

Summary
Organ formation requires coordinating signals to time proliferation, specify cell fates, and shape tissue. Tracing these events and signals remains a challenge, as intermediate states across many critical transitions are unresolvable over real time and space. Here, we designed a unique computational approach to decompose a non-linear differentiation process into key components to resolve the signals and cell behaviors that drive a rapid transition, using the hair follicle dermal condensate as a model. Combining scRNA-sequencing with genetic perturbation, we reveal that proliferative Dkk1+ progenitors transiently amplify to become quiescent dermal condensate cells by the mere spatiotemporal patterning of Wnt/β-catenin and SHH signaling gradients. Together, they deterministically coordinate a rapid transition from proliferation to quiescence, cell fate specification, and morphogenesis. Moreover, genetically re-patterning these gradients reproduces these events autonomously in “slow-motion” across more intermediates that resolve the process. This analysis unravels two morphogen gradients that intersect to coordinate events of organogenesis.
Keywords: hair follicle, dermal condensate, Sonic Hedgehog, Wnt, single-cell RNA-seq, development, dermis, niche
Graphical Abstract
eTOC Blurb:
Qu et. al. computationally decompose a dermal lineage trajectory to resolve two covarying morphogen gradients that deterministically drive a fast transition to commitment during hair follicle dermal niche genesis.
Introduction
How cell fates are determined and segregate over time and space into organized tissue is a fundamental question in development and regeneration. Decades of lineage tracing and molecular profiling efforts have identified signals required for the differentiation of many cell types. Still, recreating the events of organogenesis requires the ability to control the timing, location and levels of different signals that coordinate a precise sequence of events to form patterned tissue. Delineating these steps and the signals that regulate them has been a major challenge, as intermediate states over many critical cell state transitions are difficult to resolve over real space and time. Here, we used the hair follicle dermal condensate (DC) as a model to demonstrate how a complex non-linear differentiation process can be computationally decomposed into biologically meaningful components. We show how these components can serve as molecular handles to resolve cell state transitions and the signals responsible for them.
The hair follicle (HF) has been a tractable model to study epithelial-mesenchymal interactions that regulate organogenesis, as its constituent populations are morphologically well-defined and the stages of HF development are classic (Hardy, 1992; Millar, 2002; Paus et al., 1999; Saxena et al., 2019; Xin et al., 2016). However, in contrast to the HF epithelium, little is known about the signals and progenitors that give rise to the HF dermal niche (dermal condensate, DC) (Figure 1A). The DC is a dense cluster of quiescent niche cells that regulates HF epithelial placode growth and differentiation. It matures into the dermal papilla, which serves as the permanent signaling center that controls the cyclical regeneration of the HF throughout adulthood (Chi et al., 2013; Enshell-Seijffers et al., 2010; Mesa et al., 2015). How this cluster of specialized cells forms and regenerates has been a subject of close investigation, as it carries the coveted ability to induce new HF growth, and it determines HF size (Chi et al., 2013; Horne and Jahoda, 1992). Nonetheless, there is no distinguishable intermediate cell state prior to DC commitment that can be interrogated to identify the signals and cell behaviors that regulate its formation. This has been the major barrier to recreating DCs in adult skin or as hair-inductive “organoids”.
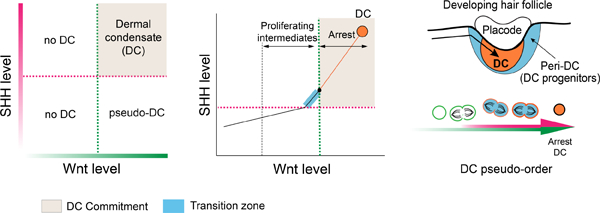
Figure 1: A selectively proliferative population transitions over a short timeframe into quiescent DC cells.
(A) HF development over time. (B) UMAP of dermal scRNA-seq data with Sox2 (left), UMAP showing ordering based on distance from DC state in transcriptome space (middle), G1/G0 fraction or gene levels across pseudo-order (right). (C) Sox2+ cell number per DC over time. (D) %EdU+ of DC population after E13.75 or E14.5 EdU pulse. (E) Expression level of indicated genes or fraction G1/G0 over pseudo-order (dashed line, highly proliferative state). (F) FISH showing Dkk1, Ccna2, or Cdkn1a with EdU (arrowheads indicating corner region of peri-DC). (G) 3D views and quantification of Tom+ Dkk1-lineage traced cells at indicated times in peri-DC or DC population; n=4. (H) Cartoon of spatial progression of DC differentiation. Data as mean± SEM (n=8 per time); *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA; ns, not significant. Scale bars, 50 µm. Related to Figure S1.
Different theories have been proposed to explain how DCs form, including placode-mediated guidance cues, self-organizing mechanisms, as well as local cell proliferation (Biggs et al., 2018; Gupta et al., 2019; Mok et al., 2019; Shyer et al., 2017; Wessells and Roessner, 1965). However, these mechanisms are neither mutually exclusive nor do they necessarily explain cell cycle exit, molecular differentiation, and cell fate patterning. A fundamental problem is the lack of tools to distinguish functional heterogeneity between cells prior to their commitment, which has limited most studies to examining events only once cells are already committed. For instance, previous studies demonstrated that committed quiescent cells condense to form a cluster (Biggs et al., 2018; Glover et al., 2017; Mok et al., 2019). However, the observation that a pool of quiescent DC cells forms rapidly raises the possibility that a latent selectively proliferative progenitor population exists to generate this reservoir and has not been comprehensively examined. Recent work showed that ablation of placode-secreted FGF20 resulted in a lack of histological or molecular evidence of DCs. Still, as many dermal signals were affected and there was no recognizable intermediate state to interrogate, its role in DC genesis remains unclear (Huh et al., 2013). As many aspects of DC genesis, including cell cycle exit, differentiation and morphogenesis are tightly coupled, attempts to resolve the signals that coordinate these complex events have been intractable to conventional techniques.
We previously used scRNA-sequencing (scRNA-seq) to measure and codify molecular differences between apparently uniform dermal cells prior to DC morphogenesis (Gupta et al., 2019). With this approach, we showed that dermal Wnt/β-catenin signaling is required for DC competency. At the same time, wes recognized that DC genesis requires dermal Wnt signaling and at least one placode signal, providing an experimental handle to study how this process occurs. In this study, we sought an unbiased approach to address this problem, considering that cells captured from a smoothly transitioning biological process usually form a continuous trajectory in high-dimensional transcriptome space. Here, we used diffusion maps, a traditional manifold-learning method, to study the inherent trajectory of the DC differentiation process and to define its constituent components using scRNA-seq data (Coifman et al., 2005).
By combining this approach with systematic genetic perturbation, we demonstrate here that a selectively proliferative population of Wnt-active (Dkk1+) cells proliferates over a short timeframe early during morphogenesis to give rise to quiescent pre-DC cells. By decomposing the DC differentiation trajectory into key components, we unbiasedly identify the placode signal that, together with dermal Wnt signaling, deterministically drives the transition to DC commitment by stimulating the proliferation and molecular differentiation of Dkk1+ progenitors, while accelerating Wnt signaling to levels that terminate this phase with cell cycle arrest. By genetically repatterning these signals we reproduce these same molecular and cellular events in “slow-motion” over more visible intermediate states that follow a linear deterministic trajectory. These intermediates resolve how two gradient signals intersect to couple molecular differentiation and morphogenesis to cell cycle exit.
A selectively proliferative population transitions over a short timeframe into quiescent DC cells
To examine the events that occur over the pre-DC-to-DC transition, we analyzed scRNA-seq data from E14.5 wildtype dermal cells and ordered cells according to their distance from the terminal DC state in transcriptome space (Figure 1B). When benchmarking this method against our previous analysis, we similarly identified a non-random proliferative state that precedes quiescence and Sox2 expression, suggesting that DCs expand from a selectively proliferative population. We performed EdU nucleotide pulse-chase experiments to determine the window of time when cells proliferate to generate committed DC cells. By E13.75-E14.0, DCs were first identified as small Sox2+ dermal clusters, which expand by incorporating new Sox2+ DC cells until approximately E15.5 (Figures 1A, 1C). EdU was first given at E13.75, and the proportion of DC or upper dermal cells that were EdU+ was quantified volumetrically after 1.5 hours (E13.75-E14.0), 14 hours (E14.5), or 38 hours (E15.5) (Figures 1D, S1A, S1C, S1D). Given that DC cells are quiescent, we found only rare EdU+ DC cells after a 1.5-hour pulse of EdU. However by E14.5, there was a significant increase in the number of EdU-traced DC cells that accounted for the majority of cells incorporated into the expanding DC over that timeframe. Similarly, the average number of EdU+ DC cells observed at E15.5 accounted for most of the increase in DC cell number during the 38-hour chase period (Figures 1C–D, S1A, S1D). Notably, the proportion of EdU+ DC cells incorporated over each interval chase period was significantly higher than that of the upper dermis (Figure S1C), indicating that DC progenitors are selectively proliferative. Interestingly, when we pulsed later at E14.5, only a low proportion of DC cells were EdU+ at E15.5 or E16.5, indicating that most pre-DC cells were already quiescent by E14.5 (Figures 1D, S1B–S1D). Collectively, these data show that a selectively proliferative population of progenitors divides to generate quiescent pre-DC cells within a short timeframe early during DC morphogenesis.
We next sought to identify the location of proliferative DC progenitors. EdU+ cells pulsed at E13.75 most frequently traced to the outer layers of DCs at E14.5, suggesting that proliferating DC progenitors transition from a region outside of the DC (Figure S1A). By the DC pseudo-order, Dkk1, a transcriptional target of Wnt signaling, is upregulated within the selectively proliferative population and downregulated in Sox2+ cells (Figures 1E, S1E). RNA fluorescent in situ hybridization (FISH) showed that Dkk1+ cells were concentrated in a region surrounding the DC (peri-DC; Figures 1F, S1F, S1H) and showed a higher rate of proliferation than interfollicular upper dermal cells, corresponding to their expression of the cell cycle regulator, Ccna2 (Figures 1F, S1E). A subset of Dkk1+ cells located closer to the DC, which we define as the corner region, showed a very low proliferation rate and expressed the cell cycle inhibitor, Cdkn1a/p21 (Figures 1E–F, S1G), suggesting that these cells represent quiescent pre-DC cells.
We next generated Dkk1CreER knock-in mice to lineage trace Dkk1+ cells as DCs form (Dkk1CreER;Rosa-LSLtdtomato) and gave tamoxifen at E12.75 and E13.25. By E13.75, we observed a low frequency of tdTomato+ (Tom+) dermal cells overall, consistent with the observation that there is a low level of Dkk1 expression in the upper dermis prior to morphogenesis (Figure 1G). By E14.5, there was a significantly increased proportion of DC cells that were Tom+. By E15.5, the proportion of Tom+ DC cells remained stable from E14.5 indicating that Dkk1+ cells continue to contribute to the expanding DC at a comparable rate. Notably, Dkk1 lineage-labeled cells also showed a significantly greater contribution to the peri-DC/perifollicular sheath at E15.5. Thus, Dkk1+ peri-DC cells selectively give rise to DC cells until E14.5 when they begin to show selective peri-DC/perifollicular potential. These collective data show that the process of DC differentiation, and thus the signals that regulate this process, follows a pattern in which selectively proliferative Dkk1+ peri-DC cells give rise to quiescent DC cells over a short timeframe during DC formation (Figure 1H).
Decomposing a non-linear DC differentiation process into key components
By the pseudo-order, Dkk1+ peri-DC cells differentiate toward a DC state in a manner that correlates with increasing levels of Wnt signaling (Figure 2A). By measuring transcript levels of a known reporter of Wnt signal transduction, Lef1, spatially at E14.5 we found a gradient of expression whose direction spans the upper interfollicular dermis, peri-DC and DC, with DC cells exhibiting the highest levels (Figures 2B–C). One interpretation of the DC trajectory is that Wnt signaling levels alone determine the probability of differentiating into a DC cell. However, we previously showed that forced activation of β-catenin in dermal cells resulted in larger DCs only where and when epidermal HF placodes normally formed (Chen et al., 2012; Gupta et al., 2019). Thus, one or more placode-dependent signals must be necessary to cooperate with dermal Wnt signaling to promote DC differentiation
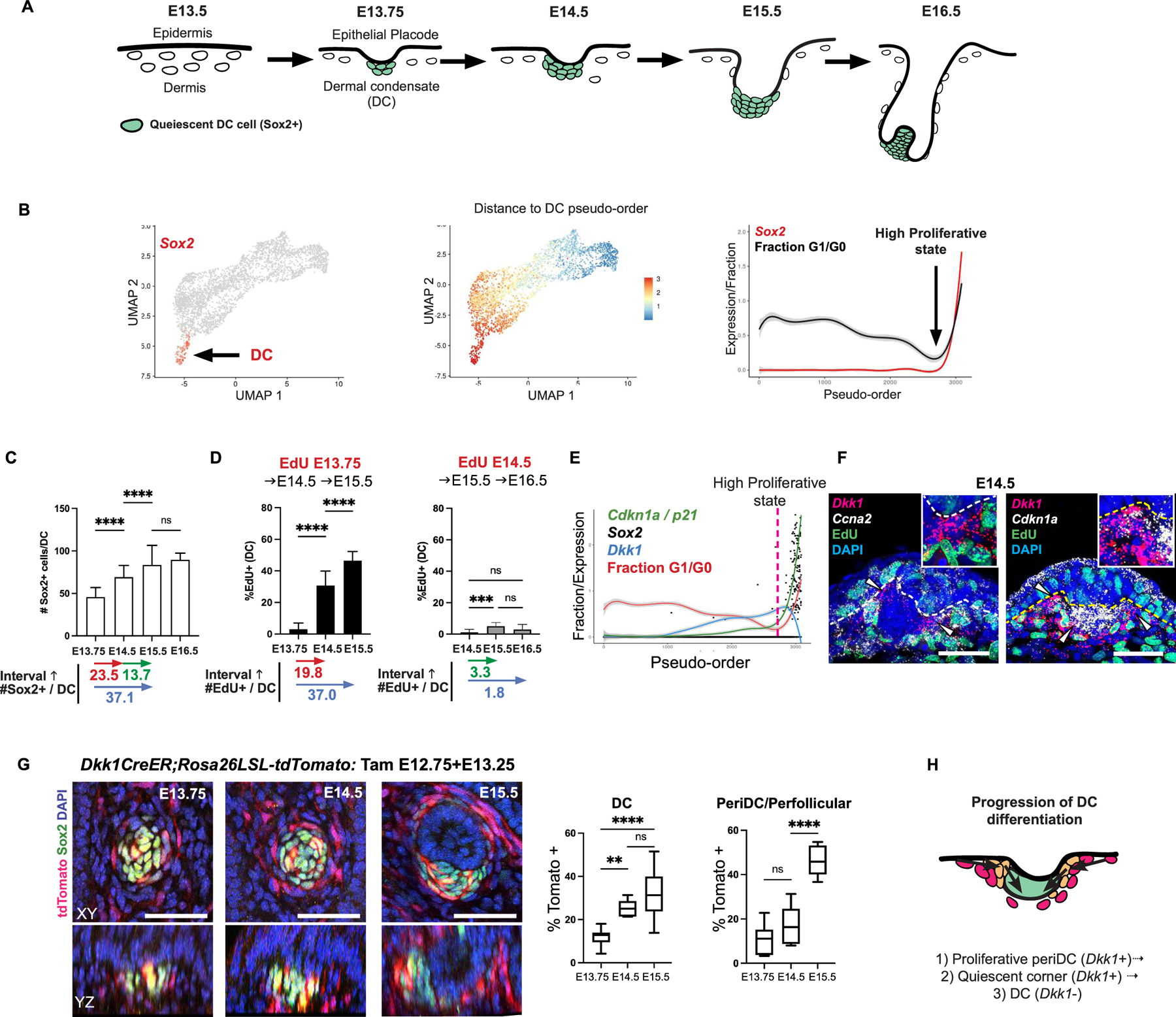
Figure 2: Decomposing a non-linear differentiation process into key components.
(A) Lef1 and Dkk1 levels by pseudo-order; dashed line, highly proliferative pre-DC state. (B, C) FISH and quantification of Lef1 transcript levels across space (n=4) with schematic of Lef1 levels over space. (D) Cartoon of predicted diffusion map by Wnt and DC components. (E) Diffusion map defined by eigenvectors that most correlate with Lef1 and Sox2. (F) Pseudo-order by Wnt component; dashed line, when Sox2 and Lef1 covary. (G) Correlation plot showing other Wnt targets and DC genes correlate with the two components. Related Figure S2.
Recognizing that the pre-DC-to-DC transition would not be accurately represented by a linear deterministic Wnt-driven trajectory, we used diffusion maps as a non-linear manifold learning method to decompose the DC differentiation process according to its related eigenvectors, which represent the primary components of this process. As one eigenvector that correlates with Wnt signaling levels would not fully capture the DC differentiation process, we reasoned that a placode-dependent signal should be contained within another eigenvector that also correlates with DC markers (Figure 2D). We computed the Pearson correlation between the expression of Lef1 or Sox2 and the coordinates of the top thirty eigenvectors. We defined the eigenvector that has the largest correlation with Lef1 as “Wnt component” and the eigenvector with the largest Sox2 correlation as “DC component” (Figures 2E–F, S2A–B). As shown by these combined perspectives, the DC differentiation trajectory progresses along both components at a certain point (Figure 2F). By examining the correlation of these two components with all significantly variable genes, we verified that these components indeed correlate with other genes known to represent Wnt signaling and DC differentiation, respectively (Figure 2G). This framework dissects the DC differentiation process into distinct components that can be used as tractable handles to interrogate signals responsible for them.
Unbiased identification of the placode factor essential for the transition to the DC component
Using this framework, we sought to identify candidate placode-dependent signals required for the pre-DC-to-DC transition. We took advantage of a mouse model that lacks placodes but still retains competent Wnt-active upper dermal cells. Wnt/β-catenin signaling is reciprocally activated in the epidermis and dermis, and previous work showed that loss of epidermal β-catenin inhibits placode initiation (Andl et al., 2002; Huelsken et al., 2001; Zhang et al., 2009). Nevertheless, epidermal Wnt ligands required for upper dermal Wnt signal transduction are maintained (Chen et al., 2012; Fan et al., 2018; Fu and Hsu, 2013). As expected, ablation of epidermal β-catenin (K14Cre;βcatfl/fl) resulted in decreased epidermal Lef1 expression by E13.5, while Lef1 transcripts within the upper dermis were preserved (Figure S3A) (Brault et al., 2001; Dassule et al., 2000). By E14.5, in contrast to control embryos, K14Cre;βcatfl/fl embryos lacked detectable placodes or DCs but retained upper dermal Lef1 expression and proliferation (Figures S3A–B). We next examined scRNA-seq data from E13.5 and E14.5 control and K14Cre;βcatfl/fl dermal cells. At E13.5, prior to placode formation, control and K14Cre;βcatfl/fl dermal populations were largely overlapping, reflecting their molecular similarity (Figures 3A, S3C). By contrast, there were significant differences when placodes formed in controls at E14.5 that were confined to Wnt-active cells (Figures 3B, S3D, S3F). Examination of E14.5 dermal diffusion maps defined by Wnt and DC components showed that dermal cells from mutant and control embryos shared a proximal region of the Wnt component (Figures 3C, S3E). However, dermal cells from mutants failed to progress along the DC component. Thus, other genes beyond those of Wnt signaling would account for the large differences between the two populations and likely contribute to the observed defect in DC differentiation.
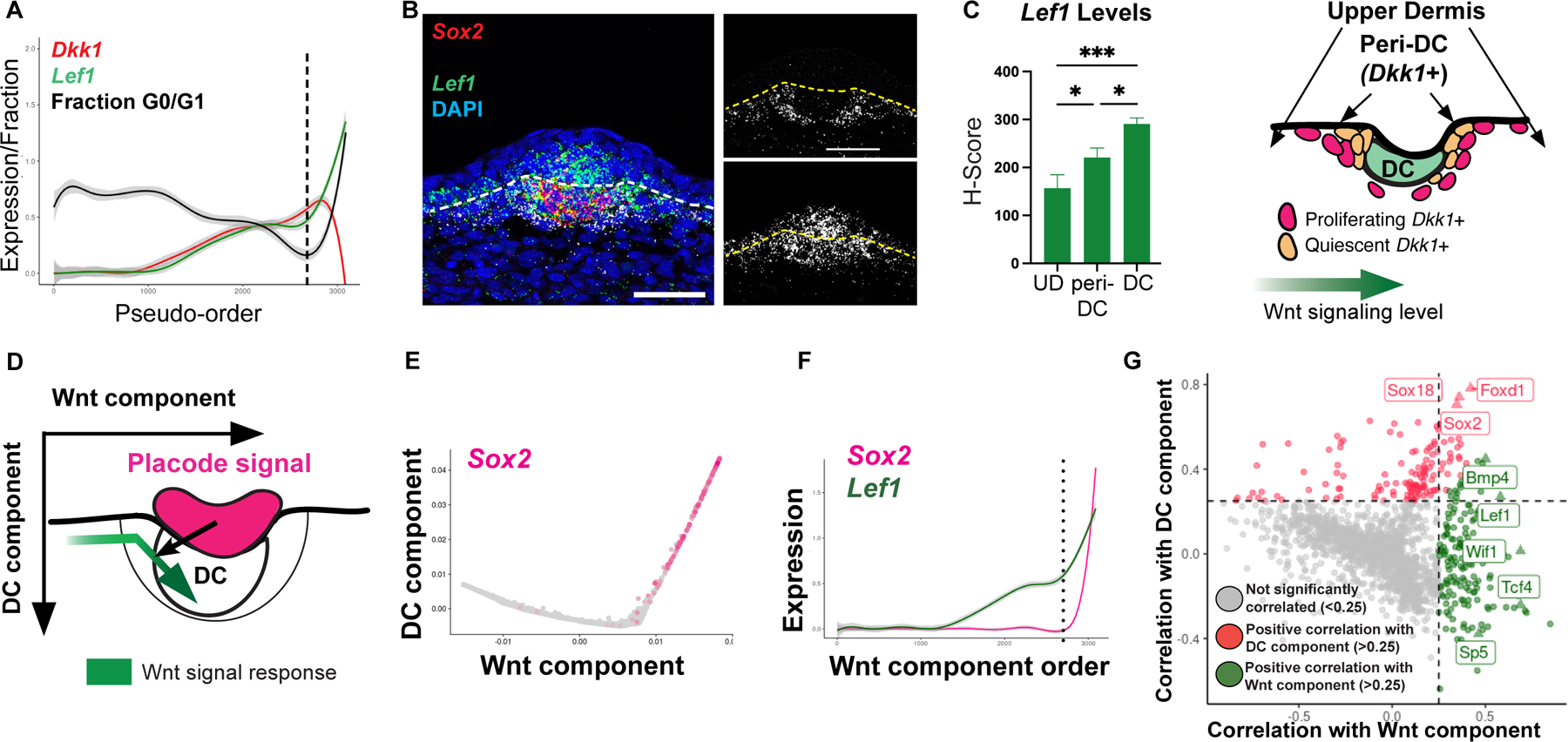
Figure 3: Unbiased identification of the placode factor essential for the transition to the DC component.
(A) UMAP E13.5 dermal cells by condition. (B) UMAP of E14.5 dermal cells by condition (left) or by Lef1 and Sox2 (right). (C) Diffusion maps by Wnt and DC components. (D) Differentially-abundant (DA) clusters of control and K14Cre;βcatfl//fl dermal cells (P < 0.01). (E) Volcano plot of control and mutant DA1 DEGs with genes (navy) that dually correlate with Wnt and DC components. (F) Violin plots of Lef1 and Ptch1 by DA clusters. (G) Pseudo-order showing covarying Ptch1 and Lef1 levels. (H) Quantification of Ptch1 levels across space (n=4). (I) Cartoon depicting SHH and Wnt signal levels by components. Data as mean± SEM; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA; ns, not significant; scale bars, 50 µm. Related to Figure S3.
To assess these molecular differences, we assumed that transcriptional changes caused by loss of epidermal β-catenin should correspond to a displacement of affected cells within transcriptome space. We used differential-abundance analysis as a statistical method to identify transcriptome regions in which cells are more abundantly represented by one condition than the other (Landa, 2020). We reasoned that Wnt-active progenitors in K14Cre;βcatfl/fl mutants lack signals required for their transition to DC status. Hence, both DC cells and DC progenitors of control and mutant dermal cells would be differentially abundant. In contrast to E13.5, there was a significant differentially abundant (DA) population at E14.5, which we subclassified into three clusters based on similarity (Figures 3D, S3C). The control DA2 cluster corresponds to the differentiated DC population, which is nearly absent in the mutant condition. The remaining control and K14Cre;βcatfl/fl DA1 clusters would include Wnt-active DC progenitors. Placode-dependent genes relevant to the pre-DC-to-DC transition should be: (1) significantly differentially expressed between the control and mutant DA1 (putative pre-DC) populations, and (2) associated with a DC differentiation process that spans the pre-DC (DA1) and DC (DA2) populations, demonstrating a dual Wnt and DC component correlation. Of all DA1 differentially expressed genes (DEGs), the most significant genes showing a dual Wnt and DC component correlation were involved in Sonic hedgehog (SHH) signaling (e.g. Ptch1, Hck; Figures 3E, S3G–H). Consistent with this, the canonical SHH target gene, Ptch1, was expressed by both the DA2 and pre-DC-containing DA1 populations from controls but not by the mutant DA1 population (Figure 3F). Further, the sparse E14.5 K14Cre;βcatfl/fl mutant outlier cells present in the control DA2 (DC) region expressed markers of SHH activation (Figure S3I). By FISH, we found that the rate of change in Ptch1 expression was greatest between the interfollicular and peri-DC regions, corresponding spatially with where higher Lef1 levels are detected (Figures 2C, 3G–H) and that Ptch1 and Lef1 were co-expressed by virtually all DC cells and the majority of Dkk1+ peri-DC cells (~62%) (Figure S3J). Based on this analysis, we hypothesized that these two co-varying signals cooperate to regulate the pre-DC-to-DC transition (Figure 3I).
SHH is essential for progression onto the DC component and genetically defines a critical transition stage of DC genesis
Previous work showed that loss of SHH ligand, which is exclusively expressed by placode cells and required for both placode and dermal SHH signal transduction, resulted in an arrest in HF development when early hair germs have formed. Yet, how dermal SHH signaling functions in DC genesis remains unclear (Chiang et al., 1999; Cui et al., 2011; St-Jacques et al., 1998; Woo et al., 2012). At E14.5, we found that K14Cre;Shhfl/fl (SHH cKO) embryos contained placodes, but Sox2+ DC cells were not detected and a morphologically distinct dermal cluster was not clearly discernible (Figure 4A) (Lewis et al., 2001). At E15.5, placodes remained arrested in growth and neither Sox2 nor the placode and DC marker, Sox9, was detected in SHH cKO embryos (Figure S4A). We next analyzed control and SHH cKO scRNA-seq samples (Figures 4B, S4B–C). Despite the presence of a placode population, SHH cKO dermal cells preserved a region along the Wnt component but failed to progress along the DC component similar to K14Cre;βcatfl/fl embryos (Figures 4C–D, S4B–D). Further, DA1 populations of SHH cKO and control dermal cells encompassed a similar region of the DC trajectory represented by K14Cre;βcatfl/fl and control DA1 regions. Hence, the cell populations affected by the lack of placodes are similar to those affected by the loss of SHH, indicating that SHH represents a major mediator of placode-dependent pre-DC changes.
Figure 4: SHH is essential for progression onto the DC component and genetically defines a critical transition stage of DC genesis.
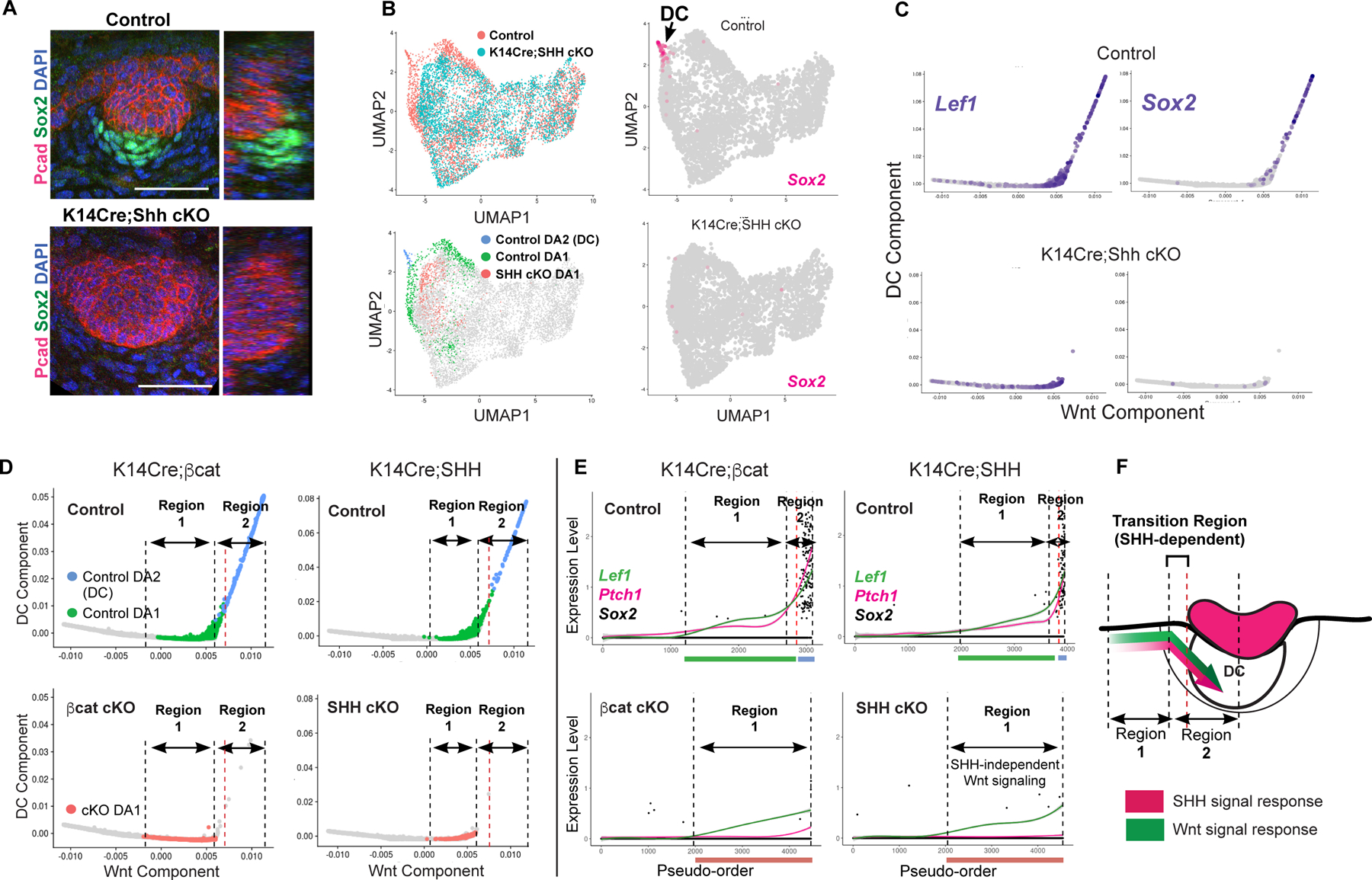
(A) Whole mount of E14.5 control and SHH cKO skin. (B) UMAP of control and SHH cKO dermal cells by condition or by DA clusters (left); Sox2 on UMAP (right). (C) Diffusion maps of SHH cKO and control showing Sox2 and Lef1. (D,E) Diffusion maps (left) and pseudo-order (right) of control and K14Cre;βcatfl//fl or control and SHH cKO color-coded by DA clusters. Dashed lines delineating DA populations corresponding to Regions 1 and 2. (F) Cartoon of Regions 1 and 2 and transition zone (bracket). Scale bars, 50 µm. Related to Figure S4.
This analysis delineates placode-dependent regions of the DC trajectory defined genetically by these mutants (Figures 4D–F): Region 1 represents cells ordered along the Wnt component that is conserved between SHH cKO and controls and characterized by a relatively slow rate of change in Lef1 and Ptch1 levels. Here, SHH does not regulate the level or rate of change of Wnt signaling levels, which we previously showed represents an intermediate stage of DC differentiation that requires dermal Wnt signal transduction. Region 2 delineates the region that lacks SHH cKO DA1 cells but includes a subset of control DA1 cells, which would correspond to transitioning pre-DC cells (delimited by red dashed line), and control DC (DA2) cells (Figure 4F). Here, Lef1 and Ptch1 covary at an accelerated rate. Thus, SHH is required for accelerated Wnt signaling and many DC genes.
Dermal SHH activation is required for the rapid transition to quiescence and mature DC differentiation.
By this analysis, dermal SHH signaling is required to induce Region 2 changes, including accelerated dermal Wnt signaling. We found that SHH cKO embryos showed expanded Wnt-active (Lef1+) placode cells at E14.5, indicating that placode Wnt ligands were preserved, but underlying dermal cells were unclustered and expressed much lower Lef1 levels than control DC cells (Figures 5A–B). By E15.5, a small cluster of dermal cells was seen in SHH cKO embryos with Lef1 levels now reaching levels similar to E14.5 control DC cells, reflecting a slower rate of increase in Lef1 expression. Using Bmp4 as a DC marker previously shown to be retained in SHH cKO DCs, we found significantly fewer Bmp4+ cells in SHH cKO DCs than control DCs (Figures 5D–F). Also, while control Bmp4+ DCs were quiescent (G0/G1 fraction=1) and surrounded by proliferating Dkk1+ cells, a significant proportion of SHH cKO Bmp4+ cells were aberrantly proliferating and coexpressed Dkk1 (Figures 5D–E, 5G). At the same time, SHH cKO Dkk1+/Bmp4-peri-DC cells showed a lower rate of proliferation than control Dkk1+ peri-DC cells, suggesting a defect in the proliferation of Dkk1+ peri-DC cells as well as their transition into quiescence (Figure 5C). Interestingly, while we did not detect a difference in apoptosis, the SHH cKO Bmp4+ population did not increase in size over time (Figures 5F, S5A). Eventually SHH cKO Bmp4+ cells formed small quiescent clusters, showing that SHH cKO DC-like (pseudo-DC) cells undergo delayed entry into quiescence (Figures 5D–G). These results were corroborated using the in vivo Fucci2 cell cycle reporter in which G1/G0 cells express mCherry (Abe et al., 2013). Only by E15.5, small quiescent mCherry+/Ki-67-dermal clusters were observed underneath SHH cKO placodes (Figure S5B).
Figure 5: Dermal SHH activation is required for the rapid transition to quiescence and mature DC differentiation.
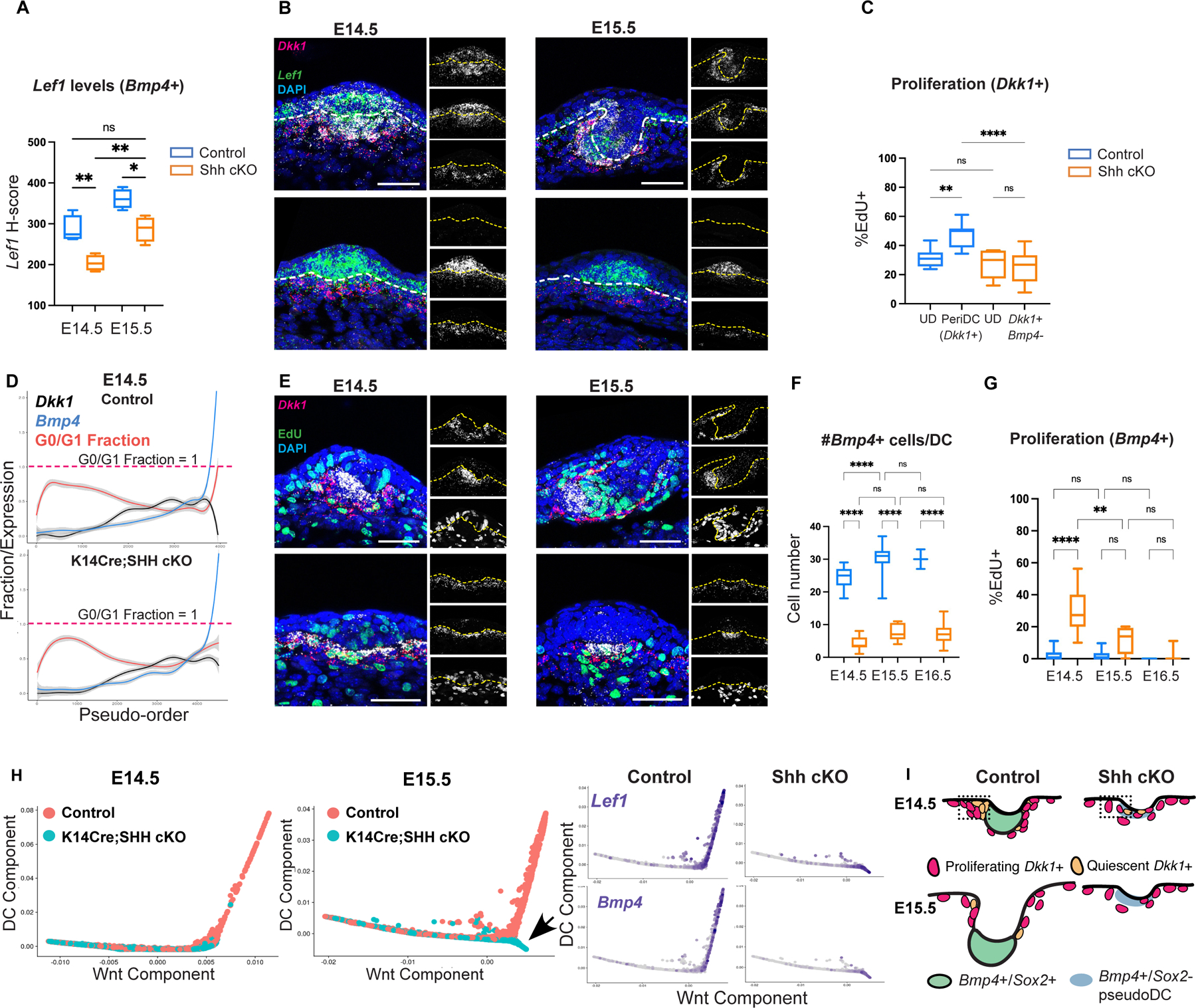
(A) Lef1 levels in Bmp4+ DCs at E14.5 and E15.5. (B) FISH showing decreased Lef1 expression in Bmp4+ SHH cKO pseudo-DCs. (C) Proliferation rate of Dkk1+ peri-DC and upper dermal cells at E14.5. (D) Pseudo-order of control and SHH cKO dermal cells at E14.5. (E) FISH showing proliferative Bmp4+ pseudo-DCs in SHH cKO at E14.5 and E15.5. (F) Number of Bmp4+ cells in SHH cKO and control over time. (G) %EdU+ of Bmp4+ population over time in control and SHH cKO. (H) Diffusion maps of E14.5 and E15.5 control and SHH cKO dermal cells showing that SHH cKO pseudo-DCs progress only on the Wnt component by E15.5. (I) Cartoon showing aberrant proliferation of SHH cKO Dkk1+ peri-DC cells and slow transition to quiescence. Data as mean± SEM; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA. Scale bars, 50 µm. Related to Figure S5.
To confirm that the defects in DC differentiation were not indirectly due to a loss of epidermal SHH signaling, we ablated the transducer of SHH signaling, Smo, in epidermal cells (K14Cre;Smofl/fl) (Long et al., 2001). At E14.5, mutant placode cells lacked SHH activity but were associated with Sox2+ DCs that showed an unexpected modest increase in the number of Sox2+ cells per DC (Figures S5C–D). Interestingly, at E15.5 and E16.5, while K14Cre;Smofl/fl placodes were stalled in growth due to the lack of epidermal SHH activation, their corresponding DCs expanded at a greater rate than control DCs. Mutant DCs remained in close approximation to the epidermis with a persistently high Lef1+/Ptch1+ co-positive peri-DC population, while controls showed a diminishing Lef1+/Ptch1+ co-positive peri-DC population that corresponded to the downward displacement of the DC from the epidermis (Figures S5C–D). Taken together with previous work (Gritli-Linde et al., 2007; Woo et al., 2012), these results show a cell-autonomous role for dermal SHH signaling in DC genesis and also support the model that SHH/Wnt co-activation promotes DC differentiation.
To address if SHH cKO pseudo-DCs may merely represent delayed progression onto the DC component. Diffusion maps of E15.5 SHH cKO and control dermal populations showed that mutant cells progressed on a Wnt component but not a DC component (Figures 5H–I, S5E–G). Specifically, E15.5 SHH cKO cells formed a divergent pseudo-DC branch that differed from controls by their lack of genes associated with the DC component, while their expression pattern was aligned with an attenuated Wnt component that terminates with quiescence (Figure S5G). Consistent with this, SHH cKO pseudo-DCs express many genes found in Wnt-active dermal cells prior to DC formation (Gupta et al., 2019). Collectively, these results reveal a previously unrecognized mechanism by which dermal SHH signaling governs DC genesis by promoting the proliferation of Wnt-active peri-DC cells and their rapid entry into quiescence and mature DC differentiation.
High dermal SHH activation in early Wnt-active cells deterministically reproduces events of DC genesis over more intermediates
Based on the trajectory alone, any other factor required for this process depicted by these components should be related to Wnt or SHH signaling, as we did not identify a third relevant eigenvector that correlates with DC changes (Figures S2A–B). To test this, we first assessed if SHH signaling is sufficient to induce DC component changes by expressing an activated form of Smo (SmoM2YFP) in Wnt-active progenitors prior to placode formation (Jeong et al., 2004). We previously showed that Axin2CreER targets dermal progenitors, and not epidermal cells, when tamoxifen is given at E11.5 (Gupta et al., 2019). Using this regimen, we examined Axin2CreER;SmoM2YFP embryos at E14.5 and noted a lack of placodes as evidenced by the lack of epidermal Edar or Ptch1 transcripts (Figures 6A, S6A). Remarkably, we found Sox2+ dermal clusters in the upper dermis that were bigger than control DCs at both E14.5 and E15.5 and that expressed eYFP and Ptch1 (Figures 6A–B, S6A–C). These clusters usually lacked a direct connection to the epidermis but were confined to the upper dermis and occasionally abutted one another (Figures S6A–D). Sox9 was also expressed by control and mutant dermal clusters, while the mutant epidermis largely lacked Sox9+ cells normally found in SHH-active placodes (Figure S6E). Thus, high dermal SHH activation is sufficient to induce Sox2+ dermal clusters in the Wnt-active upper dermis independent of placodes.
Figure 6: High dermal SHH activation in early Wnt-active cells deterministically reproduces events of DC genesis over more intermediates.
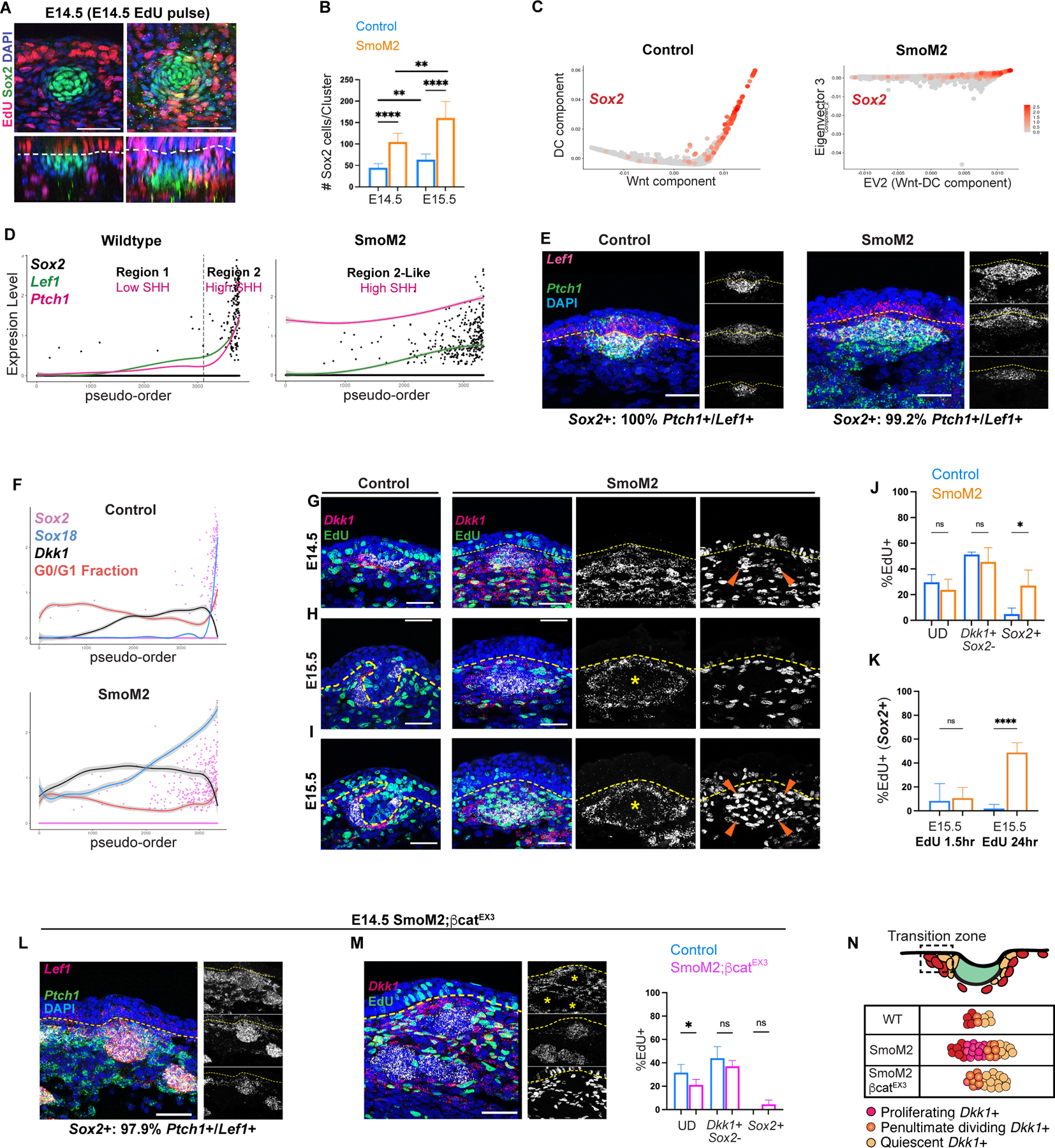
(A) E14.5 control and SmoM2YFP whole mount showing proliferative mutant Sox2+ clusters. (B) Number of Sox2+ cells per dermal cluster in control and SmoM2YFP at E14.5 and E15.5. (C) Diffusion maps of E14.5 control and SmoM2YFP cells. (D) Pseudo-order with Regions 1 and 2 demarcated. (E) FISH of E14.5 control and mutant showing virtually all Sox2+ cells co-express Ptch1 and Lef1. (F) Pseudo-order of indicated genes in mutant and control. (G) E14.5 FISH showing EdU, Sox2, and Dkk1. (H) FISH at E15.5 after EdU pulse in control and SmoM2YFP skin. (I) FISH 24 hours after EdU chase. (J) %EdU+ cells by population in E14.5 mutant and control. (K) %EdU+ of Sox2+ cells at E15.5 after EdU pulse or 24-hour chase. (L) E14.5 SmoM2;βcatfl/EX3 FISH showing Sox2+ clusters in upper and lower dermis that co-express Ptch1 and Lef1. (M) Sox2+ clusters with %EdU+ of indicated populations by condition at E14.5. (N) Depiction of transition rate and number of intermediates affected by modulating SHH and Wnt signaling. Data as mean ± SEM, one-way ANOVA. Scale bars, 50 µm. Related to Figure S6 and S7.
As high SHH activation is sufficient to induce DC differentiation in early Wnt-active cells, we hypothesized that molecular changes of Region 2 are caused by sufficient levels of SHH signaling. scRNA-seq data from E14.5 control and SmoM2YFP embryos confirmed that neither placode markers nor eYFP was detected in the epidermal population of mutants (Figure S6F). Due to the global transcriptional differences between wildtype (eYFP-) and mutant (eYFP+) dermal populations, we analyzed dermal diffusion maps of mutant cells separately (Figures 6C, S6G). Remarkably, the SmoM2YFP trajectory was represented by one eigenvector, while other eigenvectors were either not significantly correlated with DC changes or contained minimal variance (Figures 6C, S6H–J). Similar to Region 2 of controls, the mutant pseudo-order showed that Lef1 levels increased monotonically, and Sox2+ cells were concentrated near the terminus. Further, Lef1 and Ptch1 levels covaried at a nearly constant rate along the SmoM2YFP pseudo-order (Figure 6D).
By the SmoM2YFP trajectory, SHH/Wnt co-activation functions as a deterministic component that correlates with expression of DC and Wnt target genes (Figures 6C–D, 6F, S6H–J). Notably, SmoM2YFP cells transition across more Dkk1+ proliferative intermediates that gradually acquire DC genes before quiescence (Figure 6F). To verify these inferences, we first found that virtually all Sox2+ cells in mutant embryos coexpressed Ptch1 and Lef1 (Figure 6E), supporting the notion that Wnt and SHH co-activation is required for DC differentiation. As dermal Wnt signaling requires epidermal Wnt ligands, all mutant Sox2 clusters were located in the upper dermis (Figures 6E, S6A–B). At E14.5, we also found that many mutant Sox2+ cells were proliferating and coexpressed Dkk1+ (Figures 6F–G, 6J, S6K), but by E15.5, most Sox2+ cells were quiescent, lacked Dkk1 expression, and were surrounded by a ring of proliferating Dkk1+ cells similar to control DCs (Figures 6H, 6K, S6K–L). Further, E15.5 mutant cells pulsed with EdU at E14.5 showed that many quiescent Sox2+ cells originated from E14.5 proliferating cells in contrast to control DCs that expanded from a quiescent pool (Figures 1I, 6I, 6K, S6L). These data show that Wnt and SHH co-activation reproduces DC changes without placodes represented by a longer or “slower” continuum of proliferative Dkk1+ intermediate states that progressively gain DC markers prior to quiescence (Figure 6N).
Recognizing that FGF20 is a placode signal previously shown to be essential for DC differentiation and morphogenesis (Biggs et al., 2018; Huh et al., 2013; Mok et al., 2019), we sought to address the role of FGF20 in this process. We induced SmoM2YFP expression in Axin2CreER;SmoM2YFP;FGF20lacZ/lacZ embryos that lack FGF20 (Huh et al., 2012) and found mutant Sox2 clusters formed similar to SmoM2YFP embryos (Figure S6M). Similar to previous reports, FGF20lacZ/lacZ embryos largely lacked Sox2+ DCs at E14.5, suggesting that SHH may act downstream of FGF20. Consistent with this, SHH cKO placodes showed intact Fgf20 expression (Figure S6N). Further, there was a marked reduction in the number of Ptch1+ and Lef1+ cells in the dermis underlying FGF20lacZ/lacZ placodes, and Sox2+ cells that were seen coexpressed Ptch1 and Lef1 (Figure S6M). These data suggest that FGF20 modulates DC differentiation upstream of the two principal drivers of this process.
The spatial patterning of SHH and Wnt signaling gradients regulates the number of transitioning intermediates
Our data show that inducing uniformly high SHH activation across a gradient of Wnt signaling results in a gradient response of DC differentiation. Thus, DC genes that are normally restricted to the distal region of the wildtype DC trajectory are now distributed across the entire gradient of Wnt signaling and decoupled from terminal quiescent cells (Figures 6D, 6F, S6I). Based on these observations, we hypothesized that inducing high Wnt signaling in SmoM2YFP cells could bypass earlier intermediate states. We activated both β-catenin and Smo in Axin2CreER;SmoM2YFP;βcatfl/EX3 (SmoM2YFP;βcatfl/EX3) embryos (Harada et al., 1999). In contrast to SmoM2YFP embryos, Sox2+ clusters were now seen in the upper and lower dermis and surrounded by proliferating Dkk1+ cells in SmoM2YFP;βcatfl/EX3 embryos (Figures 6L–M, S7A). Additionally, most Sox2+ clusters in SmoM2YFP;βcatfl/EX3 embryos were quiescent similar to control DCs, indicating a faster transition to quiescence concurrent with molecular differentiation and morphogenesis (Figures 6M–N, S7B). Moreover at E13.5, prior to HF morphogenesis, SmoM2YFP;βcatfl/EX3 embryos demonstrated several quiescent Sox2+ clusters in the dermis (Figures 1C, 7A–C, S7C). These dermal clusters were frequently associated with overlying early placode markers not observed in control embryos (Figure 7B). As expected, Axin2CreER;βcatfl/EX3 (βcatfl/EX3) embryos largely lacked DCs at E13.5, while SmoM2YFP embryos demonstrated a variable number of small Sox2+ aggregates (Figures 7A–B, S7C) (Chen et al., 2012; Gupta et al., 2019). We conclude that sufficient levels of both SHH and Wnt can induce DC genesis autonomously.
Figure 7: The spatial patterning of SHH and Wnt signaling gradients regulates the number of transitioning intermediates.
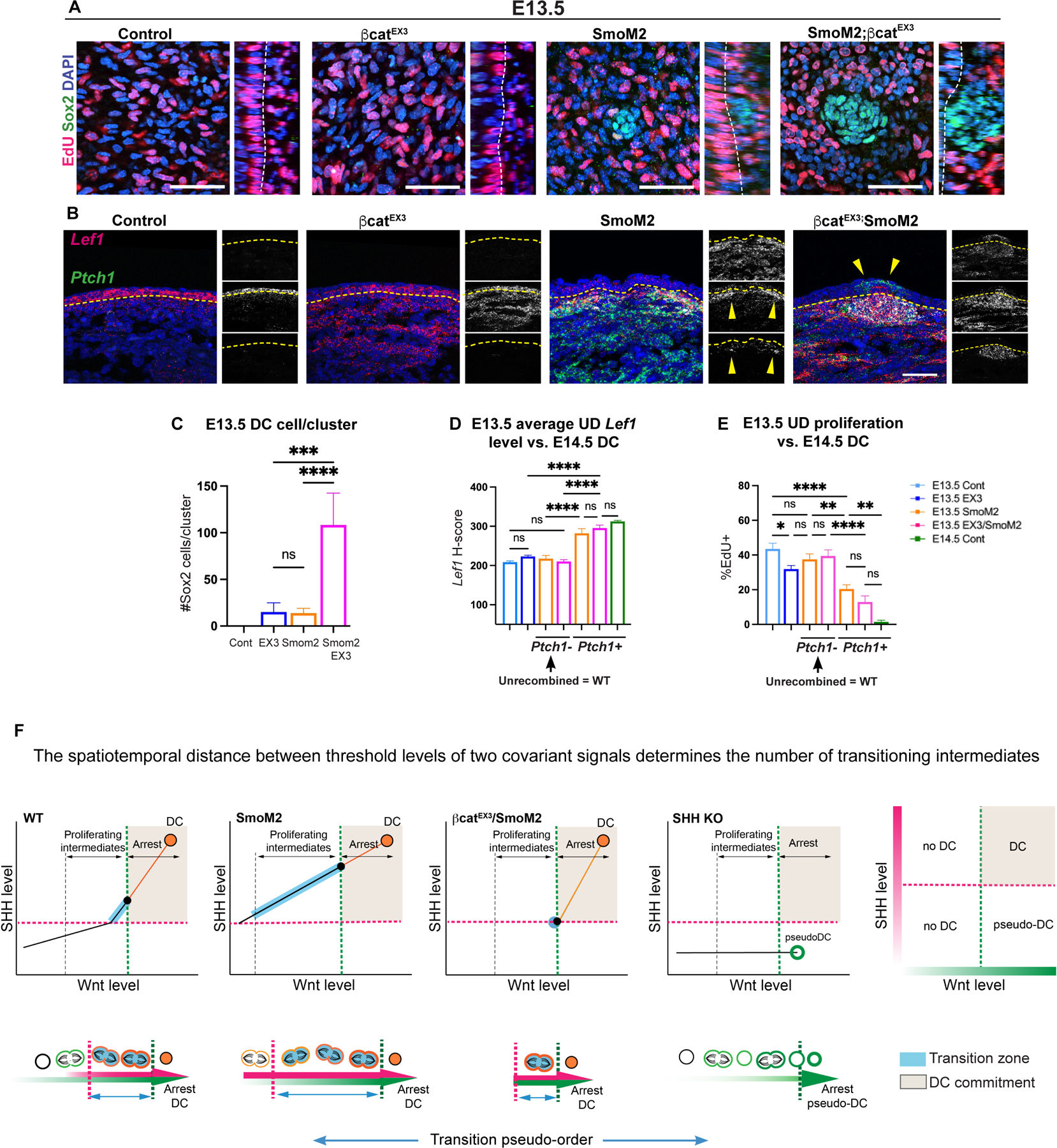
(A) E13.5 whole mounts across indicated conditions. (B) FISH of E13.5 embryos by condition showing increased Lef1 levels in SmoM2 and SmoM2YFP;βcatfl/EX3 embryos. (C) Number of Sox2+ cells per cluster by condition. (D) Average Lef1 levels by condition parsed by Ptch1 mutant status. (E) % EdU+ cells in upper dermis by condition and Ptch1 status. (F) Model plots illustrating that the spatiotemporal distance between threshold levels of Wnt and SHH signaling determines the number of transitioning intermediates and the length of transition to DC status. Bottom, depiction of the transition pseudo-order across conditions dependent on levels of SHH and Wnt signaling (orange, DC markers; blue, transitioning state; dashed lines, SHH and Wnt thresholds). Data as mean ± SEM, one-way ANOVA; scale bars, 50 µm. Related to Figure S7.
Our results show that high levels of SHH signaling can initiate Region 2 changes, including high Lef1 expression in the absence of placode Wnt ligands (Figures 6A, 7A, S6A, S6E–F), while SHH cKO pseudo-DCs show decreased Lef1 expression (Figures 5A–B). Thus, SHH may promote dermal Wnt signaling independent of placode Wnt ligands. We quantified Lef1 levels across conditions at E13.5, using E14.5 DCs as a reference for high Lef1 levels. Interestingly, E13.5 SmoM2YFP (Ptch1+) mutant cells consistently showed higher levels of Lef1 than SHH-inactive (Ptch1-) cells across all conditions and reached levels near that of E14.5 control DC cells (Figures 7B, 7D, S7D–E). As Ptch1-(unrecombined wildtype) cells of SmoM2YFP and SmoM2YFP;βcatfl/EX3 embryos expressed similar levels of Lef1 as control cells, the effect on Wnt signaling caused by the SmoM2YFP mutation is cell-autonomous (Figures 7D, S7E). Notably, Ptch1+ upper dermal cells of E13.5 SmoM2YFP embryos demonstrated significantly lower proliferation compared to E13.5 control or unrecombined Ptch1-upper dermal cells, which correlated with elevated Cdkn1a expression (Figures 7E, S7F). These results show that SHH signaling can boost Wnt signaling cell-autonomously, and these Wnt signaling levels correlate with quiescence.
Discussion:
Here, we applied a unique computational approach to identify the signals that coordinate the events that lead to DC genesis. Our data indicate that sufficient levels of SHH signaling determine when cells begin the transition to DC status, and the length of transition is delimited by a threshold level of Wnt signaling and quiescence. This transition phase is characterized by proliferation, augmented Wnt signaling, and acquisition of DC markers followed by cell cycle exit. Viewed another way, the spatiotemporal distance between threshold levels of SHH and Wnt signaling can determine the number of transitioning intermediates and a cell’s probability to undergo DC commitment (Figure 7F).
Dermal SHH and Wnt signaling coordinate events of DC genesis within a short window of time and space
Recent work showed that DCs form by the local migration of quiescent cells to form a cluster. Our work shows that a local pool of quiescent cells is generated by a selectively proliferative population. These proliferative progenitors express Dkk1 and are located in a defined transition zone in the peri-DC region. Previous seminal studies demonstrated the role of SHH in differentiated DC cells, establishing its function in DC maintenance (Woo et al., 2012). However, the essential function for dermal SHH signaling in DC differentiation and DC expansion was difficult to examine, as the progenitors and cellular processes that lead to DC differentiation were largely unknown. By leveraging scRNA-seq methods, we reveal a previously unrecognized role for SHH in cooperating with and augmenting Wnt signaling to drive the pre-DC-to-DC transition. We propose that SHH functions in Wnt-active DC progenitors to: 1) promote their proliferation, 2) induce expression of mature DC genes, 3) stimulate autonomous morphogenetic events and cell fate patterning, and 4) cause their timely arrest by augmenting Wnt signaling. In support of studies showing that high levels of Wnt signaling promote Cdkn1a expression and cell cycle exit, Wnt signaling levels correlated with quiescence across all mutants examined, including SHH cKO mutants (Choi et al., 2013; Fujimura et al., 2007; Huggins et al., 2017). Notably, genetic co-activation of SHH and Wnt signaling induces these events independent of FGF20, indicating that FGF20 likely functions upstream to modulate these two principal signals.
We hypothesize that the tight coupling of threshold levels of SHH and Wnt signaling in peri-DC cells would result in few transitioning intermediates prior to arrest, partly explaining why this transition is difficult to capture (Figure 7F). Inducing high levels of SHH signaling across the entire gradient of Wnt signaling prior to morphogenesis, and not just those in the peri-DC, would cause every cell within that gradient to augment Wnt signaling incrementally while increasing their proliferation and expression of DC genes, resulting in more transitioning intermediates proliferating until they reach a level of Wnt signaling sufficient to cause arrest. Effectively, the SmoM2YFP trajectory represents a “transition eigenvector” with more intermediates to resolve the process in “slow-motion”. In SmoM2YFP;βcatfl/EX3 mutants, both SHH and Wnt signaling levels are simultaneously induced at or above threshold levels, and mutant cells undergo that transition faster and more similar to the wildtype condition. Thus, two mitogenic signals can cooperate to regulate the length of transition between proliferation and arrest if they covary in a feed forward fashion, and one reins in this proliferation to cause arrest at a threshold level. This work suggests a mechanism by which differentiation is coordinated with proliferation and cell cycle exit, a phenomenon utilized by other somatic stem cells, including muscle and neuronal progenitors (Florio and Huttner, 2014; Yin et al., 2013). Our data suggest that the length of transition may depend upon the spatiotemporal distance between signal thresholds. This distance may define the number of divisions or duration that transit-amplifying cells divide before undergoing terminal differentiation.
The coupling of DC differentiation to DC morphogenesis and spatial patterning
Based on the SmoM2YFP trajectory SHH and Wnt signaling cooperate to deterministically drive DC differentiation, and the spatial pattern follows this order. A scenario in which differentiation is coupled to morphogenesis may have evolved to ensure that DC cells condense upon commitment to provide rapid and precise local feedback to the epidermis to regulate placode size or the spatial segregation of placode cell fates. Recent work showed that spatially segregated placode populations follow branching trajectories (Morita et al., 2021). In our case, time and space are necessarily coupled to establish a committed pool of quiescent DC cells. Interestingly, placode Wnt and SHH signaling antagonize each other to segregate placode cell fates, while the reciprocal convergence of these two signals in dermal cells results in accelerated signaling and DC differentiation (Ouspenskaia et al., 2016; Xu et al., 2015). The factors that direct cell context-specific molecular interactions in the epidermis and dermis are unclear but suggest that signal segregation may regulate cell fate trajectories.
Our data provide a mechanism to explain how dermal SHH signaling promotes adult HF neogenesis following wounding (Lim et al., 2018). At the same time, forced activation of dermal β-catenin in adult skin inhibits HF neogenesis (Hamburg and Atit, 2012; Rognoni et al., 2016). The heterogeneous cell populations involved in wound healing likely require dermal Wnt activity to be tightly regulated (Phan et al., 2020). This work uncovers the signals sufficient to reproduce a major aspect of DC genesis as well as the tunable parameters that regulate the rate of DC genesis and DC size, which can help guide biomimetic methods to recreate inductive mesenchymal condensates.
Limitations of the study:
In this study, the DC differentiation trajectory was defined by Wnt and DC components. Although our data show that the combination of these two eigenvectors represents a significant aspect of DC genesis, other methods to visualize scRNA-seq data or modalities may reveal different aspects of DC differentiation, which we did not examine here. Related to this, we recognize that HF placode maturation and growth were not fully reconstituted by genetic co-activation of these signals despite evidence of some early placode markers at E13.5 in SmoM2YFP;βcatfl/EX3 embryos. It is likely that feedback mechanisms and additional signals are necessary for epithelial-mesenchymal crosstalk. Comparative analysis between wildtype and SmoM2YFP or SmoM2YFP;βcatfl/EX3 dermal and epidermal populations along with generation of tools that dynamically regulate these signals may prove helpful. Finally, the specific biological processes to which the computational method used in this study can be generalized remains to be explored.
STAR Methods:
Resource Availability
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peggy Myung (peggy.myung@yale.edu).
Materials Availability:
Dkk1CreER knock-in mice were generated in this study and will be available from the lead contact through a Materials Transfer Agreement.
Data and Code Availability:
Genomic datasets used in this study are deposited in NCBI Gene Expression Omnibus database (GEO: GSE198487).
Experimental Model and Subject Details
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Mice
Axin2CreER (van Amerongen et al., 2012) mice were bred to Rosa-LSLSmoM2YFP (Jeong et al., 2004), β-cateninfl(Ex3/+ (Harada et al., 1999) and/or FGF20lacZ/lacZ (Huh et al., 2012) mice. K14Cre (Dassule et al., 2000) mice were bred to either β-cateninlf/fll (Brault et al., 2001), Smofl/fl (Long et al., 2001) or Shhfl/fl (Lewis et al., 2001) mice. To visualize Cre-recombination Gt(ROSA)26Sortm1(CAG-tdTomato)Hze(Madisen et al., 2010) mice were bred to Dkk1CreER knock-in mice (generated by Yale Transgenic Core). R26Fucci2aR (Abe et al., 2013) reporter mice were used to visualize cell cycle phase. A random population of both male and female mice were used for all experiments. All procedures involving animal subjects were performed under the approval of the Institutional Animal Care and Use Committee of the Yale School of Medicine.
Method Details
Tamoxifen induction of mice
Embryos were staged as days post coitum, with embryonic day E0.5 considered as noon of the day a vaginal plug was detected after overnight mating. Pregnant dams were given one or two doses of Tamoxifen dissolved in corn oil (20mg/ml, Sigma) at 40–60 µg/gm body weight by oral gavage at either E11.5 or E12.75+E13.25 as indicated.
EdU Incorporation Assay
To assess active proliferation, EdU was administered to pregnant mice intraperitoneally (25 µg/gm) and embryos were harvested after 1.5 hour. For pulse-chase experiments, 25 µg/gm EdU was administered to pregnant mice at indicated times and embryos were harvested at later times. EdU incorporation was assessed using the Click-it EdU Imaging kit Alexa 555 or Alexa 488 (Life technologies, c10338) according to manufacturer’s instructions. Briefly, skin explants were treated with a mixture of 1X Click-iT reaction buffer, CuSO4, Alexa Fluor azide dye and 1X reaction buffer additive, all provided in the Click-it EdU Imaging Kit, for 30 minutes at RT before washing in PBS.
Histology
10% formalin-fixed paraffin embedded (FFPE) whole embryos were used for histological analysis. FFPE specimens were sectioned at 10 µm thickness for FISH or 5 µm for immunofluorescence. For immunofluorescence, sections were deparaffinized and then subjected to 10 minutes antigen retrieval in citrate buffer. After washing in PBS, sections were blocked with 5% normal donkey serum, 1% bovine serum albumin and 0.2% Triton X-100 in PBS at room temperature (RT) for 1 hour. Sections were incubated overnight at 4 °C with the following antibodies: rabbit anti-Sox2 (1:400, Cell signaling 2286S), goat anti-Pcadherin (1:500, R&D Systems, AF761), mouse anti-caspase3 (1:100, BD Sciences, 610153). After washing in 0.1% Triton-X 100, sections were incubated with the following secondary antibodies: Alexa Fluor 488-donkey anti-goat (1:200, Life technologies, A11055), Alexa Fluor 488-donkey anti-goat (1:200, Life technologies, A11057), Alexa Fluor 568-donkey anti-rabbit (1:200, Life technologies, A10042) for 1 hour at RT and DAPI mounted (Vector laboratories, H-1200). Combined images of DAPI, Alexa 488, Alexa 568 and Alexa 647 were taken using a Zeiss Axio Observer.
Whole mount immunofluorescence
Dorsolateral skin from embryos were microdissected and placed on nucleopore filters (VWR, WHA 800281) and fixed overnight with 4%PFA at 4° C. Skin explants were blocked for 6 hours in 5% normal donkey serum, 1% bovine serum albumin and 0.5% Triton-X100 at RT. Explants were incubated overnight at 4° C in the following primary antibodies: chicken anti-RFP (1:250, NovusBio, NBP1–97371), rabbit anti-Sox2 (1:400, Abcam, ab97959), rabbit anti-Sox9 (1:500, Millipore, ab5535), goat anti-Pcadherin (1:500, R&D Systems, AF761), rabbit anti-RFP (1:500, Rockland, 600–401-379), Ki-67 Monoclonal Antibody (SolA15) (1:200, Invitrogen, 14–5698-82). Explants were then washed in 0.2% Tween20/PBS for 4–6 hours on a rotator and then incubated with the following secondary antibodies: Alexa Fluor 488-donkey anti-goat (1:200, Invitrogen, A11055), Alexa Fluor 568-donkey anti-rabbit (1:200, Invitrogen, A10042), Alexa Fluor 488-donkey anti-rabbit (1:200, Invitrogen, A21206), Alexa Fluor 647-goat anti-rat (1:200, Invitrogen, A-21247), Alexa Fluor 405-donkey anti-rat (1:200, Invitrogen, A-21208) overnight at RT. Washes were carried out for 6 hours with 0.2% Tween20/PBS. Hoechst (1:750 in PBS) was used for 45 min before mounting on slides with SlowFade Gold Antifade Mountant (Invitrogen, S36936).
In-situ hybridization
The RNAscope Multiplex Fluorescent Detection Kit v2 (ACDBio, 323110) was used for single-molecule fluorescence in situ hybridization according to the manufacturer’s protocol. Briefly, sections were deparaffinized, permeabilized with hydrogen peroxide followed by antigen retrieval and protease treatment before probe hybridization. After hybridization, amplification and probe detection was done using the Amp 1–3 reagents. Probe channels were targeted using the provided HRP-C1–3 reagents and TSA fluorophores: Cy3 (AkoyaBio, NEL744001KT), Cy5 (AkoyaBio, NEL745001KT), fluorescein (AkoyaBio, NEL741001KT). EdU staining was done using the Click-it EdU Imaging Kit (Invitrogen, C10337). Nuclear counter-stain was done using Hoechst 33342 (Invitrogen, H3570) before mounting with SlowFade Mountant. RNA scope probes used (ACDBio): Mm-Bmp4 (401301), Mm-Ptch1 (402811), Mm-Axin2 (400331), Mm-Dkk1 (402521), Mm-Edar (423011), Mm-Cdkn1a (408551), Mm-Lef1 (441861), Mm-Sox2 (401041), Mm-Fgf20 (526391), Mm-Shh (314361), Mm-EYFP (312131), Mm-Ccna2 (442661).
Microscopy
FISH paraffin-embedded images were acquired using the Leica TCS SP8 Gated STED 3X super-resolution confocal microscope with a 10x dry (numerical aperture (NA) 0.3) or 40x oil immersion (NA 1.3) objective lens, scanned at 5 μm thickness, 1024×1024 pixel width, 400 Hz. Widefield immunofluorescent images were acquired with a Zeiss Axio Observer Z1 equipped with a Plan APOCHROMAT dry 40x objective lens. Whole mount explants were imaged in 3 dimensions using the LaVision TriM Scope II (LaVision Biotec) microscope equipped with a Chameleon Vision II (Coherent) two-photon laser (810 −1000 nm) to acquire z-stack images ranging from 50–120 μm (2 μm serial optical sections) using a 20X water immersion lens (NA 1.0; Olympus), scanned with a field of view of 0.3–0.5 mm2 at 800 Hz.
Single-cell dissociation
Embryonic dorsolateral/flank skin was micro-dissected from littermate control and mutant embryos after genotyping (2–3 embryos pooled per condition) and dissociated into a single-cell suspension using 0.25% trypsin (Gibco, Life Technologies) for 20 minutes at 37° C. A replicate sample was prepared for both E14.5 control and SmoM2YFP samples. Single-cell suspensions were then stained with DAPI (Fisher Scientific, NBP2–31156) just prior to fluorescence-activated cell sorting.
Fluorescence-activated cell sorting
DAPI-excluded live skin cells were sorted on a BD FACS Aria II (Biosciences) sorter with a 100 µm nozzle. Cells were sorted in bulk and submitted for 10X Genomics library preparation at 0.75–1.0×106/mL concentration in 4% FCS/PBS solution.
Single-cell RNA sequencing and library preparation
Chromium Single cell 3’ Library and Gel Bead Kit v2 Chromium Single Cell 3′ Library & Gel Bead Kit v2 (PN-120237), Chromium Single Cell 3′ Chip kit v2 (PN-120236) and Chromium i7 Multiplex Kit (PN-120262) were used according to the manufacturer’s instructions in the Chromium Single Cell 3′ Reagents Kits V2 User Guide. After cDNA libraries were created, they were subjected to Novaseq 6000 (Illumina) sequencing. For each scRNA-seq experiment, each control and littermate mutant samples were prepared at the same time and pooled and sequenced on the same lane.
10X scRNA-seq data pipeline to matrix
The transcriptomes of live single cells from E13.5, E14.5 or E15.5 mouse skin samples under different biological conditions were sequenced. Raw 10X sequencing data was processed into a matrix employing the standard 10X CellRanger pipeline. Briefly, base call files were fastq format which were aligned to the mm10 reference genome followed by nUMI and barcode counting, constructing the nUMI count matrices. nUMI matrices were filtered, centered and normalized using Seurat (Butler et al., 2018; Macosko et al., 2015). Briefly, for each original run condition (i.e. E13.5 wildtype), cells were filtered to have > 2,000 genes but < 6,000, mitochondria ratio (defined by PercentageFeatureSet function in Seurat) below 0.15. Data was then log scaled, centered and normalized to nUMI.
Principal Component Calculation
Principal components (PCs) were calculated using Seurat’s RunPCA function. The top 2,000 variable genes were used for calculating PCs.
Cell type specification
For each original run, UMAP dimension reduction was performed on the normalized, centered, scaled nUMI count matrices employing the first 50 PCs. We then performed unsupervised clustering using the Seurat SNN clustering package, using a resolution of 0.5. Clusters that were positive for Col1a1 were defined as dermal, while clusters positive for Krt10 or Krt14 were defined as epidermal.
Data merge and normalization
For comparative analysis, we merged dermal cell matrices from each pair of conditions separately (i.e. E13.5 control and K14Cre;βcatfl//fl), followed by standard preprocessing and normalization using the workflow provided by Seurat. The same procedure was applied to analyze and compare epidermal cell populations between pairwise conditions. E14.5 control and SmoM2YFP data were analyzed separately due to their profound global transcriptome differences.
Diffusion Map Construction
We computed PCA of the data and retained the first 50 principal components. Let {xi} for i=1,…,N be the 50-dimensional vectors representing the cells. We constructed an N ×N sparse affinity matrix A by letting when xj is among the 50-nearest neighbors of xi , and σi is an adaptive bandwidth which is set to the distance from xi to its 10th neighbor. Next, we symmetrized the matrix, and normalized each row to sum to one, resulting in a Markov matrix. The eigenvectors of this sparse matrix corresponding to the largest eigenvalues are then computed using the R wrapper of ARPACK library and then visualized (https://cran.r-project.org/web/packages/rARPACK/index.html).
Pseudo-ordering of cells
We firstly identified the terminal DC cell state by the terminus of the DC differentiation trajectory. Cells were then ordered by their Euclidean distances to that DC cell state in transcriptome space (represented by top 50 PCs). To visualize gene expression or cell cycle phase along the pseudo order, we employed a generalized additive model to smooth the data (Hastie, 1990) using ggplot2 geom_smooth function. As the absolute values along the x-axis depend on the number of cells within each sample, comparison of expression levels should be made according to DA populations or regions in this study.
Two-component model construction and gene correlation analysis
We dissected out two major components that represent the DC differentiation process by identifying eigenvectors associated with DC differentiation. Specifically, the top 30 eigenvectors of diffusion maps were retained for analysis while eigenvectors with an index larger than 30 depicted pure noise. The eigenvector with the largest absolute correlation with Lef1 expression was denoted as “Wnt component”, while denoting the eigenvector with the largest absolute correlation with Sox2 as “DC component”. Each highlights a partial perspective of the DC trajectory and together codify this DC differentiation process. To verify the robustness of notations of “Wnt component” and “DC component”, correlations between the top 2,000 variable genes and the two components were computed and visualized. Canonical gene markers associated with Wnt signaling pathway and DC differentiation were found to be highly correlated (> 0.25) with Wnt component and DC component, respectively.
Differential abundance analysis
To identify cell populations affected by experimental perturbation, we applied a statistical method (Landa, 2020) to perform differential abundance analysis to unbiasedly identify all cell states where there are locally significantly more cells from one condition than the other (P < 0.01). DA cells were then grouped into different clusters by employing Seurat SNN clustering with a resolution of 0.075. Gene markers for each DA cluster were identified by Seurat FindAllMarkers function with default parameter settings, which were then used to infer cell types and cellular functions corresponding to DA clusters.
Differential gene expression analysis
Differential gene expression analysis between predefined DA1 regions of E14.5 control and K14Cre;βcatfl//fl dermal cells was performed by Seurat FindMarkers function with no prefiltering criteria applied. A combinatory threshold of P < 1e-20 and log2FC > 0.5 was employed to define significant DEGs for visualization. Genes that show dual correlations with Wnt and DC components were highlighted in the volcano plot of DEGs, where SHH pathway associated genes were found to be among the most significant DEGs.
Cell cycle estimation of scRNA-seq data
We employed Seurat’s cell cycle scoring (Nestorowa et al., 2016). Briefly, averaged relative expression of those cell cycle related genes were used to calculate G2/M and S scores, which were used for binning cells into G2/M, S and G1/G0 bins.
Quantification and Statistical Analysis
Image analysis
Raw image stacks acquired from whole-mount explants were imported into Fiji or Adobe Photoshop for analysis. ggplot2 and cowplot R libraries were used for graphical representation of the scRNA-seq data. Diffusion maps were generated as described above.
Quantification
For quantification of DC density, 3-dimensional whole-mount tiled mosaics of skin explants with a 1000 × 1000 µm field of view and a z-depth of 60–100 µm were used for n=3–4 embryos. For volumetric quantification of DC cell number, EdU+ or Tom+ percentage of DC or upper dermis, 3-dimensional whole mount mosaics stained with Sox2 and EdU or RFP were used to manually count positive cells, using ImageJ (Fiji) software using planar (XY) and orthogonal (YZ, XZ) views. For volumetric quantification of cell cycle phases, 3-dimensional whole mount mosaics stained with markers such as Ki67 or G0/G1 cells (based on Fucci2 reporter) were done manually by counting nuclear positive cells using ImageJ (Fiji) software. For quantification of upper dermal cells, a distance of 10 μm below the epidermis was considered upper dermis. For quantification of the peri-DC region by whole-mount, cells two cell layers out from the Sox2+ DC were counted. For quantification based on FISH, cells with 4–5 dots were considered positive (according to the RNAScope manufacturer’s instructions) and sections from a total of n=4 different embryos were examined. To measure RNA expression levels, H-scores were calculated according to ACDBio manufacturer’s instructions: a cell with 0 dot is scored 0, 1–3 dots score 1, 4–9 dots score 2, 10–15 dots and/or less than 10% clustered dots score 3, and more than 15 dots and/or more than 10% clustered dots score 4; then the final H-score of a given cell type A is calculated by summing the (% cells scored B within all cells in A)*B for score B in 0–4. For quantification of upper dermal cells, a distance of 10–20 μm below the epidermis was considered as upper dermis. For quantification of cells in the peri-DC region Dkk1+Sox2-or Dkk1+Bmp4-were considered.
Statistical analysis
All statistical values are expressed as mean ± SEM. An unpaired Student’s t-test was used to analyze data sets with two groups and *P <0.05 to, **P<0.01, ***P< 0.001 and ****P<0.00001 indicated a significant difference. When comparing more than two groups, P values were determined by one-way ANOVA with Tukey’s HSD test performed as the post hoc analysis. Statistical calculations were performed using Prism software package (GraphPad). For categorical data, a chi-square test was employed to estimate a P value.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Pcadherin | R&D Systems | Cat#AF761; (RRID:AB_355581) |
| Chicken anti-GFP | Abcam | Cat#Ab13970; (RRID:AB_300798) |
| Rabbit anti-Sox2 | Abcam | Cat#Ab97959; (RRID:AB_2341193) |
| Rabbit anti-Sox9 | Millipore | Cat#Ab5535; (RRID:AB_2239761) |
| Chicken anti-RFP | Novus Biologics | Cat#NBP1-97371 (RRID: AB_11139267) |
| Rabbit anti-RFP | Rockland | Cat# 600-401-379 (RRID: AB_2209751) |
| Ki-67 Monoclonal Antibody (SolA15) | Invitrogen | Cat#14-5698-82 (RRID: AB_10854564) |
| Goat anti-rat Alexa Fluor 647 | Invitrogen | Cat# A-21247 (RRID:AB_141778) |
| Donkey anti-rat Alexa Fluor 488 | Invitrogen | Cat#A-21208 (RRID: AB_141709) |
| Donkey anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A21206; (RRID:AB_2535792) |
| Donkey anti-goat Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A11055; (RRID:AB_2534102) |
| Donkey anti-rabbit Alexa Fluor 568 | Thermo Fisher Scientific | Cat#A10042; (RRID:AB_2534017) |
| Goat anti-rabbit Alexa Fluor 647 | Thermo Fisher Scientific | Cat#A27040; (RRID:AB_2536101) |
| Chemicals, peptides, and recombinant proteins | ||
| Hoechst | Invitrogen | H3570 |
| DAPI solution | Novus Biologicals | Cat#NBP2-31156 |
| Tamoxifen | Sigma Aldrich | Cat#T5648 |
| EdU (5-ethynyl-2-deoxyuridine) | Thermo Fisher Scientific | Cat#E10187 |
| Critical commercial assays | ||
| Click it EdU Imaging Kit 555 | Invitrogen | Cat#C10338 |
| Click it EdU Imaging Kit 488 | Invitrogen | Cat#C10337 |
| RNAscope® Multiplex Fluorescent Detection Kit v2 | ACDBio | Cat#323110 |
| Deposited data | ||
| Raw sequence data files | NCBI Gene Expression Omnibus | GEO: GSE198487 |
| Experimental models: Organisms/strains | ||
| Mouse: PDGFRαCreER | Collins et al, 2011 | N/A |
| Mouse: β-catenin EX3/+ | Harada et al, 1999 | N/A |
| Mouse: Axin2CreER | Amerongen et al, 2012 | N/A |
| Mouse: Fucci2 | Abe et al, 2013 | N/A |
| Mouse: K14Cre | Dassule et al., 2000 | N/A |
| Mouse: Shh flox/flox | Lewis et al., 2001 | N/A |
| Mouse: R26R-LSL-SmoM2YFP | Jeong et al., 2004 | N/A |
| Mouse: Smo flox/flox | Long et al, 2001 | N/A |
| Mouse: Fgf20 lacZ/lacZ | Huh et al., 2012 | N/A |
| Mouse: Dkk1CreER | PI: Yale Transgenic Core | N/A |
| Mouse: R26R-LSL-tdTomato | Madisen et al., 2010 | N/A |
| Oligonucleotides | ||
| Mouse-Bmp4 | ACDBio | Cat#401301 |
| Mouse-Axin2 | ACDBio | Cat#400331 |
| Mouse-Dkk1 | ACDBio | Cat#402521 |
| Mouse-Edar | ACDBio | Cat#423011 |
| Mouse-Cdkn1a | ACDBio | Cat#408551 |
| Mouse-Ptch1 | ACDBio | Cat#402811 |
| Mouse-Lef1 | ACDBio | Cat#441861 |
| Mouse-Sox2 | ACDBio | Cat#401041 |
| Mouse-Fgf20 | ACDBio | Cat#526391 |
| Mouse-Shh | ACDBio | Cat#314361 |
| Mouse-EYFP | ACDBio | Cat#312131 |
| Mouse-Ccna2 | ACDBio | Cat#442661 |
| Software and algorithms | ||
| Adobe photoshop CC 2020 | Adobe Systems | http://www.adobe.com/products/photoshop.html |
| Adobe Illustrator CC 2020 | Adobe Systems | http://www.adobe.com/products/illustrator/features.html |
| Fiji | National Institutes of Health | https://imagej.net/software/fiji/ |
| R | R Core Team | https://www.r-project.org/ |
| rARPACK | R Package | https://cran.r-project.org/web/packages/rARPACK/index.html |
| Differential Abundance analysis | Landa et al, 2020 | https://arxiv.org/abs/2011.03418 |
| Seurat | Macosko et al, 2015 | http://satijalab.org/seurat/ |
| GraphPad Prism 9.1.0 | GraphPad software | https://www.graphpad.com/scientific-software/prism/ |
Highlights:
Dermal progenitors transiently amplify into quiescent dermal condensate (DC) cells
Trajectory decomposition identifies signals that mediate the preDC-to-DC transition
Dermal SHH and Wnt/β-catenin signaling deterministically drive DC genesis
Repatterning SHH and Wnt signaling gradients modulates the length of transition
Acknowledgements:
We thank A. Jaffe, J. Levinsohn, and R. Coifman for their input on computational aspects and M. Ito, V. Greco, T. Xin, W. Zhong, and R. Atit for review of this manuscript. We thank D. Ornitz for providing Fgf20 null mice. This work was supported by National Institutes of Health grants R01AR076420 (P.M.), R01GM131642, P50CA121974, and UM1DA051410 (Y.K.), and American Cancer Society RSG-17-165-01-DDC (P.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References:
- Abe T, Sakaue-Sawano A, Kiyonari H, Shioi G, Inoue K, Horiuchi T, Nakao K, Miyawaki A, Aizawa S, and Fujimori T (2013). Visualization of cell cycle in mouse embryos with Fucci2 reporter directed by Rosa26 promoter. Development 140, 237–246. [DOI] [PubMed] [Google Scholar]
- Andl T, Reddy ST, Gaddapara T, and Millar SE (2002). WNT signals are required for the initiation of hair follicle development. Dev Cell 2, 643–653. [DOI] [PubMed] [Google Scholar]
- Biggs LC, Makela OJ, Myllymaki SM, Das Roy R, Narhi K, Pispa J, Mustonen T, and Mikkola ML (2018). Hair follicle dermal condensation forms via Fgf20 primed cell cycle exit, cell motility, and aggregation. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, and Kemler R (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Jarrell A, Guo C, Lang R, and Atit R (2012). Dermal beta-catenin activity in response to epidermal Wnt ligands is required for fibroblast proliferation and hair follicle initiation. Development 139, 1522–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi W, Wu E, and Morgan BA (2013). Dermal papilla cell number specifies hair size, shape and cycling and its reduction causes follicular decline. Development 140, 1676–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Robertson EK, Cooper MK, Gaffield W, Westphal H, Beachy PA, et al. (1999). Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol 205, 1–9. [DOI] [PubMed] [Google Scholar]
- Choi YS, Zhang Y, Xu M, Yang Y, Ito M, Peng T, Cui Z, Nagy A, Hadjantonakis AK, Lang RA, et al. (2013). Distinct functions for Wnt/beta-catenin in hair follicle stem cell proliferation and survival and interfollicular epidermal homeostasis. Cell Stem Cell 13, 720–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coifman RR, Lafon S, Lee AB, Maggioni M, Nadler B, Warner F, and Zucker SW (2005). Geometric diffusions as a tool for harmonic analysis and structure definition of data: diffusion maps. Proc Natl Acad Sci U S A 102, 7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui CY, Kunisada M, Childress V, Michel M, and Schlessinger D (2011). Shh is required for Tabby hair follicle development. Cell Cycle 10, 3379–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Maas R, and McMahon AP (2000). Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 127, 4775–4785. [DOI] [PubMed] [Google Scholar]
- Enshell-Seijffers D, Lindon C, Wu E, Taketo MM, and Morgan BA (2010). Beta-catenin activity in the dermal papilla of the hair follicle regulates pigment-type switching. Proc Natl Acad Sci U S A 107, 21564–21569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Wang D, Burgmaier JE, Teng Y, Romano RA, Sinha S, and Yi R (2018). Single Cell and Open Chromatin Analysis Reveals Molecular Origin of Epidermal Cells of the Skin. Dev Cell 47, 133. [DOI] [PubMed] [Google Scholar]
- Florio M, and Huttner WB (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. [DOI] [PubMed] [Google Scholar]
- Fu J, and Hsu W (2013). Epidermal Wnt controls hair follicle induction by orchestrating dynamic signaling crosstalk between the epidermis and dermis. J Invest Dermatol 133, 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura N, Vacik T, Machon O, Vlcek C, Scalabrin S, Speth M, Diep D, Krauss S, and Kozmik Z (2007). Wnt-mediated down-regulation of Sp1 target genes by a transcriptional repressor Sp5. J Biol Chem 282, 1225–1237. [DOI] [PubMed] [Google Scholar]
- Glover JD, Wells KL, Matthaus F, Painter KJ, Ho W, Riddell J, Johansson JA, Ford MJ, Jahoda CAB, Klika V, et al. (2017). Hierarchical patterning modes orchestrate hair follicle morphogenesis. PLoS Biol 15, e2002117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A, Hallberg K, Harfe BD, Reyahi A, Kannius-Janson M, Nilsson J, Cobourne MT, Sharpe PT, McMahon AP, and Linde A (2007). Abnormal hair development and apparent follicular transformation to mammary gland in the absence of hedgehog signaling. Dev Cell 12, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Levinsohn J, Linderman G, Chen D, Sun TY, Dong D, Taketo MM, Bosenberg M, Kluger Y, Choate K, et al. (2019). Single-Cell Analysis Reveals a Hair Follicle Dermal Niche Molecular Differentiation Trajectory that Begins Prior to Morphogenesis. Dev Cell 48, 17–31 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburg EJ, and Atit RP (2012). Sustained beta-catenin activity in dermal fibroblasts is sufficient for skin fibrosis. J Invest Dermatol 132, 2469–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, and Taketo MM (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 18, 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy MH (1992). The secret life of the hair follicle. Trends Genet 8, 55–61. [DOI] [PubMed] [Google Scholar]
- Hastie T.a.T., Robert (1990). Generalized additive models, Vol 43, 1st edn (Chapman and Hall/CRC; ). [Google Scholar]
- Horne KA, and Jahoda CA (1992). Restoration of hair growth by surgical implantation of follicular dermal sheath. Development 116, 563–571. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Erdmann B, Cotsarelis G, and Birchmeier W (2001). beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 105, 533–545. [DOI] [PubMed] [Google Scholar]
- Huggins IJ, Bos T, Gaylord O, Jessen C, Lonquich B, Puranen A, Richter J, Rossdam C, Brafman D, Gaasterland T, et al. (2017). The WNT target SP5 negatively regulates WNT transcriptional programs in human pluripotent stem cells. Nat Commun 8, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SH, Jones J, Warchol ME, and Ornitz DM (2012). Differentiation of the lateral compartment of the cochlea requires a temporally restricted FGF20 signal. PLoS Biol 10, e1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SH, Narhi K, Lindfors PH, Haara O, Yang L, Ornitz DM, and Mikkola ML (2013). Fgf20 governs formation of primary and secondary dermal condensations in developing hair follicles. Genes Dev 27, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, and McMahon AP (2004). Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev 18, 937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landa B, Qu R, Chang J, Kluger Y (2020). Local two-sample testing over graphs and point-clouds by random-walk distributions. arXiv 2011.03418, 1–51. [Google Scholar]
- Lewis PM, Dunn MP, McMahon JA, Logan M, Martin JF, St-Jacques B, and McMahon AP (2001). Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 105, 599–612. [DOI] [PubMed] [Google Scholar]
- Lim CH, Sun Q, Ratti K, Lee SH, Zheng Y, Takeo M, Lee W, Rabbani P, Plikus MV, Cain JE, et al. (2018). Hedgehog stimulates hair follicle neogenesis by creating inductive dermis during murine skin wound healing. Nat Commun 9, 4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, and McMahon AP (2001). Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099–5108. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa KR, Rompolas P, Zito G, Myung P, Sun TY, Brown S, Gonzalez DG, Blagoev KB, Haberman AM, and Greco V (2015). Niche-induced cell death and epithelial phagocytosis regulate hair follicle stem cell pool. Nature 522, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar SE (2002). Molecular mechanisms regulating hair follicle development. J Invest Dermatol 118, 216–225. [DOI] [PubMed] [Google Scholar]
- Mok KW, Saxena N, Heitman N, Grisanti L, Srivastava D, Muraro MJ, Jacob T, Sennett R, Wang Z, Su Y, et al. (2019). Dermal Condensate Niche Fate Specification Occurs Prior to Formation and Is Placode Progenitor Dependent. Dev Cell 48, 32–48 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita R, Sanzen N, Sasaki H, Hayashi T, Umeda M, Yoshimura M, Yamamoto T, Shibata T, Abe T, Kiyonari H, et al. (2021). Tracing the origin of hair follicle stem cells. Nature 594, 547–552. [DOI] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Gottgens B (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouspenskaia T, Matos I, Mertz AF, Fiore VF, and Fuchs E (2016). WNT-SHH Antagonism Specifies and Expands Stem Cells prior to Niche Formation. Cell 164, 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus R, Muller-Rover S, Van Der Veen C, Maurer M, Eichmuller S, Ling G, Hofmann U, Foitzik K, Mecklenburg L, and Handjiski B (1999). A comprehensive guide for the recognition and classification of distinct stages of hair follicle morphogenesis. J Invest Dermatol 113, 523–532. [DOI] [PubMed] [Google Scholar]
- Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, and Driskell RR (2020). Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognoni E, Gomez C, Pisco AO, Rawlins EL, Simons BD, Watt FM, and Driskell RR (2016). Inhibition of beta-catenin signalling in dermal fibroblasts enhances hair follicle regeneration during wound healing. Development 143, 2522–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena N, Mok KW, and Rendl M (2019). An updated classification of hair follicle morphogenesis. Exp Dermatol 28, 332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer AE, Rodrigues AR, Schroeder GG, Kassianidou E, Kumar S, and Harland RM (2017). Emergent cellular self-organization and mechanosensation initiate follicle pattern in the avian skin. Science 357, 811–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Dassule HR, Karavanova I, Botchkarev VA, Li J, Danielian PS, McMahon JA, Lewis PM, Paus R, and McMahon AP (1998). Sonic hedgehog signaling is essential for hair development. Curr Biol 8, 1058–1068. [DOI] [PubMed] [Google Scholar]
- van Amerongen R, Bowman AN, and Nusse R (2012). Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 11, 387–400. [DOI] [PubMed] [Google Scholar]
- Wessells NK, and Roessner KD (1965). Nonproliferation in dermal condensations of mouse vibrissae and pelage hairs. Dev Biol 12, 419–433. [DOI] [PubMed] [Google Scholar]
- Woo WM, Zhen HH, and Oro AE (2012). Shh maintains dermal papilla identity and hair morphogenesis via a Noggin-Shh regulatory loop. Genes Dev 26, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin T, Greco V, and Myung P (2016). Hardwiring Stem Cell Communication through Tissue Structure. Cell 164, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wang W, Jiang K, Yu Z, Huang H, Wang F, Zhou B, and Chen T (2015). Embryonic attenuated Wnt/beta-catenin signaling defines niche location and long-term stem cell fate in hair follicle. Elife 4, e10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Price F, and Rudnicki MA (2013). Satellite cells and the muscle stem cell niche. Physiol Rev 93, 23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, Birchmeier W, Paus R, Piccolo S, Mikkola ML, et al. (2009). Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev Cell 17, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic datasets used in this study are deposited in NCBI Gene Expression Omnibus database (GEO: GSE198487).