP8-Cre+ mice (purchased from Jackson Laboratory, 021614). The offspring (Adora2a+/fl MRP8-Cre+) were then crossed with Adora2afl/fl mice to obtain the experimental mice of interest: Adora2afl/fl MRP8-Cre+ and Adora2afl/fl MRP8-Cre−. The breeding scheme is described in Supplementary Figure 1.
In vivo venous thrombosis.
We used an electrolytic inferior vena cava (IVC) model that has been used previously by our group (9, 10). After exposure of the IVC, any lateral branches were ligated using 7–0 Prolene sutures. These side branches remained ligated for the duration of the experiment. Most animals had one or two side branches, but some animals had none (in which case no ligatures were placed). A 30-gauge silver-coated copper wire (KY-30–1-GRN, Electrospec) was placed inside a 25-gauge needle and inserted into the IVC. The wire was positioned against the anterior wall of the IVC where exposed copper wire at its end functioned as the anode. Meanwhile, a needle implanted subcutaneously completed the circuit and functioned as the cathode. For 15 minutes, a constant current of 250 μA was applied. The needle was then removed, and the abdomen was closed. Just before being allowed to recover from anesthesia, mice received either control or APS IgG (500 μg) by intravenous injection; the APS IgG was pooled from three patients experiencing an episode of CAPS. After 24 hours, mice were euthanized, and thrombus length was determined. Defibrotide sodium was diluted in saline and administered by retro-orbital intravenous injection. Two injections were given, the first 24 hours prior to surgery and the second at the time of thrombus induction.
Quantification of MPO-DNA complexes.
Serum was collected for MPO-DNA testing at the time of venous thrombus harvesting. MPO-DNA complexes were quantified as we described previously (9, 10). The protocol uses reagents from the Cell Death Detection ELISA kit (Roche) as well as an anti-MPO antibody (Bio-Rad0400–0002) that reacts with both human and mouse MPO.
RESULTS
Defibrotide inhibits NET formation elicited by PMA or APS patient antibodies.
We first tested the ability of defibrotide to suppress NET formation when control neutrophils were activated with PMA. We found that defibrotide significantly reduced PMA-triggered NET formation at concentrations as low as 1 μg ml−1 (Figure 1A). Beyond PMA stimulation, we reasoned that defibrotide might also prevent antiphospholipid antibody-mediated NET formation. Indeed, at concentrations as low as 1 μg ml−1, defibrotide suppressed NET formation elicited from control neutrophils by APS IgG (pooled from five patients with primary APS) (Figure 1B) or by neutrophils isolated from patients with clinical features of APS who were “triple positive” for anticardiolipin antibodies, anti-β2GPI antibodies, and lupus anticoagulant (Supplementary Figure 2). The impact of defibrotide on APS IgG-mediated NET formation was also assessed by immunofluorescence microscopy with similar results (Figure 1C). In contrast to APS IgG, IgG isolated from heterologous healthy controls did not increase NET formation by control neutrophils (Supplementary Figure 3).
Figure 1: Defibrotide suppresses NET formation in response to various stimuli through adenosine A2A receptor agonism.
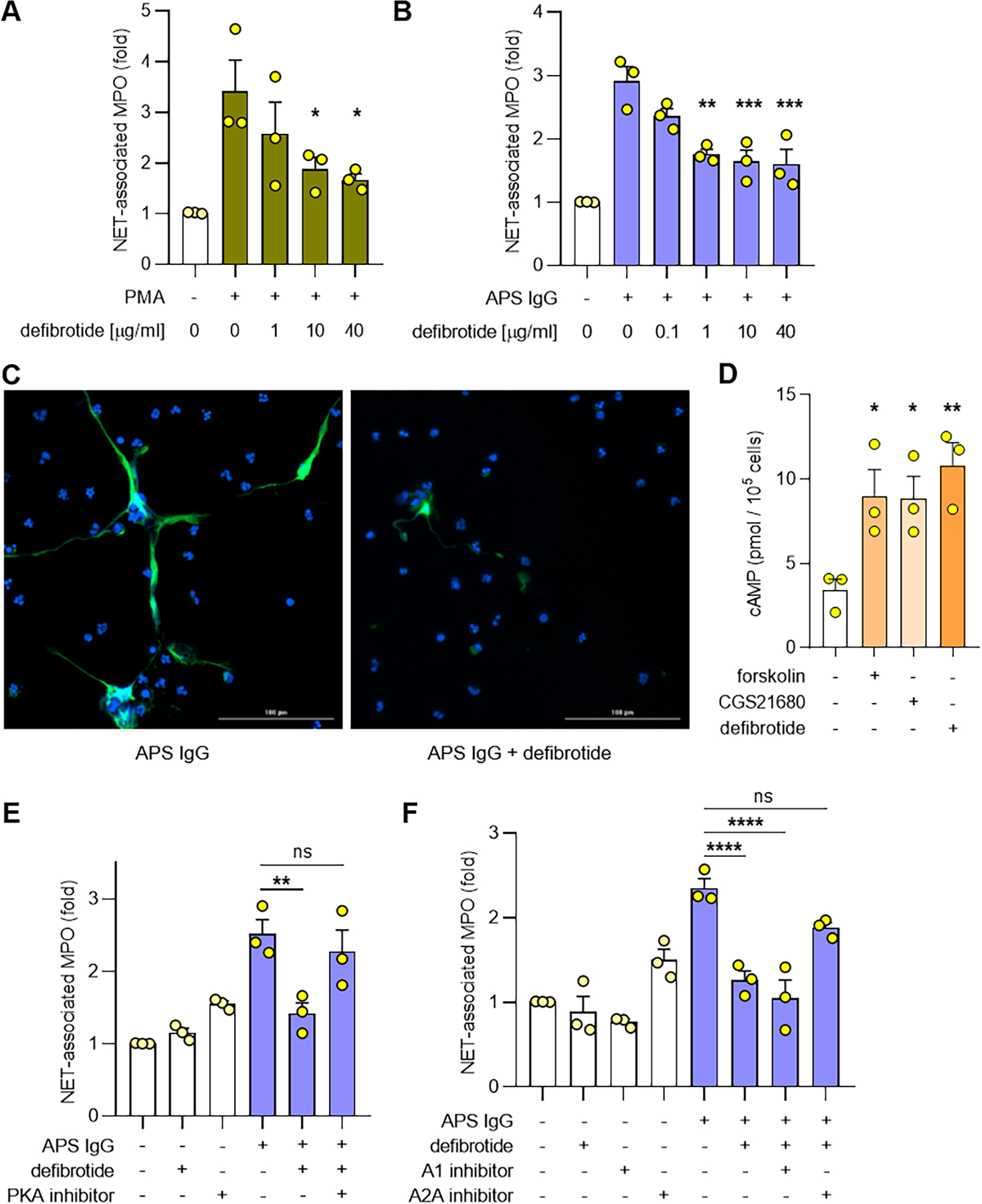
A-B, Human neutrophils were isolated from healthy volunteers and then treated with PMA (A) or APS IgG (B) for 3 hours in the presence of different concentrations of defibrotide. NET formation was quantified by measuring the enzymatic activity of nuclease-liberated myeloperoxidase (MPO). C, Representative images of NET formation assessed qualitatively by immunofluorescence microscopy. Neutrophils were treated with APS IgG in the presence or absence of defibrotide. Defibrotide=1 μg ml−1. Green=extracellular neutrophil elastase and blue=DNA. Scale bar=100 microns. D, Human neutrophils were treated with forskolin (10 minutes), CGS21680 (30 minutes), or defibrotide (30 minutes), and cyclic AMP (cAMP) levels were measured. E-F, Neutrophils were treated with APS IgG in the presence or absence of defibrotide (1 μg ml−1). Some samples were additionally treated with a protein kinase A (PKA) inhibitor (10 μM), an adenosine A1 receptor antagonist (10 μM), or an adenosine A2A receptor antagonist (10 μM). NET formation was quantified by measuring the enzymatic activity of nuclease-liberated MPO. Throughout the figure, mean and standard error of the mean (SEM) are presented for 3 independent experiments; *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 by one-way ANOVA corrected with Dunnett’s test; ns=not significant.
Defibrotide raises cAMP levels and mitigates antiphospholipid antibody-mediated NET formation through adenosine A2A receptor agonism.
Defibrotide has been reported to act as an adenosine receptor agonist in some contexts (17–19), and we recently found that adenosine receptor agonism protects against both NET formation and venous thrombosis in APS (9). We therefore hypothesized that defibrotide might mediate its action through activation of adenosine A2A receptors. In neutrophils, we found that defibrotide increased the level of intracellular cAMP in a fashion similar to the synthetic adenosine A2A receptor agonist CGS21680 and the adenylate cyclase activator forskolin (Figure 1D); defibrotide did not significantly increase cAMP levels in peripheral blood mononuclear cells (Supplementary Figure 4). We next considered that inhibiting a key cAMP-dependent kinase, protein kinase A (PKA), might reverse the effects of defibrotide. Indeed, the ability of defibrotide to suppress NET formation was neutralized by a PKA inhibitor (Figure 1E). Finally, we also found that the ability of defibrotide to suppress NET formation could be partially reversed by blocking adenosine A2A (but not adenosine A1) receptors (Figure 1F). Notably, adenosine receptor antagonists had no effect on APS IgG-mediated NET formation in the absence of defibrotide (Supplementary Figure 5). Taken together, these data demonstrate that defibrotide can suppress NET formation and that this suppression is at least in part due to activation of adenosine A2A receptors.
Defibrotide attenuates antiphospholipid antibody-mediated venous thrombosis in wild-type mice but not adenosine A2A receptor knockout mice.
Since defibrotide suppressed antiphospholipid antibody-mediated NET formation in vitro, we were interested in whether it might also mitigate antiphospholipid antibody-accelerated NET formation and thrombosis in mice. To test this, we utilized an electrolytic IVC model to induce large-vein thrombosis (Figure 2A) (9, 10). Administration of APS IgG (pooled from three patients with CAPS), but not control IgG, increased thrombus length in C57BL/6 mice, which returned to baseline levels when defibrotide was administered at doses as low as 15 mg kg−1 (Figure 2B). As expected, administration of antiphospholipid antibodies increased a surrogate marker of NETs in serum (MPO-DNA complexes), which again returned to baseline when mice were treated with defibrotide (Figure 2C). Having demonstrated in vitro that the suppressive effects of defibrotide on NET formation could be partially reversed by blocking adenosine A2A receptors, we considered that the suppressive effects of defibrotide on venous thrombosis might be reversed in myeloid-specific adenosine A2A receptor knockout mice. We first confirmed that neutrophils isolated from these mice were resistant to the ability of defibrotide to boost intracellular cAMP levels (Figure 2D). We then found that defibrotide was not able to prevent venous thrombosis (Figure 2E) or NET formation (Figure 2F) in adenosine A2A receptor knockout mice. Taken together, these data suggest that defibrotide mediates its antithrombotic effects at least partially through adenosine A2A receptors.
Figure 2: Defibrotide prevents antiphospholipid antibody-mediated venous thrombosis in wild-type mice but not in adenosine A2A receptor knockout mice.
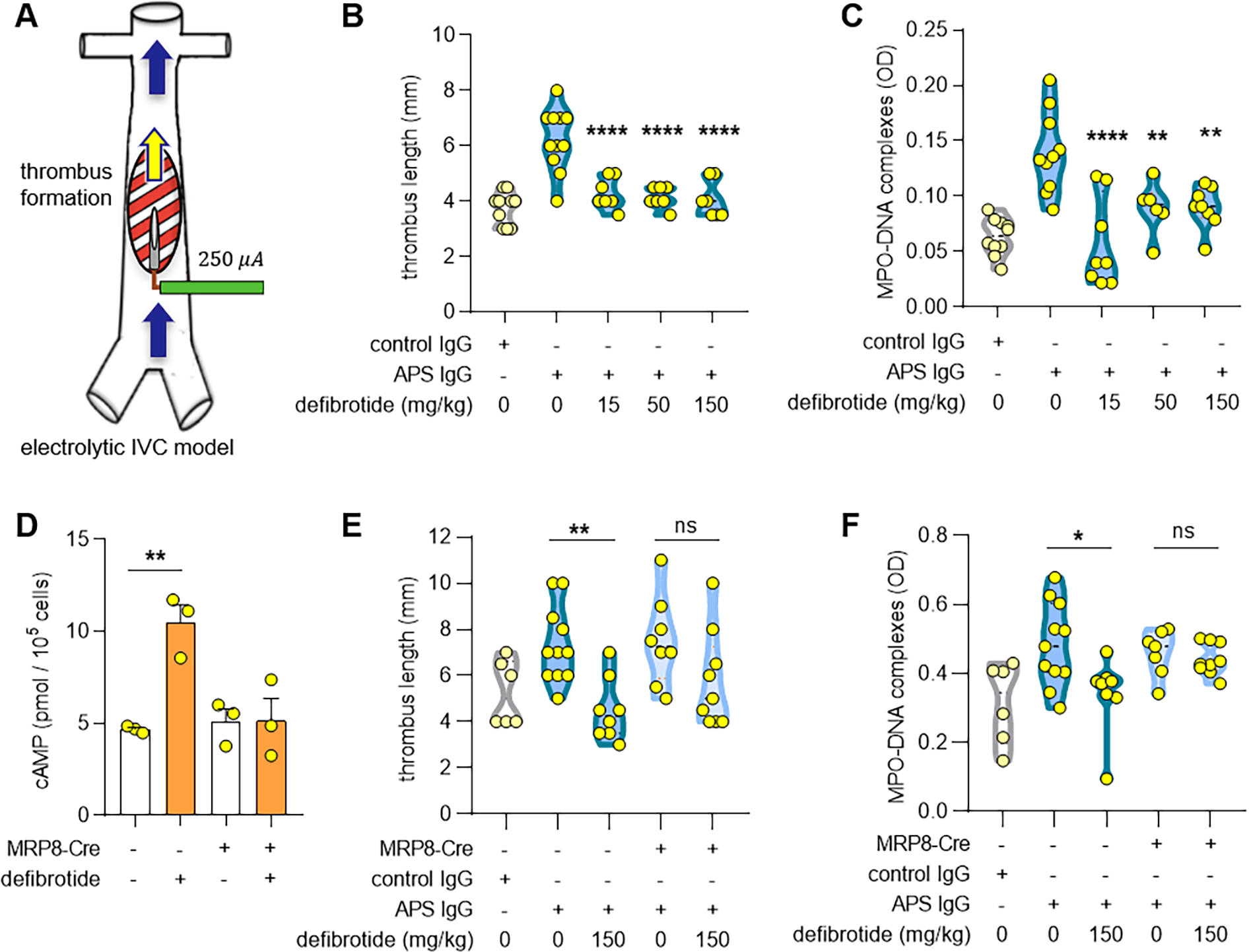
A, Schematic depiction of the electrolytic inferior vena cava model of venous thrombosis. The application of direct current to a copper wire results in the release of free radicals. This activates endothelial cells and triggers a thrombogenic environment. Blood flow remains constant. B-C, C57BL/6J mice were treated with control IgG or APS IgG in the presence or absence of defibrotide. Thrombus formation was determined after 24 hours. Thrombus length (B) and myeloperoxidase (MPO)-DNA complexes in serum (C) were quantified. D, Cyclic AMP (cAMP) levels in neutrophils isolated from Adora2afl/fl MRP8-Cre+ as compared with Adora2afl/fl MRP8-Cre− mice in the presence or absence of defibrotide (1 μg ml−1) for 30 minutes. E-F, Either Adora2afl/fl MRP8-Cre+ or Adora2afl/fl MRP8-Cre− mice were treated with control IgG or APS IgG in the presence or absence of defibrotide. Thrombus formation was assessed at 24 hours. Thrombus length (E) and MPO-DNA complexes in serum (F) were quantified. Each dot represents a unique mouse. Throughout the figure, *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 by one-way ANOVA corrected with Dunnett’s test; ns=not significant.
DISCUSSION
This study is the first to demonstrate a mechanism by which defibrotide prevents disease-relevant NET formation. Defibrotide is indicated for the treatment of patients who have VOD associated with hepatic, renal, or pulmonary dysfunction following HSCT. In those contexts, the therapeutic dose of defibrotide is 6.25 mg/kg IV every 6 hours (for a total dose of 25 mg/kg/day). The drug is typically infused over several weeks and may continue up to a maximum of 60 days. Given that the role of neutrophils in VOD has yet to receive significant attention, we can speculate that the anti-neutrophil properties of defibrotide may play a protective role in VOD. This is certainly an area that we hope to see investigated in the coming years by our group and others.
The data presented here suggest that adenosine A2A receptor agonism is at least part of the mechanism by which defibrotide reduces NET formation. Several reports (17–19) suggest that defibrotide mediates its effects by targeting multiple adenosine receptors (for example, both A1 and A2). Here, blocking adenosine A1 receptors did not interfere with defibrotide’s ability to suppress NET formation. This is similar to a pervious study in which effects of defibrotide were abolished by a dual adenosine A1/A2 receptor antagonist, but not by a selective adenosine A1 receptor antagonist (18). It is worth noting that adenosine A2A receptors are more abundantly expressed by neutrophils than are adenosine A1 receptors (20). The extent to which complementary defibrotide-mediated mechanisms may be at play in mitigating NET formation and thrombosis is certainly an area deserving of future research.
In conclusion, these preclinical data support the possibility of defibrotide as a repurposed drug candidate for APS. Given a dearth of effective therapies for patients with the microvascular variant of APS, one can consider whether defibrotide deserves systematic study in such individuals, who in many cases will be receiving therapy in the inpatient setting where administration of defibrotide would be quite feasible.
Supplementary Material
ACKNOWLEDGMENTS
Jazz Pharmaceuticals provided funding for these investigator-initiated preclinical experiments. Jazz Pharmaceuticals did not have input regarding experimental design, nor did they participate in data analysis. YZ was supported by career development grants from the Rheumatology Research Foundation, Arthritis National Research Foundation, and APS ACTION. JSK was supported by grants from the NIH (R01HL115138), Lupus Research Alliance, Rheumatology Research Foundation, and Burroughs Wellcome Fund.
Disclosures:
Jazz Pharmaceuticals provided funding for these investigator-initiated preclinical experiments. Jazz Pharmaceuticals did not have input regarding experimental design, nor did they participate in data analysis.
REFERENCES
- 1.Bertolaccini ML, Amengual O, Andreoli L, Atsumi T, Chighizola CB, Forastiero R, et al. 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmunity reviews. 2014;13(9):917–30. [DOI] [PubMed] [Google Scholar]
- 2.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. [DOI] [PubMed] [Google Scholar]
- 3.Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7(2):e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grenn RC, Yalavarthi S, Gandhi AA, Kazzaz NM, Nunez-Alvarez C, Hernandez-Ramirez D, et al. Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Ann Rheum Dis. 2017;76(2):450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015;67(11):2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, et al. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017;69(3):655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. 2019;10(1):1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali RA, Gandhi AA, Dai L, Weiner J, Estes SK, Yalavarthi S, et al. Antineutrophil properties of natural gingerols in models of lupus. JCI Insight. 2021;6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sule G, Kelley WJ, Gockman K, Yalavarthi S, Vreede AP, Banka AL, et al. Increased Adhesive Potential of Antiphospholipid Syndrome Neutrophils Mediated by β2 Integrin Mac-1. Arthritis & rheumatology (Hoboken, NJ). 2020;72(1):114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richardson PG, Elias AD, Krishnan A, Wheeler C, Nath R, Hoppensteadt D, et al. Treatment of severe veno-occlusive disease with defibrotide: compassionate use results in response without significant toxicity in a high-risk population. Blood. 1998;92(3):737–44. [PubMed] [Google Scholar]
- 13.Ho VT, Revta C, Richardson PG. Hepatic veno-occlusive disease after hematopoietic stem cell transplantation: update on defibrotide and other current investigational therapies. Bone Marrow Transplant. 2008;41(3):229–37. [DOI] [PubMed] [Google Scholar]
- 14.Pescador R, Capuzzi L, Mantovani M, Fulgenzi A, Ferrero ME. Defibrotide: properties and clinical use of an old/new drug. Vascul Pharmacol. 2013;59(1–2):1–10. [DOI] [PubMed] [Google Scholar]
- 15.Di Perri T, Laghi Pasini F, Capecchi PL, Ceccatelli L, Pasqui AL, Orrico A. Defibrotide in vitro inhibits neutrophil activation by a Ca++-involving mechanism. Int J Tissue React. 1987;9(5):399–406. [PubMed] [Google Scholar]
- 16.Burcoglu-O’Ral A, Erkan D, Asherson R. Treatment of catastrophic antiphospholipid syndrome with defibrotide, a proposed vascular endothelial cell modulator. J Rheumatol. 2002;29(9):2006–11. [PubMed] [Google Scholar]
- 17.Bianchi G, Calvani AB, Mantovani M, Prino G. Enhancement by defibrotide of prostanoid neosynthesis from arachidonic acid: an adenosine receptor-mediated effect? Semin Thromb Hemost. 1991;17(4):399–403. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi G, Barone D, Lanzarotti E, Tettamanti R, Porta R, Moltrasio D, et al. Defibrotide, a single-stranded polydeoxyribonucleotide acting as an adenosine receptor agonist. European Journal of Pharmacology. 1993;238(2):327–34. [DOI] [PubMed] [Google Scholar]
- 19.Pasini FL, Frigerio C, Capecchi PL, Ceccatelli L, Messa GL, Franchi M, et al. Modulation of venous endothelial activity and transcellular calcium transport by defibrotide: the adenosine hypothesis. Semin Thromb Hemost. 1996;22 Suppl 1:15–20. [PubMed] [Google Scholar]
- 20.van der Hoeven D, Wan TC, Auchampach JA. Activation of the A(3) adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol Pharmacol. 2008;74(3):685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.