

Abstract
Somatic hypermutation induced by activation-induced deaminase (AID) occurs at high densities between the Ig V gene promoter and intronic enhancer, which encompasses DNA encoding the rearranged V gene exon and J intron. It has been proposed that proximity between the promoter and enhancer defines the boundaries of mutation in V regions. However, depending upon the J gene used, the distance between the promoter and enhancer is quite variable and may result in differential targeting around the V gene. To examine the effect of distance in mutation accumulation, we sequenced 320 clones containing different endogenous rearranged V genes in the IgH and Igκ loci from Peyer’s patch B cells of mice. Clones were grouped by their use of different J genes. Distances between the V gene and enhancer ranged from ~2.3 kb of intron DNA for rearrangements using J1, ~2.0 kb for rearrangements using J2, ~1.6 kb for rearrangements using J3 (H) or 4 (κ), and 1.1 kb for rearrangements using J4 (H) or 5 (κ). Strikingly, >90% of intron mutations occurred within 1 kb downstream of the J gene for both H and κ clones, regardless of which J gene was utilized. Thus, there is no evidence that the intron sequence or enhancer play a role in determining the extent of mutation. The results indicate that V region intron mutations are targeted by their proximity to the promoter, suggesting they result from AID interactions with RNA polymerase II over a 1 kb region.
Introduction
Activation-induced deaminase (AID) is a B cell-specific enzyme which initiates the mechanisms of somatic hypermutation (SHM) and class switch recombination (CSR) (1, 2) through the deamination of cytosine to uracil within Ig genes (3). Uracil is then processed by error-prone base excision repair (BER) (4) and mismatch repair (MMR) (5, 6) pathways to generate mutations throughout Ig variable (V) and switch (S) regions. In the V region, AID promotes SHM by generating nucleotide substitutions in rearranged V, diversity (D), and joining (J) genes to increase affinity for foreign antigens. In the S region, AID initiates CSR by introducing double strand breaks in a donor S region (i.e., Sμ) and an acceptor S region (e.g., Sγ1) to change antibody isotype. Because of the catastrophic potential of unregulated mutations, AID is tightly targeted to these regions in H chain, κ L chain, and λ L chain loci (7).
A critical step to understanding how AID is controlled is to first define the boundaries of SHM—where it starts and where it ends. In S regions, mutations begin downstream of a transcription start site in the intronic μ enhancer (Eμ) (8). They occur at a high frequency of about 10−2 mutations/bp for 3-6 kb and then decline before the constant gene exon (9). During transcription, G:C rich nucleotide sequences form RNA-DNA hybrids (R-loops) and G-quadruplex structures (10-13), which are the basis for targeting AID because deleting the sequences reduced CSR (14-16). It has been hypothesized that these RNA/DNA structures have two functions: to promote AID recruitment through direct binding to G4-quadruplex structures (12, 17), and to pause RNA polymerase II (RNAPII), which allows co-factors to bind AID (16, 18). AID then interacts with the transcription elongation factor SPT5 (19-21), RNA exosome proteins (22, 23), and transcription initiation factor PAF1 (24, 25), which initiate mutagenesis throughout the S region. Therefore, the answer to how AID is targeted to S regions is that repetitive G-rich clusters interspersed with WGCW (W = A/T), a motif that binds AID, pause RNA polymerases and engage AID.
However, targeting of mutations to V regions is enigmatic because (a) no unique structures are formed in the 2 kb of DNA encoding V(D)J exons and flanking intron sequences, and (b) many different V gene sequences undergo SHM. Information on the location of mutations is vital to understanding how AID is recruited and functions. It has long been recognized that mutations start downstream of the transcription start site, intimating that transcription is crucial (26-33). Analogous to S regions, transcriptional co-factors such as SPT5 accumulate in the V region of germinal center B cells and delay the transition of RNAPII from initiation to elongation phases (20). However, less information is known about where mutations end. Our lab previously reported that mutations occur over a limited distance downstream of different rearranged J genes, but the study was limited to a handful of hybridomas (29), precluding an accurate assessment of AID targeting. Therefore, we set out to map mutations in a plethora of V regions using H and κ V genes rearranged to all the JH and JK genes from mice. We found that the mutation distance was surprisingly consistent, with most mutations occurring within 1 kb downstream of the J gene. The data indicate that the V gene promoter is the main driving force controlling AID activity.
Materials and Methods
Mice
C57BL/6 mice were originally obtained from Charles River, and data were collected from both sexes of mice between 4 and 6 months of age. Littermates were used. All animal protocols were reviewed and approved by the Animal Care and Use Committee of the National Institute on Aging.
Germinal center isolation and genomic DNA purification
Peyer’s patches were isolated from the small intestines of two groups of mice: 3 mice for the first set and 5 mice for the second set. Lymphocytes were collected by mechanical separation through a cell strainer, and cells were resuspended in 4 mL of SORT buffer (1x PBS, 25 mM HEPES, 1 mM EDTA, 1% FBS). Germinal center B cells were isolated by staining with antibodies for CD19+ (Biolegend, clone 6D5, labeled with PEcy7), GL7+ (Biolegend, clone GL7, labeled with Alexafluor647), and CD38− (Biolegend, clone 90, labeled with PE). Cells were sorted using a Fusion cell sorter (BD Biosciences) and collected in 1x PBS containing 50% FBS. The cells were then washed and resuspended in 500 μL of TEN buffer (100 mM Tris [pH 8.0], 10 mM EDTA, 1 M NaCl) containing 0.1 mg/mL proteinase K, and they were incubated overnight at 55°C. Genomic DNA was isolated by phenol/chloroform extraction and precipitated in 100% ethanol containing 0.3 M sodium acetate.
VH and VK amplification and mutation analysis
Intron sequences were amplified using Herculase DNA polymerase (Agilent) and nested primers for V genes and intronic enhancers (Table SI). PCR products were dA-tailed using Taq DNA polymerase (Takara) and cloned into StrataClone pSC-Amp/Kan vector (Agilent). During amplification, the smaller J-intron (JH3, JH4, Jκ4, and Jκ5) sequences were preferentially amplified. They were then extracted from the gel and cloned for sequencing. DNA from the larger PCR products (JH1, JH2, Jκ1, Jκ2) were obtained from total amplified DNA that was cloned and expressed in E. coli. Colonies were screened using 32P-end-labeled oligonucleotides specific for the intron sequences, which bound downstream of each J gene (Table SI). Plasmid inserts were sequenced by Sanger sequencing; only clones with unique VDJ and VJ joins were examined. The data was then compared to the C57BL/6 genomic sequence to identify mutations. Mutations were counted at the beginning of the J intron to avoid selection bias in the V(D)J exon. Mutational background from PCR and Sanger sequencing was previously measured as 1.8 x 10−4 mutations/bp (20). Sequences are available on request.
Results
Isolation of rearranged V(D)J clones from Peyer’s patch B cells
To get an overview of mutation in rearranged V genes from the promoter to the 3’ JH intronic region, the cumulative pattern of mutation from our data and other reports show that mutations rapidly increase from the promoter to the V(D)J exon (28, 29, 34-36). This is illustrated in Fig. 1A, which shows the frequency of mutation from the promoter to Eμ in a knockin VDJ mouse (20). The frequency is high in the VDJ exon, likely due to selection for high affinity codon changes. However, it is important to note that Alt and colleagues (37) recorded similar frequencies of mutation in cells containing both a productive VDJ allele and a nonproductive (passenger) VDJ allele with a termination codon. Thus, under non-selective conditions, the mutation mechanism is similar on both alleles. SHM continues to remain high in the unselected 3’ JH intron for about 1 kb. In this report, we sought to define whether this putative 3’ boundary changes relative to proximity of the intronic enhancer.
FIGURE 1.

PCR amplification of rearranged V genes to different J genes in germinal center B cells. (A) Frequency and distribution of SHM in the B1-8hi knockin mouse strain in the promoter (P), leader (L) exon, L intron, VDJ2 exon, and J4 intron. SHM occurs over ~1.5 kb and initiates near the L intron, increases for the next kb including the VDJ exon and proximal J intron, and declines gradually in the distal J intron. Adapted from reference 20. (B) H chain. 5’ primers for the VH1 family were paired with 3’ primers located in Eμ, and 4 distinct products of different sizes were visualized on an agarose gel. (C) κ chain. 5’ primers for the Vκ4 family were paired with 3’ primers prior to Eκ, and 4 specific bands were amplified. Triangles, placement of nested PCR primers.
In order to obtain a large, unbiased database of mutated sequences, we analyzed cells from Peyer’s patches, which accumulate high frequencies of mutations due to encounters with multiple endogenous antigens in the small intestine. Germinal center B cells were sorted, and genomic DNA was amplified using primers to identify rearrangements of different sizes. For the H chain locus, a 5’ primer in framework 3 of the large VH1 family and a 3’ primer in Eμ were used, and for the κ L chain locus, a 5’ primer in framework 3 of the large Vκ4 family and a 3’ primer located 694 bp upstream of Eκ were used (Table SI). The primers produced four bands of different sizes by gel analysis, depending on which J gene was selected. For the H genes (Fig. 1B), rearrangements to J1 produced a 2.3 kb PCR product, J2 yielded a 2.0 kb product, J3 amplified a 1.6 kb band, and J4 generated a 1.1 kb band. For the κ genes (Fig. 1C), rearrangements to J1 produced a 2.4 kb PCR product, J2 yielded a 2.1 kb product, J4 (J3 is a pseudogene on the κ locus) amplified a 1.5 kb band, and J5 generated a 1.1kb band. Primers in framework 3 allowed us to identify clones with unique VDJ or VJ joins. For the H locus, 20-62 clones were obtained, and for the κ locus, 38-53 clones were analyzed (Table I).
Table I.
Sequence data used to analyze mutational positioning in J introns
| IgH VD rearranged to: | JH1 | JH2 | JH3 | JH4 |
|---|---|---|---|---|
| Unique clones | 20 | 39 | 20 | 62 |
| Length (bp) | 2,267 | 1,954 | 1,571 | 1,001 |
| Nucleotides sequenced | 45,340 | 76,206 | 31,420 | 62,062 |
| Mutations | 309 | 618 | 347 | 741 |
| Igκ V rearranged to: | JK1 | JK2 | JK4 | JK5 |
| Unique clones | 38 | 46 | 42 | 53 |
| Length (bp) | 2,324 | 1,969 | 1,338 | 1,000 |
| Nucleotides sequenced | 88,312 | 90,574 | 56,196 | 53,000 |
| Mutations | 523 | 675 | 381 | 533 |
Data from unique VDJ and VJ clones derived from 8 mice were pooled for analysis.
Somatic mutations accumulate at higher frequencies near rearranged J genes
Intron DNA sequences were examined to obtain data from unselected mutations. Mutations for each rearrangement were collected starting with the first nucleotide 3’ of the coding J exon and continuing down to the intronic enhancer. Between 309 and 741 mutations were collected for each rearranged J gene (Table I). The data were compiled to map the density and location of mutations in rearrangements to the four JH genes (Fig. 2A) and four JK genes (Fig. 3A). Mutations in both H and κ genes were most dense in the beginning of the intron and declined further from the promoter. Furthermore, the overall trend in mutations was not influenced by WGCW hotspot density (Figs. 2B and 3B). This can be seen when analyzing VDJH1 clones, where abundant WGCW motifs are present in the downstream JH4 intron and Eμ enhancer, but mutations did not increase in this area. Additionally, in VJκ1 clones, multiple WGCW motifs in the downstream Jκ5 intron did not boost SHM frequency. The more abundant WRCH (W = A/T, R = A/G, H = T/C/A) motifs also did not directly predict SHM occurrence.
FIGURE 2.

Mutational distribution in JH intron sequences. (A) Black vertical lines represent the number of mutations/bp x 102 (y-axis) for each residue from VDJ clones utilizing JH1, JH2, JH3, and JH4 genes. X-axis depicts the distance from JH1 to Eμ (B) Triangles indicate the position of WRCH (blue) and WGCW (red) motifs relative to the JH genes (squares). Half circle shows the portion of the Eμ intronic enhancer that was sequenced.
FIGURE 3.

Mutational distribution in Jκ intron sequences. (A) Black vertical lines represent the number of mutations/bp x 102 (y-axis) for each residue from VJ clones utilizing Jκ1, Jκ2, Jκ4, and Jκ5 genes. X-axis shows the distance from Jκ1 to Eκ. (B) Triangles indicate the position of WRCH (blue) and WGCW (red) motifs relative to the Jκ genes (squares). // represents a break of 694 kb before the Eκ intronic enhancer (circle).
Intron mutation density is diminished after 1 kb from rearranged JH and Jκ genes
Comparisons of the various JH and JK data revealed a striking similarity in the distribution of mutations throughout the introns when the sequences were aligned (Figs. S1 and S2), in that mutations amass proximal to the rearranged J gene and are sparse distal to the J gene. If AID targeting is independent from the sequences being mutated, we considered that mutations should accumulate at similar frequencies for all rearranged J introns. To examine this distribution directly, we calculated mutation frequencies within the initial 1000 bp adjacent to each J gene (Fig. 4). After grouping the mutations into 200 bp increments, the data showed that although there is some variability, there are distinct similarities shared by each J-intron. The highest accumulation occurred close to the rearranged J gene, followed by a leveling off over longer distances. In JH introns, the mutational distribution showed very little differences in the four JH introns as measured by the Mann-Whitney test (Fig. 4A). In Jκ introns, the distribution was similar in Jκ1, Jκ2, and Jκ4 introns, but the Jκ5 intron was significantly different from the others as revealed by the Mann-Whitney comparison. Specifically, there are more mutations proximal to the Jκ5 gene, and they trailed off at a faster frequency than the other three segments (Fig. 4B). One potential reason for the rapid drop off may be the higher percentage of A:T residues, which reaches 63% (data not shown), in the final 400 bp of sequence before Eκ. Thus, this section of the Jκ5 intron is the exception, in that the DNA sequence of the distal 600-1000 bp may restrict G:C targets for AID deamination.
FIGURE 4.

Mutational frequency of J intron sequences. The frequency in 200 bp segments is shown for the initial 1000 bp downstream of the rearranged J genes. (A) JH introns. Mann-Whitney test to the right compares the significance of mutation in JH1 to JH2, JH3, and JH4, etc. (B) Jκ introns. Mann-Whitney test to the right compares the significance of mutation in Jκ1 to Jκ2, Jκ4, and Jκ5, etc.
To further characterize mutational proximity to the rearranged V(D)J exon, we calculated the percentage of total mutations within 500 bp and 1 kb of the rearranged J genes. Strikingly, more than 50% of all mutations analyzed were found within the first 500 bp of the JH and Jκ introns (Fig. 5). Expanding this analysis to 1000 bp revealed that for each J intron, over 75% of all mutations occurred near the rearranged V(D)J gene. This observation indicates that AID is most active close to the promoter, and mutations decrease with distance irrespective of the J-intron sequence used. Additionally, the JH and Jκ loci shared similar mutational patterns, even though the genes are found on different chromosomes and contain different promoter and enhancer elements.
FIGURE 5.

Percent mutations within 500 and 1000 bp of rearranged J genes. Mutational percentages for the JH (A) and Jκ (B) clones. Blue bars represent mutations within 500 bp and gray bars within 1000 bp.
Mutational spectrum at G:C and A:T bp remained constant over long distances
During SHM, mutations occur equally at G:C bp and A:T bp, and they are introduced by the two DNA repair pathways acting on the resulting U produced by AID deamination of C. (i) Error-prone BER removes U by uracil DNA glycosylase to produce mutations of G:C, which directly marks the site of AID activity (38). (ii) Aberrant MMR recognizes U remaining in DNA as a G:U mismatch and recruits MSH2-MSH6 and PMS2-MLH1 complexes. Exonuclease 1 creates a gap which is then filled in by DNA polymerase η to generate mutations of A:T. The length of the gap filling is unknown, but it does spread the distance for polymerase η to synthesize mutations beyond the deaminated C (39, 40). It is possible that the mutation spectra may change farther downstream from rearranged V(D)J genes to favor G:C or A:T mutations, depending on which repair pathway is used. Therefore, we analyzed the frequency of A:T mutations throughout 1400 bp of intron sequences from the heavily mutated JH2 and Jκ2 clones. Grouping mutations into 200 bp windows, A:T mutations occurred at a frequency of 50-65% throughout the length of the introns (Fig. 6). This suggests that AID is active throughout the region, albeit at a lower frequency distal to the promoter, and the deamination events are handled by both BER and MMR.
FIGURE 6.
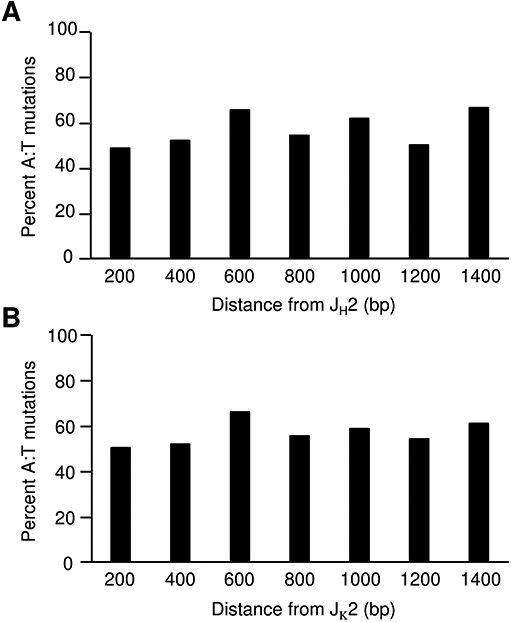
Frequency of mutations at A:T bp throughout long J-intron sequences. The percentage of A:T mutations in JH2 (A) and Jκ2 (B) clones was calculated in 200 bp increments downstream of the J gene. Percent mutations were corrected for nucleotide frequency of A and T within the sequence.
DISCUSSION
The unique targeting of SHM to rearranged V genes and their flanking sequences (29) is a conundrum that has spawned several theories about how mutations start and why they stop. Without a distinct structural mechanism such as R-loops to promote RNAPII pausing within the V region, it is feasible that pausing is caused by proximity of the promoter and intronic enhancer. These transcriptional elements may act as borders to contain AID within the V region. Thus, targeting could (1) start at the promoter, (2) occur in the unique DNA sequences of each JH and Jκ introns, and (3) stop near the Eμ enhancer. We will discuss each of these theories in the context of data in the literature.
First, it is widely accepted that the 5’ boundary of SHM is marked by the promoter in accord with the transcriptional requirement for recruiting AID (41, 42). Nonetheless, how important is promoter proximity for SHM? When comparing mutation in genes with different lengths of introns between the leader and V(D)J exon, Weber et al. (35) showed the distribution of mutations was related to the size of the leader intron. When the intron was short (150 bp), mutations were high in the V exon, and when the intron was long (500 bp), mutations were high in the intron and low in the exon. This was further confirmed by a transgene with 750 bp of foreign DNA inserted into the leader intron (31). Mutations were high in the hybrid intron and low in the exon. Furthermore, in a transgene where the promoter was separated by the leader exon by insertion of 2 kb for foreign DNA, mutations were abolished in the inserted DNA (43), suggesting that the promoter should be contiguous with the leader exon for the mutation mechanism to properly function. Overall, the literature supports promoter proximity for optimal SHM.
Second, the J introns sustain the brunt of the mutational load during SHM, with frequencies as high as those in the V(D)J exons. The exons are selected for high affinity codon changes, and it is possible that SHM in the downstream introns, which are unselected, is more robust than in V genes. For example, the intron DNA could encode palindromes or direct repeats to template mismatches (44). However, when portions of the JH or Jλ introns were deleted, the frequency of mutation in adjacent V genes was unaffected (45, 46), implying a passive role for the intron sequences to accumulate mutations. A more compelling argument against intron sequences directing AID is the similarity of mutation declines seen in the different sequences from four JH and four Jκ introns, that are reported here. This observation was also noted by Rada and Milstein (47), who concluded that the probability that a given base will mutate depends on distance from the 5’ promoter rather than local sequence environment.
Third, the 3’ boundary of mutation is murky. This is partly because the vast majority of mutational data, with some exceptions (48), has been gleaned from VDJ genes rearranged to JH4 (3, 49, 50) or from knockin mice rearranged to the JH4 intron (20, 51). Thus, most of the published data suggests that the nearby Eμ enhancer/matrix attachment regions may generate a physical wall for SHM. Such a wall might be built from the plethora of proteins associated with the enhancer (52). Further explanations of 3’ Eμ regulation have been invoked by the idea of convergent transcription of sense and antisense RNA. Antisense transcription has been shown to be controlled by the intron enhancer (53), and multiple start sites for antisense transcripts have been detected in VDJ genes and the JH introns (54, 55). Convergent transcription could de-stabilize the RNAPII/Spt5/AID axes and prevent mutations from occurring further downstream. Although antisense transcripts originating from Eμ might possibly affect the 3’ boundary in V genes rearranged to JH4, it is harder to imagine their role in enforcing limits for genes rearranged much farther away, such as JH1. Furthermore, deletion of Eμ did not reduce SHM, although its effect on distance was not examined (56, 57).
To evaluate these three theories (promoter, intron, enhancer), we extensively mapped mutations from germinal center B cells that utilized each of the JH and Jκ genes. We postulated that if the promoter and enhancer were functioning to limit mutational spread, then mutations would be distributed differently in cells with rearrangements to either an upstream J gene (JH1 or Jκ1) or a downstream J gene (JH4 or Jκ5). Furthermore, each rearrangement was adjacent to four JH and four Jκ intron sequences, which may recruit AID differently. An analysis of 282 unique clones containing 4,127 mutations revealed that mutations occurred for the same distance downstream of the V gene promoter regardless of which JH or Jκ gene was utilized. The similar trajectory of mutations also suggests an identical mechanism for AID targeting to each J loci. The results imply that AID is not associated with the distance from the intronic enhancers but is spatially associated with the promoter. This is consistent with Theory 1 where RNAPII gives AID access to DNA during the initiation phase of transcription (20), and AID dissociates as the polymerase moves away from the promoter into elongation phase.
To examine Theory 2, we interrogated the DNA sequence of the different introns comparing mutation frequency and spectra. First, we mapped mutations relative to WGCW hotspots. While WGCW density is associated with the probability of local AID deamination (58), the presence or absence of these motifs did not influence where mutations stopped in the intron. Second, an analysis of G:C and A:T mutational spectra may expose structural anomalies in the intron sequences. For example, in the Sμ sequence, the repetitive DNA sequence forms RNA-DNA hybrids, which could disrupt the G:U mismatches and not recruit DNA polymerase η to synthesize mutations opposite A:T pairs (16). Our examination of A:T mutations across the JH and Jκ intron regions showed no perturbations from the usual 50-60% distribution of A:T mutations, indicating that MMR and polymerase η are active at repairing AID-generated lesions across long intron regions.
As for Theory 3, the consistent trajectory and distance of SHM in B cells rearranged to different J genes on the H and κ loci convincingly demonstrate that the same mechanism for SHM is utilized for both loci, and proximity to the 3’ enhancer is superfluous for AID activity. In conclusion, SHM initiates at the start of transcription at the V promoter, depends on paused RNAPII, profusely extends throughout the rearranged V exon and 1 kb of 3’ flanking sequence, and diminishes with further distance. Such an extended distance for paused RNAPII is clearly unique to V regions, as paused RNAPII is mostly found within 100 bp at the start of transcription in non-Ig genes (59). The mechanism for release of AID from the paused transcription complex in the V region is unknown. It will be necessary to define the changing transcriptional landscape of the initiating, paused, and elongation states of RNAPII and associated proteins. Knockin mice with different V(D)J rearrangements will be valuable for probing the precise mechanism of AID targeting.
Supplementary Material
Key Points.
Somatic hypermutation in 4 JH and 4 Jκ introns extends for approximately 1 kb.
Intron DNA sequences and enhancers do not delimit the 3’ boundary of SHM.
V regions are likely targeted for mutation by proximity to the promoter.
ACKNOWLEDGMENTS
We thank Stormy E. Ruiz for comments on the manuscript.
This work was supported entirely through the Intramural Research Program of the National Institutes of Health, National Institute on Aging (AG000714).
Abbreviations used in this article:
- AID
activation-induced deaminase
- BER
base excision repair
- CSR
class switch recombination
- MMR
mismatch repair
- RNAPII
RNA polymerase II
- S
switch
- SHM
somatic hypermutation
REFERENCES
- 1.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563. [DOI] [PubMed] [Google Scholar]
- 2.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, and Durandy A. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 102: 565–575. [DOI] [PubMed] [Google Scholar]
- 3.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, and Neuberger MS. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12: 1748–1755. [DOI] [PubMed] [Google Scholar]
- 4.Di Noia J, and Neuberger MS. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419: 43–48. [DOI] [PubMed] [Google Scholar]
- 5.Wiesendanger M, Kneitz B, Edelmann W, and Scharff MD. 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J Exp Med 191: 579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rada C, Di Noia JM, and Neuberger MS. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell 16: 163–171. [DOI] [PubMed] [Google Scholar]
- 7.Heltzel JMH, and Gearhart PJ. 2020. What Targets Somatic Hypermutation to the Immunoglobulin Loci? Viral Immunol 33: 277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oudinet C, Braikia FZ, Dauba A, and Khamlichi AA. 2020. Mechanism and regulation of class switch recombination by IgH transcriptional control elements. Adv Immunol 147: 89–137. [DOI] [PubMed] [Google Scholar]
- 9.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, and Neuberger MS. 2008. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell 31: 474–484. [DOI] [PubMed] [Google Scholar]
- 10.Yu K, Chedin F, Hsieh CL, Wilson TE, and Lieber MR. 2003. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4: 442–451. [DOI] [PubMed] [Google Scholar]
- 11.Lennon GG, and Perry RP. 1985. Cm-containing transcripts initiate heterogeneously within the IgH enhancer region and contain a novel 5'-nontranslatable exon. Nature 318: 475–478. [DOI] [PubMed] [Google Scholar]
- 12.Qiao Q, Wang L, Meng FL, Hwang JK, Alt FW, and Wu H. 2017. AID Recognizes Structured DNA for Class Switch Recombination. Mol Cell 67: 361–373 e364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribeiro de Almeida C, Dhir S, Dhir A, Moghaddam AE, Sattentau Q, Meinhart A, and Proudfoot NJ. 2018. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol Cell 70: 650–662 e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khamlichi AA, Glaudet F, Oruc Z, Denis V, Le Bert M, and Cogne M. 2004. Immunoglobulin class-switch recombination in mice devoid of any S mu tandem repeat. Blood 103: 3828–3836. [DOI] [PubMed] [Google Scholar]
- 15.Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T, and Neuberger MS. 1994. Elements regulating somatic hypermutation of an immunoglobulin κ gene: critical role for the intron enhancer/matrix attachment region. Cell 77: 239–248. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, and Gearhart PJ. 2009. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med 206: 1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, and Chaudhuri J. 2015. Non-coding RNA Generated following lariat debranching mediates targeting of AID to DNA. Cell 161: 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Wuerffel R, Feldman S, Khamlichi AA, and Kenter AL. 2009. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med 206: 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, and Nussenzweig MC. 2010. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maul RW, Cao Z, Venkataraman L, Giorgetti CA, Press JL, Denizot Y, Du H, Sen R, and Gearhart PJ. 2014. Spt5 accumulation at variable genes distinguishes somatic hypermutation in germinal center B cells from ex vivo-activated cells. J Exp Med 211: 2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanlie A, Begum NA, Akiyama H, and Honjo T. 2012. The DSIF subunits Spt4 and Spt5 have distinct roles at various phases of immunoglobulin class switch recombination. PLoS Genet 8: e1002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, and Alt FW. 2011. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 144: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nair L, Zhang W, Laffleur B, Jha MK, Lim J, Lee H, Wu L, Alvarez NS, Liu ZP, Munteanu EL, Swayne T, Hanna JH, Ding L, Rothschild G, and Basu U. 2021. Mechanism of noncoding RNA-associated N(6)-methyladenosine recognition by an RNA processing complex during IgH DNA recombination. Mol Cell 81: 3949–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willmann KL, Milosevic S, Pauklin S, Schmitz KM, Rangam G, Simon MT, Maslen S, Skehel M, Robert I, Heyer V, Schiavo E, Reina-San-Martin B, and Petersen-Mahrt SK. 2012. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med 209: 2099–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou L, Wang Y, Liu Y, Zhang N, Shamovsky I, Nudler E, Tian B, and Dynlacht BD. 2019. Paf1C regulates RNA polymerase II progression by modulating elongation rate. Proc Natl Acad Sci U S A 116: 14583–14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Davis M, Sinn E, Patten P, and Hood L. 1981. Antibody diversity: somatic hypermutation of rearranged VH genes. Cell 27: 573–581. [DOI] [PubMed] [Google Scholar]
- 27.Pech M, Hochtl J, Schnell H, and Zachau HG. 1981. Differences between germ-line and rearranged immunoglobulin V kappa coding sequences suggest a localized mutation mechanism. Nature 291: 668–670. [DOI] [PubMed] [Google Scholar]
- 28.Gearhart PJ, and Bogenhagen DF. 1983. Clusters of point mutations are found exclusively around rearranged antibody variable genes. Proc Natl Acad Sci U S A 80: 3439–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebecque SG, and Gearhart PJ. 1990. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5' boundary is near the promoter, and 3' boundary is approximately 1 kb from V(D)J gene. J Exp Med 172: 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukita Y, Jacobs H, and Rajewsky K. 1998. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity 9: 105–114. [DOI] [PubMed] [Google Scholar]
- 31.Tumas-Brundage K, and Manser T. 1997. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. J Exp Med 185: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters A, and Storb U. 1996. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity 4: 57–65. [DOI] [PubMed] [Google Scholar]
- 33.Pham P, Malik S, Mak C, Calabrese PC, Roeder RG, and Goodman MF. 2019. AID-RNA polymerase II transcription-dependent deamination of IgV DNA. Nucleic Acids Res 47: 10815–10829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hackett J Jr., Rogerson BJ, O'Brien RL, and Storb U. 1990. Analysis of somatic mutations in kappa transgenes. J Exp Med 172: 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber JS, Berry J, Manser T, and Claflin JL. 1994. Mutations in Ig V(D)J genes are distributed asymmetrically and independently of the position of V(D)J. J Immunol 153: 3594–3602. [PubMed] [Google Scholar]
- 36.Rada C, Yelamos J, Dean W, and Milstein C. 1997. The 5' hypermutation boundary of kappa chains is independent of local and neighbouring sequences and related to the distance from the initiation of transcription. Eur J Immunol 27: 3115–3120. [DOI] [PubMed] [Google Scholar]
- 37.Yeap LS, Hwang JK, Du Z, Meyers RM, Meng FL, Jakubauskaite A, Liu M, Mani V, Neuberg D, Kepler TB, Wang JH, and Alt FW. 2015. Sequence-Intrinsic Mechanisms that Target AID Mutational Outcomes on Antibody Genes. Cell 163: 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM 3rd, and Gearhart PJ. 2011. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol 12: 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martomo SA, Yang WW, Wersto RP, Ohkumo T, Kondo Y, Yokoi M, Masutani C, Hanaoka F, and Gearhart PJ. 2005. Different mutation signatures in DNA polymerase η- and MSH6-deficient mice suggest separate roles in antibody diversification. Proc Natl Acad Sci U S A 102: 8656–8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martomo SA, Yang WW, and Gearhart PJ. 2004. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J Exp Med 200: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Prado AF, Perez-Duran P, Perez-Garcia A, Benguria A, Torroja C, de Yebenes VG, and Ramiro AR. 2018. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J Exp Med 215: 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Methot SP, Litzler LC, Subramani PG, Eranki AK, Fifield H, Patenaude AM, Gilmore JC, Santiago GE, Bagci H, Cote JF, Larijani M, Verdun RE, and Di Noia JM. 2018. A licensing step links AID to transcription elongation for mutagenesis in B cells. Nature communications 9: 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winter DB, Sattar N, Mai JJ, and Gearhart PJ. 1997. Insertion of 2 kb of bacteriophage DNA between an immunoglobulin promoter and leader exon stops somatic hypermutation in a kappa transgene. Mol Immunol 34: 359–366. [DOI] [PubMed] [Google Scholar]
- 44.Golding GB, Gearhart PJ, and Glickman BW. 1987. Patterns of somatic mutations in immunoglobulin variable genes. Genetics 115: 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kothapalli NR, Collura KM, Norton DD, and Fugmann SD. 2011. Separation of mutational and transcriptional enhancers in Ig genes. J Immunol 187: 3247–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castiblanco DP, Norton DD, Maul RW, and Gearhart PJ. 2018. JH6 downstream intronic sequence is dispensable for RNA polymerase II accumulation and somatic hypermutation of the variable gene in Ramos cells. Mol Immunol 97: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rada C, and Milstein C. 2001. The intrinsic hypermutability of antibody heavy and light chain genes decays exponentially. EMBO J 20: 4570–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, Parris T, Edelmann W, and Scharff MD. 2004. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat Immunol 5: 224–229. [DOI] [PubMed] [Google Scholar]
- 49.Jolly CJ, Klix N, and Neuberger MS. 1997. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res 25: 1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maul RW, MacCarthy T, Frank EG, Donigan KA, McLenigan MP, Yang W, Saribasak H, Huston DE, Lange SS, Woodgate R, and Gearhart PJ. 2016. DNA polymerase iota functions in the generation of tandem mutations during somatic hypermutation of antibody genes. J Exp Med 213: 1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shih TA, Roederer M, and Nussenzweig MC. 2002. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat Immunol 3: 399–406. [DOI] [PubMed] [Google Scholar]
- 52.Dinesh RK, Barnhill B, Ilanges A, Wu L, Michelson DA, Senigl F, Alinikula J, Shabanowitz J, Hunt DF, and Schatz DG. 2020. Transcription factor binding at Ig enhancers is linked to somatic hypermutation targeting. Eur J Immunol 50: 380–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolland DJ, Wood AL, Afshar R, Featherstone K, Oltz EM, and Corcoran AE. 2007. Antisense intergenic transcription precedes Igh D-to-J recombination and is controlled by the intronic enhancer Emu. Mol Cell Biol 27: 5523–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ronai D, Iglesias-Ussel MD, Fan M, Li Z, Martin A, and Scharff MD. 2007. Detection of chromatin-associated single-stranded DNA in regions targeted for somatic hypermutation. J Exp Med 204: 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perlot T, Li G, and Alt FW. 2008. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci U S A 105: 3843–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inlay MA, Gao HH, Odegard VH, Lin T, Schatz DG, and Xu Y. 2006. Roles of the Ig kappa light chain intronic and 3' enhancers in Igk somatic hypermutation. J Immunol 177: 1146–1151. [DOI] [PubMed] [Google Scholar]
- 57.Li F, Yan Y, Pieretti J, Feldman DA, and Eckhardt LA. 2010. Comparison of identical and functional Igh alleles reveals a nonessential role for Emu in somatic hypermutation and class-switch recombination. J Immunol 185: 6049–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang C, Bagnara D, Chiorazzi N, Scharff MD, and MacCarthy T. 2020. AID Overlapping and Poleta Hotspots Are Key Features of Evolutionary Variation Within the Human Antibody Heavy Chain (IGHV) Genes. Frontiers in immunology 11: 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brookes E, de Santiago I, Hebenstreit D, Morris KJ, Carroll T, Xie SQ, Stock JK, Heidemann M, Eick D, Nozaki N, Kimura H, Ragoussis J, Teichmann SA, and Pombo A. 2012. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell stem cell 10: 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.