

Abstract
Pulmonary surfactant is a mixture of lipids and proteins, consisting of 90% phospholipid, and 10% protein by weight, found predominantly in pulmonary alveoli of vertebrate lungs. Two minor components of pulmonary surfactant phospholipids, phosphatidylglycerol (PG) and phosphatidylinositol (PI), are present within the alveoli at very high concentrations, and exert anti-inflammatory effects by regulating multiple Toll like receptors (TLR2/1, TLR4, and TLR2/6) by antagonizing cognate ligand-dependent activation. POPG also attenuates LPS-induced lung injury in vivo. In addition, these lipids bind directly to RSV and influenza A viruses (IAVs) and block interaction between host cells and virions, and thereby prevent viral replication in vitro. POPG and PI also inhibit RSV and IAV infection in vivo, in mice and ferrets. The lipids markedly inhibit SARS-CoV-2 infection in vitro. These findings suggest that both POPG and PI have strong potential to be applied as both prophylaxis and post-infection treatments for problematic respiratory viral infections.
Keywords: Antiviral, Innate immunity, Pulmonary surfactant phospholipids, Toll-like receptors (TLRs), Respiratory viruses
1. Fundamentals of pulmonary surfactant
The pulmonary surfactant system of the lung is an extracellular lipid and protein complex, present at the alveolar air/tissue interface, which regulates both the biophysical properties of the alveolar compartment, and the innate immune system of the organ [1–5]. Pulmonary surfactant was initially identified as a lipoprotein mixture of 90% phospholipid and 10% protein (Fig. 1) [5–9]. The major constituents of pulmonary surfactant are phospholipids, predominantly phosphatidylcholine (PC) [6,10]. Within the phosphatidylcholine class, the most abundant phospholipid, dipalmitoyl-PC (DPPC) is responsible for the surface tension lowering properties of the surfactant complex [1,10,11]. The total phospholipid concentration in surfactant is remarkably high (~50 mg/ml) [5,10,11]. Phosphatidylglycerol (PG) and phosphatidylinositol (PI) are minor phospholipids within the surfactant complex (~10% and ~5% by weight, respectively), but their absolute concentrations within the alveolar compartment exceed that found in any other tissue or mucosal surface by more than 100-fold. The major molecular species of surfactant associated PG in humans is palmitoyl-oleoyl-phosphatidylglycerol (POPG) [5,9], and the major molecular species for PI is dioleoyl-phosphatidylinositol [6,9]. The functions of PG and PI in surfactant have long been enigmatic, but recent studies now show they play a critical role in regulating innate immunity [2]. Recent findings demonstrate that POPG and PI function as potent antagonists of TLRs 1, 2,4 and 6 [2,16,18]. The lipids complement the innate immune activity of the pulmonary surfactant proteins, SP-A and SP-D [5,11,19–21].
Fig. 1.
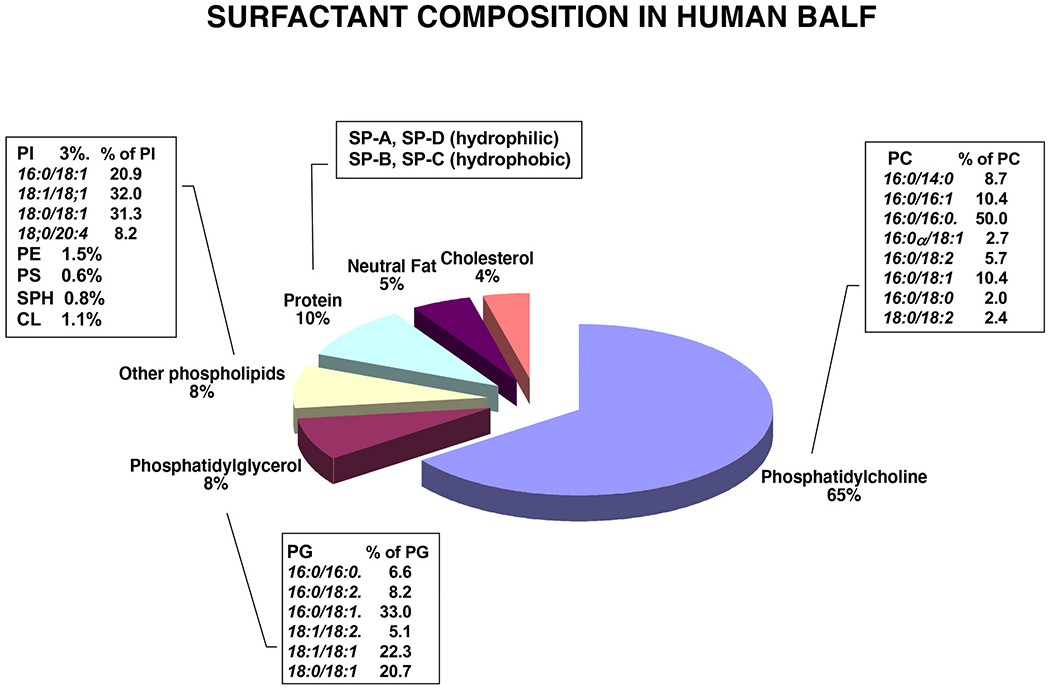
Constituents of Pulmonary Surfactant.
The surfactant complex consists mainly of phospholipids (~90%) and to a lesser extent proteins (~10%). The proteins consist of the surfactant proteins (SP-A, SP-B, SP-C and SP-D) with SP-B and SP-C being extraordinarily hydrophobic, and SP-A and SP-D being hydrophilic and highly oligomeric. The serum proteins found in the complex, consist of immunoglobulins (IgA and IgG) and albumin. Uteroglobin (CCL10) is also found associated with the complex. The lipids consist mostly of the phospholipid classes, Phosphatidylcholine (PC), phosphatidylglycerol (PG) and phosphatidylinositol (PI) and the molecular species within these classes are shown in the inset tables. Minor-Trace amounts of sphingomyelin, PE, glycosphingolipids, triacylglycerols and cholesterol are also present. In Fig. 1, the major molecular species of PC is the disaturated 16:0/16:0 species, responsible for the surface tension lowering properties of the complex. Also, from the graphic in Fig. 1, the major molecular species of PG is the 16:0/18:1 species. As shown in the Fig. 1 graphic, the 18:1/18:1 molecular species of PI predominates [9].
Pulmonary surfactant also harbors specific proteins, SP-A, SP-B, SP-C and SP-D, which are associated with extracellular surfactant lipids in the lung (Fig. 1) [1,6,10,11]. SP-B and SP-C are extraordinarily hydrophobic, and play essential roles in biophysical functions of pulmonary surfactant. Both hydrophobic proteins promote the highly efficient adsorption of extracellular surfactant lipid to the air-liquid interface, that is required to reduce alveolar surface tension [1] and prevent collapse [1,10,11]. SP-A and SP-D are hydrophilic and highly oligomeric, and recognized to mediate multiple host defense functions of surfactant [3,4,10,11,19,22]. SP-A and SP-D belong to the class of collagenous domain containing lectins (collectins). The collectins belong to a small family of soluble pattern recognition binding molecules containing collagenous regions and C-type lectin domains. The carboxy-terminal domains of the pulmonary collectins all have C-type (calcium-dependent) lectin activity. The lectin domains mediate the interaction of collectins with a wide variety of pathogens. The most well-understood consequence of this interaction is pathogen opsonization and enhanced phagocytosis [5,11].
Alveolar type II cells are the main source of the surfactant lipids and proteins in the lung, and pulmonary surfactant is uniquely synthesized, stored, and secreted by these cells [10,11]. Intracellular surfactant is packaged in specialized organelles known as the lamellar body (LB) [10,11]. Secreted LBs transform into tubular myelin (TM) before absorption of the lipids and hydrophobic proteins to the air-liquid interface of the alveolar hypophase. SP-B facilitates the formation of interfacial films enriched in dipalmitoyl- phosphatidylcholine, which reduce surface tension and prevents alveolar collapse. Pulmonary surfactant components are partially extruded into the mucus layer of conducting airways, and larger airways by mechanical processes and subsequently cleared by mucociliary escalator function [5,10,11].
In previous publications, PG and/or PI interact with surfactant proteins to play crucial roles in the pulmonary surfactant system [23–25]. PG interacts with SP-B and SP-C to support surfactant phospholipid absorption into surfactant films [23,26]. PG and PI facilitate the interactions between SP-B and surfactant lipids to stabilize surfactant films during dynamic compression of the lung [27]. PI binds to SP-D and contributes to regulate surfactant uptake by alveolar type 2 cells [23,28].
SP-A and SP-D are recognized as mediators of host defense and inhibit infections by multiple pathogens (e.g., bacteria, fungi, and viruses) [19,29,30] and regulate interaction with allergens [10,11,29]. Both proteins also interact with multiple cells of the immune system and regulate innate immune responses in the lung. SP-A and SP-D directly bind to Toll-like receptors (TLRs) and can inhibit proinflammatory responses induced by TLRs [11,31,32]. Selected surfactant components including SP-A, SP-D, and phospholipids are also present in the mucus layer of large airways [10,11].
1.1. Genetic disorders of the pulmonary surfactant system and pulmonary diseases
Genetic disorders of the pulmonary surfactant system cause diffuse alveolar diseases (e.g., idiopathic pulmonary fibrosis (IPF), pulmonary alveolar proteinosis (PAP), chronic pulmonary microlithiasis) [29,33,34]. Mutations in genes for SP-A, SP-B, and SP-C, are linked to acute respiratory distress syndrome, or interstitial lung disease in neonates, children and adults [34]. Mutations in genes encoding SP-A are associated with familial interstitial pneumonia [22,35] and idiopathic interstitial pneumonia in adults [36]. Mutations in genes encoding SP-C (SFTPC) and telomerase (TERT), also have been linked to familial interstitial pneumonia [34]. Hereditary SP-B and SP-C disorders cause respiratory distress syndrome in neonates [34]. ATP-Binding cassette A3 (ABCA3) plays important roles in intracellular lipid transport and surfactant packaging in the biogenesis of lamellar bodies [14,34,37]. The dysfunction in ABCA3 genes, results in respiratory failure in neonates and interstitial lung diseases in young children [36,38–40]. Most infants with mutations and dysfunction in the ABCA3 genes have poor prognosis, and often die within the first month of life [38].
Mutations in genes related to surfactant catabolism cause pulmonary alveolar proteinosis and alveolar pulmonary microlithiasis. Granulocyte-macrophage colony-stimulating factor (GM-CSF) promotes alveolar macrophages to clear, and/or metabolize surfactant lipids and proteins. Dysfunction of the β-chain, or the α-chain of the GM-CSF receptor, were associated with the pathogenesis of pulmonary alveolar proteinosis (PAP) [41,42]. The gene encoding solute carrier family 34 member 2 (SCL34A2), produces a sodium-dependent phosphate transporter that is highly expressed in type II cells in the lung [43,44]. Mutation in SCL34A is linked to alveolar pulmonary microlithiasis [43,44]. The dysfunction of SCL34A2 results in accumulation of calcium phosphate in alveoli and results in the aberrant degradation of surfactant phospholipids.
1.2. Collectin gene polymorphisms and host defense
Human surfactant protein polymorphisms impact host defense [45]. Human SP-A consists of SP-A1 and SP-A2 proteins that are products of SP-A1 (SFTPA1) and SP-A2 genes (SFTPA2) [23,46,47]. The products of the variants of the genes correlate with respiratory disease severity and susceptibility to infectious diseases [29,46]. SP-D levels in serum are associated with SP-D (SFTPD) gene polymorphisms and respiratory diseases [48].
SP-A1 polymorphisms correlate with risk of respiratory distress syndrome in neonates (RDS) and prognosis for IPF [33,45,46,49]. SP-A2 primary structural variants (Asn9Thr, Ala91Pro, Gln223Lys) are also linked to allergic bronchopulmonary aspergillosis (ABPA), pulmonary tuberculosis, and susceptibility to RSV infection [45]. SP-D polymorphisms (nonsynonymous, Met11Thr, Ala160Thr, Ser270Thr, and a synonymous Ala286Ala nucleotide variant) correlate with incidence of RSV infection in children, and pulmonary, allergic reactions [45].
1.3. The alteration of levels of pulmonary surfactant and pulmonary diseases
The alteration of levels of pulmonary surfactant proteins and phospholipids in bronchoalveolar lavage fluid (BALF) correlate with susceptibility, severity, and prognosis of several lung diseases including asthma [29,33,45,49], chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD) (e.g. sarcoidosis, idiopathic interstitial pneumonia (IPF), hypersensitive pneumonitis (HP)), and infectious diseases associated with acute respiratory distress syndrome (ARDS) [10,29,33]. As shown in Fig. 1, one of the minor pulmonary surfactant phospholipids, PG, is present in the lung at high concentration, and its levels strongly correlate with the prognosis of patients with pulmonary fibrosis (IPF) [33]. The level of SP-A in BALF is also predictive of survival of patients with IPF [29,33,49]. Significant changes in both lavage and serum levels of SP-A and SP-D have been described in ARDS [29,50]. SP-A and SP-D play important roles in the pathogenesis of asthma by regulating recognition of allergens, the cellular responses to them, and the clearance of allergens [29,45,51]. The levels of SP-A and SP-D are increased in the BALF of patients with asthma [10,51]. Allergen challenge induces increased amounts of SP-D in the serum of patients with asthma [52], and altered pulmonary surfactant phospholipid composition recovered in BALF [29,53].
2. Anti-inflammatory effects of pulmonary surfactant phospholipids
2.1. Structure similarity of PG and PI
PG and PI share structural similarity as shown Fig. 2. [54]. The PG molecule contains hydrophobic moieties, and a three carbon head group substituted with hydroxyl moieties [55]. PI harbors a 6-carbon cyclic headgroup and each carbon contains a hydroxyl substituent [54,56]. The PG structure is superimposable upon the structure of PI. Based on their structural similarity, we speculate that PG and PI have similar anti-inflammatory functions against TLR activation, dependent upon their polar headgroup structure [2,57].
Fig. 2.
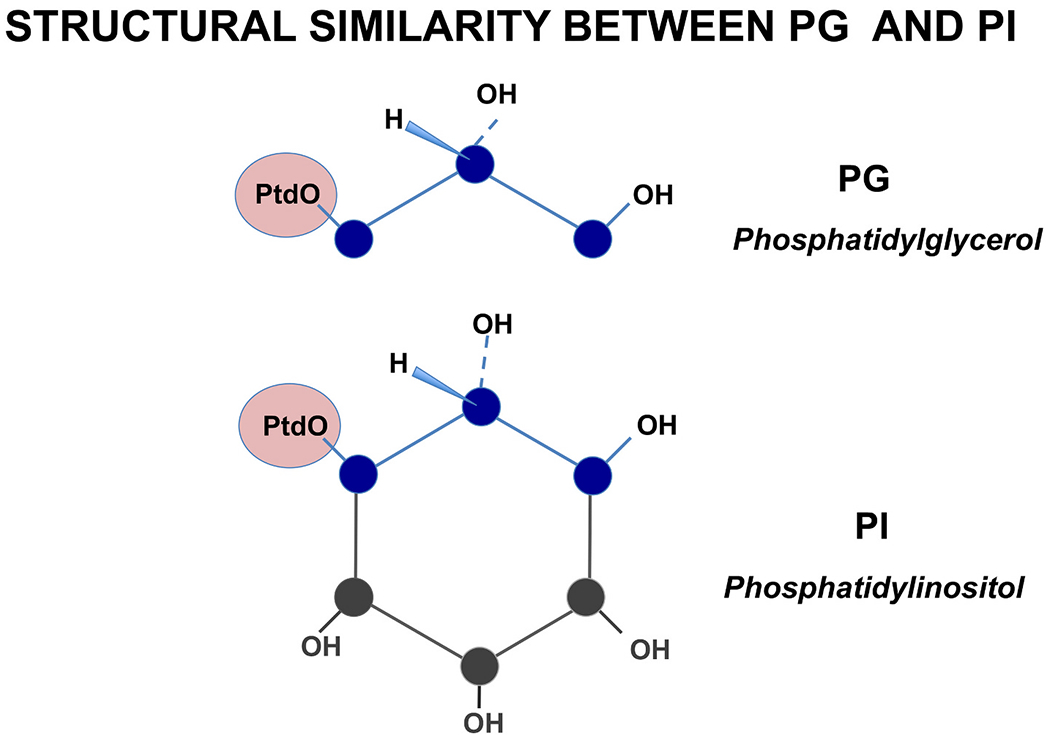
Phosphatidylglycerol (PG) and phosphatidylinositol (PI) share structural similarity.
The Schematic structures for PG and PI are shown, with emphasis placed upon the polar headgroups and their similarities. PtdO is the standard abbreviation for the phosphatidate moiety (containing a glycerolphosphate backbone to which is esterified two fatty acids) found in both the PG and PI classes [54] [55] [56].
2.2. Biological functions of pulmonary surfactant phospholipids in the lung
The major components of pulmonary surfactant are phospholipids (Fig. 1). Two minor components of pulmonary surfactant phospholipids, are phosphatidylglycerol (PG) (5–10%) and phosphatidylinositol (PI) (3–6%) (Fig. 2), and their local concentrations within the alveoli are very high compared to other organs. The concentration of PG and PI within the alveolar hypophase are 5-10 mg/ml and 2 mg/ml, respectively. But, their biological functions have long remained enigmatic. In our research, we found PG and PI can antagonize activation of multiple TLRs including TLR4, TLR1/2 and TLR2/6 [57].
2.3. POPG and PI inhibit bacterial lipopolysaccharide-induced inflammation triggered by the TLR4 pathway
In published reports, native pulmonary surfactant phospholipids, PG, PI and CL (Fig. 1) can antagonize the innate immune responses induced by TLR4 activation (e.g., Lipopolysaccharide (LPS) stimulation) [20,58].
Especially, POPG and PI strongly inhibit TLR4 activation induced by LPS [2,12,57] which is an outer membrane component of Gram-negative bacteria. It binds to LPS-binding protein (LBP) and cluster of differentiation 14 (CD14) [59,60]. TLR4 activation requires recognition of LPS by the receptor accessory proteins, CD14, and myeloid Differentiation factor 2 (MD2). Basically, LBP binds to LPS and transfers LPS to CD14 that subsequently carries LPS to MD2. A complex of CD14-LPS-MD2-TLR4 creates a conformational change to TLR4, and induces the intracellular signaling cascade [59]. The intracellular signaling pathway for TLR4 is shown in Fig. 3. The signaling cascades include interaction with Myeloid differentiation primary response 88 (MyD88), activation of IRAKs 1, 2, and 4, phosphorylation-dependent inactivation of IκBα, release of NF-kB from the complex with IκBα, nuclear import of NF-κB, initiation of transcription of pro-inflammatory genes (e.g., TNFα, IL-6, IL-8), and new transcription of the gene for MAP kinase phosphatase (MKP1). A parallel arm with the activation of NF-κB, phosphorylation of MAP kinases (ERK, p38 and JNK) leads to the activation of transcription factor AP1. The pathways for antagonism by POPG and PI are quite similar as shown in Fig. 3 [2].
Fig. 3.
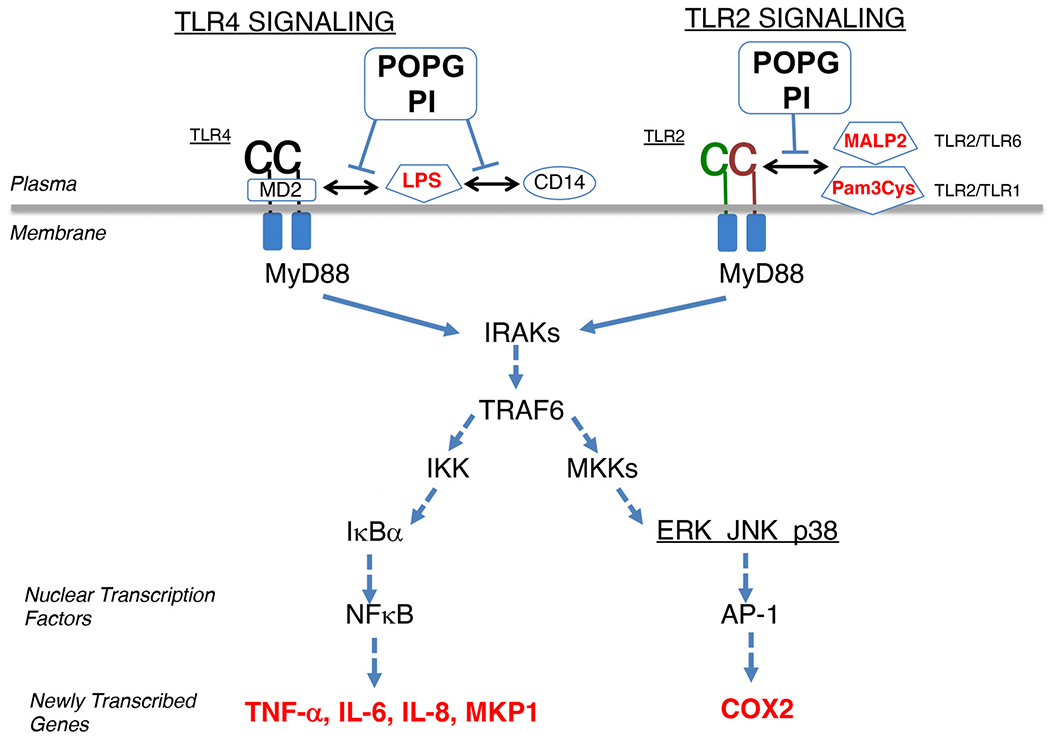
Schematic depiction of mechanism of POPG and PI disruption of TLR signaling.
As decoy ligands, POPG and PI prevent recognition of cognate ligands (e.g., LPS) by the TLR4 co-receptors CD14 and MD2. Similarly, POPG and PI disrupt the recognition of Pam3Cys and MALP2 by TLR2/1 and TLR2/6 complexes, respectively (adapted figure from ref. [57]).
POPG inhibits LPS-dependent activation of the CD14-MD2-TLR4 complex, and results in inhibition of MD2 and CD14 recognition of LPS as a ligand. In our findings [2], POPG binds to the LPS binding site of CD14 and prevents the protein from recognizing LPS. Additionally, POPG binds MD2 and inhibits the ability of this protein to recognize both LPS and TLR4. This inhibitory interaction of POPG with CD14, disrupts the conformational changes in intracellular domains of TLR4 dimers that engage intracellular signaling processes (Fig. 3).
We discovered that liposomes of POPG were potent antagonists against LPS-induced activation of TLR4 [2]. Palmitoyl-oleoyl-phosphatidylcholine (POPC), which contains the same fatty acids but a different polar head group from POPG, did not antagonize LPS-induced TLR4 activation [2]. LPS treatment induces pro-inflammatory mediators, such as TNF-α, IL-6, IL-8 and release of arachidonic acid (AA) from human primary alveolar macrophages (Fig. 4) [16,18]. These proinflammatory cytokines and AA, are sensitive markers induced by cellular response to TLR activation [16,18,61]. POPG significantly inhibits TNF-α production and AA release elicited by LPS stimulation of human primary alveolar macrophages (Fig. 4). Intratracheal instillation of POPG diminishes LPS-induced lung injury elicited in vivo, in mice [2], consistent with in vitro studies [2]. Simultaneous treatment with POPG reduces the inflammatory cytokine secretion (TNF-α, KC and MIP2) elicited by LPS challenge [2]. Hashimoto et al. also reported that POPG inhibited PAMPs-induced immune responses by blocking the binding of PAMPs to LBP and CD14. POPG decreased proinflammatory cytokine production detected in serum of LPS-injected mice and depressed abscess formation in mice infected with treponemes [62].
Fig. 4.
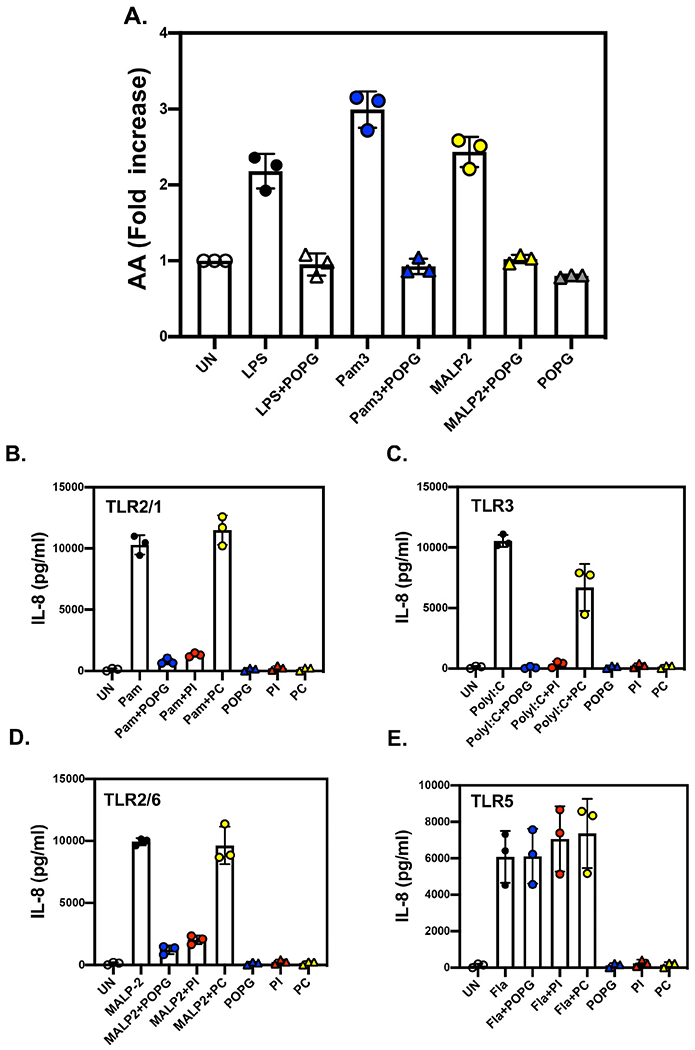
POPG and PI prevent multiple TLR activations by cognate ligands.
A) Arachidonic acid (AA) release from freshly isolated human alveolar macrophages can be used as a downstream indicator of TLR2/1, TLR4, and TLR2/6 activation. Cells prelabeled with [3H]-AA, release the AA in response to TLR activation by Pam3Cys for TLR2/1, LPS for TLR4 and MALP2 for TLR2/6. The release of [3H]-AA is inhibited by treatment with POPG liposomes, which disrupt early events in transmembrane signaling [16].
B-E) Beas2B cells were challenged with TLR agonists, B) Pam3Cysy for TLR2/1, C) Poly I:C for TLR3, D) Flagellin for TLR5, and E) MALP2 for TLR2/6, in either the absence, or presence of POPG, PI or POPC. IL-8 production was measured by ELISA at 48 h after stimulation with agonists [63].
2.4. Pulmonary surfactant phospholipids antagonize multiple TLR activations
As stated above, native pulmonary surfactant phospholipids antagonize TLR4 induced immune responses from structural inhibitory interaction with LPS [2,20,62].
We investigated the functions of pulmonary surfactant phospholipid antagonism of the activation of multiple TLRs. To determine the efficacies of POPG or PI as antagonists of multiple TLR activations, we measured IL-8 secretion induced from Beas2B cells with the TLR2/1 agonist, Pam3Cys, the TLR3 agonist, poly I:C, the TLR5 agonist, flagellin, and the TLR2/6 agonist, Mycoplasma fermentans ligand (MALP2) [63]. Both POPG and PI significantly inhibited IL-8 production by Pam3Cys, polyI:C and MALP2 (Fig. 4B,C and D). But the lipids failed to reduce IL-8 production by Flagellin (Fig. 4E), which was used as a control for non-specific effects. POPC did not affect IL-8 secretion by the TLR agonists (Fig. 4B-E). The data demonstrated that both POPG and PI acted broadly as TLR antagonists [63].
Heterodimers of TLR2/1 and TLR2/6 are expressed on the cell surface, and they recognize microbial membrane components [61]. TLR2 detects largely varieties of pathogen-associated molecular patterns (PAMPs) of Gram-positive and Gram-negative bacteria including lipoproteins and peptidoglycans (PGN) [61]. TLR2/1 and TLR2/6 heterodimers that are expressed on the cell surface, induce Toll/interleukin-1 receptor (IL-1R)/resistance protein (TIR), domain-containing adaptor proteins (TIRAP) and MyD88, following activation of interleukin-1 receptor-associated kinase (IRAK) and Tumor necrosis factor receptor associated factor 6 (TRAF6) (Fig. 3). TLR2 activation is induced by microbial ligands, including lipopeptides such as the Mycoplasma fermentans ligand (MALP2), Mycoplasma pneumoniae ligands [18], and the synthetic agonist, Pam3CysK4. Activation of TLR2 engages the same intracellular signaling pathway as TLR4 (Fig. 3). Pam3CysK4 activates TLR2/1 heterodimers and MALP2 activates TLR2/6 heterodimers, respectively (Fig. 3) [16,18,61]. In our published study, POPG inhibits proinflammatory cytokine production (TNF-alpha), eicosanoid production (including prostaglandin-D2 (PGD2), thromboxane A2 and thromboxane B2) induced by TLR2 complex activation induced by either MALP2 or Pam3CysK4, in human primary alveolar macrophages (Fig. 4A) [16,18]. We also reported that TLR2 activation by lipoproteins of Mycoplasma pneumoniae and membranes of this bacteria are inhibited by POPG [18]. M. pneumoniae binds to glycoprotein receptors on cell surfaces having terminal sialic acid residues, via the bacterial P1 adhesion protein; and α-2,3-sialyllactose supported M. pneumoniae adherence [64]. Our binding studies have shown that POPG does not disrupt the direct binding of M. pneumoniae to cell surfaces, but prevents the surface determinants on the bacteria from engaging the TLR2 receptor after the bacteria attaches to the cell surface [18].
2.5. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit TLR2 and TLR4 signaling
Our findings and evidence demonstrate that extracellular PG and PI play important roles in the lung as regulators of innate immunity. The lipids act by competitively blocking activation of multiple TLRs by cognate ligands [2,16,18,62]. Prior work has demonstrated the importance of the phospholipid headgroup of PG as the critical structural moiety for pharmacological activity against TLRs [2,18,62]. To determine the physical interactions of PG with TLRs, we synthesized PG analogs that vary in polar head group length and degree of substitution with hydroxyls and other substituents [16,18].
2.6. Phospholipid headgroup hydroxyls are important structural determinants of TLR antagonism
To understand the mechanisms of POPG antagonism for TLR4 and TLR2, we synthesized 12 POPG analogs and examined their efficacy as antagonists of TLR activation [16,18]. These compounds were altered within the head group glycerol moiety, which consists of three carbons and three hydroxyl groups, by changing 1) the number of aliphatic carbons from 0 to 5, 2) the number of hydroxyl substituents from 1 to 3, 3) the position of the hydroxyl substitutions, and 4) the branching of the head group aliphatic chain, and 5) substitution of the sn-2′-hydroxyl moiety with an amino group [18]. We determined the effects of modifying the structure of the glycerol moiety of the POPG head group, upon activity as an antagonist of TLR2 and TLR4. To determine analog potency on TLR4 activation, we used RAW264.7 cells in an in vitro system. TNF-α and AA release were measured as downstream reporters for TLR activation in this system. With regard to lipid antagonism of TLR4 activated by LPS, nearly all the analogs were as effective as POPG. However, there was an exception, for PG analogs with 3- or 4-carbon aliphatic chains and lacking hydroxyl substituents [16,18].
As stated above, POPG inhibits LPS-dependent activation of the CD14-MD2-TLR4 complex, and results in inhibition of MD2 and CD14 recognition of LPS by blocking their LPS binding sites [2]. CD14 and MD2 play a crucial role in TLR4 signaling induced by LPS [59,61]. In binding assays of POPG analogs to CD14, analogs that contain long chain aliphatic headgroups and hydroxyl-substituted aliphatic headgroups all bound to CD14 with apparent affinities comparable to, or better than POPG. Interestingly, the analog with methyl branches (POP-2′,2′-dimethylpropanediol) bound to CD14 with higher affinity than POPG [16,18,57]. In MD-2 binding assays for branched headgroup analogs, POP-(2′,2′-dimethyl)-propanediol bound to MD2 with higher affinity than POPG, but POP-2’aminopropanediol interacted very weakly. There were discrepancies, between direct binding affinities of analogs to CD14/MD2 and the potencies as antagonists of TLR4 activation. Based upon inhibition of binding studies of CD14 to Kdo2-lipid A compound, we discovered that POPG inhibited CD14 binding to the lipid by 90%, whereas analogs with a butyl headgroup inhibited binding by 45% and those with a propyl headgroup inhibited binding by 65%. These data demonstrate that the critical factor in evaluating analogs is their activity at disrupting LPS binding to the proteins. The binding activities of phospholipid analogs to CD14, or MD2, are not strongly correlated to biological potencies against TLR4 activation in intact cells.
2.7. POPG analogs antagonize TLR 2 activation
The potencies of antagonism by POPG analogs of TLR2/6 activation were determined in vitro, using RAW264.7 cells along with the TLR2/6 ligand MALP2. TNF-α and AA release were measured to determine the IC50 values for each analog [18]. POPG showed the lowest IC50, and elimination of hydroxyl groups from the head group aliphatic chain greatly reduced the efficacy of the lipid analogs. The POPG analogs exhibited higher IC50 values for TLR2/6 antagonism, requiring almost 10 times higher concentrations than found for TLR4 antagonism [18]. We performed binding studies with the proteins and POPG to determine the roles of the interaction of CD14 and phospholipids in antagonism of MALP2 signaling. CD14 did not directly bind to solid phase MALP2 that had a very weak inhibitory effect upon CD14 recognition of solid phase POPG. These data revealed that CD14 is not required in POPG antagonism of TLR2 activation.
The above findings demonstrate that POPG analogs that modify the polar headgroup, might be potent antagonists against the activation of TLR4 and TLR2 that would be equal to-, or more potent than POPG. The competition by lipid analogs for CD14 binding to solid phase LPS, or Kdo2-lipid A, are useful markers to predict molecules as antagonists of TLR4 activation at the cellular level. The structural requirements for inhibition of TLRs will be useful for development of POPG-like molecules with higher specificity and potency for individual TLRs. POPG turnover is very rapid in mouse lung, and the half-life of POPG was approximately 75 min in the lungs of mice [15]. Structural variants that exhibit a longer half-life [65], might have improved biological efficacy for inhibiting TLR4 activation in acute lung injury and ARDS [66,67] and secondary activation of TLR4 by respiratory viral infections [68,69].
3. Pulmonary surfactant phospholipids as novel anti-viral agents
We discovered that POPG antagonizes TLR4 activation by inhibiting MD2 and CD14 recognition of LPS as a ligand [2]. This finding led us to examine whether POPG can antagonize secondary activation of TLR4 by respiratory viral infections [14,57,68,69]. RSV is the most significant cause of serious lower respiratory tract infection within the first 2 years of life, and also causes serious disease in elderly individuals [70,71]. This virus has been considered a major pathogen for pediatric diseases, and currently it is also highlighted as an opportunistic pathogen causing mortality and morbidity in immunocompromised hosts [71]. There is growing concern that RSV contributes to acute exacerbations of asthma and chronic obstructive pulmonary disease (COPD) [70,71]. RSV causes significant pediatric and adult morbidity and mortality, and imposes a significant economic burden on health care systems.
Numerous clinical trials are ongoing to discover treatments for RSV. Currently there is no vaccine, and no effective treatments are available [70]. At present, aerosolized ribavirin is the licensed drug for treating RSV infection for patients at highest risk [70,72]. The beneficial effect upon clinical outcome remains unproven and its use is very limited [70]. A monoclonal antibody targeting the RSV-F fusion protein, Palivizumab, has been used for RSV prophylaxis in premature infants and newborns who also have congenital heart disease [73]. A few promising anti-RSV compounds have been developed that show efficacy for inhibiting viral replication and providing clinical benefits in patients with viral infection [70,74].
An innate immune response driven by pattern recognition receptors (PRRs) is a major line of defense in airway epithelial cells, that regulates viral replication [61]. TLRs 3,7 and 8 sense viral infections by recognizing viral replication (nucleic acid) products. TLRs are found on, and within a variety of cells [12]. TLR4 recognizes the RSV fusion protein (F-protein) and induces proinflammatory cytokine production [75] and contributes to RSV induced pulmonary pathogenesis and innate immune responses in an in vivo model in the TLR4−/− mouse [75,76]. It has also been reported that RSV induces innate immune responses through TLR2. TLR2 signaling promotes the clearance of RSV and has preventative effects against RSV-induced disease [77].
3.1. The minor anionic pulmonary surfactant phospholipids, POPG and PI, antagonize RSV infection in vitro and in vivo
POPG can inhibit RSV Infection in an in vitro system using human primary bronchial epithelial cells. POPG markedly reduced the cytopathic effects and proinflammatory cytokine production (IL-6 and IL-8) elicited by RSV infection [14]. POPG also inhibited RSV plaque progression (scored as increase in plaque diameter after infection was established in cultures of HEp2 cells [14]. To determine POPG potency against RSV infection in vivo, we used Balb/c mice as a model. POPG reduced viral replication in the mice by a factor of 103 [14]. Histopathogy revealed that RSV infection caused significant bronchial epithelial damage, alveolar wall thickening, lung collapse, massive neutrophil and lymphocyte recruitment into alveoli, indicative of pneumonia and lung injury (the panel is shown as RSV, Fig. 5B). POPG treatment prevented this inflammatory damage to the lung, elicited by RSV infections (RSV + POPG). POPG treatment alone (POPG) resulted in the same histopathology of the lung as observed for the sham treated group (CONL). In Fig. 5A, quantitative lung histopathology scores were determined for each group shown for three experiments [14], and the treatment with RSV and POPG markedly reduced the score to that of sham treated group.
Fig. 5.
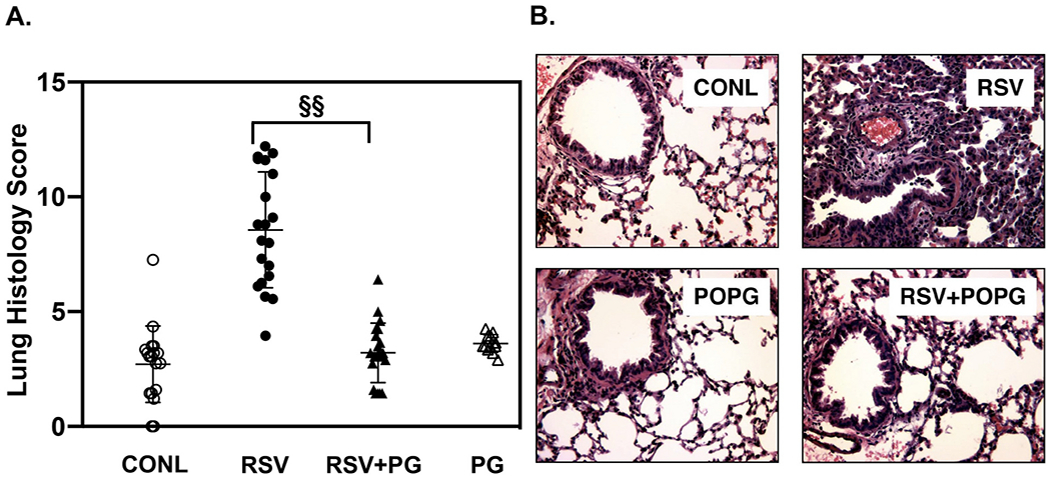
POPG inhibits RSV infection and pulmonary injury in mice.
Mice were challenged with 107 pfu of RSV by intranasal inoculation in either the absence, or presence of anionic phospholipids. Panel A) shows histopathology scores in each group, and panel B) shows histopathological images of the lungs with each condition, consisting of sham treated group (CONL), RSV group (RSV), RSV challenge supplemented with POPG (RSV + POPG), and with treatment of POPG alone (POPG). Data are from ref. [14].
3.2. POPG binds RSV with high affinity and inhibits viral binding to CD14, and host cells
To determine how POPG can prevent RSV infection, we examined the direct binding of POPG to RSV using a phospholipid solid phase assay [2,14]. RSV bound to POPG with high affinity and in a saturable manner. When the same types of experiments were performed with POPC as the solid phase, no viral attachment was observed (Fig. 6). With a CD14 solid phase [2,14], RSV bound to the protein in a dose-dependent manner; and this binding interaction was inhibited by POPG. We also examined the effect of POPG upon cell surface binding of RSV using the human epithelial cell line, HEp2. RSV attachment to HEp2 cells was determined by FACScan by quantifying the fluorescence of an anti-RSV antibody Analysis of the fluorescence and calculation of mean fluorescence intensity (M.F·I) was performed with Cell Quest software. RSV challenged HEp2 cells showed strong fluorescence signals, were 102-fold higher compared to non-RSV challenged cells, and POPG treatment reduced M.F·I by 70%. In contrast, treatment with a control phospholipid, POPC, did not reduce RSV binding signals. There was no direct viricidal effect by POPG against RSV as determined by pre-incubation followed by quantitative plaque assays [14].
Fig. 6.
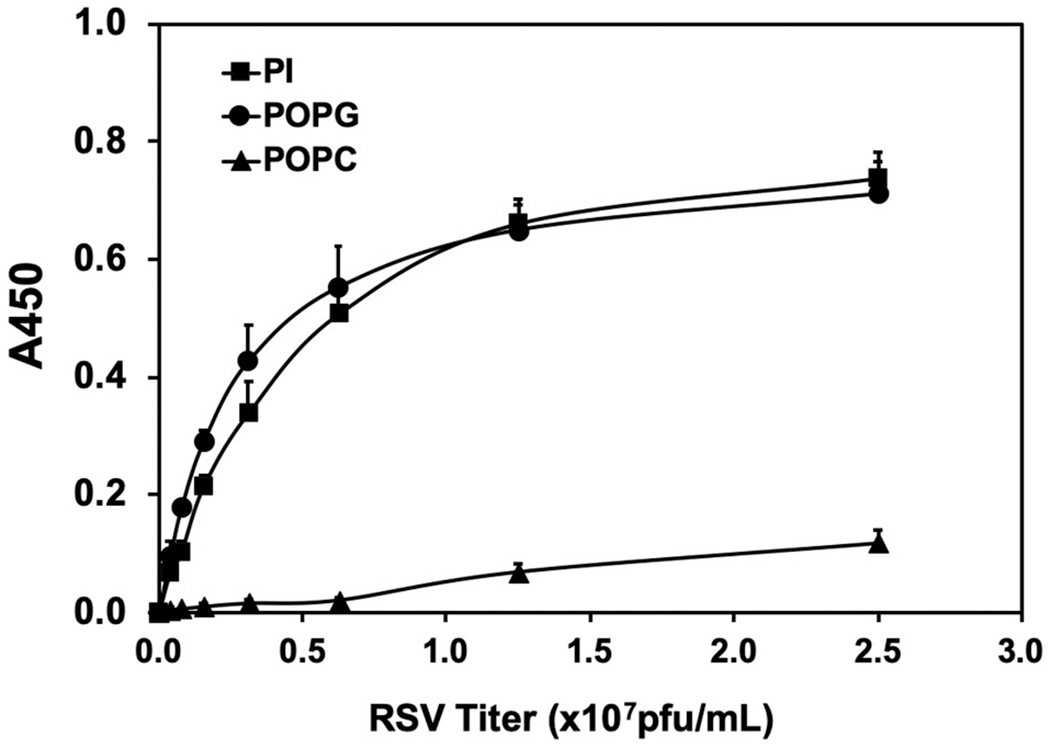
POPG and PI bind RSV with high affinity.
Solid phase phospholipids were made by applying phospholipids (POPG, PI and POPC) in ethanol to microtiter wells, followed by air drying. Various amounts of RSV were added to the solid phases and incubated at 37°C for 60 min. Viral binding was detected with a polyclonal anti-RSV antibody [16]. The figure is adapted from ref. [16].
3.3. POPG inhibits infection by clinical isolates of RSV from patients, and virulent RSV strains
In our initial experiments, we studied the anti-viral effect of POPG against an RSV-A2 laboratory strain [15]. To probe possible clinical relevance of POPG for application to patients with RSV infection, we critically tested the potency of POPG against clinical isolates of viruses from patients and recombinant strains exhibiting enhanced virulence in mice [78,79]. Using plaque assays, we tested five clinical isolates of strains from patients [15] and a recombinant strain that induced increased airway hypersensitivity and goblet cell hyperplasia (rA2-19F) in mice, as well as control recombinant strains rA2-A2F and rA2-LongF [78,79] along with a standard RSV-A2 strain. POPG significantly inhibited viral replication by 104–107 fold against clinical isolates of RSV and three different subtypes of recombinant viruses. In contrast, POPC did not have any effect upon plaque formation by these RSV subtypes [15].
In summary, POPG inhibits RSV infection by directly binding to the virus in a high affinity interaction that disrupts attachment of virions to host cells. POPG is also very potent against clinical isolates of RSV, and our findings demonstrate the lipid is a strong inhibitor of clinically relevant strains of RSV.
3.4. The minor pulmonary surfactant phospholipid, phosphatidylinositol (PI), which shares structural similarity to POPG, inhibits RSV infection in vitro
Our initial studies demonstrated inhibition of TLR4 activation by both PI and POPG. Both lipids appear to share overlapping structures and mechanisms for disrupting CD14 recognition of bacterial LPS [2]. Based on these observations, we examined PI efficacy against RSV infection using in vitro systems with the human bronchial epithelial cell line, Beas2B [17]. Both PI and POPG suppressed RSV-induced IL-8 secretion elicited from Beas2B cells. To exclude any toxic effects of POPG and PI, Beas2B cells were challenged with the TLR5 agonist flagellin (10 ng/ml) and IL-8 production by Beas2B cells was detected by ELISA. Neither POPG, nor PI, inhibited IL-8 production elicited by flagellin, thereby ruling out a non-specific mechanism of action. The antiviral activity of PI was determined by quantitative plaque assay using HEp2 cells incubated with virus, either with, or without phospholipids (PI, POPG, or POPC). Both PI and POPG markedly reduced RSV plaque numbers in a dose-dependent manner. At concentrations of 200 μg/ml, both PI and POPG reduced viral plaque numbers by a factor > 103 and a factor > 104, respectively. These data show that PI significantly inhibits RSV infection in vitro. Additionally, we tested for the antiviral efficacy of DPPC, which is the most abundant phospholipid in pulmonary surfactant. Neither DPPC nor POPC inhibited RSV plaque formation [17]. As described above, the total phospholipid concentration in surfactant is approximately ~50 mg/ml [10,11]. The concentrations of Phosphatidylglycerol (PG) and phosphatidylinositol (PI) within alveolar hypophase are ~5 mg/ml and ~ 2 mg/ml, respectively. Thus, the POPG and PI concentrations used in the in vitro experiments were very low compared to the levels of these lipids in the alveolus. Collectively, these data show that POPG and PI have very potent antiviral effects against RSV infection.
3.5. PI binds to RSV with high affinity and inhibits viral attachment to host cells
Phospholipid (PI, POPG, or POPC) binding activities to RSV were determined using phospholipid solid phases in microtiter wells [2,15,17]. PI bound RSV with high affinity, in a dose dependent manner similar to POPG (Fig. 6). Additionally, we examined whether PI can inhibit RSV binding to epithelial cells, using HEp2 cells. HEp2 cells were challenged with varying concentrations of RSV in either the absence (RSV alone) or presence of lipids, including other individual anionic phospholipids (POPG, PA, and PS), and the zwitterionic lipid, POPC [15,17]. The data showed that RSV bound to HEp2 cells dose-dependently, and both PI and POPG, strongly disrupted virus attachment to the cells by ~80%, compared to other anionic phospholipids (PA, PS), and POPC did not markedly alter viral attachment. In a previous publication [16], we reported that POPA failed to inhibit TNF-α production induced by LPS in RAW 264.7 cells, and it cannot antagonize activation of TLR4 [16]. We also published that POPG and PI significantly attenuated TNF-α and NO production in a concentration-dependent manner with the maximal inhibitory effect at 2.5μg of phospholipids/ml); and PS, was much less effective than PI and POPG [2]. These data showed that specific structural aspects of POPG and PI produce high binding affinity and specificity as antagonists of RSV infection that is not duplicated by either the anionic character, or hydrophobic constituents of other negatively charged lipids, or the hydrophobic constituents of zwitterionic phospholipids. The data further demonstrated that none of the anionic phospholipids had a direct effect upon virion integrity [15,17]. POPG prevents RSV spreading in epithelial cells [14] and also significantly inhibits RSV replication 24 h after infection is established [15]. Based on these findings, POPG and PI might prevent viral spreading by inhibiting viral entry into host cells because they can bind to RSV with high affinity.
Collectively these findings demonstrate that PI has a significant antiviral effect against RSV in vitro, similar to POPG; by directly binding to the virion and inhibiting its attachment to epithelial cells.
3.6. PI inhibits RSV infection in vivo
We examined PI efficacy against RSV infection in vivo, in mice, as previously described [14,15,17]. BALB/c mice were challenged with intranasal inoculation of RSV either with, or without PI (Fig. 7B). The treatment with PI significantly reduced viral replication in the lung by a factor of 102 (Fig. 7B). Simultaneous treatment with RSV and PI reduced the total cell numbers and inflammatory cell recruitment recovered in BALF. By histopathology, PI treatment significantly reduced inflammatory changes and damage in the lung caused by RSV infection [17]. The data demonstrated that simultaneous treatment with PI inhibited RSV infection, in vivo, in mice.
Fig. 7.
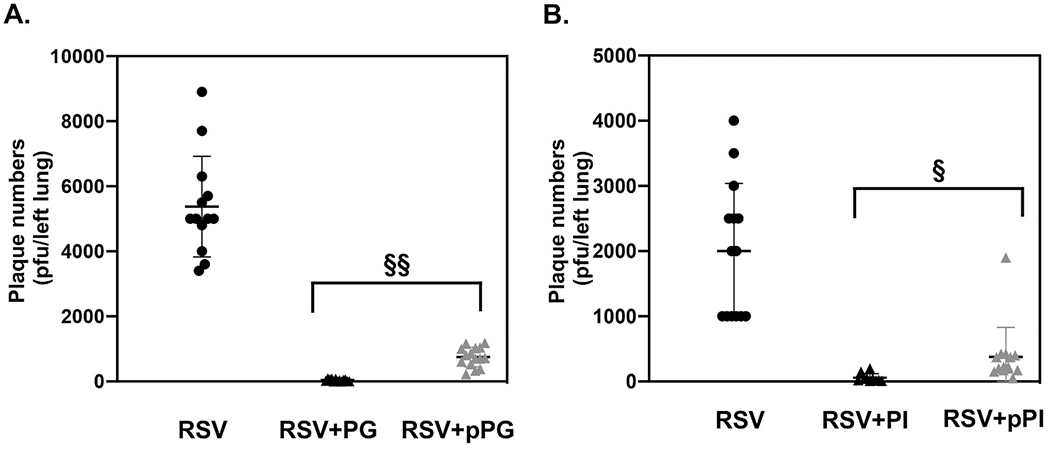
POPG and PI inhibit RSV infection as prophylaxis in mice.
Balb/c mice were infected with RSV (1 × 107 pfu/mouse) as described in Fig. 5. Panel A) shows viral burden in the lung at day 5 post infection. Virus alone (RSV), Virus + POPG simultaneous treatment (RPG), and Virus+ prophylaxis treatment with POPG (pRPG). POPG was administered to mice 45mins prior viral challenge. Panel B) shows viral burden in the lung in each group, virus alone (RSV), Virus + PI simultaneous treatment (RPI), and virus + prophylaxis treatment with PI (pRPI). Mice were treated with PI 2 h before viral challenge, The left lungs of infected animals were excised and homogenized and the viral titers of fresh homogenates were determined by plaque assay. Data are shown as, AVG ± SD, § indicates p < 0.01, §§ indicates p < 0.001. Statistical analysis by single Anova and unpaired t-test), respectively. Data are from refs [15,16].
3.7. POPG and PI inhibit RSV infection when applied as a prophylaxis treatment
Our initial findings showed that simultaneous treatments with either POPG, or PI, inhibited RSV infection in vivo, in mice. We expanded our studies to investigate whether POPG, or PI, can be effective for preventing RSV infection in vivo.
To determine the turnover of POPG in mouse lung, d5-POPG was inoculated intranasal into mice and subsequently lung lavages were performed at various time points. The cell free BALF was processed for lipid extraction and recovered material was quantified by both lipid phosphate determinations and mass spectrometry [15]. The POPG turnover kinetics in the lung had two phases, 1) in the first 60 min, there was a rapid decline of d5-POPG concentration, followed by 2) a slow phase lasting for another 360 min. The half-life of d5-POPG based upon the first rapid phase, was ~75 min. Based on the kinetics, we performed experiments examining prophylaxis with POPG (administered for 45 and 120 mins). With pretreatment for 2 h, POPG reduced RSV plaque formation by 50%. Pre-treatment with POPG for 45mins, successfully inhibited RSV replication in vivo in mouse in a dose dependent manner (Fig. 7A) [17].
PI turnover in the lung in mice was determined using the same method, as previously reported [15,17]. In contrast to POPG, PI turns over slowly [15,17] with an estimated half-life of 6 h in mouse lungs. A 2 h prophylaxis treatment with PI inhibited RSV replication by a factor of 10 (Fig. 7B) [17]. The results demonstrate that PI provides significantly longer protection than POPG against RSV infection, in vivo. Pulmonary surfactant phospholipid turnover rates are dependent upon the respiratory rate [80]. A half-life for PG reconstituted into artificial pulmonary surfactant given to human neonates has been estimated to be approximately 30 h [80]. The respiratory rate in human newborns is 30/min. The respiratory rate of mouse is very rapid (200–300/min), and we expect that the turnover rate of surfactant lipids is fast. The data suggest that PG applied to human subjects might have anti-viral effect against RSV lasting between 12 and 24 h when applied as prophylaxis.
3.8. POPG and PI inhibit secondary inflammatory pathways induced by influenza A virus infections
Our findings examining the anti-viral effects of POPG and PI against RSV, led us to expand our research to examine whether the lipids antagonize influenza A infection and the induction of inflammatory sequelae. Substantial evidence implicates TLR4 activation as a significant contributor to the pathogenesis of IAV infections [67,69]. Previous reports have shown that TLR4−/−mice were resistant to lethal challenges with mouse-adapted H1N1 (influenza A/PR/8/34) [69]. Influenza infection, elicited the secondary generation of oxidized host phospholipids, which function as ligands that activate TL4 complexes to induce a cytokine storm involving IL-1β, IL-6 and TNF-α, resulting in acute lung injury. The post IAV-infection treatment at 48 h with the TLR4 antagonist, Eritoran (E5564 by Eisai) markedly reduced lethal infection with H1N1(influenza A/PR/8/34) in mice. It was also reported that Eritoran was potent against pandemic H1N1 A/California/07/2009 used to infect mice, and a human H3N2 influenza A/Wuhan/359/95 used to infect cotton rats [69].
3.9. POPG suppresses infections by different subtypes of influenza A virus
We examined POPG effects against influenza A virus using Beas2B cells and H3N2-IAV (Philippines 82/H3N2) [12]. Beas2B cells were challenged with H3N2-IAV, and elicited IL-8 production by cells was determined by ELISA after 48 h. Virus challenge induced significant increase of IL-8 production and the treatment with POPG attenuated IL-8 secretion by H3N2-IAV by a factor of 10, but POPC was ineffective. We used a low dose of POPG, amounting to less than 10% of the PG concentration in the lung [1,10]. We determined the effect of POPG on H3N2-IAV propagation using MDCK cells infected with the virus in either the absence, or presence of POPG, or POPC. Cell layers were processed for immunoblotting to determine viral neuraminidase (NA) and matrix (MP) protein expression. POPG markedly reduced both NA and M1 protein expression in a dose-dependent manner. We also performed experiments with H1N1-PR8 in MDCK cells. Treatment with POPG attenuated MP expression by 70%, but POPC failed to alter MP expression. These data demonstrate that POPG inhibits H3N2-IAV and H1N1-PR8 protein expression and replication.
3.10. POPG strongly binds to influenza A virus and prevents virus attachment to host cells
We investigated the mechanism of POPG antagonism of infection by examining the binding interaction between IAV and the lipids) [12–15,17,81]. POPG bound to H3N2-IAV with high affinity and in a concentration-dependent manner, but POPC had very low affinity binding to H3N2-IAV (Fig. 8A) [12]. Additionally, we examined viral binding to MDCK cells. MDCK cells were challenged with H3N2-IAV at an moi of 0–10, at 19 °C, (to prevent endocytosis), either with or without, lipid treatment. Entire wells were processed for protein expression analysis by immunoblotting, to quantify the cell associated MP amounts (Fig. 8B and C). MP quantity increased with H3N2-IAV concentrations. Virus binding to the cell surface was high affinity and saturable. MP expression was reduced by treatment with POPG in a dose dependent manner (Fig. 8C). The data demonstrate that POPG binds to IAV with high affinity and prevents viral attachment to host cells The above findings clearly identify the mechanisms by which POPG inhibits H3N2-IAV and H1N1-PR8 infections.
Fig. 8.
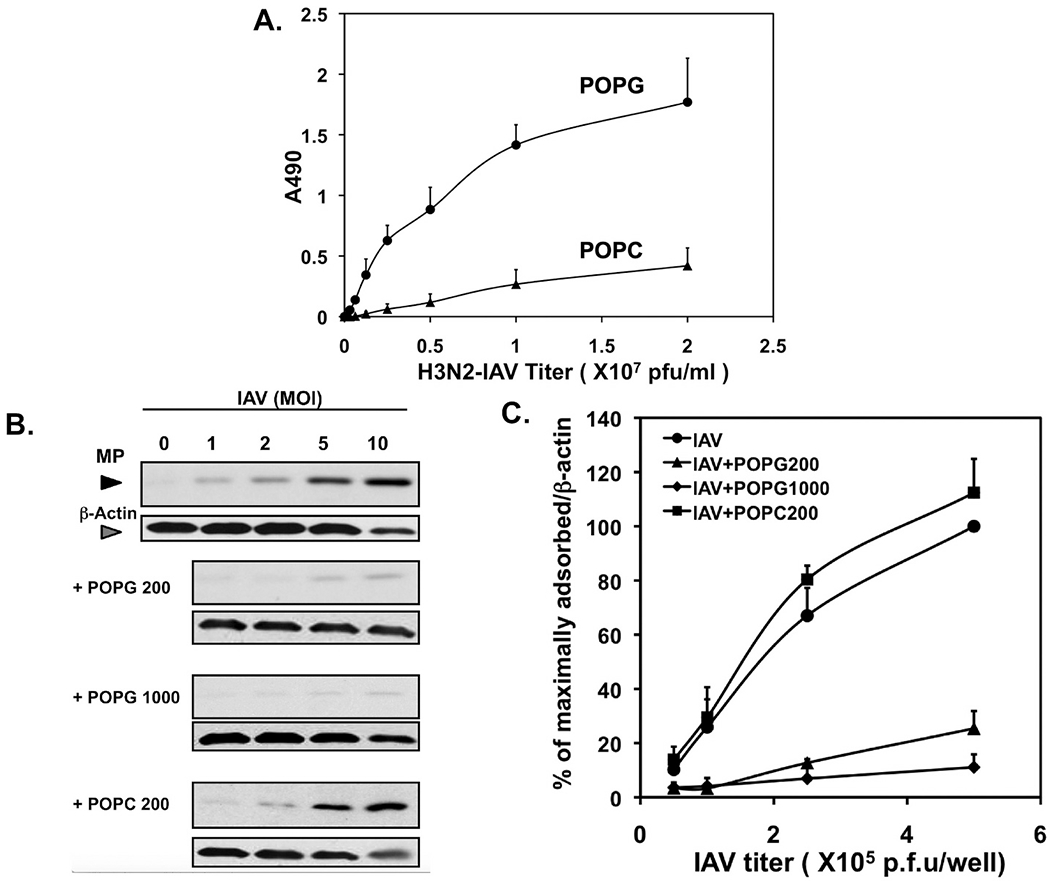
POPG inhibits H3N2-influenza A attachment to epithelial cells.
A) Solid phases of phospholipids were prepared in microtiter wells and hydrated, and subsequently defined amounts of virions were added and incubated for 1 h at 37° C. Virus attachment to phospholipids was quantified by immunodetection using HRP-conjugated polyclonal anti-IAV.
B) The attachment of H3N2-IAV to monolayers of MDCK cells was quantified by western blotting, using Image J analysis.
C) Virus attachment to cell monolayers in the absence and presence of phospholipids (POPG, POPC) was quantified by western blotting. Data are from ref. [107].
3.11. POPG markedly reduces inflammation and infection by H1N1-PR-8 in vivo
We examined POPG efficacy against H1N1-PR8 infection in vivo, in mice. BALB/c mice were challenged with H1N1-PR8 by intranasal inoculation, either with, or without POPG. Mice were euthanized at day 5 after infection. POPG treatment significantly inhibited H1N1-PR8 replication in the lung by a factor of 10 [12]. Lung histopathology scoring demonstrated POPG efficacy against H1N1-PR8 induced inflammation.
3.12. Pulmonary surfactant lipids inhibit pandemic H1N1 influenza infection
In 2009–2010, the (H1N1)pdm09 brought the greatest IAV threat to the world since 1968, and this episode highlighted the problems of the emergence of a novel virus. The (H1N1)pdm09 is a triple reassortant virus that contains unique genetic heritage derived from human, avian, and porcine influenza A sources [82] [84], that also contribute to its strong virulence and high human-to-human transmissibility. The(H1N1) pdm09 continues to reappear as a significant strain of influenza during the annual flu season [83–86]. Vaccination is a very important strategy to prevent influenza infection as a primary counter measure. Vaccine efficacies vary and depend on the successful matching of antigen and the epidemic virus [87]. Three classes of drugs are currently available for influenza. One drug class (e.g., amantadine and remantidine) inactivates the M2 membrane protein ion channel activity of the virus, but most influenza A viruses are now resistant to these drugs. The second class of anti-viral drugs consist of neuraminidase inhibitors (NAIs), (e.g., Oseltamivir and Zanamivir). However, there is already growing concern about NAI-resistance [88]. The frequency of NAI resistant influenza A virus is ~1% in adults, and 4–8% in children [89]. The oseltamivir-resistant influenza A virus mutants harboring neuraminidase [E119V] and [R292K] mutations, are more critical and frequent in children [90]. Normally this resistance results from NAI treatment coupled with relatively weak transmission of the resistant virus. However, the CDC reported the 2008 2009 seasonal oseltamivir-resistant influenza A (H1N1) viruses with the [H274Y]-NA gene mutation, circulated widely and appeared to arise independently of oseltamivir use [89]. In 2018, the novel anti-influenza drug, Baloxavir marboxil (baloxavir), a selective inhibitor of influenza cap dependent endonuclease, was approved for uncomplicated influenza A and B infections [91]. There are still further concerns that anti-influenza compounds invite the emergence and selection for resistant variants [91]. Specific amino acid substitutions in the endonuclease active site [I38T/F] are related to susceptibility to baloxavir, for representative influenza A viruses [91]. Collectively, these findings demonstrate the importance of developing new agents to block viral infections in novel ways.
3.13. POPG and PI inhibit pH1N1-IAV replication after infection is established
The antiviral effects of POPG and PI against H1N1-PR8, guided us to examine POPG and PI efficacies against a subtype of IAV designated pH1N1 [Influenza A/California/ 07/2009] using MDCK cells in an in vitro system. Cells were challenged with PH1N1-IAV and harvested at 24 h after infection to determine viral protein expression (viral neuraminidase (NA) and matrix protein (MP)). Simultaneous treatment with POPG or PI significantly reduced NA and MP expression. Cytopathic effects induced by pH1N1-IAV were prevented by POPG and PI in a dose-dependent manner, quantified at 24 h after infection [92]. We also investigated the POPG and PI efficacy against pH1N1-IAV replication after infection was established. MDCK cells were treated with 1 mg/ml of POPG, or PI, at 2 h after, pH1N1-IAV infection was established, and then allowed to progress for 24 h. Cell monolayers were collected and processed to quantify pH1N1-IAV replication by plaque assay. POPG and PI reduced plaque numbers of pH1N1-IAV. The data demonstrate that POPG and PI markedly antagonized pH1N1-IAV replication in vitro, even after viral infection was established.
We determined the direct binding of POPG or PI to pH1N1-IAV [12,17]. Both POPG and PI bound to pH1N1-IAV with very high affinities and in a dose-dependent manner as shown in Fig. 9 [92]. PI has better efficacy against pH1N1-IAV protein expression and replication in MDCK cells, compared to POPG. Interestingly, POPG has higher binding affinity to pH1N1-IAV than PI. We also examined direct viricidal effects of phospholipids (POPG, PI and POPC) against pH1N1 and none of these lipids had any direct viricidal effect against pH1N1-IAV.
Fig. 9.
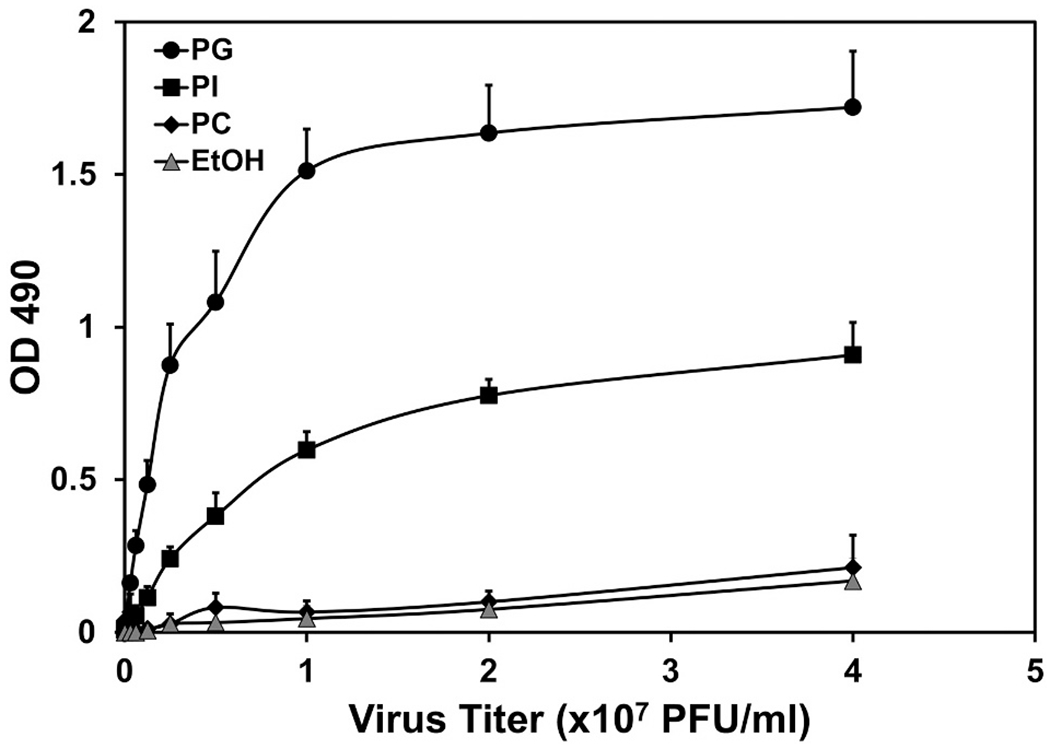
Binding studies demonstrate high affinity interactions between pH1N1-influenza A virions and phospholipids.
Phospholipid solid phases were prepared in microtiter wells and binding interactions between pH1N1-virion and phospholipids were quantified by immunodetection (A450). Figure is adapted from ref. [92].
3.14. POPG and PI inhibit pH1N1-IAV replication, and POPG prevents lethal infection by pH1N1-IAV in mice
We investigated the anti-viral effects of POPG and PI against pH1N1-IAV in mice, which were challenged with a sublethal inoculum of pH1N1-IAV alone, or with virus plus POPG or with virus plus PI administered intranasally, and subsequently harvested at day 6 post-infection. Phospholipid dosing was selected based upon preliminary studies. Left lungs were homogenized and processed for application in a quantitative plaque assay for pH1N1-IAV. POPG significantly reduced pH1N1-IAV replication in the lung by a factor of >102 (Fig. 10A). pH1N1-IAV alone, caused severe lung damage as shown in Fig. 10B. POPG treatment greatly reduced these inflammatory changes (Fig. 10B). Histological scores are also shown in Fig. 10B, and mice challenged with virus alone (pH1N1) showed a high histopathology score. Treatment with POPG, at the time of viral challenge (pH1N1 + PG) markedly reduced the score. We also determined the efficacy of PI against pH1N1-IAV in this same in vivo model. PI inhibited viral replication in the lung by a factor of 10, compared to the virus alone treatment group (Fig. 10C). Treatment with PI also prevented the tissue damage caused by pH1N1-IAV infection and improved the histopathology score (Fig. 10D). We critically investigated POPG potency against pH1N1-IAV in a lethal- challenge model in mice. Mice received a lethal dose (LD100) of pH1N1-IAV (103 pfu/mouse) with or without POPG (3 mg/mouse) in the inoculum. All mice subjected to virus alone challenge, died by day 10 post-infection. The POPG treatment, dramatically improved survival to 100% [92].
Fig. 10.
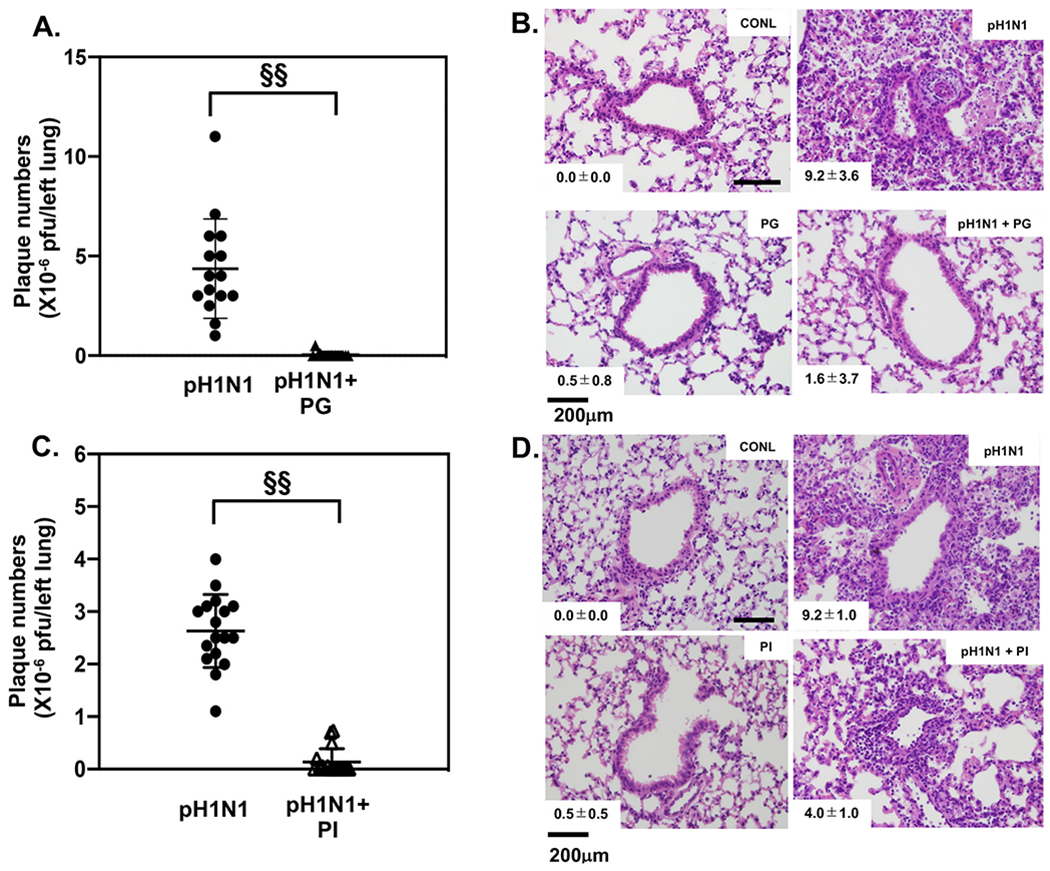
POPG and PI inhibit pH1N1-influenza A viral burdens and consequent lung damage in mice.
Panel A) Viral loads were determined by plaque assays and POPG treatment reduced viral burden. Data are shown as AVG ± SD, §§ represents p < 0.001.
Panel B) Histology of lung sections in the absence (CONL) and presence (pH1N1) of virus, or virus + PG (pH1N1 + PG). The histopathology scores are shown in the lower left insets in the micrographs.
Panel C) shows viral loads by plaque assay and antagonism by PI. Data are shown as AVG ± SD, §§ represents p < 0.001.
Panel D) shows histology of lung sections for uninfected (CONL) and pH1N1 infected mice (pH1N1) and mice treated with PI alone; or virus + PI (pH1N1 + PI). The histopathology scores are shown in the lower left hand insets in the micrographs. Data are from ref. [92].
3.15. POPG inhibits pH1N1-IAV replication and infection in ferrets
Additionally, we studied the anti-viral effect of POPG against pH1N1-IAV infection in ferrets. Ferrets are considered an ideal animal, to model human IAV infection, and horizontal transmission [93]. The animal shares similarities with human flu like symptoms and clinical signs. Ferret lungs resemble human lungs with respect to IAV receptor distribution. Ferrets were infected with pH1N1-IAV, or virus + POPG, given intranasally. Nasal and pharyngeal swabs were collected daily until day 4, when all ferrets were euthanized. Samples were used for plaque assays to quantify the pH1N1-IAV burden. POPG treatment reduced the plaque numbers recovered in samples from pharyngeal and nasal swab by ~80% and ~ 70%, respectively [92]. The histological inflammatory changes induced by pH1N1-IAV infection were markedly prevented by POPG treatment, as reflected in histopathology scores.
Collectively, POPG and PI inhibit multiple influenza A subtype infections (H3N2, H1N1-PR8, pH1N1) by preventing viral binding to host cells. The lipids antagonize influenza A virus infections and act to reduce the viral burden in vivo in mice and ferrets. Ferrets are an ideal animal model to mimic Influenza infection in humans. Especially noteworthy, is the finding that POPG treatment dramatically improved the survival rate in a lethal infection model of pH1N1-IAV in mice. The above findings demonstrate that both POPG and PI have significant potential to be applied as anti-influenza agents in humans.
3.16. Pulmonary surfactant lipids inhibit SARS-CoV-2 replication and prevent cell lysis by SARS-CoV-2 in vitro
Based on our findings regarding the anti-viral potencies of POPG and PI against multiple respiratory RNA viruses, we investigated the lipids for efficacy against SARS-CoV-2. The coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). COVID-19 is currently overwhelming health care systems in multiple countries and accounts for approximately 270 million identified cases and 5.2 million deaths worldwide [94]. Genetic variants of SARS-CoV-2 have been emerging and expanding since the end of 2020 [95]. It is noteworthy that the SARS-CoV-2 delta (B.1.617.2) variant was surging rapidly, and becoming the dominant variant worldwide [96,97]. Most recently, the SARS-CoV-2 omicron (B.1.1.529) variant has emerged that attains very high transmissibility [98]. Despite strong COVID19 vaccine efficacies [99–101], variants continue to emerge and invite vaccine breakthrough infections [102] [103–105].
We most recently examined whether POPG, or PI, can inhibit SARS-CoV-2 infection of VeroE6 cells that highly express the angiotensin I converting enzyme 2 (ACE2) receptor and differentiated human airway epithelial cells [106]. VeroE6 cells were challenged with SARS-CoV-2 (USA, WA/2020) at an m.o.i = 0.01 with or without treatments with POPG, PI or POPC (1 mg/ml). Cells were preincubated with lipids 30 min before viral inoculation. Culture media was harvested at 24 h after infection to quantify the viral burden, determined by plaque assay. The treatments with virus plus POPG and PI markedly suppressed the viral burden elicited by SARS-CoV-2 in VeroE6 cells by ~60% and 95% respectively (Fig. 11B). Neither POPG, nor PI, nor POPC elicited toxicity on VeroE6 cells as determined by cell viability assays [107]. The lipids also dramatically prevented cytopathic effects upon VeroE6 cells induced by SARS-CoV-2 infection at 72 h after infection (Fig. 11A). These data demonstrate that both POPG and PI have very strong anti-viral effect against SARS-CoV-2, in vitro.
Fig. 11.
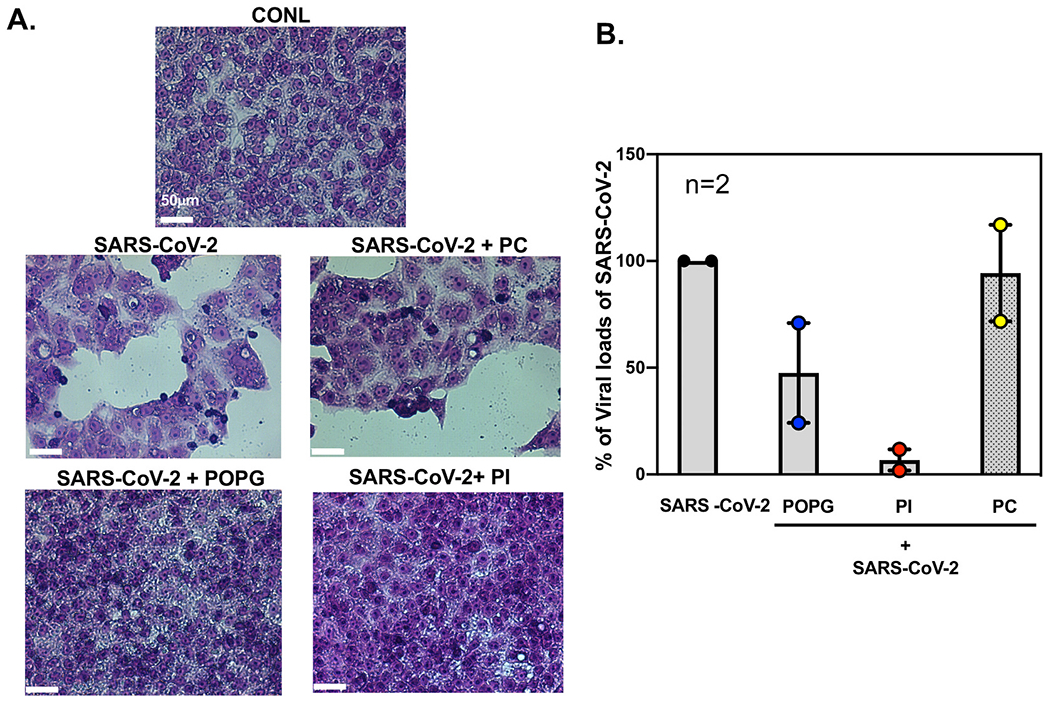
POPG and PI prevent cytopathic effect by SARS-CoV-2 and inhibit viral loads of SARS-CoV-2 in VeroE6 cells.
Panel A) Hematoxylin-Eosin staining of VeroE6 cell monolayers of untreated (CONL), or SARS-CoV-2 infected cultures (SARS-CoV-2) that were also pretreated with lipids (+PC, +POPG, +PI) 30 mins prior to the time of infection. The white bar indicates 50 μm. Viral infection causes cell lysis, cell hypertrophy and hypochromic staining. Panel B) shows quantification of viral loads by plaque assays, with POPG reducing number by ~60%, and PI reducing numbers by ~90%.
3.17. Future direction for pulmonary surfactant phospholipids as anti-inflammatory and anti-viral agents
In this article, we have focused upon the anti-inflammatory and antiviral effects of the minor anionic pulmonary surfactant phospholipids, especially against RNA containing respiratory viruses. Our earlier findings demonstrate that POPG and PI inhibit LPS induced TLR4 activation by directly interacting with CD14 and MD2. POPG also prevents LPS-induced lung injury and inhibited expression of proinflammatory mediators in mice. POPG also antagonizes TLR1/2 and TLR2/6 activation induced by mycoplasma and cognate agonists, and inhibits AA release from human alveolar macrophages. Several features of the lipids make them ideal candidate compounds to treat the cytokine storm induced during acute lung injury or acute respiratory distress syndrome (ARDS) [108]. Severe viral infections in humans by pH1N1 and SARS-CoV-2, can induce a cytokine storm including elevated levels of TNF-α, IL-6 and IL-8 [109]. These events contribute to the pathogenicity, the extensive pulmonary inflammation and the excessive lung damage [109–111]. Our preliminary data show the activities of POPG and PI as anti-viral and anti-inflammatory agents with significant potential for treating SARS-CoV-2 infections.
There are several advantages to POPG and PI for consideration as new anti-viral agents, insofar as they are: 1) of low molecular weight, 2) natural constituents of pulmonary surfactant and are non-immunogenic, 3)can be chemically synthesized in large amounts, and are inexpensive, 4)are chemically stable, and 5) easy to provide for clinical usage (either intranasally, or by inhalation). As previously stated, the respiratory rate regulates the turnover of pulmonary surfactant phospholipid [80]. For therapeutic usage of the lipids, the application should be intranasal (e.g., liquid form of nasal spray) or via a nebulizer. Based on pharmacokinetics of the lipids, we propose they can be applied at 2–3 times a day, for 5–7 days.
There are expectations that POPG and PI will provide preventatives and therapeutics for RSV [112]. In these reports, the lipids were highlighted as a possible candidate for prevention and treatment for RSV infection. The proposed mechanisms of action of the lipids, make it unlikely that they will give rise to drug-resistant variants.
3.18. Summary
Two minor constituents of pulmonary surfactant phospholipids, POPG and PI, have anti-inflammatory effects (by antagonizing activation of TLRs), and antiviral actions against multiple respiratory viruses, including RSV, Influenza A viruses, and most recently SARS-CoV-2. These same lipids also seem to hold promise as anti-inflammatory agents for acute lung injury induced by cytokine storms, which follow microbial infections.
Acknowledgements
This work was supported by CLINICAL INNOVATOR AWARD PROGRAM (CIA 160010), Flight Attendant Medical Research Institute, Inc. (FAMRI) (MN), NIH U19—A1 125357 (DRV), NIH P01 HL132821 (MN), NIH GM118819 (MN), National Emphysema Foundation (MN).
Abbreviations
- ACE2
angiotensin I converting enzyme 2
- AA
arachidonic acid
- CL
Cardiolipin
- JNK
c-Jun N-terminal kinases (JNKs)
- TIRAP
domain-containing adaptor proteins
- ERK
extracellular signal-regulated kinase
- H1N1-PR8
H1N1(influenza A/PR/8/34)
- pH1N1-IAV
H1N1 A/California/07/2009
- H3N2
H3N2-IAV (Philippines 82/H3N2)
- d5-POPG
1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-racglycerol-1′,1′,2′,3′,3′-d5) (ammonium salt)
- IL-6
Interleukin-6
- IL-8
Interluekin-8
- IRAK
interleukin-1 receptor (IL-1R) associated kinase
- KC
keratinocytes-derived chemokine
- LB
lamellar body
- MIP2
macrophage-inflammatory protein-2
- MKP1
mitogen-Activated Protein Kinase Phosphatase 1
- MD2
myeloid Differentiation factor 2
- POPG
palmitoyl-oleoyl-phosphatidylglycerol
- POPC
palmitoyl-oleoyl-phosphatidylcholine
- PAMPs
pathogen-associated molecular patterns
- PRRs
pattern recognition receptors
- PA
phosphatidic acid
- PS
phosphatidylserine
- PI
phosphatidylinositol
- PtdO
the phosphatidate moiety
- p38
p38 mitogen-activated protein kinases
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SCL34A2
solute carrier family 34 member 2
- SP-A
Surfactant protein A
- SP-B
Surfactant protein B
- SP-C
Surfactant protein C
- SP-D
Surfactant protein D
- SFTPA1
SP-A1 gene
- SFTPA2
SP-A2 gene
- SFTPC
SP-C gene
- SFTPD
SP-D gene
- COVID-19
The coronavirus disease 2019
- TLRs
Toll-like receptors
- TNFα
tumor necrosing factor-alpha
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Mari Numata has patent Surfactant Lipids, Composition Thereof and Uses Thereof. issued to National Jewish Health. Dennis. R. Voelker has patent Surfactant Lipids, Composition Thereof and Uses Thereof. issued to National Jewish Health.
References
- [1].Wright JR, Immunomodulatory functions of surfactant, Physiol Rev. 77 (4) (1997) 931–962, 10.1152/physrev.1997.77.4.931. Epub 1997/11/14. [DOI] [PubMed] [Google Scholar]
- [2].Kuronuma K, Mitsuzawa H, Takeda K, Nishitani C, Chan ED, Kuroki Y, Nakamura M, Voelker DR, Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2, J. Biol. Chem 284 (38) (2009) 25488–25500, 10.1074/jbc.M109.040832, quiz 880-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Whitsett JA, Alenghat T, Respiratory epithelial cells orchestrate pulmonary innate immunity, Nat. Immunol 16 (1) (2015) 27–35, 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kuroki Y, Takahashi M, Nishitani C, Pulmonary collectins in innate immunity of the lung, Cell Microbiol. 9 (8) (2007) 1871–1879, 10.1111/j.1462-5822.2007.00953.x. Epub 2007/05/11. [DOI] [PubMed] [Google Scholar]
- [5].Schmidt R, Meier U, Markart P, Grimminger F, Velcovsky HG, Morr H, Seeger W, Gunther A, Altered fatty acid composition of lung surfactant phospholipids in interstitial lung disease, Am. J. Physiol. Lung Cell Mol. Physiol 283 (5) (2002) L1079–L1085, 10.1152/ajplung.00484.2001. Epub 2002/10/12. [DOI] [PubMed] [Google Scholar]
- [6].Goerke J, Clements JA, Alveolar surface tension and lung surfactant, in: Mead PTM, a. J (Eds.), Handbook of Physiology—The Respiratory System III, Mechanics of Breathing, American Physiological Society, Washington, DC, 1986, pp. 247–261. [Google Scholar]
- [7].Goerke J, Pulmonary surfactant: functions and molecular composition, Biochim. Biophys. Acta 1408 (1998) 79–89. [DOI] [PubMed] [Google Scholar]
- [8].Wright SM, Hockey PM, Enhorning G, Strong P, Reid KB, Holgate ST, Djukanovic R, Postle AD, (2000) altered airway surfactant phospholipid composition and reduced lung function in asthma, J. Appl. Physiol 89 (1985) 1283–1292. [DOI] [PubMed] [Google Scholar]
- [9].Numata M, Voelker DR, Asthma and Infections, in: Martin RJ, Sutherland ER (Eds.), Lung Biology in Health and Disease, Informa Healthcare, New York, 2010. [Google Scholar]
- [10].Wright JR, Immunoregulatory functions of surfactant proteins, Nat. Rev. Immunol 5 (2005) 58–68. [DOI] [PubMed] [Google Scholar]
- [11].Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP, Activation of airway epithelial cells by toll-like receptor agonists, Am. J. Respir. Cell Mol. Biol 31 (2004) 358–364. [DOI] [PubMed] [Google Scholar]
- [12].Numata M, Kandasamy P, Voelker DR, Anionic pulmonary surfactant lipid regulation of innate immunity, Expert Rev. Respir. Med 6 (2012) 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Numata M, Chu HW, Dakhama A, Voelker DR, Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Numata M, Nagashima Y, Moore ML, Berry KZ, Chan M, Kandasamy P, Peebles RS Jr., Murphy RC, Voelker DR, Phosphatidylglycerol provides short-term prophylaxis against respiratory syncytial virus infection, J. Lipid Res 54 (2013) 2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kandasamy P, Numata M, Berry KZ, Fickes R, Leslie CC, Murphy RC, Voelker DR, Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling, J. Lipid Res 57 (2016) 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Numata M, Kandasamy P, Nagashima Y, Fickes R, Murphy RC, Voelker DR, Phosphatidylinositol inhibits respiratory syncytial virus infection, J. Lipid Res 56 (2015) 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kandasamy P, Zarini S, Chan ED, Leslie CC, Murphy RC, Voelker DR, Pulmonary surfactant phosphatidylglycerol inhibits mycoplasma pneumoniae-stimulated eicosanoid production from human and mouse macrophages, J. Biol. Chem 286 (2011) 7841–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Watson A, Madsen J, Clark HW, SP-A and SP-D: dual functioning immune molecules with antiviral and immunomodulatory properties, Front. Immunol 11 (2020), 622598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fessler MB, Summer RS, Surfactant lipids at the host-environment Interface. Metabolic sensors, suppressors, and effectors of inflammatory lung disease, Am. J. Respir. Cell Mol. Biol 54 (2016) 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ji J, Sun L, Luo Z, Zhang Y, Xianzheng W, Liao Y, Tong X, Shan J, Potential therapeutic applications of pulmonary surfactant lipids in the host defence against respiratory viral infections, Front. Immunol 12 (2021), 730022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Watson A, Phipps MJS, Clark HW, Skylaris CK, Madsen J, Surfactant proteins a and D: trimerized innate immunity proteins with an affinity for viral fusion proteins, J. Innate Immun 11 (2019) 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cañadas O, Olmeda B, Alonso A, Pérez-Gil J, Lipid—protein and protein—protein interactions in the pulmonary surfactant system and their role in lung homeostasis, Int. J. Mol. Sci 21 (2020) 3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Seaton BA, Crouch EC, McCormack FX, Head JF, Hartshorn KL, Mendelsohn R, Review: structural determinants of pattern recognition by lung collectins, Innate Immun. 16 (2010) 143–150. [DOI] [PubMed] [Google Scholar]
- [24].Piboonpocanun S, Chiba H, Mitsuzawa H, Martin W, Murphy RC, Harbeck RJ, Voelker DR, Surfactant protein a binds mycoplasma pneumoniae with high affinity and attenuates its growth by recognition of disaturated phosphatidylglycerols, J. Biol. Chem 280 (2005) 9–17. [DOI] [PubMed] [Google Scholar]
- [25].Liekkinen J, Enkavi G, Javanainen M, Olmeda B, Perez-Gil J, Vattulainen I, Pulmonary surfactant lipid reorganization induced by the adsorption of the oligomeric surfactant protein B complex, J. Mol. Biol 432 (2020) 3251–3268. [DOI] [PubMed] [Google Scholar]
- [26].Ingenito EP, Mora R, Mark L, Pivotal role of anionic phospholipids in determining dynamic behavior of lung surfactant, Am. J. Respir. Crit. Care Med 161 (2000) 831–838. [DOI] [PubMed] [Google Scholar]
- [27].Ikegami M, Grant S, Korfhagen T, Scheule RK, Whitsett JA, (2009) surfactant protein-D regulates the postnatal maturation of pulmonary surfactant lipid pool sizes, J. Appl. Physiol 106 (1985) 1545–1552. [DOI] [PubMed] [Google Scholar]
- [28].Milad N, Morissette MC, Revisiting the role of pulmonary surfactant in chronic inflammatory lung diseases and environmental exposure, Eur. Respir. Rev 30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Labarrere CA, Kassab GS, Pattern recognition proteins: first line of defense against coronaviruses, Front. Immunol 12 (2021), 652252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M, Cutting edge: the immunostimulatory activity of the lung surfactant protein-a involves toll-like receptor 4, J. Immunol 168 (2002) 5989–5992. [DOI] [PubMed] [Google Scholar]
- [31].Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, Takahashi T, Imaizumi H, Asai Y, Kuroki Y, Direct binding of toll-like receptor 2 to zymosan, and zymosan-induced NF-kappa B activation and TNF-alpha secretion are down-regulated by lung collectin surfactant protein a, J. Immunol 171 (2003) 417–425. [DOI] [PubMed] [Google Scholar]
- [32].Griese M, Pulmonary surfactant in health and human lung diseases: state of the art, Eur. Respir. J 13 (1999) 1455–1476. [DOI] [PubMed] [Google Scholar]
- [33].Whitsett JA, Wert SE, Weaver TE, Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease, Annu. Rev. Med 61 (2010) 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nogee LM, Alterations in SP-B and SP-C expression in neonatal lung disease, Annu. Rev. Physiol 66 (2004) 601–623. [DOI] [PubMed] [Google Scholar]
- [35].Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK, Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer, Am. J. Hum. Genet 84 (2009) 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Beers MF, Mulugeta S, The biology of the ABCA3 lipid transporter in lung health and disease, Cell Tissue Res. 367 (2017) 481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M, ABCA3 gene mutations in newborns with fatal surfactant deficiency, N. Engl. J. Med 350 (2004) 1296–1303. [DOI] [PubMed] [Google Scholar]
- [38].Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM, ABCA3 mutations associated with pediatric interstitial lung disease, Am. J. Respir. Crit. Care Med 172 (2005) 1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Doan ML, Guillerman RP, Dishop MK, Nogee LM, Langston C, Mallory GB, Sockrider MM, Fan LL, Clinical, radiological and pathological features of ABCA3 mutations in children, Thorax 63 (2008) 366–373. [DOI] [PubMed] [Google Scholar]
- [40].Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, Carey BC, Filippi MD, Wert SE, Denson LA, Puchalski JT, Hauck DM, Trapnell BC, GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis, N. Engl. J. Med 356 (2007) 567–579. [DOI] [PubMed] [Google Scholar]
- [41].Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC, GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1, Immunity 15 (2001) 557–567. [DOI] [PubMed] [Google Scholar]
- [42].Hashimoto M, Wang DY, Kamo T, Zhu Y, Tsujiuchi T, Konishi Y, Tanaka M, Sugimura H, Isolation and localization of type IIb Na/Pi cotransporter in the developing rat lung, Am. J. Pathol 157 (2000) 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Traebert M, Hattenhauer O, Murer H, Kaissling B, Biber J, Expression of type II Na-P(i) cotransporter in alveolar type II cells, Am. J. Phys 277 (1999) L868–L873. [DOI] [PubMed] [Google Scholar]
- [44].Haczku A, Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation, J Allergy Clin Immunol 122 (2008) 861–879, quiz 880-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Floros J, Thorenoor N, Tsotakos N, Phelps DS, Human surfactant protein SP-A1 and SP-A2 variants differentially affect the alveolar microenvironment, surfactant structure, regulation and function of the alveolar macrophage, and animal and human survival under various conditions, Front. Immunology 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sánchez-Barbero F, Rivas G, Steinhilber W, Casals C, Structural and functional differences among human surfactant proteins SP-A1, SP-A2 and co-expressed SP-A1/SP-A2: role of supratrimeric oligomerization, Biochem. J 406 (2007) 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sorensen GL, Surfactant protein D in respiratory and non-respiratory diseases, Front. Med. (Lausanne) 5 (2018) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Han S, Mallampalli RK, The role of surfactant in lung disease and host defense against pulmonary infections, Ann. Am. Thorac. Soc 12 (2015) 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Devendra G, Spragg RG, Lung surfactant in subacute pulmonary disease, Respir. Res 3 (2002) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cheng G, Ueda T, Numao T, Kuroki Y, Nakajima H, Fukushima Y, Motojima S, Fukuda T, Increased levels of surfactant protein a and D in bronchoalveolar lavage fluids in patients with bronchial asthma, Eur. Respir. J 16 (2000) 831–835. [DOI] [PubMed] [Google Scholar]
- [51].Erpenbeck VJ, Schmidt R, Gunther A, Krug N, Hohlfeld JM, Surfactant protein levels in bronchoalveolar lavage after segmental allergen challenge in patients with asthma, Allergy 61 (2006) 598–604. [DOI] [PubMed] [Google Scholar]
- [52].Hite RD, Seeds MC, Bowton DL, Grier BL, Safta AM, Balkrishnan R, Waite BM, Bass DA, Surfactant phospholipid changes after antigen challenge: a role for phosphatidylglycerol in dysfunction, Am. J. Physiol. Lung Cell Mol. Physiol 288 (2005) L610–L617. [DOI] [PubMed] [Google Scholar]
- [53].Fahy E, Sud M, Cotter D, Subramaniam S, LIPID MAPS online tools for lipid research, Nucleic Acids Res. 35 (2007) W606–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Watts A, Harlos K, Maschke W, Marsh D, Control of the structure and fluidity of phosphatidylglycerol bilayers by pH titration, Biochim. Biophys. Acta 510 (1978) 63–74. [DOI] [PubMed] [Google Scholar]
- [55].Borges-Araujo L, Fernandes F, Structure and lateral organization of phosphatidylinositol 4,5-bisphosphate, Molecules 25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Voelker DR, Numata M, Phospholipid regulation of innate immunity and respiratory viral infection, J. Biol. Chem 294 (2019) 4282–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mueller M, Brandenburg K, Dedrick R, Schromm AB, Seydel U, Phospholipids inhibit lipopolysaccharide (LPS)-induced cell activation: a role for LPS-binding protein, J. Immunol 174 (2005) 1091–1096. [DOI] [PubMed] [Google Scholar]
- [58].Medzhitov R, Toll-like receptors and innate immunity, Nat. Rev. Immunol 1 (2001) 135–145. [DOI] [PubMed] [Google Scholar]
- [59].Aderem A, Ulevitch RJ, Toll-like receptors in the induction of the innate immune response, Nature 406 (2000) 782–787. [DOI] [PubMed] [Google Scholar]
- [60].Kawai T, Akira S, Toll-like receptors and their crosstalk with other innate receptors in infection and immunity, Immunity 34 (2011) 637–650. [DOI] [PubMed] [Google Scholar]
- [61].Hashimoto M, Asai Y, Ogawa T, Treponemal phospholipids inhibit innate immune responses induced by pathogen-associated molecular patterns, J. Biol. Chem 278 (2003) 44205–44213. [DOI] [PubMed] [Google Scholar]
- [62].Nakamura M, Chu HW, Chan ED, Voelker DR, Surfactant phospholipids suppress the inflammation of bronchial epithelium induced by TLR2 and TLR3 ligands, Proc. Am. Thorac. Soc 5 (2008) 367–368. [Google Scholar]
- [63].Williams CR, Chen L, Driver AD, Arnold EA, Sheppard ES, Locklin J, Krause DC, Sialylated receptor setting influences mycoplasma pneumoniae attachment and gliding motility, Mol. Microbiol 109 (2018) 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Fickes RN, Innate Immune Activity of Xylitol-headgroup Lipid Analogs of the Anionic Pulmonary Surfactant Phospholipids, University of Colorado Denver, Anschutz Medical Campus, 2016. A thesis submitted to the Faculty of the Graduate School of the University of Colorado for the degree of Doctor of Philosophy Pharmacology Program; 2016. [Google Scholar]
- [65].Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD, Incidence and outcomes of acute lung injury, N. Engl. J. Med 353 (2005) 1685–1693. [DOI] [PubMed] [Google Scholar]
- [66].Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM, Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury, Cell 133 (2008) 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rallabhandi P, Phillips RL, Boukhvalova MS, Pletneva LM, Shirey KA, Gioannini TL, Weiss JP, Chow JC, Hawkins LD, Vogel SN, Blanco JC, Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2, mBio 3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, Chen WH, Ernst RK, Rossignol DP, Gusovsky F, Blanco JC, Vogel SN, The TLR4 antagonist eritoran protects mice from lethal influenza infection, Nature 497 (2013) 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Griffiths C, Drews SJ, Marchant DJ, Respiratory syncytial virus: infection, detection, and new options for prevention and treatment, Clin. Microbiol. Rev 30 (2017) 277–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE, Respiratory syncytial virus infection in elderly and high-risk adults, N. Engl. J. Med 352 (2005) 1749–1759. [DOI] [PubMed] [Google Scholar]
- [71].Smith DW, Frankel LR, Mathers LH, Tang AT, Ariagno RL, Prober CG, A controlled trial of aerosolized ribavirin in infants receiving mechanical ventilation for severe respiratory syncytial virus infection, N. Engl. J. Med 325 (1991) 24–29. [DOI] [PubMed] [Google Scholar]
- [72].Subramanian KN, Weisman LE, Rhodes T, Ariagno R, Sanchez PJ, Steichen J, Givner LB, Jennings TL, Top FH Jr., Carlin D, Connor E, Safety, tolerance and pharmacokinetics of a humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. MEDI-493 study group, Pediatr. Infect. Dis. J 17 (1998) 110–115. [DOI] [PubMed] [Google Scholar]
- [73].Griffin MP, Yuan Y, Takas T, Domachowske JB, Madhi SA, Manzoni P, Simoes EAF, Esser MT, Khan AA, Dubovsky F, Villafana T, DeVincenzo JP, Nirsevimab Study G, Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants, N Engl J Med 383 (2020) 415–425. [DOI] [PubMed] [Google Scholar]
- [74].Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW, Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus, Nat. Immunol 1 (2000) 398–401. [DOI] [PubMed] [Google Scholar]
- [75].Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, Vogel SN, Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent, Mucosal Immunol. 3 (2010) 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, Kurt-Jones EA, Finberg RW, Respiratory syncytial virus activates innate immunity through toll-like receptor 2, J. Virol 83 (2009) 1492–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Moore ML, Chi MH, Luongo G, Lukacs NW, Polosukhin VV, Huckabee MM, Newcomb DC, Buchholz UJ, Crowe JE Jr., Goleniewska K, Williams JV, Collins PL, Peebles RS Jr., A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction, J. Virol 83 (2009) 4185–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lukacs NW, Moore ML, Rudd BD, Berlin AA, Collins RD, Olson SJ, Ho SB, Peebles RS Jr., Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus, Am. J. Pathol 169 (2006) 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hallman M, Merritt TA, Bry K, The fate of exogenous surfactant in neonates with respiratory distress syndrome, Clin. Pharmacokinet 26 (1994) 215–232. [DOI] [PubMed] [Google Scholar]
- [80].Numata M, Grinkova YV, Mitchell JR, Chu HW, Sligar SG, Voelker DR, Nanodiscs as a therapeutic delivery agent: inhibition of respiratory syncytial virus infection in the lung, Int. J. Nanomedicine 8 (2013) 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].A.V.I.T. Novel Swine-Origin Influenza, Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM, Emergence of a novel swine-origin influenza A (H1N1) virus in humans, N. Engl. J. Med 360 (2009) 2605–2615. [DOI] [PubMed] [Google Scholar]
- [82].Epperson S, Blanton L, Kniss K, Mustaquim D, Steffens C, Wallis T, Dhara R, Leon M, Perez A, Chaves SS, Elal AA, Gubareva L, Xu X, Villanueva J, Bresee J, Cox N, Finelli L, Brammer L, N.C.f.I. Influenza Division, C.D.C. Respiratory Diseases, Influenza activity - United States, 2013-14 season and composition of the 2014-15 influenza vaccines, MMWR Morb Mortal Wkly Rep 63 (2014) 483–490. [PMC free article] [PubMed] [Google Scholar]
- [83].C. Centers for Disease and Prevention, Update: infections with a swine-origin influenza A (H1N1) virus—United States and other countries, April 28, 2009, MMWR Morb Mortal Wkly Rep 58 (2009) 431–433. [PubMed] [Google Scholar]
- [84].Morens DM, Taubenberger JK, Fauci AS, Pandemic influenza viruses—hoping for the road not taken, N. Engl. J. Med 368 (2013) 2345–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, Bi Y, Wu Y, Li X, Yan J, Liu W, Zhao G, Yang W, Wang Y, Ma J, Shu Y, Lei F, Gao GF, Origin and diversity of novel avian influenza a H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses, Lancet 381 (2013) 1926–1932. [DOI] [PubMed] [Google Scholar]
- [86].Glezen WP, Clinical practice. Prevention and treatment of seasonal influenza, N. Engl. J. Med 359 (2008) 2579–2585. [DOI] [PubMed] [Google Scholar]
- [87].Moscona A, Neuraminidase inhibitors for influenza, N. Engl. J. Med 353 (2005) 1363–1373. [DOI] [PubMed] [Google Scholar]
- [88].Dharan NJ, Gubareva LV, Meyer JJ, Okomo-Adhiambo M, McClinton RC, Marshall SA, St George K, Epperson S, Brammer L, Klimov AI, Bresee JS, Fry AM, Oseltamivir-Resistance Working G, Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States, JAMA 301 (2009) 1034–1041. [DOI] [PubMed] [Google Scholar]
- [89].Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, Kimura K, Hayden FG, Sugaya N, Kawaoka Y, Resistant influenza a viruses in children treated with oseltamivir: descriptive study, Lancet 364 (2004) 759–765. [DOI] [PubMed] [Google Scholar]
- [90].Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, Ishida T, Sekino G, Yamada K, Portsmouth S, Kawaguchi K, Shishido T, Arai M, Tsuchiya K, Uehara T, Watanabe A, G. Baloxavir Marboxil Investigators, Baloxavir marboxil for uncomplicated influenza in adults and adolescents, N. Engl. J. Med 379 (2018) 913–923. [DOI] [PubMed] [Google Scholar]
- [91].Numata M, Mitchell JR, Tipper JL, Brand JD, Trombley JE, Nagashima Y, Kandasamy P, Chu HW, Harrod KS, Voelker DR, Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models, J. Biol. Chem 295 (2020) 1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Belser JA, Eckert AM, Tumpey TM, Maines TR, Complexities in ferret influenza virus pathogenesis and transmission models, Microbiol. Mol. Biol. Rev 80 (2016) 733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Touman A, Khayat M, Bulkhi A, Khairo M, Alyamani W, Ghaleb N, Ashi H, Alsobai M, Alqurashi E, Chronic Parenchymal Lung Changes After COVID-19 Infection, 2021. [DOI] [PMC free article] [PubMed]
- [94].C.f.D. Control and Prevention, SARS-CoV-2 Variant Classifications and Definitions, Centers for Disease Control and Prevention, Atlanta, GA, 2021. [Google Scholar]
- [95].Twohig KA, Nyberg T, Zaidi A, Thelwall S, Sinnathamby MA, Aliabadi S, Seaman SR, Harris RJ, Hope R, Lopez-Bernal J, Gallagher E, Charlett A, De Angelis D, Presanis AM, Dabrera G, Consortium C-GU, Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study, Lancet Infect. Dis 22 (2022) 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Vaidyanathan G, Coronavirus variants are spreading in India - what scientists know so far, Nature 593 (2021) 321–322. [DOI] [PubMed] [Google Scholar]
- [97].Team CC-R, SARS-CoV-2 B.1.1.529 (Omicron) Variant - United States, December 1-8, 2021, MMWR Morb Mortal Wkly Rep 70 (2021) 1731–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T, Group CS, Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine, N. Engl. J. Med 384 (2021) 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Perez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW Jr., Hammitt LL Jr., Tureci O Jr., Nell H Jr., Schaefer A Jr., Unal S Jr., Tresnan DB Jr., Mather S Jr., Dormitzer PR Jr., Sahin U Jr., Jansen KU Jr., Gruber WC Jr., Group C. C. T., Safety and Efficacy of the BNT162b2 mRNA Covid 19 Vaccine, N. Engl. J. Med 383 (2020) 2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sacks HS, The single-dose J&J vaccine had 67% efficacy against moderate to severe-critical COVID-19 at >/=14 d, Ann. Intern. Med 174 (2021) JC75. [DOI] [PubMed] [Google Scholar]
- [101].Lopez Bernal J, Andrews N, Gower C, Gallagher E, Simmons R, Thelwall S, Stowe J, Tessier E, Groves N, Dabrera G, Myers R, Campbell CNJ, Amirthalingam G, Edmunds M, Zambon M, Brown KE, Hopkins S, Chand M, Ramsay M, Effectiveness of Covid-19 vaccines against the B.1.617.2 (Delta) variant, N. Engl. J. Med 385 (2021) 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, Martin-Quiros A, Caraco Y, Williams-Diaz A, Brown ML, Du J, Pedley A, Assaid C, Strizki J, Grobler JA, Shamsuddin HH, Tipping R, Wan H, Paschke A, Butterton JR, Johnson MG, De Anda C, Group MO-OS, Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients, N. Engl. J. Med 386 (2022) 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Li D, Sempowski GD, Saunders KO, Acharya P, Haynes BF, SARS-CoV-2 neutralizing antibodies for COVID-19 prevention and treatment, Annu Rev Med 73 (2021) 1–16. [DOI] [PubMed] [Google Scholar]
- [104].Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, Musser BJ, Soo Y, Rofail D, Im J, Perry C, Pan C, Hosain R, Mahmood A, Davis JD, Turner KC, Hooper AT, Hamilton JD, Baum A, Kyratsous CA, Kim Y, Cook A, Kampman W, Kohli A, Sachdeva Y, Graber X, Kowal B, DiCioccio T, Stahl N, Lipsich L, Braunstein N, Herman G, Yancopoulos GD, Trial I, REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19, N. Engl. J. Med 384 (2021) 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Harcourt J, Tamin A, Lu X, Kamili S, Sakthivel SK, Murray J, Queen K,Tao Y, Paden CR, Zhang J, Li Y, Uehara A, Wang H, Goldsmith C, Bullock HA, Wang L, Whitaker B, Lynch B, Gautam R, Schindewolf C, Lokugamage KG, Scharton D, Plante JA, Mirchandani D, Widen SG, Narayanan K, Makino S, Ksiazek TG, Plante KS, Weaver SC, Lindstrom S, Tong S, Menachery VD, Thornburg NJ, Severe acute respiratory syndrome coronavirus 2 from patient with coronavirus disease, United States, Emerg. Infect. Dis 26 (2020) 1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Numata M, Kandasamy P, Nagashima Y, Posey J, Hartshorn K, Woodland D, Voelker DR, Phosphatidylglycerol suppresses influenza a virus infection, Am. J. Respir. Cell Mol. Biol 46 (2012) 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Thompson BT, Chambers RC, Liu KD, Acute respiratory distress syndrome, N. Engl. J. Med 377 (2017) 562–572. [DOI] [PubMed] [Google Scholar]
- [108].Fajgenbaum DC, June CH, Cytokine storm, N. Engl. J. Med 383 (2020) 2255–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Stasi C, Fallani S, Voller F, Silvestri C, Treatment for COVID-19: an overview, Eur. J. Pharmacol 889 (2020), 173644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Group R. C. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T, Jeffery K, Montgomery A, Rowan K, Juszczak E, Baillie JK, Haynes R, Landray MJ, Dexamethasone in hospitalized patients with Covid-19, N. Engl. J. Med 384 (2021) 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Baas T, Giving RSV some POP(G), in: Science-Business eXchange 3, 2010, pp.4–5 [Google Scholar]
- [112].Numata-Nakamura M, Bowen R, Voelker DR, Pulmonary surfactant phospholipids as novel anti-virals against SARS-CoV-2 infection, Am. J. Respir. Crit. Care Med (2022). In press. [Google Scholar]