

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with few effective treatment options. We found a highly significant correlation between pregnancy-associated plasma protein (PAPP)-A expression in IPF lung tissue and disease severity as measured by various pulmonary and physical function tests. PAPP-A is a metalloproteinase that enhances local insulin-like growth factor (IGF) activity. We used primary cultures of normal adult human lung fibroblasts (NHLF) to test the hypothesis that PAPP-A plays an important role in the development of pulmonary fibrosis.
Treatment of NHLF with pro-fibrotic transforming growth factor (TGF)-β stimulated marked increases in IGF-I mRNA expression (> 20-fold) and measurable IGF-I levels in 72-hr conditioned medium (CM). TGF-β treatment also increased PAPP-A levels in CM 4-fold (P = 0.004) and proteolytic activity ~2-fold. There was an indirect effect of TGF-β to stimulate signaling through the PI3K/Akt pathway, which was significantly inhibited by both IGF-I-inactivating and PAPP-A inhibitory antibodies.
Induction of senescence in NHLF increased PAPP-A levels in CM 10-fold (P = 0.006) with attendant increased proteolytic activity. Thus, PAPP-A is a novel component of the senescent lung fibroblast secretome. In addition, NHLF secreted extracellular vehicles (EVs) with surface-bound active PAPP-A that were increased 5-fold with senescence.
Regulation of PAPP-A and IGF signaling by TGF-β and cell senescence suggests an interactive cellular mechanism underlying the resistance to apoptosis and the progression of fibrosis in IPF. Furthermore, PAPP-A-associated EVs may be a means of pro-fibrotic, pro-senescent communication with other cells in the lung and, thus, a potential therapeutic target for IPF.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is an age-related progressive lung disease of unknown etiology. There are no effective treatment options and patients with IPF have a median survival of less than 3 years (Raghu et al., 2011; Vancheri, 2010). Therefore, it is imperative to have a better understanding of the cellular mechanisms underlying IPF for development of novel therapeutic targets for this deadly disease.
Lung fibrosis is the result of repetitive alveolar epithelial cell injuries coupled with an unresolved process of wound healing (Vancheri et al., 2010; Venosa et al., 2020). Multiple microsites of ongoing epithelial injury induce migration and proliferation of fibroblasts and their transformation into myofibroblasts. Myofibroblasts are specialized fibroblasts expressing α-smooth muscle actin (α-SMA) and exhibit a contractile phenotype contributing to altered compliance of the lung (Phan, 2002; Scotton & Chambers, 2007). In fibroblast/myofibroblast foci there are persistent pro-survival signals and an increased response to fibrogenic factors. This results in exaggerated deposition of extracellular matrix (ECM) leading to irreversibly distorted lung architecture, respiratory failure and death (Thannickal & Horowitz, 2006).
Transforming growth factor-β1 (TGF-β) is a profibrogenic factor involved in myofibroblast differentiation and ECM production, which has been studied extensively in human IPF and mouse models of lung injury and fibrosis (Border & Noble, 1994; Broekelmann et al., 1991; Koli et al., 2008; Limper et al., 1991; Meng et al., 2016). However, clinical trials targeting TGF-β have been disappointing due, in large part, to its pleiotropic effects (Gyorfi et al., 2018; Scotton & Chambers, 2007). TGF-β appears to work with other factors to direct and promote excessive fibrosis in IPF. An important factor implicated in the fibrotic process is the insulin-like growth factor (IGF) system.
The IGF system is complex with two ligands (IGF-I, IGF-II), two receptors [type I IGF receptor (IGF-IR), type II IGF/mannose-6-phosphate receptor], high affinity IGF binding proteins (IGFBP-1 through 6), and specific IGFBP proteases (Bach, 2018; Bunn & Fowlkes, 2003; Conover, 1995; Hakuno & Takahashi, 2018). There are multiple studies linking the IGF system with normal and aberrant wound healing in several tissues, including lung. IGF-I stimulates proliferation of fibroblasts, protects myofibroblasts from apoptosis, and promotes ECM accumulation – all processes associated with fibrosis of the lung (Hung et al., 2013). IGF-I shows increased expression in lungs of patients with IPF, localizing to macrophages, alveolar epithelial cells, and interstitial fibroblastic cells (Uh et al., 1998). Therefore, IGF-I is in a key position to initiate and/or propagate lung fibrosis. Although limited in study, IGF-II expression was found to be increased in pulmonary fibroblastic foci and epithelial cells (Hsu & Feghali-Bostwick, 2008). Both IGFs bind to the IGF-IR and activate the phosphatidyl inositol-3 kinase (PI3K/Akt) and mitogen-activated protein kinase (MAPK)/Erk1,2 pathways (Hakuno & Takahashi, 2018). The PI3K/Akt pathway is highly activated in apoptosis-resistant IPF myofibroblasts, and the pro-survival effect of IGF signaling through this pathway may be important in dysfunctional resolution of the repair process (Horowitz et al., 2004; Kennedy et al., 1997; Thannickal & Horowitz, 2006).
Horowitz et al. (Horowitz et al., 2004) hypothesized that activation of the pro-survival PI3K/Akt pathway by TGF-β in lung fibroblasts was mediated by a secondary, but unidentified, secreted growth factor. Recently, Hernandez et al. (Hernandez et al., 2020) documented upregulation of IGF-I via TGF-β in fetal lung fibroblasts, and found that increased IGF-I expression correlated with decreased pulmonary function in IPF. IGF-I has been shown to stimulate TGF-β expression in fibroblasts (Ghahary et al., 1998), suggesting the possibility of a positive feedback loop promoting the progression and limiting the resolution of fibrosis. In addition, it has been shown that acute treatment of cells with IGF-I stimulates proliferation, while prolonged treatment of cells with IGF-I promotes senescence in a variety of cell types (Bitto et al., 2010; Tran et al., 2014).
Cellular senescence is a complex response of a cell to various stressors that is characterized by stable cell cycle arrest, resistance to apoptosis, and expression of a senescence-associated secretory phenotype (SASP), which includes cytokines, chemokines, proteases, inhibitors, and growth factors (Childs et al., 2015; He & Sharpless, 2017; Tchkonia et al., 2010). Importantly, the effects of the SASP can spread to nearby non-senescent cells, amplifying an adverse influence on tissue and organ function (Nelson et al., 2012). Senescent cells accumulate in IPF lung and this correlates with severity of the disease (Alvarez et al., 2017; Meiners & Lehmann, 2020; Morty & Prakash, 2019; Schafer et al., 2017). Furthermore, senescent cells have recently been reported to generate extracellular vesicles (EVs) (Basisty et al., 2019; Takasugi, 2018). In the lung, SASP-EVs increase the potential for extracellular communication with other pulmonary cells (Bartel et al. 2020; Kadota et al., 2018). The cellular origin and regulation of lung-derived EVs in IPF are unknown.
Mechanistic details of regulated IGF expression and IGF signaling in pulmonary fibrosis are sorely lacking. In particular, extracellular components that can tightly control the bioactivity of IGF ligands need to be taken into account. These include antagonistic IGFBPs and agonistic proteolytic activity. Pregnancy-associated plasma protein-A (PAPP-A) is an established IGFBP proteinase. Secreted PAPP-A tethers to the surface of cells by binding proteoglycan and thus can activate IGF by cleaving inhibitory IGFBP-4 in the pericellular environment (Conover, 2012; Conover et al., 2004; Laursen et al., 2002). Cleavage of IGFBP-4 by PAPP-A causes dissociation of bound IGF and, hence, IGF-IR activation. PAPP-A-induced enhancement of local IGF-I action through limited proteolysis of IGFBP-4 has been demonstrated in several in vitro and in vivo systems (Conover, 2012).
Based on previous studies of TGF-β, IGF-I, and senescence-related gene expression associated with human IPF (Hernandez et al., 2020; Schafer et al., 2017) and data presented herein on PAPP-A gene expression associated with human IPF, we hypothesized a mechanistic model of IPF involving coordinated interactions of TGF-β, IGF-I, PAPP-A and cell senescence, and tested it in primary cultures of normal adult human lung fibroblasts.
MATERIAL AND METHODS
Microarray.
Microarray data and clinical parameters from control subjects and patients with IPF were obtained through the Lung Tissue Research Consortium (http://www.Ltrcpublic.com), as described previously (Schafer et al., 2017).
Materials.
Cell culture components were from Gibco Life Technologies (Grand Island, NY). Bovine serum albumin (BSA) and Sm1.2 anti-IGF-I antibody were from Sigma Millipore (Temecula, CA). Recombinant human TGF-β1 and Quantikine IGF-I ELISA kits were from R&D Systems (Minneapolis, MN). Etoposide was obtained from Enzo Life Sciences (Farmingdale, NY). Total IGFBP-4 ELISA kits and monoclonal antibody generated against a unique exosite in PAPP-A (mAb-1/41) (Mikkelsen et al., 2008; Mikkelsen et al., 2014) were obtained from Ansh Labs (Webster, TX).
Cells.
Normal adult human lung fibroblasts (NHLF) were purchased from Lonza (Walkersville, MD; Cat # CC-2512). Cells were grown in DMEM supplemented with penicillin (100 IU/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum (FBS). Experiments were performed in serum-free DMEM containing 0.1% BSA. Cells were used at passages 4–9.
For induction of senescence, NHLF were treated for 48 h with 25 μM etoposide in growth media. Etoposide was removed and replaced with fresh media every three days. After six days, the cells took on a senescent morphology, which was confirmed by β-galactosidase staining (Schafer et al., 2017). Senescent and non-senescent cells were changed to serum-free media with experimental additions for the indicated number of hours (24 h for RT-qPCR and 72 h for conditioned medium).
RT-qPCR.
Total RNA was isolated from cultured cells by washing twice with cold phosphate buffered saline (PBS), lysing with 1 ml of Trizol (Ambion Life Technologies, Carlsbad, CA) and further processed as per manufacturer’s instruction. RNA (1 μg) was reversed transcribed with the SuperScript III Frist-Strand Synthesis System (Life Technologies) and evaluated by quantitative real-time PCR using the CFX Connect Real-Time System with iTAQ Universal SYBR Green Supermix (Bio-Rad, Hercules, CA). Amplification plots were analyzed using CFX Maestro Software version 4.1 (Bio-Rad). Primer pairs are presented in Supplemental Table 1. To create standard curves, designed primers were PCR’d with cDNA from known cell culture samples. Amplified PCR products were purified through QIAquick Gel Extraction Kit (Qiagen Hilden, Germany), quantified and serial diluted from 108 to 103 molecules. Relative quantification and fold changes were based on the standard curve for each gene.
ELISAs.
Determination of PAPP-A concentration in conditioned medium was carried out by ELISA essentially as previously described, using catching/detecting antibodies PA6/PA8 (Jepsen et al., 2015). In brief, 96-well MaxiSorp plates were coated with 5 μg/ml catching antibody (PA6) in PBS and incubated overnight at room temperature (RT). The wells were blocked with 2% BSA TBS (30 mm Tris-HCl, 300 mm NaCl, 2 mm CaCl2, pH 7.4) for 1 hour at RT. Samples were diluted 2x in 1% BSA TBS-T (30 mm Tris-HCl, 300 mm NaCl, 2 mm CaCl2, 0.05% Tween-20, pH 7.4) and incubated in the wells for 1 hour at RT. Diluted recombinant PAPP-A (Overgaard et al., 2000) was applied as a standard with concentrations ranging from 100 ng/ml – 0.19 ng/ml. Following incubation and washing with TBS-T, the wells were incubated with 2 μg/ml biotinylated anti-PAPP-A (PA8) for 1 hour at RT. Following washing with TBS-T, the wells were incubated with avidin-conjugated peroxidase (Sigma) diluted 1:10,000 in 1% BSA TBS-T for 1 hour at RT. Detection was carried out by adding liquid substrate TMB (Sigma) to each well before adding stop solution 0.5 M H2SO4. Absorbance was measured at 450 nm on an EnSpire Multimode Plate Reader (PerkinElmer Life Sciences). Blank values were subtracted for all measurements and data was fitted using linear regression.
Quantikine IGF-I and IGFBP-4 ELISAs were performed according to manufacturers’ instructions.
IGFBP-4 cleavage assay.
PAPP-A activity was assessed by measuring the proteolytic cleavage of radiolabeled IGFBP-4 as previously described (Gyrup & Oxvig, 2007). Conditioned medium samples were diluted (NHFLs diluted 3 times; EVs diluted 1.25 times) in the reaction mixture which included 10 nM radiolabeled IGFBP-4 (Laursen et al., 2001), 100 nM IGF-I and assay buffer (50 mM TRIS, 100 mM NaCl,1 mM CaCl2, pH 7.4). Before sample addition, IGFBP-4 and IGF-I were pre-incubated for 25 minutes at RT. Reaction mixtures were incubated at 37°C for 1, 2, 4 and, 8 hours and stopped by adding 2x SDS loading buffer supplemented with 25 mM EDTA. Recombinant PAPP-A at 100 pM was included as control. Intact IGFBP-4 and the proteolytic fragments were separated by nonreducing 12% SDS-PAGE and visualized by autoradiography, using a storage phosphor screen and a Typhoon imaging system (GE Healthcare, Denmark). Band intensities were quantified using ImageQuant TL 7 software (GE Healthcare) and the initial velocities were calculated (Gyrup & Oxvig, 2007). In some experiments, inhibitory PAPP-A mAb-1/41 (Mikkelsen et al., 2014) was added at a concentration of 40 nM.
Western blotting.
Cells were washed twice with cold PBS and lysed with 150 ul RIPA buffer containing protease and phosphatase inhibitors, as previously described (Conover et al., 2019). Briefly, cells were scraped into microfuge tubes, sonicated, centrifuged and the supernatant transferred to a new tube. Equal volumes were loaded on 12% mini-Protean TGX gels from Bio-Rad, transferred to PVDF, blocked with 3% BSA/TBS-T and incubated with primary antibodies overnight at 4°C. Next day the blots were washed, incubated with appropriate secondary antibody, treated with enhanced chemiluminescent substrate and imaged on a LiCor Odyssey imager and Image Studio software (Lincoln, NE). The sources for antibodies are presented in Supplemental Table 2.
EV isolation.
The isolation procedure for EVs was taken from Barile et al. using serial ultracentrifugation (Barile et al., 2018). Conditioned medium from T75 flasks was centrifuged 3,000 × g for 15 minutes at 4°C. The supernatant was moved to a new tube and centrifuged at 10,000 × g for 15 minutes at 4°C. The supernatant was collected into a 10 ml conical bottom ultracentrifuge tube (Beckman Coulter, Brea, CA) and centrifuged 100,000 × g for 90 minutes in an L-70 ultracentrifuge (Beckman) using an SW 41 swinging bucket. The supernatant was discarded and the pellet solubilized in either PBS or RIPA buffer. Total protein concentrations were determined by using Pierce BCA Protein Assay Kit (ThermoFisher Scientific) according to manufacturers’ instructions.
Statistical analyses.
For microarray comparisons, IPF subject data were stratified based on forced vital capacity (FVC) scores as follows: least severe (FVC > 80%), moderately severe (FVC 50–80%), most severe (FVC < 50%). PAPP-A expression values were standardized (mean = 0, sd = 1). Significance of differential expression was determined using linear regression via the functions ImFit and eBayes from the limma package (Smyth, 2004). Pearson correlation coefficients were used to summarize relationship between expression and patient characteristics. Except where indicated, data are expressed as mean ± SEM of three independent experiments. Binary variables, e.g., without and with TGF-β treatment, were compared using t-tests. For multiple comparisons, ANOVA with Tukey’s post-hoc comparison was used. Significance was set at P < 0.05
RESULTS
PAPP-A expression correlates with severity of IPF
Recent studies found that TGF-β upregulates IGF-I in IPF tissue (Hernandez et al., 2020) and that senescent biomarkers in lungs of IPF patients are increased (Schafer et al., 2017). Along with traditional markers of fibrosis, marked up-regulation of the secreted metalloproteinase, PAPP-A, and to a lesser magnitude its substrate, IGFBP-4, was also observed in IPF by RNA-seq (Schafer et al., 2017). To test whether PAPP-A is associated with IPF disease severity, we interrogated microarray data for PAPP-A gene expression in control and in mild, moderate and severe IPF lung tissues (Schafer et al., 2017). PAPP-A expression negatively correlated with lung function as measured by forced vital capacity (P < 0.001, Fig. 1A) and CO diffusion capacity (P < 0.001, Fig. 1B), and with physical function as measured by 6-minute walk distance (P = 0.008, Supplemental Fig. 1A) and self-reported physical function (P < 0.001, Supplemental Fig. 1B). Thus, PAPP-A gene expression is up-regulated in IPF lung tissue along with upregulation of TGF-β, IGF-I, and markers of senescence.
Figure 1.
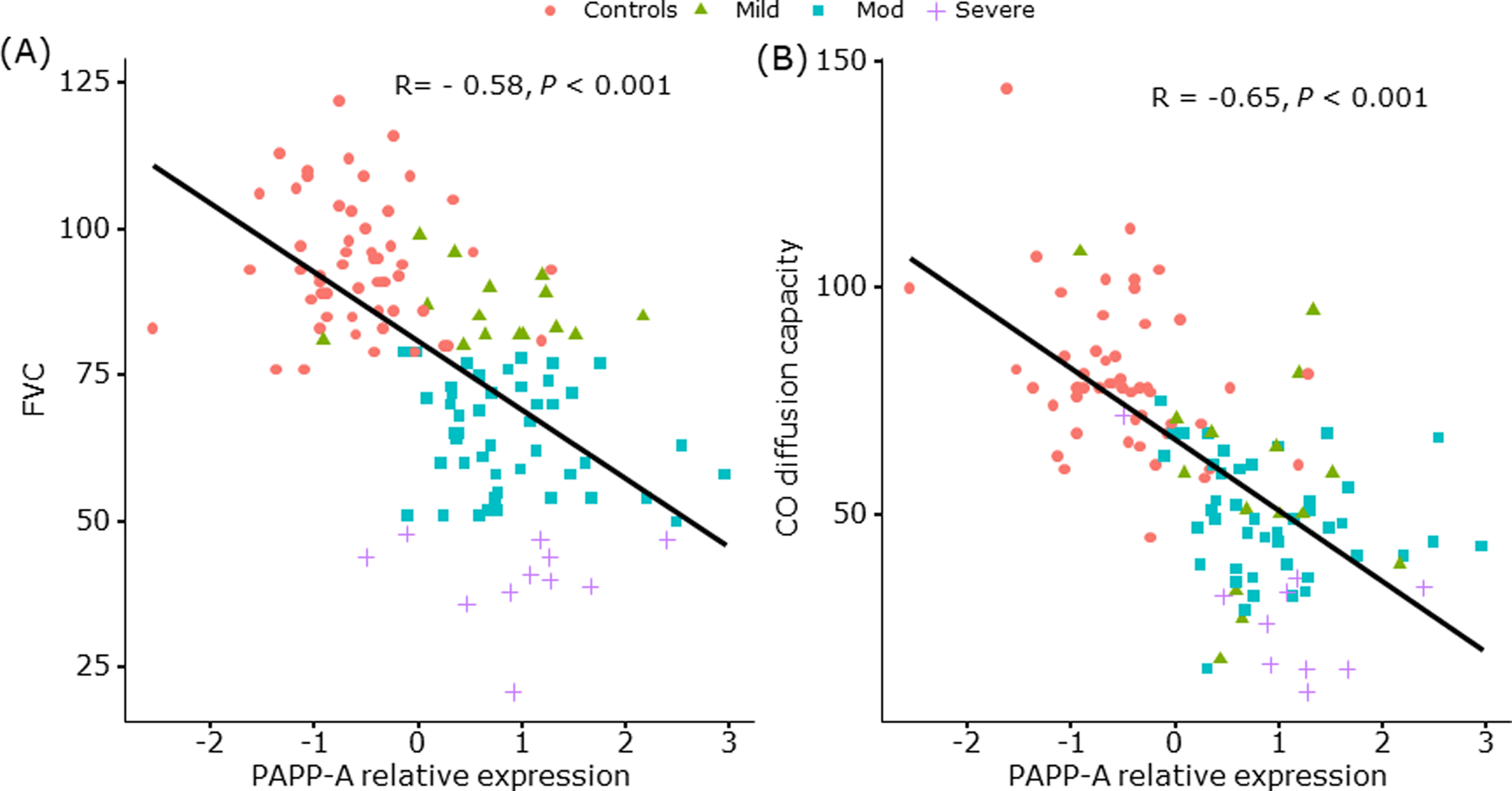
Relationship between PAPP-A gene expression in lung tissue and severity measures of pulmonary function in IPF
(A) Forced Vital Capacity
(B) Carbon Monoxide Diffusion Capacity
PAPP-A expression levels in lung specimens from control and IPF subjects were measured by microarray (Schafer et al., 2017). Significance of differential expression was determined using linear regression via the functions ImFit and eBayes from the limma package (Smyth, 2004). Pearson correlation coefficients were used to summarize relationship between expression and patient characteristics.
Pink ● Control subjects
Green ▲ Patients with mild IPF
Blue ■ Patients with moderate IPF
Purple + Patients with severe IPF
TGF-β increases IGF signaling in NHLF by multiple mechanisms
Based on these in vivo associations, we posited that regulatory interactions between TGF-β, IGF-I, PAPP-A, and cellular senescence drive fibrotic pathology, and tested this hypothesis using primary cultures of normal adult human lung fibroblasts (NHLF). NHLF were treated with TGF-β (10 ng/ml), which rapidly increased α-SMA (a marker of myofibroblasts; data not shown). For gene expression, RNA was isolated and RT-qPCR performed (Conover et al., 2010; Laursen et al., 2002) (Table 1). TGF-β treatment for 24 hours resulted in significantly increased expression of IGF-I mRNA (> 20-fold). There was no significant change in IGF-II, PAPP-A or IGFBP-4 gene expression. We next explored changes in secreted proteins in response to TGF-β treatment. NHLF were treated without and with TGF-β for 72 hours and conditioned medium was assessed by ELISAs. IGF-I increased from undetectable to 5 ± 3.2 ng/106 cells in 72-hr CM from cells treated with TGF-β. Although there was no significant increase in PAPP-A mRNA, TGF-β treatment increased PAPP-A protein levels in 72-hr CM 4-fold compared to control (155 ± 19.0 vs. 37 ± 3.4 ng/106 cells, t-test P = 0.004). Furthermore, TGF-β treatment increased specific PAPP-A enzymatic activity ~2-fold, as evidenced by IGF-dependent cleavage of IGFBP-4 that is inhibited with a neutralizing monoclonal antibody specific to PAPP-A-mediated IGFBP-4 proteolysis, mAb-1/41 (Mikkelsen et al., 2008; Mikkelsen et al., 2014), and measurement of initial velocity of the enzymatic activity (Fig. 2). Thus, TGF-β treatment of NHLF not only increases the synthesis of IGF-I but also components of the molecular machinery that allows it to become bioavailable and initiate IGF-IR signaling, i.e., PAPP-A.
Table 1.
Effect of TGF-β treatment of NHLF on IGF, PAPP-A, IGFBP-4 mRNA expression
| mRNA Expression in Control | Fold-change with TGF-β | |
|---|---|---|
| IGF-I | 1.3 × 102 | 26.0 ± 4.80* |
| IGF-II | 4.4 × 103 | 1.9 ± 0.81 |
| PAPP-A | 3.0 × 103 | 1.4 ± 0.21 |
| IGFBP-4 | 9.8 × 104 | 0.7 ± 0.07 |
NHLF were treated with SFM +/− TGF-β (10 ng/ml) for 24 hours. RNA was isolated and prepared for RT-qPCR. Results are mean ± SEM from 3 separate experiments performed in duplicate.
Significant effect of TGF-β by t-test, P < 0.05
Figure 2.
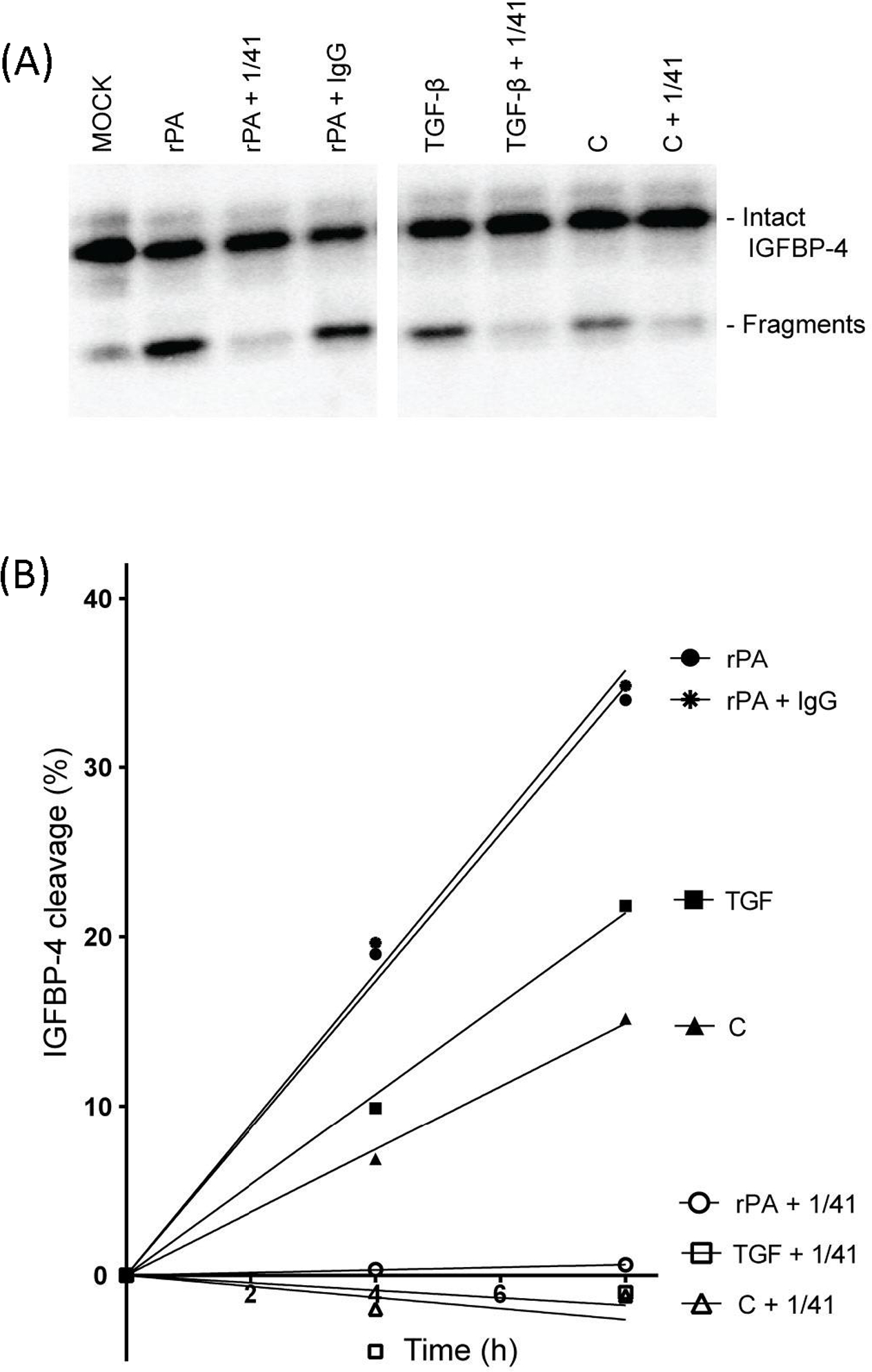
PAPP-A-mediated IGF-dependent IGFBP-4 proteolysis
72-hr CM from NHLF treated without and with TGF-β was assessed for IGF-dependent IGFBP-4 proteolysis by Western blot with radiolabeled IGFBP-4. (A) Intact and fragmented IGFBP-4 are indicated. Inhibition by mAb-1/41 confirmed specificity of PAPP-A-mediated proteolysis. (B) Measurement of initial reaction velocity.
C, control; rPA, recombinant PAPP-A
We next tested whether TGF-β activates a pro-survival pathway, PI3K/Akt. IGFs stimulate phosphorylation/activation of the Akt pathway in a variety of cell types within minutes. On the other hand, Akt phosphorylation after TGF-β treatment of fetal lung fibroblasts was shown to be delayed, occurring ~16–24 hours later (Hernandez et al., 2020). Thus, we compared serum-free medium conditioned by TGF-β (10 ng/ml) or IGF-I (10 nM) treatment of NHLF for 72 hours to unconditioned serum-free medium spiked with similar amounts of TGF-β or IGF-I. These media samples were added to fresh NHLF for 15 minutes, and then the cells harvested for Western blotting of phosphorylated and total Akt. Figure 3A indicates marked increases in phosphorylated Akt (pAkt) after treatment with IGF-I, either as spiked SFM (2.4-fold) or as 72-hr conditioned medium (CM, 2.3-fold). TGF-β was ineffective acutely but increased pAkt 2-fold when 72-hr CM from TGF-β treated cells was used. There was no effect of any treatment on total Akt. Addition of antibodies that interfere with IGF-I-stimulated IGF-IR activation (Sm1.2) significantly reduced 72-hr IGF-I CM- and 72-hr TGF-β CM-stimulated Akt phosphorylation by ~40%. Addition of antibodies that specifically inhibit PAPP-A-mediated IGFBP-4 proteolysis (mAb-1/41) significantly reduced the effect of 72-hr TGF-β CM-stimulated Akt phosphorylation by ~40%, but had no significant effect on IGF-I CM (Fig. 3B,C). In these experiments, the MAPK/Erk1,2 pathway was not stimulated (data not shown). Thus, TGF-β stimulates the PI3K/Akt pathway in NHLF indirectly through enhanced IGF-I and PAPP-A secretion and bioactivity.
Figure 3.
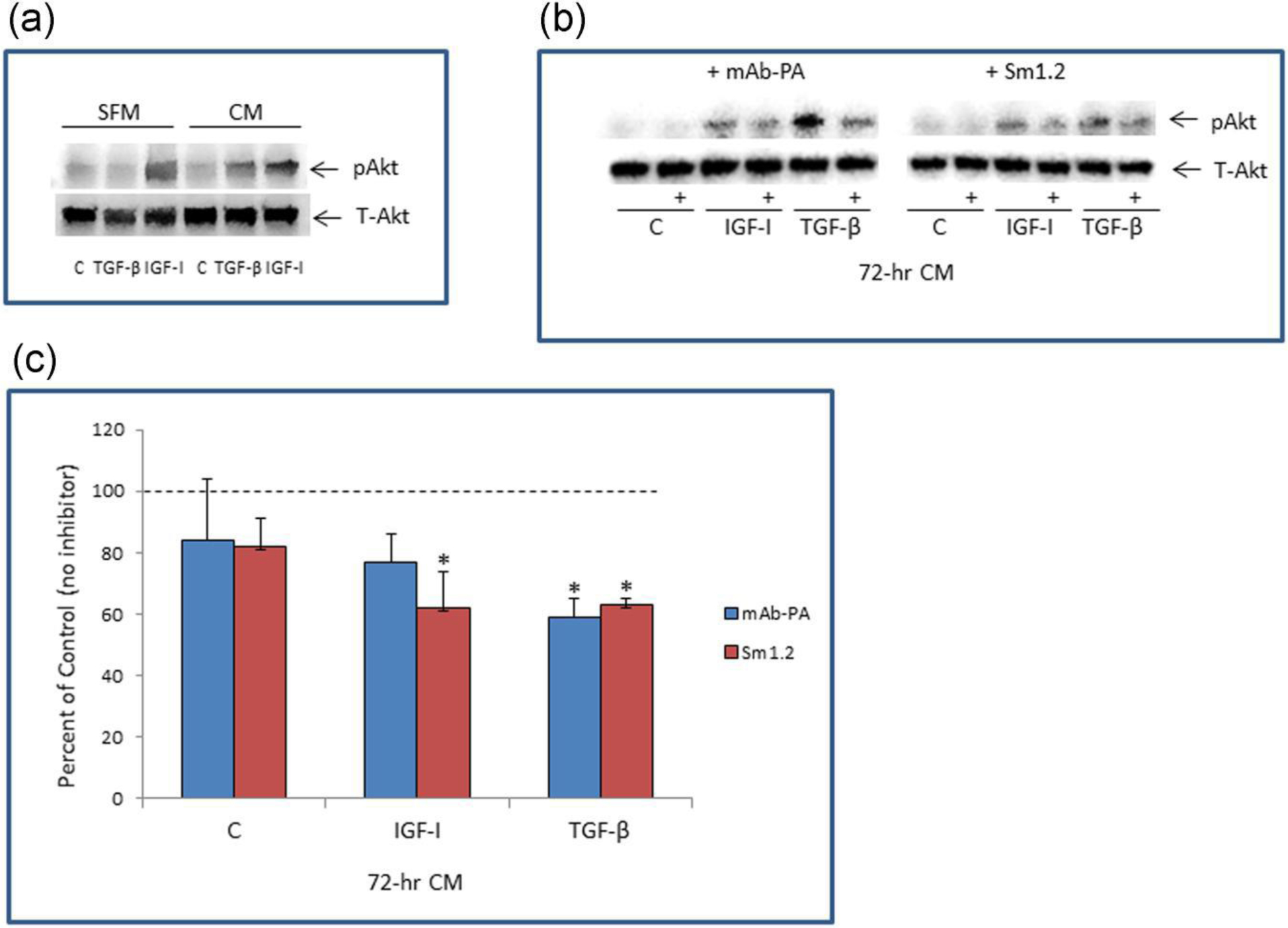
Effect of acute and chronic treatment of NHLF with TGF-β or IGF-I on Akt phosphorylation
(A) IGF-I (10 nM) and TGF-β (10 ng/ml) were incubated with NHLFs to generate 72-hr CM or directly added to unconditioned SFM. These samples were then incubated with fresh NHLF for 15 minutes and cells harvested for Western blotting of phosphorylated Akt (pAkt) and Total Akt (T-Akt).
(B) Representative Western blot of pAkt and T-Akt after treatment with 72-hr CM without or with (+) inhibitor to PAPP-A (mAb-1/41) or IGF-I (Sm1.2) added 30 minutes prior to NHLF stimulation
(C) Results (% of control) are mean ± SEM of three separate experiments. Dotted line indicates 100%, i.e., no inhibition.
*Significant effect of inhibitor by ANOVA, P < 0.05
Cellular senescence increases IGF activity independent of TGF-β
Cellular senescence was induced in NHLF by low dose etoposide (Schafer et al., 2017). An enlarged flattened morphology with many protrusions and β-galactosidase staining confirmed that the cells were in a senescent state six days following treatment (data not shown). TGF-β was added to senescent and non-senescent NHLF for 24 hours (mRNA) or 72 hours (CM and cell number). RT-qPCR data are presented in Table 2. Senescence, per se, increased mRNA expression of PAPP-A (~3-fold) and IGFBP-4 (~2-fold) but had no effect on IGF-I or IGF-II expression. There was no hyper-response to TGF-β in senescent cells compared to non-senescent cells. However, TGF-β-induced increases in IGF-I and IGF-II were retained in senescent cells. Acute TGF-β treatment increased collagen (Col1A1) and fibronectin (FN1) gene expression ~3- to 4-fold in non-senescent cells, and this was not altered in the senescent state (Table 2).
Table 2.
Effect of Cell Senescence on IGF, PAPP-A, IGFBP, ECM mRNA expression Fold-change (relative to non-senescent control)
| Senescent | TGF-β | Senescent + TGF-β | |
|---|---|---|---|
| IGF-I | 0.8, 1.0 | 37.4, 32.3 | 20.7, 13.8 |
| IGF-II | 0.8, 0.8 | 2.0, 1.7 | 2.1, 1.9 |
| PAPP-A | 3.7, 2.6 | 1.4, 1.2 | 2.4, 2.6 |
| IGFBP-4 | 2.4, 2.1 | 0.7, 0.5 | 2.1, 1.6 |
| Col1A1 | 0.8, 1.0 | 3.8, 2.9 | 2.6, 1.8 |
| FN1 | 0.8, 0.9 | 3.3, 5.6 | 2.8, 2.5 |
Non-senescent and senescent-induced NHLF were treated with SFM ± TGF-β (10ng/ml) for 24h. RNA was isolated and prepared for RT-qPCR. All samples for each gene were run together in the same assay (in duplicate) and expressed relative to non-senescent control = 1.0. Results presented are from two separate experiments and separated by a comma.
Col1A1 - collagen
FN1 - fibronectin
The increased mRNA transcription observed after induction of cell senescence also resulted in a marked (10-fold) increase in PAPP-A levels in 72-hr CM compared to non-senescent CM and increased total IGFBP-4 levels ~4-fold (Table 3). This increased concentration of PAPP-A antigen correlated with increased PAPP-A activity. Senescence resulted in a 4-fold increase in its proteolytic activity, although some variability was observed (Table 3). TGF-β treatment added little to the major influence of senescence on PAPP-A expression or activity. As in non-senescent NHLF, PAPP-A proteolytic activity was completely inhibited by mAb-1/41 in all samples in senescent NHLF (data not shown). Thus, cellular senescence has a marked effect on PAPP-A expression and proteolytic cleavage of IGFBP-4, which is independent of TGF-β.
Table 3.
Effect of cell senescence on PAPP-A protein expression and proteolytic activity in NHLF
| PAPP-Aa | IGFBP-4b | PAPP-A proteolytic activityc | |
|---|---|---|---|
| Non-senescent | |||
| Control | 23 ± 4.3 | 17.5 ± 1.98 | 3.1 ± 0.61 |
| TGF-β | 49 ± 4.9‡ | 11.1 ± 0.55‡ | 3.6 ± 0.28 |
| Senescent | |||
| Control | 252 ± 42* | 64.1 ± 9.48* | 13.4 ± 9.0 |
| TGF-β | 276 ± 53* | 61.7 ± 7.40* | 14.1 ± 9.7 |
Non-senescent and senescence-induced NHLF were treated with SFM ± TGF-β (10 ng/ml) for 72 h. CM was collected and assayed for PAPP-A protein, total IGFBP-4 protein, and PAPP-A-mediated proteolysis of IGFBP-4. Results are mean ± SEM from three separate experiments.
PAPP-A protein; ng/106 cells
Total IGFBP-4; ng/105 cells
PAPP-A mediated IGFBP-4 proteolysis; Initial velocity,
by ANOVA
Significant effect of senescence, P < 0.05
Significant effect of TGF-β, P < 0.05
Increased PAPP-A activity is present on extracellular vesicles from senescent lung fibroblasts
Non-senescent NHLF and senescent NHLF were incubated with serum-free medium without or with TGF-β (10 ng/ml) for 72 hours, and conditioned medium collected and processed for extracellular vesicles (EVs). EVs solubilized in RIPA buffer were assayed for both PAPP-A protein and proteolytic activity. Non-senescent NHLF secreted low levels of PAPP-A associated with EVs. EV-PAPP-A expression was increased ~5-fold with senescence from 51 ± 5 to 240 ± 40 ng/mg protein (t-test P = 0.006). EV-PAPP-A proteolytic activity was increased ~4-fold with senescence from 0.8 ± 0.01 to 2.9 ± 1.0 % initial velocity but did not reach statistical significance (P = 0.11). By assaying EVs in PBS, we could show that PAPP-A activity was associated with the surface membrane of intact EVs. We conclude that the EVs generated by senescent NHLF exhibit more PAPP-A and thus have an increased potential to stimulate IGF signaling in recipient cells.
DISCUSSION
Individually, TGF-β, IGF-I and senescent biomarkers have been shown to correlate with severity of IPF in human lung tissue (Hernandez et al., 2020; Schafer et al., 2017). Herein we show that PAPP-A, a metalloproteinase that enhances local IGF bioactivity, is highly significantly correlated with IPF disease severity as assessed by lung function and overall physical function tests in patients. Based on these in vivo data, we set out to develop an interactive cellular model whereby normal human adult lung fibroblasts can be converted to IPF-like cells in vitro (Fig. 4). The major findings on which this model is built are: 1) TGF-β treatment significantly increases IGF-I and PAPP-A secretion and PAPP-A activity, which promotes IGF-IR signaling through the PI3K/Akt pathway; 2) Cellular senescence further increases PAPP-A expression (mRNA and protein) and proteolytic activity; 3) Senescent cells secrete EVs with active PAPP-A associated with the outer membrane.
Figure 4.
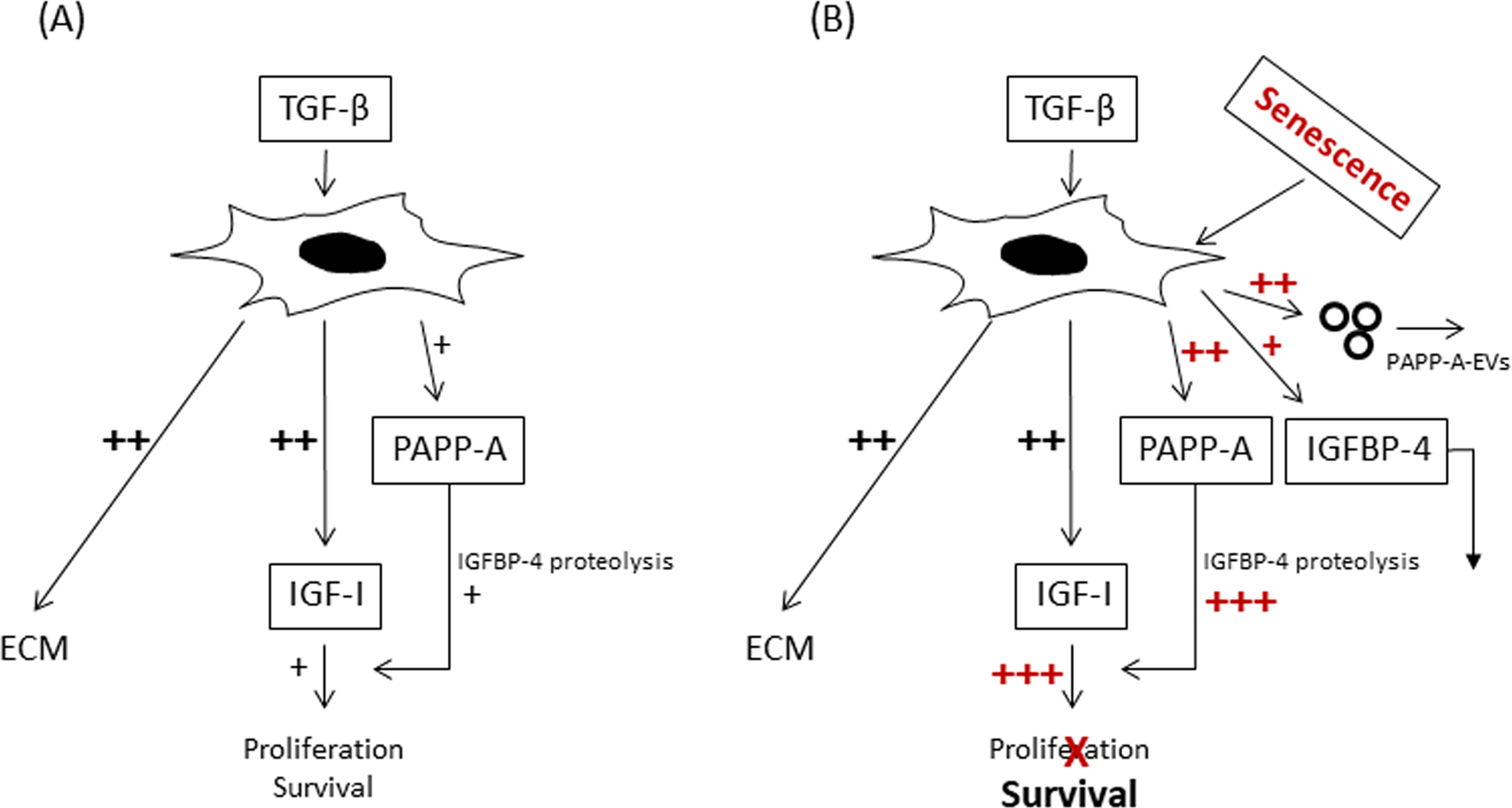
Interactive model of TGF-β, IGF, PAPP-A and cellular senescence in IPF
(A) TGF-β-induced IGF signaling in early stages of fibrosis through increases in IGF-I and PAPP-A expression promotes proliferation and survival of myofibroblasts, the major producer of ECM.
(B) Senescence markedly augments PAPP-A expression and activity in lung SASP while TGF-β-induced expression of IGF-I is retained. Thus, TGF-β, IGF, PAPP-A and senescence appear to interact to promote a pro-fibrotic, anti-apoptotic environment, which could contribute to unresolved pulmonary fibrosis in IPF. In addition, active PAPP-A associated with EVs that are produced by senescent lung fibroblasts and released into the surrounding environment has the potential to participate in crosstalk with other cells in the lung.
TGF-β had a profound effect on IGF-I expression, with 20-fold or more increases in mRNA expression, similar to the findings of Hernandez et al. (Hernandez et al., 2020). In control cultures, there was virtually no IGF-I peptide expression whereas TGF-β treatment for 72 hr resulted in detectable levels in the CM that could initiate IGF-IR signaling through the PI3K/Akt pathway. PI3K/Akt is considered an anti-apoptotic intracellular signaling pathway (Horowitz et al., 2004; Kennedy et al., 1997). TGF-β also stimulated PAPP-A expression in NHLF, with 4-fold increases in PAPP-A protein in 72-hr CM. This secreted PAPP-A was enzymatically active i.e., increased IGF-dependent IGFBP-4 proteolysis, which results in freed IGF for receptor activation. The actions of TGF-β are highly cell-specific and TGF-β stimulation of PAPP-A expression has been shown in human osteoblasts, but not in human dermal fibroblasts (Durham et al., 2003). Our data suggest that TGF-β enhances local IGF bioavailability in NHLF through direct effects on IGF-I and PAPP-A expression. In our working model, TGF-β modulation of IGF signaling in early stages of fibrosis promotes both cell proliferation and survival (Fig. 4A). Proliferation of fibroblasts increases the fibroblastic mass that could be converted to myofibroblasts by TGF-β. The anti-apoptotic effects of TGF-β-induced IGF-I signaling through the PI3K/Akt pathway acts to prevent the clearance of myofibroblasts, the major producer of ECM. In these studies, TGF-β directly stimulated collagen and fibronectin expression in NHLF but IGF-I did not (data not shown).
Accumulation of senescent cells with age appears to have adverse consequences on tissue and organ function, including lung (Alvarez et al., 2017; He & Sharpless, 2017; Meiners & Lehmann, 2020; Morty & Prakash, 2019; Nelson et al., 2012; Schafer et al., 2017). SASP can reinforce the senescent phenotype and spread senescence to non-senescent cells. Here we present data suggesting that PAPP-A is a newly recognized SASP component. A 10-fold increase in PAPP-A protein, 4-fold increase in IGFBP-4 substrate, and 4-fold increase in PAPP-A-mediated proteolytic activity was seen in CM from senescent versus non-senescent NHLF. It is interesting that the relative magnitude of the effect of cellular senescence on PAPP-A and IGFBP-4 expression in NHLF is similar to RNA-seq data of Schafer et al. in IPF lung tissue biopsies (Schafer et al., 2017). SASP PAPP-A may play a significant role in promoting the spread of cellular senescence to non-senescent cells in IPF through its ability to enhance IGF action. In non-proliferating senescent cells, TGF-β-induced expression of IGF-I and ECM was retained with treatment of senescent cells. Thus, TGF-β, IGFs, PAPP-A and senescence appear to interact to promote a pro-fibrotic, anti-apoptotic environment, which could contribute to unresolved pulmonary fibrosis in IPF (Phan, 2002; Scotton & Chambers, 2007) (Fig. 4B).
EVs appear to have a distinct role as regulatory mediators beyond that of traditional soluble factors that are generally considered the main players in SASP. EVs arise from the outward budding and fission of plasma membrane and reflect the membrane composition of the donor cell. EV production increases in multiple cell types following a variety of senescent-inducing stimuli, and EVs from senescent cells appear to play a role in pathogenesis of IPF (Bartel et al., 2020; Kadota et al., 2018). Thus, our finding of active PAPP-A associated with EVs that are produced by lung fibroblasts, especially senescent lung fibroblasts, and released into the surrounding environment would have the potential to participate in crosstalk with other cells involved in the wound healing response in the lung (Fig. 4B).
One proposed therapeutic approach for IPF is to target networks/pathways of anti-apoptosis regulation to promote resolution of the healing process. IGF-I is a potent pro-survival factor for cells (Kooijman, 2006; Kurmasheva & Houghton, 2006). Since PAPP-A can enhance IGF-I action, the results of these experiments provide rationale for potential PAPP-A-targeted therapies. One such therapeutic approach would be to target PAPP-A’s ability to cleave IGFBP-4 with a novel neutralizing monoclonal antibody generated against a unique exosite in PAPP-A (mAb-1/41) (Mikkelsen et al., 2008; Mikkelsen et al., 2014). We have shown that mAb-1/41 works in vivo as well as in vitro (Conover, 2012; Conover et al., 2016; Kashyap et al., 2020; Mikkelsen et al., 2014). Advantages of targeting PAPP-A with this antibody is selectivity of context (high PAPP-A expression) and specificity (no effect on other enzymes and no secondary endocrine effects). We will need in vivo models of IPF to test the hypothesis built upon this cell model that SASP-PAPP-A may be an effective target for prevention or reversal of the fibrogenic state.
Supplementary Material
AKNOWLEDGEMENTS
This study utilized biological specimens and data provided by the Lung Tissue Research Consortium (LTRC) supported by the National Heart, Lung, and Blood Institute (NHLBI).
The authors would like to thank Pernile Noer for excellent technical assistance and Hanne Lucier for her help in formatting this manuscript.
Funding Sources:
This work was supported by a grant from the National Institute on Aging (NIA AG65143, CAC), the Novo Nordisk Foundation (CO), and a generous gift from the John E. and Virginia H. Kunkel family (NKL).
Footnotes
Disclosure Statement: The authors have no conflicts of interests to disclose
DATA AVAILABILITY STATEMENT
Data available on request from the authors
References:
- Alvarez D, Cardenes N, Sellares J, Bueno M, Corey C, Hanumanthu VS, … Rojas M. (2017). IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol, 313(6), L1164–L1173. doi: 10.1152/ajplung.00220.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach LA (2018). IGF-binding proteins. J Mol Endocrinol, 61(1), T11–T28. doi: 10.1530/JME-17-0254 [DOI] [PubMed] [Google Scholar]
- Barile L, Cervio E, Lionetti V, Milano G, Ciullo A, Biemmi V, … Vassalli G. (2018). Cardioprotection by cardiac progenitor cell-secreted exosomes: role of pregnancy-associated plasma protein-A. Cardiovasc Res, 114(7), 992–1005. doi: 10.1093/cvr/cvy055 [DOI] [PubMed] [Google Scholar]
- Bartel S, Deshane J, Wilkinson T, & Gabrielsson S (2020). Extracellular Vesicles as Mediators of Cellular Cross Talk in the Lung Microenvironment. Front Med (Lausanne), 7, 326. doi: 10.3389/fmed.2020.00326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basisty N, Kale A, Jeon O, Kuehnemann C, Payne T, Rao C, … Schilling B. (2019). A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. bioRxiv, 604306. doi: 10.1101/604306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto A, Lerner C, Torres C, Roell M, Malaguti M, Perez V, … Sell C. (2010). Long-term IGF-I exposure decreases autophagy and cell viability. PloS one, 5(9), e12592. doi: 10.1371/journal.pone.0012592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Border WA, & Noble NA (1994). Transforming growth factor beta in tissue fibrosis. N Engl J Med, 331(19), 1286–1292. doi: 10.1056/NEJM199411103311907 [DOI] [PubMed] [Google Scholar]
- Broekelmann TJ, Limper AH, Colby TV, & McDonald JA (1991). Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proceedings of the National Academy of Sciences of the United States of America, 88(15), 6642–6646. doi: 10.1073/pnas.88.15.6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn RC, & Fowlkes JL (2003). Insulin-like growth factor binding protein proteolysis. Trends Endocrinol Metab, 14(4), 176–181. doi: 10.1016/s1043-2760(03)00049-3 [DOI] [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ, & van Deursen JM (2015). Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med, 21(12), 1424–1435. doi: 10.1038/nm.4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover CA (1995). Insulin-like growth factor binding protein proteolysis in bone cell models. Prog Growth Factor Res, 6(2–4), 301–309. doi: 10.1016/0955-2235(95)00032-1 [DOI] [PubMed] [Google Scholar]
- Conover CA (2012). Key questions and answers about pregnancy-associated plasma protein-A. Trends Endocrinol Metab, 23(5), 242–249. doi: 10.1016/j.tem.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover CA, Bale LK, Frye RL, & Schaff HV (2019). Cellular characterization of human epicardial adipose tissue: highly expressed PAPP-A regulates insulin-like growth factor I signaling in human cardiomyocytes. Physiol Rep, 7(4), e14006. doi: 10.14814/phy2.14006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover CA, Bale LK, Mader JR, Mason MA, Keenan KP, & Marler RJ (2010). Longevity and age-related pathology of mice deficient in pregnancy-associated plasma protein-A. Journals of Gerontology Series A-Biological Sciences & Medical Sciences, 65(6), 590–599. doi: 10.1093/gerona/glq032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover CA, Bale LK, Overgaard MT, Johnstone EW, Laursen UH, Fuchtbauer E-M, … van Deursen J. (2004). Metalloproteinase pregnancy-associated plasma protein A is a critical growth regulatory factor during fetal development. Development, 131(5), 1187–1194. doi: 10.1242/dev.00997 [DOI] [PubMed] [Google Scholar]
- Conover CA, Bale LK, & Oxvig C (2016). Targeted inhibition of pregnancy-associated plasma protein-A activity reduces atherosclerotic plaque burden in mice. Journal of cardiovascular translational research, 9(1), 77–79. doi: 10.1007/s12265-015-9666-9 [DOI] [PubMed] [Google Scholar]
- Durham SK, Riggs BL, & Conover CA (1994). The insulin-like growth factor-binding protein-4 (IGFBP-4)-IGFBP-4 protease system in normal human osteoblast-like cells: regulation by transforming growth factor-beta. J Clin Endocrinol Metab, 79, 1752–1758. doi: 10.1210/jcem.79.6.7527411 [DOI] [PubMed] [Google Scholar]
- Ghahary A, Shen Q, Shen YJ, Scott PG, & Tredget EE (1998). Induction of transforming growth factor beta 1 by insulin-like growth factor-1 in dermal fibroblasts. J Cell Physiol, 174(3), 301–309. doi: [DOI] [PubMed] [Google Scholar]
- Gyorfi AH, Matei AE, & Distler JHW (2018). Targeting TGF-beta signaling for the treatment of fibrosis. Matrix Biol, 68–69, 8–27. doi: 10.1016/j.matbio.2017.12.016 [DOI] [PubMed] [Google Scholar]
- Gyrup C, & Oxvig C (2007). Quantitative analysis of insulin-like growth factor-modulated proteolysis of insulin-like growth factor binding protein-4 and −5 by pregnancy-associated plasma protein-A. Biochemistry, 46(7), 1972–1980. doi: 10.1021/bi062229i [DOI] [PubMed] [Google Scholar]
- Hakuno F, & Takahashi SI (2018). IGF1 receptor signaling pathways. J Mol Endocrinol, 61(1), T69–T86. doi: 10.1530/JME-17-0311 [DOI] [PubMed] [Google Scholar]
- He S, & Sharpless NE (2017). Senescence in Health and Disease. Cell, 169(6), 1000–1011. doi: 10.1016/j.cell.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DM, Kang JH, Choudhury M, Andrianifahanana M, Yin X, Limper AH, & Leof EB (2020). IPF pathogenesis is dependent upon TGFbeta induction of IGF-1. FASEB J, 34(4), 5363–5388. doi: 10.1096/fj.201901719RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, … Thannickal VJ. (2004). Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem, 279(2), 1359–1367. doi: 10.1074/jbc.M306248200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu E, & Feghali-Bostwick CA (2008). Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via Jun N-terminal kinase- and phosphatidylinositol-3 kinase-dependent pathways. Am J Pathol, 172(6), 1580–1590. doi: 10.2353/ajpath.2008.071021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CF, Rohani MG, Lee SS, Chen P, & Schnapp LM (2013). Role of IGF-1 pathway in lung fibroblast activation. Respir Res, 14, 102. doi: 10.1186/1465-9921-14-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen MR, Kloverpris S, Mikkelsen JH, Pedersen JH, Fuchtbauer EM, Laursen LS, & Oxvig C (2015). Stanniocalcin-2 inhibits mammalian growth by proteolytic inhibition of the insulin-like growth factor axis. J Biol Chem, 290(6), 3430–3439. doi: 10.1074/jbc.M114.611665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, & Ochiya T (2018). Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: Insights into the pathophysiology of lung diseases. Mol Aspects Med, 60, 92–103. doi: 10.1016/j.mam.2017.11.005 [DOI] [PubMed] [Google Scholar]
- Kashyap S, Hein KZ, Chini CC, Lika J, Warner GM, Bale LK, … Chini EN. (2020). Metalloproteinase PAPP-A regulation of IGF-1 contributes to polycystic kidney disease pathogenesis. JCI Insight, 5(4). doi: 10.1172/jci.insight.135700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, & Hay N (1997). The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes & development, 11(6), 701–713. doi: 10.1101/gad.11.6.701 [DOI] [PubMed] [Google Scholar]
- Koli K, Myllarniemi M, Keski-Oja J, & Kinnula VL (2008). Transforming growth factor-beta activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid Redox Signal, 10(2), 333–342. doi: 10.1089/ars.2007.1914 [DOI] [PubMed] [Google Scholar]
- Kooijman R (2006). Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev, 17(4), 305–323. doi: 10.1016/j.cytogfr.2006.02.002 [DOI] [PubMed] [Google Scholar]
- Kurmasheva RT, & Houghton PJ (2006). IGF-I mediated survival pathways in normal and malignant cells. Biochimica et biophysica acta, 1766(1), 1–22. doi: 10.1016/j.bbcan.2006.05.003 [DOI] [PubMed] [Google Scholar]
- Laursen LS, Overgaard MT, Soe R, Boldt HB, Sottrup-Jensen L, Giudice LC, … Oxvig C. (2001). Pregnancy-associated plasma protein-A (PAPP-A) cleaves insulin-like growth factor binding protein (IGFBP)-5 independent of IGF: implications for the mechanism of IGFBP-4 proteolysis by PAPP-A. FEBS letters, 504, 36–40. doi: 10.1016/s0014-5793(01)02760-0 [DOI] [PubMed] [Google Scholar]
- Laursen LS, Overgaard MT, Weyer K, Boldt HB, Ebbesen P, Christiansen M, … Oxvig C. (2002). Cell surface targeting of pregnancy-associated plasma protein A proteolytic activity. Reversible adhesion is mediated by two neighboring short consensus repeats. J Biol Chem, 277(49), 47225–47234. doi: 10.1074/jbc.M209155200 [DOI] [PubMed] [Google Scholar]
- Limper AH, Broekelmann TJ, Colby TV, Malizia G, & McDonald JA (1991). Analysis of local mRNA expression for extracellular matrix proteins and growth factors using in situ hybridization in fibroproliferative lung disorders. Chest, 99(3 Suppl), 55S–56S. doi: 10.1378/chest.99.3_supplement.55s [DOI] [PubMed] [Google Scholar]
- Meiners S, & Lehmann M (2020). Senescent Cells in IPF: Locked in Repair? Frontiers in Medicine, 7(1002). doi: 10.3389/fmed.2020.606330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Nikolic-Paterson DJ, & Lan HY (2016). TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol, 12(6), 325–338. doi: 10.1038/nrneph.2016.48 [DOI] [PubMed] [Google Scholar]
- Mikkelsen JH, Gyrup C, Kristensen P, Overgaard MT, Poulsen CB, Laursen LS, & Oxvig C (2008). Inhibition of the proteolytic activity of pregnancy-associated plasma protein-A by targeting substrate exosite binding. J Biol Chem, 283(24), 16772–16780. doi: 10.1074/jbc.M802429200 [DOI] [PubMed] [Google Scholar]
- Mikkelsen JH, Resch ZT, Kalra B, Savjani G, Kumar A, Conover CA, & Oxvig C (2014). Indirect targeting of IGF receptor signaling in vivo by substrate-selective inhibition of PAPP-A proteolytic activity. Oncotarget, 5(4), 1014–1025. doi: 10.18632/oncotarget.1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morty RE, & Prakash YS (2019). Senescence in the lung: is this getting old? Am J Physiol Lung Cell Mol Physiol, 316(5), L822–L825. doi: 10.1152/ajplung.00081.2019 [DOI] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, & von Zglinicki T (2012). A senescent cell bystander effect: senescence-induced senescence. Aging Cell, 11(2), 345–349. doi: 10.1111/j.1474-9726.2012.00795.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz CO, Chen BK, Bale LK, Overgaard MT, Oxvig C, & Conover CA (2003). Transforming growth factor-beta regulation of the insulin-like growth factor binding protein-4 protease system in cultured human osteoblasts. J Bone Miner Res, 18(6), 1066–1072. doi: 10.1359/jbmr.2003.18.6.1066 [DOI] [PubMed] [Google Scholar]
- Overgaard MT, Haaning J, Boldt HB, Olsen IM, Laursen LS, Christiansen M, … Oxvig C. (2000). Expression of recombinant human pregnancy-associated plasma protein-A and identification of the proform of eosinophil major basic protein as its physiological inhibitor. J Biol Chem, 275(40), 31128–31133. doi: 10.1074/jbc.M001384200 [DOI] [PubMed] [Google Scholar]
- Phan SH (2002). The myofibroblast in pulmonary fibrosis. Chest, 122(6 Suppl), 286S–289S. doi: 10.1378/chest.122.6_suppl.286s [DOI] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, … Fibrosis A. E. J. A. C. o. I. P. (2011). An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med, 183(6), 788–824. doi: 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, Miller JD, & LeBrasseur NK (2017). Cellular senescence: Implications for metabolic disease. Molecular & Cellular Endocrinology, 455, 93–102. doi: 10.1016/j.mce.2016.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, … LeBrasseur NK. (2017). Cellular senescence mediates fibrotic pulmonary disease. Nature communications, 8, 14532. doi: 10.1038/ncomms14532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotton CJ, & Chambers RC (2007). Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest, 132(4), 1311–1321. doi: 10.1378/chest.06-2568 [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol, 3, Article3. doi: 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- Takasugi M (2018). Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell, 17(2). doi: 10.1111/acel.12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, … Kirkland JL. (2010). Fat tissue, aging, and cellular senescence. Aging Cell, 9(5), 667–884. doi: 10.1111/j.1474-9726.2010.00608.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, & Horowitz JC (2006). Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc, 3(4), 350–356. doi: 10.1513/pats.200601-001TK [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran D, Bergholz J, Zhang H, He H, Wang Y, Zhang Y, … Xiao ZX. (2014). Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell, 13(4), 669–678. doi: 10.1111/acel.12219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uh ST, Inoue Y, King TE Jr., Chan ED, Newman LS, & Riches DW (1998). Morphometric analysis of insulin-like growth factor-I localization in lung tissues of patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med, 158(5 Pt 1), 1626–1635. doi: 10.1164/ajrccm.158.5.9804025 [DOI] [PubMed] [Google Scholar]
- Vancheri C, Failla M, Crimi N, & Raghu G (2010). Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J, 35(3), 496–504. doi: 10.1183/09031936.00077309 [DOI] [PubMed] [Google Scholar]
- Venosa A (2020). Senescence in Pulmonary Fibrosis: Between Aging and Exposure. Front. Med, 7. doi: 10.3389/fmed.2020.606462 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request from the authors