

Abstract
The orexin receptors (OXRs) have been involved in multiple physiological and neuropsychiatric functions. Identification of PET imaging probes specifically targeting OXRs enables us to better understand the OX system. Seltorexant (JNJ-42847922) is a potent OX2R antagonist with the potential to be an OX2R PET imaging probe. Here, we describe the synthesis and characterization of [18F]Seltorexant as an OX2R PET probe. The ex vivo autoradiography studies indicated the good binding specificity of [18F]Seltorexant. In vivo PET imaging of [18F]Seltorexant in rodents showed suitable BBB penetration with the highest brain uptake of %ID/cc = 3.4 at 2 minutes post-injection in mice. The regional brain biodistribution analysis and blocking studies showed that [18F]Seltorexant had good binding selectivity and specificity. However, pretreatment with unlabelled Seltorexant and P-gp competitor CsA observed significantly increased brain uptake of [18F]Seltorexant, indicating [18F]Seltorexant could interact P-gp at the blood-brain barrier. Our findings demonstrated that [18F]Seltorexant is a potential brain OX2R PET imaging probe, which paves the way for new OX2R PET probes development and OX system investigation.
Keywords: orexin receptors, PET, radiotracer, imaging
Graphical Abstract
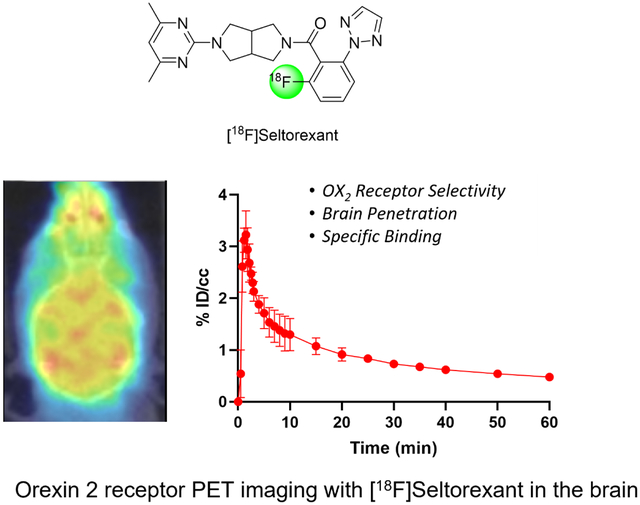
Introduction
Orexins (also known as hypocretins), which are endogenous neuropeptides secreted by the hypothalamus, contains two subtypes of orexin-A and orexin-B.1, 2 Orexin-A and orexin-B have been found to interact with G protein-coupled receptors orexin 1 receptor (OX1R) and orexin 2 receptor (OX2R) respectively to regulate a variety of physiological functions, including energy homeostasis, sleep-wake cycle, stress response, and brain reward mechanisms.2–4 Studies have shown that abnormalities in orexin signaling have a close relationship with many diseases, especially sleep disorders.5 Further research revealed that OX1R and OX2R have different expressions and distributions in the brain, suggesting their distinct physiological functions. For example, OX1Rs are mainly located in the limbic system, paraventricular thalamic nucleus, and locus coeruleus regulate emotion responsible for reward and autonomic regulation.6 Conversely, OX2Rs are exclusively expressed in the controlling arousal regions, which play a critical role in regulating sleep and wakefulness.7
Several lines of evidence indicate that the blockade of orexin receptors shows therapeutic potential for various diseases, primarily for insomnia treatment.4, 8–12 Subsequently, the development of OXR antagonists has aroused widespread interest in this field of research. In the past decade, a number of small-molecule OXR antagonists have been discovered. One of the most representative OXR antagonists is Suvorexant (MK-4305, Figure 1), a dual OXR antagonist (DORA), which has been approved by the U.S. Food and Drug Administration (FDA) for insomnia treatment.13, 14 Notably, an increasing number of selective OX1R or OX2R antagonists have been reported in recent years (Figure 1).15 Compared with dual OXR antagonists, selective OXR antagonists exhibited advantages such as better efficacy and fewer adverse effects. However, to date, the biological mechanism of OX1R and OX2R in CNS-related diseases has not been fully elucidated.15, 16
Figure 1.
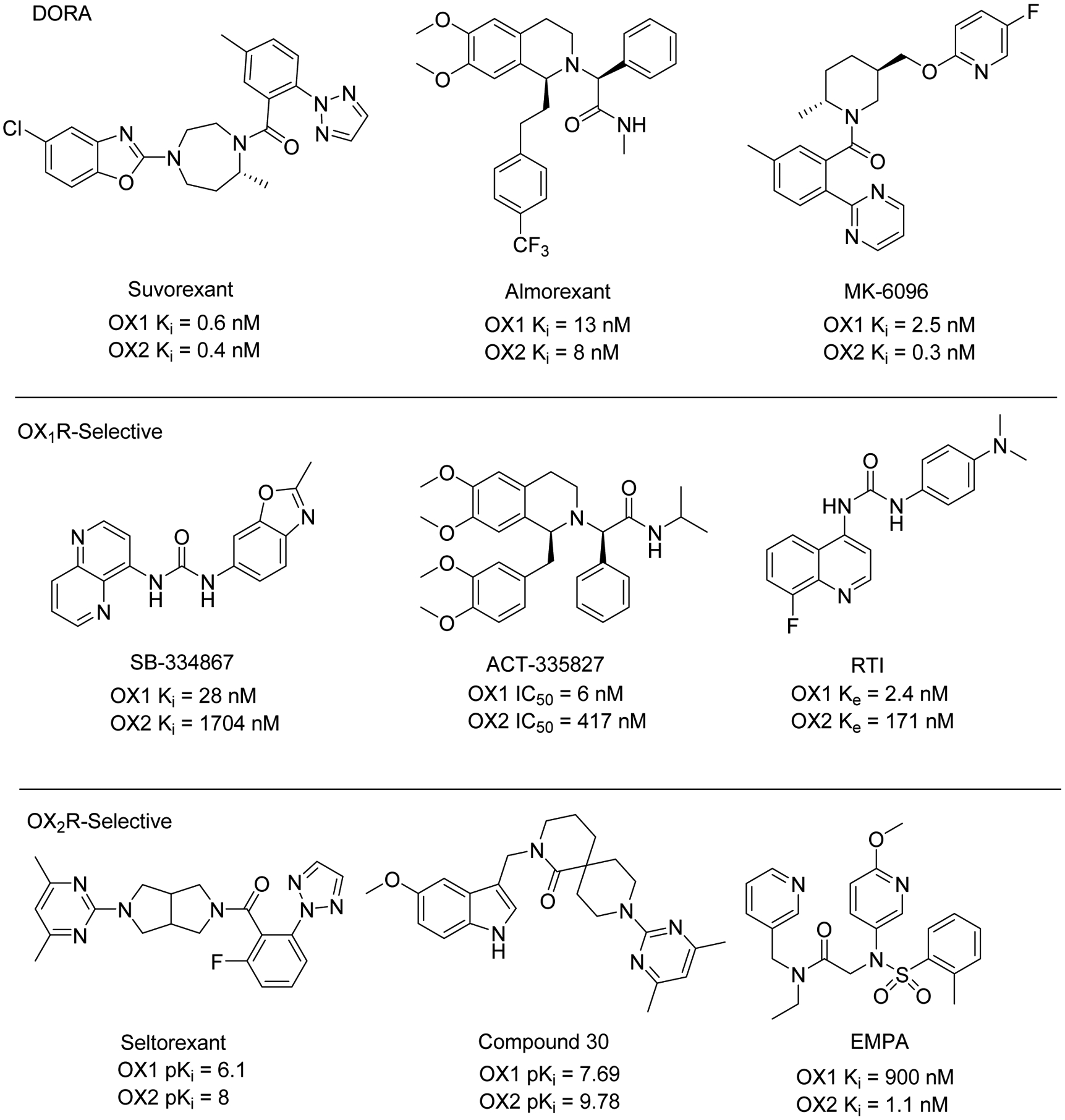
Chemical structures and binding affinities of representative dual orexin receptor antagonists (DORA) and selective orexin 1/2 receptor antagonists.
Molecular imaging is an advanced technique that enables us to obtain valuable biological information at the molecular level.17 For instance, in vitro autoradiography of [3H]EMPA, a selective OX2R radioligand, has indicated the biodistribution and density of OX2R in the rat brain.18 Positron emission tomography (PET) imaging provides a powerful tool for visualization and quantification of pharmacological targets in living subjects in a non-invasive way.19 PET imaging of orexin receptors with a suitable OXR radioligand could contribute to elucidate the biological function of the orexin system as well as facilitate novel selective OXR antagonists discovery. Although a series of OXR PET probes were synthesized and characterized in the past few years, limited success has been achieved. For example, Watanabe et al. reported F18-labeled tetrahydroisoquinoline derivatives OX1R PET tracers with low brain uptake in the brain of mice.20 The carbon-11 labeled OX2R PET radioligands, including [11C]CW421, [11C]EMPA22, and [11C]9a23 exhibited either limited brain uptake or unexpected non-specific binding in rodents and non-human primates (NHP) studies, which impeded their further translation for OX2R imaging. Recently, we have reported a non-selective OXRs PET radioligand [11C]CW24 with good brain uptake in rodents and NHP.24 However, the moderate binding affinity and specificity of [11C]CW24 for OXRs still need to be optimized. As a result, so far, no PET radioligand specifically targets OX2R for neuroimaging in preclinical or clinical application.
Radiolabeling the potent known OX antagonist with short half-life isotopes (e.g., carbon-11 (t1/2 = 20 minutes) and fluorine-18 (t1/2 = 109 minutes)) is a practical and effective strategy to develop PET imaging probes specifically targeting OXR, such as [11C]MK-106425, [11C]CW4, and [11C]EMPA. As part of our continuing effort for the OXRs PET radioligand development, we screened for the known selective OXR antagonists that can be labeled with radioisotopes. There are some criteria that we use to choose OXR antagonist candidates for radiolabelling. First, the binding affinity and selectivity for OXR must be good enough to measure the target precisely in vivo. Additionally, as our interested organ is the brain, the candidates should possess appropriate physicochemical properties to penetrate the blood-brain barrier (BBB). Seltorexant (JNJ-42847922), a selective OX2R antagonist, was first discovered by Janssen Pharmaceutical Research & Development, LLC and is being in clinical studies for the treatment of insomnia and major depressive disorder.26 In the preclinical studies26–28, Seltorexant exhibited favorable pharmacological properties with good binding affinity and selectivity for OX2R, good brain uptake, and suitable pharmacokinetic properties, which is an ideal candidate for radiolabeling and OX2R PET imaging in the brain.
In this study, we describe the synthesis of the precursor for [18F]Seltorexant preparation. Comprehensive evaluations of [18F]Seltorexant, including binding specificity and brain permeability, were carried out by in vitro autoradiography and in vivo dynamic PET imaging in rodents, which provide significant information for future OX2R PET radiotracers’ development.
Materials and methods
All commercially available chemical reagents and solvents were of ACS–grade purity or higher and directly used without further purification. The unlabelled Seltorexant, orexin 2 antagonist compound 30 and competitive P-gp inhibitor Cyclosporin A were purchased from MedChemExpress. Anhydrous Dimethylsulfoxide (DMSO) was purchased from Acros Organics. No-carrier-added 18F-fluoride was produced from water 97% enriched in 18O (Sigma-Aldrich®) by the nuclear reaction 18O(p, n)18F with a Siemens Eclipse HP cyclotron and a silver-bodied target at Athinoula A. Martinos Center for Biomedical Imaging.
All animal studies were carried out at Massachusetts General Hospital (PHS Assurance of Compliance No. A3596–01). The Subcommittee on Research Animal Care (SRAC) serves as the Institutional Animal Care and Use Committee (IACUC) for the Massachusetts General Hospital (MGH). SRAC reviewed and approved all procedures detailed in this paper.
Chemistry
2-iodo-6-nitrobenzoic acid (2) To a solution of Compound 1 (1.0 g, 5.49 mmol) in concentrated HCl (3 mL) and water (3 mL) was added NaNO2(0.46 g, 6.59 mmol) in H2O (3 mL) below 5 °C. Then the mixture was stirred at 5°C for 0.5 h and followed by adding KI (1.83 g, 10.98 mmol) in H2O (3 mL), heated to 90°C for 16 h. The reaction mixture was quenched with saturated NaHSO3 (50 mL), extracted with ethyl acetate (25 mL × 3). The aqueous layer was adjusted to pH = 2 by HCl aqueous (2 N), extracted with ethyl acetate (25 mL × 3). The combined organic layer was dried over Na2SO4, filtered, and concentrated afford the crude product. The crude product was purified using CombiFlash (0%–10% MeOH in DCM) to yield compound 2 (0.9 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ 14.00 (br, 1H), 8.352 − 8.330 (dd, J = 0.80, 8.80 Hz, 1H), 8.233 − 8.210 (dd, J = 0.80, 8.00 Hz, 1H), 7.495 − 7.454 (t, J = 8.4, 8.0 Hz, 1H). LC-MS [M+H]+ : 293.9. 2-nitro-6-(2H-1,2,3-triazol-2-yl)benzoic acid (3) To a mixture of compound 2 (0.9 g, 3.07 mmol) and K2CO3 (1.06 g, 7.68 mmol) in THF (15 mL) was added CuI (0.06 g, 0.30 mmol) and N1,N2-Dimethylethane-1,2-diamine (0.06 g, 0.61 mmol). 2H-1,2,3-triazole (0.43 g, 6.14 mmol) in THF (2 mL) was added. The reaction mixture was stirred at 65 °C for 16 h. After the complete consumption of compound 2, the solvent was removed, the residue was acidified to pH = 3 before 200 mL water was added. The aqueous layer was extracted with ethyl acetate (25 mL × 2). The combined organic layers were dried over Na2SO4 and evaporated. The crude product was purified using CombiFlash (0%–10% MeOH in DCM) to obtain compound 3 (0.35 g, crude, 49%). LC-MS [M+H]+: 235.1. (5-(4,6-dimethylpyrimidin-2-yl)hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)(2-nitro-6-(2H-1,2,3-triazol-2-yl)phenyl)methanone (4) To a solution of Compound 3 (350 mg, 1.49 mmol) in DMF (5 mL) was added DIPEA (580 mg, 4.48 mmol), HATU (853 mg, 2.24 mmol). The reaction mixture was stirred at room temperature for 30 min. Then 2-(4,6-dimethylpyrimidin-2-yl)octahydropyrrolo[3,4-c]pyrrole (358 mg, 1.64 mmol) was added. Then the reaction mixture was stirred at room temperature for 16 h. The resulting mixture was quenched with brine (15 mL) and extracted with ethyl acetate (25 mL × 2). The combined organic layers were dried over Na2SO4, filtered, concentrated in vacuum. The residue was purified by column chromatography eluting with DCM / MeOH (20 : 1) to yield the precursor 4 (120 mg, 18.5%). 1H NMR (400 MHz, CDCl3) δ 8.320 − 8.239 (m, 3H), 8.003 (s, 1H),7.931 − 7.885 (m, 1H), 6.417 − 6.407 (d, J = 4.00 Hz, 1H), 3.764 − 3.319 (m, 6H), 3.147 − 3.055 (m, 1H), 3.046 − 2.690 (m, 3H), 2.259 − 2.231(m, 6H). LC-MS [M+H]+: 435.2.
Log D Determination.
The general procedure for Log D determination assays as described in our previous literature.29, 30 Briefly, 10 μL Seltorexant DMSO solution (10 mM) was mixed with 100 μL octanol in PBS buffer (pH 7.4) and followed by shaking in a rotator for 1 hour at 30 rpm. After incubations, the concentrations of Seltorexant in octanol and water were measured by LC-MS (Agilent 6310 ion trap mass spectrometer). Then the Log D7.4 was calculated by Log [the ratio between the amount of test compound in n-octanol and PBS]. The assay was performed in triplicate.
Radiochemistry
The 18F-Fluoride solution (97% 18O enriched water, Sigma-Aldrich) was generated from the cyclotron (Siemens Eclipse HP) and directly loaded onto a QMA anion exchange cartridge (Chromafix® 30-PS-HCO3−). The cartridge with [18F]fluoride was washed with water and eluted into a vial with 3.5 mg/ml Kryptofix 2.2.2 (K222) solution in MeCN (1.2 mL) and 2.5 mg/ml K2CO3 solution in water (0.3 mL). Removal of the water was accomplished by azeotropic evaporation with anhydrous MeCN (1 ml each) under N2 three times. After the [18F]fluoride solution dried down, precursor 4 (2 mg) in DMSO (1 mL) was added to the vial and it was heated to 150 °C for 20 min. After adding 1.0 mL water, it was purified by injecting the solution to a semi-preparative HPLC (Agilent Eclipse XDB-C18 (5 μm, 250 mm × 9.4 mm), with a mobile phase of 68% H2O + 0.1% TFA/32% CH3CN, at the flow rate of 5.0 mL/min). [18F]Seltorexant was further reformulated by loading onto a solid-phase exchange (SPE) C-18 SepPak cartridge, rinsed with 5 mL water, 1 mL ethanol, and diluted by saline solution (0.9%, 9 mL). The radiochemical yield of [18F]Seltorexant was 7 – 9% (non-decay corrected, n = 6) and purity > 97% (measured with HPLC equipped with a UV detector and a gamma detector), and the specific activity (As) of [18F]Seltorexant was 110 – 130 GBq/μmol (EOS).
In vitro Autoradiography
The in vitro autoradiography assay has been described in our previous report.31 Briefly, the mice brain sections (sagittal, 20 μM) were pre-incubated with Tris-HCl buffer (50 mM) solution for 20 min, followed by incubation with [18F]Seltorexant (1 mCi/L, 50 mM Tris-HCl buffer). For blocking studies, unlabeled Seltorexant (10 μM) or compound 30 (10 μM) with radiotracer mixed in the incubation solution. Following incubation, mice brain sections were washed in ice-cold buffer and then dipped in ice-cold distilled water. Then the mice brain sections were dried at ambient temperature. An imaging plate (BAS-MS2025, GE Healthcare, NJ, USA) was exposed to the dried brain sections. Autoradiograms were obtained, and ROIs were carefully drawn with the reference of naked-eye observation. The gray value of images was measured by ImageJ software.
Rodent PET/CT acquisition
The general procedure for rodent PET/CT imaging studies was described previously.30, 32 Briefly, male C57BL/6 mice (6-month, n = 4 for each test group) were arranged in a Triumph PET/CT scanner (Gamma Medica, Northridge, CA) with inhalational isoflurane anesthesia (2%, Patterson Vet Supply, Inc., Greeley, CO, USA). The mice were administrated with 0.2 mL of saline with 5% [18F]Seltorexant via a lateral tail vein catheter at the beginning of PET acquisition. For the blocking study, mice were pretreatment with unlabeled Seltorexant (1.0 mg/kg) or compound 30 (1.0 mg/kg), or Cyclosporin A (0.5 mg/kg) before [18F]Seltorexant injection. Each PET scan lasted for 60 minutes and was followed by computed tomography (CT). PET data were reconstructed using a 3D-MLEM method resulting in full width at a half-maximum resolution of 1 mm.
Rodent PET/CT Image Analysis.
The reconstructed PET and CT images were processed by PMOD software (PMOD Technologies Ltd., Zurich, Switzerland). Anatomical volumes of interest (VOIs) were generated manually in spheres under the guide of high-resolution CT structural images. Time-activity curves (TACs) were exported as decay-corrected activity per unit volume. The TACs were expressed as percent injected dose per unit volume (%ID/cc) for analysis.
Results and discussion
Pharmacological and Physicochemical Properties of Seltorexant
For a CNS PET radiotracer, binding potential (BP, the ratio of receptor’s density (Bmax) to ligand-receptor binding affinity (Ki)) is crucial for visualization and quantitation of the objective target. Generally, the BP of an ideal CNS PET radioligand needs to be greater than 5.33 Seltorexant has been reported to exhibit high binding affinity and selectivity against OX2R, with Ki value of 10 nM and 80-fold selectivity for OX2R over OX1R.28 The Bmax of OX2R in the rat brain was tested within 40–140 nM,21 suggesting Seltorexant can be a potential PET imaging agent for OX2R imaging. Several physicochemical properties such as molecular weight, total polar surface area (tPSA), and lipophilicity can be used to predict the brain permeability of a candidate compound prior to radiolabeling. The tPSA and cLogP values of Seltorexant were determined in silico prediction with ChemBioDraw 16.0, and the Log D value was evaluated by means of the “shake flask method” (Figure 2). These physicochemical properties of Seltorexant are within the favorable range for sufficient brain permeability. Indeed, the brain permeability and binding specificity of Seltorexant were also evaluated by Bonaventure et al. by in vitro autoradiography study with [3H]EMPA.26 After orally administrated 30 mg/kg Seltorexant, high brain concentration and OX2R occupancy were observed in rat brain. As such, the pharmacokinetic and physicochemical properties of Seltorexant make it a potential PET probe candidate for OX2R imaging in the brain.
Figure 2.
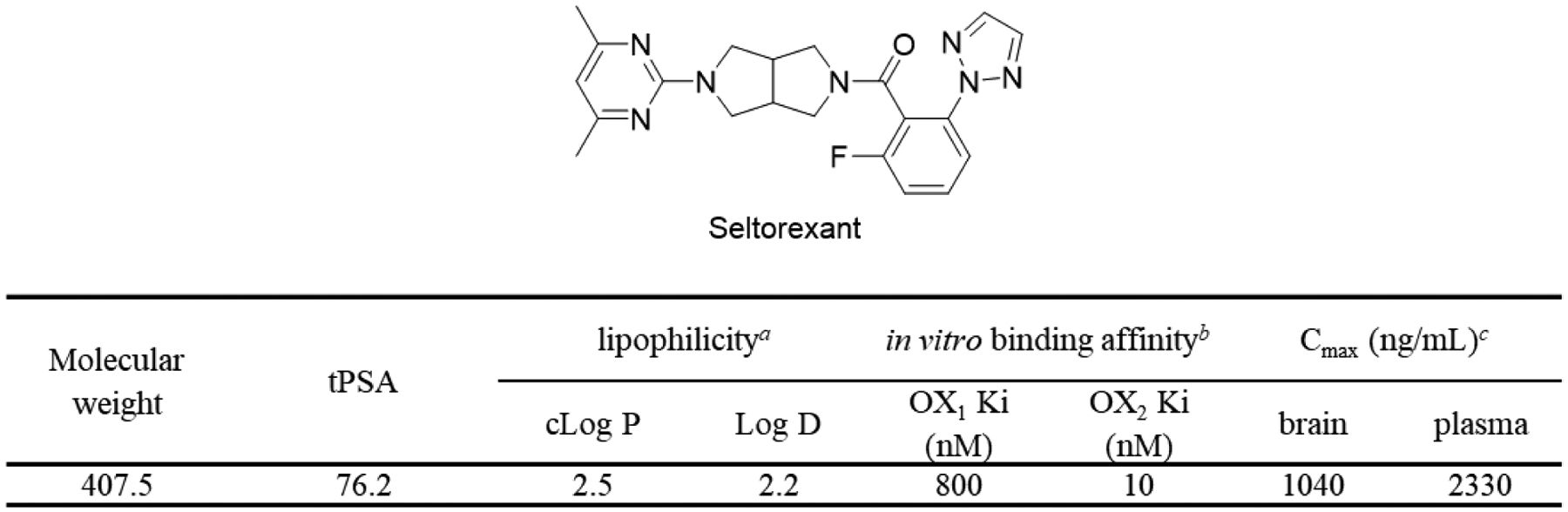
The pharmacological and physicochemical properties of Seltorexant. acLogP value of Seltorexant is calculated by ChemBioDraw 16.0; Log D values were quantified in n-octanol/phosphate buffer (pH 7.4) by the shake-flask method. bLetavic et al. reported the in vitro binding affinity of Seltorexant.28 cBonaventure et al. reported the pharmacokinetics of Seltorexant; single oral administration (p.o.) of Seltorexant at 30 mg/kg showed high concentration in rat brain.26
Chemistry and Radiochemistry
Aromatic nucleophilic radiofluorination is a commonly used method for the preparation of fluorine-18 labeled PET radiotracers.34, 35 The presence of fluorine atom in the structure of Seltorexant indicated it could be directly labeled with fluorine-18 without altering its molecular structure. Hence, for the radiosynthesis of [18F]Seltorexant, the radiolabeling precursor was synthesized by substituting the native fluorine atom of Seltorexant with a nitro group (as the leaving group), with amenability for fluorine-18 nucleophilic substitution. Precursor 4 was prepared by three steps (Scheme 1): 1) intermediate 2 was obtained by diazotization and iodization of starting material 1 in the presence of hydrochloric acid, sodium nitrite, and potassium iodide in 56% yield; 2) intermediate 2 was then reacted with 2H-1,2,3-triazole to give compound 3; 3) the nitro substitution precursor 4 for [18F]Seltorexant labeling was synthesized by condensation of intermediate 3 with 2-(4,6-dimethylpyrimidin-2-yl)octahydropyrrolo[3,4-c]pyrrole.
Scheme 1.
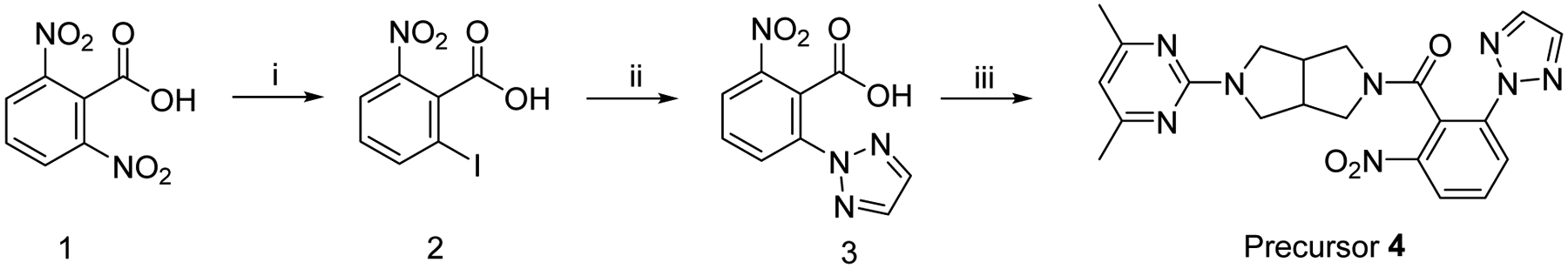
Synthesis of precursor 4. Reagents and conditions: (i) NaNO2, KI, in H2O, 90 °C, 16 h; (ii) 2H-1,2,3-triazole, N1,N2-Dimethylethane-1,2-diamine, K2CO3, CuI, THF, 90 °C, 16 h; (iii) 2-(4,6-dimethylpyrimidin-2-yl)octahydropyrrolo[3,4-c]pyrrole, DIPEA, HATU, DMF, room temperature, 16 h.
As outlined in Scheme 2, radiosynthesis of [18F]Seltorexant was carried out via reacting precursor 4 with [18F]fluoride in the presence of K2CO3 and Kryptofix K222 and DMSO at 150 °C for 20 minutes. The reaction mixture was then injected into a reverse semi-preparative HPLC for purification. The desired fraction [18F]Seltorexant was collected and reformulated for animal injection. The validation of [18F]Seltorexant was performed by coinjection with the unlabeled Seltorexant (purchased for MedChemEpxress) on an analytical HPLC (Figure S1). The radiochemical yield (RCY) was 7 – 9% (decay corrected to end of cyclotron bombardment (EOB), n = 6) after 90 minutes of average synthesis time; the molar activity was 110 – 130 GBq/μmol, the radiochemical purity was ≥97%.
Scheme 2.
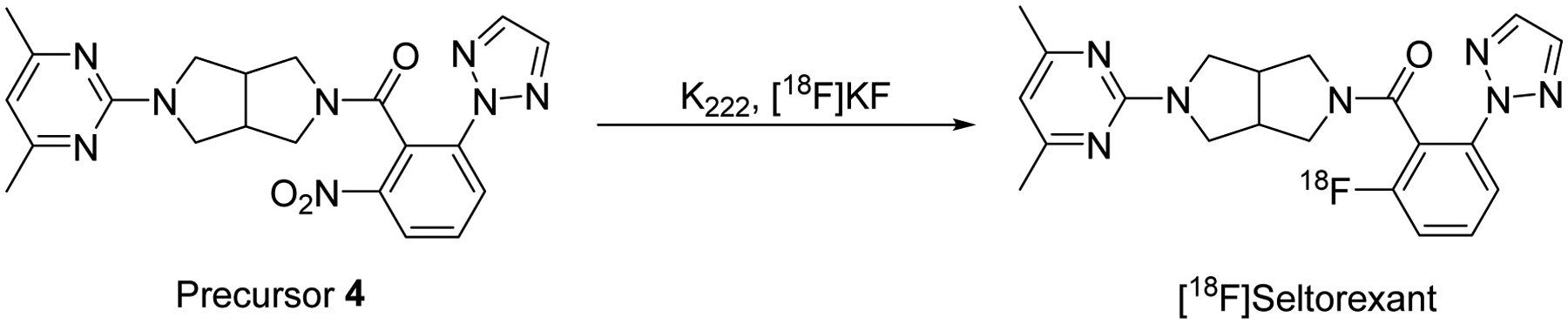
Radiosynthesis of [18F]Seltorexant. Radiolabeling condition: 4 (precursor, 3.0 mg), DMSO, K222, K2CO3, [18F]F−, 150°C, 20 min.
Ex Vivo Autoradiography Study
The ex vivo autoradiography studies of [18F]Seltorexant for mice brain sections were carried out to evaluate the OX2R binding specificity of [18F]Seltorexant. The structurally distinct OX2R antagonist, 9-(4,6-dimethylpyrimidin-2-yl)-2-((5-methoxy-1H-indol-3-yl)methyl)-2,9-diazaspiro[5.5]undecan-1-one (Compound 30, the structure showed in Figure 1), was used for the blocking study. The representative baseline and blocking (co-incubation with 10 μM compound 30) autoradiography images of sagittal mice brain sections with [18F]Seltorexant are outlined in Figure 3. Heterogeneous distribution of [18F]Seltorexant signal in the baseline was detected. In the whole brain, the binding was decreased by 27% in the presence of OX2R antagonist compound 30. Notably, a significant decrease in specific binding of [18F]Seltorexant in the cortical layer, hippocampus, thalamus, and striatum in the blocking was observed, indicating the specific binding sites for [18F]Seltorexant in these brain regions. The promising ex vivo results prompted us to move [18F]Seltorexant forward to an in vivo PET investigation.
Figure 3.
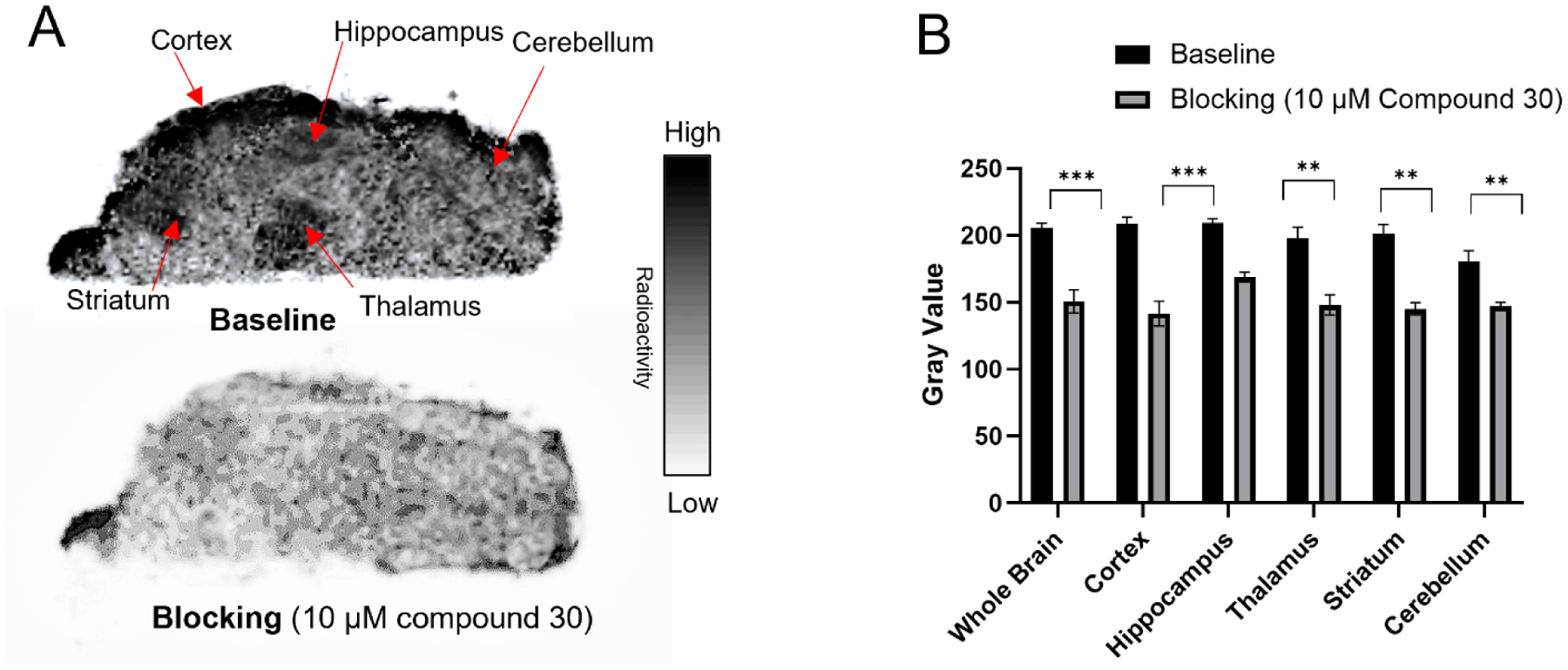
(A) Representative in vitro autoradiographic images of mice brains (sagittal); (B) the relative radioactive uptake in baseline and blocking. The baseline mice brain sections were treated with [18F]Seltorexant only, and the blocking mice brain sections were co-incubation with OX2R antagonist compound 30 (10 μM). The gray value data were expressed as mean ± SD, n = 4; Asterisks indicate statistical significance. *p < 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Rodents PET-CT imaging of [18F]Seltorexant
Next, PET-CT imaging in mice (C57BL/6 male mice, 6-month old) was conducted to evaluate the in vivo properties of [18F]Seltorexant as an OX2R PET probe. 100–150 μCi (0.1–0.15 mL, 31–46 pmol) of [18F]Seltorexant was administered by intravenous bolus injection and then followed a 60 minutes dynamic PET imaging scanning and 10 minutes computed tomography (CT). We first examined the brain permeability of [18F]Seltorexant. In general, for a CNS PET radiotracer, the required brain uptake (determined by injected dose per cc (%ID/cc)) within 5 min of injection in rodents is at least 0.1%.33 In vivo PET imaging of [18F]Seltorexant in mice demonstrated that [18F]Seltorexant had a suitable BBB penetration with a maximum %ID/cc of 3.4 at 2 minutes post-injection in the whole brain (Figure 4B). The time−radioactivity curve (TAC) showed that [18F]Seltorexant can rapidly penetrate the brain, bind with OX2R, and wash out gradually, suggesting reasonable brain kinetics. Additional biodistribution analysis of [18F]Seltorexant in mice brain was performed with FUSION module in image analysis software PMOD (PMOD 4.01, PMOD Technologies Ltd., Zurich, Switzerland). Relatively high radioactivity uptake was observed in OX2R-rich regions, including the cortex and hippocampus. In contrast, low radioactivity uptake was found in the cerebellum, where OX2R was less expressed (Figure 4C), corresponding with previous results on OX2R imaging.23 The regional brain biodistribution analysis results of [18F]Seltorexant demonstrated the OX2R biodistribution in the brain regions.
Figure 4.
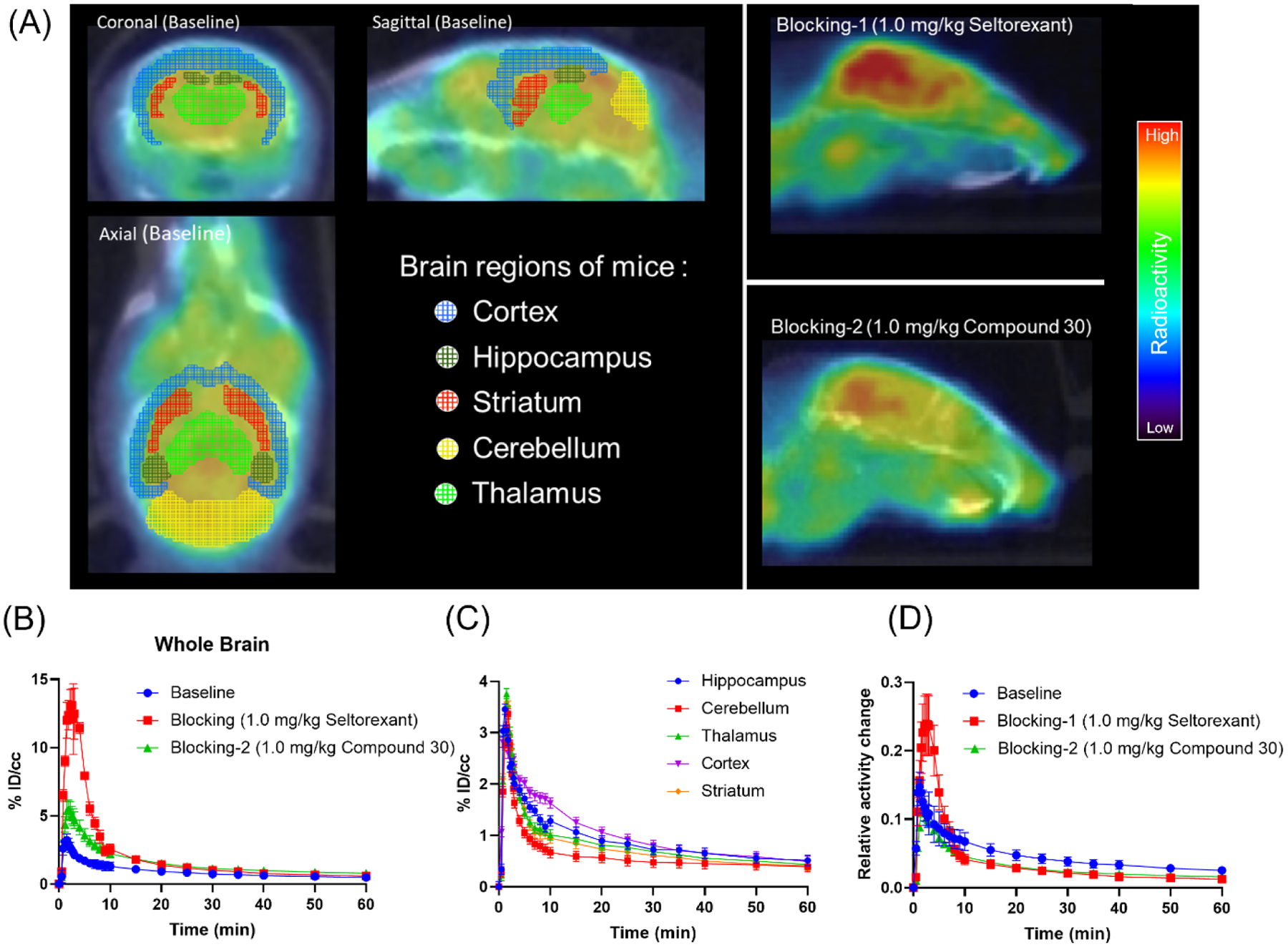
(A) Representative baseline and blocking PET/CT images of [18F]Seltorexant focus on the mice brain (0–60 min, the baseline is shown as horizontal, coronal, and sagittal plane; the blockings are shown as a sagittal plane); (B) the baseline and blocking time-activity curves of [18F]Seltorexant in the mice whole brain; (C) Time-radioactivity curves for [18F]Seltorexant in the hippocampus, cerebellum, thalamus, cortex, and striatum (n = 3); (D) the normalized baseline and blocking time-activity curves [18F]Seltorexant (normalized the brain uptake curves with the highest radioactivity in the blood at each time point).
In the blocking studies, mice were pre-administrated unlabeled Seltorexant (1.0 mg/kg) and compound 30 (1.0 mg/kg) 5 minutes before radiotracer administration to confirm the specific binding of [18F]Seltorexant. The TACs showed a remarkably increased brain uptake of [18F]Seltorexant when pretreated with blocking agents (Figure 4B). The blocking effects may cause the increased free radiotracer in plasma, resulting in increased total uptake of radiotracer in the brain compared with baseline. Consequently, we normalized the brain uptake with the highest radioactivity in the blood at each time point. From the normalized TAC curves, blocking effects were observed in both Seltorexant and compound 30 pretreated mice groups (Figure 4D). Together with in vitro autoradiography studies, these results indicated the binding specificity of [18F]Seltorexant for OX2R.
The significantly increased brain uptake of [18F]Seltorexant in the self-blocking mice may be attributed to its interaction with efflux transporters, such as the P-glycoprotein (P-gp), located at the BBB. To verify this hypothesis, we carried out PET imaging studies of [18F]Seltorexant in mice pretreatment with the competitive P-gp inhibitor Cyclosporin A (CsA, 0.5 mg/kg). As we assumed, compared with baseline, significantly increased uptake of [18F]Seltorexant was observed in the CsA pretreatment mice brain, which indicated that Seltorexant could interact with P-gp at the BBB (Figure 5). Of the previously reported OXR PET probes, most had low brain penetration and failed to further translation for OX2R imaging due to their interaction with P-gp, such as [11C]EMPA22 and [11C]9a23. Though [18F]Seltorexant appears to interact with P-gp, it maintained good brain uptake and binding specificity, which could guide further OX2R PET probes optimization and development.
Figure 5.
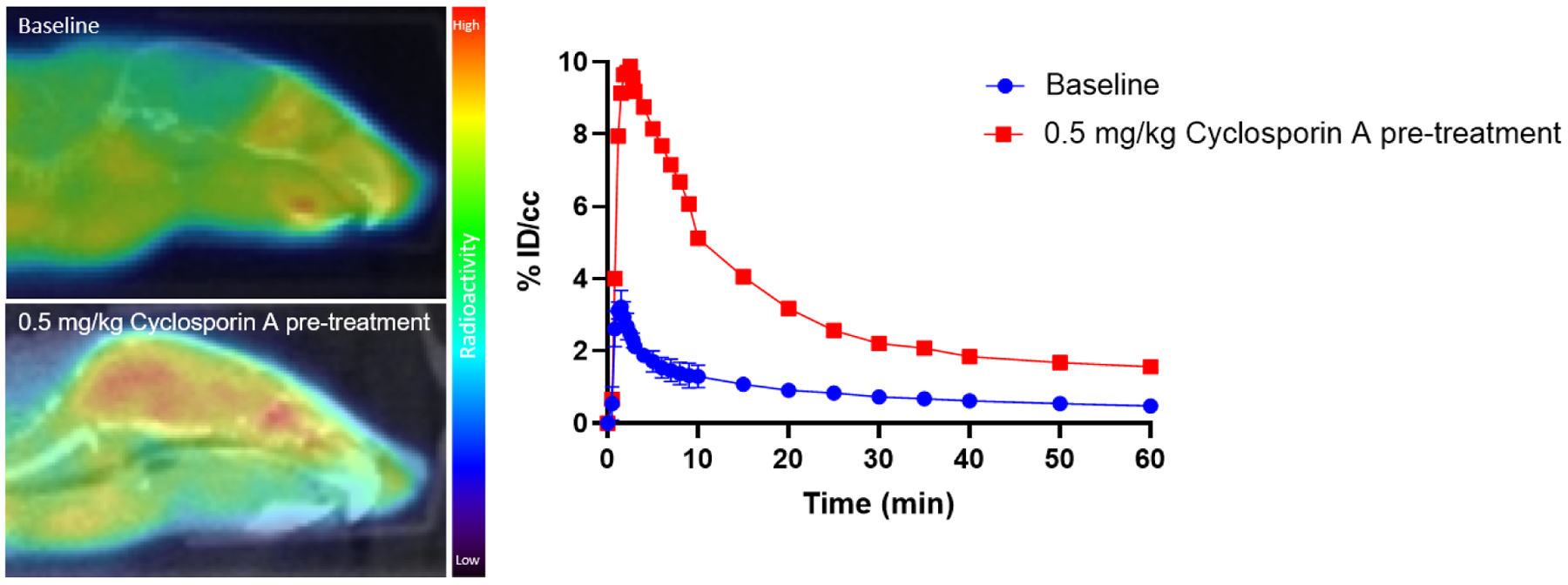
The representative baseline and Cyclosporin A pretreatment PET/CT images with [18F]Seltorexant focused on mice brain and time-activity curves of [18F]Seltorexant in the whole brain of mice.
Conclusion
In this work, we screened known selective OX2R antagonists and found that Seltorexant has the potential to be an OX2R imaging probe. We successfully synthesized the labeling precursor and obtained [18F]Seltorexant. Ex vivo autoradiography studies for mice brain sections indicated the good binding specificity of [18F]Seltorexant toward OX2R. In vivo PET imaging in rodents showed that [18F]Seltorexant had a suitable BBB penetration for OX2R imaging in the brain. The regional brain biodistribution analysis and blocking studies showed that [18F]Seltorexant had good binding selectivity and specificity. Pretreatment with competitive P-gp inhibitor CsA increased the brain uptake of [18F]Seltorexant, suggesting that [18F]Seltorexant might be the substrate of the efflux transporter. In conclusion, our preliminary results showed [18F]Seltorexant is a potential PET probe for OX2R imaging in the brain, which could assist further OX2R PET probes development.
Supplementary Material
OX2R PET imaging probe [18F]Seltorexant was successfully synthesized and characterized.
[18F]Seltorexant exhibited good binding specificity for OX2R in ex vivo autoradiography studies.
In vivo PET imaging of [18F]Seltorexant in rodents showed good brain uptake and specificity for OX2R.
Acknowledgments
This work was supported by NIH DA048123 (C.W.). The imaging studies were carried out at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Regional Resource supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program; specifically, grant numbers: S10OD025234, S10RR017208, S10RR026666, S10RR022976, S10RR019933, S10RR023401 and S10OD023517. We are grateful to Athinoula A. Martinos Center staff for their technical support and assistance with cyclotron operation and radionuclide production.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
We have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Sakurai T; Amemiya A; Ishii M; Matsuzaki I; Chemelli RM; Tanaka H; Williams SC; Richarson JA; Kozlowski GP; Wilson S; Arch JR; Buckingham RE; Haynes AC; Carr SA; Annan RS; McNulty DE; Liu WS; Terrett JA; Elshourbagy NA; Bergsma DJ; Yanagisawa M, Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 1998, 92 (5), 573–585. [DOI] [PubMed] [Google Scholar]
- 2.Sakurai T, Orexins and orexin receptors: implication in feeding behavior. Regul Pept 1999, 85 (1), 25–30. [DOI] [PubMed] [Google Scholar]
- 3.Nishino S, Hypothalamus, hypocretins/orexin, and vigilance control. Handb Clin Neurol 2011, 99, 765–782. [DOI] [PubMed] [Google Scholar]
- 4.Roecker AJ; Cox CD; Coleman PJ, Orexin Receptor Antagonists: New Therapeutic Agents for the Treatment of Insomnia. J Med Chem 2016, 59 (2), 504–530. [DOI] [PubMed] [Google Scholar]
- 5.Brisbare-Roch C; Dingemanse J; Koberstein R; Hoever P; Aissaoui H; Flores S; Mueller C; Nayler O; van Gerven J; de Haas SL; Hess P; Qiu C; Buchmann S; Scherz M; Weller T; Fischli W; Clozel M; Jenck F, Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med 2007, 13 (2), 150–155. [DOI] [PubMed] [Google Scholar]
- 6.Trivedi P; Yu H; MacNeil DJ; Van der Ploeg LH; Guan XM, Distribution of orexin receptor mRNA in the rat brain. FEBS Lett 1998, 438 (1–2), 71–75. [DOI] [PubMed] [Google Scholar]
- 7.Marcus JN; Aschkenasi CJ; Lee CE; Chemelli RM; Saper CB; Yanagisawa M; Elmquist JK, Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol 2001, 435 (1), 6–25. [DOI] [PubMed] [Google Scholar]
- 8.Gatfield J; Brisbare-Roch C; Jenck F; Boss C, Orexin receptor antagonists: a new concept in CNS disorders? ChemMedChem 2010, 5 (8), 1197–1214. [DOI] [PubMed] [Google Scholar]
- 9.Coleman PJ; Schreier JD; Roecker AJ; Mercer SP; McGaughey GB; Cox CD; Hartman GD; Harrell CM; Reiss DR; Doran SM; Garson SL; Anderson WB; Tang C; Prueksaritanont T; Winrow CJ; Renger JJ, Discovery of 3,9-diazabicyclo[4.2.1]nonanes as potent dual orexin receptor antagonists with sleep-promoting activity in the rat. Bioorg Med Chem Lett 2010, 20 (14), 4201–4205. [DOI] [PubMed] [Google Scholar]
- 10.Bingham MJ; Cai J; Deehan MR, Eating, sleeping and rewarding: orexin receptors and their antagonists. Curr Opin Drug Discov Devel 2006, 9 (5), 551–559. [PubMed] [Google Scholar]
- 11.Andrews SP; Aves SJ; Christopher JA; Nonoo R, Orexin Receptor Antagonists: Historical Perspectives and Future Opportunities. Curr Top Med Chem 2016, 16 (29), 3438–3469. [DOI] [PubMed] [Google Scholar]
- 12.Perrey DA; Zhang Y, Therapeutics development for addiction: Orexin-1 receptor antagonists. Brain Res 2020, 1731, 145922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herring WJ; Snyder E; Budd K; Hutzelmann J; Snavely D; Liu K; Lines C; Roth T; Michelson D, Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology 2012, 79 (23), 2265–2274. [DOI] [PubMed] [Google Scholar]
- 14.Winrow CJ; Gotter AL; Cox CD; Doran SM; Tannenbaum PL; Breslin MJ; Garson SL; Fox SV; Harrell CM; Stevens J; Reiss DR; Cui D; Coleman PJ; Renger JJ, Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet 2011, 25 (1–2), 52–61. [DOI] [PubMed] [Google Scholar]
- 15.Lebold TP; Bonaventure P; Shireman BT, Selective orexin receptor antagonists. Bioorg Med Chem Lett 2013, 23 (17), 4761–4769. [DOI] [PubMed] [Google Scholar]
- 16.Dugovic C; Shelton JE; Aluisio LE; Fraser IC; Jiang X; Sutton SW; Bonaventure P; Yun S; Li X; Lord B; Dvorak CA; Carruthers NI; Lovenberg TW, Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J Pharmacol Exp Ther 2009, 330 (1), 142–151. [DOI] [PubMed] [Google Scholar]
- 17.Willmann JK; van Bruggen N; Dinkelborg LM; Gambhir SS, Molecular imaging in drug development. Nat Rev Drug Discov 2008, 7 (7), 591–607. [DOI] [PubMed] [Google Scholar]
- 18.Malherbe P; Borroni E; Gobbi L; Knust H; Nettekoven M; Pinard E; Roche O; Rogers-Evans M; Wettstein JG; Moreau JL, Biochemical and behavioural characterization of EMPA, a novel high-affinity, selective antagonist for the OX(2) receptor. Br J Pharmacol 2009, 156 (8), 1326–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phelps ME, PET: the merging of biology and imaging into molecular imaging. J Nucl Med 2000, 41 (4), 661–681. [PubMed] [Google Scholar]
- 20.Watanabe H; Fukui K; Shimizu Y; Idoko Y; Nakamoto Y; Togashi K; Saji H; Ono M, Synthesis and biological evaluation of F-18 labeled tetrahydroisoquinoline derivatives targeting orexin 1 receptor. Bioorg Med Chem Lett 2019, 29 (13), 1620–1623. [DOI] [PubMed] [Google Scholar]
- 21.Wang C; Wilson CM; Moseley CK; Carlin SM; Hsu S; Arabasz G; Schroeder FA; Sander CY; Hooker JM, Evaluation of potential PET imaging probes for the orexin 2 receptors. Nucl Med Biol 2013, 40 (8), 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C; Moseley CK; Carlin SM; Wilson CM; Neelamegam R; Hooker JM, Radiosynthesis and evaluation of [11C]EMPA as a potential PET tracer for orexin 2 receptors. Bioorg Med Chem Lett 2013, 23 (11), 3389–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oi N; Suzuki M; Terauchi T; Tokunaga M; Nakatani Y; Yamamoto N; Fukumura T; Zhang MR; Suhara T; Higuchi M, Synthesis and evaluation of novel radioligands for positron emission tomography imaging of the orexin-2 receptor. J Med Chem 2013, 56 (16), 6371–6385. [DOI] [PubMed] [Google Scholar]
- 24.Bai P; Bai S; Placzek MS; Lu X; Fiedler SA; Ntaganda B; Wey HY; Wang C, A New Positron Emission Tomography Probe for Orexin Receptors Neuroimaging. Molecules 2020, 25 (5), 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao M; Wang M; Zheng QH, Synthesis of [(11)C]MK-1064 as a new PET radioligand for imaging of orexin-2 receptor. Bioorg Med Chem Lett 2016, 26 (15), 3694–3699. [DOI] [PubMed] [Google Scholar]
- 26.Bonaventure P; Shelton J; Yun S; Nepomuceno D; Sutton S; Aluisio L; Fraser I; Lord B; Shoblock J; Welty N; Chaplan SR; Aguilar Z; Halter R; Ndifor A; Koudriakova T; Rizzolio M; Letavic M; Carruthers NI; Lovenberg T; Dugovic C, Characterization of JNJ-42847922, a Selective Orexin-2 Receptor Antagonist, as a Clinical Candidate for the Treatment of Insomnia. J Pharmacol Exp Ther 2015, 354 (3), 471–82. [DOI] [PubMed] [Google Scholar]
- 27.Brooks S; Jacobs GE; de Boer P; Kent JM; Van Nueten L; van Amerongen G; Zuiker R; Kezic I; Luthringer R; van der Ark P; van Gerven JM; Drevets W, The selective orexin-2 receptor antagonist seltorexant improves sleep: An exploratory double-blind, placebo controlled, crossover study in antidepressant-treated major depressive disorder patients with persistent insomnia. J Psychopharmacol 2019, 33 (2), 202–209. [DOI] [PubMed] [Google Scholar]
- 28.Letavic MA; Bonaventure P; Carruthers NI; Dugovic C; Koudriakova T; Lord B; Lovenberg TW; Ly KS; Mani NS; Nepomuceno D; Pippel DJ; Rizzolio M; Shelton JE; Shah CR; Shireman BT; Young LK; Yun S, Novel Octahydropyrrolo[3,4-c]pyrroles Are Selective Orexin-2 Antagonists: SAR Leading to a Clinical Candidate. J Med Chem 2015, 58 (14), 5620–5636. [DOI] [PubMed] [Google Scholar]
- 29.Bai P; Bai S; Placzek MS; Lu X; Fiedler SA; Ntaganda B; Wey HY; Wang C, A New Positron Emission Tomography Probe for Orexin Receptors Neuroimaging. Molecules 2020, 25 (5), 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai P; Lu X; Lan Y; Chen Z; Patnaik D; Fiedler S; Striar R; Haggarty SJ; Wang C, Radiosynthesis and in vivo evaluation of a new positron emission tomography radiotracer targeting bromodomain and extra-terminal domain (BET) family proteins. Nucl Med Biol 2020, 84 (2), 96–101. [DOI] [PubMed] [Google Scholar]
- 31.Bai P; Wey HY; Patnaik D; Lu X; Lan Y; Rokka J; Stephanie F; Haggarty SJ; Wang C, Positron emission tomography probes targeting bromodomain and extra-terminal (BET) domains to enable in vivo neuroepigenetic imaging. Chem Commun 2019, 55 (86), 12932–12935. [DOI] [PubMed] [Google Scholar]
- 32.Bai P; Lan Y; Wang H; Chen Z; Fiedler S; Striar R; Lu X; Wang C, Development of a Novel Positron Emission Tomography (PET) Radiotracer Targeting Bromodomain and Extra-Terminal Domain (BET) Family Proteins. Front Mol Biosci 2020, 7 (2), 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van de Bittner GC; Ricq EL; Hooker JM, A philosophy for CNS radiotracer design. Acc Chem Res 2014, 47 (10), 3127–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z; Conti PS, Radiopharmaceutical chemistry for positron emission tomography. Adv Drug Deliv Rev 2010, 62 (11), 1031–1051. [DOI] [PubMed] [Google Scholar]
- 35.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH, Chemistry for Positron Emission Tomography: Recent Advances in (11) C-, (18) F-, (13) N-, and (15) O-Labeling Reactions. Angew Chem Int Ed Engl 2019, 58 (9), 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.