

Abstract
Advances in CRISPR-Cas9 genome editing technology have strengthened the role of zebrafish as a model organism for genetics and development biology. These tools have led to a significant increase in the production of loss-of-function mutant zebrafish lines. However, the generation of precisely edited knock-in lines has remained a significant challenge in the field due to the decreased efficiency of homology directed repair (HDR). In this study, we overcame some of these challenges by combining available design tools and synthetic, commercially available CRISPR reagents to generate a knock-in line carrying an in-frame MYC epitope tag at the sox11a locus. Zebrafish Sox11a is a transcription factor with critical roles in organogenesis, neurogenesis, craniofacial, and skeletal development; however, only a few direct molecular targets of Sox11a have been identified. Here, we evaluate the knock-in efficiency of various HDR donor configurations and demonstrate the successful expression and localization of the resulting knock-in allele. Our results provide an efficient, streamlined approach to knock-in experiments in zebrafish, which will enable expansion of downstream experimental applications that have previously been difficult to perform. Moreover, the MYC-Sox11a line we have generated will allow further investigation into the function and direct targets of Sox11a.
Keywords: zebrafish, Sox11, knock-in, retina, CRISPR
Introduction
The zebrafish (Danio rerio) is a powerful model organism for genetics and developmental biology research [1]. Recently, the widespread adoption of CRISPR-Cas9 genome editing has enabled the generation of numerous zebrafish knockout lines by exploiting error-prone DNA repair of Cas9 induced double strand breaks via non-homologous end joining (NHEJ) [2]. This method has greatly facilitated zebrafish reverse genetics approaches [3,4].
The CRISPR-Cas9 system can also be used to harness homology-dependent repair (HDR) mechanisms. This approach involves providing a donor template that contains homology to the CRISPR target site. Repair of the resulting double strand break is accompanied by insertion of the donor sequence at the site of interest. This method allows for the precise alteration of nucleotides in a coding region [5,6], or the addition of sequences that encode fluorescent markers or epitope tags [8,9]. These additions can help overcome the lack of reliable antibodies in zebrafish and can facilitate live imaging approaches.
While using the CRISPR system to produce knockouts is highly efficient, the success rate of HDR remains very low in zebrafish [4]. Efforts to improve HDR efficiency have included varying the type of donor sequence, the length and ratio of homology arms, and the use of synthetic and chemically modified donors and gRNAs [9–14]. These experiments have produced widely variable results, and there is little consensus in the field as to what produces the highest efficiency of sequence integration. Additionally, germline transmission, expression, and function of the knock-in allele are not always demonstrated after successful donor sequence integration [16].
In this work, we sought to establish a knock-in zebrafish line in which an in-frame MYC epitope tag is incorporated into the coding region of the developmentally important transcription factor sox11a. Loss of Sox11a expression in zebrafish has previously been associated with numerous developmental defects; however, the current lack of a reliable Sox11a antibody has limited further investigation into its function and downstream transcriptional targets [16,17].
We determined the HDR insertion efficiency of various chemically modified donor templates. An established founder was then evaluated for germline transmission, functional integration, and off-target effects. The streamlined and efficient approach we describe should be easily adoptable for other researchers. Moreover, our MYC-Sox11a transgenic line provides a useful new reagent for further studies of its role in development.
Material and Methods
Zebrafish Lines and Maintenance
Zebrafish were bred, raised, and housed at 28.5°C on a 14-hour light:10-hour dark cycle in compliance with established protocols for zebrafish husbandry [18]. All animal procedures were carried out in accordance with guidelines established by the University of Kentucky Institutional Animal Care and Use Committee and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Wildtype zebrafish (AB strain) were obtained from the Zebrafish International Resource Center (ZIRC, Eugene, OR).
CRISPR Design and Microinjections
The sox11a target site and donor template were designed using the Integrated DNA Technologies Alt-R™ CRISPR HDR Design Tool (https://www.idtdna.com/pages/tools/alt-r-crispr-hdr-design-tool; IDT, Coralville, IA). The target site for genome editing was selected in the 5’UTR of sox11a, 16 base pairs (bp) upstream of the start codon. The donor template sequences were designed with the MYC tag sequence placed just after the start codon and included 40 bp of left arm and 80 bp of right arm homology (Figure 1). The donor templates were synthesized and supplied by IDT as two Alt-R HDR Donor Blocks (Donor A and Donor B), an unmodified double stranded template (Donor C), and two single stranded Alt-R HDR Donor Blocks (forward strand Donor D and reverse strand Donor E).
Figure 1. Knock-in design for MYC-sox11a.
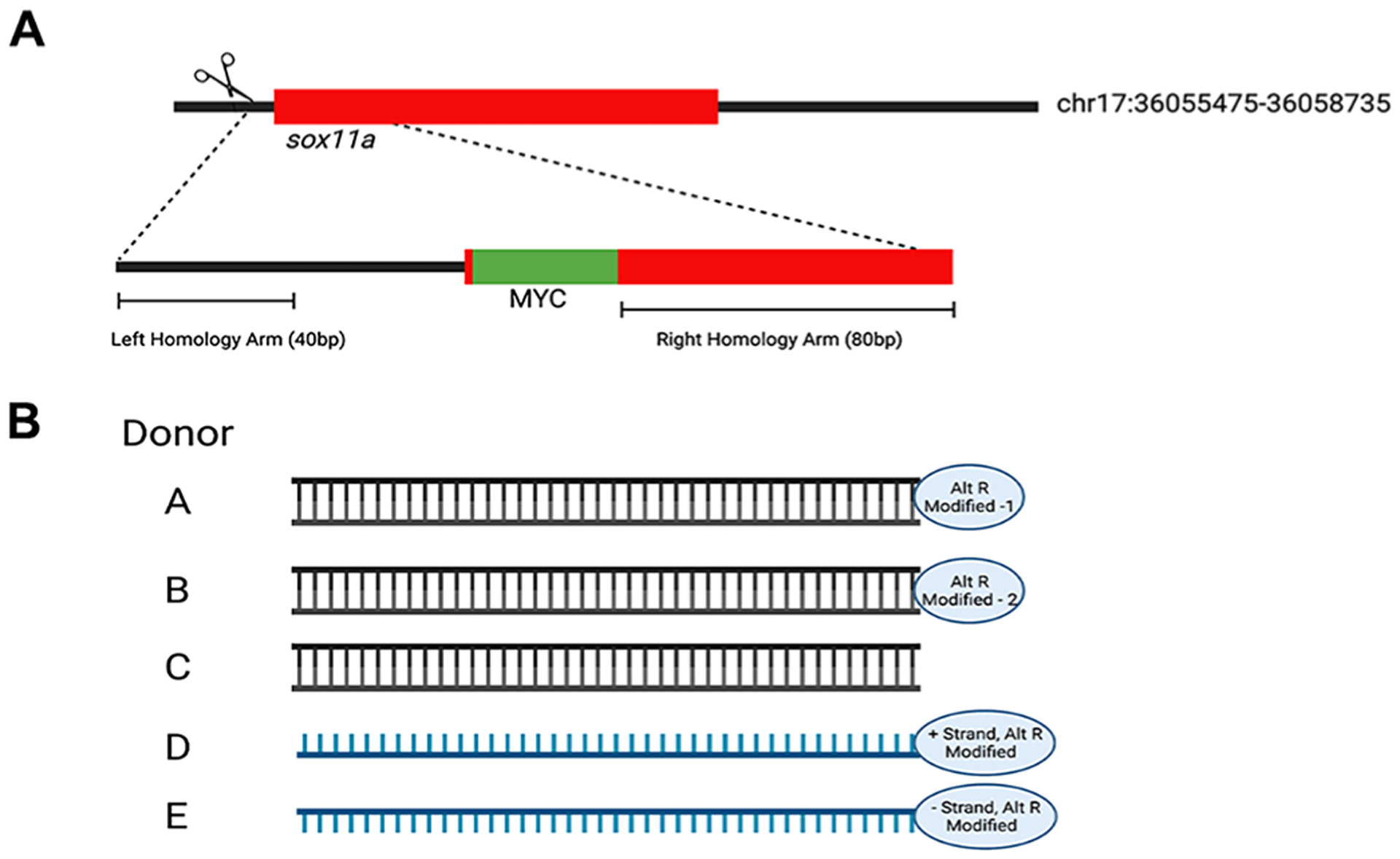
A. Schematic of sox11a locus, including Cas9 target site (scissors) and MYC-donor template. B. Depiction of five different donors (A-E). Donor A, Double Stranded Alt-R Modified #1; Donor B, Double Stranded Alt-R Modified #2; Donor C, Double Stranded unmodified; Donor D, Forward single strand Alt-R Modified; Donor E, Reverse single strand Alt-R Modified.
The Alt-R CRISPR-Cas9 crRNA was synthesized by IDT and was duplexed with Alt-R CRISPR-Cas9 tracrRNA (IDT: 1073190) in a Eppendorf Mastercycler with the following program: 95°C for 5 minutes, cooling to 25°C at a rate of 0.1°C/second, (Eppendorf, Enfield, CT). Cas9 protein was purchased as Alt-R® S.p. Cas9 Nuclease V3 (IDT: 1081058). One-cell stage embryos were injected with 250 pg gRNA, 500 pg Cas9, 37.5 pg HDR template, and Dextran red.
Insertion Screening
Genomic DNA (gDNA) was extracted from whole embryos or tails of injected adult fish or their progeny. Tissue was placed in 50mM sodium hydroxide and incubated at 95°C for digestion. The solution was neutralized with 1 M Tris-HCl, pH 8.0. The gDNA was PCR amplified using primers described in Supplemental Table 1. Products were visualized on a 10% polyacrylamide gel. Products that matched the expected insertion size were extracted from the gel into dialysis tubing (Thermo Scientific: 88243, Rockford, IL), ligated into pGEM-T Easy (Promega: A1360, Madison, WI), and sequenced with universal T7 and Sp6 primers (Eurofins Genomics Services, Louisville, KY).
Immunohistochemistry and RNAscope
Embryos were fixed overnight in 4% paraformaldehyde, then incubated overnight in 10% followed by 30% sucrose at 4°C. 10μm transverse cryosections were taken on Leica CM1900 crysostat (Leica Biosystems, Buffalo Grove, IL).
Immunohistochemistry was performed on cryosections as previously described with slight modifications for indirect antibody detection [19]. We used an anti-MYC primary antibody (9B11, 1:1000; Cell Signaling #2276, Danvers, MA) followed by signal amplification with goat anti-mouse IgG horseradish peroxidase (1: 500) (Perkin Elmer Inc: NEF822001EA, Waltham, MA). The TSA plus Cy3 Kit (1:1500) (Perkin Elmer Inc: NEL744001KT, Waltham, MA) was used for detection. The sections were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI, 1:10,000 dilution, Sigma-Aldrich). Images were obtained on a Nikon Eclipse Ti-U inverted fluorescent microscope (Nikon Instruments, Melville, NY)
The sox11a RNAscope probe was obtained from Advanced Cell Diagnostics (ACD: 590461, Newark, CA) and labelling was carried out on cryosections according to RNAscope™ Multiplex Fluorescent V2 Assay protocol.
At least 10 embryos were analyzed per timepoint, and 3 separate biological replicates were performed for each experiment.
RT-PCR
Total RNA was extracted from whole sox11aMYC embryos at 48 hpf using TRIzol reagent (Invitrogen, GrandIsland, NY). First strand cDNA was synthesized from 0.5 μg of the extracted RNA using GoScript Reverse Transcriptase System (Promega, Madison, WI). PCR primers were designed to amplify a unique region of the MYC tag and the junction of sox11a-MYC (Eurofins Genomics, Supplemental Table 1). PCR products were visualized on a 10% polyacrylamide gel.
Off-Target Screening
The top 50 potential off-target sites were compiled using CRISPOR (http://crispor.tefor.net/), Cas-OFFinder (http://www.rgenome.net/cas-offinder/) and CRISPR-Cas9 guide RNA design checker (IDT, https://www.idtdna.com/site/order/designtool/index/CRISPR_SEQUENCE) [20,21]. These targets were used to design an rhAMP Seq Amplicon Sequencing panel (IDT). Genomic DNA was extracted from tails of sox11aMYC and wildtype fish with Monarch Genomic DNA Purification Kit (NEB: T3010S, Ipswich, MA). Targeted amplicons and rhAMP Seq library were produced according to the rhAmpSeq library kit manual. The prepared libraries were then sequenced on an Novaseq S4 Illumina platform (Novogene, Sacramento, CA). Sequencing data were analyzed with rhAmpSeq™ CRISPR Analysis Tool (IDT).
Data Analysis and Figure Construction
Schematics in Figures 1, 2, and 4 were created using BioRender (biorender.com). All figures were constructed using Photoshop (Adobe version 22.0.0).
Figure 2. Detection of HDR to generate MYC-sox11a.
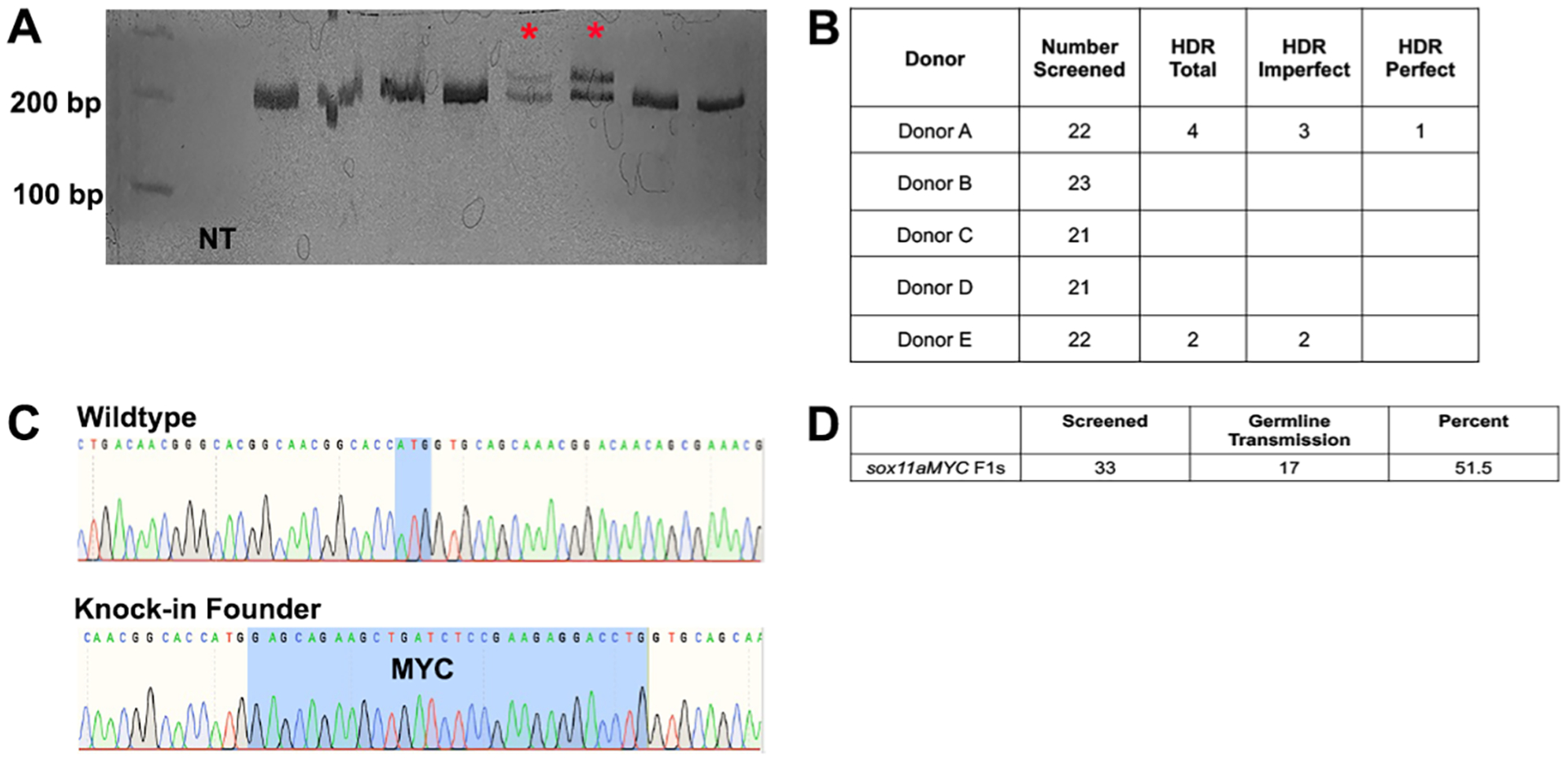
A. Example of polyacrylamide gel used to observe insertion events in injected zebrafish. Asterisks represent samples that display an upper band of the correct size for MYC insertion; NT, no template. B. Efficiency of homology directed repair (HDR) for the five different donors in adult founder zebrafish and whether the HDR events included additional indels (Imperfect) or were precise (Perfect). C. Chromatograms from Sanger sequencing of wildtype and knock-in founders from Donor A. D. Percent of germline transmission of the knock-in allele to F1 progeny.
Figure 4. rhAMP Sequencing analysis for confirmation of insertion and screening for off-target events.
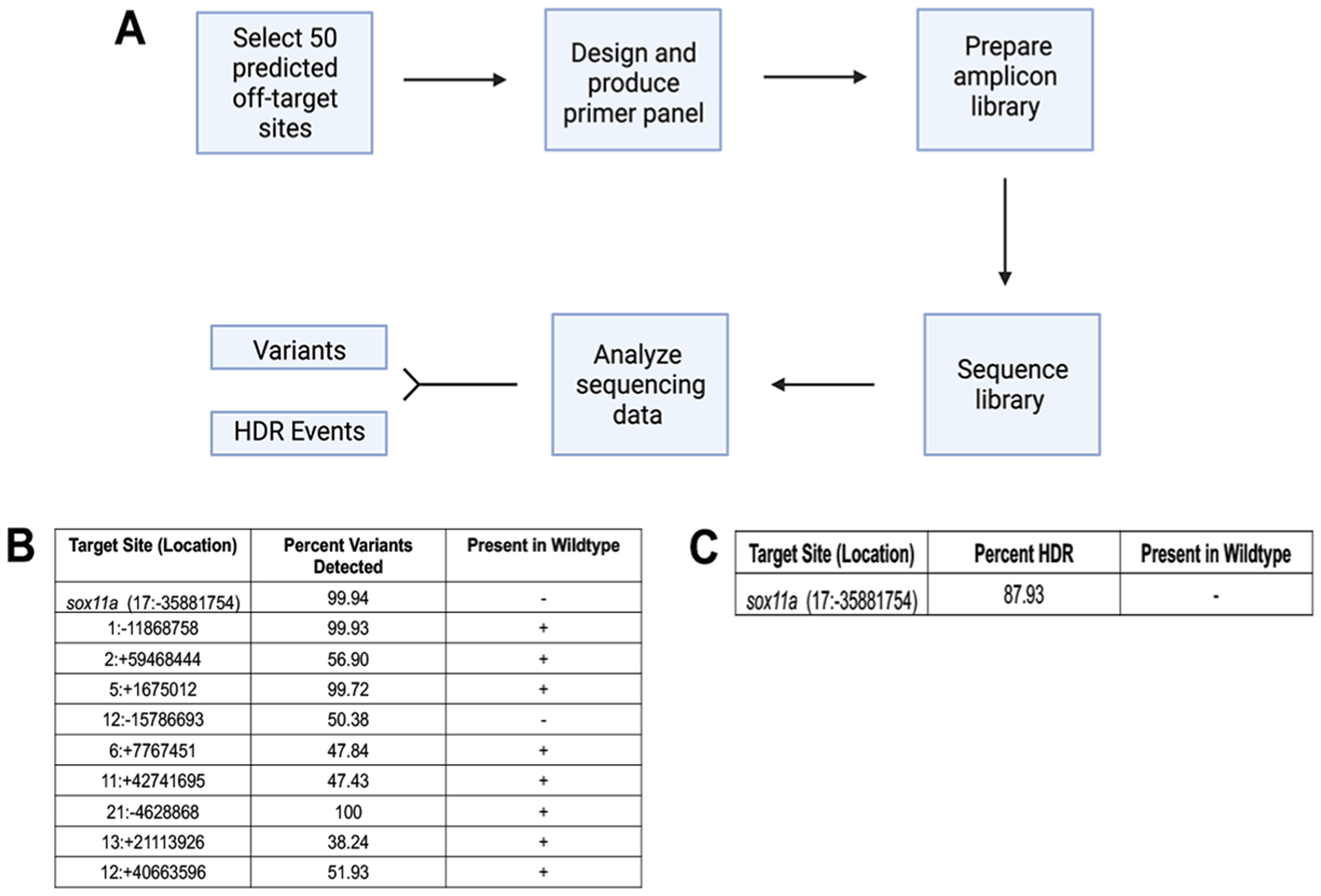
A. Workflow schematic for design, production, and analysis of amplicon sequencing. B. Table characterizing detected variants in knock-in MYC-sox11a versus and wildtype genomic DNA. C. Chart depicting percent of HDR events in knock-in MYC-sox11a and in wildtype fish.
Results
Knock-in design and donor template production
To develop an efficient and direct method to knock-in an epitope tag (MYC) at the 5’ end of the coding sequence of zebrafish sox11a, we utilized a synthetic crRNA-tracrRNA and Cas9 ribronucleoprotein (RNP) complex developed by Integrated DNA Technologies (IDT), which has previously been shown to produce a high rate of mutagenesis in zebrafish [14,22]. We used IDT’s HDR Design Tool to choose the target site and donor sequence for the knock-in experiment. We selected a crRNA corresponding to a site 16 base pairs (bp) upstream of the sox11a transcription start site (Supplementary Table 1), which had high on-target and low off-target scores. Our HDR donor design included 40 bp left homology and 80 bp right homology arms, based on previous work showing that homology arm asymmetry provides a slightly higher efficiency for HDR [14]. This design resulted in a 169 bp donor template that includes the MYC epitope sequence in-frame and just downstream of the sox11a start codon (Figure 1A).
Given that there is currently no consensus on which type of donor sequence (single or double stranded) produces the highest efficiency for HDR we decided to test five different donor templates. In collaboration with IDT, we obtained an unmodified double-stranded template (Donor C), two double-stranded donors, each with distinct chemical modifications (Donors A and B), a forward strand oriented single-stranded chemically modified donor (Donor D), and a reverse strand oriented single-stranded chemically modified (Donor E), (Figure 1B). The specific chemical modifications of Donor A are now incorporated into a commercially available product and are referred to as Alt-R HDR Donor Blocks.
Screening for knock-in efficiency by donor and germline transmission
Wildtype zebrafish embryos were microinjected with the sox11a targeting crRNA-tracrRNA duplex and Cas9 protein complex along with one of the donors at the one cell stage. Injected embryos were raised to adulthood and tail clipped for genomic DNA extraction. We analyzed twenty fish for each donor by polymerase chain reaction (PCR) with primers located on either side of expected insertion site (Supplementary Table 1). Based on the length of the MYC epitope, successful HDR would result in a 30 bp insertion, producing an amplicon of 224 bp compared to the wildtype size of 194 bp (Figure 2A). We sequenced DNA purified from bands of the correct size, and analyzed the sequences for evidence of HDR-mediated insertion of the MYC epitope. Insertions were characterized as perfect or imperfect, depending on whether any additional indels were detected at the target site. Donors A and E were the only two donors that showed evidence of HDR. For each donor, the proportion of injected individuals that harbored the donor sequences was 18% and 9%, respectively. However, only Donor A produced a perfect integration resulting in the complete insertion of MYC at the correct site with no other indels (Figure 2B, 2C).
The Donor A founder fish was then outcrossed to wildtype fish and the offspring were analyzed for MYC insertion as described above. Seventeen out of 33 F1 progeny carried the MYC epitope, a germline transmission rate of over 50% (Figure 2D). These results indicate that an epitope knock-in can be generated with use of CRISPR-Cas9 complex in combination with an Alt-R double-stranded HDR donor block.
Validation of MYC-Sox11a expression in knock-in line
To determine whether MYC-Sox11a mRNA and protein were detectable in our knock-in line, we performed RT-PCR with three different primer sets on RNA extracted from heterozygous MYC-sox11a embryos. Primer pair 1 was outside insertion site, primer pair 2 has a forward primer anchored in MYC, and primer pair 3 has a forward primer at the junction of MYC and sox11a. All three primer pairs produced the expected PCR product sizes, indicating that MYC is indeed present in the mRNA transcribed from the sox11a locus of the knock-in animals (Figure 3A).
Figure 3. Validation of MYC-sox11a expression.
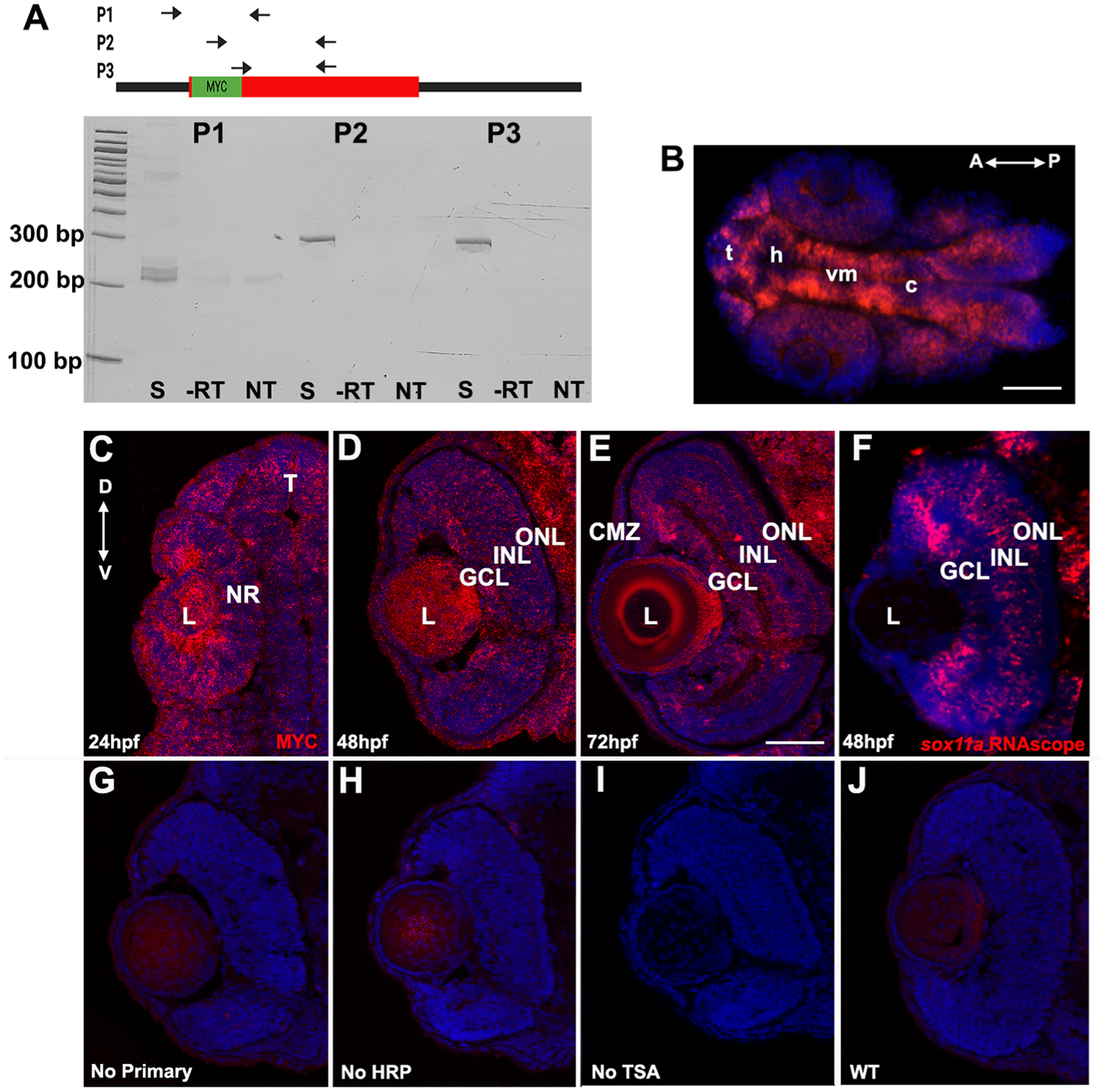
A. RT-PCR of MYC-sox11a mRNA. Location of primer sets P1, P2, and P3 are indicated in the schematic. S, sample; -RT, no reverse transcriptase; NT, no template B. Immunohistochemistry of 24 hpf brain section for MYC-Sox11a confirms expected expression pattern in the telencephalon (t), posterior diencephalon, and ventral midbrain (vm). (C-F) Immunohistochemistry for MYC-sox11a in the developing retina at 24 hpf (C), 48 hpf (D), and 72 hpf (E); MYC-Sox11a expression is detected throughout the neural retina at 24 hpf, and in the ganglion cell layer, the inner nuclear layer, and the ciliary marginal zone at 48 and 72 hpf. F. Expression pattern of sox11a at 48 hpf detected by RNAscope for comparison. G-J. Control sections at 48 hpf: G, no primary antibody; H, no horseradish peroxidase (HRP) amplification; I, no Tyramide signal amplification (TSA); and J, wildtype uninjected retina. L, lens; NR, neural retina; GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; CMZ, ciliary marginal zone; t, telencephalon; h, hypothalamus; vm, ventral midbrain; c, cerebellum. Scale Bar, 100 uM.
To confirm that MYC-tagged Sox11a is translated and detectable we completed a series of immunohistochemistry (IHC) experiments. Retinal and brain sections were obtained from embryos at 24, 48, and 72 hours post fertilization (hpf) and IHC was performed with an anti-MYC epitope antibody (Figure 3C–F). Fluorescence detection with a fluorescently labeled secondary antibody did not produce a detectable signal (data not shown). Therefore, we performed IHC followed by indirect detection with horseradish peroxidase catalyzed tyramide signal amplification (TSA; Figure 3C–E). Consistent with previous work from our lab describing sox11a expression in the developing retina [18], at 48 hpf anti-MYC positive cells were detected in the ganglion cell layer (GCL), and at 72hpf MYC+ cells were detectable in the GCL, in some cells of the inner nuclear layer (INL), and in the persistently neurogenic ciliary marginal zone (CMZ). Control sections (no primary antibody, no TSA, and uninjected) had no signal (Figure 3G–J). Interestingly, MYC expression was also detected throughout the developing retina at 24 hpf and in some INL cells at 48 hpf, which was not detected by in situ hybridization in our previous study [16]. This could be due to the increased sensitivity of TSA amplification or to differences in Sox11a protein versus sox11a mRNA stability. Using the more sensitive technique of RNAscope, we found that the MYC-Sox11a expression pattern was similar to sox11a mRNA expression in the retina at 48 hpf (Figure 3F). In the brain, we detected MYC-Sox11a expression in the telencephalon, posterior ventral diencephalon, and ventral midbrain at 24 hpf, all of which are consistent with published descriptions of sox11a expression in this region (Figure 2B) [23]. Taken together, these results confirm that the MYC-tagged Sox11a protein is detectable and is expressed in a similar manner to endogenous, untagged sox11a.
Sequencing screen for off-target and on-target integration
Double stranded DNA donors have been reported to integrate at off-target locations in the genome [24,25], therefore we investigated whether there were any off-target insertions or other editing events in our MYC-Sox11a knock-in line. We utilized targeted amplicon sequencing of a DNA library containing 50 potential off-target sites identified from three different off-target prediction programs (compiled by the IDT rhAmp Seq CRISPR Analysis System); DNA libraries were then prepared from wildtype zebrafish and the MYC-Sox11a knock-in line [20,21]. The libraries were sequenced, and the sequencing data were analyzed in two different ways (Figure 4A).
First, using rhAmpSeq CRISPR Analysis Tool we analyzed our sequencing data for all variants present by comparing the sequencing data to the zebrafish genome assembly, GRCz11. There were 10 sites in our knock-line that displayed a significant percentage of variants in the sequencing reads compared to the reference genome. However, 8 of the 10 sites also contained the same variant in the wildtype DNA (Figure 4B), indicating that these are likely single nucleotide polymorphisms (SNPs) present in our laboratory stocks of wildtype zebrafish and thus are not Cas9-mediated editing events. For the two variants that were not present in wildtype sequence, the first is the intended target site of sox11a. The second, 12:−15786693, is located in the intronic region of the gene supervillind. While this may represent a true off-target editing event, the number of sequence reads that mapped to this site was very low (only 397 total reads, compared to an average of 344,953 reads for the other sites); therefore, we suspect this result is an artifact due to poor amplicon production.
Next, we used the rhAmpSeq CRISPR Analysis Tool to scan for all instances of HDR integration using the sequence of the donor template as a guide. The only site detected as an HDR event was the on-target site of sox11a (Figure 4C). This HDR event was present in about 88% of the sequencing reads from the knock-in line and as expected was not detected in the wildtype line. Taken together, these results confirm that MYC was inserted at the target site but not at any potential off-target sites.
Discussion
In this study, we describe a direct and simplified method for establishing a zebrafish transgenic knock-in line that introduces a MYC epitope tag at the N-terminus of Sox11a. We utilized readily available tools from Integrated DNA Technologies (IDT) to design our reagents, including the synthetic crRNA for sox11a and the donor template containing the MYC tag sequence. We tested different chemical modifications of the donor template which we screened for efficiency of successful HDR. A chemically modified dsDNA donor, Alt-R HDR Donor Block, successfully introduced MYC in frame after the start site of sox11a and this founder fish has produced a stable line by germline transmission. This knock-in line was further validated for detection of MYC and establishes a tool for downstream experiments which we previously did not have.
Similar to some previous studies, our approach relied on commercially synthesized in-vitro donors[14,26] allowing for more time spent on injecting and screening, resulting in an increased number of potential founders. The caveats to this approach are that synthetic and commercial synthesis of the donor could lead to the introduction of errors into the template and there may be limitations in the production of complex sequences.
While we tested five different donors only two produced any evidence of HDR and of these only one donor produced a perfect HDR event with no other mutations resulting in an efficiency of 4.5%. However, we note that one of the three imperfect HDR events for Donor A did have MYC inserted in the right position but contained an indel in the untranslated region upstream of the start site. This founder fish may also have been a suitable line after further functional studies, in which case the efficiency of HDR could be as high as 9%. Nevertheless, even 4.5% falls within the range of previously reported incorporation of epitope tags through HDR and is consistent with the rate of previously reported synthetic dsDNA donors [14,27].
One striking finding of our approach is that it appears to have significantly improved germline transmission (~52%) compared to previous attempts (10–30%) [28,29]. Indeed, another group reported successful knock-in of a composite HBH-3xFLAG tag at the same position as our MYC tag in sox11a. Using a long single-stranded DNA donor (lssDNA), this group reported a 12% correct integration of the 3X FLAG, but only 13–25% germline transmission in F1 fish [15]. In addition to the increased germline transmission, we validated MYC-Sox11a mRNA and protein expression. MYC-Sox11a expression was not detected by conventional IHC, perhaps because of the low endogenous expression of Sox11a, or the insertion of only a single copy of the MYC epitope; in the future a 3xMYC could potentially address this problem. Nevertheless, ours is the first demonstration of successful protein expression of an epitope-tagged sox11a, and allows for this knock-in line to be used for downstream functional and target site detection experiments.
Efficient methods for functional integration of an epitope tag in a gene of interest are critical for the field because they permit researchers to circumvent the absence of reliable antibodies for zebrafish proteins [30]. With the successful integration of MYC into sox11a we can continue our research investigating its role in the development of the retina. Specifically, the knock-in line will allow us to determine the precise retinal cell types expressing Sox11a, and will facilitate experiments like CUT&RUN to further our understanding of the Sox11a target genes in the retina. Furthermore, this approach should be applicable to any gene that can be targeted by CRISPR/Cas9, which will improve the value of zebrafish as genetic and developmental model organism.
Supplementary Material
Highlights.
Commercially available CRISPR knock-in reagents allow for ease of use and application.
Increased germline transmission of knock-in allele compared to previous reports.
Detection and validation of MYC-Sox11a expression patterns in brain and retina.
Acknowledgments
We thank Brandi Bolton, Evelyn Turnbaugh, and Lucas Vieira Francisco for exceptional zebrafish care. We also thank members of the Morris lab for valuable input and technical assistance. This work was supported by grants from the NIH National Eye Institute (R01EY021769, to A.C.M.; F30EY031545, to L.A.K.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Lieschke GJ, Currie PD, Animal models of human disease: Zebrafish swim into view, Nature Reviews Genetics. 8 (2007) 353–367. 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- [2].Hwang WY, Fu Y, Reyon D, et al. , Efficient genome editing in zebrafish using a CRISPR-Cas system, Nature Biotechnology. 31 (2013) 227–229. 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Talbot JC, Amacher SL, A streamlined CRISPR pipeline to reliably generate zebrafish frameshifting alleles, Zebrafish. 11 (2014) 583–585. 10.1089/zeb.2014.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jao LE, Wente SR, Chen W, Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system, Proceedings of the National Academy of Sciences of the United States of America. 110 (2013) 13904–13909. 10.1073/pnas.1308335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tessadori F, Roessler HI, Savelberg SMC, et al. , Effective CRISPR/Cas9-based nucleotide editing in zebrafish to model human genetic cardiovascular disorders, DMM Disease Models and Mechanisms. 11 (2018). 10.1242/dmm.035469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Prykhozhij S. v., Fuller C, Steele SL, et al. , Optimized knock-in of point mutations in zebrafish using CRISPR/Cas9, Nucleic Acids Research. 46 (2018). 10.1093/nar/gky512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burg L, Zhang K, Bonawitz T, et al. , Internal epitope tagging informed by relative lack of sequence conservation, Scientific Reports. 6 (2016). 10.1038/srep36986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ota S, Taimatsu K, Yanagi K, et al. , Functional visualization and disruption of targeted genes using CRISPR/Cas9-mediated eGFP reporter integration in zebrafish, Scientific Reports. 6 (2016). 10.1038/srep34991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Auer TO, Duroure K, de Cian A, et al. , Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair, Genome Research. 24 (2014) 142–153. 10.1101/gr.161638.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Burg L, Palmer N, Kikhi K, et al. , Conditional mutagenesis by oligonucleotide-mediated integration of loxP sites in zebrafish, PLoS Genetics. 14 (2018). 10.1371/journal.pgen.1007754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hisano Y, Sakuma T, Nakade S, et al. , Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish, Scientific Reports. 5 (2015). 10.1038/srep08841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kimura Y, Hisano Y, Kawahara A, et al. , Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering, Scientific Reports. 4 (2014). 10.1038/srep06545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ota S, Taimatsu K, Yanagi K, et al. , Functional visualization and disruption of targeted genes using CRISPR/Cas9-mediated eGFP reporter integration in zebrafish, Scientific Reports. 6 (2016). 10.1038/srep34991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].DiNapoli SE, Martinez-McFaline R, et al. , Synthetic CRISPR/Cas9 reagents facilitate genome editing and homology directed repair, Nucleic Acids Research. (2020). 10.1093/nar/gkaa085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ranawakage DC, Okada K, Sugio K, et al. Efficient CRISPR-Cas9-Mediated Knock-In of Composite Tags in Zebrafish Using Long ssDNA as a Donor, Frontiers in Cell and Developmental Biology. 8 (2021). 10.3389/fcell.2020.598634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pillai-Kastoori L, Wen W, Wilson SG, et al. , Sox11 Is Required to Maintain Proper Levels of Hedgehog Signaling during Vertebrate Ocular Morphogenesis, PLoS Genetics. 10 (2014). 10.1371/journal.pgen.1004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jia S, Wu X, Wu Y, et al. , Multiple developmental defects in sox11a mutant zebrafish with features of coffin-siris syndrome, International Journal of Biological Sciences. 16 (2020) 3039–3049. 10.7150/ijbs.47510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Westerfield M, The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio)., 4th ed., Univ. of Oregon Press, Eugene, 2000. [Google Scholar]
- [19].Forbes-Osborne MA, Wilson SG, Morris AC, Insulinoma-associated 1a (Insm1a) is required for photoreceptor differentiation in the zebrafish retina, Developmental Biology. 380 (2013) 157–171. 10.1016/j.ydbio.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bae S, Park J, Kim JS, Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases, Bioinformatics. 30 (2014) 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Concordet JP, Haeussler M, CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens, Nucleic Acids Research. 46 (2018) W242–W245. 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hoshijima K, Jurynec MJ, Klatt Shaw D, et al. , D.J. Grunwald, Highly Efficient CRISPR-Cas9-Based Methods for Generating Deletion Mutations and F0 Embryos that Lack Gene Function in Zebrafish, Developmental Cell. 51 (2019) 645–657.e4. 10.1016/j.devcel.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].de Martino S, Yan Y-L, Jowett T, et al. , Expression of sox11 Gene Duplicates in Zebrafish Suggests the Reciprocal Loss of Ancestral Gene Expression Patterns in Development, 2000. http://www.informatics.jax.org/. [DOI] [PubMed]
- [24].Roth TL, Puig-Saus C, Yu R, Shifrut E, et al. , Reprogramming human T cell function and specificity with non-viral genome targeting, Nature. 559 (2018) 405–409. 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Canaj H, Hussmann JA, Li H, et al. , Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in experiments, BioRxiv. (2019). 10.1101/841098. [DOI] [Google Scholar]
- [26].Sá nchez Alvarado A, Arturo Gutierrez-Triana J, Tavhelidse T, et al. , Efficient single-copy HDR by 5’ modified long dsDNA donors, (2018). 10.7554/eLife.39468.001. [DOI] [PMC free article] [PubMed]
- [27].Hruscha A, Krawitz P, Rechenberg A, et al. , Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish, Development (Cambridge). 140 (2013) 4982–4987. 10.1242/dev.099085. [DOI] [PubMed] [Google Scholar]
- [28].Bai H, Liu L, An K, et al. , CRISPR/Cas9-mediated precise genome modification by a long ssDNA template in zebrafish, BMC Genomics. 21 (2020). 10.1186/s12864-020-6493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Levic DS, Yamaguchi N, Wang S, et al. , Knock-in tagging in zebrafish facilitated by insertion into non-coding regions, Development (Cambridge). 148 (2021). 10.1242/DEV.199994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Partridge EC, Watkins TA, Mendenhall EM, Every transcription factor deserves its map: Scaling up epitope tagging of proteins to bypass antibody problems, BioEssays. 38 (2016) 801–811. 10.1002/bies.201600028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.