

Abstract
Purpose
Long non-coding RNAs (lncRNAs) control gene expression at multiple levels. By interacting with microRNAs (miRNAs), they regulate their mRNA targets creating dynamic regulatory networks involved in different cellular processes. Their role in follicle development and oocyte maturation has recently emerged. lncRNA deregulation has been found associated with different pathological conditions. In this study, we identified differentially expressed lncRNAs in cumulus cells (CCs) isolated from MII oocytes of advanced maternal age women and proposed ceRNA-networks involved in signaling pathways crucial in ovarian folliculogenesis and female germ cell maturation.
Methods
We performed a high-throughput analysis of the expression profile of 68 lncRNAs from CCs of aged and young women by using NanoString technology. By miRNet, TarPmiR, miRTarBase, OKdb, and KEGG we predicted some ceRNA-networks involving the differentially expressed (DE) lncRNAs, miRNA interactors, and their mRNA target genes.
Results
We identified 28 lncRNAs down-regulated in CC samples from aged women. The analysis revealed that the miRNAs binding 11 of the DE lncRNAs and their mRNA targets are included in ceRNA-networks involved in the regulation of the PI3K-Akt, FOXO, and p53 signaling pathways.
Conclusion
We proposed that the lncRNA down-regulation in CCs from aged women could influence the expression of genes encoding proteins deregulated in reproductive aging. A better understanding of the interplay of lncRNA-miRNA-mRNA networks in human CCs could increase our knowledge about the mechanisms of regulation of gene expression involved in aging, lead to the development of novel therapeutics, and improve reproductive outcomes in aged women.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10815-022-02446-8.
Keywords: Regulation of gene expression, lncRNAs, Cumulus cells, Reproductive aging, ceRNA-networks
Introduction
In the last few decades, numerous studies have reported with convincing evidence the pervasive transcription across 70–90% of the human genome. Considering that less than 2% of the total genome encodes protein-coding genes, non-coding RNAs (ncRNAs) represent most of the human transcriptome. The term “non-coding RNAs” is referred to RNA molecules that do not encode proteins but this does not mean that they do not have any functions or do not contain any important information [1].
ncRNAs are classified into two major classes based on their size: small non-coding RNAs, smaller than 200 nucleotides, comprising transfer RNAs, microRNAs (miRNAs), small-interfering RNAs, small nuclear RNAs, nucleolar RNAs, PIWI-interacting RNAs and transcription initiation RNAs, promoter upstream transcripts, and promoter-associated small RNAs, and long non-coding RNAs (lncRNAs) ranging in length from 200 nucleotides to 100 kilobases or more [2].
lncRNAs represent the most heterogeneous class of ncRNAs involved in a large spectrum of molecular mechanisms regulating genome functions and the generation of complex networks of RNA-RNA competitive interaction [3]. Although lncRNAs were initially considered to not have any functions, recent studies have revealed that they cooperate in gene regulation at multiple levels, from transcriptional and post-transcriptional levels to translational and post-translational levels, up to epigenetic and cell signaling modulation [1, 4–6]. Based on their multiple functions, it is not surprising that lncRNA dysregulation may affect the genesis and/or progression of pathological conditions [1] and, in the last few years, it was also observed in the development and maturation of the ovarian follicle [7]. It has been demonstrated that specific expression patterns of lncRNAs could be used as promising biomarkers of diseases [8], and, in general, it appears clear that the non-coding transcriptome is of crucial importance in determining the greater complexity of higher eukaryotes and disease pathogenesis [9].
Numerous studies have demonstrated that several interactions among RNA molecules, such as lncRNAs and miRNAs, miRNAs and mRNAs, and lncRNAs and mRNAs, exist [10]. These molecules collaborate to create a dynamic regulatory network of lncRNAs acting as competing endogenous RNAs (ceRNAs). In fact, it is known that lncRNAs are considered the main component of ceRNA-networks that regulate mRNA expression by sponging miRNAs and thus reducing their availability to bind mRNA target [11, 12].
Instances of ceRNA crosstalk are widespread in a very large number of contexts and they have been observed in both normal and pathological conditions, demonstrating that the spread of the mechanism, together with all the other layers of gene regulation, could allow understanding molecular and cellular mechanisms [3]. There is also a good deal of evidence of ceRNA-networks in embryonic stem cells [13]. However, despite studies describing ceRNA interactions, little is known about the molecular conditions of the optimal activity related to their intracellular expression [3].
Over the last few years, human oocytes and cumulus cells (CCs) have been widely studied; however, in this context, lncRNA molecular functions are still not fully understood [14, 15]. Mature and competent oocytes are generated by ovarian folliculogenesis. In this context, a precise and coordinated dialogue exists between somatic follicle cells (mural granulosa cells and CCs) and germinal cells. CCs are in physical contact with the oocyte creating a specialized structure, the cumulus-oocyte complex (COC). Communication between the oocyte and the surrounding CCs is necessary for follicular compartment development and the acquisition of oocyte competence [16].
It is known that maternal age can influence not only the expression patterns of mRNAs, proteins, and miRNAs in human CC samples but also those of lncRNAs [17–23]. Distinctive miRNA expression profiles, correlating with age and ovarian stimulation protocols, were recently characterized and higher concentrations and increased numbers of miRNAs were recorded in younger than in advanced maternal age patients, regardless of treatment [17, 24]. Functional and expression studies, performed to retrieve common miRNome profiles, have found an enrichment of biological functions in oocyte growth and maturation, embryo development, steroidogenesis, ovarian hyperstimulation, apoptosis and cell survival, glucagon and lipid metabolism, and cell trafficking. The highest scored pathways of target genes of the commonly expressed miRNAs were associated with MAPK signaling pathway [17].
It has been demonstrated that some lncRNAs are involved in processes associated with oocyte maturation and follicle development and some of them are under-expressed in CC samples from advanced maternal age women [7]. Specifically, these altered lncRNAs are involved in many functional processes mostly associated with angiogenesis, lipid transport, cell cycle regulation, and transcriptional control [7]. Despite these findings, little is known about the interaction among the different RNA molecules in CCs and about the ceRNA-networks regulating folliculogenesis.
The identification of ceRNA-networks dysregulated in CCs of MII oocytes from advanced maternal age women could represent a promising tool to deepen the biology of aging oocytes and to evaluate the efficacy of different stimulation protocols. The CC transcriptomic analysis and the ceRNA-network studies could allow us to better understand the regulation of the dialogue between oocyte and CCs and to propose candidate biomarkers to evaluate IVF (in vitro fertilization) success in aged couples [25]. Here, we analyzed the expression of 68 lncRNAs in CC samples from aged and young women by high-throughput analysis (NanoString technology). Then, we computationally predicted the miRNAs binding the differently expressed (DE) lncRNAs, and we selected the validated miRNA target genes, filtering them by their expression in CCs, their involvement in antral follicle growth, and their expression regulated by both FSH and LH/hCG. Finally, we identified the potential ceRNA-networks involved in molecular pathways altered in aging. In this way, we provided a start point for a better understanding of reproductive aging, for the study of future applications in infertility treatments, and the improvement of the reproductive outcome in aged women.
Materials and methods
Patient population
Participants enrolled in this study were women having undergone ovarian stimulation protocol and intracytoplasmatic sperm injection (ICSI) at IVF Center Cannizzaro Hospital Catania (Italy). The women had been treated with gonadotrophin-releasing hormone agonists (triptorelin 0.1 mg/day or buserelin 1 mg/day) until human chorionic gonadotropin (HCG) trigger to induce multiple follicular development; ovarian stimulation was then carried out using recombinant FSH (follitropin alfa) in combination with human menopausal gonadotrophin (HMG). Stimulation was monitored using serum estradiol concentrations and ultrasound measurement of follicle number and diameter. Ovulation was induced with 10,000 IU uHCG only when follicles had reached a diameter greater than 18 mm and serum estradiol concentration per follicle reached 150–200 ng/L.
Healthy women whose primary infertility was caused by male factors were included in the study; pathologies that could influence oocyte quality, such as endometriosis, polycystic ovary, and ovarian insufficiency, were excluded from the study as well as heavy smokers and overweight women. Our research followed the tenets of the Declaration of Helsinki and the patients gave their informed consent to use the CCs for the study.
We enrolled 12 women grouped by age; six women are aged between 27 and 33 years (young women) and six women are aged between 40 and 44 years (advanced maternal age women). We excluded women between the ages of 34 and 38, as they represent the category with the greatest variability. Even if chronological age represents the most important factor influencing the female reproductive potential, reproductive aging, determined by both genetic and environmental factors, varies considerably among individuals. Basic and clinical information for all participants are presented in Table 1.
Table 1.
Clinical parameters of women enrolled in the study
| Parameters | Young women | Advanced maternal age women | p-Value |
|---|---|---|---|
| Age (years) | 29.3 ± 2.2 | 42.2 ± 1.5 | < 0.00001 |
| r-hFSH (IU) HMG (IU) | 1904.9 ± 838.4 320 ± 227.3 | 4037.5 ± 1230.9 637.5 ± 240.7 | 0.004 0.03 |
| N COCs retrieved | 12.3 ± 5.9 | 8.7 ± 3.7 | 0.1 |
| N MII oocytes | 10.2 ± 5.2 | 6 ± 2.3 | 0.05 |
| N CCs processed | 4.3 ± 1.5 | 4.3 ± 0.5 | 0.5 |
Values are reported as mean ± standard deviation. The p-values are based on two-sample t-test
Cumulus cell collection
Three to seven CC samples per patient were removed from the oocytes mechanically, using needles, and set aside individually. The complete removal of the cumulus and corona cells continued, always individually, using a combination of enzymatic (hyaluronidase) and mechanical (pipetting) methods, in order to identify oocytes in metaphase II (MII). Only those from the MII oocytes were washed in the culture medium and immediately frozen at − 20 °C. A total of 52 CC samples were collected from 52 MII oocytes.
RNA extraction
Total RNA isolation and purification was performed by TRIzol® (Thermo Fisher Scientific), according to the manufacturer’s instructions and quantified through a spectrophotometer. Samples showing a 260–280 ratio absorbance of approximately 2.0 were considered.
High-throughput lncRNA expression analysis
The NanoString nCounter system was used to analyze the expression profile of 68 lncRNAs from CC samples by using NanoString technology and customized nCounter XT CodeSet gene expression assay (NanoString Technologies, Seattle, WA, USA), according to the manufacturer’s instructions. In particular, 68 lncRNAs and 4 housekeeping genes (B2M, GAPDH, HPRT1, and PPIA) were tested. lncRNA expression profiling was performed on 6 CC samples from advanced maternal age women and 6 from young women (Table 1). Approximately 100 ng of RNA in a final volume of 3 μL was used. Briefly, a master mix comprising hybridization buffer and reporter CodeSet was prepared and added to each sample in the initial steps of the hybridization reaction. The capture ProbeSet was added to conclude hybridization. The processing of samples was performed by the automated nCounter Prep Station. At this point, samples were purified and immobilized on a sample cartridge for quantification and data collection by using the nCounter Digital Analyzer. Raw data normalization was performed using nSolver 3.0 software and applying the normalization method according to the user manual instructions (https://www.nanostring.com/products/analysis-software/nsolver). More in detail, raw data normalization was performed combining the geometric mean of positive controls and the mean of B2M, HPRT1, and PPIA housekeeping genes. We did not consider GAPH since it did not pass the data quality control check. In addition, for the same reason, one sample from the group of aged women and one sample from the group of young women were excluded from the analysis.
Selection of lncRNAs
The customized nCounter XT CodeSet gene expression assay comprising 68 lncRNAs and 4 housekeeping genes was used for expression data normalization. lncRNAs and reference genes were selected according to data reported in the literature and bioinformatic analysis. More in detail, 18 lncRNAs were selected through accurate reading of the most recent reviews and experimental studies reported in the literature about lncRNAs associated with reproduction, female infertility, CCs, and epigenetics [7, 17–19, 25–30]. The remaining 50 lncRNAs were chosen after bioinformatic analysis of 3 different array-based expression datasets present in the GEO DataSets repository (GSE86427, GSE113239, and GSE81579).
In silico analyses
In order to propose ceRNA-networks comprising the differentially expressed (DE) lncRNAs, their miRNA targets, and miRNA target genes, many prediction analyses of interactions were performed by using different online databases and tools. The applied workflow is shown in Fig. 1. First, the interactions between the DE lncRNAs and their miRNA targets were predicted by using miRNet 2.0 (https://www.mirnet.ca/). The identified lncRNA-miRNA interactions were further confirmed by the identification of the miRNA binding sites within the specific isoform of each lncRNA sequence by using TarPmiR software (http://hulab.ucf.edu/research/projects/miRNA/TarPmiR/). lncRNA-miRNA interactions reported by miRNet and also found in TarPmiR involving at least 3 or four binding sites were selected. Successively, the list of miRNA target genes of Homo sapiens was obtained by querying the miRTarBase database (https://mirtarbase.cuhk.edu.cn/~miRTarBase/miRTarBase_2022/php/index.php) and filtered based on the expression of genes in cumulus cells, their involvement in antral follicle growth, and their expression regulated by both FSH and LH/hCG as reported by the Ovarian Kaleidoscope Database (OKdb; http://ovary.stanford.edu/). Lastly, the KEGG database (Encyclopedia of Genes and Genomes) (https://www.genome.jp/kegg/) was queried to obtain the list of genes involved in specific signaling pathways that are altered in aging and it was matched with the list of miRNA target genes previously selected from the miRTarBase and OKdb. ceRNA-networks were built by using Cytoscape 3.8.2 software.
Fig. 1.

Workflow to identify the ceRNA-networks involved in molecular pathways altered in reproductive aging
Statistical analysis
Statistical analysis of high-throughput lncRNA expression profiling was performed by applying significance analysis of microarray (SAM) statistical test using MeV (Multi experiment Viewer) software v4.8.1. All statistical SAM tests were computed by applying a two-class unpaired test among log2FC and using a q-value based on all unique permutations: imputation engine: K-nearest neighbors and number of K-nearest neighbors: 10 neighbors and false discovery rate (FDR) < 0.05.
Results
Expression profiling of lncRNAs in CC samples
High-throughput lncRNA expression analysis revealed 28 DE lncRNAs down-regulated in CC samples from women of advanced maternal age vs young women. The statistical significance of their differential expression was assessed by SAM statistical analysis (Fig. 2).
Fig. 2.
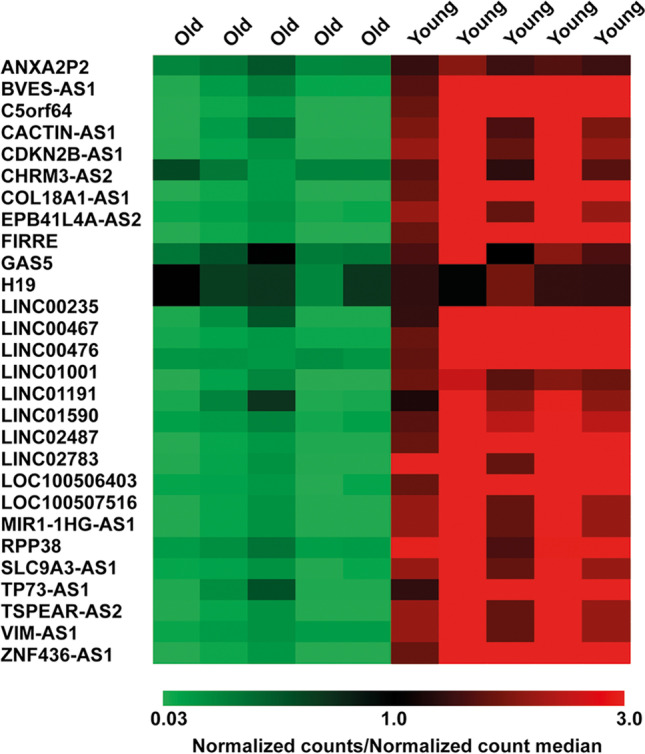
lncRNA expression profiling. Heatmap of DE lncRNAs. DE lncRNAs were down-regulated in advanced maternal age vs young CC samples. Values are reported as the ratio between the normalized counts of each lncRNA in every CC sample and the median value of normalized counts of each lncRNA in all samples
Functional enrichment analyses
Functional enrichment analyses showed that most of the validated target genes of miRNAs interacting with DE lncRNAs are expressed in CCs, that they are involved in the regulation of follicle development, and that their expression is regulated by FSH or LH/hCG treatments (Tables 2, 3, and 4), as reported by OKdb. In addition, some of these miRNA-target genes are involved in the regulation of signaling pathways altered in aging, such as the PI3K-Akt signaling pathway, p53 signaling pathway, and FOXO signaling pathway as reported by KEGG database data analysis (Tables S1–S3).
Table 2.
DE lncRNAs interact with miRNAs whose target genes are expressed in CCs, as reported by OKdb
| Cumulus cell localization | ||
|---|---|---|
| lncRNA | miRNA | Target gene |
| CDKN2B-AS1 | let-7b-5p | CCND2, TGFBR1, TLR4 |
| let-7c-5p | MTOR, STAT3, TGFBR1 | |
| let-7e-5p | AURKB | |
| let-7i-5p | AURKB, TLR4 | |
| H19 | miR-193a-3p | MCL1, TGFB2 |
| miR-93-5p | TGFBR2 | |
| LINC00467 | miR-92a-3p | RAD21, STAT3 |
| LINC01001 | let-7c-5p | MTOR, STAT3, TGFBR1 |
| miR-128-3p | MAPK14, MTOR, TGFBR1 | |
| miR-433-3p | CREB1 | |
| miR-98-5p | CCND2, CYP19A1 | |
| SLC9A3-AS1 | miR-134-5p | ITGB1 |
| miR-214-3p | CTNNB1, PAPPA, XBP1 | |
| miR-34a-5p | INHBB, LDHA | |
| ZNF436-AS1 | let-7 g-5p | TGFBR1 |
Table 3.
DE lncRNAs interact with miRNAs whose target genes are involved in follicle development, as reported by OKdb
| Follicle development | ||
|---|---|---|
| lncRNA | miRNA | Target gene |
| CDKN2B-AS1 | let-7b-5p | CCND2, HMGA2, IRS2, LGR4, TGFBR1 |
| let-7c-5p | HMGA2, IL6, MTOR, RICTOR, TGFBR1 | |
| let-7e-5p | FASLG, HMGA2, IGF1 | |
| let-7i-5p | IGF1 | |
| miR-302a-3p | AKT1, CDKN1A, GAB2, PTEN | |
| GAS5 | miR-138-5p | AKT1, RELN, SIRT1, TERT, YAP1 |
| miR-532-5p | RUNX3, TERT | |
| H19 | miR-138-5p | AKT1, RELN, SIRT1, TERT, YAP1 |
| miR-193a-3p | PTEN, TGFB2 | |
| miR-93-5p | CDKN1A, CXCL8, PTEN, TGFBR2, TP53INP1 | |
| LINC00235 | miR-423-3p | CDKN1A |
| LINC00467 | miR-92a-3p | ESR2, HDAC2, PTEN, RAD21, SIRT1 |
| LINC00476 | miR-409-3p | AKT1, MET |
| LINC01001 | let-7c-5p | HMGA2, IL6, MTOR, RICTOR, TGFBR1 |
| let-7d-5p | HMGA2 | |
| miR-128-3p | EGFR, IGF1, MTOR, PTEN, RELN, RICTOR, SIRT1, TGFBR1 | |
| miR-28-5p | CDKN1A, IGF1 | |
| miR-345-5p | CDKN1A | |
| miR-433-3p | MET | |
| miR-98-5p | CCND2, CYP19A1, HMGA2, HMGA2, IGF1, IL6 | |
| SLC9A3-AS1 | miR-134-5p | ITGB1 |
| miR-214-3p | CTNNB1, PAPPA, PTEN | |
| miR-34a-5p | AKT1, AR, BMP7, FOS, HDAC1, MET, NOTCH1, RICTOR, SIRT1, SMAD4 | |
| miR-449a | FOS, HDAC1, MET, NOTCH1, SIRT1 | |
| TSPEAR-AS2 | miR-708-5p | AKT1, SMAD3 |
| ZNF436-AS1 | let-7 g-5p | GAB2, HMGA2, TGFBR1, THBS1 |
Table 4.
DE lncRNAs interact with miRNAs whose target gene expression is regulated by treatment with FSH or LH/hCG, as reported by OKdb
| Expression regulated by FSH and LH/hCG | ||
|---|---|---|
| lncRNA | miRNA | Target gene |
| CDKN2B-AS1 | let-7b-5p | CCND2 |
| let-7c-5p | CASP3, IL6, MTOR, MYC | |
| let-7e-5p | FASLG, IGF1, MMP9 | |
| let-7i-5p | IGF1 | |
| miR-302a-3p | AKT1, CDK4 | |
| GAS5 | miR-138-5p | AKT1, CASP3, HIF1A, YAP1 |
| H19 | miR-138-5p | AKT1, CASP3, HIF1A, YAP1 |
| miR-193a-3p | MCL1, MMP14, PLAU, TGFB2 | |
| miR-93-5p | CXCL8, VEGFA | |
| LINC00467 | miR-92a-3p | KLF2 |
| LINC00476 | miR-409-3p | AKT1 |
| LINC01001 | let-7c-5p | CASP3, IL6, MTOR, MYC |
| miR-128-3p | BAX, EGFR, IGF1, MTOR, VEGFC | |
| miR-28-5p | IGF1, MAPK1 | |
| miR-433-3p | CFTR, CREB1, HIF1A | |
| miR-98-5p | CASP3, CCND2, CYP19A1, EDN1, IGF1, IL6, MYC | |
| SLC9A3-AS1 | miR-134-5p | VEGFA |
| miR-214-3p | BAX, MAPK1, PAPPA, PIM1, POR | |
| miR-34a-5p | AKT1, BAX, CDK4, FOS, MYC, PDGFRB, POU5F1 | |
| miR-449a | CDK4, FOS, MYC | |
| miR-744-5p | MYC | |
| TSPEAR-AS2 | miR-708-5p | AKT1, CASP2 |
| VIM-AS1 | miR-197-3p | MAPK1 |
| ZNF436-AS1 | let-7 g-5p | CASP3, MYC, THBS1 |
ceRNA-networks in aging
Among the 28 DE lncRNAs in advanced maternal age women, we found 11 deregulated lncRNAs annotated on miRNet database that we used for building the ceRNA-networks. All the DE lncRNAs are part of 3 ceRNA-networks involved in the PI3K-Akt, FOXO, and p53 signaling pathway control (Figs. 3, 4, and 5). The 3 ceRNA-networks shared 7 lncRNAs: CDKN2B-AS1, LINC00235, LINC00467, LINC01001, H19, SLC9A3-AS1, and ZNF436-AS1. lncRNAs GAS5, LINC00476, and TSPEAR-AS2 are shared by 2 out of the 3 ceRNA-networks. In addition, some miRNA-target genes are common to 3 (CCND2, IGF1, PTEN, and CDKN1A) or two (IL6, FASLG, THBS1, EGFR, and AKT1) of signaling pathways we analyzed.
Fig. 3.

ceRNA-network in the PI3K-Akt signaling pathway. Interaction between DE lncRNAs, miRNAs, and miRNA-target genes involved in the PI3K-Akt signaling pathway. Green ellipses, white rectangles, and red hexagons represent DE lncRNAs, miRNAs, and miRNA-target genes, respectively
Fig. 4.

ceRNA-network in the FOXO signaling pathway. Interaction between DE lncRNAs, miRNAs, and miRNA-target genes involved in the FOXO signaling pathway. Green ellipses, white rectangles, and red hexagons represent DE lncRNAs, miRNAs, and miRNA-target genes, respectively
Fig. 5.

ceRNA-network in the p53 signaling pathway. Interaction between DE lncRNAs, miRNAs, and miRNA-target genes involved in the p53 signaling pathway. Green ellipses, white rectangles, and red hexagons represent DE lncRNAs, miRNAs, and miRNA-target genes, respectively
Discussion
Folliculogenesis is a very complex process involving intra-follicular and oocyte-derived signals to create the most appropriate microenvironment for oocyte development and competence [16]. This proper environment is achieved through the fine-tuned transcriptional and post-transcriptional expression of a plethora of genes and ncRNAs involved in a large spectrum of molecular mechanisms regulating the genome functions [16]. The essential equilibrium in signal production defines sophisticated molecular networks that govern successful fertilization and embryo development. In this regard, communication between the oocyte and its companion CCs is crucial in the acquisition of the development competency of the oocyte [27]. In the last few years, the physio-pathological role of lncRNAs in the development and maturation of ovarian follicles has come to light [7].
In this study, using NanoString technology, we performed the expression analysis of 68 lncRNAs in CCs, in order to associate their expression profiles with reproductive aging. We found 28 down-regulated lncRNAs in CC samples from advanced maternal age vs young women (Fig. 2). The lncRNAs can regulate gene expression as ceRNAs or “sponges” of miRNAs, reducing miRNA availability to bind mRNA targets as shown in Fig. 6 [6]. According to this role, by computational analysis, we identified the miRNAs binding the lncRNAs deregulated in reproductive aging and the mRNA targets expressed in CCs, involved in follicle development and regulated by FSH and LH/hCG (Tables 2, 3, and 4). The computational investigation allowed us to pinpoint 11 lncRNAs whose dysregulation could be related to the altered expression of miRNAs and mRNAs already described in the scientific literature as involved in reproductive aging [17, 28, 29]. In Tables 2, 3, and 4 we showed 11 lncRNAs, their miRNA, and mRNA interactors, these last sorted according to their localization (expressed in CCs), biological role (follicle development), and their regulation (regulated by FSH and LH/hCG).
Fig. 6.

Model of ceRNA-network regulation. General regulation mechanism of ceRNA-network involving lncRNA, miRNA, and mRNA molecules (A). The down-regulation of lncRNAs in CCs from aged women could determine a major abundance of miRNAs that are able to bind to their mRNA target genes causing their down-regulation and the decrease of protein expression. On the contrary, in CCs from young women, the up-regulation of lncRNAs could determine an increase in the expression miRNA-target genes and their related proteins (B)
Interestingly, these lncRNAs are part of 3 ceRNA-networks that can act inside of the PI3K-Akt, FOXO, and p53 signaling pathways (Figs. 3, 4, and 5). These signaling pathways have been well characterized in follicular development and oocyte maturation acting in different moments of folliculogenesis. In fact, they control cellular growth and proliferation, oxidative stress, cellular senescence, and apoptosis. Alterations in their regulation have been extensively demonstrated in reproductive aging [31–33].
The PI3K-Akt pathway is involved in different steps of follicular maturation both in the gonadotropin-independent phase and in the gonadotropin-responsive phase. In granulosa cells, its activation contributes to the acquisition of oocyte maturation during follicle development. PI3K catalyzes the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) and converts it to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits Akt. Akt is inhibited by PTEN (phosphatase and tensin homolog 10), a negative regulator of PI3K, that converts PIP3 back to PIP2 [31]. The pathway activation promotes cell survival by inhibiting pro-apoptotic proteins, such as BAX, BAD, and TP53; activating anti-apoptotic proteins, such as BCL2; and stimulating protein synthesis and cell growth by the mammalian target of rapamycin (mTOR). The PI3K-Akt pathway activation negatively controls the FOXO signaling pathway; in fact, Akt can phosphorylate FOXO3a inactivating it. FOXO3a is a transcription factor and induces the expression of genes involved in cell death and cellular growth triggering apoptosis and inhibiting proliferation [34]. FOXO3a up-regulation maintains the pool number of primordial follicles and the physiological functions of the ovarian reserve. Furthermore, FOXO3 protein can regulate follicle growth and atresia by promoting apoptosis of the granulosa cells and oocytes [34]. Its inactivation, mediated by Akt, inhibits oocyte apoptosis and contrasts the effects of reactive oxygen species (ROS) [33]. A high level of oxidative stress activates a specific p53 transcriptional response, which can lead to cell cycle arrest, apoptosis, or senescence. Alternatively, as we proposed in a recent paper, the increase of vesicle secretion in follicular fluid, mediated by p53, alters miRNA and lncRNA concentration inside the cells. This could represent a stress-response mechanism, implemented by follicular cells, to repress p53 expression and the apoptosis [20].
In order to identify the most significant lncRNAs whose down-regulation can influence oocyte quality, we focused our attention on the lncRNAs able to regulate the higher number of hubs and competing with mRNAs encoding key proteins for follicular maturation and whose deregulation has been extensively studied in aging. CDKN2B-AS1, LINC00235, LINC00467, LINC01001, H19, SLC9A3-AS1, and ZNF436-AS1 are located in the 3 networks analyzed. GAS5, LINC00476, and TSPEAR-AS2 are located in 2 out of 3 networks, inside the PI3K-Akt and FOXO signaling pathways (Figs. 3, 4, and 5).
The lncRNA H19, down-regulated in CCs from aged women, is shared by the 3 networks. H19 belongs to a highly conserved, imprinted gene cluster involved in embryonic development and growth control. It is expressed during embryogenesis and repressed in most adult tissues, except for the ovary, uterus, skeletal muscle, and heart [35]. H19 acts as proto-oncogene in different tumors increasing cell viability, growth, invasion, and epithelial-mesenchymal transition [36]. H19 has also been found to take part in the miRNA-mediated network of gene regulation by influencing the activity of the downstream mRNAs that facilitate the aggressive phenotypes of these tumors [36]. However, in pituitary adenomas, H19 is down-regulated showing tumor suppressor activity and it has been demonstrated that its down-regulation in CCs is closely associated with decreased ovarian reserve [30, 37].
Among the miRNAs interacting with H19, we found miR-93-5p and miR-193a-3p targeting PTEN and miR-138-5p targeting SIRT1. Another down-regulated lncRNA, able to control PTEN and SIRT1, is LINC01001 (long intergenic non-protein coding RNA 1001), a little-known gene, centrally located in the 3 networks controlling different mRNAs. Among these, it is able to regulate PTEN and SIRT1 sequestering different miRNAs, including some members of the let7 family. The ceRNA-networks involving H19 and LINC01001 could determine the down-regulation of PTEN and SIRT1, already described in reproductive aging. In fact, the down-regulation of the lncRNAs could cause the up-regulation of the miRNA targets which in turn could determine the down-regulation of PTEN and SIRT1 mRNAs. The tumor-suppressor PTEN represses cell survival, proliferation, and metastasis through controlling different pathways, and its down-regulation has been related to cancer and other diseases as obesity and autism spectrum disorders [38]. In female reproduction, PTEN, regulating AKT signaling pathways, is involved in different steps of follicle maturation and oocyte-specific PTEN deletion increases primordial follicle activation and may be associated with accelerated clearance of follicles leading to primordial follicle pool exhaustion [33]. PTEN expression can be regulated by various events, including genomic mutation or deletion, transcriptional, epigenetic, post-transcriptional, and post-translational modulation. It has recently been demonstrated that some lncRNAs epigenetically or post-transcriptionally regulate PTEN expression [38]. It is known that SIRT1 regulates proliferation and apoptosis in granulosa cells and its down-regulation is associated with a reduced ovarian reserve. In addition, SIRT1 senses DNA damage with the scope to preserve telomere integrity from oxidative stress and it was demonstrated as biomarker of ovarian aging [39]. It has also been demonstrated that loss of SIRT1 is associated with premature aging [33, 39].
Inside these pathways, the relative expression levels of cellular brakes, apoptotic proteins, and survival factors will establish the fate of the ovarian follicle, determining also the oocyte quality. The expression levels are controlled by the networks involving the regulatory ncRNAs. These networks are extremely complex and redundant: a single mRNA can be targeted by different miRNAs, which in turn could interact with different lncRNAs, and a single lncRNA could sponge several miRNAs regulating different mRNAs (Figs. 3, 4, and 5). Even if this complexity makes difficult to understand if the down-regulation of the lncRNAs is the cause or the effect of mRNA altered expression, we suggest that some lncRNA down-regulation in CCs from advanced maternal age women, causing the altered expression of the proteins involved in the follicular maturation, could explain the poor oocyte quality in reproductive aging (Fig. 6).
Conclusion
In conclusion, we identified some lncRNAs down-regulated in CCs from aged women and proposed some ceRNA-networks involved in the control of signaling pathways critical in follicular maturation. Certainly, to confirm the truthfulness of computational predictions, experimental validation by biochemical techniques (miRNA pull-down, immunoprecipitation, microRNA capture affinity technology) and functional assays (loss-of-function and gain-of-function assays) will be needed. Despite it all, we believe that this work represents an important starting point for further investigation, in order to understand the complex mechanisms of regulation of gene expression involved in reproductive aging. Moreover, a better understanding of the interplay of lncRNA-miRNA-mRNA networks in human CCs could lead to the development of novel therapeutics to treat infertility and improve reproductive outcomes in advanced maternal age women.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank the Scientific Bureau of the University of Catania for the language support, the Centro Servizi B.R.I.T. of the University of Catania and Fondi di Ateneo 2020–2022, University of Catania, Open Access line.
Author contribution
A.C. performed the experiments, analyzed and interpreted the data, and wrote the manuscript. R.B. and C.F. performed the experiments and analyzed and interpreted the data. M.E.V., P.B., and M.P. collected the CC samples and clinical data. P.S. and M.P. revised critically the manuscript. S.L. and T.D.H. participated in the study design and revised critically the manuscript. D.V. participated in the study design and coordination. C.D.P. participated in the study design, coordination, data analysis, and interpretation and wrote the manuscript. All authors have read and approved the manuscript.
Funding
This study was funded by Merck KGaA, Darmstadt, Germany.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest except S.L. and T.D.H. who are fully employed by Merck KGaA.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Angela Caponnetto, Email: angela.caponnetto@unict.it.
Rosalia Battaglia, Email: rosalia.battaglia@unict.it.
Carmen Ferrara, Email: carmen.ferrara@phd.unict.it.
Maria Elena Vento, Email: mrln.vento@gmail.com.
Placido Borzì, Email: pla.borzi@gmail.com.
Marianna Paradiso, Email: marpar121284@gmail.com.
Paolo Scollo, Email: paolo.6giugno@gmail.com.
Michele Purrello, Email: purrello@unict.it.
Salvatore Longobardi, Email: salvatore.longobardi@merckgroup.com.
Thomas D’Hooghe, Email: thomas.dhooghe@merckgroup.com.
Domenico Valerio, Email: dvalgen@gmail.com.
Cinzia Di Pietro, Email: dipietro@unict.it.
References
- 1.Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018;109(7):2093–2100. doi: 10.1111/cas.13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dahariya S, Paddibhatla I, Kumar S, Raghuwanshi S, Pallepati A, Gutti RK. Long non-coding RNA: classification, biogenesis and functions in blood cells. Mol Immunol. 2019;112:82–92. doi: 10.1016/j.molimm.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Tay Y, Rinn J, Pandolfi PP. 2014 The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52. 10.1038/nature12986. [DOI] [PMC free article] [PubMed]
- 4.Zhang X, Wang W, Zhu W, Dong J, Cheng Y, Yin Z, et al. 2019 Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int J Mol Sci. 2019;20(22). 10.3390/ijms20225573. [DOI] [PMC free article] [PubMed]
- 5.Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21(5):542–551. doi: 10.1038/s41556-019-0311-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouckenheimer J, Fauque P, Lecellier CH, Bruno C, Commes T, Lemaitre JM, et al. Differential long non-coding RNA expression profiles in human oocytes and cumulus cells. Sci Rep. 2018;8(1):2202. doi: 10.1038/s41598-018-20727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolha L, Ravnik-Glavac M, Glavac D. Long noncoding RNAs as biomarkers in cancer. Dis Markers. 2017;2017:7243968. doi: 10.1155/2017/7243968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lekka E, Hall J. Noncoding RNAs in disease. FEBS Lett. 2018;592(17):2884–2900. doi: 10.1002/1873-3468.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen R, Ghosal S, Das S, Balti S, Chakrabarti J. Competing endogenous RNA: the key to posttranscriptional regulation. ScientificWorldJournal. 2014;2014:896206. doi: 10.1155/2014/896206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niu ZS, Wang WH, Dong XN, Tian LM. Role of long noncoding RNA-mediated competing endogenous RNA regulatory network in hepatocellular carcinoma. World J Gastroenterol. 2020;26(29):4240–4260. doi: 10.3748/wjg.v26.i29.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22(2):96–118. doi: 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42(12):1113–1117. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu XF, Li J, Cao YX, Chen DW, Zhang ZG, He XJ, et al. Differential expression of long noncoding RNAs in human cumulus cells related to embryo developmental potential: a microarray analysis. Reprod Sci. 2015;22(6):672–678. doi: 10.1177/1933719114561562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Battaglia R, Vento ME, Borzi P, Ragusa M, Barbagallo D, Arena D, et al. Non-coding RNAs in the ovarian follicle. Front Genet. 2017;8:57. doi: 10.3389/fgene.2017.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutt KJ, Albertini DF. An oocentric view of folliculogenesis and embryogenesis. Reprod Biomed Online. 2007;14(6):758–764. doi: 10.1016/s1472-6483(10)60679-7. [DOI] [PubMed] [Google Scholar]
- 17.Dell’Aversana C, Cuomo F, Longobardi S, D’Hooghe T, Caprio F, Franci G, et al. Age-related miRNome landscape of cumulus oophorus cells during controlled ovarian stimulation protocols in IVF cycles. Hum Reprod. 2021;36(5):1310–1325. doi: 10.1093/humrep/deaa364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Edani T, Assou S, Ferrieres A, Bringer Deutsch S, Gala A, Lecellier CH, et al. Female aging alters expression of human cumulus cells genes that are essential for oocyte quality. Biomed Res Int. 2014;2014:964614. doi: 10.1155/2014/964614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McReynolds S, Dzieciatkowska M, McCallie BR, Mitchell SD, Stevens J, Hansen K, et al. 2012 Impact of maternal aging on the molecular signature of human cumulus cells. Fertil Steril. 2012;98(6):1574–80 e5. 10.1016/j.fertnstert.2012.08.012. [DOI] [PubMed]
- 20.Battaglia R, Musumeci P, Ragusa M, Barbagallo D, Scalia M, Zimbone M, et al. 2020 Ovarian aging increases small extracellular vesicle CD81(+) release in human follicular fluid and influences miRNA profiles. Aging (Albany NY). 2020;12(12):12324–41. 10.18632/aging.103441. [DOI] [PMC free article] [PubMed]
- 21.Guglielmino MR, Santonocito M, Vento M, Ragusa M, Barbagallo D, Borzi P, et al. TAp73 is downregulated in oocytes from women of advanced reproductive age. Cell Cycle. 2011;10(19):3253–3256. doi: 10.4161/cc.10.19.17585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battaglia R, Vento ME, Ragusa M, Barbagallo D, La Ferlita A, Di Emidio G, et al. MicroRNAs are stored in human MII oocyte and their expression profile changes in reproductive aging. Biol Reprod. 2016;95(6):131. doi: 10.1095/biolreprod.116.142711. [DOI] [PubMed] [Google Scholar]
- 23.Santonocito M, Guglielmino MR, Vento M, Ragusa M, Barbagallo D, Borzi P, et al. The apoptotic transcriptome of the human MII oocyte: characterization and age-related changes. Apoptosis. 2013;18(2):201–211. doi: 10.1007/s10495-012-0783-5. [DOI] [PubMed] [Google Scholar]
- 24.Andrei D, Nagy RA, van Montfoort A, Tietge U, Terpstra M, Kok K, et al. Differential miRNA expression profiles in cumulus and mural granulosa cells from human pre-ovulatory follicles. Microrna. 2019;8(1):61–67. doi: 10.2174/2211536607666180912152618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng L, Teerds K, Tang Z, Zuo B, Hong L. Editorial: non-coding RNAs in reproductive biology. Front Cell Dev Biol. 2021;9:712467. doi: 10.3389/fcell.2021.712467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yerushalmi GM, Salmon-Divon M, Yung Y, Maman E, Kedem A, Ophir L, et al. Characterization of the human cumulus cell transcriptome during final follicular maturation and ovulation. Mol Hum Reprod. 2014;20(8):719–735. doi: 10.1093/molehr/gau031. [DOI] [PubMed] [Google Scholar]
- 27.Liu K, Mao X, Chen Y, Li T, Ton H. Regulatory role of long non-coding RNAs during reproductive disease. Am J Transl Res. 2018;10(1):1–12. [PMC free article] [PubMed] [Google Scholar]
- 28.Bouckenheimer J, Assou S, Riquier S, Hou C, Philippe N, Sansac C, et al. Long non-coding RNAs in human early embryonic development and their potential in ART. Hum Reprod Update. 2016;23(1):19–40. doi: 10.1093/humupd/dmw035. [DOI] [PubMed] [Google Scholar]
- 29.Ernst EH, Nielsen J, Ipsen MB, Villesen P, Lykke-Hartmann K. Transcriptome analysis of long non-coding RNAs and genes encoding paraspeckle proteins during human ovarian follicle development. Front Cell Dev Biol. 2018;6:78. doi: 10.3389/fcell.2018.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Wang J, Fan Y, Qin C, Xia X, Johnson J, et al. Absence of the long noncoding RNA H19 results in aberrant ovarian STAR and progesterone production. Mol Cell Endocrinol. 2019;490:15–20. doi: 10.1016/j.mce.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Felici M, Klinger FG. 2021 PI3K/PTEN/AKT signaling pathways in germ cell development and their involvement in germ cell tumors and ovarian dysfunctions. Int J Mol Sci. 2021;22(18). 10.3390/ijms22189838. [DOI] [PMC free article] [PubMed]
- 32.Makker A, Goel MM, Mahdi AA. PI3K/PTEN/Akt and TSC/mTOR signaling pathways, ovarian dysfunction, and infertility: an update. J Mol Endocrinol. 2014;53(3):R103–R118. doi: 10.1530/JME-14-0220. [DOI] [PubMed] [Google Scholar]
- 33.Maidarti M, Anderson RA, Telfer EE. 2020 Crosstalk between PTEN/PI3K/Akt signalling and DNA damage in the oocyte: implications for primordial follicle activation, oocyte quality and ageing. Cells. 2020;9(1). 10.3390/cells9010200. [DOI] [PMC free article] [PubMed]
- 34.Zhang H, Lin F, Zhao J, Wang Z. Expression regulation and physiological role of transcription factor FOXO3a during ovarian follicular development. Front Physiol. 2020;11:595086. doi: 10.3389/fphys.2020.595086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia X, Burn MS, Chen Y, Karakaya C, Kallen A. The relationship between H19 and parameters of ovarian reserve. Reprod Biol Endocrinol. 2020;18(1):46. doi: 10.1186/s12958-020-00578-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Qi M, Fei X, Wang X, Wang K. lncRNA H19: a novel oncogene in multiple cancers. Int J Biol Sci. 2021;17(12):3188–3208. doi: 10.7150/ijbs.62573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu ZR, Yan L, Liu YT, Cao L, Guo YH, Zhang Y, et al. Inhibition of mTORC1 by lncRNA H19 via disrupting 4E-BP1/Raptor interaction in pituitary tumours. Nat Commun. 2018;9(1):4624. doi: 10.1038/s41467-018-06853-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Zhang T, Guo L, Huang L. Regulation of PTEN expression by noncoding RNAs. J Exp Clin Cancer Res. 2018;37(1):223. doi: 10.1186/s13046-018-0898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carafa V, Nebbioso A, Altucci L. Sirtuins and disease: the road ahead. Front Pharmacol. 2012;3:4. doi: 10.3389/fphar.2012.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.