

Abstract
Both MLL-AF9 and MLL-ENL leukemia fusion proteins drive oncogenic transformation of hematopoietic cells through their N-terminal DNA/histone binding mixed-lineage leukemia 1 domain and their C-terminal fragment of AF9 or ENL containing a unstructured linker region and the ANC1 homology domain, which recruits transcription factors. Despite of their structural similarity, acute myeloid leukemia (AML) patients bearing MLL-ENL show more adverse outcomes compared to those with MLL-AF9. We recapitulated the clinical patterns of these two MLL-fusions driven AMLs using murine models and found that MLL-ENL AML cells showed slower cell cycle progression and more resistance to standard chemotherapy than MLL-AF9 cells. These phenotypes were primarily controlled by the linker regions of ENL and a highly conserved lysine residue K469 within. Substitution of K469 with an acetylated mimic glutamine abolished the ability of MLL-ENL to suppress proliferation and promote chemo-resistance. We showed that deacetylase Sirt2 might act as an upstream regulator of MLL-ENL. Deletion of Sirt2 promoted proliferation of AML cells with either MLL fusions. Importantly, loss of Sirt2 greatly enhanced the sensitivity of the MLL-ENL AML cells to chemo-treatment. Taken together, our study uncovered a unique regulatory role of Sirt2 in leukemogenesis and suggested targeting SIRT2 as a new way to sensitize MLL-ENL AML patience for chemotherapy.
INTRODUCTION
The mixed-lineage leukemia 1 (MLL1) protein is a member of lysine [K]-specific methyltransferase family that regulates hematopoietic development and regeneration through histone modification on target gene promotors. MLL1 deletion causes severe defects in hematopoietic generation in early embryo and self-renewal of hematopoietic stem cells (HSCs) in adult [1,2]. MLL1 localizes on chromosome 11q23 and the chromosome rearrangements of the region is accounted for approximately 10% of all acute leukemias [3]. MLL-AF9 and MLL-ENL fusions are the two most common types of MLL-fusions in acute myeloid leukemia (AML) [4]. AF9 and ENL are highly homologous, and both contains a conserved ANC1 homology domain (AHD) [5]. AHD of AF9 and ENL interacts with the same set of factors including AF4, DOT1L, CBX8 and BCOR, which in turn recruit corresponding chromatin regulatory complexes, such as super elongation complex (SEC), the DOT1L complex and PRC1 etc to influence transcription [6–12]. Notably, all above complexes are critical for MLL fusions-induced leukemia transformation and deletion of any one impairs leukemogenesis. Likewise, AHD of both MLL-AF9 and MLL-ENL fusion proteins interacts with the same partner proteins and recruits the same regulatory complexes, thus they are critical to induce leukemia development. Intriguingly, despite of the structural similarity between MLL-ENL and MLL-AF9, their leukemogenic capacities are different as shown in clinical studies. MLL-AF9 is mostly associated with AML, while MLL-ENL occurs in both acute lymphoid leukemias (ALL) and AML [4,13]. Importantly, MLL-AF9 AML patients have an intermediate outcome, while patients with MLL-ENL display an adverse outcome [14]. However, the underlying mechanism remain largely unknown.
Histone deacetylases play important roles in MLL fusion protein driven AML [15]. Sirtuin-2 (SIRT2) is one of the NAD+-dependent protein lysine deacetylase family proteins. Unlike other sirtuins which normally function in either nucleus (SIRT1, 6 and 7) or mitochondria (SIRT3, 4 and 5), SIRT2 is the only sirtuin that is primarily localized in cytosol and regulates cell cytoskeleton and migration [16,17]. Notably, SIRT2 may serve as either tumor suppressor or oncogenic driver depending on different cancer types. SIRT2 expression is downregulated in many solid tumors such as gliomas, breast cancers and hepatocellular cancers, suggesting that SIRT2 inhibits tumor formation [18]. However, in AML, SIRT2 mRNA expression in CD34+ cells are upregulated [19]. Based on a cohort of 167 AML patients, SIRT2 also shows increased expression in AML with MLL-fusions. Clinically, SIRT2 high-expression patients show significantly shorter overall and event-free survival compared to those with low SIRT2 expression, making it a potential unfavorable biomarker for AML risk stratification [20].
To study mechanisms underlying poorer prognosis of MLL-ENL AML patients, we first recapitulated the clinical patterns of both MLL-ENL and MLL-AF9 driven AMLs using murine models. We revealed that MLL-ENL AML cells showed retard cell cycle progression and are resistant to chemotherapy. We further mapped the unstructured linker of ENL and identified a highly conserved lysine residue K469 that is critical for MLL-ENL functions. To find the upstream regulator of ENL linker region, we surveyed a pool of histone deacetylase inhibitors and identified deacetylase Sirt2 as a candidate. Deletion of SIRT2 promoted proliferation of AML cells with either MLL fusions. Importantly, loss of Sirt2 enhanced the sensitivity of MLL-ENL AML to chemo-treatment. Taken together, our study uncovered a unique regulatory role of Sirt2 in leukemogenesis and we propose that targeting SIRT2 may be a novel strategy to sensitize MLL-ENL AML patience for chemotherapy.
MATERIALS AND METHODS
Animals.
All animals used in this study were raised in individually ventilated cages and under a protocol approved by Shanghai Laboratory Animal Science Association. The C57Bl6/J mice were purchased from Shanghai SLAC Laboratory Animal Co. Sirt2−/− mice were purchased from Jackson laboratory and maintained in C57Bl6/J background in our facility.
Generation of murine AML cells.
Phoenix-ECO cells were co-transfected with retroviral vector containing MLL-fusion gene and package vector. MLL-fusion gene expressing virus were collected from the supernatant and used for HSPC transduction. c-Kit+ HSPCs were isolated from WT or Sirt2−/− mice using mouse cKIT Positive Selection Kit (Stemcell Technologies). c-Kit+ HSPCs were infected with indicated MLL-fusion gene expressing retrovirus. Infected cells were selected with G418 for 3 weeks in MethoCult™ GF M3434 medium for colony assay with weekly replating. Cells were then collected from colonies assay or transplanted into sub-lethally irradiated (500rad) recipient mice by tail vein injection to generate leukemic mice. Each mouse was received 1x106 cells. All mice were monitored daily for leukemia development. AML development was verified by examining AML blasts in peripheral blood. After AML developed, mice were sacrificed and AML cells were isolated from spleens and BM of the corresponding leukemic mice.
The Clone forming assay and proliferation assay.
MLL-fusion gene transduced HSPCs or AML cells were seeded in MethoCult™ GF M3434 medium for colony formation ability studies. To ensure reproducible, we used MoFlo XDP Cell sorter directly sorted 3000 cells into 3.5ml methylcellulose medium. After quick mixing, cells with methylcellulose medium were equally aliquoted into three 35mm dishes. Colony number in each dish was counted on day 5 of culture. To study the proliferation of the MLL-fusion gene transduced HSPCs or AML cells, 1000 cells were sorted into 1ml RPMI 1640 medium supplied with 10% FBS, 100ng/ml SCF 50ng/ml IL-6 30ng/ml IL-3 30ng/ml GM-CSF. For cytarabine sensitivity experiments, 4x104 cells in 200μl medium were used in 96-well plates and treated for 48 hours. Cell numbers were measured every day for 1 week using the MTT assy.
In vivo chemotherapy treatment.
Sub-lethally irradiated C57Bl6/J mice were transplanted with 1x106 MLL-fusion gene transduced HSPCs or 2x104AML cells. After 20 days of transplantation, the mice were randomly divided into two groups with 8 mice in each group. One group was treated with vehicle as control and another one was treated with Doxorubicin 3mg/kg for 3 days and Ara-C 100mg/kg for 5 days. All mice were monitored for leukemia development daily. The AML related death of the mice was summarized in Kaplan-Meier curve. AML development in all mice were verified immediately after the death by examining the AML blasts in BM and peripheral blood and as well as AML blasts infiltration in livers and lungs.
Cell cycle analysis:
2x105/ml of MLL-fusion gene transduced AML cells were seeded into RPMI 1640 medium supplied with 10% FBS, 100ng/ml SCF 50ng/ml IL-6 30ng/ml IL-3 30ng/ml GM-CSF. After cultured for 24 hours, cells were collected and fixed in cold 75% ethanol for 24hour at 4°C, stained with Propidium iodide/RNase Staining Buffer Solution, they subjected to FACS analysis.
Statistical analysis.
Results are given as mean ± standard error of the mean or percentages. Differences in continuous variables were calculated using unpaired 2-tailed Student t test or analysis of variance, as appropriate. *, **, and ** stand for P < 0.05, P <0.01 and P<0.001 respectively.
RESULTS:
The linker region of ENL is critical for regulating proliferation, clone forming capacity and leukemogenic ability of MLL-ENL leukemia.
To compare the leukemogenic capacity of MLL-AF9 and MLL-ENL fusion at the cellular level, we isolated hematopoietic stem/progenitor cells (HSPCs) from gene-transduced AML mouse models commonly used in studying the pathogenesis of MLL-related leukemia. We first compared the survival ability of in vitro cultured MLL-AF9 and MLL-ENL transduced HSPCs and consistently observed that the growth of MLL-ENL-transduced HSPCs to be significantly slower than that of MLL-AF9 -transduced ones (Figure 1c). In addition, colonies from MLL-ENL-transduced HSPCs are smaller after 5-day culture (Figure 1d). Next, we transplanted MLL-AF9 and MLL-ENL transduced HSPCs into sub-lethally irradiated recipient mice by intravenous injection and monitor the AML development (Figure 1b). Consistent with cellular results, MLL-ENL-transduced HSPCs required significant longer latency period for AML development compared to MLL-AF9-transduced HSPCs (Figure 1e), which nicely mirrors the clinical outcomes of patients with these two types of MLL-fusions.
Figure 1. The linker region of ENL represses the proliferation, clone forming capacity and leukemogenic ability of MLL-ENL leukemia.
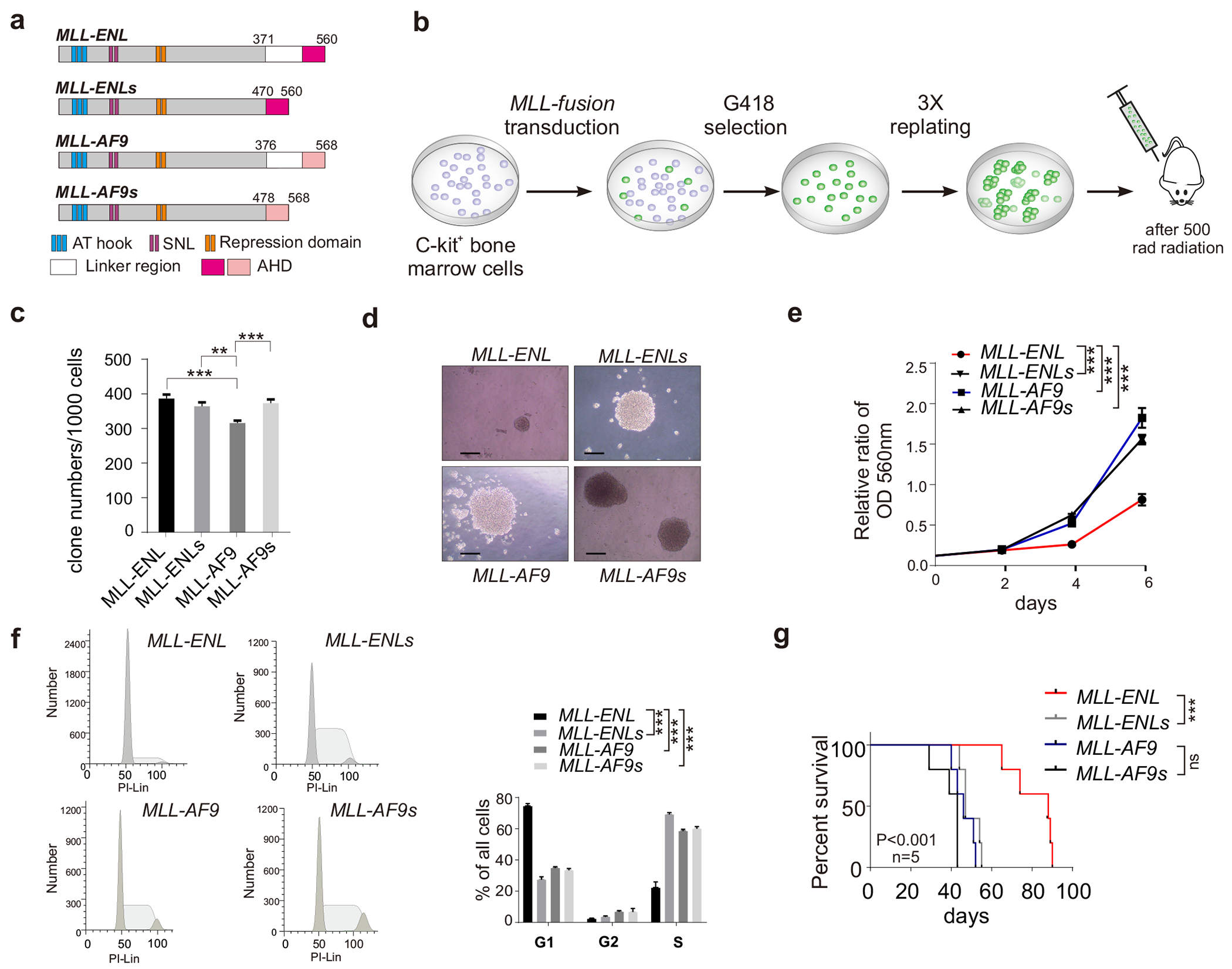
(a) The schematic diagram of MLL-AF9, MLL-AF9s, MLL-ENL and MLL-ENLs fusion proteins; (b) Schematic outline for generating AML mouse model; (c) Clone forming capacity of MLL-fusion AML cells; (d) Representative clone images were presented. The bar scale represents 200μM; (e) Growth curves of the MLL-fusion AML cells were measured by the MTT assay; (f) FACS analysis of MLL-fusion AML cells; (g) Kaplan-Meier survival curve of mice received HSPCs transduced with indicated MLL-fusion genes.
MLL-AF9 and MLL-ENL fusion protein share the same C-terminal MLL fragment and the AF9/ENL fusion part both contain a ~100aa unstructured linker region and an AHD domain. To explore different phenotypes caused by these two fusions, we focused on the linker region and AHD. We constructed two shorter versions of MLL-AF9 and MLL-ENL by removing the linker region, which were referred thereafter as MLL-AF9 short (or MLL-AF9s) and MLL-ENL short (or MLL-ENLs), respectively (Figure 1a) and generated HSPCs lines that stably expressing shorter forms of fusion proteins in Figure 1a. We next created AML mice of with these fusions and AML cells were then collected from bone marrow and spleen of these leukemic mice for MTT proliferation assay. We found that MLL-ENL AML cells show significantly (p<0.001) slower proliferation compared to the other three forms of MLL fusion (Figure 1e).
To further characterize slower proliferation of MLL-ENL cells, we performed cell cycle analysis and revealed that MLL-ENL AML cells exhibit retard cell cycle progression. Comparing to MLL-AF9 AML cells, FACS analysis showed that MLL-ENL AML cells were found mostly in G1/G0 phase and not in G2/M-S phase (Figure 1f). In line with this result, mice that received MLL-ENL AML cells had significant longer latency for AML development compared to those transduced with MLL-AF9 AML cells (Figure 1g). Importantly, removing the linker-region from MLL-ENL restore the proliferation and colony forming capacity of these AML cells to similar levels as MLL-AF9 AML cells (Figure 1e-1g). The colony forming capacity, proliferation, cell cycle and in vivo leukemogenic ability of MLL-ENLs AML cells are all comparable to MLL-AF9 and MLL-AF9s AML cells. Collectively, these data suggested that the linker region in MLL-ENL is critical for its leukemogenic ability.
A lysine residue is critical for the activity of the MLL-ENL linker region.
To understand the unique regulatory function of the linker region of MLL-ENL, we aligned the sequence of the linker regions of ENL and AF9 and found three ENL-specific lysine residues that may be the potential target for post-translationally modification such as acetylation, ubiquitination and methylation. These lysines are highly conserved in all species including rats and mice. However, the corresponding residues in AF9 linker-region are substituted with asparagine (N), leucine (L) and glutamine (Q) respectively (Figure 2a). To dissect the function of K469 in the ENL linker region, we generated a corresponding MLL-ENL-K469Q mutant by replacing this lysine residue with a glutamine, an effective mimic of acetylated lysine [21]. We transduced the mutant retrovirus to c-Kit+ HSPCs and obtained MLL-ENL-K469Q stable expressing lines. Colony formation and MTT assays showed that MLL-ENL-K469Q -transduced HSPCs exhibited markedly increased survival (Figure 2b) as well as cell proliferation (Figure 2c), to a similar level of those of the MLL-ENLs -transduced HSPCs. In line with elevated proliferation, we observed increased population of MLL-ENL-K469Q cells in G2/M phase (Figure 2d). Using transplantation, we found that the leukemogenic ability of MLL-ENL-K469Q -transduced HSPCs was also significantly enhanced compared to MLL-ENL-transduced HSPCs (Figure 2e). The latency time of AML development for mice received MLL-ENL-K469Q -transduced HSPCs was similar to mice received MLL-ENLs-transduced HSPCs (Figure 2e). In summary, our results indicate that the K469 of ENL within the linker-region of MLL-ENL plays a critical role in regulating MLL-ENL functions in vivo.
Figure 2. A lysine residue is critical for the activity of the MLL-ENL linker region.
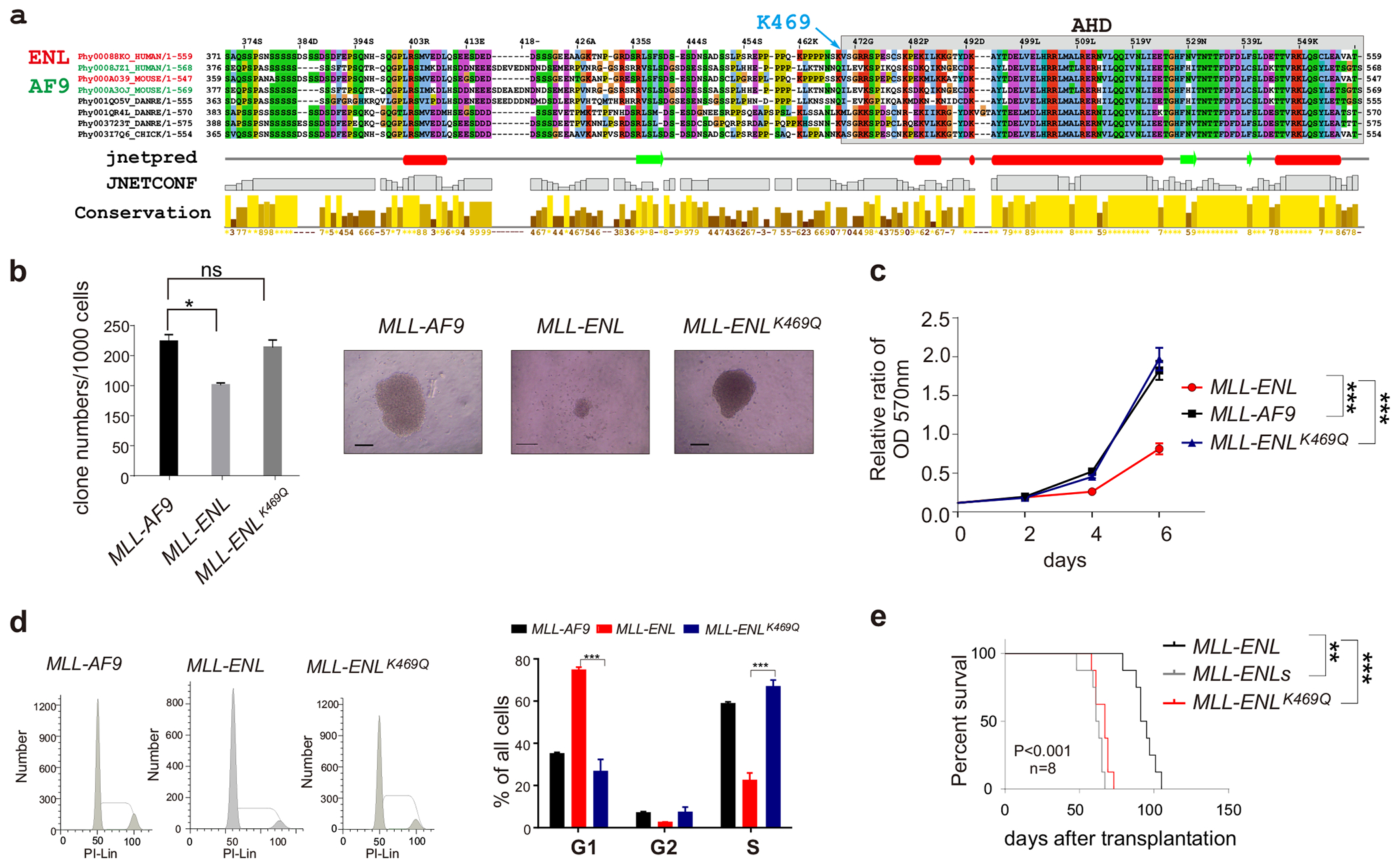
(a) Sequence alignment of the linker region and AHD of human, mouse and rat AF9 and ENL; (b) Clone forming capacity of indicated MLL-fusion AML cells. Representative clone images were presented. The bar scale represents 200μM.; (c) MTT analysis of MLL-fusion AML cells. (d) FACS analysis; (e) Kaplan-Meier survival curve of mice with indicated MLL-fusion genes.
SIRT2 regulates proliferation, clone forming capacity and leukemogenic ability of MLL-ENL leukemia.
We next sought to investigate how the ENL linker-region regulates MLL-ENL leukemogenic ability. Given essential roles of HDAC in leukemia and the critical K469 residue is acetylatable, we thought that acetylation of ENL-linker region might be an important regulatory mechanism. Thus, we tested a small pool of deacetylases inhibitors on their efficacy in inhibiting MLL-ENL AML cells. We treated MLL-fusion AML cells with pan-HDAC inhibitor Vorinostat (SAHA) and Trichostatin A (TSA), selective SIRT1 inhibitor Selisistat (EX 527), selective SIRT1/3 inhibitor 3-TYP and selective SIRT2 inhibitors Thiomyristoyl (TM) and AGK2 and monitored cell survival. We found that the two pan-HDAC inhibitors decreased the survival of both MLL-AF9 and MLL-ENL AML cells, whereas EX 527 and 3-TYP show only minor effects. Interestingly, both SIRT2 inhibitors significantly increased the colony forming capacity of MLL-ENL AML cells but had little effect on MLL-AF9 AML cells (Figure 3a and 3b), suggesting a MLL-ENL specific effect of Sirt2. To directly test the effect of SIRT on MLL fusion-driven AMLs, we generated Sirt2 knock-out AML mouse models, including SIRT2−/− MLL-AF9, Sirt2−/− MLL-AF9s, Sirt2−/− MLL-ENL and Sirt2−/− MLL-ENLs. Deletion of SIRT2 in each AML cells was verified by western blotting (Figure 3c). Sirt2 deletion AML cells showed significantly increased colony forming capacity (Figure 3d) and cell proliferation (Figure 3e) compared to wild-type AML cells. This is in line with increased cell percentage in G2/M-S phase (Figure 3f and 3g). Since deletion of Sirt2 elevated the proliferation, colony forming capacity and leukemogenic ability of MLL-ENL AML cells to a level comparable to MLL-AF9 cells, we speculated that the ENL-linker region associated proliferation repression might be controlled by the SIRT2-mediated deacetylation. We also noted that depletion of SIRT2 promoted the proliferation and survival of MLL-AF9, MLL-AF9s, and MLL-ENLs AML (Figure 3b–f), suggesting that SIRT2 can function through a linker-region-independent mechanism to regulate AML cell proliferation as well.
Figure 3. Sirt2 regulates proliferation, clone forming capacity and leukemogenic ability of MLL-ENL leukemia.
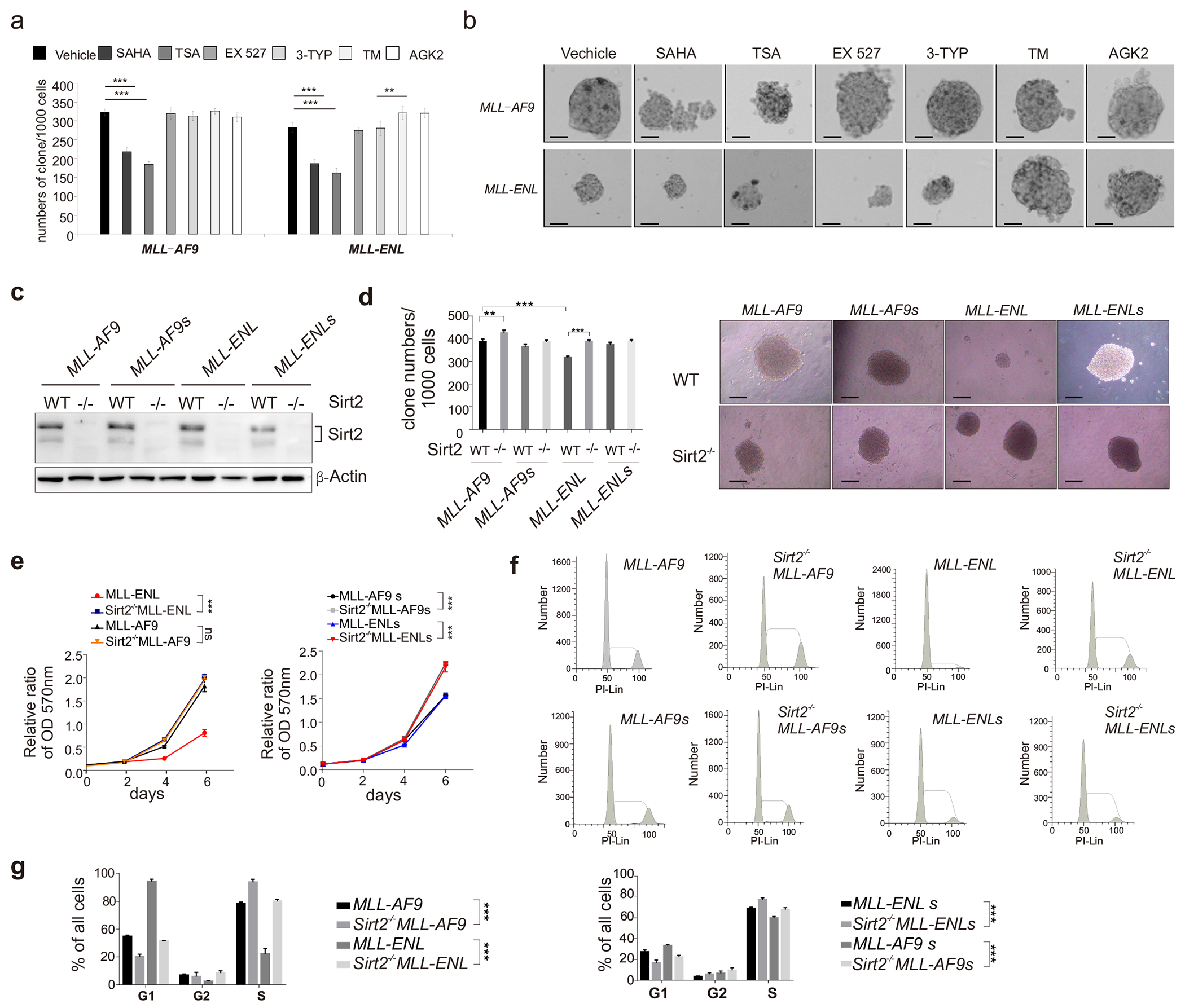
(a-b) Drug treatment of MLL-fusion AML cells: (a) Colony formation; (b) Representative images. The bar scale represents 200μM; (c) Sirt2 knockout was verified in AML cells by western blotting; (d) clone formation of Sirt2 WT and Sirt2−/− AML cells; (e) MTT analysis MLL-fusion AML cells; (f-g) FACS analysis.
Sirt2-mediated deacetylation promotes drug-resistance of MLL-ENL AML cells.
Clinical studies suggest that AML patients with MLL-ENL do not respond to conventional chemotherapies as well as MLL-AF9 patients. Given the essential role of Sirt2 played in MLL-ENL leukemogenesis, we next aim to test if SIRT2 regulates AML drug resistance. We compared the sensitivity of MLL-AF9 and MLL-ENL AML cells to two standard chemotherapy drugs: cytarabine and daunorubicin. MTT assay showed that MLL-ENL AML cells were more resistant to cytarabine than MLL-AF9 cells (Figure 4a). Interestingly, removing the linker region makes MLL-ENL cells more sensitive but have no effect on MLL-AF9 cells (Figure 4a). Next, we generated MLL-AF9 and MLL-ENL transplanted mice, and tested the in vivo response of daunorubicin and cytarabine combination (DA) in these murine AML models. Consistently, we found that the MLL-ENL AML mice were relatively resistant to DA treatment (Figure 4b). DA treatment significantly prolonged the survival of MLL-AF9 transplanted mice, but did not affect the survival of MLL-ENL mice (Figure 4b). Importantly, MLL-ENL AML cells depleted of Sirt2 showed increased sensitive both in vitro (Figure 4c) and in vivo (Figure 4d and 4e) to cytarabine or DA treatment, respectively, underlying the importance of SIRT2 in regulating drug resistance of MLL-ENL AML cells. Remarkably, MLL-ENL-K469Q–transduced HSPCs showed increased sensitive to cytarabine treatment in vitro compared to MLL-ENL–transduced HSPCs (Figure 4f), suggesting that the Sirt2-K469 axis might collectively regulate ENL drug-resistance. Taken together, our study revealed dual regulatory roles of SIRT2 in repressing MLL-ENL AML proliferation while promoting resistance to chemotherapy.
Figure 4. SIRT2 regulates the response of MLL-ENL AML cells to chemotherapy treatment.
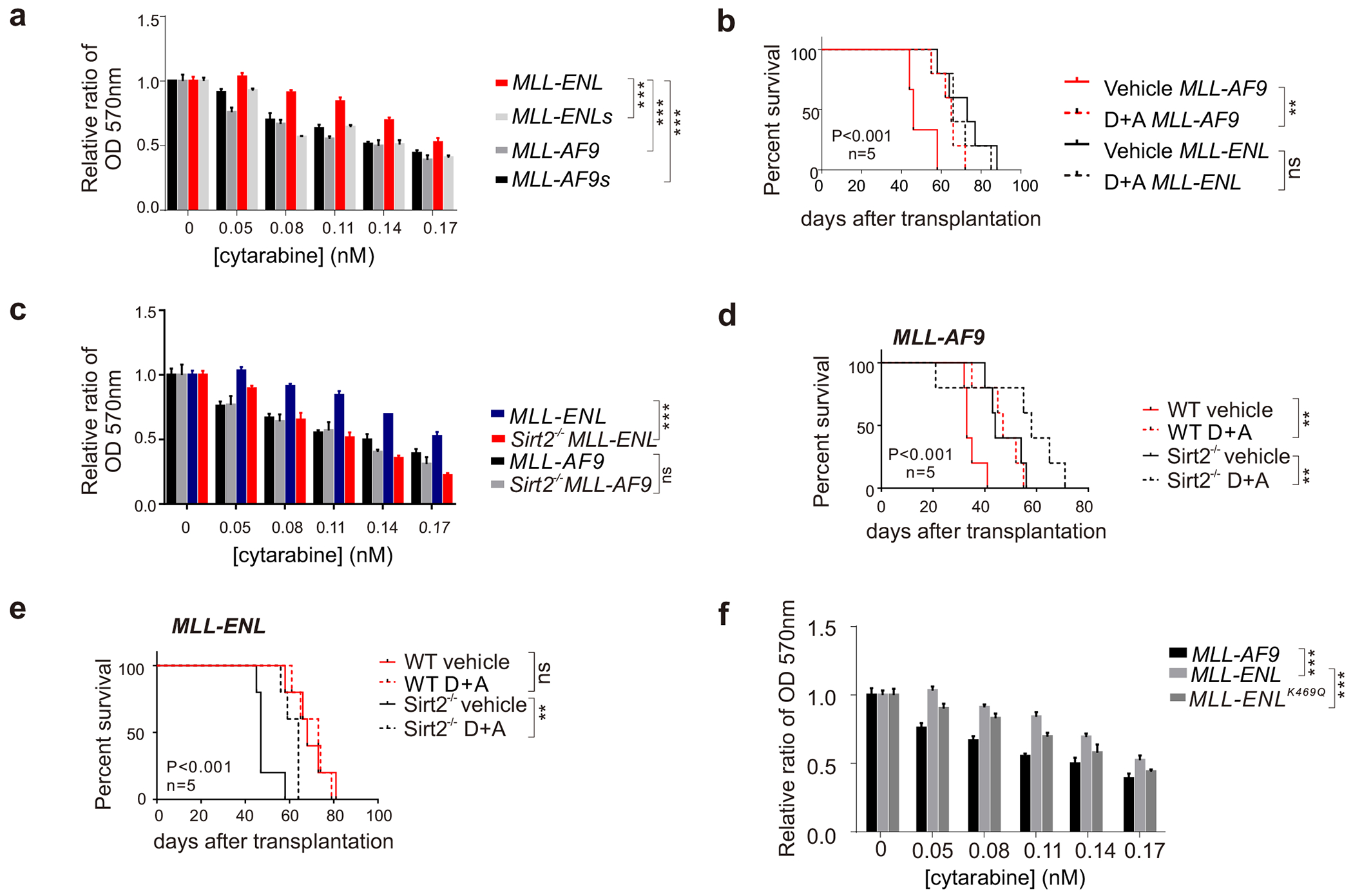
(a) MTT analysis of MLL-fusion AML cells treated with cytarabine; (b) Kaplan-Meier survival curve for AML-related death of the mice with DA treatment; (c) MTT analysis of SIRT2 depleted MLL-fusion AML cells treated with cytarabine; (d-e) Kaplan-Meier survival curve for AML-related death of the mice with DA treatment; (f) MTT analysis of MLL-fusion AML cells treated with cytarabine.
DISCUSSION
Despite the structural and biochemical similarities of AF9 and ENL proteins, these two proteins show distinct functions in normal tissue regeneration and cancer pathogenesis [22,23]. Several mechanisms have been proposed to explain this discrepancy including differential expression patterns in tissues, differential chromatin targeting regulation. Consistently, clinical features of MLL-AF9 and MLL-ENL AML patients are also different. In this study, we discovered that different clinical outcomes of MLL-ENL and MLL-AF9 patients are partially attributed to the linker regions of ENL. We identified an important regulatory switch-K469 within this region and showed that it is critical for both tumor growth regulation and chemo-therapy resistance. However, due to the low expression levels of almost all MLL-fusion proteins in AML cells and the lack of robust antibody to detect acetylated K469, we currently cannot determine whether fusion proteins are acetylated and whether acetylation states of ENL-linker-region regulates the proliferation of MLL-ENL AML cells. It is also possible that this critical residue within the linker region directly influences the binding of the partner complexes to AHD or alter the MLL-fusion conformation so that MLL portion might influence factor binding to AHD. In addition, acetylation states of MLL-ENL can also affect its stability by inhibiting poly-ubiquitination.
SIRT2 is a multifaceted deacetylase. Even within tumorigenesis context, it can act as a tumor suppressor or tumor promoter dependent upon the grade of each cancers [15,24]. SIRT2 can also influence tumor immune environment by directly regulating T cell metabolism [25]. In this study, we added another new layer of SIRT2 functions that is to regulate chemo-resistance of MLL-ENL-driven AML. Deletion of Sirt2 or treating cells with Sirt2 inhibitors can both manifest strong effects on AML development, underscoring the importance of its catalytic activity. Either inhibition of SIRT2 or deletion of the ENL-linker-region can revert MLL-ENL driven slow cell proliferation phenotype and sensitize these AML cells to chemotherapy treatment, suggesting that Sirt2 might directly mediate the deacetylation of MLL-ENL. Future studies aiming to determine whether SIRT2-mediated deacetylation regulates the stability of MLL-ENL proteins and/or association of their partner complexes in both physiological and pathological contexts are of great importance.
ACKNOWLEDGMENT
We thank the Zhang and Li Laboratory members for insightful discussion. This work was supported in part by National Key R&D Program of China (2021YFA1300100, 2018YFC1004500 to B.L.) and the National Natural Science Foundation of China (32030019, 31872817 to B.L.; 32000413 to J.S.; 81802773 to Y.L.); This work was also supported by NIH grants R01 HL133560-01 and R01 CA223194-01 through Loyola University Chicago, as well as Loyola program development funds to Jiwang Zhang.
REFERENCES
- [1].McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ, Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal, Cell Stem Cell 1 (2007) 338–345. [DOI] [PubMed] [Google Scholar]
- [2].Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P, Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors, Cell Stem Cell 1 (2007) 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].de Boer J, Walf-Vorderwulbecke V, Williams O, In focus: MLL-rearranged leukemia, Leukemia 27 (2013) 1224–1228. 10.1038/leu.2013.78. [DOI] [PubMed] [Google Scholar]
- [4].Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, Pombo de Oliveira M, Renneville A, Villarese P, Macintyre E, Cave H, Clappier E, Mass-Malo K, Zuna J, Trka J, De Braekeleer E, De Braekeleer M, Oh SH, Tsaur G, Fechina L, van der Velden VH, van Dongen JJ, Delabesse E, Binato R, Silva ML, Kustanovich A, Aleinikova O, Harris MH, Lund-Aho T, Juvonen V, Heidenreich O, Vormoor J, Choi WW, Jarosova M, Kolenova A, Bueno C, Menendez P, Wehner S, Eckert C, Talmant P, Tondeur S, Lippert E, Launay E, Henry C, Ballerini P, Lapillone H, Callanan MB, Cayuela JM, Herbaux C, Cazzaniga G, Kakadiya PM, Bohlander S, Ahlmann M, Choi JR, Gameiro P, Lee DS, Krauter J, Cornillet-Lefebvre P, Te Kronnie G, Schafer BW, Kubetzko S, Alonso CN, zur Stadt U, Sutton R, Venn NC, Izraeli S, Trakhtenbrot L, Madsen HO, Archer P, Hancock J, Cerveira N, Teixeira MR, Lo Nigro L, Moricke A, Stanulla M, Schrappe M, Sedek L, Szczepanski T, Zwaan CM, Coenen EA, van den Heuvel-Eibrink MM, Strehl S, Dworzak M, Panzer-Grumayer R, Dingermann T, Klingebiel T, Marschalek R, The MLL recombinome of acute leukemias in 2013, Leukemia 27 (2013) 2165–2176. 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schulze JM, Wang AY, Kobor MS, YEATS domain proteins: a diverse family with many links to chromatin modification and transcription, Biochem Cell Biol 87 (2009) 65–75. 10.1139/O08-111. [DOI] [PubMed] [Google Scholar]
- [6].Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W, Li H, Shi X, AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation, Cell 159 (2014) 558–571. 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McLean CM, Karemaker ID, van Leeuwen F, The emerging roles of DOT1L in leukemia and normal development, Leukemia 28 (2014) 2131–2138. 10.1038/leu.2014.169. [DOI] [PubMed] [Google Scholar]
- [8].Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, Doench JG, Xu H, Chu SH, Qi J, Wang X, Delaney C, Bernt KM, Root DE, Hahn WC, Bradner JE, Armstrong SA, DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia, Nat Med 21 (2015) 335–343. 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, Armstrong SA, MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L, Cancer Cell 20 (2011) 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen CW, Armstrong SA, Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond, Exp Hematol 43 (2015) 673–684. 10.1016/j.exphem.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Grigsby SM, Friedman A, Chase J, Waas B, Ropa J, Serio J, Shen C, Muntean AG, Maillard I, Nikolovska-Coleska Z, Elucidating the Importance of DOT1L Recruitment in MLL-AF9 Leukemia and Hematopoiesis, Cancers (Basel) 13 (2021). 10.3390/cancers13040642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu A, Zhi J, Tian T, Cihan A, Cevher MA, Liu Z, David Y, Muir TW, Roeder RG, Yu M, DOT1L complex regulates transcriptional initiation in human erythroleukemic cells, Proc Natl Acad Sci U S A 118 (2021). 10.1073/pnas.2106148118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Winters AC, Bernt KM, MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches, Frontiers in pediatrics 5 (2017) 4. 10.3389/fped.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz M, Sierra J, Tallman MS, Tien HF, Wei AH, Lowenberg B, Bloomfield CD, Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel, Blood 129 (2017) 424–447. 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang J, Gao X, Yu L, Roles of Histone Deacetylases in Acute Myeloid Leukemia With Fusion Proteins, Front Oncol 11 (2021) 741746. 10.3389/fonc.2021.741746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Inoue T, Hiratsuka M, Osaki M, Oshimura M, The molecular biology of mammalian SIRT proteins: SIRT2 in ell cycle regulation, Cell Cycle 6 (2007) 1011–1018. [DOI] [PubMed] [Google Scholar]
- [17].Nakagawa T, Guarente L, Sirtuins at a glance, J Cell Sci 124 (2011) 833–838. 10.1242/jcs.081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX, SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity, Cancer Cell 20 (2011) 487–499. 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dan L, Klimenkova O, Klimiankou M, Klusman JH, van den Heuvel-Eibrink MM, Reinhardt D, Welte K, Skokowa J, The role of sirtuin 2 activation by nicotinamide phosphoribosyltransferase in the aberrant proliferation and survival of myeloid leukemia cells, Haematologica 97 (2012) 551–559. 10.3324/haematol.2011.055236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Deng A, Ning Q, Zhou L, Liang Y, SIRT2 is an unfavorable prognostic biomarker in patients with acute myeloid leukemia, Scientific reports 6 (2016) 27694. 10.1038/srep27694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mishra LN, Pepenella S, Rogge R, Hansen JC, Hayes JJ, Acetylation Mimics Within a Single Nucleosome Alter Local DNA Accessibility In Compacted Nucleosome Arrays, Scientific reports 6 (2016) 34808. 10.1038/srep34808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, Souza AL, Bradner JE, Shen L, Li W, Li H, Allis CD, Armstrong SA, Shi X, ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia, Nature 543 (2017) 265–269. 10.1038/nature21687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, Sanjana NE, Shalem O, Nabet B, Zeid R, Offei-Addo NK, Dhe-Paganon S, Zhang F, Orkin SH, Winter GE, Bradner JE, Transcription control by the ENL YEATS domain in acute leukaemia, Nature 543 (2017) 270–274. 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McGlynn LM, Zino S, MacDonald AI, Curle J, Reilly JE, Mohammed ZM, McMillan DC, Mallon E, Payne AP, Edwards J, Shiels PG, SIRT2: tumour suppressor or tumour promoter in operable breast cancer?, Eur J Cancer 50 (2014) 290–301. 10.1016/j.ejca.2013.10.005. [DOI] [PubMed] [Google Scholar]
- [25].Hamaidi I, Zhang L, Kim N, Wang MH, Iclozan C, Fang B, Liu M, Koomen JM, Berglund AE, Yoder SJ, Yao J, Engelman RW, Creelan BC, Conejo-Garcia JR, Antonia SJ, Mule JJ, Kim S, Sirt2 Inhibition Enhances Metabolic Fitness and Effector Functions of Tumor-Reactive T Cells, Cell Metab 32 (2020) 420–436 e412. 10.1016/j.cmet.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]