

Abstract
Background:
The glycolytic enzyme pyruvate kinase muscle 2 (PKM2) is upregulated in monocytes/macrophages of patients with atherosclerotic coronary artery disease. However, the role of cell type-specific PKM2 in the setting of atherosclerosis remains to be defined. We determined whether myeloid cell-specific PKM2 regulates efferocytosis and atherosclerosis.
Methods:
We generated myeloid cell-specific PKM2−/− mice on Ldlr-deficient background (PKM2mye-KOLdlr−/−). Controls were littermate PKM2WTLdlr−/− mice. Susceptibility to atherosclerosis was evaluated in whole aortae and cross sections of the aortic sinus in male and female mice fed a high fat “Western” diet for 14 weeks, starting at eight weeks.
Results:
PKM2 was upregulated in macrophages of Ldlr−/− mice fed a high-fat “Western” diet compared with chow diet. Myeloid cell-specific deletion of PKM2 led to a significant reduction in lesions in the whole aorta and aortic sinus despite high cholesterol and triglyceride levels. Furthermore, we found decreased macrophage content in the lesions of myeloid cell-specific PKM2−/− mice associated with decreased MCP-1 levels in plasma, reduced transmigration of macrophages in response to MCP-1, and impaired glycolytic rate. Macrophages isolated from myeloid-specific PKM2−/− mice fed the Western diet exhibited reduced expression of pro-inflammatory genes, including MCP-1, IL-1β, and IL-12. Myeloid cell-specific PKM2−/− mice exhibited reduced apoptosis concomitant with enhanced macrophage efferocytosis and upregulation of LRP1 in macrophages in vitro and atherosclerotic lesions in vivo. Silencing LRP1 in PKM2-deficient macrophages restored inflammatory gene expression and reduced efferocytosis. As a therapeutic intervention, inhibiting PKM2 nuclear translocation using a small molecule reduced glycolytic rate, enhanced efferocytosis, and reduced atherosclerosis in Ldlr−/− mice.
Conclusion:
Genetic deletion of PKM2 in myeloid cells or limiting its nuclear translocation reduces atherosclerosis by suppressing inflammation and enhancing efferocytosis.
Keywords: PKM2, atherosclerosis, efferocytosis, macrophage, Basic Science Research, Vascular Biology
Graphical Abstract
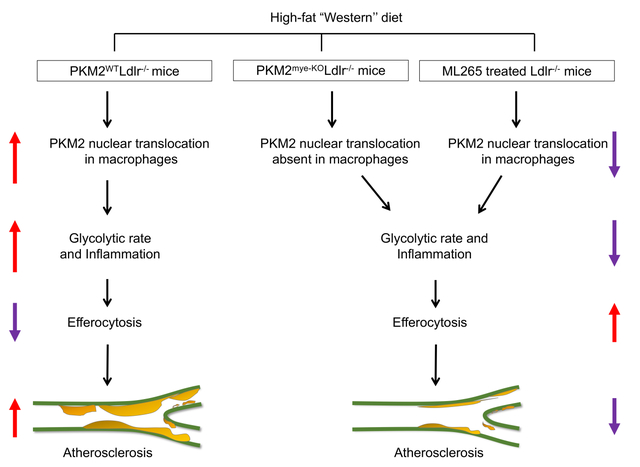
Introduction
Atherosclerosis is initiated by low-density lipoprotein (LDL) depositing in the sub-endothelium of the artery. Despite several therapies that target inflammation or lipoprotein metabolism, there is still an unmet need for better treatment options to treat atherosclerosis. Therefore, understanding the causal molecular mechanisms involved in the progression of atherosclerosis and associated chronic inflammation in hyperlipidemia is essential to develop novel targets toward better management of patients at high risk for acute coronary events.
Mature atherosclerotic lesions contain numerous immune cell types, including monocytes, neutrophils, natural killer T cells, B cells, dendritic cells, mast cells, and innate-lymphocyte-like cells. Among all cell types, macrophages remain the most important cell type in atherosclerosis. Recent advances from single-cell sequencing suggest the presence of five subsets of macrophages in mouse atherosclerotic lesions.1 These subsets include resident, interferon-inducible cell, cavity, inflammatory, and Trem 2 (triggering receptor expressed on myeloid cells-2) foamy macrophages. The role of the resident, interferon-inducible cell, cavity macrophages in atherosclerotic disease progression is not clearly understood. Inflammatory macrophages, most likely derived from monocytes, are present in lesions and show a pro-inflammatory gene profile. Trem2 foamy macrophages, the predominant subtype found in lesions, take up oxidized-LDL (ox-LDL) and become engorged with lipids resulting in foam cell formation.1 Over a period, these foam cells contribute to necrotic core formation due to uncleared dead cells and debris from the subendothelium. Efferocytosis is an intricate process of phagocytosis of apoptotic cells within atherosclerotic lesions. Prolonged accumulation of foam cells results in apoptosis, and if the apoptotic cells are not cleared by efficient efferocytosis, they undergo necrosis and aggravate inflammation. Studies have shown that enhanced efferocytosis inhibits foam cell accumulation, reduces the release of pro-inflammatory mediators from apoptotic cells, and limits the progression of atherosclerosis.2, 3 Genetic targeting or proteolytic cleavage of efferocytosis receptor LDL-receptor related protein 1 (LRP1) during atherosclerosis compromises the ability of plaque macrophages to clear dead cells within atherosclerotic lesions.4, 5
Mammalian cells contain two pyruvate kinase (PK) genes: (1) the Pkrl gene encodes for PKR and PKL, which are expressed in red blood cells and liver, respectively, (2) the Pkm gene encodes for PKM1 and PKM2, which are expressed in many cell types, including monocytes/macrophages. PKM1 has high constitutive enzymatic activity, whereas PKM2 is less active and can be allosterically activated by the upstream glycolytic metabolite fructose-1,6-bisphosphate (FBP).6 Unlike other isoforms of PK that function only as tetramers, PKM2 exists in tetramer or dimeric forms composed of same monomers with different biological activities. Nuclear dimeric PKM2 can act as a coactivator for transcription factors implicated in the activation of the HIF-1α, STAT3, which regulate pro-inflammatory genes.7 Pharmacological inhibition of nuclear PKM2 translocation reduces LPS-induced pro-inflammatory cytokine secretion by macrophages and protects mice from lethal endotoxemia and polymicrobial sepsis.7–9 Gene profiling experiments have suggested that monocytes and macrophages from patients with coronary artery disease (CAD) over-utilize glucose and overexpress several genes, including PKM2.10 Nuclear PKM2 expression levels were increased in LPS/IFN-γ-stimulated macrophages from CAD patients when compared with healthy controls.10 Together, these studies suggest that nuclear PKM2 regulates inflammatory responses in monocytes and macrophages. Several studies have shown the role of PKM2-mediated inflammation in other cell types, including T cells and smooth muscle cells that are known to contribute to atherosclerosis.11–14 Recently, we demonstrated that PKM2 regulates platelet function.15 and contributes to neutrophil hyperactivation and cerebral thrombo-inflammation.16 The role of myeloid-specific PKM2 in the setting of atherosclerosis has not yet been elucidated.
Herein, we sought to determine the contribution of myeloid cell-specific PKM2 to atherosclerosis. We demonstrate that genetic ablation of PKM2 in myeloid cells of the low-density lipoprotein receptor-deficient (Ldlr−/−) mice significantly reduces atherosclerotic lesion progression despite high levels of cholesterol and triglyceride levels. Unexpectedly, we found that reduced atherosclerosis in myeloid cell-specific PKM2−/− mice was associated with enhanced efferocytosis and upregulation of LRP1 in vitro and in vivo. Finally, we demonstrate that inhibiting PKM2 nuclear translocation using a small molecule reduced atherosclerosis in Ldlr−/− mice as a therapeutic intervention.
Methods
Data Availability.
A detailed description of materials and methods is available in Supplemental Materials. Representative images were selected based on the mean value of each experimental group across all available data. The data that support the study are available upon reasonable request from the first authors and corresponding authors.
Human samples
Human coronary artery tissue samples from autopsies of patients with a history of CAD were obtained from the University of Iowa Decedent Center per guidelines established by the University of Iowa Institutional Review Board. The presence of atherosclerosis was confirmed by gross pathology. Type VI category human plaque samples were used.17 For immunohistochemistry, samples were fixed in formalin and processed for paraffin embedding. Antigen retrieval with 10mM Sodium Citrate, pH 6.0 was performed prior to immunohistochemical staining. Sections were then processed for immunohistochemical staining.
Animal model
PKM2fl/fl strain were genotyped by PCR as described.18 All the mice strains are on a C57BL/6J background (backcrossed >15 times). To generate myeloid cell-specific PKM2- deficient mice (PKM2fl/flLysMCre+/−), PKM2fl/fl mice were crossed with LysMCre+/− mice. To generate myeloid-specific PKM2−/− mice on Ldlr −/− background (PKM2fl/flLysMCre+/−Ldlr−/−), PKM2fl/flLysMCre+/− mice were crossed with PKM2fl/flLdlr −/− mice. Littermates PKM2fl/flLysMCre−/−Ldlr −/− mice were used as controls. Mice were kept in a regular 12 h dark/light cycle with controlled temperature and humidity and had ad libitum access to standard laboratory diet (NIH-31 modified mouse diet: 7913) and water. Male and female mice (8–10 weeks) were utilized. The University of Iowa Animal Care and Use Committee approved all the procedures and studies performed according to the Animal Research: Reporting of In Vivo Experiment guidelines (https://www.nc3rs.org.uk/arriveguidelines).
Results
PKM2 expression is upregulated in macrophages during atherosclerosis progression
Previous studies have shown that quiescent macrophages upregulate PKM2 expression when stimulated with the toll-like receptor 4 agonist LPS.9, 10, 23 However, none have examined whether PKM2 expression is upregulated in macrophages under chronic progressive inflammatory condition such as atherosclerosis. To determine whether PKM2 was upregulated in macrophages during atherosclerosis, peritoneal macrophages were isolated from Ldlr−/− mice fed a Western diet for 14 weeks and compared with Ldlr−/− mice fed a chow diet. Using Western blotting, we found that PKM2 expression was upregulated in macrophages of Ldlr−/− mice (P<0.05 versus Ldlr−/− mice fed a chow diet; Figure 1A). These findings were confirmed by immunofluorescence (Figure 1B). Similarly, we found that PKM2 expression was upregulated in ox-LDL-stimulated macrophages (Figure S1A). In line with these observations, increased PKM2-positive staining, which colocalized with macrophage (CD68-positive) staining, was observed in atherosclerotic plaques from both humans and mice (Figure S1B).
Figure 1. Myeloid cell-specific PKM2−/− mice exhibit reduced atherosclerosis.
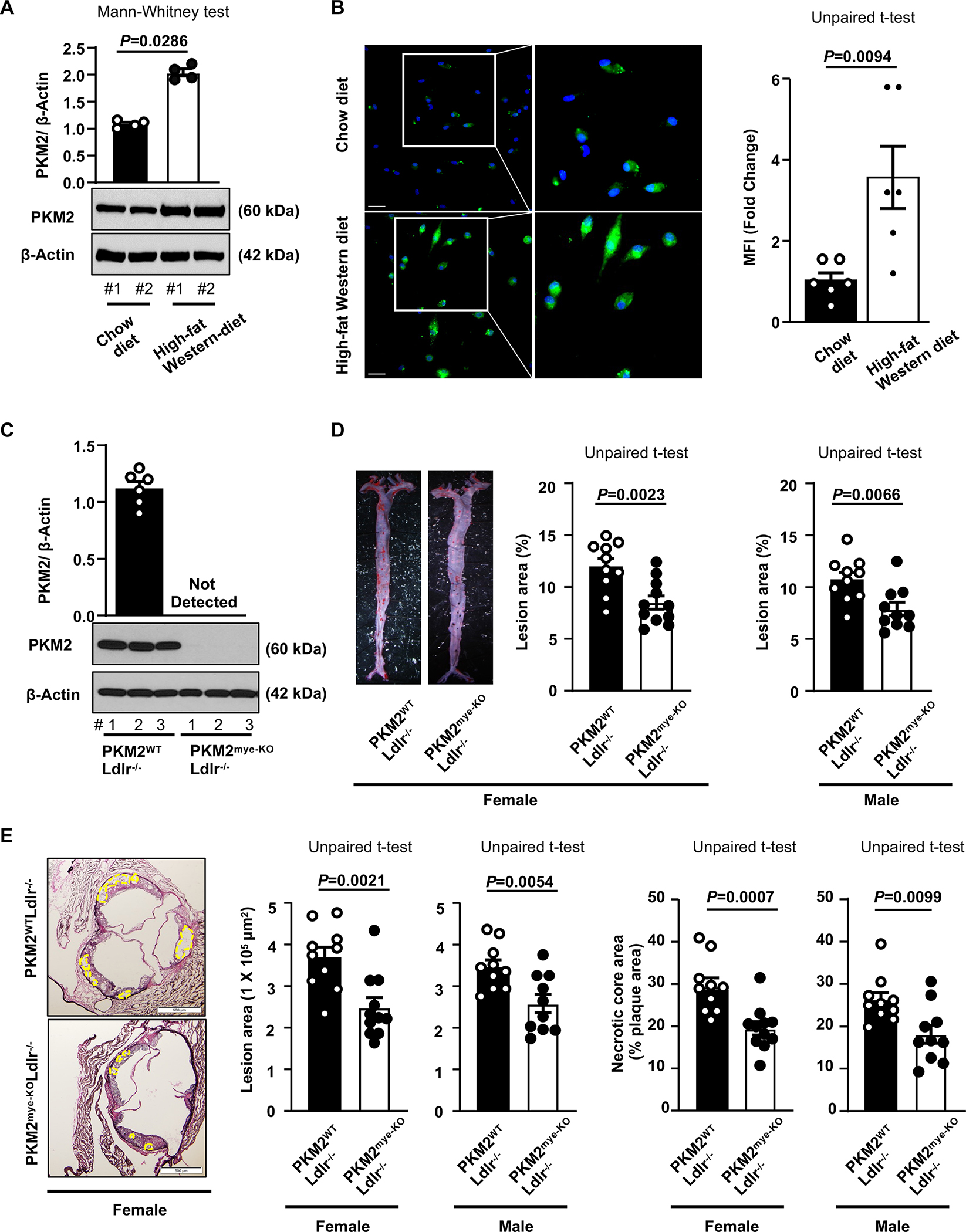
A, Representative Western blot and quantification of PKM2 levels in peritoneal macrophages from Ldlr−/− mice fed a chow diet (n=4) or high-fat “Western” diet (n=4) for 14 weeks. Loading control:β-actin. #1, 2, represents individual mouse. B, Left panels show representative immunofluorescence staining of PKM2 (green) from peritoneal macrophages of Ldlr−/− mice fed a chow diet (n=6) or high-fat “Western” diet (n=6) for 14 weeks. Nuclei are counterstained with Hoechst (blue). The boxed region is magnified. The right panel shows the mean fluorescence intensity (MFI). Scale bars, 50 μm. C, Western blot analysis of PKM2 from the peritoneal macrophages (n=6, 6). #1, 2, 3 represents individual mouse. D, Left panels show representative photomicrographs and right panels show quantification of en face lesion area in the whole aortae of female (n=10, 11) or male (n=10, 10) mice fed a high-fat Western-diet for 14 weeks. E, Left panels show representative photomicrographs, middle panels show quantification of cross-sectional lesion area in aortic sinuses, and right panels show acellular (demarcated) necrotic area. Female (n=10, 11), Male (n=10, 10). Scale bar, 500 μm. Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
Genetic deletion of PKM2 in myeloid cells reduces atherosclerotic lesion progression
To determine a definitive role of myeloid cell-specific PKM2 in atherosclerosis pathogenesis, we generated PKM2fl/flLysMCre+/− (hereafter will be referred to as PKM2mye-KO) and littermate control PKM2fl/flLysMCre−/− (hereafter will be referred to as PKM2WT) mice. Next, we generated PKM2mye-KO mice on the Ldlr-deficient background (Figure S2A). Controls were littermate PKM2WTLdlr−/− mice. Genomic PCR confirmed the presence of the LysMCre gene in the PKM2mye-KOLdlr−/− mice (Figure S2B). By Western blotting, we confirmed the absence of PKM2 expression in macrophages from PKM2mye-KOLdlr−/−mice (Figure 1C). Male and female mice were placed on the Western diet for 14 weeks to rule out sex-based differences, starting at eight weeks of age. We compared the extent of atherosclerotic lesion progression in whole aortae and cross-sectional area of atherosclerotic lesions in the aortic sinus. Irrespective of gender, we found significantly reduced atherosclerosis progression in the aortae and aortic sinuses of PKM2mye-KOLdlr−/− mice characterized by smaller necrotic core area (P<0.05 versus littermate control PKM2WTLdlr−/− mice; Figure 1D and E). Bodyweight, metabolic parameters including plasma glucose and insulin, and plasma cholesterol and triglycerides were comparable (Table S1–4).
To determine whether the observed phenotype is reproducible in another model, lethally irradiated 6-week-old (male & female) Ldlr−/− mice were reconstituted with bone marrow from either PKM2mye-KO or littermate PKM2WT mice. PCR confirmed successful engraftment of the bone marrow after four weeks in peripheral myeloid cells (not shown). Mice were fed a high-fat “Western” diet for 12 weeks at ten weeks of age. Bodyweight, total blood cell counts, total cholesterol, and triglyceride levels were comparable among groups (Table S5–8). Irrespective of sex, we found that the mean lesion area in the aortae and aortic sinuses was significantly decreased in PKMmye-KO BM➔Ldlr−/− mice compared with PKM2WT-BM➔Ldlr−/− mice (Figure 2A&B). These findings show that myeloid-specific PKM2 contributes to atherosclerotic lesion progression irrespective of sex or atherosclerotic model.
Figure 2. Chimeric mice lacking PKM2 in myeloid cells exhibit reduced atherosclerosis.

Lethally irradiated 6-week-old Ldlr−/− mice were reconstituted with bone marrow from either PKM2mye-KO or littermate PKM2WT mice. Mice were placed on a high-fat “Western” diet for additional 12 weeks beginning at the 10 weeks of age. A, Representative photomicrographs and quantification of en face lesion area in the whole aortae. Female (n=10, 12). Male (n=10, 10). B, Representative photomicrographs and quantification of cross-sectional lesion area in aortic sinuses. The acellular necrotic area is marked (yellow dotted line). Scale bar, 500 μm. Female (n=10, 12). Male (n=10, 10). Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
Myeloid cell-specific PKM2-deficient mice exhibit reduced macrophage accumulation in atherosclerotic lesions
Given that inflammatory cytokines contribute to atherogenesis, we measured plasma levels of monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-α (TNF-α), which are known to promote endothelial activation, monocyte adhesion and migration. We found decreased levels of MCP-1 and TNF-α in PKM2Mye-KOLdlr−/− mice fed a high-fat “Western” diet (P<0.05 versus PKM2WTLdlr−/− mice; Figure 3A). We next examined the expression levels of key adhesion molecules that are upregulated following endothelial activation due to elevated inflammatory cytokines in plasma and are known to contribute to atherosclerosis. We found that ICAM-1, VCAM-1, and E-selectin mRNA expression levels were significantly decreased in aortic samples isolated from PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/−mice; Figure 3B). We next examined whether the reduced MCP-1 and TNF-α levels in the plasma result in a decreased content of lesional macrophages in the aortic root. Using immunohistochemistry, we found that PKM2mye-KOLdlr−/− mice exhibited significantly less macrophage content (mac3-positive area) within plaques than PKM2WTLdlr−/− mice (Figure 3C). In parallel, using flow cytometry, we confirmed reduced macrophages in the aortic lesions of PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/− mice; Figure S3). In line with these observations, decreased macrophage content was observed in another atherosclerotic model; PKM2mye-KOBM➔LDlr−/− mice compared with PKM2WTBM➔Ldlr−/− mice (Figure S4). Because we found reduced MCP-1 levels in the plasma of PKM2mye-KOLdlr−/− mice associated with decreased macrophage content within the lesions, we examined the effect of PKM2 deletion on macrophage migration when stimulated with MCP-1. In a chemotaxis assay, we found that PKM2-deficient macrophages had a significantly reduced migration rate compared with control (Figure 3D), suggesting PKM2 regulates macrophage migration in response to MCP-1. Because PKM2mye-KOLdlr−/− mice lesions had smaller necrotic areas (Figure 1E), we measured other histological features of plaque stability, including interstitial collagen content and fibrous cap thickness. Histological features revealed higher collagen content and fibrous cap thickness in PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/− mice; Figure 3E and F).
Figure 3. Deletion of PKM2 in myeloid cells suppresses inflammation, chemotaxis, accumulation of macrophages in lesions, and promotes collagen deposition.
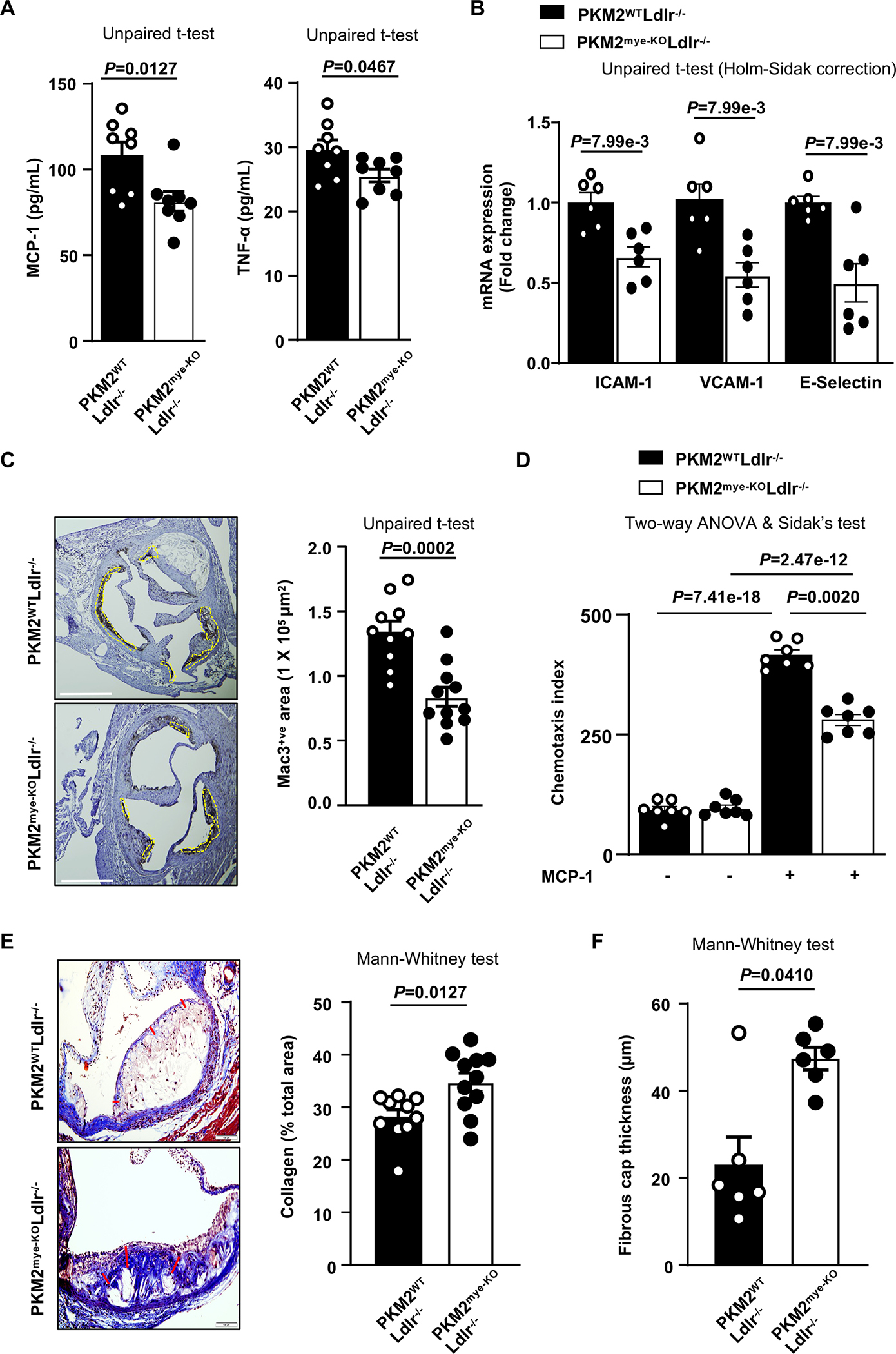
All the mice were females. A, Quantification of pro-inflammatory cytokines in plasma (n=8, 8). B, Real-time quantitative PCR analysis of ICAM-1, VCAM-1 and E-Selectin genes from the RNA isolated from whole aortae (n=6, 6). C, Representative photomicrographs and mac3-positive cells quantification in aortic sinuses (n=10, 11). Scale bar, 500 μm. D, Chemotaxis index for transmigrated bone marrow-derived macrophages (n=7, 7). E, Representative photomicrographs and quantification of collagen deposition (Masson’s Trichrome staining) in aortic sinuses (n=10, 11). Scale bar, 100 μm. F, Fibrous cap (demarcated by the red line) was calculated as the thickness of fibrous tissue overlying the necrotic core (n=6, 6). Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
Deficiency of PKM2 in macrophages suppresses inflammatory gene expression and reduces glycolytic rate
Since we observed decreased macrophage content in the lesions of PKM2mye-KOLdlr−/− mice compared with PKM2WTLdlr−/− mice, we next examined the inflammatory gene expression signature in macrophages under the conditions of high-fat “Western” diet feeding. We observed reduced mRNA expression levels of pro-inflammatory genes, including MCP-1, IL-1β, IL-12, and iNOS, in elicited peritoneal macrophages of PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/− mice; Figure 4A). On the other hand, the anti-inflammatory genes Arg1 and IL-10 were increased in PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/− mice; Figure 4A). To determine whether the observed macrophage inflammatory gene expression phenotype is reproducible in vitro, we stimulated bone marrow-derived macrophages with LPS to model inflammatory macrophages in atherosclerosis. In line with the in vivo results, we observed reduced mRNA expression levels of MCP-1, IL-1β, and IL-12, cytokines MCP-1 and IL-β and increased anti-inflammatory IL-10 levels in macrophages from PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/− mice; Figure S5). Next, we examined nuclear PKM2 expression levels in macrophages under inflammatory and anti-inflammatory conditions. We found that, nuclear PKM2 expression was increased in LPS-stimulated macrophages (P<0.05 versus naïve macrophages; Figure 4B). Conversely, the nuclear PKM2 expression level was comparable between IL-4-stimulated macrophages and naïve macrophages (Figure 4B). Nuclear PKM2 has been shown to bind STAT3 and promote STAT3 phosphorylation, thereby regulating inflammatory gene expression in several cell types, including macrophages.7, 10, 24 We examined whether deletion of PKM2 in macrophages reduces phosphorylation of STAT3 in the nucleus. Western blotting revealed reduced phospho-STAT3 in macrophages from PKM2mye-KOLdlr−/− mice when stimulated with LPS (P<0.05 versus PKM2WTLdlr−/− mice; Figure 4C). Similar results were obtained in macrophages isolated from PKM2mye-KO mice on a WT (Ldlr+) background, ruling out the effect of Ldlr- background on phospho-STAT3 levels (Figure S6). To assess the role of STAT3-mediated inflammation, we silenced STAT3 in BMDMS from PKM2WT mice. Scrambled siRNA was used as a negative control. The macrophages were then treated with LPS in vitro. We achieved good silencing with STAT3 siRNA; however, STAT3 silencing reduced expression of MCP-1 mRNA, but not IL-1β and IL12 mRNA (Figure S7). Next, we determined the effect of PKM2 deletion on glycolytic proton efflux rate (glycoPER) in basal and LPS-treated bone-derived macrophages utilizing Seahorse extracellular flux analyzer. We observed that macrophages from the PKM2mye-KOLdlr−/− mice exhibited reduced glycoPER at baseline and when stimulated with LPS (Figure 4D).
Figure 4. PKM2 deficient macrophages exhibit reduced pro-inflammatory response and reduced glycolytic activity.

All the mice were females. A, Real-time quantitative PCR analysis of pro-inflammatory and anti-inflammatory genes in primary elicited macrophages of mice fed a high-fat “Western” diet (n=7, 7). B, Bone marrow-derived macrophages (BMDMs) from wild-type Ldlr−/− mice were unstimulated (n=7) or stimulated with 100 ng/mL LPS (n=7) or 10 ng/mL IL-4 (n=7) for 24 hrs and nuclear and cytosolic PKM2 protein levels were analyzed by immunoblotting. Loading control: Lamin-B1 and GAPDH. C, BMDMs were stimulated with LPS for 4 hours. Representative immunoblots and densitometric analysis of phospho and total STAT3 in nuclear proteins fraction. Loading control: Lamin-B1. Unstimulated (n=7, 7). LPS-treated (n=7, 7). D, Glycolytic proton efflux rate (glycoPER) levels in untreated and LPS-treated (100 ng/ml) BMDMs (100,000 cells per well) were assessed by Seahorse extracellular flux analyzer. The basal respiration values were noted for 15 mins before injecting the Rot/AA (0.5 μM). Finally, 2-DG was injected, and values were measured for 40 mins. Left panel shows glycoPER curve, whereas right panel shows basal and compensatory glycolysis rate. Unstimulated, (n=10, 10). LPS-treated (n=10, 10). Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
Deficiency of PKM2 in macrophages suppresses foam cell formation, reduces apoptosis, and enhances efferocytosis
Since PKM2 deficiency did not reduce elevated cholesterol or triglyceride levels in Ldlr−/− mice, we speculated that reduced atherosclerotic lesion progression in PKM2mye-KOLdlr−/− mice might be due to decreased uptake of lipids by macrophages within lesions. We, therefore, examined the role of PKM2-deficiency on foam cell formation in vitro. Treatment of BM-derived macrophages with ox-LDL resulted in the reduction of foam cell formation in PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/− mice; Figure 5A), suggesting that PKM2 potentiates foam cell formation.
Figure 5. Deletion of PKM2 in macrophages suppresses foam cell formation and enhances macrophage efferocytosis in vitro and in lesions.
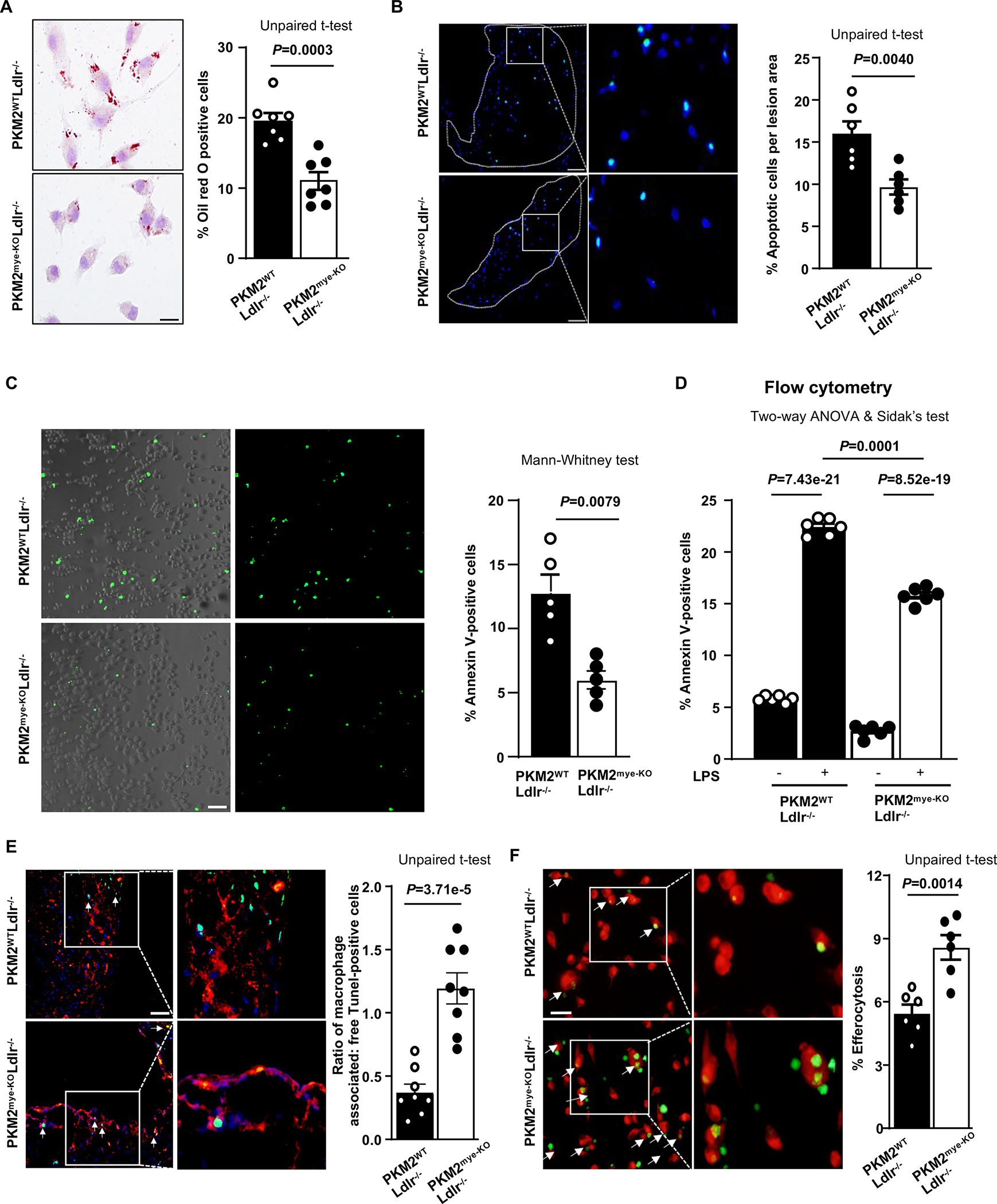
All the mice were females. A, Representative photomicrographs and quantification of ox-LDL (oxidized low-density lipoprotein; 40 μg/mL, 24 hours)-treated BMDMs, stained with Oil Red O (n=7, 7). Scale bar, 50 μM. B, The left panels show representative TUNEL-positive cells (green) counterstained with Hoechst (blue) per plaque area. The right panel shows the quantification of percentage TUNEL-positive cells to a total number of cells per plaque area (n=6, 6). The boxed region is magnified. Scale bars, 50 μm. C, Annexin V staining using BMDMs determined the percentage of apoptotic cells (n=5, 5). Scale bar, 100 μm. D, Flow cytometry analysis of percent of apoptotic macrophages positive for Annexin V after 24 hours in the presence or absence of LPS (100 ng/mL) (n=6, 6). E, Quantification of the ratio of macrophage-associated TUNEL-positive versus free TUNEL-positive cells in aortic sections (white arrows) (n=8, 8). The boxed region is magnified. Scale bar, 100 μm. F, Fluorescent images of labeled macrophages (red) with apoptotic thymocytes (green). Percent efferocytosis was quantified as the number of macrophages with engulfed apoptotic cells as a percentage of total macrophages (n=6, 6). The boxed region is magnified. Scale bar, 20 μM. Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
It is known that prolonged accumulation of foam cells results in apoptosis, and if not cleared by efferocytosis, they undergo necrosis and aggravate inflammation. We, therefore, determined whether PKM2 deficiency suppresses apoptosis. We observed reduced apoptotic cells within the lesions of PKM2mye-KOLdlr−/− mice compared with PKM2WTLdlr−/− mice (Figure 5B). At basal and inflammatory conditions, we found that PKM2mye-KO BMDMs were less apoptotic (% of Annexin V-positive cells) when compared to PKM2WT mice, in vitro (Figure 5CD). Since apoptotic cell density within lesions can arise from increased apoptosis and reduced efferocytosis, we next determined the effect of PKM2 deficiency on efferocytosis in vitro and in vivo. By immunohistochemistry, we found that the ratio of macrophage-associated TUNEL-positive cells to free TUNEL-positive cells was increased in the lesions of PKM2mye-KOLdlr−/− mice (P<0.05 versus PKM2WTLdlr−/−; Figure 5E), suggesting that PKM2 deficiency enhances efferocytosis in vivo. In parallel, apoptotic thymocytes (CFDA SE, green tracer-labeled) were cocultured with elicited peritoneal macrophages (Red tracer CMTPX-labeled) to determine whether PKM2 deficiency enhances efferocytosis in vitro. We found that PKM2 deficiency in macrophages led to enhanced efferocytosis (Figure 5F). These findings were further confirmed by FACS analysis (Figure S8A). To analyze efferocytosis ex vivo, green-labeled apoptotic thymocytes from PKM2WTLdlr−/− mice were injected into the peritoneum of PKM2mye-KOLdlr−/− or PKM2WTLdlr−/− mice. After two hours, peritoneal macrophages were isolated and stained with the red tracer CMTPX and analyzed by FACS. We found enhanced efferocytosis of apoptotic thymocytes in PKM2mye-KOLdlr−/− macrophages compared with PKM2WTLdlr−/− macrophages (Figure S8B). Similar results were obtained in the efferocytosis assay in the presence of foam cells (Figure S9).
PKM2 deficiency in macrophages facilitates efferocytosis by upregulating LRP-1
Prior studies have suggested that LRP1 regulates both inflammatory response and efferocytosis in macrophages.5, 25 Deletion of LRP1 in macrophages was shown to exacerbate atherosclerosis.4 Since deletion of PKM2 in macrophages exhibited enhanced efferocytosis in vitro and in vivo, we examined whether PKM2 deletion was associated with LRP1 upregulation in lesions and in vitro. PKM2mye-KOLdlr−/− mice exhibited increased LRP1 expression within atherosclerotic lesions compared with PKM2WTLdlr−/− mice (Figure 6A). In line with these observations, LRP1 expression was increased in PKM2-deficient macrophages (Figure 6B). To examine whether increased LRP1 could be one of the molecular mechanisms by which PKM2 deficiency suppresses inflammation and enhances efferocytosis, LRP1 was silenced in BMDMs and scrambled siRNA was used as negative control. The BMDMs were treated with LPS in vitro then assayed for pro-inflammatory gene expression and efferocytosis. We achieved very good silencing with LRP1 siRNA (Figure 6C). For BMDMs under inflammatory conditions, LRP1 silencing increased inflammatory gene expression in PKM2-deficient macrophages but not in WT macrophages except MCP1(Figure 6D). Similarly, LRP1 silencing reduced efferocytosis in PKM2-deficient macrophages but not in WT macrophages (Figure 6E). We speculate that the lack of effect of LRP1 silencing in WT macrophages could be due to reduced basal levels of LRP1 compared to PKM2-deficient macrophages.
Figure 6. PKM2 deficiency in macrophages suppresses inflammation and enhances efferocytosis by upregulating LRP-1.
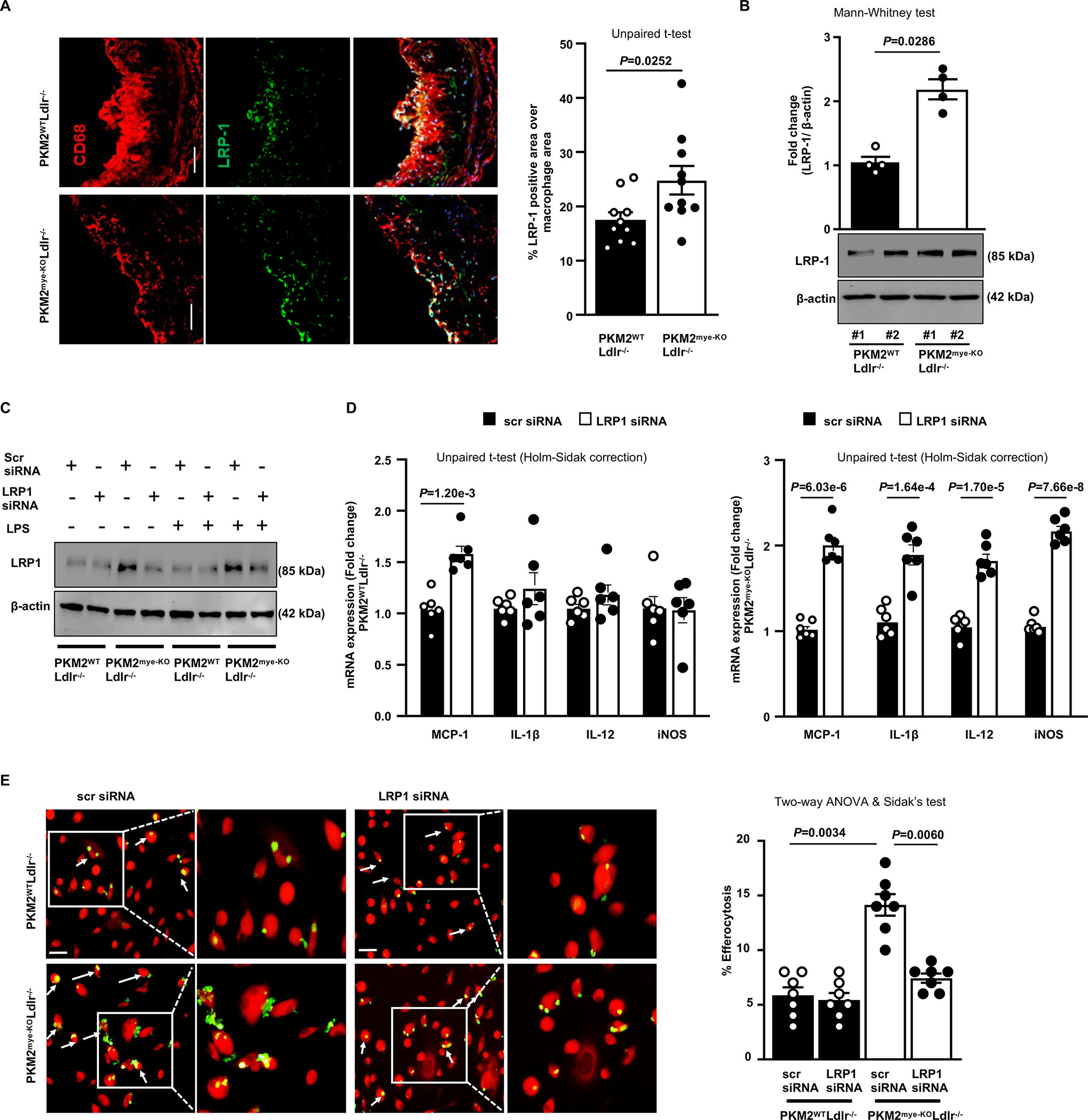
All the mice were females and fed a high-fat western diet for 14 weeks. A, The left panels show representative double immunostaining for macrophage (CD68; Red) and LRP-1 (green) in aortic sinuses. The right panel shows quantification of LRP-1 positive area in the macrophages (n=10, 10). Scale bar, 50 μm. B, Representative immunoblots and densitometric analysis of LRP-1 and β-actin in whole-cell lysates from macrophages (n=4, 4). C, BMDMs were transfected with either scrambled siRNA (10 nM) or LRP1 siRNA (10 nM). Representative Western blot of LRP1 levels in BMDMs upon LRP1 siRNA transfection in the presence or absence of LPS (100 ng/mL). Scrambled siRNA was used as a negative control. D, Real-time quantitative PCR analysis of pro-inflammatory genes in BMDMs respectively upon LPS (100 ng/mL) treatment for 24 hours (n=6, 6). E, Fluorescent images of CMTPX-labeled macrophages (red) with CFDA SE-labeled apoptotic thymocytes (green). Percent efferocytosis was quantified as the number of macrophages with engulfed apoptotic cells as a percentage of total macrophages (n=7, 7). The boxed region is magnified. Scale bar, 20 μM. Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
Inhibiting PKM2 nuclear translocation with ML265 suppresses inflammatory cytokines, decreases chemotaxis, and reduces efferocytosis
To evaluate the therapeutic significance of targeting nuclear PKM2 to reduce atherosclerosis, we examined whether ML265, a small molecule that inhibits PKM2 nuclear translocation by inducing PKM2 tetramerization,26 limits inflammation and enhances efferocytosis. We first confirmed the inhibition of nuclear PKM2 expression upon ML265 treatment in LPS-stimulated macrophages (Figure 7A). As expected, ML265 inhibited nuclear PKM2 translocation. Furthermore, ML265 treatment reduced secretion of inflammatory cytokines MCP-1 and IL-1β (Figure 7B), transmigration of macrophages in response to MCP-1 (Figure 7C) and enhanced efferocytosis in vitro (Figure 7D).
Figure 7. Limiting PKM2 nuclear translocation inhibits pro-inflammatory cytokines, reduces chemotaxis and enhances efferocytosis.
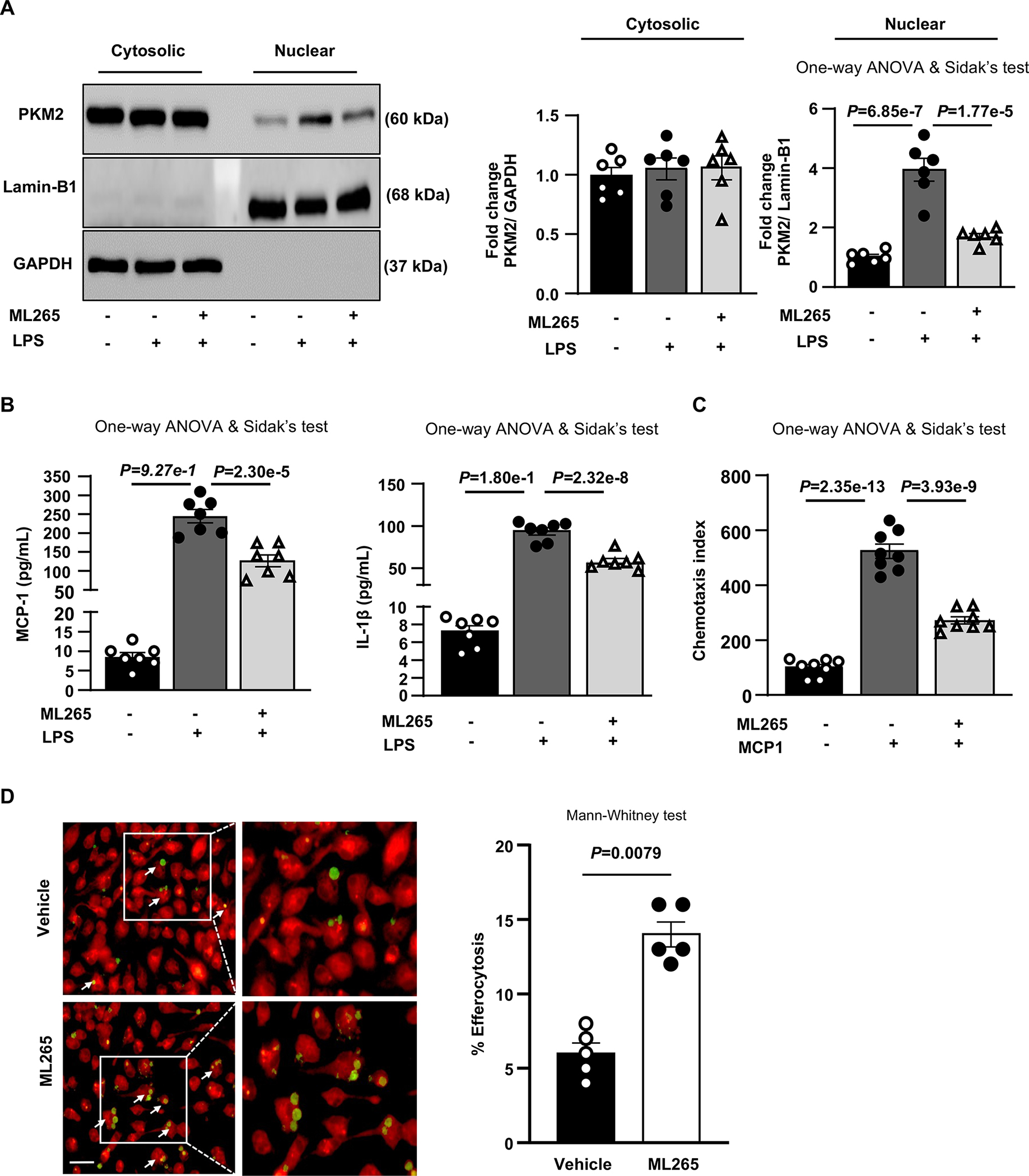
BMDMs were pretreated with vehicle or ML-265 (100 μM) for 1 hour and further stimulated with LPS (100 ng/ml; 24 hours) or MCP-1 (50 ng/mL; overnight) as indicated. A, Representative immunoblots and densitometric analysis of PKM2, Lamin-B1, and GAPDH in cytosolic and nuclear protein fractions (n=6, 6). B, Quantification of MCP-1 and IL-1β levels in cell culture supernatants (n=7, 7). C, Chemotaxis index for migrated cells upon MCP-1 treatment (n=8, 8). D, Elicited peritoneal macrophages were pretreated with vehicle (n=5) or ML-265 (n=5) for 1 hour and then cocultured with CFDA SE labeled (green) apoptotic wild-type mouse thymocytes and labeled with CMTPX (red) for the next 2 hours. Percent efferocytosis was quantified as the number of macrophages with engulfed apoptotic cells as a percentage of total macrophages. Scale bar, 20 μM. The boxed region is magnified. Results were presented with mean ± SEM. Statistical analysis are mentioned in the figure panels.
ML265-treated mice exhibited reduced atherosclerosis
Next, we determined the therapeutic potential of targeting PKM2 in vivo. Female Ldlr−/− mice were randomized to receive either ML265 (50 mg/kg) or vehicle orally once daily and placed on the Western diet for 14 weeks, starting at eight weeks. The individuals quantifying the atherosclerotic lesions, efferocytosis and glycolysis were blinded to the treatment. Bodyweight and triglycerides were comparable between the groups (Table S9–10). First, we determined the effect of ML265 treatment on macrophage function in a subset of mice fed the Western diet for 4 weeks. We found that BMDMs isolated from ML265-treated mice transmigrated less in a chemotaxis assay (P<0.05 versus vehicle-treated mice; Figure S10). After 14 weeks on the Western diet, a significant reduction in atherosclerotic lesions in the aortae and aortic sinuses was observed in ML265-treated mice (P<0.05 versus vehicle-treated mice; Figure 8AB) concomitant with reduced MCP-1 and IL-1β cytokines levels in plasma (Figure 8C). To determine whether reduced atherosclerosis in the ML265-treated mice was associated with enhanced efferocytosis, in a subset of mice, green-labeled apoptotic thymocytes from Ldlr−/− mice were injected into the peritoneum of ML265-treated Ldlr−/− or vehicle-treated Ldlr−/− mice fed a Western diet for 14 weeks. After two hours, peritoneal macrophages were isolated and stained with the red tracer CMTPX. We found enhanced efferocytosis of apoptotic thymocytes in ML265-treated Ldlr−/− mice compared to vehicle-treated Ldlr−/− mice (Figure 8D). In another subset of mice, we determined the effect of ML265 treatment on the glycolytic rate. Elicited macrophages were isolated from vehicle-treated and ML265-treated mice fed a Western diet for 14 weeks, and glycolytic proton efflux rate (glycoPER) was determined using Seahorse extracellular flux analyzer. We observed that macrophages from the ML-265-treated mice exhibited reduced glycoPER (P<0.05 versus vehicle-treated mice, Figure 8EF). Together, these studies suggest that ML265 treatment reduces atherosclerosis in association with reduced inflammation, decreased glycolytic rate, and enhanced efferocytosis.
Figure 8. ML265 treatment reduces atherosclerosis.
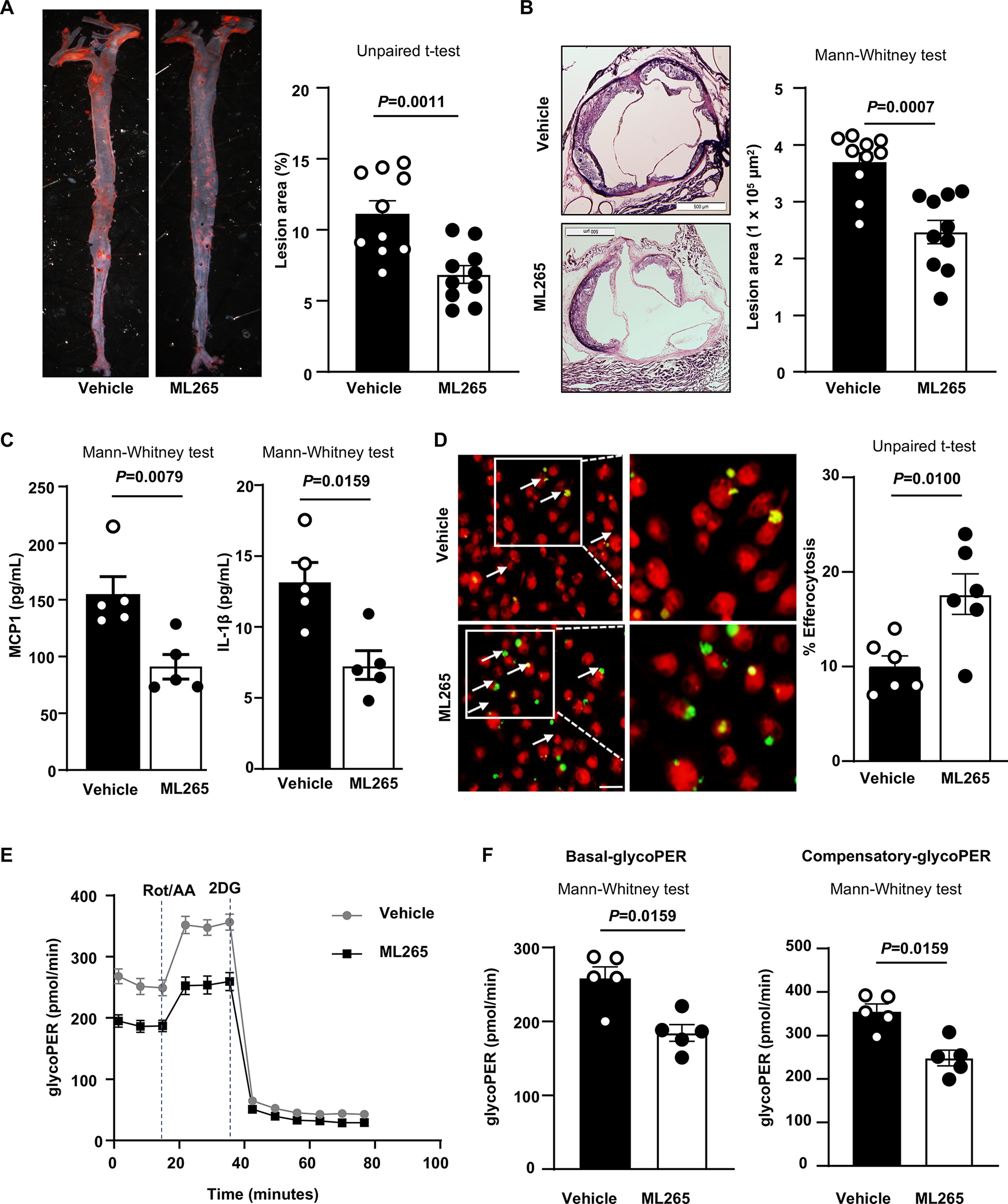
Female Ldlr−/− mice were randomized to receive either vehicle or ML265 (50 mg/kg) orally daily and placed on the Western diet for 14 weeks, starting at 8 weeks of age. A, Left panels show representative photomicrographs, and right panels show quantification of en face lesion area in the whole aortae (n=10, 10). B, Left panels show representative photomicrographs and right panels show quantification of cross-sectional lesion area in aortic sinuses (n=10, 10). Scale bar, 500 μm. C, Quantification of pro-inflammatory cytokines in plasma (n=5, 5). D, In a subset of Ldlr−/− mice that received either vehicle (n=6) or ML265 (n=6) and fed a Western diet for 14 weeks, wild-type CFDA-SE labeled thymocytes (green) were injected 2 hours prior to harvesting elicited peritoneal macrophages. Harvested cells were labeled with CMTPX (red), and percent efferocytosis was quantified as the number of macrophages with engulfed apoptotic cells as a percentage of total macrophages. Scale bar, 20μM. E, Elicited macrophages were isolated from a vehicle and ML265-treated mice fed a WTD for 14 weeks. Cells (100,000 per well) were plated and treated for 30 minutes in seahorse media. The curve shows glycolytic proton efflux rate (glycoPER). F, The basal and compensatory glycoPER rate derived from E (n=5, 5). Results were presented with mean ± SEM. Statistical analysis as indicated in the figure panels.
Discussion
PKM2 regulates inflammatory responses in macrophages stimulated with LPS or oxidized-LDL,7, 10, 27 and PKM2 is upregulated in monocytes/macrophages of patients with atherosclerotic coronary artery disease.10 Evidence has been lacking whether PKM2 potentiates inflammatory responses in macrophages in response to high-fat “Western diet” and thereby exacerbates atherosclerosis. The current study provides in vivo evidence that PKM2 is upregulated in macrophages in murine atherosclerotic lesions and that genetic deletion of myeloid cell-specific PKM2 reduces atherosclerosis. Unexpectedly, we found that deletion of PKM2 in macrophages increased LRP1 expression and enhanced efferocytosis in vitro and in vivo. Our results suggest a previously unreported link between PKM2-mediated inflammatory response, macrophage efferocytosis, and LRP1 that may regulate macrophage inflammatory responses within the atherosclerotic lesion microenvironment. To assess translational potential, we demonstrate that limiting PKM2 nuclear translocation with ML265 treatment reduces atherosclerosis, decreases inflammation and glycolytic rate, and enhances efferocytosis.
We found that PKM2 expression was upregulated in macrophages during atherosclerosis progression. Our findings align with a previous study that demonstrated PKM2 overexpressed in ex-vivo generated macrophages from CAD patients.10 The overexpression of PKM2 in macrophages during mouse atherosclerosis or in CAD patients could be attributed to increased glucose uptake needed for high energy consumption required for macrophage activation, which further correlates with overexpression of GLUT1 transcripts in CAD macrophages.10 PK is a rate-limiting enzyme that catalyzes the final step of glycolysis by converting phosphoenolpyruvate to pyruvate and plays a crucial role in controlling the glycolytic flux. Since activation of macrophages is accompanied by increased glucose uptake, the upregulation of PKM2 may prevent the complete conversion of glucose to pyruvate, which enables the upstream glycolytic metabolic intermediates to accumulate, thus shifting the metabolism towards the anabolic phase for amino acids, lipids, and nucleotides, all required for macrophage activation.
We next showed that PKM2mye-KO mice were less susceptible to atherosclerosis. Reduced atherosclerosis in PKM2mye-KO mice was independent of changes in plasma lipid levels, a finding that is in agreement with other studies that have shown that the extent of atherosclerosis does not always correlate with plasma cholesterol and LDL levels.20, 28, 29 A similar phenotype was observed in female mice, ruling out sex-based differences. These results suggest that myeloid cell-specific PKM2 exacerbates atherosclerosis independently of plasma lipid levels. We found that myeloid cell-derived PKM2 potentiates macrophage accumulation in atherosclerotic lesions by promoting foam cell formation and aggravating inflammatory responses. Genetic ablation of PKM2 in macrophages decreased uptake of lipids by macrophages. When our studies were in progress, another study demonstrated a role for PKM2 in macrophage foam cell formation.27 Driven primarily by glycolysis, inflammatory macrophages express transcription factors and secrete pro-inflammatory cytokines such as IL-1β and TNF-α.30 We found that genetic deletion of PKM2 in macrophages of mice fed a Western diet led to a reduced mRNA expression of several pro-inflammatory genes, including MCP-1, IL-1β, IL-12, and iNOS, and increased expression of the anti-inflammatory genes Arg1 and IL-10, suggesting a role for PKM2 in macrophage polarization.
Additionally, we found that deletion of PKM2 in myeloid cells reduced plasma levels of MCP-1 and TNF-α and decreased endothelial activation and macrophage migration in response to MCP-1. Elevated plasma levels of MCP-1 are associated with incident of CAD,31 and mice deficient in MCP-1 or its receptor chemokine receptor 2 develop fewer and smaller lesions than control mice due to decreased monocyte recruitment to atheroprone sites.32, 33 We demonstrated in vitro that dimeric PKM2 regulates pro-inflammatory gene expression in LPS-stimulated macrophages. Dimeric PKM2 was shown to promote STAT3 phosphorylation, thereby regulating inflammatory gene expression in macrophages.7, 10 Based on these studies we, speculated that reduced phospho-STAT3 could be the mechanism by which PKM2 deficiency suppresses inflammatory cytokine secretion. However, siRNA experiments showed that STAT3 silencing reduced MCP1 gene expression in macrophages from PKM2WT mice, but not expression of IL-1β and IL12. These findings suggest that reduced phospho-STAT3 is only a part of the mechanism by which PKM2 deficiency may suppress inflammatory cytokine secretion in the setting of atherosclerosis.
Defective efferocytosis results in uncleared apoptotic cells that may undergo post-apoptotic necrosis and aggravate inflammatory responses within plaques.34–36 Herein, unexpectedly, we found that deletion of PKM2 in myeloid cells enhanced efferocytosis in vitro and in vivo associated with reduced necrosis, suggesting that PKM2 in macrophages impairs efferocytosis. Multiple mechanisms can impair efferocytosis. First, because of the competition for apoptotic cell binding. As atherosclerosis progresses, oxidized phospholipids continue to accumulate in the lesions. Studies suggest that oxidized phospholipids can bind to efferocytosis receptors and compete for apoptotic cells recognition, thereby inhibiting phagocytosis of apoptotic cells by elicited macrophages.37, 38 Indeed, we found that PKM2 regulates the uptake of oxidized lipids that in turn might inhibit phagocytosis of apoptotic cells by macrophages. Second, as a consequence of pro-inflammatory microenvironment within lesions, which causes downregulation of key efferocytosis molecules including LRP1.5 Studies have shown that deletion of LRP1 in macrophages impairs efferocytosis and exacerbates atherosclerosis.4, 5, 35 Notably, we found that deletion of PKM2 in macrophages enhances efferocytosis of apoptotic cells in vitro and in vivo that was associated with upregulation of LRP1. Silencing LRP1 in PKM2-deficient macrophages increased the inflammatory gene expression of MCP-1, IL-1β, IL-12, and iNOS in association with reduced efferocytosis. Together, these results suggest that increased LRP1 is most likely the underlying mechanism by which PKM2 deficiency suppresses inflammatory cytokine secretion and enhances efferocytosis. The precise mechanism how PKM2 regulates LRP1 and compromise efferocytosis remains unclear and continue to be an area of future investigation. Importantly, in preclinical experiments with translational potential, we demonstrated that inhibiting PKM2 nuclear translocation with a small molecule ML265 reduces atherosclerosis, glycolytic rate, and inflammation, and enhances efferocytosis. A summary of the proposed pathway is provided in the Figure S11.
Currently, patients at risk for CVD are treated mainly with therapies that target hypertension and hyperlipidemia, as well as aspirin, which has anti-platelet and mild anti-inflammatory effects. While these approaches have efficacy, they do not adequately address the pro-inflammatory mechanisms that promote atherosclerosis independently of elevated lipid levels. The CANTOS trial proved that limiting inflammation by administering an anti-IL-1β antibody reduced recurrent cardiovascular events independently of hyperlipidemia.39 The strength of our study is that targeting PKM2 nuclear translocation might limit inflammation and reduce foam cell formation and promote clearance of apoptotic cells within lesions by enhancing efferocytosis via up regulating LRP1. Despite this strength, our study has limitations. PKM2 is expressed by other immune cell types, including neutrophils, B cells, T cells, and platelets, that are present in atherosclerotic lesions, all of which are known to contribute to disease progression. Thus, a possible role for PKM2 in these other cell types in atherosclerosis cannot be completely ruled out.
Conclusions:
Our studies unequivocally support a mechanistic role of myeloid-specific PKM2 in regulating macrophage inflammation, efferocytosis most likely via LRP1, and thereby, atherosclerosis. Targeting nuclear PKM2 may offer a promising approach to limit atherosclerotic lesion progression in patients at high risk for coronary artery disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The glycolytic enzyme pyruvate kinase muscle 2 (PKM2) is upregulated in the monocytes and macrophages of patients with atherosclerotic coronary artery disease.
PKM2 regulates inflammatory phenotype in several cell types, including monocytes, when stimulated with LPS.
The role of myeloid-specific PKM2 in the setting of atherosclerosis is not yet elucidated.
What New Information Does This Article Contribute?
Efferocytosis is a process by which apoptotic cells are removed by phagocytic cells. We report a previously unidentified link between PKM2-mediated inflammatory response and LRP1-mediated efferocytosis in macrophages.
Genetic deletion of PKM2 in myeloid cells reduces atherosclerosis by limiting inflammation and enhancing efferocytosis by upregulating macrophage LRP1.
Limiting PKM2 nuclear translocation with ML265 treatment reduces atherosclerosis in Ldlr−/− mice fed a Western diet, suggesting the translational potential of this approach.
Although lipid-lowering drugs are the mainstay of therapies to reduce atherosclerosis in patients at high risk for cardiovascular diseases, they do not effectively inhibit chronic inflammation, which is a key contributor to atherosclerosis exacerbation. Moreover, lipid-lowering drugs do not reduce lipid levels to such an extent in patients at risk to completely suppress atherosclerosis. Thus, new complementary treatments are required that can be combined with lipid-lowering drugs to inhibit chronic inflammation as well promote macrophage efferocytosis during inflammation resolution. In the current study, we demonstrated the in vivo role of myeloid-specific PKM2 in the pathophysiology of atherosclerosis. Importantly, using in vitro and in vivo approaches, we demonstrate that targeting PKM2 in myeloid cells reduces atherosclerosis by promoting macrophage efferocytosis by upregulating LRP1. Based on murine studies, we propose PKM2 as a novel therapeutic target to suppress atherosclerosis in patients at risk of cardiovascular diseases that are not benefitted from lipid-lowering therapies.
Sources of funding
The A.K.C. lab is supported by grants from the National Institutes of Health grant (R35HL139926, R01NS109910 & U01NS113388) and by Established Investigator Award 18EIA33900009 from American Heart Association. P.D. is supported by Career Development Award (20CDA35310580) from the American Heart Association.
Non-standard Abbreviations and Acronyms
- PKM2
Pyruvate Kinase M2
- Ldlr
Low-density lipoprotein receptor
- MCP1
Monocyte chemoattractant protein-1
- TNF-α
Tumor necrosis factor-α
- Trem 2
Triggering receptor expressed on myeloid cells-2
- LRP1
LDL-receptor related protein 1
- HIF-1
Hypoxia-inducible factor 1
- STAT3
Signal transducer and activator of transcription 3
- LPS
Lipopolysaccharides
- IFNγ
Interferon gamma
- ICAM1
Intracellular adhesion molecule 1
- VCAM1
Vascular cell adhesion molecule 1
- BMDMs
Bone marrow-derived macrophages
Footnotes
Disclosures
None
SUPPLEMENTAL MATERIALS
Detailed Methods
References
- 1.Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, Robbins CS, Monaco C, Park I, McNamara CA, Binder CJ, Cybulsky MI, Scipione CA, Hedrick CC, Galkina EV, Kyaw T, Ghosheh Y, Dinh HQ, Ley K. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127:402–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brophy ML, Dong Y, Tao H, Yancey PG, Song K, Zhang K, Wen A, Wu H, Lee Y, Malovichko MV, Sithu SD, Wong S, Yu L, Kocher O, Bischoff J, Srivastava S, Linton MF, Ley K, Chen H. Myeloid-specific deletion of epsins 1 and 2 reduces atherosclerosis by preventing lrp-1 downregulation. Circ Res. 2019;124:e6–e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351 [DOI] [PubMed] [Google Scholar]
- 4.Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage ldl receptor-related protein increases atherogenesis in the mouse. Circ Res. 2007;100:670–677 [DOI] [PubMed] [Google Scholar]
- 5.Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, Linton MF, Fazio S. Macrophage lrp-1 controls plaque cellularity by regulating efferocytosis and akt activation. Arterioscler Thromb Vasc Biol. 2010;30:787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda Y, Noguchi T. Allosteric regulation of pyruvate kinase m2 isozyme involves a cysteine residue in the intersubunit contact. J Biol Chem. 1998;273:12227–12233 [DOI] [PubMed] [Google Scholar]
- 7.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O’Neill LA. Pyruvate kinase m2 regulates hif-1alpha activity and il-1beta induction and is a critical determinant of the warburg effect in lps-activated macrophages. Cell Metab. 2015;21:65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang H, Cao L, Jiang J, Tang D. Pkm2-dependent glycolysis promotes nlrp3 and aim2 inflammasome activation. Nat Commun. 2016;7:13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H, Cao L, Tang D. Pkm2 regulates the warburg effect and promotes hmgb1 release in sepsis. Nat Commun. 2014;5:4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, Goronzy JJ, Weyand CM. The glycolytic enzyme pkm2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X, Tan F, Cao X, Cao Z, Li B, Shen Z, Tian Y. Pkm2-dependent glycolysis promotes the proliferation and migration of vascular smooth muscle cells during atherosclerosis. Acta Biochim Biophys Sin (Shanghai). 2020;52:9–17 [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Dang G, Lu S, Liu H, Ma X, Han L, Deng J, Miao Y, Li X, Shao F, Jiang C, Xu Q, Wang X, Feng J. T-cell-derived extracellular vesicles regulate b-cell igg production via pyruvate kinase muscle isozyme 2. FASEB J. 2019;33:12780–12799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu S, Deng J, Liu H, Liu B, Yang J, Miao Y, Li J, Wang N, Jiang C, Xu Q, Wang X, Feng J. Pkm2-dependent metabolic reprogramming in cd4(+) t cells is crucial for hyperhomocysteinemia-accelerated atherosclerosis. J Mol Med (Berl). 2018;96:585–600 [DOI] [PubMed] [Google Scholar]
- 14.Jain M, Dhanesha N, Doddapattar P, Nayak MK, Guo L, Cornelissen A, Lentz SR, Finn AV, Chauhan AK. Smooth muscle cell-specific pkm2 (pyruvate kinase muscle 2) promotes smooth muscle cell phenotypic switching and neointimal hyperplasia. Arterioscler Thromb Vasc Biol. 2021;41:1724–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nayak MK, Ghatge M, Flora GD, Dhanesha N, Jain M, Markan KR, Potthoff MJ, Lentz SR, Chauhan AK. The metabolic enzyme pyruvate kinase m2 regulates platelet function and arterial thrombosis. Blood. 2021;137:1658–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhanesha N, Patel RB, Doddapattar P, Ghatge M, Flora GD, Jain M, Thedens D, Olalde H, Kumskova M, Leira E, Chauhan AK. Pkm2 promotes neutrophil activation and cerebral thrombo-inflammation: Therapeutic implications for ischemic stroke. Blood. 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275 [DOI] [PubMed] [Google Scholar]
- 18.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC, Vander Heiden MG. Pkm2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doddapattar P, Dev R, Jain M, Dhanesha N, Chauhan AK. Differential roles of endothelial cell-derived and smooth muscle cell-derived fibronectin containing extra domain a in early and late atherosclerosis. Arterioscler Thromb Vasc Biol. 2020;40:1738–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME, Lentz SR, Chauhan AK. Fibronectin splicing variants containing extra domain a promote atherosclerosis in mice through toll-like receptor 4. Arterioscler Thromb Vasc Biol. 2015;35:2391–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doddapattar P, Jain M, Dhanesha N, Lentz SR, Chauhan AK. Fibronectin containing extra domain a induces plaque destabilization in the innominate artery of aged apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2018;38:500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasikara C, Schilperoort M, Gerlach B, Xue C, Wang X, Zheng Z, Kuriakose G, Dorweiler B, Zhang H, Fredman G, Saleheen D, Reilly MP, Tabas I. Deficiency of macrophage phactr1 impairs efferocytosis and promotes atherosclerotic plaque necrosis. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, O’Neill LA. Pyruvate kinase m2 is required for the expression of the immune checkpoint pd-l1 in immune cells and tumors. Front Immunol. 2017;8:1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang P, Li Z, Fu R, Wu H, Li Z. Pyruvate kinase m2 facilitates colon cancer cell migration via the modulation of stat3 signalling. Cell Signal. 2014;26:1853–1862 [DOI] [PubMed] [Google Scholar]
- 25.Mantuano E, Brifault C, Lam MS, Azmoon P, Gilder AS, Gonias SL. Ldl receptor-related protein-1 regulates nfkappab and microrna-155 in macrophages to control the inflammatory response. Proc Natl Acad Sci U S A. 2016;113:1369–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang J, Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, Hong BS, Tempel W, Dimov S, Veith H, Yang H, Kung C, Yen KE, Dang L, Salituro F, Auld DS, Park HW, Vander Heiden MG, Thomas CJ, Shen M, Boxer MB. Ml265: A potent pkm2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. Probe reports from the nih molecular libraries program. Bethesda (MD); 2010. [PubMed] [Google Scholar]
- 27.Kumar A, Gupta P, Rana M, Chandra T, Dikshit M, Barthwal MK. Role of pyruvate kinase m2 in oxidized ldl-induced macrophage foam cell formation and inflammation. J Lipid Res. 2020;61:351–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappab-mediated inflammation in macrophages. Circ Res. 2014;114:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in ccr2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897 [DOI] [PubMed] [Google Scholar]
- 30.Tabas I, Bornfeldt KE. Intracellular and intercellular aspects of macrophage immunometabolism in atherosclerosis. Circ Res. 2020;126:1209–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoogeveen RC, Morrison A, Boerwinkle E, Miles JS, Rhodes CE, Sharrett AR, Ballantyne CM. Plasma mcp-1 level and risk for peripheral arterial disease and incident coronary heart disease: Atherosclerosis risk in communities study. Atherosclerosis. 2005;183:301–307 [DOI] [PubMed] [Google Scholar]
- 32.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281 [DOI] [PubMed] [Google Scholar]
- 33.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yurdagul A Jr., Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yancey PG, Ding Y, Fan D, Blakemore JL, Zhang Y, Ding L, Zhang J, Linton MF, Fazio S. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory ly-6chigh monocytosis: Evidence that the effects are not apolipoprotein e dependent. Circulation. 2011;124:454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yurdagul A Jr., Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, Kolluru GK, Rymond CC, Gerlach BD, Zheng Z, Kuriakose G, Kevil CG, Koomen JM, Cleveland JL, Muoio DM, Tabas I. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 2020;31:518–533 e510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang MK, Bergmark C, Laurila A, Horkko S, Han KH, Friedman P, Dennis EA, Witztum JL. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci U S A. 1999;96:6353–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw PX, Horkko S, Tsimikas S, Chang MK, Palinski W, Silverman GJ, Chen PP, Witztum JL. Human-derived anti-oxidized ldl autoantibody blocks uptake of oxidized ldl by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol. 2001;21:1333–1339 [DOI] [PubMed] [Google Scholar]
- 39.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of materials and methods is available in Supplemental Materials. Representative images were selected based on the mean value of each experimental group across all available data. The data that support the study are available upon reasonable request from the first authors and corresponding authors.
Human samples
Human coronary artery tissue samples from autopsies of patients with a history of CAD were obtained from the University of Iowa Decedent Center per guidelines established by the University of Iowa Institutional Review Board. The presence of atherosclerosis was confirmed by gross pathology. Type VI category human plaque samples were used.17 For immunohistochemistry, samples were fixed in formalin and processed for paraffin embedding. Antigen retrieval with 10mM Sodium Citrate, pH 6.0 was performed prior to immunohistochemical staining. Sections were then processed for immunohistochemical staining.
Animal model
PKM2fl/fl strain were genotyped by PCR as described.18 All the mice strains are on a C57BL/6J background (backcrossed >15 times). To generate myeloid cell-specific PKM2- deficient mice (PKM2fl/flLysMCre+/−), PKM2fl/fl mice were crossed with LysMCre+/− mice. To generate myeloid-specific PKM2−/− mice on Ldlr −/− background (PKM2fl/flLysMCre+/−Ldlr−/−), PKM2fl/flLysMCre+/− mice were crossed with PKM2fl/flLdlr −/− mice. Littermates PKM2fl/flLysMCre−/−Ldlr −/− mice were used as controls. Mice were kept in a regular 12 h dark/light cycle with controlled temperature and humidity and had ad libitum access to standard laboratory diet (NIH-31 modified mouse diet: 7913) and water. Male and female mice (8–10 weeks) were utilized. The University of Iowa Animal Care and Use Committee approved all the procedures and studies performed according to the Animal Research: Reporting of In Vivo Experiment guidelines (https://www.nc3rs.org.uk/arriveguidelines).