

Abstract
Native mass spectrometry (MS) involves the analysis and characterization of macromolecules, predominantly intact proteins and protein complexes, whereby as much as possible the native structural features of the analytes are retained. As such, native MS enables the study of secondary, tertiary, and even quaternary structure of proteins and other biomolecules. Native MS represents a relatively recent addition to the analytical toolbox of mass spectrometry and has over the past decade experienced immense growth, especially in enhancing sensitivity and resolving power but also in ease of use. With the advent of dedicated mass analyzers, sample preparation and separation approaches, targeted fragmentation techniques, and software solutions, the number of practitioners and novel applications has risen in both academia and industry. This review focuses on recent developments, particularly in high-resolution native MS, describing applications in the structural analysis of protein assemblies, proteoform profiling of—among others—biopharmaceuticals and plasma proteins, and quantitative and qualitative analysis of protein–ligand interactions, with the latter covering lipid, drug, and carbohydrate molecules, to name a few.
1. Introduction and Scope of This Review
Biological mass spectrometry comes in many flavors. This diversity originates not only from the many different biomolecules that can be analyzed and investigated, such as proteins, peptides, lipids, DNA, RNA, carbohydrates, and metabolites, but also from the wide assortment of tools available to characterize them. Focusing on proteins and peptides, to date the dominant portion of MS-based analysis is performed by peptide-centric proteomics. Herein proteins are identified and quantified following enzymatic digestion into easily amenable smaller peptides, whose sequences can be determined by different fragmentation methods and matched by well-developed search algorithms against protein, RNA, and DNA databases. MS-based proteomics provides a means to measure proteome-wide protein abundances and monitor them upon perturbation of a system. Additionally, it can also be used to chart proteome-wide protein–protein interactions and various post-translational modifications (PTMs). Several excellent reviews are available covering all of these distinct flavors of MS-based proteomics.1,2
Beyond MS-based proteomics, mass spectrometry’s role in biochemistry and biology has over the past decades expanded to cover many aspects of structural and molecular biology.3 For MS-based structural biology—as is the case for conventional proteomics—quite a few different approaches contribute highly complementary information. Surface labeling techniques, involving either hydrogen–deuterium exchange4,5 or chemical labeling with radicals or other small molecules,6 coupled with mass spectrometry can provide information about structural changes and interaction interfaces. Chemical cross-linking approaches coupled to mass spectrometry facilitate structural investigation by providing distance restraints between identified cross-linked amino acids and offer information about new protein–protein and even protein–DNA interactions.7−9 These technologies still primarily use peptide-centric proteomics approaches and involve a proteolytic digestion step prior to LC-MS analysis.
In contrast to the technologies outlined above, native mass spectrometry (native MS), the core focus of this review, analyzes intact proteins and their non-covalent complexes, as well as other biomolecules, in a native-like folded state. Although the birth of native MS can be traced back to the early 1990s,10,11 just a few years after the introduction of electrospray ionization mass spectrometry (ESI-MS),12 MS-based technologies involving the analysis of intact proteins and protein complexes are still not as mature as their peptide-centric counterparts, mainly because of the challenges behind efficient ionization and detection of the larger intact protein ions. In native MS, the aim is to bring the analyte into the mass analyzer while retaining its original native structure and inter- and intramolecular interactions as much as possible.13−17 This task is not trivial, as the biomolecule is charged in the ionization process and stripped of all solvent molecules before mass analysis can occur under (ultra)high-vacuum conditions. Thus, a fully native state can never be retained. However, through a plethora of experimental work over the past decades, it has become apparent that when conditions are carefully managed, electrospray ionization may provide gas-phase ions of proteins and protein complexes that retain many of their native features.18,19 The first examples of that focused on the analysis of intact non-covalent complexes, which remained intact and largely retained their quaternary structures throughout their transfer from the solvent into the gas phase up until they hit the mass analyzer’s detector. In the early years of this century, the term “native MS” was coined to describe this area of biomolecular mass spectrometry.14 Native MS has matured substantially since then, and now many groups are applying this technology to study all sorts of proteins, their assemblies, and the interactions between proteins and ligands, including small-molecule drugs, cofactors, lipids, nucleotides, DNA, and RNA (Figure 1). Although native MS was initially primarily used to study soluble protein assemblies, membrane protein complexes have recently entered the realm of native MS through electrospraying of these assemblies from detergent micelles, nanodiscs, or even native lipid membranes.20,21
Figure 1.

Native MS has over the past decade experienced immense advances, primarily in enhancement of sensitivity and resolving power of mass analyzers but also in its ease of use. These advances have enabled the use of high-resolution native MS to analyze and characterize a wide range of macromolecules, including protein and ribonucleoprotein assemblies (section 7) and proteoforms of intact biopharmaceuticals and plasma proteins (section 8), and binding between proteins and small-molecule ligands such as lipids, drugs, and carbohydrates (section 9).
Significant advances have been made in native MS over the past decades, yet some analytical challenges remain to be overcome. Although we will focus on instrumentation in this review (section 3), one of the most critical challenges in native MS lies in sample preparation or, more specifically, making the analytes amenable to native electrospray ionization. This process is quite sample-dependent, and therefore, no universal guidelines can suffice. Nevertheless, it is crucial to recognize the important role of volatile salt solutions, such as ammonium acetate, that not only create a “native-like” environment for biomolecules but also minimize adduct formation and enable facile solvent removal at low activation energies, consequently resulting in clean native spectra.22 Although the principles of electrospray ionization are long known and have been discussed in detail,23,24 the mechanism of electrospray ionization of biomolecules has remained relatively elusive.25 Alternative ionization methods and designs are also emerging, with desorption electrospray ionization being a notable example, enabling ionization of biomolecules directly from biological tissues.26
Key instrumentational challenges today relate to the growing size and mass heterogeneity of the protein assemblies analyzed. The field of native MS is moving toward analyzing larger and larger protein assemblies, some with masses of several megadaltons or more. Moreover, increasing (micro)heterogeneity resulting from a plethora of PTMs (e.g., phosphorylation, acetylation, glycosylation, lipidation) is introduced as the field moves away from bacterially produced recombinant proteins toward more endogenous analytes of eukaryotic origin. These shifts in the nature of the samples analyzed pose new challenges to the instruments used for native MS.
Large protein assemblies ionized by native electrospray attain relatively fewer charges than those ionized under denaturing conditions, thus requiring analysis using mass analyzers with extended m/z ranges (up to m/z = 20 000 Th and beyond). As a result, in the first decades, the field of native MS was highly dependent on time of flight (ToF)-based mass analyzers that have a theoretically unlimited m/z range. However, most standard mass analyzers are optimized for the transmission of relatively small molecules (e.g., peptides), and thus, ToF-based instruments also needed to be modified for the dedicated mass analysis of protein complexes. Besides ToF-based mass analyzers, ion-trap-based instruments have also been modified and optimized for native MS, with developments on both Fourier transform ion cyclotron resonance (FT-ICR) and Orbitrap mass analyzers.27−30 The development of commercial Orbitrap mass analyzers with extended mass ranges has given a strong impetus to the field of native MS, whereby next to academic laboratories, pharmaceutical and biotechnological companies have adopted the technology.30,31
The other challenge, which is inherent to almost any mass spectrometric approach, is the mass resolving power, a central subject in section 2: the higher the mass resolving power, the more precise the mass analysis. This is as true for native MS as for any other form of mass spectrometry. However, the attainable mass resolving power in native MS is not only dependent on the instrumental mass resolution limits but also heavily affected by the ionization process and especially by the ability (or the lack thereof) to completely desolvate the protein or protein complex, stripping off all solvent molecules and salt adducts during the transfer from the electrospray solution into the (ultra)high vacuum of the mass analyzer.32 Only when all of these small molecules are removed can the mass and m/z of the detected ions correspond to the analyte’s exact mass. For large proteins and protein complexes, such complete desolvation upon electrospray ionization is not trivial. Even if all of the water molecules can be removed, salt cations (e.g., Na+ or K+) may remain attached because of their stronger binding, leading to ion signals that are broadened because they originate not only from the multiply protonated analytes but also from analytes carrying metal cations, which slightly increase the mass and m/z of the ions. The presence of these small-molecule adducts depends heavily on sample preparation, and extensive desalting is often a prerequisite for obtaining highly resolved mass spectra. However, this often comes at the expense of sample loss. On the other hand, desolvation can be promoted during the ionization process and while the ions are being transferred through the mass analyzer. This is often achieved via ion activation, either by heating the ion source or by letting the ions collide with inert gas molecules in a balanced manner, whereby the analyte ions lose their attached solvent adducts but do not undergo dissociation.
This review focuses on high-resolution native MS. In section 3 we describe how technological innovations in mass analyzers have advanced native MS by extending the achievable mass (or m/z range), enhancing the sensitivity by optimizing the ion transmission, and improving the achievable mass resolving power. In particular, we focus on the latter and describe how different factors are optimized to enhance the mass resolving power, including improved desolvation by ion activation. In section 6, we describe different methods of tandem mass spectrometry (i.e., collision-, electron-, and photon-induced) used in native MS to obtain more structural information on the analyzed protein complexes but also to improve desolvation and the precision of the mass measurement. Sections 4 and 5 focus on parallel developments in advancing spectral data processing for native MS, which are geared toward the more efficient and improved analysis of the native mass spectra. Automated data processing becomes of the utmost importance when the analytes are very heterogeneous in mass and when the resolution in native mass spectra is hampered by imperfect desolvation or binding of various small-molecule cofactors.
In the remainder of the review, we describe various studies highlighting the recent advances and diverse applications of high-resolution native MS. Broadly, this part of the review is separated into several sections. Section 7 includes an overview of studies on protein complexes by high-resolution native MS, wherein the generic aim is to determine the stoichiometry, structural features, subcomplexes, and possible binding of functionally important ligands (e.g., lipids, DNA, and glycans). Next, in section 8 we describe studies using high-resolution native MS to characterize intact single proteins, whereby the aim is to assess qualitatively and quantitatively all of the proteoforms present. Here we primarily focus on the analyses of protein biotherapeutics and plasma glycoproteins. In section 9 we highlight progress in the field of characterizing protein–small molecule interactions by high-resolution native MS. We end the review with a future outlook.
2. Critical Measures of Performance in High-Resolution Native MS
With the ever-increasing size and complexity of the analytes, native MS needs to continually improve its performance regarding critical metrics such as mass accuracy, detection sensitivity, upper mass boundary, tandem MS capabilities, and mass resolution. High mass accuracy and sensitivity are essential for quality control analyses of large therapeutic molecules produced by the biopharmaceutical industry, as described in more detail in section 8.33 Mass range boundaries for native MS are constantly pushed by the desire to investigate larger macromolecules, such as viral particles and nanocontainers with molecular weights (MWs) in the megadalton mass range.34 Finally, aside from detecting distinct charge states of large ionized macromolecular assemblies, mass resolution in native MS must distinguish between species with minute mass differences that stem from salt adducts, small non-covalently bound ligands, or PTMs. Such demands are continuously tackled by the research community and have led to the modern age of high-resolution native MS with the emergence of exciting new technologies and novel instruments, as further described below.
2.1. A Closer Look at Resolution and Resolving Power
Although it is an essential measure of performance in mass spectrometry, mass resolution is still often ill-defined as a term by parts of the community. According to the International Union of Pure and Applied Chemistry (IUPAC), mass resolution is defined as m/Δm, where m is typically the mass-to-charge ratio of a singly charged ion and Δm is determined either as the width of a single peak at a fraction of its height (e.g., 50%) or as a mass difference between two equally abundant peaks with a valley between them not exceeding 10% of their heights.35 On the other hand, resolving power is the ability of an instrument to distinguish between two peaks differing by a small m/z value and is defined as the peak width, Δm.36 The misperception quite often observed in the literature arises because mass resolving power is defined similarly to the mass resolution as m/Δm.35 The similarity between the definitions of these two terms has resulted in their somewhat interchangeable usage in the literature. In addition, different definitions of resolution and resolving power have been proposed,37−39 although they are not widespread. In the literature on mass spectrometric instrumentation, mass resolution is often defined as m/Δm, where Δm is determined at 50% peak height, also called the full width at half-maximum (fwhm).40−42 Here we adhere to defining both the mass resolution and mass resolving power using the fwhm since it is the most adopted definition in the native MS literature and is advised in the IUPAC terminology recommendations for mass spectrometrists.36 While we use the term mass resolution throughout this review, we use the term mass resolving power to characterize the mass analyzer performance, as the latter is very commonly used in such context. It should be noted that it is important to take into account the mass value at which the resolution or resolving power is defined, as—for many instruments, most notably in FT-ICR and Orbitrap instruments—the resolution does not scale up well with increasing m/z.
2.2. High Resolution in the Context of Native MS
In the field of small-molecule and peptide-centric mass spectrometry, high resolution is primarily used for the disentanglement of isotope patterns, enabling the determination of accurate masses and—in the case of small molecules—even exact chemical formulas, with sub-ppm mass accuracies achieved on modern FT MS instruments.44−46 The absolute highest resolution in the range of several million (at m/z = 100–200 Th) was achieved with FT MS, especially FT-ICR, with reported resolutions far beyond 106 at m/z = 200 Th. This is closely followed by another representative of FT MS, the Orbitrap mass analyzer. Although it features lower resolution at low m/z, its resolution still surpasses 106 when measured at m/z = 200 Th. With increasing acceptance, such high resolution is called ultrahigh resolution, and such measurements have led to significant advancements in the fields of petroleomics,47 proteomics,48 environmental analysis,49 forensics,50 and space exploration.51 However, such ultrahigh resolution is not necessarily beneficial for native MS, as outlined below.
Although measuring isotopically resolved spectra seems beneficial, above certain molecular weights (∼150 kDa) the isotopic distributions of various codetected species, e.g., ions carrying salt adducts or additional solvent molecules, start to overlap and superimpose, leading to distortions of the acquired signal (Figure 2A). This effect is increasingly more pronounced for larger molecules, whereby the detected peaks of globular proteins are substantially broader than expected (Figure 2B). In 2014 Lössl et al. argued that mass-resolving small buffer molecules or salt adducts is impossible for proteins larger than 65 kDa.32 Moreover, in the case of large multiply charged biomolecules, PTM-related microheterogeneities result in only minute m/z differences, e.g., oxidation of a 1 MDa molecule carrying 80 charges would lead to a shift of only m/z = 0.2 Th, while the width of the isotopic distribution is above 0.5 Th.34 The current methods for desalting and complete desolvation of ions of large macromolecular assemblies are only partially effective for analytes in the megadalton range. Furthermore, the technical requirements for achieving sufficient resolution are often incompatible with large macromolecules. For example, in Orbitrap MS, the resolution is inversely proportional to the square root of m/z, requiring a stable image current signal to be recorded in the range of several seconds to gain isotopic resolution for larger macromolecular assemblies. These limitations hamper the analysis of large multiply charged protein assemblies with conventional native MS, whereby they are more prone to suffer from space-charge effects or decay due to field imperfections. As a result, in native MS, most researchers aim to resolve distinct charge states rather than individual isotope peaks while still endeavoring to disentangle microheterogeneities introduced by PTMs52 and co-occurring stoichiometries.53
Figure 2.
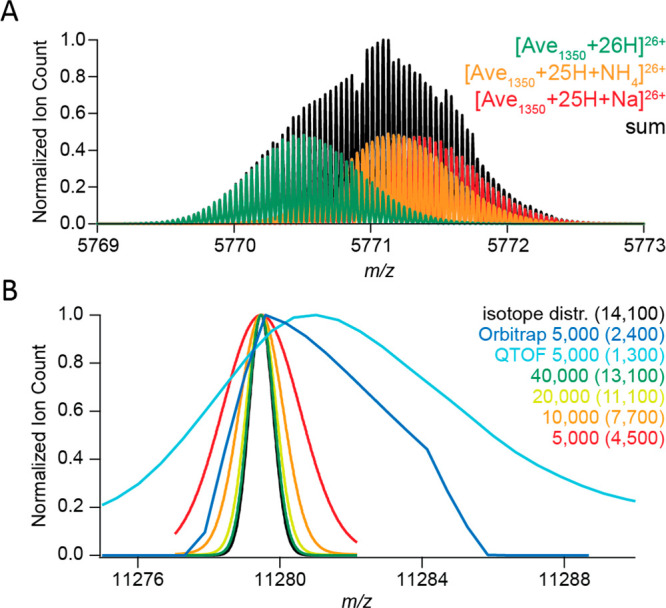
Challenges for resolving isotopologues with high-resolution native MS. (A) Adduct ions affect the mass resolving power. Baseline isotope mass resolution does not permit bare, sodium-bound, and ammonium-bound ions of a 150 kDa averagine protein to be distinguished using native ESI-MS. According to the empirical charging behavior of globular proteins in native MS, 26+ is the most abundant charge state for a molecular weight of 150 kDa. Therefore, the peaks of 26-fold-charged cations of a 150 kDa averagine protein (1350 averagine residues) were generated with MassLynx ver. 4.1, assuming baseline isotope mass resolution (R = 500 000). The isotope distributions of unmodified (green), ammonium-bound (orange), and sodium-bound (red) protein ions were simulated individually and subsequently summed to produce their combined mass spectrum (black). (B) Experimental peaks of globular protein complexes are substantially broader than simulated peaks of their molecular ions. The apparent mass resolution depends on the preset instrumental resolution and the efficiency of adduct removal. Shown are mass spectra of a GroEL ion with a charge of 71+. These were measured on Orbitrap Exactive Plus (blue) and a QToF (cyan) instruments, both operating at an instrument mass resolution of 5000, or simulated with MassLynx ver. 4.1 at mass resolutions of 5000 (red), 10 000 (orange), 20 000 (yellow), and 40 000 (green). The black curve represents the natural isotope envelope of GroEL. Numbers in parentheses correspond to the apparent mass resolutions Rnat determined by measuring the experimental peak widths. Reproduced from ref (32). Copyright 2014 American Society for Mass Spectrometry.
Similar to how narrow charge state distributions in native MS improve sensitivity, recording the average mass of an isotopic envelope instead of all of the peaks within the fine isotope distribution can provide further gains in sensitivity and signal-to-noise ratio (S/N), as the signal is binned over fewer channels. As a bonus, in conventional FT-based native MS, the signals of large ions decay less within the shorter acquisition durations provided by intermediate instrumental resolutions.54 Although ultrahigh mass resolution is thus not essential, native MS still requires reasonably high instrumental resolving power to detect the individual charge states and mass shifts induced by PTMs on macromolecules or coexisting stoichiometries. Taken together, the results indicate that the best attainable resolution in native MS is analyte-specific and often pragmatically determined. Therefore, “High-Resolution” in the title of this review reflects the best apparent (empirical) resolution attainable for the specific analyte rather than the inherently achievable resolving power of the instrument.
2.3. Mass Accuracy in the Detection of Macromolecules
Another crucial metric of performance, which is highly dependent on instrumental resolution, is the attainable mass accuracy. Mass accuracy is typically defined as the mass error as a fraction of the theoretical mass and is often expressed in parts per million or parts per billion.55 For small molecules (MW < 500 Da), depending on the attainable mass accuracy, a mass spectrometer can either unambiguously identify the molecule or point at a multitude of putative structures of similar yet distinct elemental compositions.56 At the dawn of mass spectrometry, with mass analyzers having limited resolution and mass accuracy such as the quadrupole mass analyzer,57 it was only feasible to determine nominal masses, which are calculated by rounding masses of the most abundant isotopes.55 However, with the advent of more advanced mass analyzers, such as ToF and, more notably, FT-ICR, it became feasible to measure masses incredibly close to the theoretical values, with sub-ppm or even ppb accuracy, albeit mostly at the cost of long observation periods.58 While for the analysis of small molecules or for peptide/protein sequencing it is essential to determine monoisotopic masses, either directly or by inferring them from fine isotope distributions, in native MS average mass profiles are recorded. Not only is it challenging to obtain isotopically resolved spectra of pure macromolecular species (see section 2.2), but also the increasing probabilities of heavier isotopes in mass profiles of macromolecules result in the near absence of monoisotopic mass peaks.59 As is typical in native MS, recording average masses leads to more pronounced deviations from theoretical values, although these are typically still in the range of 2–30 ppm.
From a technical perspective, high mass accuracy requires a good mass calibration and elimination of all analyzer-specific systematic errors (discussed below). Calibration can be performed either internally or externally, with internal calibration, whereby the internal standard is present in every spectrum, yielding the best mass accuracies.46 Calibrating the mass analyzer for native MS internally is challenging, and therefore, external calibration is more common. For that, most typically, a high-concentration aqueous CsI solution (about 10–100 mg/mL) is used, whereby the generated singly charged virtually monoisotopic CsI clusters can cover quite a wide m/z range and require very similar experimental conditions for gas-phase transmission and detection as large protein assemblies.60 As for systematic errors of mass analyzers, they vary significantly since different mass analyzers adhere to different principles of mass analysis and detection. For example, Orbitrap mass analyzers, which are becoming very popular in native MS, can suffer from space-charge effects61 or electric field imperfections.62 When all such systematic errors are eliminated, the mass accuracy is equal to mass measurement precision, reflecting random errors in multiple data points acquired for the same species.55
The resolving power of the instrument usually defines the attainable mass accuracy. However, the empirical widths of peaks detected in native MS often exceed the theoretical widths defined by the instrumental resolution.32 Although careful sample preparation and dedicated ion cooling63 in the mass analyzer can help to reduce the peak widths, it is increasingly challenging to achieve this for large macromolecules. Since instrumental resolution is not the only limiting factor for accurate mass analysis of large ions with native MS, developments in mass analyzers have focused not only on aspects of pure mass detection but also on efficient manipulation of large macromolecular ions in the gas phase, maximizing analyte desolvation and adduct removal.
3. Features of Mass Analyzers Optimized for High-Mass Measurements
Although native MS has benefited massively from the general advances made in mass spectrometry over the past decades, the mass analyzers used for native MS still require distinctive characteristics, primarily because of the high molecular weights of the analyzed particles, their relatively lower charges, and the distinctive dynamics of ion motion within the mass analyzers. Initially, mass analyzers used for native MS were standard instruments designed for small molecules that were modified to enable the analysis of high-mass particles.64 At the advent of native MS, modified ToF instruments were predominantly used, but more recently FT-ICR and Orbitrap mass analyzers have been adapted for high-resolution native MS.13,65
Many modern implementations of these instruments feature a lower-resolution secondary mass analyzer, e.g., a linear ion trap or a quadrupole, which is primarily used for ion selection, fragmentation, or other adjacent functions (Figure 3). Although essential for modern mass spectrometry, such secondary mass analyzers are often built-in along the transmission path of a mass spectrometer and may limit the attainable mass range, as their resolution and ion transmission efficiencies at high m/z values are not ideal. Nevertheless, modern implementations of quadrupoles with lower RF frequencies66 or frequency-scanning (as opposed to amplitude-scanning) design67 enable the extension of the attainable m/z range up to or even above 100 kTh.
Figure 3.
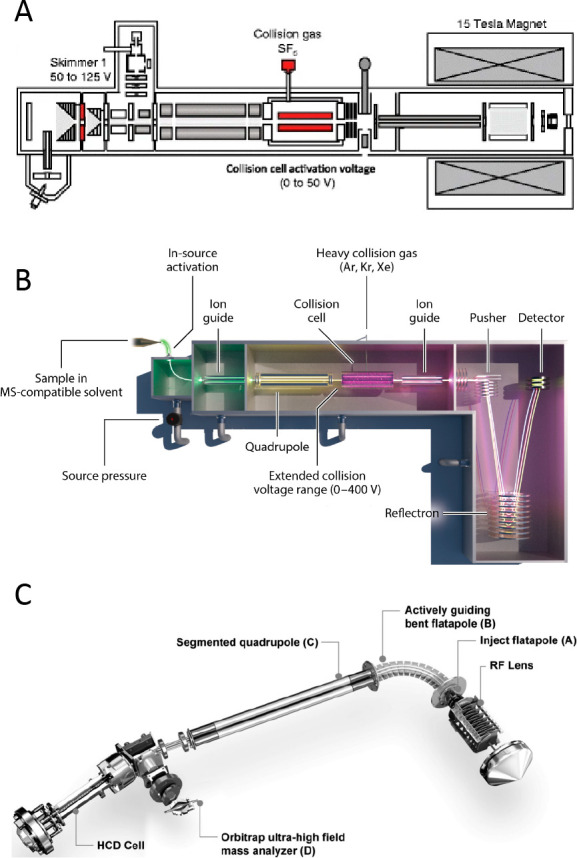
Schematics of mass spectrometers commonly used for native MS: (A) 15 T FT-ICR. Adapted from ref (76). Copyright 2017 American Society for Mass Spectrometry. (B) Synapt G2 instrument featuring a Q-ToF mass analyzer. Reproduced with permission from ref (77). Copyright 2013 Nature Publishing Group. (C) Orbitrap Q-Exactive UHMR instrument with extended mass range. Reproduced with permission from ref (66). Copyright 2017 Nature Publishing Group.
High-resolution mass spectrometers provide an array of useful and complementary sets of features that facilitate native MS analysis of large macromolecular assemblies. For example, ToF instruments enable fast scanning and a theoretically unlimited mass range,68 while FT-based mass spectrometers, although suffering from relatively slower signal acquisition, provide exceptional mass resolution, primarily in the low m/z range.38 Novel Orbitrap-based mass spectrometers have been specifically tailored for sensitive measurements in the high-m/z region and provide increased practical resolution through superior desolvation.27,30,31,69 Recently, with native MS increasing in popularity, a number of new ToF and FT-ICR instruments have emerged, improving upon initial limitations of speed and transmission in high-mass measurements.70−73 Advances in attainable sensitivity, accuracy, resolution, and mass range over the past decade have empowered high-resolution native MS to unravel heterogeneity and structural features of very complex systems like membrane proteins,74 ribosomal particles,53 and crystalline oligomers,75 to name just a few.
3.1. Time-of-Flight Mass Analyzers
ToF mass analyzers have proven to be extremely powerful in the analysis of large macromolecular assemblies and have been used to set mass records in native MS still unmatched by other instruments today, for instance, providing a charge-resolved mass spectrum of an intact 18 MDa bacteriophage assembly.34 At its core, ToF mass analyzers allow for the simultaneous detection of masses in a very wide mass range, and furthermore, ToF-based analysis is inherently sensitive and fast.68 Because in ToF mass analysis m/z is proportional to the square of the time of flight of the ions through the analyzer, the resolution (m/Δm) of the ToF mass analyzer is equivalent to t/2Δt, which means that the resolution remains nearly constant over the whole m/z range (Figure 4). This feature is very distinctive from FT-based analyzers, where the resolution decreases drastically with increasing m/z.
Figure 4.
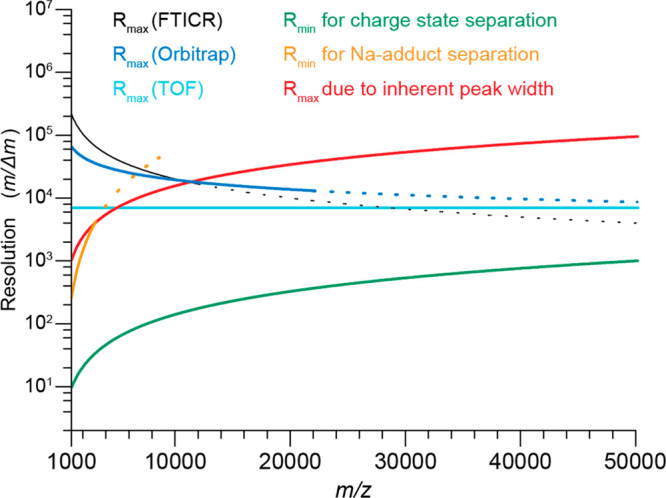
Theoretical and experimental mass resolutions (m/Δm) in the m/z range from 1000 to 50 000 Th for the common mass analyzers used in native MS. Shown are plots of R vs m/z for the three mass analyzers (FT-ICR, Orbitrap, and ToF) based on their respective Rmax values reported at m/z = 5000–6000 Th (FTICR, 40 000; Orbitrap; 25 000; ToF, 7000) and the theoretically achievable relations between analyzer mass resolution and m/z (FT-ICR (black line), R ∼ z/m; Orbitrap (blue line), R ∼ (z/m)0.5; ToF (cyan line), R = constant). Dotted lines in the FT-ICR and Orbitrap graphs represent theoretical values for m/z ranges that were not accessible with these mass analyzers at the time of the publication. Reproduced from ref (32). Copyright 2014 American Society for Mass Spectrometry.
Although the history of ToF analyzers goes back to 1948, when they appeared under the name “Velocitron”,78 major developments important for native MS came much later. In the 1970s Mamyrin et al. introduced the ToF-reflectron, which increased the resolution of ToF instruments by effectively extending the flight path and compensating for ion energy drift.79 In the early 1990s, the advent of orthogonal-acceleration ToF (oa-ToF)80,81 following innovations in fast digitizing electronics further improved the resolution of ToF MS by accumulating ions in the acceleration region and thus unifying their position and velocity prior to acceleration. Shortly after the introduction of oa-ToF, the addition of an ion-selecting quadrupole (Q) compartment82,83 enabled the conversion of ToF analyzers into so-called Q-ToF mass analyzers. The first Q-ToF mass spectrometers used for native MS featured nano-ESI (nESI) sources and were modified to enable an increased pressure regime in the instrument’s front end. While nESI was essential for soft ionization of macromolecules, high pressure was crucial for ion cooling through collisions with neutral gas molecules, which is absolutely necessary for electrostatic focusing of large ions.63 Various solutions for efficient ion cooling were explored by both the Heck and Robinson groups between 2006 and 2009, including restriction of pumping in the front end of the mass spectrometer84,85 and the use of heavy gases in the collision cell.86 For instance, several important modifications to employ Q-ToFs for the analysis of macromolecular assemblies were implemented by Van den Heuvel et al. in 2006.87 They modified a first-generation Q-ToF by introducing high-transmission grid ion optics, a low-frequency quadrupole, a low-repetition pusher, and a high-pressure collision cell. These developments enabled the pioneering mass measurements of large viral particles88−90 and membrane protein assemblies,91,92 exposing—in addition to the MW—their structural features, such as composition, protein–ligand stoichiometry, and binding stability.
In the past decade, developments in ToF instruments have primarily focused on tailoring the electronics for ion-selecting or ion-routing optics and further gas regime control to improve the transmission of high-mass ions. Since high resolution requires increasing the flight distance of ions between the pusher and detector, multiple solutions have recently emerged to improve the mass resolution, including multipass and multireflection instrumental setups.93−97 These recent experimental improvements have led to significant improvements in resolution (>10 000) and mass accuracy (∼5–10 ppm) while simultaneously improving the transmission of high-mass ions and spectral quality in ToF-based native MS. Still, on such instruments the ion signals recorded by native MS remained much broader than would be expected if they were purely limited by the mass resolution of the instrument, resulting often in an overestimation of molecular weights by up to a few percent.32 As discussed above, this limitation originates primarily from imperfect desolvation of the ions in transmission-type Q-ToF instruments. In addition, the extended ion flight paths in multireflection and multipass instruments additionally imply lower sensitivity in detecting macromolecular assemblies, which may dissociate prior to reaching the detector. If such hurdles can be addressed, ToF-based mass analyzers would further consolidate and could possibly see increased usage in native MS.
3.2. FT-ICR Mass Analyzers
Since its introduction back in 1974,98 FT-ICR MS has developed into a wonderful tool for mass measurements that enables ultrahigh mass resolution (>106 at m/z = 200 Th)99 and ultrahigh mass accuracy, even reaching into the ppb range,58 significantly advancing the fields of proteomics, metabolomics, petroleomics, and environmental analysis, among others. Nevertheless, for a long time the application of FT-ICR for native MS was somewhat limited and only started to be substantially explored around a decade ago with the emergence of instruments using magnets with strong fields (e.g., 12 or even 15 T).100
FT-ICR instruments represent a variation of a Penning trap device, which traps ions along the axis of the ICR cell within a spatially uniform magnetic field (B) that provides radial ion trapping and governs their cyclotron motion.38 This ion cyclotron motion in turn induces an image current in the two opposing electrodes when the ions are excited to higher orbits. Signal detection in FT-ICR can happen only after the ion excitation step, enabling ions with identical m/z to rotate coherently with a locked phase close to the detector electrodes. Facilitating mass detection, ion cyclotron resonance is independent of the ion energy, and the recorded signal is linearly proportional to the rotational amplitude of ion motion. Such linearity enables the time-domain signal to be recorded and subsequently converted into a frequency-domain spectrum by Fourier transformation,101 adapting FT principles developed within the rich field of Fourier spectroscopy and NMR spectroscopy.102 Since the ion “image current” is induced by ions rotating at the cyclotron frequency fc = B/[2π(m/z)], the resolution of FT-ICR is directly dependent on the number of cyclotron rotations and therefore the length of the ion flight path. This holds true only when ions of the same m/z are coherently locked in phase within a perfectly homogeneous magnetic field.
The advantages of FT-ICR for high-resolution measurements compared to ToF become clear when their corresponding lengths of ion flight paths are compared. For example, ions with m/z = 1000 Th analyzed by FT-ICR with a 9.4 T magnet in 1 s undergo 144 346 rotations, which is equivalent to a flight path of 9 km,103 an incredible leap compared with the 2–5 m ion flight path in a typical ToF instrument. However, the outstanding resolving power comes at a high cost. Since the cyclotron frequency is proportional to (m/z)−1, the attainable instrumental resolution drops rapidly with increasing m/z, which somewhat limits the usability of FT-ICR in native MS of larger protein assemblies. The direct dependence of the resolution on the duration of the time-domain signal also hampers high-resolution detection of large macromolecules. Their ions signals are often susceptible to decay due to magnetic field imperfections, collisions with neutral background gas molecules, and coherence-disrupting ion–ion interactions.
Since ion spatial coherence is of key importance in FT-ICR MS, it was crucial to develop methods for pulsed rather than continuous introduction of ions into the ICR cell. Therefore, in the 1990s, FT-ICR instruments were equipped with a multipole, which enabled ions to be accumulated and then simultaneously injected as a packet into the ICR cell.104 This modification enabled the efficient excitation of ions and phase locking. Another substantial improvement of ICR-type instruments was achieved by increasing the magnet conductivity. It was shown that with stronger magnetic fields multiple metrics of FT-ICR performance could be enhanced, with some (e.g., the upper mass range) scaling quadratically.105 Modern FT-ICR instruments can be equipped with very powerful 15 T or even >20 T magnets. These higher magnetic field strengths not only increase the attainable resolution, accuracy of mass measurement,58 and stability of ion cyclotron motion in the ICR cell but also initiated the application of FT-ICR for the analysis of large biomolecules, especially for the analysis of intact antibodies.100,106,107 Apart from using more conductive magnets, multiple ICR cell designs have been proposed to reduce magnetic and electric field imperfections and increase the mass resolving power. Some of the recent advances were reported by Campuzano et al.,108 who described high-resolution native MS analyses of antibody–drug conjugates (ADCs) using a powerful 15 T solariX FT-ICR instrument (Figure 5).
Figure 5.

High-resolution native MS analyses of antibody–drug conjugates using a 15 T FT-ICR instrument. Shown are the native MS spectra of (a) 5 equiv (biotin) lysine conjugate, (b) 10 equiv (biotin) lysine conjugate, (c) 5 equiv tris(2-carboxyethyl)phosphine (TCEP) and 10 equiv (biotin) cysteine conjugate, and (d) 10 equiv TCEP and 10 equiv (biotin) cysteine conjugate. Drug-to-antibody ratio (DAR) values are annotated on selected charge states and all of the deconvoluted spectra. Asterisks (*) represent a +162 Da glycation mass increase. Data are displayed in magnitude mode using a symmetric Hann apodization function. The annotated DAR values in insets III and IV of (c) and (d) are consistent with the numbers of biotin moieties covalently attached to the specific species and not the full intact initial monoclonal antibody conjugate molecule. Peak widths at half-height for z = 28+ are annotated. The peak widths at half-height for the equivalent deconvoluted data range from 39.2 to 41.1 Th. Reproduced from ref (108). Copyright 2017 American Chemical Society.
The high-resolution capabilities of modern ICR mass spectrometers make them well-suited for native top-down MS analysis of proteins and their assemblies, whereby the isotopic envelopes of low-charge dissociation products can still be resolved. Because of the required ultrahigh-vacuum operation, FT-ICR instruments are less compatible with conventional collision-based ion fragmentation methods, such as collisional dissociation (CID). Therefore, alternative fragmentation methods have been introduced, featuring—among others—photon-induced and electron-induced fragmentation techniques, which also were shown to have benefits over CID in the structural investigation of larger protein assemblies.28,76
Overall, high-field FT-ICR instruments represent excellent mass analyzers for native MS, although their advantages come at the expense of a relatively high cost, which—together with their relatively large size and high maintenance—likely has hampered somewhat the wide usage of these instruments in the field to date.
3.3. Orbitrap Mass Analyzers
Despite being the youngest among the mass analyzers frequently used for native MS, the Orbitrap has experienced rapid developments and currently represents one of the most prominent technologies for the mass analysis of large and heterogeneous macromolecular assemblies.30,69,74,109 The Orbitrap typically forms the end-stage detector of a hybrid mass spectrometer that is further equipped with other mass analyzers such as quadrupoles and ion traps. Through the combination of these mass analyzers, Orbitrap-based instruments can build upon several strengths of more mature mass analyzing/manipulating devices while eliminating some of their drawbacks.110 Ultimately, Orbitraps inherited many features, such as pulsed ion injection from the earlier developed oa-ToF, trapping principles from radiofrequency ion traps, and signal detection and processing from FT-ICR. Circumventing some of the shortcomings, the Orbitrap mass analyzer does not require superconductive magnets and benefits from significantly smaller dimensions compared with ICR cells while enabling “ion excitation by injection”.62 In addition, Orbitraps experience a less steep drop in resolution with increasing m/z, as the oscillation frequency—in contrast with the cyclotron frequency in FT-ICR—scales proportionally to (m/z)−1/2 (see Figure 4). Similar to FT-ICR and as an improvement upon the ToF mass analyzers, the recorded axial ion oscillation frequency in an Orbitrap is independent of the ion energy, making the Orbitrap very suitable for mass detection.
Physically, the Orbitrap can be represented as a two-electrode device consisting of an inner spindle-like electrode and an outer curved electrode, which together create a quadrologarithmic field.111 Although the Orbitrap was introduced in 1999 and commercially released as a linear ion trap Orbitrap (LTQ Orbitrap) instrument in 2005, the principles of orbital ion trapping were established by Kingdon about a century ago.112 In an Orbitrap, ions oscillate around the central electrode with m/z-specific axial frequencies, inducing a current on the split-in-half outer electrode. Prior to mass detection, ions are first stored and confined in a linear ion trap adjacent to the Orbitrap, known as the C-trap, and then injected into the mass analyzer by applying a voltage onto the back plate of the C-trap.41 In contrast to FT-ICR, such external excitation is sufficient to achieve spatial coherence of the ions without additional oscillating or rotating electric fields, although dipolar excitation has been explored for selective ion ejection and motion stabilization.113,114
Since its introduction, the Orbitrap mass analyzer has undergone several developments, including the introduction of a high-field Orbitrap mass analyzer and enhanced methods for processing of the time-domain waveform signal,115 but arguably the most important modifications for native MS were those targeting the ion optics and electronics. High-field Orbitrap mass analyzers feature a more compact design with improved ion trapping and enhanced resolution and dynamic range.116 The utility of the Orbitrap for native MS was first demonstrated in 2012 by Rose et al., who modified a benchtop Orbitrap instrument by lowering the radiofrequencies of the ion transition optics and implemented a gas control system for the higher-energy collisional dissociation (HCD) cell.27 With these modifications, the Orbitrap instrument demonstrated efficient transmission and collisional cooling of large (high m/z) ions, outperforming Q-ToF instruments available at that time in terms of resolving power but not yet in mass range. The high resolution and sensitivity of the Orbitrap mass analyzer provided a means for detailed analysis of viral particles117 and biopharmaceuticals with native MS.31,118 A schematic of a modern Orbitrap-based mass spectrometer for native MS is provided in Figure 3C.
Shortly after this first demonstration, commercial Orbitrap mass spectrometers were introduced, geared toward analysis in the extended mass range (Orbitrap-EMR). This first generation of instruments lacked possibilities for mass selection, which is essential for tandem MS analysis of large analytes in native MS. This problem was overcome by Belov et al., who equipped the instrument with an EMR quadrupole operating at lower radiofrequencies119 to enable selection of high-mass ions. Dyachenko et al. shortly thereafter demonstrated the utility of these innovations by measuring stoichiometries and localizations of drug moieties in ADCs and antibody–antigen complexes exhibiting masses in the MDa range.120 More recently, an Orbitrap with ultrahigh mass range (UHMR) capabilities that was developed for native MS was commercially released, closing the gap in upper mass limit with ToF MS and providing even more powerful desolvation and ion cooling capabilities in the front end of the instrument.30,66 The Heck group has used such instruments to stretch the boundaries of accurate high-resolution mass analysis up to m/z = 50 000 Th by studying intact Flock House viruses.66 The ultrahigh mass range also allowed van de Waterbeemd et al. to study compositional variants and to unravel heterogeneity within ribosomal particles extracted from three different organisms.53 Multiple groups also utilized the strong desolvating capabilities of the instrument to analyze membrane proteins21,121 and even to perform triple-stage tandem MS analysis, wherein fragmentation in the front end is followed by product isolation using the quadrupole and secondary fragmentation either by conventional HCD69,74 or custom fragmentation methods (e.g., surface-induced dissociation by the Wysocki lab122 or UV photodissociation by the Brodbelt123 and Heck124 groups). Among the latest advances, Orbitraps were utilized for ion mobility analysis, providing an extra dimension to reduce sample complexity and further advance structural analysis of macromolecules.125,126 These recent developments demonstrate that the future may still bring many more exciting possibilities for Orbitrap-based native MS, some of which will be discussed further below.
4. Novel Solutions for Enhancing Resolution and Disentangling Heterogeneous Macromolecules by Native MS
The demand for mass analyses of heterogeneous macromolecules with ever-increasing masses is continually expanding, bringing additional challenges to native MS. Since, as described in the earlier sections, the instrumental mass resolution is not the major practical limiting factor for accurate mass detection in native MS, most of the recent advances in resolving high-mass and high-heterogeneity samples are focused on other factors such as improving gas-phase transmission and desolvation (see section 3), advancing algorithms for data analysis, and implementing separation in dimensions orthogonal to m/z. Additionally, newer approaches such as single-ion mass spectrometry can boost empirical resolution, circumventing high sample microheterogeneity issues, especially for high-mass analytes,127 by the use of charge detection mass spectrometry (CD-MS).128 In this section, we further review these developments in more detail.
4.1. Advanced Algorithms for FT MS
In FT MS, high resolution comes at the expense of the need for long ion observation times. This is exacerbated by imperfect magnetic and electric fields in FT-based mass analyzers, which result in gradual dephasing or loss of coherence in the trajectories of the ions. Moreover, while ion cooling is beneficial, this is typically performed by increasing the pressure along the transmission path, but the presence of gas molecules is detrimental in the acquisition of long transients. To tackle such limitations, it is desirable to reduce the ion observation time without decreasing the resolving power. In 2013, for Orbitrap MS, this was partly achieved by improving the initial data processing with the advent of enhanced Fourier transform (eFT), with benefits of both absorption-mode and magnitude-mode signal processing,115 whereby absorption-mode FT provides a resolution nearly twice as high as that of magnitude-mode FT, which in turn results in more robust amplitude spectra. Absorption-mode processing was also implemented for FT-ICR instruments, although relatively late, as in FT-ICR it is challenging to achieve phase coherence for all of the analyzed ions.40,129 Even more recently, a new method for FT MS data processing was introduced, named ΦSDM, which is based on deconvolution of FT spectra beyond the FT uncertainty, enabling high resolution by the use of just a small fraction of the transient length compared with eFT.130 Although it has been implemented only on Orbitrap mass analyzers and optimized primarily for the low-m/z region, for instance as used in TMT labeling experiments,131 ΦSDM also has the potential to improve the detection of short-lived signals characteristic of ensembles of large macromolecules as measured in native MS, thus providing improved resolution even when short transients are recorded. However, this still needs to be demonstrated in practice.
4.2. Additional Dimensions for Gas-Phase Separation in Native MS
An alternative way to reduce sample complexity and increase resolving power is by separating ions on the basis of their mobility in the gas phase. We will not discuss ion mobility MS (IM-MS) here extensively since it is covered in depth by another review in this issue and has already been reviewed in earlier publications.132−134 However, it is worth mentioning that IM-MS is a powerful and rapidly evolving branch of mass spectrometry in general, with fruitful features also for native MS. It not only provides a means to separate complex mixtures of proteins but can also be used to probe structural features. To illustrate the latter, IM-MS was recently used in a wide range of structural applications, including investigations of amyloid formation,135 conformational landscapes of the ubiquitin protein,136 and structural features of DNA/RNA telomeric G-quadruplexes.137 Moreover, combining ion mobility with tandem mass spectrometry has proven to be useful for fingerprinting of biopharmaceuticals138 and studies of protein interactions139 and conformational dynamics of protein assemblies.140
Spatial separation of ions was recently explored by Mathew et al.,141 who coupled a modified ToF instrument to a Timepix pixelated detector, which enabled them to unravel distinctive trajectories of ions in the mass spectrometer upon excitation. Such a hybrid instrument provided a first example of single-ion imaging of protein assemblies in native MS and distinguished between the signal of protein ions and secondary electrons produced by ion–surface collisions. The time- and space-sensitive detection with the Timepix detector has the potential to facilitate the disentangling of dissociation products of protein assemblies in native top-down MS.
4.3. Single-Molecule and Charge Detection for Analysis of Large and Heterogeneous Assemblies
Aside from tackling instrumental and processing resolution limits, alternative experimental designs have also been explored to improve the performance of native MS, most notably by measuring single ions as opposed to ensemble ion detection. Single-ion signals can be statistically postprocessed, enabling the data to be filtered to remove aberrant ion signals, thereby improving the S/N, mass resolution, and accuracy. This is possible because each ion is analyzed separately, and its detection is thus void of space-charge effects, ion coalescence, or high signal damping, which makes it more similar to time-resolved ion detection using time-to-digital converters in ToF MS. However, compared with ToF MS, for which single-molecule detection in native MS was already demonstrated in 2000,142 FT MS approaches provide much higher resolution and mass accuracy.
Single-molecule analysis in native MS can be performed using an Orbitrap mass analyzer. Single-molecule detection was initially demonstrated for intact myoglobin143 and readily showed superior resolution and mass accuracy compared with conventional ensemble measurement. Shortly after, a method utilizing multiple single-molecule scans to enhance resolution and accuracy was proposed by Makarov.144 As a proof of principle for measuring single ions in native MS, Rose et al.27 demonstrated single-molecule detection of the intact 800 kDa GroEL protein complex on the Orbitrap-EMR. More recently, using single-molecule analysis and postacquisition spectral filtering to remove decaying ion signals, Kafader et al.127 achieved a 10- to 20-fold resolution improvement for intact proteins with masses of up to 150 kDa on an Orbitrap instrument. Overall, single-molecule analysis extends far beyond conventional native MS and has been implemented using different ion detection principles, which are further described elsewhere.128
In addition to improved mass resolution and accuracy, single-molecule analysis allows researchers to probe the charge states of proteins that are typically too large or too heterogeneous to analyze with the conventional native MS approach. Early on, CD-MS experiments by the Smith group145,146 on FT-ICR instruments enabled the detection of extremely large ions (>100 MDa) with a huge number of carried charges (>30 000), such as T4 DNA particles.147 Since then, CD-MS has become a useful tool that is complementary to native MS for measuring large ions and has been implemented on specialized platforms, often featuring an electrostatic linear ion trap (ELIT) with a detecting cylinder inside.148 For a long time, resolution in CD-MS devices was lagging. However, recent advances have shown significant improvements, as exemplified by Jarrold’s group, who combined dynamic calibration and advanced ELIT designs.149 The lower limits typically restrict CD-MS to 250 charges and 1 MDa for mass. However, most recently, several exciting CD-MS applications in the mass range similar to that in conventional native MS have been achieved by using Orbitrap-UHMR mass analyzers, wherein single-ion signal amplitudes are proportional to the charge. Such data were reported simultaneously by the groups of Heck109 and Kelleher,150 whereby the new charge dimension allowed them to disentangle extremely heterogeneous viral particles and large immunoglobulin oligomers with high charge and m/z accuracy.
5. Spectral Deconvolution in High-Resolution Native MS
Native MS spectra can be very dense and congested because of both heterogeneity of the samples and often co-occurring ion signals of multiple charge states of the same species. In isotopically resolved spectra, overlapping ion signals of different ions and charging by charge carriers other than protons may further complicate the spectra. Therefore, the development of spectral deconvolution tools has become crucial for the interpretation of such native MS data. In this section we review these developments, primarily focusing on deconvolution in native MS but also addressing the deconvolution of isotopically resolved mass spectra.
5.1. Spectral Deconvolution for Native Mass Spectra
In ESI-based MS, biomolecules are detected as gaseous ions with a distribution of charge states, represented by adjacent peaks in the m/z space. Provided that the mass resolution is sufficient to record charge-resolved spectra, mass determination requires that these distributions of multiply charged ion peaks are converted into a monodispersed zero-charge format. Over the past three decades, multiple algorithms have been developed to address this either through specific peak assignment in the charge distributions or by simulation and fitting of a hypothetical spectrum to the entire raw spectrum or parts thereof. While the former category provides speed and robustness for the deconvolution of relatively simple spectra, the latter excels at addressing challenges in the mass analysis of heterogeneous samples, although these algorithms are generally more computationally intense.
The first algorithm for automatic charge state assignments and mass deconvolution of multiply charged mass spectra was developed by Mann et al.151 at the advent of electrospray ionization more than 30 years ago. In essence, the algorithm achieved mass determination by iteratively calculating the cumulative charge state abundance as a function of a hypothetical mass in a defined range. Although it was efficient for deconvolution of uncomplicated spectra of pure proteins, this algorithm suffered from some artifacts and was less useful for the deconvolution of spectra of mixtures. The next generation of deconvolution algorithms appeared in the early 1990s and utilized a metric to ensure the best probability-based fit between the predicted and experimental charge distributions.152,153 Maximum-entropy-based algorithms, in particular, resulted in a more reliable deconvolution. However, some drawbacks persisted, especially regarding speed, quantitation, spectral complexity, and deconvolution artifacts. Over the next two decades, various implementations of the algorithms, which combined advanced peak picking and charge assignment, culminated in overall faster and more accurate deconvolution algorithms.154−156 In parallel, novel approaches based on finding the best fit to raw data among multiple simulations of mass and charge distributions were pushing the boundaries of disentangling spectral crowdedness,157,158 as exemplified by the robust tool Massign developed by the Robinson group.159 The difficulty of deconvolving overlapping charge distributions, which is common for multimeric heterogeneous protein assemblies, was further optimized by using the minimax theorem originating from game theory (AutoMass)160 and—to an extent—resolved by second-derivative-based peak detection (PeakSeeker).161 Although direct peak detection provided significant advantages for disentanglement of complex native mass spectra, it often failed to use all of the peaks in the experimental mass spectra, which became a large focus of the more recent emerging tools for spectral deconvolution in high-resolution native MS.
In 2015, Marty et al. released UniDec (short for Universal Deconvolution), a software tool based on a Bayesian deconvolution algorithm that enables fast and quantitative processing of native MS data.162 Although the approach relied on some user input for correct mass deconvolution of complex mass spectra, it provided significant improvements in speed and accuracy compared with earlier methods, chiefly by applying a customized Richardson–Lucy deconvolution algorithm with separate charge and mass smoothing. In addition, UniDec allowed for the processing of native ion mobility mass spectra along with native MS data of varying complexity. Unlike many other academic deconvolution tools, UniDec has been well-maintained and further developed over the years. It is worth mentioning a few later additions, among which are an extension for batch processing of large data sets (MetaUniDec)163 and the SoftMax function for removal of artifacts in deconvolution of extraordinarily congested and charge-overlapping spectra164 (Figure 6). In 2020, Marty presented a universal scoring system for quality assessment of each deconvoluted mass feature,165 further improving the spectral deconvolution reliability in native MS.
Figure 6.

Spectral deconvolution of native MS spectra using UniDec software. The native mass spectra of AqpZ with bound POPC (a) and HSP17.7 (d) show satellite artifacts (red) when deconvolved (b, e) that are removed by addition of the SoftMax function (c, f). POPC nanodiscs with mixed heavy and light belts (g) show harmonic artifacts (orange) after deconvolution (h) that are removed by the SoftMax function (i). The triplet peaks from the mixed belts are preserved by the SoftMax function, as shown in the inset. Reproduced from ref (164). Copyright 2019 American Society for Mass Spectrometry.
Commercial software solutions for deconvolution seem to be somewhat biased, being primarily geared for the biopharmaceutical industry166 and showcasing robust performance for the analysis and quality control of biotherapeutic molecules such as antibodies and ADCs. Two of the currently most prominent algorithms include ReSpect, which uses an implementation of the maximum-entropy algorithm and is available in the BioPharma Finder software (Thermo Scientific), and another commercial deconvolution tool called PMI Intact (Protein Metrics).167 Within the BioPharma Finder suite, ReSpect is primarily used for the deconvolution of intact protein LC-MS data. On the other hand, in a recent study by Campuzano et al., PMI Intact has been shown to cope exceptionally well with very complex high-resolution native mass spectra of common biopharmaceuticals and polydisperse nanodiscs166 (Figure 7). In addition to advanced peak picking and smoothing of deconvoluted data, the PMI Intact algorithm uses a “parsimonious” criterion, which ensures that the resultant zero-charged spectrum has the minimum number of peaks necessary to explain the underlying experimental MS data.
Figure 7.

Spectral deconvolution by PMI Intact software. Shown is the deconvolution of the highly polydisperse native mass spectrum of an empty MSP1D1 nanodisc containing the phospholipid DMPC analyzed on an Orbitrap-EMR. The inset displays the initial native mass spectrum. The Comb filter, available in PMI Intact, results in the successful deconvolution of this complex spectrum. Reproduced from ref (166). Copyright 2019 American Chemical Society.
Two alternative recent approaches for the deconvolution of complicated mass spectra have emerged from the Prell and Kohlbacher laboratories. Using a fast Fourier transform-based algorithm allowed the Prell lab to deconvolve heavily populated spectra of heterogeneous ion populations while practically eliminating the need for prior knowledge about the putative mass and charge ranges and subunit composition.168 Potentially, this promises to be a more parameter-free method compared with existing deconvolution tools. By showcasing the algorithm’s strength on complex spectra of “empty” nanodiscs (Figure 8), Cleary et al. demonstrated how overtone peaks in Fourier spectra could complement Bayesian deconvolution as implemented in UniDec.169 It is worth noting that successful analysis with this approach requires repeating mass units to be present in the spectrum. Another novel deconvolution algorithm was recently published by the Kohlbacher lab, whereby they achieved speedy and robust charge assignment by transforming the observed raw peaks into the logarithm of m/z, taking advantage of the resultant charge-specific and mass-independent constant patterns that could be easily identified.170 It is exciting to see all of these parallel developments in spectral deconvolution that, combined with advances in mass resolution and mass range, promise to improve mass identification in high-resolution native MS, potentially extending its applications to previously uncharted areas of research.
Figure 8.

Native mass spectra of DPPC-MSP1E3D1 nanodiscs acquired on a Q-ToF mass analyzer and corresponding Fourier spectra for (a) the fundamentals and (b) the second harmonics. Inverse Fourier transforms of the charge-state-specific peaks in Fourier spectra (insets) are shown as overlaid envelope functions of the same color in the mass spectra. (c) Harmonic-averaged reconstruction of envelope functions. (d) Zero-charge spectrum (black) calculated from harmonic-averaged spectra for all charge states. Reproduced from ref (169). Copyright 2018 American Society for Mass Spectrometry.
5.2. Spectral Deconvolution for Isotopically Resolved Mass Spectra
Most of this review is focused on the analysis of large proteins and protein complexes by native MS, whereby the analytes cannot be isotopically resolved (yet). However, for sufficiently small proteins, typically below 50 kDa, isotopically resolved spectra can already be acquired using high-resolution mass analyzers, especially those based on FT MS and some of the more recently introduced high-resolution ToF-based instruments. A few groups are currently aiming to extend the reachable range for isotopically resolved spectra up to 150–200 kDa either by single-ion measurements127 or by long-transient acquisitions as are possible on high-end FT-ICR instruments.71 Advantageously, in isotopically resolved spectra, the charge can be directly extracted from the m/z differences between consecutive peaks in an isotope envelope. Moreover, this approach allows for either explicit or implicit detection of the monoisotopic masses, whereby implicit detection using robust computational approaches is essential for large molecules, for which monoisotopic mass peaks are nearly undetectable (e.g., 0.04% relative abundance for ∼17 kDa species).59 The application of spectral deconvolution methods in native (top-down) MS is exceptionally pertinent in protein fragmentation, whereby some of the backbone fragments are often detected in a single charge state, making high-resolution acquisition (>50 000 at m/z = 200 Th) and therefore deconvolution of isotopically resolved spectra a necessity.171 As native MS is more and more combined with tandem MS,28,172 we also include here a brief discussion about progress in spectral deconvolution of isotopically resolved mass spectra. Such deconvolution has been extensively developed in parallel with algorithms for unresolved mass spectra, with tools like Zscore,156 ProMex,173 and FLASHDeconv170 supporting both types of data as an input. Specific algorithms have been explicitly tailored to handle highly charged and congested spectra of isotopically resolved protein fragments, including MS-Deconv,174 YADA,175 THRASH,176 and its commercial implementation Xtract available in the BioPharma Finder software suite (Thermo Scientific). More recently, the Liu group released EnvCNN, a statistical artificial-intelligence-based model for scoring identified isotopic envelopes, whereby they demonstrated superior performance compared with existing scoring functions.177 While these tools are primarily used in conventional top-down MS analysis, most of them can also be applied to the deconvolution of native top-down mass spectra.28,172
Overall, there are many tools available for spectral deconvolution of high-resolution native mass spectra, which have substantially helped the field make the advances described in this review. Notwithstanding this huge progress, quite a few challenges remain ahead. For instance, deconvolution of spectra on the fly (in real time), which has been achieved to an extent on some Orbitrap-based instruments, will significantly facilitate the optimization of data acquisition methods. Another challenge for the upcoming deconvolution methods is to handle incomplete desolvation and salt adducts, which are the most common sources of errors in mass determination in native MS. As not yet widely implemented, deconvolution can be improved by better leveraging of the expected repeating mass differences in native mass spectra, stemming, for instance, from the infamous salt adducts. Finally, with the advent of CD-MS, there is a huge potential in utilizing additional layers of information about single ions to guide deconvolution algorithms for more accurate results.
6. Tandem Mass Spectrometry and Ion Activation in Native MS
Tandem mass spectrometry, or MS/MS, provides a means to obtain an additional layer of structural information about the analytes and is used widely to sequence peptides and small proteins. In such experiments, the precursor ions are mass-selected and submitted to a particular type of ion activation with the aim to fragment them. Tandem MS thus requires the deposition of energy into the precursor ions, and in the field of mass spectrometry there are currently quite a few different ion activation methods available.178−180 Although these methods have not been developed with native MS applications in mind, they are often complementary, with some being more applicable to native MS than others, also depending on the aim of the ion activation. Often in native MS, ion activation is not necessarily used to generate dissociation products but rather is employed to improve desolvation,63 enhancing the attainable mass resolving power. This section briefly describes first the different ion activation methods available and subsequently their particular applications in native MS.
Ion activation techniques can be broadly categorized into collision-based methods, irradiation-based methods, and methods based on ion–ion interactions.181 Collisions with inert gas molecules (collision-induced dissociation (CID) or collisional-activation dissociation (CAD)) represent the most frequently used ion activation methods in mass spectrometry, including native MS. This dominance likely originates from their widespread availability on nearly every mass spectrometer as well as their simplicity and robustness.182 Although displaying excellent fragmentation efficiency, collision-based activation is mainly hampered by slow and stepwise energy deposition that primarily dissociates the most labile bonds, often leading to small neutral losses and other less informative dissociation products. Over the years, alternative ion activation techniques have been introduced that complement CID/CAD and provide better access to high-energy dissociation pathways. These alternative activation methods include methods based on collisions with surfaces (surface-induced dissociation (SID)122), interactions with electrons (e.g., electron-capture dissociation (ECD)183 and electron-transfer dissociation (ETD)184), or absorption of either low-energy photons (infrared multiphoton dissociation (IRMPD)) or high-energy photons (ultraviolet photodissociation (UVPD)).185 Because of the cross-complementarity of different ion activation methods, multiple hybrid techniques have recently emerged, with some getting widely adopted by the mass spectrometric community, e.g., electron-transfer higher-energy collisional dissociation (EThcD), in which ETD is combined with supplemental low-energy collisional activation.186,187 We refer readers interested in a more detailed overview of ion activation technologies in mass spectrometry to excellent reviews from the Brodbelt group.180,188
When applied to protein complexes studied by native MS, ion activation methods are used to (1) enhance desolvation and improve mass resolving power, (2) eject protein subunits from non-covalent protein complexes to provide structural information, and (3) fragment polypeptide chains of individual subunits to determine amino acid sequences used to identify the protein. While the slow, stepwise, and low-energy activation provided by collisional activation works well for desolvating ions and removing adducts (1), faster and more energetic photon- or electron-induced methods are more suitable for fragment formation and sequencing of subunits from within native complexes (3). Non-covalent dissociation (2) can benefit from various techniques, as distinct mechanisms of protein complex partitioning provide very complementary results.
6.1. Improving Mass Resolution and Accuracy by Ion Activation through Enhanced Desolvation
Incomplete removal of solvent adducts (i.e., water, buffer, and salt molecules) is a major limiting factor for obtaining high resolution and mass accuracy in native MS, as this increases the peak width of the detected ion signals and artificially increases the measured mass.32,189 Carefully balancing the amount of ion activation allows residually bound small molecules to be removed, whereby the internal energy should remain below the thresholds for fragmentation or dissociation of protein assemblies. Collisions with background inert small gas molecules (He, N2, Ar, Xe) are widely used for this purpose, as this approach is characterized by its ability for a slow and gradual buildup of internal energy.86,190,191 In particular, for native MS of large macromolecules, desolvation can be promoted by using heavier gas molecules (e.g., Kr, Xe), as they generally deposit more energy into the precursor molecule with each collision.86 Since ion activation by CID is m/z- and charge-state-dependent, the ions with lower charge within a charge state envelope of a particular precursor generally display substantially broader peaks due to unequal levels of desolvation.34 Particularly large protein assemblies measured at high m/z can therefore benefit from the development of alternative desolvation methods that are less charge-state-dependent. Recent work demonstrated that in particular IRMPD could also be used to enhance desolvation, possibly even more effectively than CID,192−194 and should therefore possibly be more explored for native MS.195
Effective desolvation in native MS is a balancing act in removing unwanted adducts while preventing the loss of specific interactions and labile PTMs. In native MS of membrane proteins, for example, sufficient desolvation is essential for stripping proteins of detergent molecules or other solubilizing agents while retaining specifically bound lipids or other small molecules (e.g., drugs).196,197 On the other hand, there are also protein assemblies that require small molecules or specific cations to stabilize their native structures.198 Furthermore, labile PTMs that are often present on native protein assemblies, e.g., glycosylation and phosphorylation, are known to be susceptible to dissociation upon harsher desolvation conditions. Thus, in native MS, optimal desolvation must always be balanced to avoid inducing fragmentation or the loss of small molecules or labile PTMs that are essential for the structure and stability of the studied protein complexes. As each protein complex exhibits its own biochemical and biophysical features, there are unfortunately no general rules to determine the best way to achieve this.
6.2. Revealing Structural Features of Protein Assemblies by Gas-Phase Ion Activation
Perhaps the most intriguing benefit of ion activation in native MS is its ability to provide information about the quaternary structures of protein assemblies. In tandem MS experiments, the precursor ions are mass-selected and subsequently activated, which may lead to the specific ejection of individual subunits87,199 or other modes of complex partitioning.188,200,201 The resulting mass measurement of the monomeric dissociation products and corresponding concomitant higher-mass dissociation products can reveal the compositional buildup of the precursor. Often the order of subunit ejection and the observed dissociation pathways can provide information on the arrangement of subunits within the protein complex.199 Moreover, additional fragmentation of the ejected monomeric subunits can be used to reveal the identity of the subunits. However, from all of the available tandem MS studies on protein assemblies, it also has become apparent that the smaller peripheral subunits often display a preference to be ejected and also that the structures of the activated protein complexes can drastically change when transferred into the gas phase and even more so upon activation.202 To summarize, although it should be used with care, tandem MS studies can provide essential structural information that is not easily accessible by other means, as briefly illustrated below with a few prominent examples.
6.2.1. Collisional Activation in the Gas Phase
Collisional activation in the gas phase has been used most extensively in native MS for protein complex dissociation, and it is featured by a wide range of instruments. Recently, Wang et al.202 demonstrated that although collisional dissociation (CID/CAD/HCD) preferentially results in ejection of peripheral subunits, less exposed nonperipheral subunits also can be ejected from certain protein assemblies. Studying the 20S core proteasome of Thermoplasma acidophilum, engineered antibody complexes, and elongated complement protein complexes, they identified two major pathways by which nonperipheral subunits can be released. First, sequential dissociation of highly charged peripheral subunits may transfer residual energy back to neighboring subunits and increase their exposure. Alternatively, nonperipheral subunits may be ejected either directly from complexes stabilized by chemical charge reduction or from elongated complexes that may undergo compaction upon transfer into the gas phase. Several other examples of studies that combine native and tandem MS using CID/CAD/HCD will be described in more detail below, particularly in section 7.
Generally, the mechanism of collisional dissociation of protein complexes involves the release of weakly bound single subunits that take away a large portion of charges carried by the precursor. Several groups have tried to come up with an explanation for this frequently observed behavior, which is known as asymmetric charge partitioning. In addition to available empirical evidence, the Konermann group performed molecular dynamics simulations that provide a more detailed fundamental understanding of how multimeric complexes behave upon collisional dissociation.203 In brief, because of the slow buildup of internal energy inherent to collisional activation methods, extensive structural rearrangements and unfolding can take place prior to dissociation.204−207 This is attributed to a microsecond-time-scale process in which the to-be-ejected single subunit unfolds, leading to Coulombically favored charge redistribution. Consequently, the high-charge monomer subunit departs from the concomitantly formed charge-stripped high-mass fragment.207,208 Thus, although widely used, protein complex partitioning in collision-based native tandem MS is a complicated process, and further studies are needed to understand abnormal dissociation pathways observed for complex heteromeric assemblies.
6.2.2. Surface-Induced Dissociation
SID combined with native MS has been pioneered primarily by the group of Wysocki as an alternative approach that, in contrast to dissociation methods based on collisions with gas molecules, largely avoids unfolding of individual subunits and asymmetric charge partitioning.200,201,209−211 Collision with a surface is a relatively fast activation process that provides access to higher-energy dissociation pathways and occurs on the picosecond time scale, and it can lead to the direct ejection of folded subunits even from stably bound assemblies resistant to CID.204,212 Such a mode of partitioning is mostly accompanied by symmetric charge redistribution between the dissociation products, which allows SID to retain a high level of structural information that, when combined with IM-MS, is sensitive enough to reveal differences between precursor conformations.213,214 Moreover, ejected subunits largely preserve non-covalent interactors because of their folded nature, enabling SID to probe stoichiometries of ligand binding. In a study of structurally and energetically well-characterized protein complexes, it was shown that the strength of the interaction interface in a given protein assembly could be correlated with the amount of energy required for its dissociation by SID.215 Illustratively, Vimer et al.216 recently utilized these approaches to probe structural conservation and variation among orthologous 20S proteasome complexes purified from five different organisms (described in more detail in section 7.2). Structural information obtained from SID and covalent labeling studies also enabled Song et al.217 to model and refine the structure of the hexameric toyocamycin nitrile hydratase.
6.2.3. Ultraviolet Photodissociation
UVPD was initially exploited in mass spectrometry for applications in bottom-up and top-down fragmentation.185,188,218−220 Its applicability to dissociate native protein complexes has more recently been explored. Using high-energy 193 nm photons, UVPD is able to deposit substantial amounts of energy on a time scale close to that of SID, giving access to higher-energy dissociation pathways.221−223 Two recent studies have shown that high-energy laser pulses (≥1.5 mJ) can eject subunits from protein complexes with more symmetrical charge partitioning, in some cases resembling SID-like behavior.224,225 Contrary to SID, however, UVPD can provide additional backbone fragment ions, yielding sufficient sequence coverage for the identification of individual subunits.226 The efficiency of UVPD also exhibits little dependence on the precursor charge compared with collision-based approaches,227 making this approach well-suited also to study large native protein assemblies that are detected in multiple charge states. As an example, this allowed Greisch et al.124 to eject intact 16 kDa subunits from a 1 MDa AaLS virus-like particle while simultaneously providing diagnostic fragment ions that covered around 60% of the subunit’s sequence.28
6.2.4. Infrared Multiphoton Photodissociation
The use of powerful infrared lasers for the fragmentation of proteins and protein assemblies has also been explored in native MS. IRMPD, although very similar to collisional dissociation, has several advantages. First, it does not require elevated gas pressures, which is especially important in FT-ICR and Orbitraps, which need a high vacuum for optimal performance. In addition, IRMPD provides a high level of control over the amount of energy deposited by photon irradiation. These advantages prompted researchers to implement IRMPD on the various instruments used for native MS, including Q-ToF-,228 FT-ICR-,6 and Orbitrap-based instruments.195 The groups of Loo28 and Gross229 have extensively explored the application of IRMPD for native top-down MS on ICR platforms, primarily using this method for supplemental activation of proteins and protein complexes along with fragmentation using more energetic and prompt activation techniques like ECD. In an attempt to study membrane protein complexes, Mikhailov et al.228 implemented IRMPD on a Q-ToF instrument, whereby IR irradiation provided great results for liberating nonsoluble protein assemblies from detergent micelles. Finally, in a very recent study combining IRMPD with native MS analysis, Greisch et al.195 explored the dissociation of protein complexes with masses up to 1 MDa, revealing that the increasing cross sections of larger protein assemblies enable more efficient irradiation and fragmentation.
6.2.5. Electron-Based Dissociation Techniques
Electron-based fragmentation techniques include a range of methods that vary in the way electrons are delivered to the proteins. For instance, in electron-capture dissociation (ECD) the analyte is directly irradiated with electrons, while in electron-transfer dissociation (ETD) the fragmentation is induced via ion–ion interactions between positively charged protein ions and the negatively charged electron-transferring reagent anions. Because electron-based activation on its own does not fully induce disruption of non-covalent interactions, observed fragments primarily stem from the most exposed regions on the protein surface. Thus, for high sequence coverages, it is often necessary to use supplemental activation to release the formed fragments. By using ETD on a Q-ToF instrument for fragmentation of protein complexes, Lermyte and Sobott230 established that, by balancing the supplemented activation it is possible to probe surface-exposed regions and gain insights into protein behavior upon collision-induced unfolding. Leveraging the ability of ETD to preserve labile modifications, Tamara et al.231 applied ETD in MS3 on an Orbitrap Lumos instrument to position phosphate transfer events within the binding interface between Pin1 and its phosphorylated binding partner. ECD implemented on an FT-ICR mass spectrometer was successfully applied for structural investigation of various protein assemblies, exposing flexible regions229 and binding interfaces in protein–ligand complexes.232 Native ECD, which is induced by the generation of endogenous electrons in the areas adjacent to metal-binding sites through infrared excitation, allowed researchers to map iron-binding pockets in a ferritin complex.233 Using yet another electron-based activation method, electron ionization dissociation (EID), in their studies of metal-binding complexes, Li et al.234 could reflect structural differences between apo, Zn-, and Cu,Zn-SOD1 dimeric complexes and probe the structural stability of carbonic anhydrase I.
6.3. Higher-Order MSn Methods in Native MS
Characterization of proteoforms and subcomplexes can be achieved by pseudo-MS3 analysis, whereby the protein complexes are interrogated in a two-step process.119 Such experiments have been implemented on Orbitrap-UHMR instruments and have shown powerful capabilities in providing structural information even when collisional activation is used in both steps.69 For example, van de Waterbeemd et al.53 employed collisional dissociation to investigate the stalk complexes in chloroplastic ribosomes. First, these subcomplexes were ejected from intact ribosomal particles by collisional activation in the front end of the mass spectrometer and then dissociated further with HCD to determine their exact stoichiometry. By combining complementary fragmentation methods in two-tier ion activation on an Orbitrap-UHMR, first using collisional activation in the front end of the instrument and then UVPD in the back end, Mehaffey et al. were able to sequence ejected subunits and position the non-covalently bound cofactors on human mitochondrial BCAT2.123 In another approach, Li et al.28 combined native MS with top-down sequencing on a high-resolution FT-ICR instrument. They demonstrated that assemblies with masses of up to 1.8 MDa can be resolved in charge states similar to those observed in ToF or Orbitrap experiments. Next, they employed various ion activation techniques to investigate the compositional makeup of various protein complexes. In-source dissociation (ISD) or CAD was used to eject individual subunits, followed by IRMPD to reveal information on the backbone sequence and PTMs, thereby characterizing proteoforms in various assemblies, including β-galactosidase. On the other hand, ECD fragmentation was used to investigate the topology of the intact complex. This activation method largely preserves non-covalent interactions, meaning that when N- and C-terminal fragments are observed, they likely originate from the surface of the complex. The integrated platform presented by Li et al. thus shows great promise for native MS as a bridge between proteomics and structural biology, providing new insights into protein structure and function. Higher-order tandem MS methods have also been used to study the interactions of membrane proteins with endogenous lipids, peptides, and small molecules. In such an application, Ro et al.235 interrogated the membranous metalloenzyme pMMO embedded into nanodiscs (Figure 9). In the first collisional activation step, they released the complex from a solubilizing nanodisc or, by elevating the collisional activation energies, even dissociated the complex into distinct pMMO subunits. Subsequently, the ejected subunits were further fragmented in the HCD cell of the Orbitrap instrument. Overall, this approach allowed the researchers to determine the stoichiometry of copper binding and the presence of several PTMs in the pMMO complex.
Figure 9.

Example of a higher-order native top-down MS approach used for the characterization of membrane proteins such as pMMO. Nanodiscs embedded with pMMO were subjected to CID in the source region for ejection of the protein complex from the nanodisc, resulting in the stabilization of the pMMO protomer species (MS1). Increasing collisional activation breaks up the protomer into individual pMMO subunits (MS2). Further collisional activation in the HCD cell of the Orbitrap-UHMR enables backbone fragmentation of each subunit (MS3). From ref (235). CC BY 4.0.
Recently, the Robinson group introduced the concept of “nativeomics” as a multilevel native top-down MS approach to identify endogenous ligands of membrane protein assemblies, distinguishing them from other copurified lipids.74,236 The first step of the workflow introduced by Gupta et al.236 involves stepwise delipidation of the protein assembly using preservation of the oligomeric state as a measure for conditions that still allow key ligands to remain bound. The extracted lipid fractions are then subjected to LC-MS analysis, providing a library of potential ligands. High-energy native MS measurements were performed on a ToF instrument modified for dissociation of detergent micelles in the front end of the instrument. This enabled mass selection of a single charge state of the protein–ligand complex followed by dissociation of the ligand in the collision cell to determine its intact mass. Database searches of the mass uncovered in this manner were then performed to determine the identity of the ligand. In a follow-up study, Gault et al.74 utilized a modified Orbitrap instrument to enable systematic selection and fragmentation (up to pseudo-MS6), providing further confidence in ligand assignments and expanding the approach to more complex samples. Studying endogenous lipids binding to aquaporin Z (AqpZ), they identified interactions with various lipid families, with AqpZ showing a preference for shorter and unsaturated chains. Similarly, they were able to identify previously unknown ligands bound to dimeric outer mitochondrial membrane translocator protein (TSPO) (Figure 10), demonstrating the utility of tandem MS for identification of potential regulators of this critical drug target.
Figure 10.

Identification of unknown ligands bound to TSPO and subsequent fitting of PE 16:0/18:1 lipids into unresolved electron density in the X-ray structure. (a) Schematic showing the release of the TSPO dimer from detergent micelles (the subunits are shown as red and blue cartoons and the detergent micelle as orange spheres). (b) Native mass spectrum of the TSPO dimer (pseudo-MS2 step (pMS2)) and isolation of the 7– charge state (red box) (inset). (c) Collisional activation (pseudo-MS3 step (pMS3)) yields dissociated monomer, apo dimer produced from neutral ligand loss, and multiple ligands at low m/z (blue box). (d) Zoom of the low-m/z region showing two peak series corresponding to multiple lipids: PE (red) and PG (orange). (e–g) Isolation and subsequent fragmentation of released lipids (pseudo-MS4 step (pMS4)) define the hydrocarbon chain length and extent of unsaturation. (h) Position of the most abundant PE lipid identified, PE (16:0/18:1) (red sticks), in the TSPO A139T structure (PDB entry 4UC1). (i) Critical protein–lipid interactions (zoom box). Reproduced with permission from ref (74). Copyright 2020 Gault et al.
6.4. Combining Tandem MS with Native Separation in Native Top-Down Proteomics
When samples contain multiple protein assemblies with similar masses, it is quite challenging to characterize the constituent assemblies with direct-infusion native MS, and therefore, a separation step, akin to liquid chromatography used in shotgun proteomics, is required. Native top-down proteomics is a relatively new direction in the field that aims to analyze proteomes at the level of endogenous protein assemblies.237 This approach utilizes a mild (native) separation technique such as size-exclusion chromatography (SEC), ion-exchange chromatography (IEX), or capillary electrophoresis (CE) to first fractionate complex biological samples, followed by native tandem MS to analyze intact protein assemblies and their constituents.172,238,239 In a recent study by Skinner et al.,172 a two-step off-line fractionation method was used to characterize 125 protein assemblies and 217 distinct proteoforms from human cell lines and mouse tissue. An alternative approach by Shen et al.238 combined SEC prefractionation with online capillary zone electrophoresis (CZE), providing a high-throughput platform that identified 144 proteins, 672 proteoforms, and 23 protein complexes from Escherichia coli.
In section 6, we have summarized how native MS can be combined with tandem mass spectrometry and how this combination can help to improve the desolvation of the produced ions, resulting in more precise mass measurements. Tandem MS can also be used to gain insights into the quaternary structures of the studied protein assemblies. The latter significantly benefits from the monitoring of distinctive dissociation pathways, which provide information about the connectivity of loosely bound peripheral subunits, subunit topologies, proteoform identities, and sometimes even cofactor binding. More examples of the combination of native MS and tandem MS will be described later in the review, especially in section 7, wherein we highlight recent work on different classes of proteins and protein complexes as studied by high-resolution native MS.
7. Application of High-Resolution Native MS in the Characterization of Protein Assemblies
As an analytical technique, native MS has been around now for several decades. Although it was initially used to study small proteins and protein–ligand interactions, the applications of native MS have expanded and by now cover a huge variety of protein assemblies involved in a plethora of different biological processes. Although we aim to provide an extensive review, we cannot be all-inclusive in describing all of the reported applications of high-resolution native MS in the characterization of protein assemblies. Instead, we provide and review a selection of applications categorized by the biological processes in which these assemblies are involved, notably protein synthesis and degradation, complement activation, and light harvesting. Additionally, we review the application of native MS to characterize virus(-like) particles and membrane-embedded protein assemblies. The focus and scope of this review necessitate that we leave out some other exciting applications, so we apologize especially to the authors of these studies for not being fully comprehensive in this section.
7.1. Protein Assemblies Involved in Protein Synthesis: Ribosomal Particles
Ribosomes are very large macromolecular machines found within all living cells that are responsible for protein synthesis (i.e., mRNA translation). Ribosomes consist of two major components: the small and large ribosomal subunits. Each subunit consists of one or more large ribosomal RNA (rRNA) molecules and many ribosomal proteins (RPs). As such, ribosomes represent rather challenging samples for native MS. First, they are very large and multicomponent ribonucleoprotein assemblies that display multiple putative PTMs and composition-related heterogeneities. Additionally, significantly negatively charged nucleic acids, originating from the rRNA, result in a decreased total positive charge of the ribosomal particles, leading to detection at elevated m/z compared with merely proteinaceous assemblies of similar mass. Perhaps because of these aspects, the application of native MS to the analysis of ribosomes has been somewhat limited. Structural analysis of ribosomal particles has been primarily achieved with crystallography and now more and more by cryogenic electron microscopy (cryo-EM), sometimes supplemented by chemical cross-linking mass spectrometry.241,242 Some novel detailed insights into ribosome composition can also be obtained via bottom-up mass spectrometry.243,244 Recently, the earlier view that the ribosome is a static system has been challenged on the basis of multiple lines of evidence for a more heterogeneous and dynamic nature that likely has implications for translation and protein synthesis.245 The latest studies have shown that the current state of high-resolution native MS can provide a high level of insight into compositional and structural heterogeneity of ribosomal particles, especially when combined with other modes of mass spectrometry or other structural biology tools.53,66
In the early 2000s, the limited mass resolution and desolvation capabilities of native-MS-capable mass spectrometers hampered accurate mass detection, leading to blindness toward the substoichiometric or low-abundance variants of ribosomal particles.246,247 However, the Robinson group demonstrated the potential of native MS to provide useful information on ribosome composition by unraveling the stoichiometry and proteoforms of the stalk complex within the 50S ribosomal particle derived from Thermus thermophilus.246 These findings complemented more standard high-resolution structural techniques such as crystallography, which were limited in providing high-resolution structural information for the more flexible parts of the ribosomes. Although they were unable to obtain accurate masses of ribosome assemblies because of large peak widths and low overall resolution, in 2006 McKay et al. reported that the major bottlenecks for solving this issue were caused by incomplete desolvation of the produced ions and potentially co-occurring components within ribosomal complexes.248 Notably, by modeling the average mass increase as a function of peak width for standard non-covalent assemblies, they could better explain the observed mass distributions of the ribosomal particles. Tandem MS experiments further confirmed that multiple ion populations did coexist in their experiments.
About 10 years later, the advanced desolvation and transmission capabilities of Orbitrap instruments with ultrahigh mass range (Orbitrap-UHMR)30 enabled the recoding of more detailed high-resolution mass spectra, revealing altered ribosomal particles and substoichiometric subunits (Figure 11). Such spectra were achieved mainly as a result of improved instrumental sensitivity and desolvation in both the front and back ends of the instrument and by the use of charge-reducing conditions, i.e., addition of triethylammonium acetate to the spraying solvent.66 The mass accuracy for detection of the E. coli 70S ribosome was within 0.7% of the theoretical mass, an excellent result for a particle with a mass of more than 2 MDa. By lowering the concentration of Mg2+ ions essential for 70S complexation, smaller and better-resolved spectra of the 30S and 50S ribosomes could be recorded, revealing their intrinsic heterogeneities, which partly also explained the relatively less resolved spectra of the intact 70S ribosome. In the follow-up study by van Waterbeemd et al., several ribosomal particles originating from human cells and spinach chloroplasts were analyzed using multiple mass spectrometric modes.53 Ribosomal proteoforms detected in complementary top-down analysis allowed for a more confident assignment of the native mass spectra and provided exact compositions of the substoichiometric subunits. Additionally, high-resolution native MS was used to investigate the binding and stoichiometry of viral internal ribosome entry site (IRES) RNA elements of hepatitis C virus and cricket paralysis virus to the 40S human ribosome particles (Figure 12).
Figure 11.

High-resolution native MS of ribosomal particles. (a) (left) Structures of the E. coli 70S ribosome consisting of the 50S (proteins shown in blue and rRNA in green) and 30S (proteins shown in red and rRNA in orange) particles. (right) High-resolution native mass spectra of the 70S, 30S, and 50S ribosomes. (b) Distinct particles of the 30S ribosome, with annotations provided at the top. Masses are shown as mean ± SD. (c) Distinct particles of the 50S ribosome, with annotations provided at the top. In most of the particles, the pentameric stalk complex is absent. Reproduced with permission from ref (66). Copyright 2017 Nature Publishing Group.
Figure 12.

High-resolution native mass spectra of free and internal ribosome entry site (IRES) RNA-bound human 40S subunits. (a) Native mass spectrum of the human 40S subunit acquired with an Orbitrap-UHMR mass analyzer. The well-resolved charge states of three distinct forms of the ribosomal subunit could be detected. The most abundant fully assembled 1.2 MDa 40S particles are labeled in blue, while lower-abundance particles lacking either the S25 or S10 protein are labeled in magenta and red, respectively. (b) Monitoring of the formation of a complex containing human 40S ribosomes and IRES RNA fragments of cricket paralysis virus (CrPV) and hepatits C virus (HCV). The mass spectra of the RNA fragments alone (insets) provide the accurate masses of the IRES elements. Structures of the free 40S ribosomes (PDB entry 5A2Q) and particles bound by CrPV (PDB entry 4V91) and HCV IRES (PDB entry 5A2Q) are shown, with the ribosomal proteins in blue, the rRNA in gray, and the IRES elements in yellow and green, respectively. From ref (53). CC BY 4.0.
In a recent study, Abdillahi et al.249 applied anion–cation reactions to study the attachment of high-mass myoglobin ions [hMb – 11H]11– to positively charged 30S ribosomal particles prepared by native MS. They demonstrated that these ion–ion reactions can facilitate obtaining well-separated ion signals that can yield confident charge-state and mass assignments from otherwise poorly resolved signals. Only upon attachment of multiple myoglobin anions could the intact 30S particles and the particles missing the S1 protein be resolved. This is a promising new development that is somewhat similar to the more well-established proton transfer reaction (PTR)250 and especially can help to disentangle poorly resolved ion signals on instruments that have inherently lower resolution.
7.2. Protein Assemblies Involved in Protein Degradation: Proteasomal Particles
Proteasomes are ubiquitously present in all eukaryotes and archaea and also in some bacteria, where they regulate the abundance of particular proteins and remove misfolded or damaged proteins by degradation. This degradation process yields peptides, which can then be further degraded for use in the synthesis of new proteins or presented to immune cells by major histocompatibility complexes (MHCs) on the cell surface. The full 26S proteasome holoenzyme is built from two subcomplexes, the 20S catalytic core particle and one or two 19S regulatory particles that recruit, select, and unfold ubiquitinated substrates, preparing them for degradation in the 20S catalytic core particle. The 26S complex is a multimeric assembly with a mass of approximately 1.5 MDa, whereas the 30S complex contains the 20S core with two 19S lids. In the case of the 26S proteasome, progress in elucidating its structure has for a long time been hampered by the complexity of the system and its variability and fragility. Through advances in cryo-EM, several structures of the full 26S proteasomal particles are nowadays available.251−253 As far as we know, no native mass spectra have been reported to date for the full 26S proteasome, but several groups have been working on the 20S core particle and the 19S regulatory lid.
The overall structure of the 20S core proteasome is highly conserved, forming an ∼700 kDa barrel-shaped compartment whose proteolytic active sites are restricted to its interior so that only proteins entering the chamber are degraded. The 20S proteasome is composed of 28 subunits, arranged in a cylindrical structure consisting of four heptameric rings: two outer α-type subunit rings embracing two central β-type subunit rings (α7β7β7α7). The two outer α rings function as a gate that regulates the entry into the proteolytic chamber confined by the two β rings. Although the 20S proteasome is ubiquitously present in archaea and eukaryotes, a substantial increase in complexity and diversification of the complex is observed with the evolution of different organisms. Prokaryotic 20S proteasomes (e.g., from Thermoplasma acidophilum) are generally composed of 14 identical α subunits and 14 β subunits, while in eukaryotic proteasomes, the α and β subunits each differentiate into seven distinct subtypes, accounting for a total of 14 unique subunits. Moreover, in mammalian systems, dedicated distinct proteasome particles co-occur next to the standard (or constitutive) proteasome, such as the immunoproteasome (which contains distinctive βi subunits), the thymoproteasome, or the testis-specific proteasome.
Early in the era of native MS, several studies reported spectra of 20S proteasome core particles originating from different species. These studies readily provided highly charge-resolved spectra for archaeal and yeast 20S proteasomes, but the native MS spectra became more cluttered in the case of more heterogeneous particles originating from mammalian cells.18,27,254,255 These initial studies were often combined with tandem mass spectrometry to define the masses of the subunits. Later studies, apart from structural analysis of the main proteasome assemblies, also focused on the binding and stoichiometries of the different proteasomal inhibitors and substrates.254
A large body of the work performed by native MS to study the 20S proteasome has recently been reviewed by Ben-Nissan et al.256 They described several studies on how native MS was used to reveal the composition, intrinsic heterogeneity, stoichiometry, subunit architecture, and topology of the 20S particles. Also, they provided protocols on how to extract endogenous 20S proteasomes from yeast, rat liver, and human cells that are menable for analysis by native MS. In a subsequent elegant study, Vimer et al.216 used these enrichment protocols for the purification and subsequent analysis by native MS combined with IM-MS and top-down MS to probe structural and functional conservation between 20S proteasome particles extracted from archaeal Thermoplasma acidophilum, yeast (Saccharomyces cerevisiae), and mammals, including rat (Rattus norvegicus), rabbit (Oryctolagus cuniculus), and human (i.e., from HEK293 cells) (see Figure 13). Using native IM-MS, they observed higher collision cross sections (CCSs) for the eukaryotic proteasomes compared with those of the archaeal 20S complex, which they related to the increased complexity of eukaryotic 20S particles. Distinctively, the eukaryotic 20S proteasome particles exhibited several PTMs, extending their diversification. As these latter proteoform variabilities could not be directly resolved from the native MS spectra alone, their elucidation required the combination of native MS with top-down proteomics using both HCD and ECD as fragmentation methods.
Figure 13.

20S proteasomal particles analyzed by surface-induced dissociation (SID) combined with ion mobility mass spectrometry (IM-MS). SID-IM-MS spectra of the rabbit 20S proteasome complex reflect the cylindrical topology of the complex. (A) Charge-reduced rabbit 20S proteasome precursor ions were measured on a Synapt G2 instrument equipped with an SID cell (top), and the 43+ and 44+ charge states were isolated (bottom) and accelerated into the surface at 150 V. (B) IM-MS plot of the SID spectra of the rabbit 20S proteasome. The separation in drift time (vertical axis) assists in the discrimination of species that overlap in m/z (horizontal axis), as shown in (C). The major populations of the dissociation products are designated by dashed lines in (B) and labeled with symbols that are graphically depicted in (D–J). The extracted m/z spectra from the underlined regions in the IM-MS plot (D–J) show the identified dissociation products. Reproduced from ref (216). Copyright 2020 American Chemical Society.
Although a large body of reported work has been focused on the 20S core particles, Sharon et al.257 did report on analyzing the structural organization of the yeast 19S proteasomal lid by native MS. Their native MS data on the intact lid complex demonstrated that eight of the nine subunits were present stoichiometrically (with the Rpn6 subunit missing), and they observed a stable tetrameric subcomplex. Tandem MS was used to infer details on the subunit architecture at a time when little structural information was available for the lid.
A challenge in the analysis of proteasomes is to detect the 26S and 30S proteasome complexes by native MS, which has remained difficult because of the instability of these holo enzymes. Using mass photometry in parallel with native MS may be of assistance here. In this context, Sonn-Segev et al.258 studied the stability of the proteasome purified from bovine heart tissue by mass photometry and observed distinct 2.4 MDa 30S particles (two regulatory particles and one core particle) and 1.5 MDa 26S particles (one regulatory particle and one core particle) as well as the 700 kDa core particle and 800 kDa regulatory particle. In line with earlier literature, their data reveal that the holo enzyme dissociates at ionic strengths above 50 mM. Screening sample stability under varying conditions by mass photometry may help to interrogate distinct proteasomal assembly states with native MS.
7.3. Virus(-like) Particles
Even in the early years of native MS, because of their biophysical stability, viruses and virus-like particles (VLPs) became targets for mass analysis, primarily to probe whether such large assemblies could be transferred intact into the mass spectrometer and subsequently mass-analyzed.259,260 For detection, either early-generation charge-detection-based devices,260 electrophoretic mobility (i.e., GEMMA),261−263 or Q-ToF mass analyzers were employed, with the latter allowing the most accurate mass analysis via (partly) resolved charge states.142 Some of the first high-resolution data were obtained for intact capsids of hepatitis B virus (HBV) by making use of a Q-ToF mass analyzer modified for high-mass analysis.88,264 HBV capsids are quite unique in being able to exhibit two distinct icosahedral morphologies in vivo, composed of 90 or 120 dimers with masses of roughly 3 and 4 MDa, respectively. These early native mass spectra displayed well-separated charge state distributions for both types of morphologies, enabling mass assignments within 0.1% error. The HBV capsids were surprisingly stable during transfer into the gas phase and through the vacuum of the mass spectrometer. Measuring the CCS of the capsids by ion mobility mass spectrometry allowed an estimation of the capsid radii that was in good agreement with the particle dimensions as observed by EM, suggesting a largely retained native capsid morphology in the gas phase.264
Since this early work, native MS has contributed substantially to the field of structural virology, particularly in regard to our understanding of capsid assembly, virus maturation, genome packaging, viral protein subunit stoichiometries, and virus stability. This work has been summarized in several excellent review articles, to which we refer readers with a deeper interest in mass-spectrometry-based structural virology.128,265−268 We divide this subsection into applications focusing on empty viral capsids, endogenous genome-packed viruses, and virus-like assemblies, including bacterial compartments and natural as well as designed synthetic protein nanocages. The breadth of these applications already demonstrates that high-resolution native MS has become an integral biophysical tool for studying these viral assemblies.
7.3.1. Empty Capsids and Virus-like Particles
When a virus capsid is built up from a single type of subunit that carries no PTMs and is not affected by truncations due to processing, the resulting native mass spectra for the empty capsid can often be nicely charge-resolved by native MS, even when the whole assembly has a molecular weight of several megadaltons. This has been demonstrated in recent years for a variety of viral assemblies such as the capsid of MS2,142 the HBV capsid,66,88 norovirus subparticles,90,269 portal oligomers of the phages P22, Phi-29, and SPP1,270 and the 18 MDa bacteriophage HK97.34,271
Many virus capsids are built up from multiple distinct subunits with unique intrinsic structures, sequences, and masses. A quite interesting example of that is the adeno-associated virus (AAV), which nowadays is receiving considerable attention because it is one of the most explored viruses for gene delivery and therapy. AAVs are small (20–25 nm in diameter), non-enveloped viruses with a T = 1 icosahedral capsid built up of 12 pentamers. Thus, the AAV capsid consists of a total of 60 subunits made up of three distinct viral proteins originating from the same cap gene (VP1, VP2, and VP3), which vary only in their N-terminal sequences. All three VPs play crucial and specific roles in cell entry and transduction. The exact stoichiometry and organization within AAV capsids have been ambiguous for a long time, although generally it has been assumed that VP3 is dominant, with smaller amounts of VP1 and VP2 present as well. On the basis of gel, LC-MS, or CE-MS analysis of the VP monomers formed by denaturing the capsid, a VP1:VP2:VP3 ratio of around 5:5:50 is often quoted. However, a 5:5:50 capsid monomer ratio does not necessarily mean that each particle contains exactly those numbers of VP proteins. In contrast, using high-resolution native MS, first Snijder et al.117 and more recently Wörner et al.272 demonstrated that AAVs assemble quite randomly from the VP capsid proteins available in the host cell. This results in an ensemble of particles, all having a different distribution of subunits, with theoretically 1891 possible co-occurring capsid stoichiometries with different masses! High-resolution native mass spectra of intact AAV capsids (MW ≈ 3.8 MDa) displayed both highly resolved regions and regions wherein spectral interferences did occur (Figure 14). Through extensive spectrum simulation, Wörner et al. were able to resolve and annotate this spectral complexity, assessing the VP stoichiometries in a panel of AAV serotypes from different production platforms. They argued that by systematic scoring of the stochastic assembly model against experimental high-resolution native MS data, a sensitive and accurate method was created to characterize these exceptionally heterogeneous gene-delivery vectors.
Figure 14.
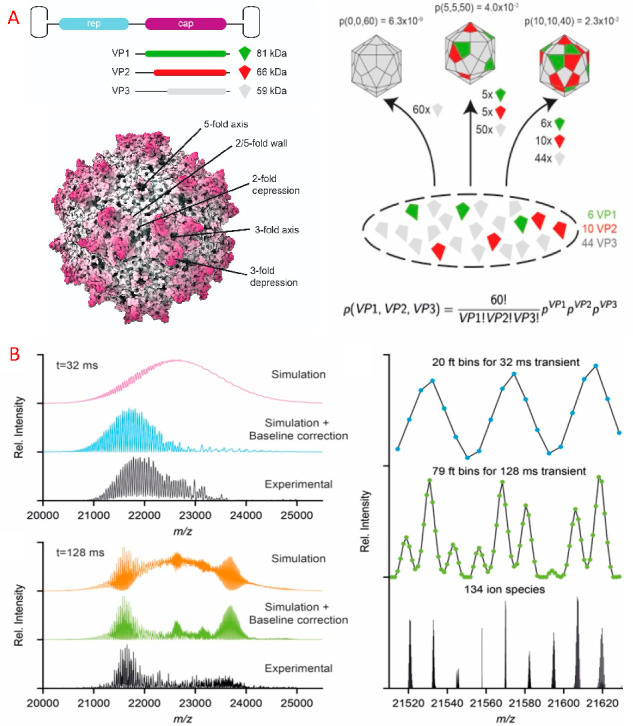
Analysis of adeno-associated virus (AAV) by high-resolution native MS: (A) AAV (structure from PDB entry 3NG9) is composed of 60 copies of a combination of VP1, VP2, and VP3, encoded within the same cap open-reading frame, sharing a common C-terminal sequence. AAV capsid assembly follows a stochastic model, where VP proteins are taken randomly from the pool of expressed VP proteins. The probability for each stoichiometry is determined by the ratio of the expressed VP proteins. (B) Simulated and experimental high-resolution native mass spectra of AAV at mass resolutions corresponding to transient times of 32 and 128 ms. Each plot shows (top to bottom) the simulated mass spectra before and after baseline correction and their experimental counterparts. On the right are depicted zoomed-in parts of these spectra. Adapted from ref (272). CC BY 4.0.
7.3.2. Genome-Loaded Authentic Viruses
Most of the high-resolution MS studies reported to date have been on empty viral capsids. Studies on genome-packed viruses are scarce, partly because analyzing such viruses typically requires special biosafety measures. Moreover, heterogeneity in the packed genome can lead to mass spectra with reduced resolving power. Consequently, most of the data reported to date have been on viruses that are not infectious for humans. The viruses studied harbored a homogeneous RNA- or DNA-based genome. For instance, Snijder et al. used high-resolution native MS to investigate the structure and assembly of the picorna-like triatoma virus.273 Interestingly, the genome-packed 8.3 MDa virus could not be charge-state-resolved, but when the genome was released, triggered by a pH jump in vitro, the empty capsid proteins reassembled into 5.4 MDa particles that could be partly charge-state-resolved using a modified Q-ToF mass analyzer. Genome loading within the plant viruses cowpea chlorotic mottle virus (CCMV) and brome mosaic virus (BMV) was studied by native MS by van de Waterbeemd et al., who compared data obtained with Q-ToF and Orbitrap-EMR instruments.274 Both CCMV and BMV have masses of approximately 4.6 MDa, of which approximately 1 MDa originates from the genomic content of the virion. These viruses pose an analytical challenge because they both represent mixtures of three particles carrying different genome segments that vary in mass by approximately 0.1 MDa (∼2%). They showed that by manipulation of the charging of the particles (using additives in the electrospray spray solution), the mass spectra could be mass-resolved, enabling correct assignment of the charge states and demonstrating that native MS of these endogenous multipartite virions is possible. Although the spectra obtained by the Q-ToF and Orbitrap-EMR were alike, the improved desolvation obtained by using the Orbitrap instrument generally produced better-resolved mass spectra. In contrast, when CCMV was loaded not with its genome but rather with small water-soluble phthalocyanine molecules, the mass and small-molecule content could only be estimated from a nonresolved “bump” in the mass spectra.275
Some of the most well-resolved high-resolution native mass spectra of authentic viruses were obtained for the Flock House virus (FHV).66 FHV is a small, non-enveloped, icosahedral T = 3 virus containing a single-stranded RNA genome comprising two genes: RNA1 (3.1 kb) and RNA2 (1.4 kb). RNA2 encodes capsid precursor α, 180 copies of which form the initial viral capsid. Upon maturation, α undergoes an autocatalytic cleavage in its C-terminus to form β, the main structural capsid component, and a short hydrophobic g peptide required for endosome penetration that remains associated with the viral capsid but can be released at lower pH. The intact FHV virus particles, containing both strains of RNA and 180 copies of both the β and g chains, were analyzed by native MS on an Orbitrap-UHMR, both under normal aqueous ammonium acetate conditions and under conditions where charging was reduced. Under both sets of conditions, clear baseline-resolved spectra could be obtained, at m/z ≈ 42 000 and 57 000 Th, respectively, enabling an accurate mass determination at 9.3 MDa.66,109 Interestingly, as these ions still carry around 200 or 160 charges, they produce sufficient image current in the Orbitrap to be detected as single ions, as demonstrated by Orbitrap-based charge-detection single-particle MS.109
7.3.3. Viral-like Assemblies, Bacterial Compartments, and Synthetic Protein Nanocages
One of the better known viral-like particles of bacterial origin is encapsulin, whose virus-like structure was originally reported by the Ban group.276 Since then, encapsulins have been found to be present in multiple microorganisms.277 The encapsulins from Thermotoga maritima and Brevibacterium linens are formed from 60 copies of the same monomer that assemble into an icosahedral shell with a diameter of around 25 nm. They naturally encapsulate the enzyme dye-decolorizing peroxidase (DyP). DyP itself is assembled as a trimer of dimers, i.e., a 240 kDa hexamer. The specific encapsulation mechanism of DyP is mediated by its C-terminal end, which interacts with a defined region of the encapsulin inner surface. Snijder et al. used native MS to confirm that encapsulin encapsulated just a single hexameric DyP and used native MS, atomic force microscopy, and multiscale computational modeling to demonstrate that cargo encapsulation has a substantial mechanical impact on the shell.240 Encapsulin nanocompartments are gaining considerable interest from bioengineers because of their potential to allow the targeted delivery of drugs, proteins, and mRNAs to specific cells of interest.278,279 For instance, Rurup et al. fused the DyP C-terminal end to the teal fluorescent protein (TFP), which by coexpression could be packaged within the encapsulin. High-resolution native MS was subsequently used to determine the cargo load and reveal the distribution of around 8–12 TFP molecules per encapsulin.500
Another protein that naturally self-assembles is lumazine synthase (termed AaLS-WT), which forms a 60-mer assembly composed of 12 pentamers (T = 1, ∼1 MDa).280,281 The group of Hilvert programmed electrostatic effects by incorporating designed mutations to tailor this protein, induce larger compartments, and optimize its assembly and cargo loading efficiency.30,282 They achieved a stepwise expansion of the natural protein shell, giving rise to thermostable ∼3 and ∼6 MDa assemblies containing 180 and 360 subunits, named AaLS-neg and AaLS-RR, respectively. High-resolution native (tandem) MS was used to probe the stoichiometry and topology of all of these variants. The tandem CID-MS data for AaLS-neg are depicted in Figure 15. Increasing the collision energy revealed the consecutive elimination of up to 10 monomeric subunits. As these ejected monomers were highly charged, the resulting concomitant high-mass fragments became substantially less charged and even reached m/z = 70 000 Th.30 Remarkably, as revealed by parallel analysis with cryo-EM, these expanded AaLS-neg and AaLS-RR particles retained tetrahedrally and icosahedrally symmetric structures constructed entirely from pentameric units,282 a relatively rare topology for virus-like particles. In a follow-up study, the T = 1, ∼1 MDa AaLS-WT particle was also investigated by native top-down mass spectrometry. Its dissociation was induced by UVPD, with the loss of both monomeric subunits and sequence tags thereof, which could be used to identify the subunits.124
Figure 15.

High-resolution native tandem mass spectra of the designed charge-reduced AaLS-neg particles. (top) The charge-reduced intact Cp180 shows a well-resolved charge-state envelope centered at m/z ≈ 30 000 Th. (middle, bottom) The charge-state envelope was subjected to increasing HCD collision energies of 250 and 300 V. At maximal HCD the mass spectrum revealed the consecutive elimination of monomers from the Cp180-mer with fragment ions up to m/z = 70 000 Th. The obtained mass resolution enabled baseline resolution of these different dissociation products. At the highest m/z, the mass resolution was still greater than 500. The figure is adapted from ref (30). Copyright 2017 Royal Society of Chemistry. The inset structure in the top panel is from ref (282). CC BY 4.0.
In recent years, the groups of King283 and Baker284 have explored protein engineering and molecular self-assembly to generate de novo sophisticated self-assembling protein structures that even can be designed and used as nanoparticle vaccines against COVID-19.285 For quality control, it is important to have the appropriate analytical tools to monitor their correct assembly and stability, whereby native MS can be a technique of choice. For instance, Wargacki et al.286 combined high-resolution native MS with several complementary biophysical methods to monitor in detail the assembly of two of such computationally designed 120-subunit icosahedral protein cages that resemble VLPs. They used native MS to understand the assembly process and particularly its robustness toward perturbation. Also, for these designed protein cages, the assembly process was highly cooperative and exclusively yielded complete 120-subunit complexes except in one nonstoichiometric regime for one of the materials. The latter could be uniquely monitored only by making use of the high-resolution native MS data. Related to this work, the group of Wysocki used high-resolution native MS to screen a library of designed building blocks for their propensity to form higher oligomers, defining their stoichiometry and topology.284,287,288
Cumulatively, all of these data reveal that high-resolution native MS is an excellent tool to characterize viruses and VLPs. Viruses (and bacterial encapsulins) not only are important as some of the most critical pathogens threatening (human) health but also are structurally very stable and therefore attractive biomolecular tools to be used in nanotechnology and bioengineering with applications in gene delivery and vaccine development. Their biochemical and biophysical properties are also fundamentally interesting, and native MS provides an ideal means to investigate their stability and assembly mechanisms.
Future developments in native MS will hopefully address the remaining challenges in structural virology, which are mainly posed by the mass heterogeneity of enveloped viruses, larger viruses containing a dozen different subunits (e.g., adenovirus), or viruses containing extensively glycosylated spike proteins (e.g., HIV and SARS-CoV-2). Such systems may readily be analyzed by CD-MS,109,128,150,289 although to date it lacks the resolving power obtainable by conventional native MS as described here.
7.4. Protein Complexes Involved in Complement Activation
In this section, we focus on contributions that native MS has made to our understanding of some of the molecular pathways underlying immunity, in particular through the classical pathway of complement activation.290 When initiated by target-bound antibodies, complement ensues as a cascade of proteolytic reactions that generate an array of bioactive compounds, including chemoattractants, opsonins, and anaphylatoxins. Together, these molecules provide signals to form ultimately membrane attack complexes (MACs) and to recruit immune cells for the clearance of pathogens and damaged host cells. The complement recognition complex C1 comprises six low-affinity binding arms that require antibody clustering to enable stronger multivalent binding, making it structurally compatible with naturally occurring antibody oligomers such as immunoglobulin M (IgM) pentamers and hexamers.291 Although researchers have long known that IgGs, which occur mostly as monomers in serum, can also activate complement, the exact mechanism by which they do so remained elusive until just a few years ago.
In a landmark study, Diebolder et al.292 showed that IgG activates complement by preferentially forming hexameric rings on antigenic surfaces. On the basis of the observation that some crystal structures of IgG show hexameric packing through Fc interactions, they used targeted mutations in the Fc–Fc interface that could enhance oligomerization and thus also complement activation. Native MS proved to be a key tool in this context, enabling them to confirm that a triple mutant, termed IgG1-RGY, readily forms non-covalent hexamers already in solution. Using cryo-EM, they next investigated the structure of target-bound (IgG1-RGY)6:C1 complexes, showing hexagonal rings of monovalent antigen-bound antibodies with C1 bound on top of the IgG platform. Encouraged by these observations, de Jong et al.503 explored the use of hexamerization-enhanced IgGs as a therapeutic platform. Different constructs were screened using various methodologies, including native MS, to identify IgG mutants that remain in their monomeric inactive state in solution but display increased hexamerization and complement activation upon target binding.
Follow-up studies investigated the molecular mechanisms by which complement activation complexes are assembled. Using tandem MS on an Orbitrap Q Exactive EMR to study heterogeneous antibody–antigen complexes, Dyachenko et al.120 showed that solution-phase IgG1-RGY hexamers can still bind up to 12 copies of their antigens. Interestingly, however, Wang et al.293 revealed that antigen binding was not a requirement for C1 recruitment, as it binds readily with IgG hexamers in either the presence or absence of antigen. The role of antigen binding was rather related to activation of C1, likely by a conformational change induced through the IgG Fab arms. Finally, Wang et al. reconstituted a complex of C1 bound to an antigen-saturated IgG hexamer and used native MS to visualize the complete 2.1 MDa complement initiation complex (Figure 16). In a recent study by Strasser et al.,294 native MS was also used to corroborate high-speed atomic force microscopy (HS-AFM) studies into the mechanisms of IgG hexamerization on antigenic surfaces. Two main assembly pathways were identified, the more important of which was a “vertical pathway” in which target-bound IgG recruits additional IgG monomers from solution, followed by monovalent antigen binding. Although intermediate oligomers were also observed, at least five binding sites were required for C1 binding and activation. These reports have improved our mechanistic view into the initial steps of the complement cascade and highlight the complementary and rather unique role of native MS in hybrid structural biology.
Figure 16.

Native-MS-assisted analysis of the initial steps in complement activation: stepwise reconstitution of the 2.1 MDa C1:IgG:Ag complexes, comprising 40 protein subunits, monitored by native MS. (A) Native MS analysis of reconstituted C1, C1:IgG, and C1:IgG:Ag complexes. The signals shown in dashed boxes are color-coded according to the stoichiometries of C1 assembly, as specified to the left of the corresponding spectra. The bottom spectrum demonstrates the successful reconstitution of C1, containing C1q, two copies of C1r, and two copies of C1s. The middle and top spectra show that the latter assembly successfully binds to (IgG)6 and subsequently to 12 copies of the antigen (Ag). (B) Model summarizing molecular determinants contributing to IgG-mediated activation of the classical complement pathway. The assembly of the complement initiation complex is proposed to be dependent on (1) the availability of specific antigens recognized by IgG antibodies, (2) Ag compatibility with clustering at the cell surface or in solution, (3) the Fc-domain conformation, (4) the high-avidity binding site for hexavalent C1q, (5) the composition of the IgG glycan chains, (6) the presence of Fab arms, and (7) Ag binding by the Fab arms. Reproduced with permission from ref (293). Copyright 2016 Elsevier Inc.
7.5. Protein Assemblies Involved in Light Harvesting
The conversion of light into energy is one of the most fundamental biological processes on earth, and through billion years of evolution it has been optimized to great efficiency. The most efficient light-to-energy conversion occurs within the outer membranes of some microbes and plants and is mostly mediated by very large photosynthetic protein assemblies. These photosynthetic protein complexes are mediated by well-structured and protein-mediated networks of light-absorbing pigment molecules. Studying their structures remains a key prerequisite to understanding better the process of light-energy harvesting, which is of tremendous fundamental importance and crucial for the development of novel artificial light-harvesting machineries.295 The main advantage of utilizing native MS in the analysis of protein assemblies involved in photosynthesis is its ability to preserve non-covalent interactions, which mediate not only interactions of protein subunits within a photosynthetic complex but also the covalent and non-covalent binding of various cofactors and multiple pigments (e.g., chlorophylls). Even when these cofactors are bound covalently, as in the case of phycobiliproteins, the bonds are typically weaker than the covalent bonds in the proteins, making soft ionization conditions very important in the analysis of photosynthetic protein assemblies. Because of the varying occupancy of cofactors or pigments bound per single photosynthetic complex, high resolution is often required to discern between complex variants with native MS. Over the past decade, native MS alone and as a complementary tool was successfully employed to analyze various photosynthetic protein complexes to study pigment occupancies, oligomeric states, and stabilities and even to define photoprotective and light-harvesting mechanisms.
A large amount of the earliest work that leveraged the structural analysis of photosynthetic proteins with native MS was performed by the Blankenship group.29,296−303 Their first attempt at using native MS to characterize the well-studied Fenna–Matthews–Olson (FMO) complex, the first pigment–protein complex with a solved crystal structure,304,305 complemented available structural data by determining the exact number of pigment molecules bound within the complex. In their work, Wen et al. took advantage of native MS to provide a snapshot of complex heterogeneity and confirmed that one of the pigments missing in hitherto available structures demonstrated partial occupancy due to surface exposure.303 In a subsequent study, Zhang et al. employed ECD on an FT-ICR mass spectrometer under native conditions to determine flexible regions within the FMO protein subunits.29 Further work was done on the orange carotenoid protein (OCP), which protects cyanobacteria from photodamage by close interaction with the light-harvesting phycobilisome (PBS).302 The sensitivity of native MS allowed them to detect the protein in a complex with its pigments and investigate the light-dependent oligomerization of OCP, which is essential for its photoprotective function. In these studies, some of the inherent limitations of native MS in providing high-resolution structural information were acknowledged. However, they also demonstrated the prospects of combining multiple mass spectrometric modalities (e.g., cross-linking MS and native MS) to analyze these complex multicomponent light-harvesting assemblies. In the follow-up studies, native MS was further used in combination with collisional unfolding in the analysis of OCP, revealing structural features of its functional domains and compensating for the absence of high-resolution structures.300
The oligomeric state, pigment content, and complex stabilities were also determined for other photoprotective proteins, like peridinin–chlorophyll–protein301 and fluorescent recovery protein (FRP).298 By combining native MS with cross-linking MS and site-specific mutations, Lu et al. proposed a novel mechanism of function for dimeric FRP, which participates in photoquenching by dissociating OCP from the PBS.299 When native MS matured for the analysis of membrane-embedded proteins, it was applied to characterize membrane photosynthetic reaction center (RC) complexes.296,297 Although membrane complexes are electrosprayed in the presence of solubilizing detergents and require additional collisional activation to be released from the micelle, RC complexes could still be detected with multiple non-covalent cofactors attached, further demonstrating the ability of native MS to retain even labile non-covalent interactions (Figure 17).
Figure 17.
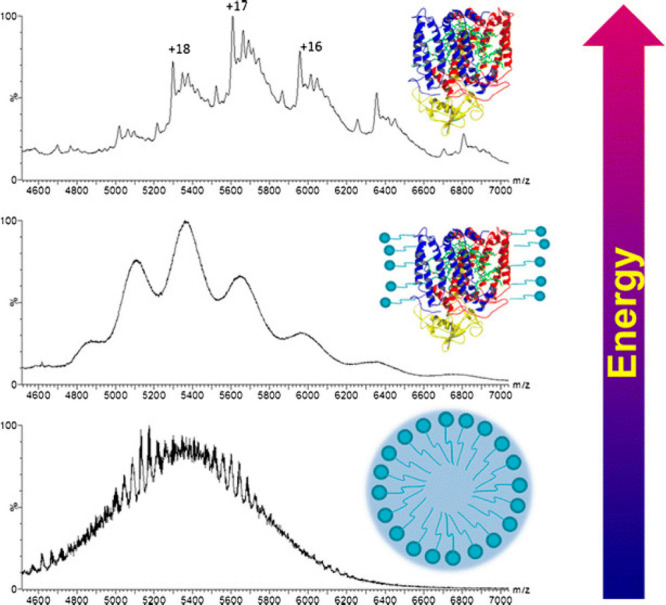
Native MS analysis of a membrane-embedded photosynthetic reaction center: release of the photosynthetic reaction center complex from detergent micelles by collisional activation. At low collisional energy (bottom spectrum), the broad peak represents ions from the micelles. The detergent micelles and extra bound detergent can be removed by increasing the collision energy at the trap region of a Synapt G2 mass spectrometer. Reproduced from ref (299). Copyright 2017 American Chemical Society.
More recently, with increasing interest in designing even more efficient light-harvesting proteins, various components of the main light-harvesting machinery in cyanobacteria and red algae, called phycobiliproteins, have been investigated. Although in the last years cryo-EM—following the so-called “resolution revolution”—has yielded exciting and detailed structural studies of entire PBS systems from different origins,306,307 native MS has provided a unique angle, exposing heterogeneities observed in these complexes and exploring complexation dynamics under various conditions. Leney et al. investigated the stabilities and fluorescence of heptameric B-phycoerythrin (BPE), the most abundant PBS subcomplex in red algae.308 Since phycoerythrins purified from red algae represent some of the brightest fluorophores on the planet and can be commercially obtained in large quantities, it is also important to determine their stabilities upon environmental perturbations. By taking snapshots of BPE complexes with native MS under different pH conditions, Leney et al. tracked the complex dissociation and assembly pathways and determined that the complete heptameric form is essential for its distinctive photochemical properties. In the past few years, several other studies employing native MS as the core technique for analyzing phycobiliproteins have been reported.124,309−312
Although the use of native MS by itself has provided great insight into the biology and light-harvesting mechanisms of several photosynthetic assemblies, the technique is more and more complemented by other MS modalities, especially top-down and bottom-up MS and IM tandem MS. For instance, to explain the anomalous heterogeneity of BPE observed by high-resolution native MS that could not be explained by the expected pigment content and oligomeric state, Tamara et al. opted for top-down analysis of BPE proteoforms to chart the masses of all of the assembly variants, ultimately assigning the observed heterogeneity to the numerous co-occurring forms of the core g subunit.310 Furthermore, they discerned the isobaric yet distinctive pigment molecules via bottom-up HCD-MS/MS analysis and subsequently assessed each pigment’s total content in the native complexes. A high degree of sequence information and partial sequence positioning was achieved for entire BPE assemblies with native top-down MS via UVPD.124 In another example, native MS combined with IM-MS allowed the stabilizing role of the critical subunits in the phycobilisome proteins to be distinguished.309 In a study on UVR8 plant photoreceptors, Camacho et al.313 explored the functional role of the oligomeric state in the photoprotection mechanism with native IM-MS and collisional unfolding (Figure 18). Such strategies, supplemented by improvements in desolvation, transmission, and resolution of instruments for native MS, can open new avenues for analyzing other photosynthetic protein complexes of ever-increasing size and complexity, such as Photosystem II supercomplexes.314
Figure 18.
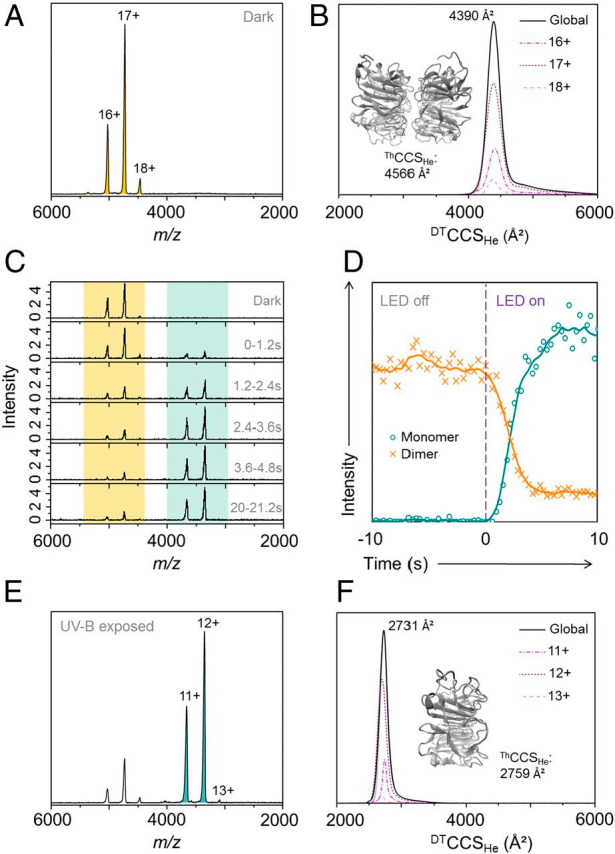
Native MS and IM-MS of the UVR8 photoreceptor core domain (UVR812–381). (A) Native mass spectrum of the UVR812–381 dimer (pale-orange peaks) in the absence of UV-B light. (B) Monodisperse drift tube collision cross section in helium (DTCCSHe) of the UVR8 dimer. The inset shows the energy-minimized structure of the UVR8 dimer and associated theoretical CCS (ThCCSHe). (C) Mass spectra of UVR812–381 as a function of illumination time (280 nm, 25 mW, 350 mA). Mass spectra were combined over a period of 1.2 s after the light-emitting diode (LED) was switched on at t0. Dimer signal, pale orange; monomer signals, pale green. (D) Ion chromatograms extracted for the UVR812–381 dimer (orange) and monomer (green) as functions of acquisition time. Data to the left of the purple dashed line are from when the ion source tip was in the dark, and the data to the right are from when the tip was illuminated. (E) Native mass spectrum of UVR812–381 following illumination in the source tip with the 280 nm LED for 10 s (to ensure maximum conversion). The spectrum is now dominated by the UVR812–381 monomer (pale-green peaks). (F) Monodisperse DTCCSHe of the UVR812–381 monomer. The inset shows the energy-minimized structure of the UVR812–381 monomer and associated ThCCSHe. From ref (313). CC BY 4.0.
7.6. Membrane-Embedded Protein Complexes
In recent years, native MS has emerged as a versatile and highly sensitive technique to study membrane proteins and their interactions, almost becoming a separate discipline. For a long time, membrane proteins were thought to be intractable by electrospray native MS because of their intrinsic hydrophobic nature, which makes them incompatible with the aqueous solutions used in standard electrospray. Now that the solutions to circumvent this incompatibility have been developed (see below), the application of native MS to the investigation of membrane proteins has been greatly expanding. Several excellent and recent reviews extensively cover the development in this field,20,196,197,315,316 showing the specific set of challenges mass spectrometrists face when analyzing membrane proteins and their complexes. Membrane proteins and their interactions are of major biological importance, as they contribute roughly 30% to the human proteome and are targeted by as many as 50% of all currently used drugs. Although they have key functions in signaling, transport, metabolism, and the respiratory chain, this class of proteins remains understudied in vitro compared with their soluble counterparts. This is the case because membrane proteins require membranes or mimetics thereof to remain in a functional native-like state, making them not only harder to produce recombinantly but also more challenging to tackle analytically using traditional biophysical techniques. For native MS analysis, much effort has gone into the development of MS-compatible membrane mimetics to transfer membrane proteins into the gas phase as well as instrumental modifications that enable more extensive ion activation to strip away resulting unwanted adducts. In keeping with the scope of this review, we will briefly highlight some of the most recent advances.
Partly driven by skepticism about the biological relevance of using detergent micelles, advances in nanodisc technology have enabled researchers to solubilize membrane proteins in a more native-like environment. Nanodiscs are lipid bilayers of which the hydrophobic edge is covered by two copies of amphipathic membrane scaffolding protein (MSP), providing a homogeneous and monodisperse model system. However, initial native MS studies required exceptionally high levels of collisional activation to release the “naked” membrane proteins,317 while the stability of the nanodiscs was too low to measure them intact.318,319 To overcome these challenges, Keener et al.320 explored the use of chemical charge modulation additives to tune the gas-phase stability of the used nanodiscs. Using macromolecular mass defect analysis to assign stoichiometries, they determined the oligomeric states of ammonium transport protein (AmtB) and AqpZ in intact nanodiscs without potential distortions introduced by collisional activation. In two follow-up studies, Walker et al.321,322 utilized the same method to characterize the interactions of antimicrobial peptides (AMPs) with nanodiscs modeling mammalian or bacterial membranes. AMPs were titrated into preformed nanodiscs to promote insertion into the membrane, followed by direct measurement via native MS. Unique stoichiometries were observed for each AMP depending on the lipid type used and the AMP concentration. Moreover, collisional activation experiments could distinguish between the formation of specific membrane-destabilizing complexes and nonspecific binding, demonstrating the potential of native MS to provide key insights into AMP mechanisms.
In two recent papers, Chorev et al.323,324 demonstrated the mass analysis of protein complexes from lipid vesicles derived directly from native cellular membranes. By sonication of large membrane fragments isolated from cells, they were able to produce small vesicles of similar lipid and protein composition without the need for recombinant overexpression or further purification. Native mass spectra were obtained using a modified Orbitrap-UHMR, although the heterogeneity of the proteins and bound lipids presented a significant challenge for mass assignment. They therefore employed small-molecule MS, lipidomics, proteomics, and molecular dynamics simulations to gain more confidence in their mass assignments. This combination of methods enabled them to report on multiple protein assemblies present in the E. coli membrane (Figure 19) and those present in bovine mitochondrial membranes, some of which had not been previously described. Although some skepticism remained about their analysis and in particular their mass assignments,325,326 this advancement shows that native MS has the potential to provide pivotal data when it comes to membrane proteins in a very native-like context.
Figure 19.

Native MS of membrane-embedded protein complexes analyzed directly from their native environment. Regions of the mass spectrum recorded for inner membranes from E. coli yield cytochromes, the Ton complex multidrug transporters, and the intact ATP synthase in complex with the SecYEG translocon. (A, B) Expanded regions of the spectrum assigned to cytochrome bo3 and cytochrome bd oxidase, showing peak splitting due to binding of quinol and heme groups. The pentameric ExbB complex (with one copy of ExbD in the center of the pore) that forms part of the TonB complex is also observed (yellow). (C) High-m/z region of the mass spectrum assigned to the multidrug efflux pumps AcrAB and MdtAB and the intact ATP synthase. Expansion of peaks assigned to the ATPase reveals binding of the SecY (blue), SecYG (green), and SecYEG (orange) charge states 52+, 53+, and 54+. Complexes observed in mass spectra are shown schematically, with subunits that have dissociated shown in gray. Reproduced with permission from ref (323). Copyright 2018 Chorev et al.
8. Proteoform Profiling by High-Resolution Native MS
In recent years, high-resolution native MS has also emerged as a powerful technology for proteoform profiling, exposing the microheterogeneity induced by protein glycosylation and additional PTMs. The rapidly expanding field of mass-spectrometry-based glycoproteomics relied until recently primarily on two levels of analysis, targeting either released N-glycans or analyzing proteolytically formed and enriched glycopeptides.327−329 Although very powerful, these approaches also have limitations, mainly caused by the enormous structural microheterogeneity that most glycoproteins exhibit.330 Therefore, there is a dire need for additional approaches that are more protein-centric.
In the analysis of intact proteins, native MS has some well-defined advantages compared with denaturing (conventional) MS also when it comes to proteoform profiling. Native MS provides an improved view of sample heterogeneity through the characteristically increased spacing between adjacent charge states and can even boost sensitivity by minimizing the number of charge states over which the signal is distributed.54 Moreover, less charged and more globular molecules measured under native MS conditions are less prone to decay during longer signal acquisition times, which for FT MS instruments are essential to achieve high mass resolution.40 These features make high-resolution native MS an excellent method for revealing the glycoform profiles of glycoproteins qualitatively and quantitively.331
In this section, we begin by describing how native MS first contributed to the analysis of intact glycoproteins. We then discuss the emerging role of native MS in characterizing microheterogeneities in therapeutic antibodies and other biopharmaceuticals. Next, we discuss advancements in MS-based profiling of plasma glycoproteins and finally review work on proteins harboring other PTMs, such as phosphorylation.
8.1. Native MS of Intact Glycoproteins
The enormous structural heterogeneity of intact glycoproteins is the major bottleneck in their effective characterization also when mass spectrometry is used. Early studies analyzing intact glycoproteins, such as ovalbumin, by denaturing mass spectrometry suffered from poorly resolved ion signals, hampering accurate mass assignments and therefore in-depth glycoprofile analysis.332 Not surprisingly, as a widely available model system for analytical technology development, ovalbumin was also one of the first glycoproteins analyzed by high-resolution native MS shortly after the introduction of the Orbitrap-EMR. Using high-resolution native MS, Yang et al.333 revealed that ovalbumin exhibits a diverse glycoproteoform profile even though it harbors only a single N-glycosylation site. This is the case because two additional phosphorylation sites present on ovalbumin create quite a few additional proteoforms. Therefore, Yang et al. also used native MS to analyze ovalbumin treated with a phosphatase and/or an endoglycosidase to selectively cleave off the phosphate and glycan moieties, providing simplified proteoform profiles that facilitated the annotation of around 60 distinguishable proteoforms. The detected repertoire of ovalbumin proteoforms was recently extended even further by Füssl et al.,334 who subjected intact ovalbumin to online anion-exchange chromatography using a pH gradient elution coupled to mass spectrometric detection under native conditions and reported more than 150 different proteoforms. Already these data highlight certain advantages of analyzing intact glycoproteins compared with peptide-centric approaches, as the latter are hampered by simultaneous detection of unmodified and glycosylated peptides as well as peptides decorated with other PTMs (e.g., phosphorylation).
Another glycoprotein that has often been tackled by new analytical approaches and therefore evidently also by high-resolution native MS is bovine fetuin.335,336 The glycosylation profile of bovine fetuin is quite well understood, as it has been exposed by a plethora of glycan- and glycopeptide-centric studies. Intriguingly, Lin et al.337 used high-resolution native MS to directly compare the glycoproteome profiles of bovine serum fetuin, human serum fetuin, and recombinant human fetuin expressed in HEK-293 cells. The native MS data for these three similar proteins displayed considerable differences in their proteoform profiles due to differences in protein maturation, phosphorylation, and N- and O-glycosylation. Although the two N-glycosylation sites, the O-glycosylation site, and the phosphorylation site are conserved from bovine to human, the stoichiometry of the modifications and the specific glycan structures they harbor were found to be quite distinct. By comparison of serum and recombinant human fetuin, it was observed that fetuin from serum exhibited a much simpler proteoform profile, indicating that the recombinant protein is not ideally engineered to mimic human serum fetuin, as is likely true for many more recombinant glycoproteins currently in use in various biochemical studies. As these glycoproteoform profiles may have functional consequences, such studies should therefore be evaluated carefully.
Compared with ovalbumin and even fetuin, nature has created many glycoproteins that are much more complicated. Notably, several viruses contain spike proteins that are heavily glycosylated in the form of a glycan shield, which directly impacts antibody binding and interactions with the host cell.338,339 Struwe et al.340 used high-resolution native MS to probe the N-glycan site occupancy of the HIV-1 gp120 glycoprotein. Using gp120 expressed in HEK293T cells in the presence of the α-mannosidase inhibitor kifunensine to reduce the heterogeneity in glycosylation, they were able to determine the occupancy of several glycosylation sites. However, it remains a future challenge to perform such analyses on wild-type gp120, ideally extracted from endogenous viral particles. As protein glycosylation has such an impact on the virus–host interaction, studies that target viral glycoproteins will likely become more prominent in the future.
8.2. Therapeutic Antibodies
Over the past decades, biopharmaceutical therapies have rapidly emerged to dramatically change the treatment of diseases, with monoclonal antibodies (mAbs) currently dominating biopharmaceutical approvals and sales.341 As complex biomolecules, biopharmaceuticals are amenable to a variety of PTMs, including complex glycosylation, glutamine cyclization, and C-terminal lysine clipping. Many of these modifications are considered critical quality attributes (CQAs) because they can influence the safety and efficacy profile of the drug. Notably, protein N-glycosylation is known to modulate the function of biopharmaceuticals in a variety of ways. For instance, antibody-dependent cell-mediated cytotoxicity is significantly increased when the N-glycans in the Fc domain of IgG molecules lack a fucose residue.342 Additionally, for therapeutic antibodies, galactosylation and sialylation play a role in complement-dependent cytotoxicity343 and anti-inflammatory activity,344 respectively. As protein glycosylation is very much dependent on the particular host cell and the specific conditions employed for production, its pattern provides a means to distinguish the original patented drug from biosimilars.345−347
8.2.1. High-Resolution Native MS Provides a Snapshot of Antibody Microheterogeneity
High-resolution native MS has become an integral technology in the toolbox for exposing the microheterogeneity of antibody products.348−350 With the first demonstrations of the Orbitrap platform for native MS, Rose et al.27 and Rosati et al.31 immediately noted that the ToF instruments used up until that point could be outperformed substantially when it comes to resolving antibody microheterogeneity. From a single mass measurement, different glycoforms of intact ∼150 kDa IgG1 antibodies could be baseline-resolved with molecular weights differing by 162 Da, representing distinct numbers of hexose units incorporated into N-linked glycan structures. Mass differences down to 25 Da could be sufficiently resolved, opening the door to the identification of smaller PTMs and amino acid mutations.
The methods by which native MS can provide detailed analysis of microheterogeneity in antibodies were extensively described in two papers by Rosati et al.118,351 One of the key advantages of using native MS for this purpose is that it allows for increased charge spacing compared with denaturing MS, extending the level of heterogeneity and sample complexity that can be studied. The first step in analyzing monoclonal antibodies by native MS is generally to determine their backbone mass profile after enzymatic removal of N-linked glycans using PNGase F. Alternatively, neuraminidase and β1,4-galactosidase may be used to selectively remove sialic acid and free galactose moieties, reducing the complexity of glycans and therefore the resulting native MS spectra. This step is essential to discern double fucosylation from sialylation since these modifications amount to similar mass shifts (292.3 and 291.3 Da, respectively). The total contribution of glycosylation to the molecular weight is readily assessable by comparison of the mass spectra of antibodies before and after complete deglycosylation. Rosati et al.118 showed that their workflow enables both qualitative and quantitative analysis of composite glycosylation profiles and other sources of microheterogeneity, in some cases identifying over 30 proteoforms of a single antibody.
The simplicity of sample preparation and speed of analysis has made high-resolution native MS a method of choice for initial screening of antibody batches during development and production. Using protein chemistry for in vitro glycoengineering of trastuzumab, Parsons et al.352 utilized high-resolution native MS to study the reaction products. After selective endoglycosidase-catalyzed glycosylation in the presence of an oxazoline donor, native MS revealed not only the expected product but also the occurrence of unwanted nonselective glycation reactions. Native MS enabled them to direct the optimization of their strategy, producing highly pure trastuzumab samples with specific natural or non-natural glycans.
In another antibody engineering approach, van der Schoot et al.353 recently presented a CRISPR/HDR platform to rapidly engineer immunoglobulin constant domains and to produce recombinant hybridomas that excrete these antibodies in the preferred format, species, and isotype. High-resolution native MS enabled them to rapidly characterize antibodies with complex microheterogeneities, confirming for example that an Fc-silent N297A mutant IgG2 indeed lacked N-glycosylation, diminishing its ability to recruit effector functions through FcγR binding.
In another study, Thompson et al.354 characterized oligoclonic mixtures of antibodies produced in a single production platform, demonstrating that up to 15 unique IgGs could be identified and quantified unambiguously and simultaneously. Native MS can even be used to characterize overproduced proteins directly from crude samples from expression systems, as was shown by Vimer et al.355 By comparison of the growth media of HEK293F cells for nonexpressing and expressing conditions, peaks corresponding to a secreted antibody could be assigned, thereby exposing its glycosylation profile. Combined, these studies highlight the promising role of native MS as a screening tool in optimizing expression conditions and studying batch-to-batch variations in therapeutic antibodies.
When native MS is being used for glycoform “fingerprinting” of antibodies, care should be taken regarding the occurrence of hard-to-discern isobaric variants. The most common of such modifications is glycation, which can reduce the efficacy and can even render the antibody immunogenic. Non-enzymatic attachment of a hexose to a backbone residue results in a mass shift identical to that for a mannose or antennal galactose extension of an N-glycan, making it indiscernible on the intact-protein level. Recently, Esser-Skala et al.356,357 presented a method to assign possible PTM compositions and to eliminate hexosylation bias in the analysis of N-glycosylation patterns by native MS. When measuring the antibody in its N-deglycosylated form, satellite peaks at intervals of 162 Da indicated the presence of nonspecifically glycated isoforms. N-Glycoform abundances in the native mass spectrum of the original product could then be computationally corrected by construction of a glycation graph that gathers all possible abundance transfers between proteoforms. Esser-Skala et al. validated their approach on various antibody samples, demonstrating for example that although the apparent N-glycan abundances in native mass spectra of two batches of denosumab were highly similar, different levels of glycation led to substantial differences in their corrected N-glycan profiles (Figure 20).
Figure 20.

Eliminating hexosylation bias in glycoproteoform profiling by high-resolution native MS. Glycation obscures differences between the glycoform profiles of two batches of Prolia (old vs new). (a, b) Raw mass spectra of the (a) intact and (b) de-N-glycosylated mAb, corresponding to 2 kDa sections of the respective zero-charge spectra (the secondary x axes indicate the respective masses). (c) Fractional glycoform abundances before and after correction for the effects of glycation. (d) Interbatch differences of glycoform abundances as derived from (c). Lines connect points denoting identical glycoforms. The three most common glycoforms are labeled. pp, percentage points. (e) Absolute deviations of observed and actual glycoform abundances from simulated abundances based on released N-glycan data. In each batch, those five glycoforms for which correction leads to the largest decrease in deviation are highlighted (thick line: most pronounced decrease). Point areas are proportional to observed/actual glycoform abundance. Error bars represent (propagated) 95% confidence intervals from five technical replicates. RMSD, root-mean-square deviation. From ref (356). CC BY 4.0.
8.2.2. Hybrid Approaches Localize and Detail the Sources of Microheterogeneity
While native MS can rapidly provide a snapshot of the ensemble of complex proteoforms, especially when combined with bias correction algorithms, more information is needed to gain an in-depth understanding of glycan structures and PTM localization. Hybrid mass spectrometry approaches therefore combine native MS with additional information at the level of released glycans or glycopeptides. Combining native MS with released glycan analysis by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD), Rosati et al.118 studied the effects of CH3 domain mutations on N297 glycosylation in the distant CH2 domain of IgG4 half molecules (IgG4Δhinge). In particular, mutations of Y407 by charged residues or glutamine stood out, as these mutants displayed a dramatic increase in galactosylation, chain branching, and sialylation. Translating the Y407E mutation to IgG1 resulted in similar changes in the glycosylation profile of this subclass, with HDX experiments revealing that structural changes may be responsible for this effect. In another hybrid approach, Yang et al.331 profiled released glycans by nanoLC-Chip-Q-ToF MS, followed by mass and retention-time matching to a previously reported human serum N-glycan structural library. An in silico-constructed zero-charge mass spectrum of the antibody was then compared to the real native MS data qualitatively and quantitatively. Although the approaches showed good agreement for the glycosylation profiles of IgG4Δhinge Y407 mutants, their combination proved to provide the most reliable and thorough characterization of mAbs. Other types of modifications could also be revealed. By measuring N-deglycosylated mAbs, Yang et al. showed that trastuzumab has a relatively monodisperse backbone, whereas infliximab displays variable degrees of C-terminal lysine clipping and bevacizumab is extensively glycated.
Hybrid approaches incorporating glycopeptide analysis can complement native MS analysis by localizing glycans and other PTMs, determining site occupancy, and revealing the compositional makeup of glycans. Recently, Brücher et al.358 applied such an approach to investigate the potentially distinctive glycosylation profile of antibodies produced in malignant tissues via gene delivery. To this end, they expressed two therapeutic mAbs with different modes of action in various cancer cell lines, both responsive and nonresponsive to the mAb produced, and then compared the glycosylation profiles to that of the same mAb expressed in standard HEK293 and CHO-S producer cells. They found that both the producer tissue and the antibody isotype influence sialylation and fucosylation and therefore the efficacy of the antibody, enabling them to identify optimal cell types according to the desired mode of action. Furthermore, they found that relatively high amounts of non-glycosylated antibodies were produced by cell lines responsive to the antibody, potentially decreasing the therapeutic efficacy of antibodies that function through antibody-dependent cell-mediated cytotoxicity (ADCC) because they depend on glycosylation for FcγR engagement. Native MS thus revealed substantial differences in microheterogeneity between producer cells that affect antibody efficacy, highlighting the importance of in vivo glycoengineering for the development of improved anticancer antibodies that are produced at the target tissue through gene delivery.
8.2.3. Online Separation Enables More Extensive Characterization of mAb Charge Variants
Because many PTMs affect the surface charge distribution of proteins, antibodies frequently exhibit a variety of charge variants that differ from one another in terms of efficacy and half-life. Therefore, antibody proteoforms can often be separated by charge-sensitive methods, which improves the sensitivity and the ability to resolve (nearly) isobaric proteoforms.349,359 Although the elution buffers traditionally used in these approaches are incompatible with MS, limiting them to offline fractionation, recent efforts exploring volatile alternatives have enabled direct hyphenation to high-resolution native MS. By coupling pH-gradient-based weak cation-exchange chromatography (WCX) directly to an Orbitrap instrument, Bailey et al.360 demonstrated effective native separation of antibody charge variants based on minimal differences in their isoelectric points. This enabled them to characterize heterogeneities that result in minimal mass shifts (e.g., deamidation, ΔM = 1 Da) and even isobaric proteoforms (e.g., due to aspartate isomerization) in both a qualitative and quantitative manner. In a follow-up study by Phung et al.,361 a similar approach was used to study mispairing in bispecific IgGs produced by a single-cell host, providing a unique solution to resolve isobaric species. An alternative approach, hyphenation of strong cation-exchange chromatography (SCX) to native MS, was presented by Füssl et al.,362 who characterized charge variants of several therapeutic antibodies using tailored gradient slopes for each mAb. Similarly, Yan et al.501 extensively studied microheterogeneities in the NISTmAb. Notably, due to the high sensitivity of the approach, a nonconsensus Fab glycosylated isoform could be identified at <0.1% abundance. In another approach, Ma et al.502 complemented native SCX-MS with middle-up proteomics for subdomain analysis of several commercially available mAbs, exposing an array of microheterogeneities and changes in their abundance upon stressing of the sample. Although ion-exchange chromatography shows particular promise for exposing antibody microheterogeneity, alternative LC separation methods are also being explored for hyphenation to native MS. This includes in particular hydrophobic interaction chromatography (HIC)363 and SEC.364,365
Because of its ability to achieve fast separation paired with low sample consumption, capillary zone electrophoresis has emerged as an attractive MS-compatible alternative to LC separation.366 Although CZE was originally used under denaturing conditions, two papers by Belov et al.367,368 demonstrated one of the first implementations of CZE hyphenated to native MS. Using a neutral polyacrylamide-coated capillary coupled to an Orbitrap-EMR, they analyzed various native proteins and protein complexes, including antibodies. By combining this approach with middle-down and intact CZE-MS analysis under denaturing conditions, they were able to identify and quantify multiple antibody charge variants and confirm PTM site localizations. Using a commercially available microfluidic CE device, Füssl et al.369 and Carillo et al.370 recently characterized charge variants of various therapeutic antibodies by native MS. Notably, they compared the performance of the CE platform to cation-exchange chromatography (CEX) in the analysis of cetuximab, an exceptionally heterogeneous IgG1 that harbors additional glycosylation sites in the Fab region. Using glycopeptide mapping to facilitate peak annotation, they identified and quantified over 200 proteoforms of cetuximab by CE-MS, twice as many as by CEX-MS (Figure 21). Combined with inherently low sample consumption, this makes CE-MS a promising tool for the analysis of therapeutic antibodies, also in the early stages of drug development when less material is available.
Figure 21.

Comparison between cation-exchange chromatography MS (CEX-MS) and capillary electrophoresis MS (CE-MS) in the analysis of cetuximab charge variants. (A) Cetuximab chromatogram and electropherogram, with peaks annotated in red and the corresponding net charges in blue. (B) Deconvoluted native mass spectra corresponding to the peaks in (A). Spectra from the two separation approaches containing the same charge variant species are aligned horizontally. Modifications that cause differences in the net charge are indicated in blue, whereas only the most probable combination of modifications is given. The most abundant species corresponding to biantennary Fab glycan pairs are shown in orange, whereas the most abundant forms carrying triantennary Fab glycans are labeled in gray. (C) Deconvoluted spectra of the peak front, center, and tail for peaks 5 and 3 of the CEX and CE separations, respectively. Associated spectra are aligned horizontally and are shown in the same color. Reproduced from ref (369). Copyright 2020 American Chemical Society.
8.3. Antibody–Drug Conjugates
Next to the more conventional therapeutic antibodies described in the previous section, ADCs have recently become another important class of cancer biotherapeutics.371 Some ADCs approved for clinical use (mostly in cancer) include Adcetris, Kadcyla, and Mylotarg. ADCs are typically composed of a monoclonal antibody to which a potent cytotoxic drug is attached via a cleavable linker. The specificity of the mAb component of the ADC to cancer antigens expressed on the cell surface ensures that the cytotoxic drug is targeted to the tumor cells only. The drug is typically coupled to specific amino acids in the mAb chain, with cysteine or lysine conjugation used most frequently. As most mAbs have a molecular weight of around 150 kDa and the drug molecule coupled with a linker moiety has a mass of a few hundred daltons, it is an analytical challenge to perform quality control, especially as one likes to know how many drug molecules are bound per mAb and whether they are equally distributed over all mAb molecules.
Cysteine-linked ADCs are composed of antibodies with drugs conjugated to cysteine residues involved in interchain disulfide links, i.e., those between the two heavy chains (HCs) or those connecting the two light chains (LCs) to the HCs. Cysteine conjugation is performed via a prior partial reduction of the interchain disulfides, which implies that the HC–HC and LC–HC associations in the ADC monomer become a mixture of covalent and non-covalent associations. The potency of an ADC is determined in large part by the average number of drugs attached to the mAb, i.e., the drug-to-antibody ratio (DAR). With each reduced disulfide bridge, two drug molecules can be coupled to the mAb, and thus, a typical cysteine-linked ADC carries two, four, six, or eight drug molecules. Valliere-Douglas et al.372 used native MS early on to determine the intact masses of the non-covalently associated antibody HCs and LCs that result from the attachment of drug conjugates to interchain cysteine residues, focusing on IgG1 mAbs conjugated with maleimidocaproyl-monomethyl auristatin F (mcMMAF) or valine-citrulline-monomethyl auristatin E (vcMMAE). By using native MS on a high-resolution Bruker MaXis II Q-ToF mass spectrometer, they could indeed retain the intact bivalent structure of the ADC, which ordinarily would decompose as a consequence of the denaturing chromatographic conditions typically used for LC-MS analysis. In a follow-up study, the same group benchmarked SEC hyphenated to native MS versus analytical HIC to determine drug loads on a variety of ADCs.373 The accurate results obtained were sufficient to support the use of native SEC-MS as a quantitative DAR method for all three tested ADCs. Similarly, Debaene et al.374 explored the potential of native MS and IM-MS compared with HIC for quality control of the interchain cysteinyl-linked ADC brentuximab vedotin and obtained results in line with earlier data from Rosati et al.118
Dyachenko et al.120 combined native MS with tandem MS for the characterization of brentuximab vedotin. Tandem MS allowed them to selectively fragment isolated precursors carrying a specific number of drug molecules. This enabled localization of cysteines to which the drug molecules were bound, revealing that drug conjugation took place nonhomogeneously to cysteine residues on both the LCs and HCs of the ADC when coupling was not saturated.
Next to cysteine-conjugated ADCs, lysine-conjugated ADCs have also been explored. Antibodies contain many more lysine residues than cysteines in their primary sequence, and this conjugation strategy therefore results in an even more heterogeneous ADC with potentially a much higher DAR. Conjugation to the free amines of the Lys residues depends on the solvent accessibility and reactivity of each individual Lys residue in the mAb. The overall lysine conjugation properties in human IgGs were studied by Gautier et al.,375 integrating high-resolution native MS and bottom-up proteomics. High-resolution Orbitrap native MS enabled monitoring of the sequential incorporation of up to 70 molecules, each attached to a different Lys, into human IgGs. Complementary bottom-up proteomics facilitated the identification of the most reactive “hotspot” conjugation sites. Such data are important in controlling the drug load and specificity in lysine-linked ADCs.
For the next generation of ADCs, strategies that produce less heterogeneous drug loads are being explored. Illustratively, Botzanowski et al.376 used native MS to investigate a site-specific DAR4 ADC generated through aldehyde-specific bioconjugation, whereby reactive formylglycine (fGly) amino acids are produced by formylglycine-generating enzyme (FGE) via highly selective oxidation of a cysteine residue found within a specific pentapeptide consensus sequence. The resulting mAb, containing four fGly residues, is then further modified using aldehyde-specific chemistries. The ADCs generated using these methods possess increased therapeutic indices and activities. By using online SEC hyphenated to native MS on a Q-ToF instrument, they observed a single species corresponding to a mass of 152 773 ± 1 Da, in agreement with the expected mass of the mAb with four conjugated drug molecules. Still, also a minor species (less than 10% of the total signal) was observed, corresponding to the binding of three drug molecules. These data clearly displayed the relatively high structural homogeneity of this mAb. Benchmarking their data, they analyzed in parallel the cysteine-conjugated mAb brentuximab vedotin and the lysine-conjugated mAb trastuzumab emtansine, and the native MS data clearly revealed the heterogeneity levels of these different classes of ADCs.376
8.4. Other Biopharmaceuticals
Moving beyond antibody-based biotherapeutic molecules, high-resolution native MS has also been applied to study microheterogeneity in other biopharmaceuticals, also providing a means to distinguish between biosimilars and the original drug. Below we briefly highlight recent work on three commonly used non-antibody biopharmaceuticals: erythropoietin (EPO), etanercept, and human chorionic gonadotropin.
8.4.1. Erythropoietin
EPO is a human endogenous glycoprotein cytokine that is secreted mainly by the kidney in response to cellular hypoxia to stimulate red blood cell production (erythropoiesis). Recombinant EPO (rhEPO) is widely used in the treatment of anemia, for example in chronic kidney disease or myelodysplasia or as a result of cancer chemotherapy. Also, it is known that glycosylation of EPO affects its function in a variety of ways. For instance, sialylation and increased branching of EPO N-glycans increase its serum half-life,377 while EPO lacking sialylation exhibits a neuroprotective role in vivo.378 Thus, a well-defined view of the detailed glycosylation profile of EPO is essential, also in the context of the fact that for rhEPO many biosimilars are reaching the market and clinic.379
In one of the first studies using the Orbitrap-EMR for detailed analysis of non-antibody-based biopharmaceuticals, Yang et al.380 analyzed EPO. Compared with the relatively simple native MS spectra of therapeutic IgGs that typically display only a dozen of different glycoforms, high-resolution native mass spectra of EPO exhibited hundreds of different glycoproteoforms380,381 arising from the heterogeneity on its one O- and three N-glycosylation sites, making the compositional analysis of these spectra significantly more challenging. Treatment of EPO with a sialidase, which cleaves off all sialic acids present on both the N- and O-glycans on rhEPO, reduced the complexity of the spectra tremendously.380 Combining the native MS analysis with a glycopeptide-centric MS approach331 allowed them to assign most of the glycoforms. Additionally, they used the semiquantitative glycopeptide data to reconstruct the native mass spectra and looked for the overlap between the experimental and reconstructed native MS spectra.380 Moreover, they analyzed the glycoproteoform profiles of three recombinant EPO products obtained from different manufacturers. These rhEPO therapeutics all had an identical protein backbone sequence, although they were found to be ornamented with differential glycosylation patterns, likely as a result of different conditions during their production. By directly comparing the glycoproteoform profiles obtained from the native mass spectra, Yang et al. introduced a biosimilarity score to describe the level of structural similarity between the three rhEPO therapeutics.380
In a follow-up story, Čaval et al.381 used a similar method for the structural characterization of 24 engineered variants of EPO with diverse N-glycosylation patterns. To this end, they employed a set of glycoengineered CHO cells to express EPO with different glycosylation features and degrees of heterogeneity.382 Additionally, in that study the glycosylation profiles of the glycoengineered EPOs monitored by native MS revealed hundreds of co-occurring variants. Pleasingly, the chosen glycoengineering approach could be validated, revealing a stepwise decrease in heterogeneity from tetra-antennary polyLacNAc-elongated N-glycans all the way to homogeneous, biantennary, non-elongated, and non-sialylated N-glycans. Similar to Yang et al.,380 they employed a classification scheme based on the acquired native MS spectral fingerprints to create a product similarity matrix (Figure 22).381 Using hierarchical clustering, they were able to differentiate and classify glycoengineered EPO clones, extracting the overall structural differences. This classification scheme, termed biosimilarity scoring, could benefit clonal selection in cell line development and be used for batch-to-batch quality control of the glycoproteoform profiles and assessment of structural aspects linked to biosimilarity.
Figure 22.

Characterization of wild-type EPO and 23 glycoengineered EPO variants by high-resolution native MS. (A) Illustrative deconvoluted native mass spectra of WT EPO (top) and the knockout-based glycoengineered clones C23 (middle) and C21 (bottom). (B) Comparison of native mass spectra of EPO from two biological replicate clones C22 (black) and C24 (orange) by correlation scoring. (C) Clustering of the glycoengineered EPO clones based on the correlation between their native MS spectra. From ref (381). CC BY 4.0.
8.4.2. Etanercept (Enbrel)
Combining high-resolution mass spectrometry with a sophisticated strategy of enzymatic digestions, Wohlschlager et al.383 tackled the molecular complexity of the fusion protein etanercept (Enbrel). Etanercept consists of two copies of the tumor necrosis factor-α receptor (TNFR) and the Fc portion of human IgG1. This biopharmaceutical, which is used to treat autoimmune diseases such as rheumatoid arthritis, contains four N-glycosylation sites and 26 O-glycosylation sites in its dimeric TNFR domain and two N-glycosylation sites in the IgG Fc portion. Wohlschlager et al. used the protease IdeS to cleave etanercept at the hinge region of its IgG Fc portion, enabling separate analyses of the N-glycosylation on the TNFR and Fc domains independently. Next, the heterogeneity in glycosylation was trimmed by using in parallel and consecutively the three enzymes sialidase, PNGase F, and O-glycosidase. Moreover, glycopeptide-centric approaches were used concomitantly, and all of the data were integrated with advanced software357 to achieve a comprehensive characterization of this extremely complex glycoprotein biopharmaceutical (Figure 23).
Figure 23.

Molecular structure and high-resolution native mass spectra of etanercept. (a) Structure of dimeric etanercept consisting of a TNFR and an Fc domain. Some of the cleavage sites of IdeS, PNGase F, and sialidase are indicated. (b–d) Native mass spectra of (b) intact etanercept and etanercept treated with (c) sialidase, (d) PNGase F, or (f) a combination of PNGase F and sialidase. (f, g) Annotation of the etanercept O-glycoforms after treatment with (f) PNGase F and sialidase or (g) PNGase F alone. Adapted from ref (383). CC BY 4.0.
8.4.3. Human Chorionic Gonadotropin (Ovitrelle)
In an elegant combination of glycoproteoform profiling and non-covalent mass spectrometry, Lebede et al.384 used high-resolution native MS to analyze recombinant human chorionic gonadotropin (hCG, Ovitrelle). The protein hormone hCG is involved in early embryo–maternal communication and maintenance of pregnancy. The two non-covalently interacting subunits, hCGα and hCGβ, both contain several N- and O-glycosylation sites, making the characterization of the proteoform profiles of the heterodimer even more challenging. Still, by integrating the native MS data with data from released glycans and glycopeptide analysis as well as intact mass analysis of the monomeric subunits and the heterodimer, they were able to provide an in-depth analysis of the heavily glycosylated non-covalent hCG heterodimer, suggesting that hCG is built up from more than 1000 distinct glycoforms. This is another example revealing that a single biopharmaceutical can in fact represent a highly diverse molecular landscape.
8.5. Proteoform Diversity of Selected Plasma Glycoproteins
At the molecular level, human plasma is a highly complicated biofluid. Its proteome exhibits an extraordinary dynamic range, making it hard to monitor lower-abundance proteins. About a dozen proteins represent 90% of the serum proteome in total protein concentration, and adding a dozen more increases this number to about 99%. In high-throughput proteomics experiments, these highly abundant proteins are often immune-depleted from plasma or serum, benefiting proteome coverage,385 although this depletion may also induce biases due to off-target codepletion. Staggering improvements in MS-based proteomics technologies over the past decade have renewed the interest in plasma proteomics, whereby now without depletion simultaneous quantitative monitoring of over 1000 plasma proteins can be achieved.386,387 Interestingly, many established or claimed plasma protein biomarkers, such as α-1-antitrypsin, C-reactive protein, ceruloplasmin, and haptoglobin, belong to the category of highly abundant proteins in plasma388 and do not directly require these advances in proteomic depth.
In recent years, a few groups have started to use high-resolution native MS to especially target the most abundant plasma proteins with the aim of determining the variation in proteoforms present in individual donors. Such analyses provide a new dimension to the diversity of the plasma proteome.52 Many of these abundant plasma proteins are glycoproteins (e.g., α-1-antitrypsin, α-1-antichymotrypsin, ceruloplasmin, hemopexin, haptoglobin, α2-HS-glycoprotein, and various complement factors). They are predominantly expressed in the liver and subsequently secreted into the bloodstream. Changes in the glycosylation machinery of the liver cells that produce these proteins will directly translate to changes in the glycoproteome profiles of the resulting serum proteins, and therefore, plasma glycan and glycosylation analysis has become a rich source of potential biomarkers for diseases such as cancer389,390 and chronic obstructive pulmonary disease (COPD).391 Although the glycosylation pattern of a plasma glycoprotein is largely determined by the glycosylation machinery of the (liver) cells in which they are produced, other factors may influence these proteoform profiles as well. For instance, liver cells may have biases in the glycoproteoforms that they secrete, and the lifetimes of plasma glycoproteins may be different for different glycoforms. Therefore, it is essential to monitor the plasma glycoproteoform profiles directly from plasma.
In some of the earlier work on human plasma, the proteins were purified from pooled plasma from multiple donors or obtained from commercial sources. However, already in those studies it was revealed that various plasma glycoproteins can exhibit a plethora of proteoforms, all having different molecular weights and potentially different functions. For instance, for complement protein C9, a striking number of at least 50 different proteoforms were exposed that had variations in N-, O-, and C-glycosylation.380,392 Although this may suggest the presence of a very diverse glycoproteome profile, Franc et al.392 concluded almost the opposite and stated that C9 exposed strong preferences for the amount and nature of glycans attached. Similar data were reported for the related proteins properdin380 and C8, although the latter is somewhat more complicated because it is built up from three different chains (i.e., C8α, C8β, and C8γ).393
8.5.1. Acute-Phase Proteins
Wu et al.394,395 used high-resolution native MS to analyze purified α1-acid glycoprotein (AGP). AGP is a highly abundant acute-phase plasma protein that functions as a carrier for many hormones, lipids, and exogenous drugs and is proposed to deliver drug molecules to cells. The sources of the proteins were commercial and therefore likely originated from more than one donor. They were able to nicely disentangle the microheterogeneity of AGP, even though it harbors five N-glycosites. As the native mass spectra of sialylated AGP turned out to be very complex because of the varying degree of sialylation across all sites, they analyzed primarily asialo-AGP (neuraminidase-treated AGP) to facilitate glycan assignments. The resulting well-resolved and glycan-annotated native MS spectra of AGP are shown in Figure 24. Subsequently, they used native MS to measure binding of warfarin to AGP, which revealed that different glycoproteoforms of AGP exhibit distinct affinities for the drug. Most notably, an increase both in fucosylation and N-glycan branching/elongation reduced the binding to warfarin. Similarly, N-glycan branching and elongation also decreases binding of asialo-AGP to warfarin.
Figure 24.
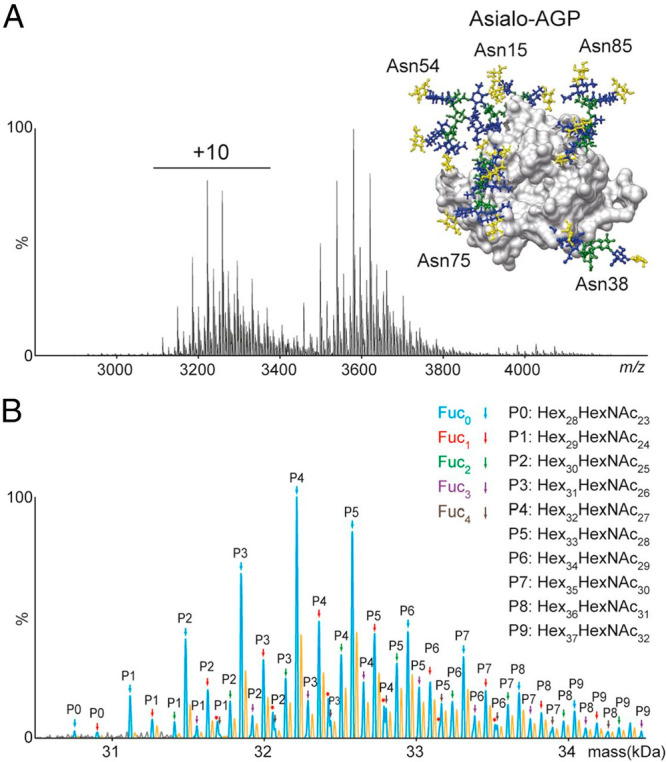
Glycan-annotated high-resolution native mass spectra of α1-acid glycoprotein (AGP). (A) High-resolution native mass spectrum and (B) zero-charged and glycan annotated deconvoluted mass spectrum of asialo-AGP. The inset in (A) shows the AGP structure with five highly branched N-glycans at Asn15, Asn38, Asn54, Asn75, and Asn85. From ref (394). CC BY-NC-ND 4.0.
A breakthrough toward personalized plasma proteome profiling came when Lin et al.396 demonstrated that roughly the top 30 most abundant plasma proteins can be efficiently purified from individual donors using only 50 μL of plasma for analysis by high-resolution native MS. They subjected serum samples to various forms of prefractionation, notably SEC and various forms of IEX. The purification of individual proteins from individual donors opened up the interesting question whether each donor exhibits a unique proteoform profile for each serum glycoprotein.396 This question was first tackled by analyzing α2-HS-glycoprotein (AHSG, fetuin) extracted from serum from 20 different donors, of which 10 had experienced a septic episode. Lin et al.396 observed that the proteoform profile of fetuin for each of these donors turned out to be unique and thus highly personalized. This diversity could be explained to a great extent by the presence of two dominant alleles of fetuin in the population (AHSG1 and AHSG2), which could be either homozygous or heterozygous. The mutations present in AHSG also affect an important O-glycosylation site, with mutation of a Ser into a Thr leading to a highly abundant Thr O-glycosylation, whereas the Ser was found to be mostly unmodified. Although the proteomic differences between donors of different genotypes were already interesting, also donors of the same genotype exhibited striking differences in phosphorylation and fucosylation, whereby the latter seemed to be enhanced in the older patients and especially those that had experienced a septic episode.
Using partly the same cohort of donors, Čaval et al.397 used high-resolution native MS to profile the proteoforms of intact alpha-1-antichymotrypsin (AACT). This protein was purified from individual plasma samples that were longitudinally collected from 10 patients at four time points over the duration of a septic episode. As determined by standard proteomics, AACT, a positive acute-phase protein, followed a similar abundance profile as C-reactive protein, being elevated during sepsis but returning back to baseline when the patients were dismissed. More interestingly, the proteoform profiles of AACT from the 10 patients gradually increased in mass over the monitored period, which could be ascribed to increased levels of fucosylation, antennary branching, and LacNAc moieties in response to the septic episode (Figure 25). This glycoproteoform remodeling thus extended over a much longer period than the septic episode. Although several trends in AACT abundance and extent of glycosylation were alike in all 10 monitored patients, it was apparent that each donor exhibited a unique glycoproteoform profile.
Figure 25.

Native-MS-derived proteome profiles of serum SerpinA3 (AACT) from a donor before and after a septic episode revealing an extensive increase in glycosylation.397
8.5.2. Haptoglobin
Both the Robinson and Heck groups studied the abundant plasma protein haptoglobin (Hp) by high-resolution native MS. Haptoglobin has been implicated as a biomarker for several pathological conditions,398,399 but in most studies, merely the abundance level of haptoglobin is considered as the biomarker. In humans, there are two frequently occurring allelic forms of Hp, resulting in three major genotypes: homozygous Hp 1-1 and Hp 2-2 and heterozygous Hp 2-1. This genetic polymorphism has an intriguing effect on the quaternary structure of Hp. The simplest genotype, Hp 1-1, yields dimers consisting of two α1β units connected by disulfide bridges. Hp 2-1 forms mixtures of linear (α1)2(α2)n−2(β)n oligomers (n > 1) while Hp 2-2 occurs in cyclic (α2)n(β)n oligomers (n > 2). The main function of Hp in plasma is that it scavenges toxic hemoglobin (Hb) leaked into the bloodstream from erythrocytes. It has been shown that different Hp genotypes bind Hb with different affinities, with Hp 2-2 being the weakest binder.
Hp also harbors four N-glycosylation sites on the β-chain, located at Asn184, Asn207, Asn211, and Asn241, all occupied by complex type N-glycans with a varying number of antennas, which may be fucosylated and sialylated.400,401 Wu et al. combined high-resolution native MS and glycoproteomics to investigate Hp 1-1 glycosylation microheterogeneity and examine its impact on the interactions with Hb and lectins.394,395 Already the ∼80 kDa Hp 1-1 dimers exhibited quite complex glycoproteoform profiles. Using affinity chromatography, they established that N-glycan branching attenuates Hp–Hb binding and, on the contrary, that fucosylation stabilizes Hp–Hb binding. Overall, these studies indicated that N-glycosylation fine-tunes Hp–Hb interactions. Building further upon these findings, Tamara et al.402 analyzed Hp from all three genotypes: homozygous Hp 1-1 and Hp 2-2 and heterozygous Hp 2-1. They first combined native MS and mass photometry to study the oligomer distributions in all three genotypes qualitatively and quantitatively. Subsequently, they dissected the glycoproteoform profiles of individual oligomer states using a combination of SEC and IEX with high-resolution native MS and obtained well-resolved glycoproteoform profiles for—among others—the 138, 188, and 237 kDa Hp 2-1 trimer, tetramer, and pentamer, respectively (Figure 26). These high-resolution native mass spectra were matched with simulations based on quantitative intact-mass LC-MS data for distinct Hp subunits generated in parallel, ultimately facilitating glycoproteoform annotations. Strikingly, these high-resolution native MS spectra revealed that each oligomer displayed distinct glycosylation patterns. Subsequently, also the hemoglobin binding propensities of these distinct oligomers were probed by affinity chromatography, revealing that Hb binding is tightly and finely regulated by both Hp oligomerization and Hp glycosylation. Overall, these studies clearly revealed that a wealth of genotype-specific proteoforms fine-tunes hemoglobin scavenging for haptoglobin in plasma, which should also be considered when designing cell-free haptoglobin-based therapeutics.403
Figure 26.

Comparison of deconvoluted high-resolution native mass spectra with simulated mass spectra reconstructed from intact-mass hydrophilic interaction chromatography (HILIC) LC-MS data. (A, C, E) Mirror plots for Hp 2-1 multimers: (A) (α1)2α2β3 trimer, (C) (α1)2(α2)2β4 tetramer, and (E) (α1)2(α2)3β5 pentamer. The zero-charged deconvoluted native MS spectrum is depicted at the top and the in silico-reconstructed spectrum at the bottom. The Pearson correlation coefficient (r) for each pair of native and in silico-reconstructed spectra is given in the top left corner of each panel, and a schematic drawing of each Hp 2-1 multimer is shown in the bottom left corner of each panel. (B, D, F) Annotation of peaks detected in the native deconvoluted MS spectra for the (B) trimer, (D) tetramer, and (F) pentamer of Hp 2-1. From ref (402). CC BY-NC-ND 4.0.
These initial studies focusing on just a single serum glycoprotein have provided a new dimension of quantitative plasma proteome profiling. For almost every plasma glycoprotein, the glycosylation profile seems to be unique for each individual donor in terms of proteoforms and their abundances. This striking observation opens the way to a next level of personalized proteome profiling in which the unique proteoform profiles are used to stratify patient cohorts and to monitor their adaptation to physiological changes. More research is needed to find out which plasma protein(s) will be the best biomarker(s) for each particular disease, but it is apparent that glycoproteoform profiles need to be considered in future plasma proteomics.
8.6. High-Resolution Native MS of Intact Phosphoproteins
In the previous section, we described how high-resolution native MS can be very powerful for the analysis of protein glycosylation. However, this technology is equally applicable to the analysis of intact proteins decorated with other PTMs. Protein phosphorylation is an important modification that is involved in signal transduction, may regulate protein–protein interactions, and (de)activates kinases and phosphatases, among others. The ability of native MS to investigate protein–protein interactions and protein phosphorylation concomitantly was exploited by Kleppe et al.404 to study the phosphorylation dependence and stoichiometry of the complex formed by tyrosine hydroxylase and 14-3-3γ. Tyrosine hydroxylase (TH) is a high-affinity binding partner of 14-3-3γ protein, but this interaction is tightly regulated by phosphorylation. Kleppe et al. performed native MS analyses of human TH (non-phosphorylated or phosphorylated on Ser19 (TH-pS19) or Ser40 (TH-pS40) alone and together with 14-3-3γ. Whereas tetrameric TH-pS19 (224 kDa) bound 14-3-3γ (58.3 kDa) with high affinity (Kd = 3.2 nM), generating complexes containing either one (282.4 kDa) or two (340.8 kDa) dimers of 14-3-3γ, no complex formation between non-phosphorylated TH or TH-pS40 and 14-3-3γ was observed, highlighting the crucial role of pS19.
Similarly, van de Waterbeemd et al.405 used a combination of high-resolution native MS and bottom-up proteomics to monitor the effects of protein phosphorylation on complex formation. More in-depth, in this study the phosphorylation and cyclic nucleotide binding of dimeric 150 kDa cGMP-dependent protein kinase (PKG) were simultaneously monitored by high-resolution native MS, which showed that binding of cAMP or cGMP causes different PKG phosphorylation kinetics. In a second example, it was demonstrated that the binding and phosphorylation of the mitotic regulator Bora by the cycle kinase Aurora A proceed independently. Interestingly, the three investigated proteins—Aurora A, Bora, and PKG—existed in different phosphorylation states. The relative abundances of these phosphoisoforms could be accurately monitored by native MS, whereas complementary peptide-centric MS experiments were done to localize the phosphorylated residues.405 In an extension of those experiments, high-resolution native MS was also used by Lössl et al.406 to decipher the phosphoproteoforms of Bora resulting from phosphorylation by either Aurora A or Polo-like kinase 1 (Plk1), showing that the two kinases target different Bora residues and generate distinct phosphorylation cascades. The tripartite Aurora A/Bora/Plk1 interplay modulates the cell’s recovery from DNA-damage-induced cell cycle arrest, and it is characterized by numerous mutual phosphorylation events with various implications for protein structure, interactions, and function. Lössl et al. simultaneously probed these often temporarily occurring effects by integrating native MS, cross-linking-MS, IM-MS, top-down sequencing, and bottom-up proteomics.406 In a somewhat related study, Abdul Azeez et al.407 utilized native MS and IM-MS to determine the phosphorylation-dependent formation of a complex between Aurora C and the inner centromere protein (INCENP). They introduced mutations on different Ser residues of the TSS motif, which weakened the interaction substantially. IM-MS data revealed that the phosphorylated Aurora C–INCENP complex exhibited higher cross sections, hinting at a more flexible structure containing partly disordered regions.
In another elegant study, Ben-Nissan et al.69 used high-resolution native (tandem) MS to analyze the yeast homotetrameric ∼155 kDa FBP1 complex. FBP1 is the rate-limiting enzyme in gluconeogenesis. The data revealed that each of the subunits in the FBP1 tetramer is differently phosphorylated when expressed under different growth conditions, whereby the subsequent incorporation of each of the four phosphate moieties (80 Da each) could be nicely resolved. In this manner, they were able to determine the stoichiometry, the kinetics, and by top-down proteomics the exact position of the phosphorylations. Similarly, Potel et al.408 used high-resolution native (tandem) MS to monitor the stoichiometry and phosphorylation in heterohexamers of the tumor metastasis suppressor Nm23. Human Nm23 is present in various isoforms, of which Nm23-H1 and Nm23-H2 are by far the most dominant. Extracting these hexameric assemblies from different compartments of different cells, they demonstrated cell- and compartment-specific stoichiometries of this abundant complex. Subsequently, they combined native and top-down MS to investigate the histidine autophosphorylation activity of the purified Nm23 assemblies.
9. Non-covalent Interactions of Proteins with Small Ligands Probed by High-Resolution Native MS
Interactions between proteins and small ligands are fundamental to many cellular activities and processes. A wide variety of small molecules, including metal ions, heme groups, metabolites, hormones, drugs, (oligo)nucleotides, and peptides, can non-covalently bind to proteins to regulate their structure and function. Understanding and quantifying such protein–ligand interactions is therefore of key importance for biological research and the discovery and design of effective therapeutics. The affinities and kinetics of protein–ligand interactions are typically assessed by various biochemical and biophysical techniques, the most common of which are surface plasmon resonance (SPR),409,410 circular dichroism (CD),411 and isothermal titration calorimetry (ITC).412 Using native MS to study protein–ligand interactions seems to be a very attractive prospect because of the technique’s inherent simplicity and sensitivity, as well as the richness of the information that can be obtained. Native MS does not require chemical labeling or immobilization, and sample consumption is relatively low. With direct mass measurements, one can also resolve multiple coexisting species, determine the stoichiometry of ligand binding, and investigate cooperativity.
The first studies of protein–ligand interactions by native MS were initiated in the early 1990s with the works of Ganem, Li, and Henion10,413 and Katta and Chait11 shortly after the introduction of ESI. Attempts at quantitative assessment of affinities quickly followed suit, with Loo and co-workers providing some of the first examples of such studies.414 Because of the unique ability of native MS to monitor the abundance of coexisting species simultaneously, several different strategies emerged. Affinities could be determined from a titration experiment by following the intensity ratio between the protein and the protein–ligand complex.415−417 Alternatively, a secondary ligand of known affinity was used as a reference point in competitive binding experiments,415−417 with Jørgensen et al.415 being among the first to report absolute values for dissociation constants as determined by native MS. The field received another impulse with the advent of automation,418 as Zhang et al.419 introduced the NanoMate, a combination of an autosampler with a chip-based nESI source that could be used to drastically increase the throughput, making the technology ready for application in the pharmaceutical industry. By the early 2000s, several reviews discussed this new role of native MS as a tool for quantitative studies of protein–ligand interactions.420−422 It was expected that measurements of dissociation constants would have been routine by now, especially with the availability of automation. However, a lack of consistency in early literature led to a “hit or miss” perception, limiting the use to a few specialist laboratories. For native MS to become a more widespread technique for studies of protein–ligand binding, several questions needed to be answered, both fundamental and practical in nature, as discussed in the following sections.
9.1. Optimizing Native MS for the Quantitative Determination of Binding
With appropriate testing and optimization, quantitative information about ligand binding can be obtained from native MS experiments, even though it is not an inherently quantitative technique. This relates mostly to the ESI process but also to artifacts that may be introduced by ion transfer and detection in the gas phase. One of the main questions raised early on was whether the ESI process would distort the solution-phase equilibrium. Measurement times should be kept short, as slow acidification of the sample may occur through electrochemical water oxidation in positive ion mode.423,424 Acidification also occurs within the ESI droplets themselves, but this is thought to happen on a time scale that is too short to impact the equilibrium substantially.425,426 Aqueous ammonium acetate solutions only partially alleviate this issue, as their buffering capacity becomes meaningful only when the pH drops toward 4.75, the pKa of acetic acid.427 According to the charge residue model, evaporation also leads to a short-lived increase in analyte concentrations. Although too short to affect the equilibrium substantially, this can still introduce nonspecific binding, especially when high concentrations of protein and ligands are used. However, it has been shown that such weakly bound species can often easily be removed further downstream in the mass spectrometer.421
Several studies have focused on how ionization efficiency, ion transfer, and detection may differentially affect different analytes. Although an equal response in ionization would be a reasonable assumption when the species have similar sizes and biochemical properties (e.g., non-covalent protein complexes that differ by a single bound small ligand), this is often not the case for metal-binding, DNA/RNA-binding, and larger protein complexes. The ionization efficiency depends not only on the charge but also on access to the surface of the ESI droplet, which has been shown to vary with the size, conformation, and hydrophobicity of the analyte.421,428 Furthermore, complexes stabilized by ionic and/or polar interactions are much more stable in the gas phase than those stabilized by hydrophobic interactions.429−431 A response factor accounting for ionization efficiency and instrumental bias was introduced by Gabelica et al.,432 and Tjernberg et al.433 shortly thereafter also addressed gas-phase dissociation. Alternatively, the approximation of response factors can be avoided altogether by including a reference complex with a known Kd value, for instance as described by Kempen and Brodbelt.434
Native MS also differs from solution-based techniques in that it is known to be quite incompatible with solvents containing nonvolatile salts. High metal salt concentrations, which are often desirable to preserve protein–ligand equilibria, result in protein ions carrying multiple adducts, which not only dilutes the ion signal but also complicates the detection of low-abundance protein–ligand complexes. Partly as a result of this, researchers have often struggled to detect full ligand binding by native MS for interactions dependent on the presence of metal ions.419 However, in recent studies435−437 this problem has been largely circumvented using nanoscale ion emitters. In contrast with micrometer spraying capillaries, the narrower-diameter capillaries produce even smaller droplets that carry fewer nonvolatile components. In a comprehensive work, Nguyen et al.435 demonstrated that a narrower tip diameter (∼250 nm) can help to improve the determination of ligand–protein binding affinities. For example, they obtained quite accurate Kd values for binding of an inhibitor to the metalloenzyme carbonic anhydrase (Figure 27).
Figure 27.
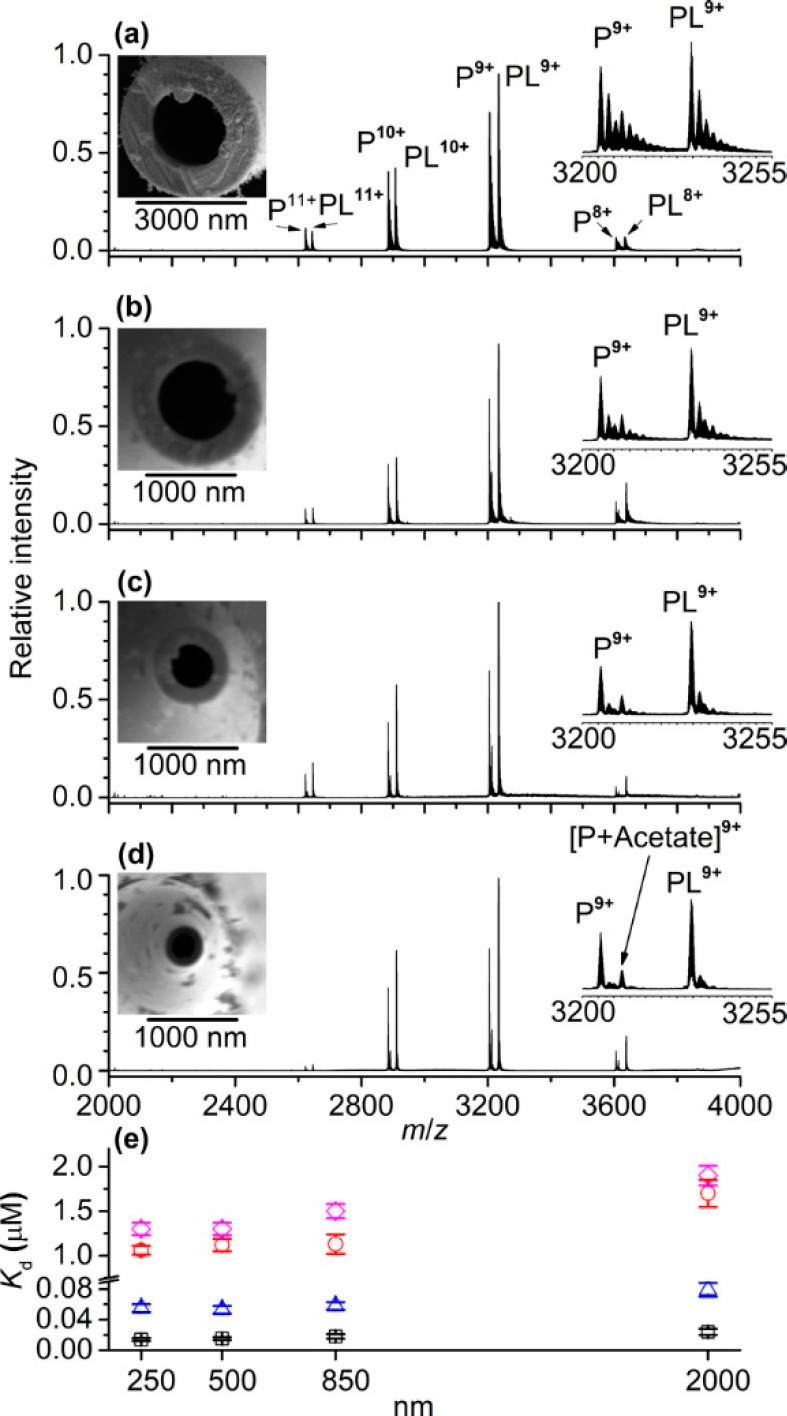
Narrow-bore nanoelectrospray ionization emitters with inner tip diameters less than 1000 nm can be used to obtain more accurate Kd values for carbonic anhydrase (P) inhibitors (L). (a–d) nESI mass spectra of aqueous solutions containing 5 μM human carbonic anhydrase I, 2 μM ethoxzolamide, and 70 mM ammonium acetate obtained using emitter tips with inner diameters of (a) ∼2000, (b) ∼850, (c) ∼500, and (d) ∼250 nm. (e) Kd values measured using nESI as a function of the emitter tip diameter for the binding of ethoxzolamide (squares), brinzolamide (circles), furosemide (triangles), and dichlorphenamide (diamonds) to human carbonic anhydrase I. Reproduced from ref (435). Copyright 2019 American Chemical Society.
Although native MS can thus be used for the quantitative assessment of protein–ligand binding constants, there are limitations in the range of Kd values that can be measured, also depending on the type of experiment. With the most standard titration assays, the ion intensity ratio between the free protein and the protein–ligand complex is measured. By deducing the corresponding free ligand concentration or by measuring it directly using a calibration curve, one can make a Scatchard plot, from which the Kd value can be extracted. The dynamic range of this approach is limited, however, as the target protein concentration should ideally be below the expected Kd value while the ligand concentration is titrated through this range. In practice, this means that only affinities above 100 nM can be measured reliably by this native MS approach. On the other hand, competitive binding experiments can be applied in which a secondary ligand of known affinity is used to compete with a high-affinity ligand of interest. This effectively shifts the binding curve toward higher concentrations that can be measured by native MS. In this manner, nanomolar-range affinities can be measured, even though typical native MS concentrations in the micromolar range are employed in these experiments. By focusing on the free ligand in the lower m/z range of the instrument, Wortmann et al.438 showed that the lower limit of Kd measurements could be extended even below the picomolar range.
With careful optimization of experimental parameters to avoid biases and artifacts, native MS can thus provide reliable quantitative data on protein–ligand binding. Systematic comparisons between ESI-MS and other methods by the groups of Klassen, Zenobi, and others have shown that native MS has a unique position within the toolbox for quantitative assessment of protein–ligand interactions.439−445 Notably, Jecklin et al.444 investigated a set of eight well-characterized inhibitors of human carbonic anhydrase I (hCAI) with Kd values spanning over 4 orders of magnitude (low nM to high μM). While good agreement was seen for four inhibitors, limitations of the different techniques became apparent when the others were analyzed. While the assessment by native MS was found to be hampered by gas-phase dissociation of hydrophobic ligands and nonspecific adducts at higher concentrations, native MS was able to overcome issues related to fast kinetics (hampering SPR) and poor solubility (hampering ITC). This study by Jecklin et al. clearly reveals that every method may provide false positives and false negatives, and thus, cross-validation of approaches should always be encouraged.
9.2. Pharmaceutical Applications of Protein–Ligand Screening by Native MS
Mass spectrometry is nowadays used in the early stages of drug discovery to screen compounds or fragment libraries against a protein target in fairly high-throughput approaches.446−451 The role of native MS in fragment-based lead discovery (FBLD) was pioneered by Swayze et al.,452 who identified structure–activity relationships (SARs) to generate a lead compound binding the 1061 region of bacterial 23S rRNA. Hits from a first fragment screen revealed two interesting SAR trends, and when these hits were combed, ternary complexes observed in native MS readily showed that these two trends involved different binding sites. Competitive binding experiments on ligand derivatives provided additional information on how the two binding sites may be combined in rationally synthesized fused compounds. The resulting lead compounds displayed up to a 20-fold increase in affinity as well as increased functional activity. An automated approach was presented a few years later by Maple et al.,453 who used the NanoMate platform to screen a library of 157 compounds against an apoptotic protein target. Although extensive optimization of the instrumental parameters was required, both the throughput and results proved to be comparable to those of NMR- or ITC-based library screening. A similar strategy employed by Woods et al.454 using a 720-member fragment library also showed a very good correlation between native MS and traditional screening setups based on SPR and X-ray crystallography. Moreover, these and other studies455 showed that sample consumption and the required fragment concentrations were lower in the MS-based approach, widening accessibility to poorly soluble compounds that would otherwise have been discarded. More recently, an alternative automation approach was presented by Ren et al.,456 who used an autosampler and SEC to remove unbound compounds from a target protein incubated with pools of small molecules followed by native MS to improve the sensitivity (Figure 28).
Figure 28.

Automated screening of protein–ligand interactions using a compound library and native SEC-MS. (a–c) Native mass spectra of the enzyme indoleamine-pyrrole 2,3-dioxygenase (IDO1) alone, IDO1 with compound 1, and IDO1 with compound 2, respectively. The left panels show the charge-state distributions, and the right panels focus on the enlargements of the +14 charge state for identification. For all of the spectra shown, the concentration of IDO1 was 5 μM (2 μM for IDO1 w/heme), whereas the concentration of compound 1 was 3.5 μM in (b) and that of compound 2 was 10 μM in (c). (d) Saturation curves from titration experiments. The concentration of the IDO1 sample was 13 μM (9 μM for IDO1 w/heme). The concentrations of compounds 1 and 2 ranged from 0.5 to 100 μM. (e) In-source dissociation profiles for the IDO1–compound 1 and IDO1–compound 2 complexes. Dashed lines indicate the corresponding Vc50 values. The concentration of the IDO1 sample was 12 μM, and the concentrations of compounds 1 and 2 were 89 μM. (f) Plot of Vc50 against initial complex percentage at different compound 1/compound 2 to IDO1 ratios. (g) Competition experiment for evaluation of relative binding affinities. IDO1 (total 13 μM, IDO1 w/heme 9 μM) was incubated with compounds 1 and 2 (each at 10 μM). Each precursor experiences a different lab-frame energy based on its charge state. All of the data points in (a) and (b) were replicated three times, with error bars shown on the graph. Adapted from ref (456). Copyright 2018 American Chemical Society.
The advantages and complementarity of ligand screening by native MS also become apparent in the later stages of drug development, including the analysis of high-affinity compounds in the hit-to-lead (H2L) and lead optimization (LO) processes.446,449,451 Chip-based native MS can be used in competitive binding experiments for affinity ranking of optimized compounds. Bovet et al.457 demonstrated this principle using different ligands binding the human estrogen receptor, although poor reproducibility prevented them from providing reliable Kd values for hydrophobic interactions, which are typically weakened in the gas phase. Another competitive binding study by Jecklin et al.458 compared a set of clinical kinase inhibitors and introduced a chip-based method focusing solely on the unbound ligands. Better sensitivity and resolution in the low-m/z range circumvented the need for harsher ion transmission conditions—and therefore dissociation. Both applied approaches, focused on either intact ligand–protein complexes or unbound ligands, led to results that were in good agreement with known IC50 values. Evidently, for the latter approach, focused solely on analyzing the free ligand, high resolution is easily achieved even on conventional mass spectrometers.
9.3. Binding of Small Molecules to Heterogeneous Proteins Analyzed by High-Resolution Native MS
Outside of a few laboratories equipped with high-resolution FT-ICR mass spectrometers, studying protein–ligand binding was often limited by the mass resolution of the typically employed ToF instruments. With the advent of the Orbitrap mass spectrometers as a more accessible high-resolution MS platform, protein–ligand binding studies became possible for smaller and smaller ligands in interaction with more heterogeneous larger proteins. In a follow-up study to their earlier work, Maple et al.459 were among the first to employ the Orbitrap-EMR instrument for high-throughput ligand screening. Baseline separation of glycoforms enabled the approach to be extended toward glycoproteins not tractable by traditional ToF-based approaches. Furthermore, the sensitivity advantage of the Orbitrap instrument enabled Maple et al. to measure proteins at lower concentrations, assess stronger interactions, and reduce sample consumption.
Following up on their pioneering work in making native MS amenable for the analysis of membrane proteins,91 the Robinson group started to use Orbitraps with extended mass range to probe small-molecule and lipid binding to this important class of proteins in more detail. In a breakthrough study, Gault et al.21 measured a wide range of membrane proteins on an Orbitrap instrument, using the HCD cell to strip off residually bound detergent molecules. Because of the high resolving power of the instrument, unambiguous distinctions could be made between lipids from the same class with different chain lengths and degrees of saturation when tightly bound to the trimeric OmpF assembly (Figure 29). Moreover, analysis of the interaction between OmpF and a small peptide revealed three successive binding events for which the Kd could be determined individually. A follow-up study by Mehmood et al.460 used the same approach on the human intramembrane zinc metalloprotease ZMPSTE24 to reveal and quantitate the off-target binding and activity of several HIV protease inhibitors. In 2018, Gupta et al.236 released a protocol for identifying lipids that tightly bind to membrane protein complexes and explored how delipidation affects the disruption of oligomeric interactions.
Figure 29.
Native MS spectrum (main panel) of trimeric OmpF bound to an equimolar ratio of DMPG, DPPG, and POPG lipids (cartoon inset). The 22+ charge state is shown in an expanded view (right), with peaks showing up to three bound lipids. Theoretical distributions corresponding to different combinations of lipids are shown by colored lines and correlate with the spectrum. Reproduced with permission from ref (21). Copyright 2016 Nature Publishing Group.
9.4. Resolving Multiple Binding Events to Oligomeric Protein Assemblies and DNA/RNA Molecules
Whereas concomitant binding to multisubunit complexes cannot be accurately assessed by standard approaches for probing protein–ligand binding (e.g., ITC, SPR), native MS can distinguish by mass and thus reveal the entire distribution of ligand-bound states. Gavriilidou et al.461 demonstrated this unique advantage of native MS in their study of the dimer–tetramer equilibrium of M2 pyruvate kinase (PKM2), a regulatory enzyme that is often inactive in the glycolytic pathway in tumor cells. An allosteric activator, fructose-1,6-bisphosphate (FBP), was found to shift the dimer–tetramer equilibrium toward the active tetramer, with the 4:4 stoichiometry of FBP binding to the tetramer only. Another study by Root et al.462 investigated the isoprenoid biosynthesis enzyme IspF from Arabidopsis thaliana, a homotrimeric assembly that binds multiple ligands, including a metal cofactor and a synthetic inhibitor. Whereas standard biophysical techniques failed to reveal the mode of action of recently discovered inhibitors, native MS enabled researchers to propose a mechanism that involves competition with the substrate and extraction of the Zn2+ from the active site. Using native MS, El-Baba et al.463 recently also investigated the ability of various small molecules to modulate the activity of dimeric SARS-CoV-2 main protease, including a compound proposed to disrupt the oligomeric interface.
Apart from analyzing protein–ligand interactions, native MS provides a great means for in-depth investigation of the interactions between DNA/RNA and small molecules. In a comprehensive screening study, Gülbakan et al.464 captured all major aspects of aptamer–ligand interactions for three DNA aptamers, showing the stoichiometry, selectivity, and cooperativity of various interactions. They supplemented MS data with SPR, ITC, and CD measurements and highlighted the unique strengths of native MS for aptamer–ligand analysis, whereby the different techniques complemented each other. Recently, Nguyen et al.465 applied native MS and its ability to distinguish between multiple binding modes to study how netrospin, a potent antibiotic and anticancer agent precursor, interacts with hairpin and duplex DNA molecules. Using nanoscale emitters, they were able to spray DNA–ligand samples with high salt concentrations, enabling them to simultaneously determine binding affinities for five ligand–DNA and DNA–DNA complexes.
9.5. Investigating the Kinetics and Thermodynamics of Ligand Binding
An additional attractive feature of native MS for the assessment of protein–ligand interactions is that the abundance of different species can be followed over the course of the measurement, enabling the investigation of kinetics and thermodynamics.466,467 Using a temperature-controlled ESI source, Cong et al.468 investigated the thermodynamics of lipid binding to AmtB, an integral membrane protein of Escherichia coli. Their approach allowed them to determine the thermodynamics of individual binding events for lipids with variable chain length, resolving unique thermodynamic signatures. In another report, Moghadamchargari et al.469 demonstrated that high-resolution native MS could be used to study the intrinsic inactivation rates of the oncoprotein K-RAS and mutants thereof. This was done by monitoring the hydrolysis of non-covalently bound GTP, as identified by a mass shift corresponding to the loss of a phosphate group (Figure 30). The inactivation rates were in good agreement with complementary methods measuring the concentration of released organic phosphate. However, native MS unexpectedly revealed that some oncogenic mutants have a higher intrinsic hydrolysis rate for 2′-deoxy-GTP than for GTP, which are separated in mass by only 16 Da. Another approach explored by Marchand et al.470 determined the entropic and enthalpic contributions to the binding equilibrium of G-quadruplex nucleic acid structures and their ligands using a temperature-controlled nESI source.
Figure 30.
Determination of the guanosine triphosphate (GTP) hydrolysis rate (khyd,GTP) for K-RAS. (A) Representative native mass spectra recorded at different time points for K-RAS (2 μM) loaded with GTP incubated at 25 °C. The abundance of K-RAS bound to guanosine diphosphate (GDP) increases as GTP is hydrolyzed. Asterisks denote species with bound sodium or magnesium adducts. (B) The concentration of K-RAS bound to GDP and GTP was determined from deconvolution of the native MS data (dots) and fitted to a first-order rate constant model (solid lines). (C) Plot of inorganic phosphate concentration determined for K-RAS·GTP using a malachite green (MG) assay. The inset shows the similar rate constant values determined by native MS and the MG assay. Reproduced from ref (469). Copyright 2019 American Chemical Society.
9.6. Determination of the Ligand-Binding Site Using Native Top-Down ECD and UVPD Fragmentation
High-resolution native MS provides additional advantages for the study of protein–ligand interactions in that it can probe the ligand-binding interfaces. Using fast and high-energy ion activation techniques, the protein backbone can be fragmented, and this may occur without breaking the non-covalent interactions. Such dissociation results in a mass shift corresponding to the ligand’s mass, ideally for both the N- and C-terminal fragments. The idea of probing interaction sites in this manner was pioneered by Xie et al.,471 who used ECD on α-synuclein in complex with polycationic spermine. This allowed them to localize the binding site to residues 106–138, consistent with data obtained by NMR spectroscopy. The approach is applicable to both electrostatic interactions that are stable in the gas phase472,473 and hydrophobic interactions that are more prone to dissociation.474 Several of the first applications of UVPD to native protein–ligand complexes also revealed that ligands could remain bound to the fragments.224,226,475,476 For example, O’Brien et al.226 demonstrated that the C-terminal fragment ions of peptidyl-prolyl cis–trans isomerase 1 (Pin1) that retained a peptide ligand were formed by UVPD within the functionally relevant WW domain. The only N-terminal fragments that retained the peptide originated from the PPIase domain. This was in agreement with the reported crystal structure. These studies demonstrate some of the seemingly ever-expanding opportunities that are open for researchers by bridging native MS with a multitude of activation methods, both established and just emerging in the field of native top-down MS.
10. Conclusions and Outlook
Since its introduction by John Fenn, electrospray ionization has impacted biomolecular mass spectrometry way beyond the early high expectations. Whole new research fields such as proteomics and metabolomics now use mass spectrometry as their core technology and combine advanced separation technologies with electrospray ionization. Deservedly, Fenn was awarded the Nobel Prize for this invention in 2002, which he accepted with a Nobel Lecture entitled “Electrospray Wings for Molecular Elephants”.477 At that time, early on in this century, it was slowly becoming apparent that these flying molecular elephants could be partly kept “alive” while flying through the vacuum of the mass spectrometer on their way to the detector for mass analysis. To achieve this, the solvent conditions in the electrospray process needed to be adjusted, avoiding organic solvents and acidification, typically used in LC-MS.15 Primarily by using volatile buffer-mimicking solutions, such as aqueous ammonium acetate, primary, secondary, tertiary, and even quaternary structures of proteins and protein complexes could be largely retained, leading to the advent of what is now known as native mass spectrometry.14 By now, about 20 years later, native MS has matured into a versatile analytical method with widespread applications in molecular and structural biology and beyond in all areas of the life sciences.3,20 In this review, we have broadly categorized the applications into (1) analysis of protein assemblies, (2) analysis of proteoforms, and (3) analysis of protein–ligand interactions, but evidently, many of the applications reported here and in other literature cross over these three categories. Furthermore, many other applications are emerging and maturing, such as the analysis of DNA and RNA molecules, ribonucleoprotein complexes, and membrane-embedded protein complexes.
In this review, we have presented published work describing what is already possible by high-resolution native MS, but at the end of this review it is tempting to ask what is not yet possible? Even more, what do we dream of?
A dream likely shared by many would be to perform “sample-preparation-free” native MS analysis. We have described here the laborious efforts often needed to purify protein complexes of interest, aiming to desalt these analytes as much as possible, for instance by several rounds of dialysis, while simultaneously avoiding the copurification of detrimental detergent molecules and polymers. However, with the increased sensitivity and capabilities to desolvate ions within the latest mass analyzers,66,69,74 several pioneering attempts have been made to analyze proteins and protein complexes directly from cellular broths478 or cellular membranes,323 and reasonable success has already been achieved. Such attempts will certainly expand and make native MS hopefully even more feasible also for nonexperts.
Another futuristic dream is to be able to count and measure the mass of every molecule and molecular assembly within a cell. This is quite a challenge, as it has been estimated that there are ∼109 protein molecules479 in a given human cell, each with its own specific function, interactions, and potential decoration with a unique set of PTMs. Charting individual molecules requires single-molecule approaches. Several single-molecule analytical approaches have been developed over the past decades,480−482 but mass spectrometry has lagged behind and largely remained an ensemble-based method. However, several single-molecule and single-particle mass spectrometry approaches are now emerging109,128,150 and may go hand-in-hand with other technologies that can be used to measure masses of single particles, such as mass photometry258 and nanomechanical-systems-based mass analysis.483 Single-particle approaches enhance not only the sensitivity but also the specificity, as each molecule can be analyzed by itself, revealing its own unique features. This will surely help to chart unknown territories, such as those presented by the wide variety of proteoforms co-occurring within a cell. Potentially, one day we will be able to assert that each protein within the cell is indeed unique. Ideally, these two dreams can be combined, enabling each molecule within the cell to be measured with minimal distortions and sample preparation.
High-resolution mass spectrometry, as powerful as it is, delivers even more biological information when used in concert with other technologies. It has taken some adjustments, but as reviewed also here, native MS has now become an integral part of many structural biology studies, contributes to the exciting fields of protein design and engineering, and has already contributed to COVID-19-related science.338,339 Native mass spectrometry has benefited from the resolution revolution in electron microscopy,484,485 but also, vice versa, sample preparation for electron microscopy can be assisted by prior native MS analysis.486−489 While high-resolution structural methods provide unprecedented detail toward structure and function, native mass spectrometry is very well suited to reveal the molecular heterogeneity inherent in each and every biological sample. Within the discipline of mass spectrometry, native MS is more and more conjugated with hydrogen–deuterium exchange MS, cross-linking MS, chemical labeling, and interaction proteomics.3 From a niche technology, it has now become a key technology embedded in the structural biology toolbox.
The field of high-resolution native mass spectrometry still has many challenges ahead. Through a concerted effort with parallel future developments in mass analyzers, sample preparation, separation, and data analysis, we can dream on and expect fast evolution of native MS in the near future.
Acknowledgments
Since its conception about 30 years ago, the research field of native MS has grown steadily, which by now has led to a rather substantial and lively research community. We acknowledge all involved, making this a scientifically competitive but very friendly and cooperative community. We acknowledge all of the researchers whose work has made it possible for us to write this comprehensive review. We apologize for missing contributions made by others; the field has grown so much that it is rather difficult to be all-inclusive. We thank all of the members of the Heck lab for their support in writing this review and for executing some of the work described in it, and we also thank all of our collaborators over the years for supplying interesting research questions and precious samples. A.J.R.H. acknowledges support from The Netherlands Organization for Scientific Research (NWO) through the Spinoza Award (SPI.2017.028). M.A.d.B. and A.J.R.H. were additionally supported by NWO NACTAR Project 16442.
Glossary
Abbreviations
- nESI
nano electrospray ionization
- fwhm
full width at half-maximum
- eFT
enhanced Fourier transform
- ToF
time of flight
- FT-ICR
Fourier transform ion cyclotron resonance
- IM-MS
ion mobility mass spectrometry
- SEC-MS
size-exclusion chromatography–mass spectrometry
- CD-MS
charge detection mass spectrometry
- CID/CAD
collision-induced dissociation/collision-activated dissociation
- HCD
higher-energy collisional dissociation
- ETD
electron transfer dissociation
- ECD
electron capture dissociation
- SID
surface-induced dissociation
- UVPD
ultraviolet photodissociation
- mAb
monoclonal antibody
- ADC
antibody–drug conjugate
- IgG
immunoglobulin G
Biographies
Sem Tamara (1992) is a postdoctoral researcher at Utrecht University in The Netherlands. He gained experience with mass spectrometry at the Proteomics and Metabolomics group of Novosibirsk’s International Tomography Center in Russia. Expanding on this, in 2019 he obtained a Ph.D. at Utrecht University, focusing on integrative top-down MS analysis of large and heterogeneous protein assemblies. He is mainly interested in developing new analytical approaches using alternative gas-phase fragmentation methods in bottom-up, top-down, and native MS for the characterization and sequencing of intact protein complexes and biopharmaceuticals.
Maurits A. den Boer (1991) is currently a Ph.D. candidate at Utrecht University. He has a background in biotechnology and obtained degrees from TU Delft (B.Sc.) and Leiden University (M.Sc., cum laude). His research combines method development for de novo sequencing of antibodies with native MS studies into immunoprotein complexes. Together, these approaches are applied with the aim of finding therapeutic antibodies against multidrug-resistant bacteria.
Albert J. R. Heck (1964) is distinguished faculty professor at Utrecht University, chairing the Biomolecular Mass Spectrometry and Proteomics group since 1998. His research focuses on the development and applications of mass-spectrometry-based proteomics and structural biology. Heck introduced several innovative proteomics technologies for phospho-enrichment, the use of alternative proteases, and hybrid peptide fragmentation techniques. He is also known for his expertise in structural biology, being a pioneer in native MS and cross-linking mass spectrometry. He has received several awards, including the Field and Franklin Award (ACS), The Thomson Medal Award (IMSF), the Krebs Medal (FEBS), and the Spinoza Award (NWO). He is member of the Royal Netherlands Academy of Sciences and Arts (KNAW) and EMBO.
Author Contributions
§ S.T. and M.A.d.B. contributed equally.
The authors declare no competing financial interest.
References
- Altelaar A. F.; Munoz J.; Heck A. J. Next-Generation Proteomics: Towards an Integrative View of Proteome Dynamics. Nat. Rev. Genet. 2013, 14, 35–48. 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass-Spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537, 347–355. 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Lössl P.; van de Waterbeemd M.; Heck A. J. R. The Diverse and Expanding Role of Mass Spectrometry in Structural and Molecular Biology. EMBO J. 2016, 35, 2634–2657. 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson G. R.; Burke J. E.; Ahn N. G.; Anand G. S.; Borchers C.; Brier S.; Bou-Assaf G. M.; Engen J. R.; Englander S. W.; Faber J.; et al. Recommendations for Performing, Interpreting and Reporting Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) Experiments. Nat. Methods 2019, 16, 595–602. 10.1038/s41592-019-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Strutzenberg T.; Pascal B. D.; Griffin P. R. Protein Dynamics and Conformational Changes Explored by Hydrogen/Deuterium Exchange Mass Spectrometry. Curr. Opin. Struct. Biol. 2019, 58, 305–313. 10.1016/j.sbi.2019.06.007. [DOI] [PubMed] [Google Scholar]
- Li K. S.; Shi L.; Gross M. L. Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (Fpop) for Higher Order Structure Characterization. Acc. Chem. Res. 2018, 51, 736–744. 10.1021/acs.accounts.7b00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly F. J.; Rappsilber J. Cross-Linking Mass Spectrometry: Methods and Applications in Structural, Molecular and Systems Biology. Nat. Struct. Mol. Biol. 2018, 25, 1000–1008. 10.1038/s41594-018-0147-0. [DOI] [PubMed] [Google Scholar]
- Liu F.; Heck A. J. Interrogating the Architecture of Protein Assemblies and Protein Interaction Networks by Cross-Linking Mass Spectrometry. Curr. Opin. Struct. Biol. 2015, 35, 100–108. 10.1016/j.sbi.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Iacobucci C.; Gotze M.; Sinz A. Cross-Linking/Mass Spectrometry to Get a Closer View on Protein Interaction Networks. Curr. Opin. Biotechnol. 2020, 63, 48–53. 10.1016/j.copbio.2019.12.009. [DOI] [PubMed] [Google Scholar]
- Ganem B.; Li Y. T.; Henion J. D. Observation of Noncovalent Enzyme-Substrate and Enzyme-Product Complexes by Ion-Spray Mass Spectrometry. J. Am. Chem. Soc. 1991, 113, 7818–7819. 10.1021/ja00020a085. [DOI] [Google Scholar]
- Katta V.; Chait B. T. Observation of the Heme-Globin Complex in Native Myoglobin by Electrospray-Ionization Mass Spectrometry. J. Am. Chem. Soc. 1991, 113, 8534–8535. 10.1021/ja00022a058. [DOI] [Google Scholar]
- Fenn J. B.; Mann M.; Meng C. K.; Wong S. F.; Whitehouse C. M. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246, 64–71. 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Leney A. C.; Heck A. J. R. Native Mass Spectrometry: What Is in the Name?. J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. 10.1007/s13361-016-1545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel R. H. H.; Heck A. J. R. Native Protein Mass Spectrometry: From Intact Oligomers to Functional Machineries. Curr. Opin. Chem. Biol. 2004, 8, 519–526. 10.1016/j.cbpa.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Loo J. A. Studying Noncovalent Protein Complexes by Electrospray Ionization Mass Spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. . [DOI] [PubMed] [Google Scholar]
- Heck A. J. Native Mass Spectrometry: A Bridge between Interactomics and Structural Biology. Nat. Methods 2008, 5, 927–933. 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Sharon M.; Robinson C. V. The Role of Mass Spectrometry in Structure Elucidation of Dynamic Protein Complexes. Annu. Rev. Biochem. 2007, 76, 167–193. 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- Sharon M.; Witt S.; Glasmacher E.; Baumeister W.; Robinson C. V. Mass Spectrometry Reveals the Missing Links in the Assembly Pathway of the Bacterial 20 S Proteasome. J. Biol. Chem. 2007, 282, 18448–18457. 10.1074/jbc.M701534200. [DOI] [PubMed] [Google Scholar]
- Bakhtiari M.; Konermann L. Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures. J. Phys. Chem. B 2019, 123, 1784–1796. 10.1021/acs.jpcb.8b12173. [DOI] [PubMed] [Google Scholar]
- Robinson C. V. Mass Spectrometry: From Plasma Proteins to Mitochondrial Membranes. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 2814–2820. 10.1073/pnas.1820450116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J.; Donlan J. A. C.; Liko I.; Hopper J. T. S.; Gupta K.; Housden N. G.; Struwe W. B.; Marty M. T.; Mize T.; Bechara C.; et al. High-Resolution Mass Spectrometry of Small Molecules Bound to Membrane Proteins. Nat. Methods 2016, 13, 333–336. 10.1038/nmeth.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebarle P.; Verkerk U. H. Electrospray: From Ions in Solution to Ions in the Gas Phase, What We Know Now. Mass Spectrom. Rev. 2009, 28, 898–917. 10.1002/mas.20247. [DOI] [PubMed] [Google Scholar]
- Wilm M. Principles of Electrospray Ionization. Mol. Cell. Proteomics 2011, 10, M111.009407. 10.1074/mcp.M111.009407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Mazumdar S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information Beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012, 2012, 282574. 10.1155/2012/282574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Ahadi E.; Rodriguez A. D.; Vahidi S. Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85, 2–9. 10.1021/ac302789c. [DOI] [PubMed] [Google Scholar]
- Garza K. Y.; Feider C. L.; Klein D. R.; Rosenberg J. A.; Brodbelt J. S.; Eberlin L. S. Desorption Electrospray Ionization Mass Spectrometry Imaging of Proteins Directly from Biological Tissue Sections. Anal. Chem. 2018, 90, 7785–7789. 10.1021/acs.analchem.8b00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R. J.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. High-Sensitivity Orbitrap Mass Analysis of Intact Macromolecular Assemblies. Nat. Methods 2012, 9, 1084–1086. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- Li H.; Nguyen H. H.; Ogorzalek Loo R. R.; Campuzano I. D. G.; Loo J. A. An Integrated Native Mass Spectrometry and Top-Down Proteomics Method That Connects Sequence to Structure and Function of Macromolecular Complexes. Nat. Chem. 2018, 10, 139–148. 10.1038/nchem.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Wen J.; Blankenship R. E.; Gross M. L. Native Electrospray and Electron-Capture Dissociation Fticr Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal. Chem. 2011, 83, 5598–5606. 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort K. L.; van de Waterbeemd M.; Boll D.; Reinhardt-Szyba M.; Belov M. E.; Sasaki E.; Zschoche R.; Hilvert D.; Makarov A. A.; Heck A. J. R. Expanding the Structural Analysis Capabilities on an Orbitrap-Based Mass Spectrometer for Large Macromolecular Complexes. Analyst 2018, 143, 100–105. 10.1039/C7AN01629H. [DOI] [PubMed] [Google Scholar]
- Rosati S.; Rose R. J.; Thompson N. J.; van Duijn E.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. Exploring an Orbitrap Analyzer for the Characterization of Intact Antibodies by Native Mass Spectrometry. Angew. Chem., Int. Ed. 2012, 51, 12992–12996. 10.1002/anie.201206745. [DOI] [PubMed] [Google Scholar]
- Lössl P.; Snijder J.; Heck A. J. R. Boundaries of Mass Resolution in Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25, 906–917. 10.1007/s13361-014-0874-3. [DOI] [PubMed] [Google Scholar]
- Rathore D.; Faustino A.; Schiel J.; Pang E.; Boyne M.; Rogstad S. The Role of Mass Spectrometry in the Characterization of Biologic Protein Products. Expert Rev. Proteomics 2018, 15, 431–449. 10.1080/14789450.2018.1469982. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Rose R. J.; Veesler D.; Johnson J. E.; Heck A. J. R. Studying 18 Mda Virus Assemblies with Native Mass Spectrometry. Angew. Chem., Int. Ed. 2013, 52, 4020–4023. 10.1002/anie.201210197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught A. D.; Wilkinson A.. Compendium of Chemical Terminology: IUPAC Recommendations (The “Gold Book”), 2nd ed.; Blackwell Scientific Publications: Oxford, U.K., 1997. [Google Scholar]
- Murray K. K.; Boyd R. K.; Eberlin M. N.; Langley G. J.; Li L.; Naito Y. Definitions of Terms Relating to Mass Spectrometry (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1515–1609. 10.1351/PAC-REC-06-04-06. [DOI] [Google Scholar]
- Xian F.; Hendrickson C. L.; Marshall A. G. High Resolution Mass Spectrometry. Anal. Chem. 2012, 84, 708–719. 10.1021/ac203191t. [DOI] [PubMed] [Google Scholar]
- Marshall A. G.; Hendrickson C. L.; Jackson G. S. Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: A Primer. Mass Spectrom. Rev. 1998, 17, 1–35. . [DOI] [PubMed] [Google Scholar]
- Pelander A.; Decker P.; Baessmann C.; Ojanperä I. Evaluation of a High Resolving Power Time-of-Flight Mass Spectrometer for Drug Analysis in Terms of Resolving Power and Acquisition Rate. J. Am. Soc. Mass Spectrom. 2011, 22, 379–385. 10.1007/s13361-010-0046-z. [DOI] [PubMed] [Google Scholar]
- Tsybin Y. O.; Nagornov K. O.; Kozhinov A. N.. Chapter 5 - Advanced Fundamentals in Fourier Transform Mass Spectrometry. In Fundamentals and Applications of Fourier Transform Mass Spectrometry; Elsevier, 2019; pp 113–132. 10.1016/B978-0-12-814013-0.00005-3. [DOI] [Google Scholar]
- Makarov A.; Denisov E.; Kholomeev A.; Balschun W.; Lange O.; Strupat K.; Horning S. Performance Evaluation of a Hybrid Linear Ion Trap/Orbitrap Mass Spectrometer. Anal. Chem. 2006, 78, 2113–2120. 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- Guilhaus M.; Selby D.; Mlynski V. Orthogonal Acceleration Time-of-Flight Mass Spectrometry. Mass Spectrom. Rev. 2000, 19, 65–107. . [DOI] [PubMed] [Google Scholar]
- Bowman A. P.; Blakney G. T.; Hendrickson C. L.; Ellis S. R.; Heeren R. M. A.; Smith D. F. Ultra-High Mass Resolving Power, Mass Accuracy, and Dynamic Range MALDI Mass Spectrometry Imaging by 21-T FT-ICR MS. Anal. Chem. 2020, 92, 3133–3142. 10.1021/acs.analchem.9b04768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuth C.; Vogel A. L.; Ockenfeld S.; Huesmann R.; Hoffmann T. Ultrahigh-Resolution Mass Spectrometry in Real Time: Atmospheric Pressure Chemical Ionization Orbitrap Mass Spectrometry of Atmospheric Organic Aerosol. Anal. Chem. 2018, 90, 8816–8823. 10.1021/acs.analchem.8b00671. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; de Godoy L. M. F.; Li G.; Macek B.; Mortensen P.; Pesch R.; Makarov A.; Lange O.; Horning S.; Mann M. Parts Per Million Mass Accuracy on an Orbitrap Mass Spectrometer via Lock Mass Injection into a C-Trap. Mol. Cell. Proteomics 2005, 4, 2010–2021. 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Marshall A. G.; Rodgers R. P. Petroleomics: Chemistry of the Underworld. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18090–18095. 10.1073/pnas.0805069105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. D.; Anderson G. A.; Lipton M. S.; Pasa-Tolic L.; Shen Y.; Conrads T. P.; Veenstra T. D.; Udseth H. R. An Accurate Mass Tag Strategy for Quantitative and High-Throughput Proteome Measurements. Proteomics 2002, 2, 513–523. . [DOI] [PubMed] [Google Scholar]
- Lebedev A. T. Environmental Mass Spectrometry. Annu. Rev. Anal. Chem. 2013, 6, 163–189. 10.1146/annurev-anchem-062012-092604. [DOI] [PubMed] [Google Scholar]
- Orr A.; Stotesbury T.; Wilson P.; Stock N. L. The Use of High-Resolution Mass Spectrometry (Hrms) for the Analysis of DNA and Other Macromolecules: A How-to Guide for Forensic Chemistry. Forensic Chemistry 2019, 14, 100169. 10.1016/j.forc.2019.100169. [DOI] [Google Scholar]
- Selliez L.; Maillard J.; Cherville B.; Gautier T.; Thirkell L.; Gaubicher B.; Schmitz-Afonso I.; Afonso C.; Briois C.; Carrasco N. High-Resolution Mass Spectrometry for Future Space Missions: Comparative Analysis of Complex Organic Matter with Lab-Cosmorbitrap and Laser Desorption/Ionization Fourier Transform Ion Cyclotron Resonance. Rapid Commun. Mass Spectrom. 2020, 34, e8645. 10.1002/rcm.8645. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Franc V.; Heck A. J. R. Glycoproteomics: A Balance between High-Throughput and in-Depth Analysis. Trends Biotechnol. 2017, 35, 598–609. 10.1016/j.tibtech.2017.04.010. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Tamara S.; Fort K. L.; Damoc E.; Franc V.; Bieri P.; Itten M.; Makarov A.; Ban N.; Heck A. J. R. Dissecting Ribosomal Particles Throughout the Kingdoms of Life Using Advanced Hybrid Mass Spectrometry Methods. Nat. Commun. 2018, 9, 2493. 10.1038/s41467-018-04853-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafader J. O.; Melani R. D.; Schachner L. F.; Ives A. N.; Patrie S. M.; Kelleher N. L.; Compton P. D. Native vs Denatured: An in Depth Investigation of Charge State and Isotope Distributions. J. Am. Soc. Mass Spectrom. 2020, 31, 574–581. 10.1021/jasms.9b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenton A. G.; Godfrey A. R. Accurate Mass Measurement: Terminology and Treatment of Data. J. Am. Soc. Mass Spectrom. 2010, 21, 1821–1835. 10.1016/j.jasms.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Gross M. L. Accurate Masses for Structure Confirmation. J. Am. Soc. Mass Spectrom. 1994, 5, 57. 10.1016/1044-0305(94)85036-4. [DOI] [PubMed] [Google Scholar]
- Quadrupole Mass Spectrometry and Its Applications; Dawson P. H., Ed.; Elsevier, 1976. [Google Scholar]
- Savory J. J.; Kaiser N. K.; Mckenna A. M.; Xian F.; Blakney G. T.; Rodgers R. P.; Hendrickson C. L.; Marshall A. G. Parts-Per-Billion Fourier Transform Ion Cyclotron Resonance Mass Measurement Accuracy with a “Walking” Calibration Equation. Anal. Chem. 2011, 83, 1732–1736. 10.1021/ac102943z. [DOI] [PubMed] [Google Scholar]
- Senko M. W.; Beu S. C.; McLafferty F. W. Determination of Monoisotopic Masses and Ion Populations for Large Biomolecules from Resolved Isotopic Distributions. J. Am. Soc. Mass Spectrom. 1995, 6, 229–233. 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- Hernández H.; Robinson C. V. Determining the Stoichiometry and Interactions of Macromolecular Assemblies from Mass Spectrometry. Nat. Protoc. 2007, 2, 715–726. 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Gorshkov M. V.; Good D. M.; Lyutvinskiy Y.; Yang H.; Zubarev R. A. Calibration Function for the Orbitrap FTMS Accounting for the Space Charge Effect. J. Am. Soc. Mass Spectrom. 2010, 21, 1846–1851. 10.1016/j.jasms.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Makarov A.; Grinfeld D.; Ayzikov K.. Chapter 2 - Fundamentals of Orbitrap Analyzer. In Fundamentals and Applications of Fourier Transform Mass Spectrometry; Elsevier, 2019; pp 37–61. 10.1016/B978-0-12-814013-0.00002-8. [DOI] [Google Scholar]
- Chernushevich I. V.; Thomson B. A. Collisional Cooling of Large Ions in Electrospray Mass Spectrometry. Anal. Chem. 2004, 76, 1754–1760. 10.1021/ac035406j. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Heck A. J. R. Analytical Approaches for Size and Mass Analysis of Large Protein Assemblies. Annu. Rev. Anal. Chem. 2014, 7, 43–64. 10.1146/annurev-anchem-071213-020015. [DOI] [PubMed] [Google Scholar]
- Schachner L. F.; Ives A. N.; McGee J. P.; Melani R. D.; Kafader J. O.; Compton P. D.; Patrie S. M.; Kelleher N. L. Standard Proteoforms and Their Complexes for Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 1190–1198. 10.1007/s13361-019-02191-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Fort K. L.; Boll D.; Reinhardt-Szyba M.; Routh A.; Makarov A.; Heck A. J. R. High-Fidelity Mass Analysis Unveils Heterogeneity in Intact Ribosomal Particles. Nat. Methods 2017, 14, 283. 10.1038/nmeth.4147. [DOI] [PubMed] [Google Scholar]
- Shinholt D. L.; Anthony S. N.; Alexander A. W.; Draper B. E.; Jarrold M. F. A Frequency and Amplitude Scanned Quadrupole Mass Filter for the Analysis of High M/Z Ions. Rev. Sci. Instrum. 2014, 85, 113109. 10.1063/1.4900627. [DOI] [PubMed] [Google Scholar]
- Mamyrin B. A. Time-of-Flight Mass Spectrometry (Concepts, Achievements, and Prospects). Int. J. Mass Spectrom. 2001, 206, 251–266. 10.1016/S1387-3806(00)00392-4. [DOI] [Google Scholar]
- Ben-Nissan G.; Belov M. E.; Morgenstern D.; Levin Y.; Dym O.; Arkind G.; Lipson C.; Makarov A. A.; Sharon M. Triple-Stage Mass Spectrometry Unravels the Heterogeneity of an Endogenous Protein Complex. Anal. Chem. 2017, 89, 4708–4715. 10.1021/acs.analchem.7b00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano I. D. G.; Nshanian M.; Spahr C.; Lantz C.; Netirojjanakul C.; Li H.; Wongkongkathep P.; Wolff J. J.; Loo J. A. High Mass Analysis with a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: From Inorganic Salt Clusters to Antibody Conjugates and Beyond. J. Am. Soc. Mass Spectrom. 2020, 31, 1155–1162. 10.1021/jasms.0c00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Lin T.-Y.; Leach F. E.; Tolmachev A. V.; Tolić N.; Robinson E. W.; Koppenaal D. W.; Paša-Tolić L. 21 T Fourier Transform Ion Cyclotron Resonance Mass Spectrometer Greatly Expands Mass Spectrometry Toolbox. J. Am. Soc. Mass Spectrom. 2016, 27, 1929–1936. 10.1007/s13361-016-1507-9. [DOI] [PubMed] [Google Scholar]
- Mallis C. S.; Zheng X.; Qiu X.; McCabe J. W.; Shirzadeh M.; Lyu J.; Laganowsky A.; Russell D. H. Development of Native MS Capabilities on an Extended Mass Range Q-Tof MS. Int. J. Mass Spectrom. 2020, 458, 116451. 10.1016/j.ijms.2020.116451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach T.; Kemper P. R.; Baykut G.; Park M. A.; Bowers M. T. A New Instrument with High Mass and High Ion Mobility Resolution. Int. J. Mass Spectrom. 2018, 434, 108–115. 10.1016/j.ijms.2018.09.008. [DOI] [Google Scholar]
- Gault J.; Liko I.; Landreh M.; Shutin D.; Bolla J. R.; Jefferies D.; Agasid M.; Yen H.-Y.; Ladds M. J. G. W.; Lane D. P.; et al. Combining Native and ‘Omics’ Mass Spectrometry to Identify Endogenous Ligands Bound to Membrane Proteins. Nat. Methods 2020, 17, 505–508. 10.1038/s41592-020-0821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R.; Takata T.; Fujii N.; Ishii K.; Uchiyama S.; Sato N.; Oba Y.; Wood K.; Kato K.; Fujii N.; et al. New Insight into the Dynamical System of Ab-Crystallin Oligomers. Sci. Rep. 2016, 6, 29208. 10.1038/srep29208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippens J. L.; Nshanian M.; Spahr C.; Egea P. F.; Loo J. A.; Campuzano I. D. G. Fourier Transform-Ion Cyclotron Resonance Mass Spectrometry as a Platform for Characterizing Multimeric Membrane Protein Complexes. J. Am. Soc. Mass Spectrom. 2018, 29, 183–193. 10.1007/s13361-017-1799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Hopper J. T.; Robinson C. V. Mass Spectrometry of Intact Membrane Protein Complexes. Nat. Protoc. 2013, 8, 639–651. 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron A. E.; Eggers D. F. An Ion “Velocitron”. Rev. Sci. Instrum. 1948, 19, 605–607. 10.1063/1.1741336. [DOI] [Google Scholar]
- Mamyrin B. A.; Karataev V. I.; Shmikk D. V.; Zagulin V. A. The Mass-Reflectron. A New Nonmagnetic Time-of-Flight High Resolution Mass-Spectrometer. Zh. Ehksp. Teor. Fiz. 1973, 64, 82–89. [Google Scholar]
- Mirgorodskaya O. A.; Shevchenko A. A.; Chernushevich I. V.; Dodonov A. F.; Miroshnikov A. I. Electrospray-Ionization Time-of-Flight Mass Spectrometry in Protein Chemistry. Anal. Chem. 1994, 66, 99–107. 10.1021/ac00073a018. [DOI] [Google Scholar]
- Dawson J. H. J.; Guilhaus M. Orthogonal-Acceleration Time-of-Flight Mass Spectrometer. Rapid Commun. Mass Spectrom. 1989, 3, 155–159. 10.1002/rcm.1290030511. [DOI] [Google Scholar]
- Krutchinsky A. N.; Zhang W.; Chait B. T. Rapidly Switchable Matrix-Assisted Laser Desorption/Ionization and Electrospray Quadrupole-Time-of-Flight Mass Spectrometry for Protein Identification. J. Am. Soc. Mass Spectrom. 2000, 11, 493–504. 10.1016/S1044-0305(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Morris H. R.; Paxton T.; Dell A.; Langhorne J.; Berg M.; Bordoli R. S.; Hoyes J.; Bateman R. H. High Sensitivity Collisionally-Activated Decomposition Tandem Mass Spectrometry on a Novel Quadrupole/Orthogonal-Acceleration Time-of-Flight Mass Spectrometer. Rapid Commun. Mass Spectrom. 1996, 10, 889–896. . [DOI] [PubMed] [Google Scholar]
- Sobott F.; Hernández H.; Mccammon M. G.; Tito M. A.; Robinson C. V. A Tandem Mass Spectrometer for Improved Transmission and Analysis of Large Macromolecular Assemblies. Anal. Chem. 2002, 74, 1402–1407. 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- Tahallah N.; Pinkse M.; Maier C. S.; Heck A. J. R. The Effect of the Source Pressure on the Abundance of Ions of Noncovalent Protein Assemblies in an Electrospray Ionization Orthogonal Time-of-Flight Instrument. Rapid Commun. Mass Spectrom. 2001, 15, 596–601. 10.1002/rcm.275. [DOI] [PubMed] [Google Scholar]
- Lorenzen K.; Versluis C.; Van Duijn E.; Van Den Heuvel R. H. H.; Heck A. J. R. Optimizing Macromolecular Tandem Mass Spectrometry of Large Non-Covalent Complexes Using Heavy Collision Gases. Int. J. Mass Spectrom. 2007, 268, 198–206. 10.1016/j.ijms.2007.06.012. [DOI] [Google Scholar]
- Van Den Heuvel R. H. H.; Van Duijn E.; Mazon H.; Synowsky S. A.; Lorenzen K.; Versluis C.; Brouns S. J. J.; Langridge D.; Van Der Oost J.; Hoyes J.; et al. Improving the Performance of a Quadrupole Time-of-Flight Instrument for Macromolecular Mass Spectrometry. Anal. Chem. 2006, 78, 7473–7483. 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Versluis C.; Watts N. R.; Roos W. H.; Wuite G. J. L.; Wingfield P. T.; Steven A. C.; Heck A. J. R. High-Resolution Mass Spectrometry of Viral Assemblies: Molecular Composition and Stability of Dimorphic Hepatitis B Virus Capsids. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 9216–9220. 10.1073/pnas.0800406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C.; Watts N. R.; Stahl S. J.; Wingfield P. T.; Steven A. C.; Heck A. J. R. Subunit Exchange Rates in Hepatitis B Virus Capsids Are Geometry- and Temperature-Dependent. Phys. Chem. Chem. Phys. 2010, 12, 13368. 10.1039/c0cp00692k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker G. K.; Van Duijn E.; Crawford S. E.; Uetrecht C.; Baclayon M.; Roos W. H.; Wuite G. J. L.; Estes M. K.; Prasad B. V. V.; Heck A. J. R. Norwalk Virus Assembly and Stability Monitored by Mass Spectrometry. Mol. Cell. Proteomics 2010, 9, 1742–1751. 10.1074/mcp.M900620-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera N. P.; Robinson C. V. Advances in the Mass Spectrometry of Membrane Proteins: From Individual Proteins to Intact Complexes. Annu. Rev. Biochem. 2011, 80, 247–271. 10.1146/annurev-biochem-062309-093307. [DOI] [PubMed] [Google Scholar]
- Barrera N. P.; Isaacson S. C.; Zhou M.; Bavro V. N.; Welch A.; Schaedler T. A.; Seeger M. A.; Miguel R. N.; Korkhov V. M.; Van Veen H. W.; et al. Mass Spectrometry of Membrane Transporters Reveals Subunit Stoichiometry and Interactions. Nat. Methods 2009, 6, 585–587. 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer P.; Schweikhard L. Multiple-Ion-Ejection Multi-Reflection Time-of-Flight Mass Spectrometry for Single-Reference Mass Measurements with Lapping Ion Species. Rev. Sci. Instrum. 2020, 91, 023201. 10.1063/1.5131582. [DOI] [PubMed] [Google Scholar]
- Knauer S.; Fischer P.; Marx G.; Müller M.; Rosenbusch M.; Schabinger B.; Schweikhard L.; Wolf R. N. A Multi-Reflection Time-of-Flight Setup for the Improvement and Development of New Methods and the Study of Atomic Clusters. Int. J. Mass Spectrom. 2019, 446, 116189. 10.1016/j.ijms.2019.116189. [DOI] [Google Scholar]
- Dziekonski E. T.; Johnson J. T.; Lee K. W.; McLuckey S. A. Fourier-Transform MS and Closed-Path Multireflection Time-of-Flight MS Using an Electrostatic Linear Ion Trap. Anal. Chem. 2017, 89, 10965–10972. 10.1021/acs.analchem.7b02797. [DOI] [PubMed] [Google Scholar]
- Chernushevich I. V.; Merenbloom S. I.; Liu S.; Bloomfield N. A W-Geometry Ortho-Tof MS with High Resolution and up to 100% Duty Cycle for MS/MS. J. Am. Soc. Mass Spectrom. 2017, 28, 2143–2150. 10.1007/s13361-017-1742-8. [DOI] [PubMed] [Google Scholar]
- Richardson K.; Hoyes J. A Novel Multipass Oa-Tof Mass Spectrometer. Int. J. Mass Spectrom. 2015, 377, 309–315. 10.1016/j.ijms.2014.08.031. [DOI] [Google Scholar]
- Comisarow M. B.; Marshall A. G. Fourier Transform Ion Cyclotron Resonance Spectroscopy. Chem. Phys. Lett. 1974, 25, 282–283. 10.1016/0009-2614(74)89137-2. [DOI] [Google Scholar]
- Hendrickson C. L.; Quinn J. P.; Kaiser N. K.; Smith D. F.; Blakney G. T.; Chen T.; Marshall A. G.; Weisbrod C. R.; Beu S. C. 21 T Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: A National Resource for Ultrahigh Resolution Mass Analysis. J. Am. Soc. Mass Spectrom. 2015, 26, 1626–1632. 10.1007/s13361-015-1182-2. [DOI] [PubMed] [Google Scholar]
- Li H.; Wolff J. J.; Van Orden S. L.; Loo J. A. Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 Kda Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2014, 86, 317–320. 10.1021/ac4033214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comisarow M. B.; Marshall A. G. Theory of Fourier Transform Ion Cyclotron Resonance Mass Spectroscopy. I. Fundamental Equations and Low-Pressure Line Shape. J. Chem. Phys. 1976, 64, 110–119. 10.1063/1.431959. [DOI] [Google Scholar]
- Bartholdi E.; Ernst R. R. Fourier Spectroscopy and the Causality Principle. J. Magn. Reson. 1973, 11, 9–19. 10.1016/0022-2364(73)90076-0. [DOI] [Google Scholar]
- Marshall A. G.; Hendrickson C. L. High-Resolution Mass Spectrometers. Annu. Rev. Anal. Chem. 2008, 1, 579–599. 10.1146/annurev.anchem.1.031207.112945. [DOI] [PubMed] [Google Scholar]
- Senko M. W.; Hendrickson C. L.; Emmett M. R.; Shi S. D. H.; Marshall A. G. External Accumulation of Ions for Enhanced Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1997, 8, 970–976. 10.1016/S1044-0305(97)00126-8. [DOI] [Google Scholar]
- Marshall A. G.; Guan S. Advantages of High Magnetic Field for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1819–1823. . [DOI] [PubMed] [Google Scholar]
- Marty M. T.; Zhang H.; Cui W.; Blankenship R. E.; Gross M. L.; Sligar S. G. Native Mass Spectrometry Characterization of Intact Nanodisc Lipoprotein Complexes. Anal. Chem. 2012, 84, 8957–8960. 10.1021/ac302663f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Wen J.; Blankenship R. E.; Gross M. L. Native Electrospray and Electron-Capture Dissociation in Fticr Mass Spectrometry Provide Top-Down Sequencing of a Protein Component in an Intact Protein Assembly. J. Am. Soc. Mass Spectrom. 2010, 21, 1966–1968. 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano I. D. G.; Netirojjanakul C.; Nshanian M.; Lippens J. L.; Kilgour D. P. A.; Van Orden S.; Loo J. A. Native-MS Analysis of Monoclonal Antibody Conjugates by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2018, 90, 745–751. 10.1021/acs.analchem.7b03021. [DOI] [PubMed] [Google Scholar]
- Wörner T. P.; Snijder J.; Bennett A.; Agbandje-McKenna M.; Makarov A. A.; Heck A. J. R. Resolving Heterogeneous Macromolecular Assemblies by Orbitrap-Based Single-Particle Charge Detection Mass Spectrometry. Nat. Methods 2020, 17, 395–398. 10.1038/s41592-020-0770-7. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Makarov A. Orbitrap Mass Spectrometry. Anal. Chem. 2013, 85, 5288–5296. 10.1021/ac4001223. [DOI] [PubMed] [Google Scholar]
- Makarov A. Electrostatic Axially Harmonic Orbital Trapping: A High-Performance Technique of Mass Analysis. Anal. Chem. 2000, 72, 1156–1162. 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- Kingdon K. H. A Method for the Neutralization of Electron Space Charge by Positive Ionization at Very Low Gas Pressures. Phys. Rev. 1923, 21, 408–418. 10.1103/PhysRev.21.408. [DOI] [Google Scholar]
- Hu Q.; Cooks R. G.; Noll R. J. Phase-Enhanced Selective Ion Ejection in an Orbitrap Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2007, 18, 980–983. 10.1016/j.jasms.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Makarov A. A.; Cooks R. G.; Noll R. J. Resonant Ac Dipolar Excitation for Ion Motion Control in the Orbitrap Mass Analyzer. J. Phys. Chem. A 2006, 110, 2682–2689. 10.1021/jp054485v. [DOI] [PubMed] [Google Scholar]
- Lange O.; Damoc E.; Wieghaus A.; Makarov A. Enhanced Fourier Transform for Orbitrap Mass Spectrometry. Int. J. Mass Spectrom. 2014, 369, 16–22. 10.1016/j.ijms.2014.05.019. [DOI] [Google Scholar]
- Makarov A.; Denisov E.; Lange O. Performance Evaluation of a High-Field Orbitrap Mass Analyzer. J. Am. Soc. Mass Spectrom. 2009, 20, 1391–1396. 10.1016/j.jasms.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Snijder J.; van de Waterbeemd M.; Damoc E.; Denisov E.; Grinfeld D.; Bennett A.; Agbandje-McKenna M.; Makarov A.; Heck A. J. R. Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry. J. Am. Chem. Soc. 2014, 136, 7295–7299. 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati S.; van den Bremer E. T. J.; Schuurman J.; Parren P. W. H. I.; Kamerling J. P.; Heck A. J. R. In-Depth Qualitative and Quantitative Analysis of Composite Glycosylation Profiles and Other Micro-Heterogeneity on Intact Monoclonal Antibodies by High-Resolution Native Mass Spectrometry Using a Modified Orbitrap. MAbs 2013, 5, 917–924. 10.4161/mabs.26282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov M. E.; Damoc E.; Denisov E.; Compton P. D.; Horning S.; Makarov A. A.; Kelleher N. L. From Protein Complexes to Subunit Backbone Fragments: A Multi-Stage Approach to Native Mass Spectrometry. Anal. Chem. 2013, 85, 11163–11173. 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- Dyachenko A.; Wang G.; Belov M.; Makarov A.; de Jong R. N.; van den Bremer E. T. J.; Parren P. W. H. I.; Heck A. J. R. Tandem Native Mass-Spectrometry on Antibody–Drug Conjugates and Submillion Da Antibody–Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Anal. Chem. 2015, 87, 6095–6102. 10.1021/acs.analchem.5b00788. [DOI] [PubMed] [Google Scholar]
- Campuzano I. D. G.; Li H.; Bagal D.; Lippens J. L.; Svitel J.; Kurzeja R. J. M.; Xu H.; Schnier P. D.; Loo J. A. Native MS Analysis of Bacteriorhodopsin and an Empty Nanodisc by Orthogonal Acceleration Time-of-Flight, Orbitrap and Ion Cyclotron Resonance. Anal. Chem. 2016, 88, 12427–12436. 10.1021/acs.analchem.6b03762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder D. T.; Panczyk E. M.; Somogyi A.; Kaplan D. A.; Wysocki V. Simple and Minimally Invasive Sid Devices for Native Mass Spectrometry. Anal. Chem. 2020, 92, 11195–11203. 10.1021/acs.analchem.0c01657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehaffey M. R.; Sanders J. D.; Holden D. D.; Nilsson C. L.; Brodbelt J. S. Multistage Ultraviolet Photodissociation Mass Spectrometry to Characterize Single Amino Acid Variants of Human Mitochondrial BCAT2. Anal. Chem. 2018, 90, 9904–9911. 10.1021/acs.analchem.8b02099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greisch J.-F.; Tamara S.; Scheltema R. A.; Maxwell H. W. R.; Fagerlund R. D.; Fineran P. C.; Tetter S.; Hilvert D.; Heck A. J. R. Expanding the Mass Range for UVPD-Based Native Top-Down Mass Spectrometry. Chem. Sci. 2019, 10, 7163–7171. 10.1039/C9SC01857C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltash M. L.; McCabe J. W.; Shirzadeh M.; Laganowsky A.; Russell D. H. Native IM-Orbitrap MS: Resolving What Was Hidden. TrAC, Trends Anal. Chem. 2020, 124, 115533. 10.1016/j.trac.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltash M. L.; McCabe J. W.; Shirzadeh M.; Laganowsky A.; Clowers B. H.; Russell D. H. Fourier Transform-Ion Mobility-Orbitrap Mass Spectrometer: A Next-Generation Instrument for Native Mass Spectrometry. Anal. Chem. 2018, 90, 10472–10478. 10.1021/acs.analchem.8b02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafader J. O.; Melani R. D.; Senko M. W.; Makarov A. A.; Kelleher N. L.; Compton P. D. Measurement of Individual Ions Sharply Increases the Resolution of Orbitrap Mass Spectra of Proteins. Anal. Chem. 2019, 91, 2776–2783. 10.1021/acs.analchem.8b04519. [DOI] [PubMed] [Google Scholar]
- Keifer D. Z.; Jarrold M. F. Single-Molecule Mass Spectrometry. Mass Spectrom. Rev. 2017, 36, 715–733. 10.1002/mas.21495. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Barrow M. P.; Li H.; Meier J. E.; Van Orden S. L.; Thompson C. J.; O’Connor P. B. Absorption-Mode: The Next Generation of Fourier Transform Mass Spectra. Anal. Chem. 2012, 84, 2923–2929. 10.1021/ac3000122. [DOI] [PubMed] [Google Scholar]
- Grinfeld D.; Aizikov K.; Kreutzmann A.; Damoc E.; Makarov A. Phase-Constrained Spectrum Deconvolution for Fourier Transform Mass Spectrometry. Anal. Chem. 2017, 89, 1202–1211. 10.1021/acs.analchem.6b03636. [DOI] [PubMed] [Google Scholar]
- Kelstrup C. D.; Aizikov K.; Batth T. S.; Kreutzman A.; Grinfeld D.; Lange O.; Mourad D.; Makarov A. A.; Olsen J. V. Limits for Resolving Isobaric Tandem Mass Tag Reporter Ions Using Phase-Constrained Spectrum Deconvolution. J. Proteome Res. 2018, 17, 4008–4016. 10.1021/acs.jproteome.8b00381. [DOI] [PubMed] [Google Scholar]
- Allison T. M.; Landreh M. Ion Mobility in Structural Biology. Compr. Anal. Chem. 2019, 83, 161–195. 10.1016/bs.coac.2018.10.001. [DOI] [Google Scholar]
- Göth M.; Pagel K. Ion Mobility-Mass Spectrometry as a Tool to Investigate Protein-Ligand Interactions. Anal. Bioanal. Chem. 2017, 409, 4305–4310. 10.1007/s00216-017-0384-9. [DOI] [PubMed] [Google Scholar]
- López A.; Tarragó T.; Vilaseca M.; Giralt E. Applications and Future of Ion Mobility Mass Spectrometry in Structural Biology. New J. Chem. 2013, 37, 1283–1289. 10.1039/c3nj41051j. [DOI] [Google Scholar]
- Österlund N.; Moons R.; Ilag L. L.; Sobott F.; Graslund A. Native Ion Mobility-Mass Spectrometry Reveals the Formation of B-Barrel Shaped Amyloid-B Hexamers in a Membrane-Mimicking Environment. J. Am. Chem. Soc. 2019, 141, 10440–10450. 10.1021/jacs.9b04596. [DOI] [PubMed] [Google Scholar]
- May J. C.; Jurneczko E.; Stow S. M.; Kratochvil I.; Kalkhof S.; McLean J. A. Conformational Landscapes of Ubiquitin, Cytochrome C, and Myoglobin: Uniform Field Ion Mobility Measurements in Helium and Nitrogen Drift Gas. Int. J. Mass Spectrom. 2018, 427, 79–90. 10.1016/j.ijms.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Atri V.; Gabelica V. DNA and RNA Telomeric G-Quadruplexes: What Topology Features Can Be Inferred from Ion Mobility Mass Spectrometry?. Analyst 2019, 144, 6074–6088. 10.1039/C9AN01216H. [DOI] [PubMed] [Google Scholar]
- Hernandez-Alba O.; Wagner-Rousset E.; Beck A.; Cianférani S. Native Mass Spectrometry, Ion Mobility, and Collision-Induced Unfolding for Conformational Characterization of IgG4 Monoclonal Antibodies. Anal. Chem. 2018, 90, 8865–8872. 10.1021/acs.analchem.8b00912. [DOI] [PubMed] [Google Scholar]
- Rabuck-Gibbons J. N.; Lodge J. M.; Mapp A. K.; Ruotolo B. T. Collision-Induced Unfolding Reveals Unique Fingerprints for Remote Protein Interaction Sites in the Kix Regulation Domain. J. Am. Soc. Mass Spectrom. 2019, 30, 94–102. 10.1007/s13361-018-2043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Vasiljevic S.; Allen J. D.; Seabright G. E.; Duyvesteyn H. M. E.; Doores K. J.; Crispin M.; Struwe W. B. Signature of Antibody Domain Exchange by Native Mass Spectrometry and Collision-Induced Unfolding. Anal. Chem. 2018, 90, 7325–7331. 10.1021/acs.analchem.8b00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew A.; Buijs R.; Eijkel G. B.; Giskes F.; Dyachenko A.; Van Der Horst J.; Byelov D.; Spaanderman D.-J.; Heck A. J. R.; Porta Siegel T.; et al. Ion Imaging of Native Protein Complexes Using Orthogonal Time-of-Flight Mass Spectrometry and a Timepix Detector. J. Am. Soc. Mass Spectrom. 2021, 32, 569–580. 10.1021/jasms.0c00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tito M. A.; Tars K.; Valegard K.; Hajdu J.; Robinson C. V. Electrospray Time-of-Flight Mass Spectrometry of the Intact MS2 Virus Capsid. J. Am. Chem. Soc. 2000, 122, 3550–3551. 10.1021/ja993740k. [DOI] [Google Scholar]
- Makarov A.; Denisov E. Dynamics of Ions of Intact Proteins in the Orbitrap Mass Analyzer. J. Am. Soc. Mass Spectrom. 2009, 20, 1486–1495. 10.1016/j.jasms.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Makarov A. A.Method of Generating a Mass Spectrum Having Improved Resolving Power. US 9043164 B2, May 26, 2015.
- Smith R. D.; Cheng X.; Brace J. E.; Hofstadler S. A.; Anderson G. A. Trapping, Detection and Reaction of Very Large Single Molecular Ions by Mass Spectrometry. Nature 1994, 369, 137–139. 10.1038/369137a0. [DOI] [Google Scholar]
- Bruce J. E.; Cheng X.; Bakhtiar R.; Wu Q.; Hofstadler S. A.; Anderson G. A.; Smith R. D. Trapping, Detection, and Mass Measurement of Individual Ions in a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. J. Am. Chem. Soc. 1994, 116, 7839–7847. 10.1021/ja00096a046. [DOI] [Google Scholar]
- Chen R.; Cheng X.; Mitchell D. W.; Hofstadler S. A.; Wu Q.; Rockwood A. L.; Sherman M. G.; Smith R. D. Trapping, Detection, and Mass Determination of Coliphage T4 DNA Ions by Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 1995, 67, 1159–1163. 10.1021/ac00103a004. [DOI] [Google Scholar]
- Benner W. H. A Gated Electrostatic Ion Trap to Repetitiously Measure the Charge Andm/Zof Large Electrospray Ions. Anal. Chem. 1997, 69, 4162–4168. 10.1021/ac970163e. [DOI] [Google Scholar]
- Todd A. R.; Barnes L. F.; Young K.; Zlotnick A.; Jarrold M. F. Higher Resolution Charge Detection Mass Spectrometry. Anal. Chem. 2020, 92, 11357–11364. 10.1021/acs.analchem.0c02133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafader J. O.; Melani R. D.; Durbin K. R.; Ikwuagwu B.; Early B. P.; Fellers R. T.; Beu S. C.; Zabrouskov V.; Makarov A. A.; Maze J. T.; et al. Multiplexed Mass Spectrometry of Individual Ions Improves Measurement of Proteoforms and Their Complexes. Nat. Methods 2020, 17, 391–394. 10.1038/s41592-020-0764-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M.; Meng C. K.; Fenn J. B. Interpreting Mass Spectra of Multiply Charged Ions. Anal. Chem. 1989, 61, 1702–1708. 10.1021/ac00190a023. [DOI] [Google Scholar]
- Ferrige A. G.; Seddon M. J.; Green B. N.; Jarvis S. A.; Skilling J.; Staunton J. Disentangling Electrospray Spectra with Maximum Entropy. Rapid Commun. Mass Spectrom. 1992, 6, 707–711. 10.1002/rcm.1290061115. [DOI] [Google Scholar]
- Reinhold B. B.; Reinhold V. N. Electrospray Ionization Mass Spectrometry: Deconvolution by an Entropy-Based Algorithm. J. Am. Soc. Mass Spectrom. 1992, 3, 207–215. 10.1016/1044-0305(92)87004-I. [DOI] [PubMed] [Google Scholar]
- Tseng Y.-H.; Uetrecht C.; Heck A. J. R.; Peng W.-P. Interpreting the Charge State Assignment in Electrospray Mass Spectra of Bioparticles. Anal. Chem. 2011, 83, 1960–1968. 10.1021/ac102676z. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Ojha P. C.; McClean S.; Black N. D.; Hughes J. G.; Shaw C. Heuristic Charge Assignment for Deconvolution of Electrospray Ionization Mass Spectra. Rapid Commun. Mass Spectrom. 2003, 17, 429–436. 10.1002/rcm.927. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Marshall A. G. A Universal Algorithm for Fast and Automated Charge State Deconvolution of Electrospray Mass-to-Charge Ratio Spectra. J. Am. Soc. Mass Spectrom. 1998, 9, 225–233. 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- Stengel F.; Baldwin A. J.; Bush M. F.; Hilton G. R.; Lioe H.; Basha E.; Jaya N.; Vierling E.; Benesch J. L.P. Dissecting Heterogeneous Molecular Chaperone Complexes Using a Mass Spectrum Deconvolution Approach. Chem. Biol. 2012, 19, 599–607. 10.1016/j.chembiol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Breukelen B.; Barendregt A.; Heck A. J. R.; Van Den Heuvel R. H. H. Resolving Stoichiometries and Oligomeric States of Glutamate Synthase Protein Complexes with Curve Fitting and Simulation of Electrospray Mass Spectra. Rapid Commun. Mass Spectrom. 2006, 20, 2490–2496. 10.1002/rcm.2620. [DOI] [PubMed] [Google Scholar]
- Morgner N.; Robinson C. V. Massign: An Assignment Strategy for Maximizing Information from the Mass Spectra of Heterogeneous Protein Assemblies. Anal. Chem. 2012, 84, 2939–2948. 10.1021/ac300056a. [DOI] [PubMed] [Google Scholar]
- Tseng Y.-H.; Uetrecht C.; Yang S.-C.; Barendregt A.; Heck A. J. R.; Peng W.-P. Game-Theory-Based Search Engine to Automate the Mass Assignment in Complex Native Electrospray Mass Spectra. Anal. Chem. 2013, 85, 11275–11283. 10.1021/ac401940e. [DOI] [PubMed] [Google Scholar]
- Lu J.; Trnka M. J.; Roh S. H.; Robinson P. J. J.; Shiau C.; Fujimori D. G.; Chiu W.; Burlingame A. L.; Guan S. Improved Peak Detection and Deconvolution of Native Electrospray Mass Spectra from Large Protein Complexes. J. Am. Soc. Mass Spectrom. 2015, 26, 2141–2151. 10.1007/s13361-015-1235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty M. T.; Baldwin A. J.; Marklund E. G.; Hochberg G. K. A.; Benesch J. L. P.; Robinson C. V. Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles. Anal. Chem. 2015, 87, 4370–4376. 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid D. J.; Diesing J. M.; Miller M. A.; Perry S. M.; Wales J. A.; Montfort W. R.; Marty M. T. MetaUniDec: High-Throughput Deconvolution of Native Mass Spectra. J. Am. Soc. Mass Spectrom. 2019, 30, 118–127. 10.1007/s13361-018-1951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty M. T. Eliminating Artifacts in Electrospray Deconvolution with a SoftMax Function. J. Am. Soc. Mass Spectrom. 2019, 30, 2174–2177. 10.1007/s13361-019-02286-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty M. T. A Universal Score for Deconvolution of Intact Protein and Native Electrospray Mass Spectra. Anal. Chem. 2020, 92, 4395–4401. 10.1021/acs.analchem.9b05272. [DOI] [PubMed] [Google Scholar]
- Campuzano I. D. G.; Robinson J. H.; Hui J. O.; Shi S. D.-H.; Netirojjanakul C.; Nshanian M.; Egea P. F.; Lippens J. L.; Bagal D.; Loo J. A.; et al. Native and Denaturing MS Protein Deconvolution for Biopharma: Monoclonal Antibodies and Antibody-Drug Conjugates to Polydisperse Membrane Proteins and Beyond. Anal. Chem. 2019, 91, 9472–9480. 10.1021/acs.analchem.9b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M.; Čaval T.; Kil Y. J.; Tang W.; Becker C.; Carlson E.; Kletter D.; Sen K. I.; Galy N.; Hagemans D.; et al. Parsimonious Charge Deconvolution for Native Mass Spectrometry. J. Proteome Res. 2018, 17, 1216–1226. 10.1021/acs.jproteome.7b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary S. P.; Thompson A. M.; Prell J. S. Fourier Analysis Method for Analyzing Highly Congested Mass Spectra of Ion Populations with Repeated Subunits. Anal. Chem. 2016, 88, 6205–6213. 10.1021/acs.analchem.6b01088. [DOI] [PubMed] [Google Scholar]
- Cleary S. P.; Li H.; Bagal D.; Loo J. A.; Campuzano I. D. G.; Prell J. S. Extracting Charge and Mass Information from Highly Congested Mass Spectra Using Fourier-Domain Harmonics. J. Am. Soc. Mass Spectrom. 2018, 29, 2067–2080. 10.1007/s13361-018-2018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong K.; Kim J.; Gaikwad M.; Hidayah S. N.; Heikaus L.; Schlüter H.; Kohlbacher O. Flashdeconv: Ultrafast, High-Quality Feature Deconvolution for Top-Down Proteomics. Cell Systems 2020, 10, 213–218. 10.1016/j.cels.2020.01.003. [DOI] [PubMed] [Google Scholar]
- Toby T. K.; Fornelli L.; Kelleher N. L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner O. S.; Haverland N. A.; Fornelli L.; Melani R. D.; Do Vale L. H. F.; Seckler H. S.; Doubleday P. F.; Schachner L. F.; Srzentić K.; Kelleher N. L.; et al. Top-Down Characterization of Endogenous Protein Complexes with Native Proteomics. Nat. Chem. Biol. 2018, 14, 36–41. 10.1038/nchembio.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Piehowski P. D.; Wilkins C.; Zhou M.; Mendoza J.; Fujimoto G. M.; Gibbons B. C.; Shaw J. B.; Shen Y.; Shukla A. K.; et al. Informed-Proteomics: Open-Source Software Package for Top-Down Proteomics. Nat. Methods 2017, 14, 909–914. 10.1038/nmeth.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Inbar Y.; Dorrestein P. C.; Wynne C.; Edwards N.; Souda P.; Whitelegge J. P.; Bafna V.; Pevzner P. A. Deconvolution and Database Search of Complex Tandem Mass Spectra of Intact Proteins. Mol. Cell. Proteomics 2010, 9, 2772–2782. 10.1074/mcp.M110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P. C.; Xu T.; Han X.; Cociorva D.; Barbosa V. C.; Yates J. R. 3rd. Yada: A Tool for Taking the Most out of High-Resolution Spectra. Bioinformatics 2009, 25, 2734–2736. 10.1093/bioinformatics/btp489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn D.; Zubarev R.; McLafferty F. Automated Reduction and Interpretation of High Resolution Electrospray Mass Spectra of Large Molecules. J. Am. Soc. Mass Spectrom. 2000, 11, 320–332. 10.1016/S1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- Basharat A. R.; Ning X.; Liu X. EnvCNN: A Convolutional Neural Network Model for Evaluating Isotopic Envelopes in Top-Down Mass-Spectral Deconvolution. Anal. Chem. 2020, 92, 7778–7785. 10.1021/acs.analchem.0c00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleno L.; Volmer D. A. Ion Activation Methods for Tandem Mass Spectrometry. J. Mass Spectrom. 2004, 39, 1091–1112. 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- Brodbelt J. S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2016, 88, 30–51. 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias L. A.; Santos I. C.; Brodbelt J. S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2020, 92, 227–251. 10.1021/acs.analchem.9b04859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey S. A.; Mentinova M. Ion/Neutral, Ion/Electron, Ion/Photon, and Ion/Ion Interactions in Tandem Mass Spectrometry: Do We Need Them All? Are They Enough?. J. Am. Soc. Mass Spectrom. 2011, 22, 3–12. 10.1007/s13361-010-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch J. L. P. Collisional Activation of Protein Complexes: Picking up the Pieces. J. Am. Soc. Mass Spectrom. 2009, 20, 341–348. 10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Fort K. L.; Cramer C. N.; Voinov V. G.; Vasil’ev Y. V.; Lopez N. I.; Beckman J. S.; Heck A. J. R. Exploring Ecd on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2018, 17, 926–933. 10.1021/acs.jproteome.7b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S. Photodissociation Mass Spectrometry: New Tools for Characterization of Biological Molecules. Chem. Soc. Rev. 2014, 43, 2757–2783. 10.1039/C3CS60444F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Anal. Chem. 2015, 87, 7109–7116. 10.1021/acs.analchem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S.; Morrison L. J.; Santos I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120, 3328–3380. 10.1021/acs.chemrev.9b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Gross M. L.; Blankenship R. E. Native Mass Spectrometry of Photosynthetic Pigment-Protein Complexes. FEBS Lett. 2013, 587, 1012–1020. 10.1016/j.febslet.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobott F.; Robinson C. V. Characterising Electrosprayed Biomolecules Using Tandem-MS—the Noncovalent Groel Chaperonin Assembly. Int. J. Mass Spectrom. 2004, 236, 25–32. 10.1016/j.ijms.2004.05.010. [DOI] [Google Scholar]
- Benesch J. L.; Robinson C. V. Mass Spectrometry of Macromolecular Assemblies: Preservation and Dissociation. Curr. Opin. Struct. Biol. 2006, 16, 245–251. 10.1016/j.sbi.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Freitas M. A.; Hendrickson C. L.; Marshall A. G.; Rostom A. A.; Robinson C. V. Competitive Binding to the Oligopeptide Binding Protein, Oppa: In-Trap Cleanup in an Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2000, 11, 1023–1026. 10.1016/S1044-0305(00)00180-X. [DOI] [PubMed] [Google Scholar]
- Speir J. P.; Senko M. W.; Little D. P.; Loo J. A.; McLafferty F. W. High-Resolution Tandem Mass Spectra of 37–67 Kda Proteins. J. Mass Spectrom. 1995, 30, 39–42. 10.1002/jms.1190300108. [DOI] [Google Scholar]
- Gardner M. W.; Brodbelt J. S. Reduction of Chemical Noise in Electrospray Ionization Mass Spectrometry by Supplemental Ir Activation. J. Am. Soc. Mass Spectrom. 2009, 20, 2206–2210. 10.1016/j.jasms.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Greisch J.-F.; van der Laarse S. A. M.; Heck A. J. R. Enhancing Top-Down Analysis Using Chromophore-Assisted Infrared Multiphoton Dissociation from (Phospho)Peptides to Protein Assemblies. Anal. Chem. 2020, 92, 15506–15516. 10.1021/acs.analchem.0c03412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin C.; Reid D. J.; Marty M. T.; Landreh M. Scratching the Surface: Native Mass Spectrometry of Peripheral Membrane Protein Complexes. Biochem. Soc. Trans. 2020, 48, 547–558. 10.1042/BST20190787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keener J. E.; Zhang G.; Marty M. T. Native Mass Spectrometry of Membrane Proteins. Anal. Chem. 2021, 93, 583–597. 10.1021/acs.analchem.0c04342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeke J.; Robinson C. V.; Ruotolo B. T. Residual Counter Ions Can Stabilise a Large Protein Complex in the Gas Phase. Int. J. Mass Spectrom. 2010, 298, 91–98. 10.1016/j.ijms.2009.08.001. [DOI] [Google Scholar]
- Benesch J. L. P.; Aquilina J. A.; Ruotolo B. T.; Sobott F.; Robinson C. V. Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblies. Chem. Biol. 2006, 13, 597–605. 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Stiving A. Q.; VanAernum Z. L.; Busch F.; Harvey S. R.; Sarni S. H.; Wysocki V. H. Surface-Induced Dissociation: An Effective Method for Characterization of Protein Quaternary Structure. Anal. Chem. 2019, 91, 190–209. 10.1021/acs.analchem.8b05071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Wysocki V. H. Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas Phase. Acc. Chem. Res. 2014, 47, 1010–1018. 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- Wang G.; Chaihu L.; Tian M.; Shao X.; Dai R.; de Jong R. N.; Ugurlar D.; Gros P.; Heck A. J. R. Releasing Nonperipheral Subunits from Protein Complexes in the Gas Phase. Anal. Chem. 2020, 92, 15799–15805. 10.1021/acs.analchem.0c02845. [DOI] [PubMed] [Google Scholar]
- Popa V.; Trecroce D. A.; McAllister R. G.; Konermann L. Collision-Induced Dissociation of Electrosprayed Protein Complexes: An All-Atom Molecular Dynamics Model with Mobile Protons. J. Phys. Chem. B 2016, 120, 5114–5124. 10.1021/acs.jpcb.6b03035. [DOI] [PubMed] [Google Scholar]
- Beardsley R. L.; Jones C. M.; Galhena A. S.; Wysocki V. H. Noncovalent Protein Tetramers and Pentamers with “n” Charges Yield Monomers with n/4 and n/5 Charges. Anal. Chem. 2009, 81, 1347–1356. 10.1021/ac801883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanasundara S. N.; Thachuk M. Toward an Improved Understanding of the Dissociation Mechanism of Gas Phase Protein Complexes. J. Phys. Chem. B 2010, 114, 11646–11653. 10.1021/jp103576b. [DOI] [PubMed] [Google Scholar]
- Pagel K.; Hyung S.-J.; Ruotolo B. T.; Robinson C. V. Alternate Dissociation Pathways Identified in Charge-Reduced Protein Complex Ions. Anal. Chem. 2010, 82, 5363–5372. 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- Sciuto S. V.; Liu J.; Konermann L. An Electrostatic Charge Partitioning Model for the Dissociation of Protein Complexes in the Gas Phase. J. Am. Soc. Mass Spectrom. 2011, 22, 1679–1689. 10.1007/s13361-011-0205-x. [DOI] [PubMed] [Google Scholar]
- Jurchen J. C.; Williams E. R. Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J. Am. Chem. Soc. 2003, 125, 2817–2826. 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. M.; Beardsley R. L.; Galhena A. S.; Dagan S.; Cheng G.; Wysocki V. H. Symmetrical Gas-Phase Dissociation of Noncovalent Protein Complexes via Surface Collisions. J. Am. Chem. Soc. 2006, 128, 15044–15045. 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- VanAernum Z. L.; Gilbert J. D.; Belov M. E.; Makarov A. A.; Horning S. R.; Wysocki V. H. Surface-Induced Dissociation of Noncovalent Protein Complexes in an Extended Mass Range Orbitrap Mass Spectrometer. Anal. Chem. 2019, 91, 3611–3618. 10.1021/acs.analchem.8b05605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder D. T.; Panczyk E.; Stiving A. Q.; Gilbert J. D.; Somogyi A.; Kaplan D.; Wysocki V. Design and Performance of a Second-Generation Surface-Induced Dissociation Cell for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry of Native Protein Complexes. Anal. Chem. 2019, 91, 14049–14057. 10.1021/acs.analchem.9b03746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki V. H.; Joyce K. E.; Jones C. M.; Beardsley R. L. Surface-Induced Dissociation of Small Molecules, Peptides, and Non-Covalent Protein Complexes. J. Am. Soc. Mass Spectrom. 2008, 19, 190–208. 10.1016/j.jasms.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Dagan S.; Wysocki V. H. Protein Subunits Released by Surface Collisions of Noncovalent Complexes: Nativelike Compact Structures Revealed by Ion Mobility Mass Spectrometry. Angew. Chem., Int. Ed. 2012, 51, 4336–4339. 10.1002/anie.201108700. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Huang C.; Wysocki V. H. Surface-Induced Dissociation of Ion Mobility-Separated Noncovalent Complexes in a Quadrupole/Time-of-Flight Mass Spectrometer. Anal. Chem. 2012, 84, 6016–6023. 10.1021/ac300810u. [DOI] [PubMed] [Google Scholar]
- Harvey S. R.; Seffernick J. T.; Quintyn R. S.; Song Y.; Ju Y.; Yan J.; Sahasrabuddhe A. N.; Norris A.; Zhou M.; Behrman E. J.; et al. Relative Interfacial Cleavage Energetics of Protein Complexes Revealed by Surface Collisions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8143–8148. 10.1073/pnas.1817632116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimer S.; Ben-Nissan G.; Morgenstern D.; Kumar-Deshmukh F.; Polkinghorn C.; Quintyn R. S.; Vasil’ev Y. V.; Beckman J. S.; Elad N.; Wysocki V. H.; et al. Comparative Structural Analysis of 20S Proteasome Ortholog Protein Complexes by Native Mass Spectrometry. ACS Cent. Sci. 2020, 6, 573–588. 10.1021/acscentsci.0c00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Nelp M. T.; Bandarian V.; Wysocki V. H. Refining the Structural Model of a Heterohexameric Protein Complex: Surface Induced Dissociation and Ion Mobility Provide Key Connectivity and Topology Information. ACS Cent. Sci. 2015, 1, 477–487. 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly L.; Antoine R.; Broyer M.; Dugourd P.; Lemoine J. Specific Uv Photodissociation of Tyrosyl-Containing Peptides in Multistage Mass Spectrometry. J. Mass Spectrom. 2007, 42, 818–824. 10.1002/jms.1222. [DOI] [PubMed] [Google Scholar]
- Madsen J. A.; Kaoud T. S.; Dalby K. N.; Brodbelt J. S. 193-Nm Photodissociation of Singly and Multiply Charged Peptide Anions for Acidic Proteome Characterization. Proteomics 2011, 11, 1329–1334. 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Li W.; Holden D. D.; Zhang Y.; Griep-Raming J.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc. 2013, 135, 12646–12651. 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly T.; Julian R. R. Ultraviolet Photodissociation: Developments Towards Applications for Mass-Spectrometry-Based Proteomics. Angew. Chem., Int. Ed. 2009, 48, 7130–7137. 10.1002/anie.200900613. [DOI] [PubMed] [Google Scholar]
- Reilly J. P. Ultraviolet Photofragmentation of Biomolecular Ions. Mass Spectrom. Rev. 2009, 28, 425–447. 10.1002/mas.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey S. A.; Goeringer D. E. Special Feature: Tutorial Slow Heating Methods in Tandem Mass Spectrometry. J. Mass Spectrom. 1997, 32, 461–474. . [DOI] [Google Scholar]
- Tamara S.; Dyachenko A.; Fort K. L.; Makarov A. A.; Scheltema R. A.; Heck A. J. R. Symmetry of Charge Partitioning in Collisional and Uv Photon-Induced Dissociation of Protein Assemblies. J. Am. Chem. Soc. 2016, 138, 10860–10868. 10.1021/jacs.6b05147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison L. J.; Brodbelt J. S. 193 Nm Ultraviolet Photodissociation Mass Spectrometry of Tetrameric Protein Complexes Provides Insight into Quaternary and Secondary Protein Topology. J. Am. Chem. Soc. 2016, 138, 10849–10859. 10.1021/jacs.6b03905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J. P.; Li W.; Zhang Y.; Brodbelt J. S. Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2014, 136, 12920–12928. 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe S. N.; Brodbelt J. S. Impact of Charge State on 193 Nm Ultraviolet Photodissociation of Protein Complexes. Phys. Chem. Chem. Phys. 2019, 21, 9265–9276. 10.1039/C9CP01144G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov V. A.; Liko I.; Mize T. H.; Bush M. F.; Benesch J. L. P.; Robinson C. V. Infrared Laser Activation of Soluble and Membrane Protein Assemblies in the Gas Phase. Anal. Chem. 2016, 88, 7060–7067. 10.1021/acs.analchem.6b00645. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cui W.; Wecksler A. T.; Zhang H.; Molina P.; Deperalta G.; Gross M. L. Native MS and ECD Characterization of a Fab-Antigen Complex May Facilitate Crystallization for X-ray Diffraction. J. Am. Soc. Mass Spectrom. 2016, 27, 1139–1142. 10.1007/s13361-016-1398-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lermyte F.; Sobott F. Electron Transfer Dissociation Provides Higher-Order Structural Information of Native and Partially Unfolded Protein Complexes. Proteomics 2015, 15, 2813–2822. 10.1002/pmic.201400516. [DOI] [PubMed] [Google Scholar]
- Tamara S.; Scheltema R. A.; Heck A. J. R.; Leney A. C. Phosphate Transfer in Activated Protein Complexes Reveals Interaction Sites. Angew. Chem. 2017, 129, 13829–13832. 10.1002/ange.201706749. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Loo R. R. O.; Loo J. A. Structural Characterization of a Thrombin-Aptamer Complex by High Resolution Native Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1815–1822. 10.1007/s13361-017-1751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner O. S.; McAnally M. O.; Van Duyne R. P.; Schatz G. C.; Breuker K.; Compton P. D.; Kelleher N. L. Native Electron Capture Dissociation Maps to Iron-Binding Channels in Horse Spleen Ferritin. Anal. Chem. 2017, 89, 10711–10716. 10.1021/acs.analchem.7b01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Sheng Y.; McGee W.; Cammarata M.; Holden D.; Loo J. A. Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal. Chem. 2017, 89, 2731–2738. 10.1021/acs.analchem.6b02377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro S. Y.; Schachner L. F.; Koo C. W.; Purohit R.; Remis J. P.; Kenney G. E.; Liauw B. W.; Thomas P. M.; Patrie S. M.; Kelleher N. L.; et al. Native Top-Down Mass Spectrometry Provides Insights into the Copper Centers of Membrane-Bound Methane Monooxygenase. Nat. Commun. 2019, 10, 2675. 10.1038/s41467-019-10590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Li J.; Liko I.; Gault J.; Bechara C.; Wu D.; Hopper J. T. S.; Giles K.; Benesch J. L. P.; Robinson C. V. Identifying Key Membrane Protein Lipid Interactions Using Mass Spectrometry. Nat. Protoc. 2018, 13, 1106–1120. 10.1038/nprot.2018.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman A. D.; Skinner O. S.; Kelleher N. L. Top Down Proteomics: Facts and Perspectives. Biochem. Biophys. Res. Commun. 2014, 445, 683–693. 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Kou Q.; Guo R.; Yang Z.; Chen D.; Liu X.; Hong H.; Sun L. Native Proteomics in Discovery Mode Using Size-Exclusion Chromatography-Capillary Zone Electrophoresis-Tandem Mass Spectrometry. Anal. Chem. 2018, 90, 10095–10099. 10.1021/acs.analchem.8b02725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani R. D.; Skinner O. S.; Fornelli L.; Domont G. B.; Compton P. D.; Kelleher N. L. Mapping Proteoforms and Protein Complexes from King Cobra Venom Using Both Denaturing and Native Top-Down Proteomics. Mol. Cell. Proteomics 2016, 15, 2423–2434. 10.1074/mcp.M115.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber B. J.; Boehringer D.; Leitner A.; Bieri P.; Voigts-Hoffmann F.; Erzberger J. P.; Leibundgut M.; Aebersold R.; Ban N. Architecture of the Large Subunit of the Mammalian Mitochondrial Ribosome. Nature 2014, 505, 515–519. 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- Greber B. J.; Boehringer D.; Leibundgut M.; Bieri P.; Leitner A.; Schmitz N.; Aebersold R.; Ban N. The Complete Structure of the Large Subunit of the Mammalian Mitochondrial Ribosome. Nature 2014, 515, 283–286. 10.1038/nature13895. [DOI] [PubMed] [Google Scholar]
- Samir P.; Browne C. M.; Rahul; Sun M.; Shen B.; Li W.; Frank J.; Link A. J. Identification of Changing Ribosome Protein Compositions Using Mass Spectrometry. Proteomics 2018, 18, 1800217. 10.1002/pmic.201800217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baßler J.; Grandi P.; Gadal O.; Leßmann T.; Petfalski E.; Tollervey D.; Lechner J.; Hurt E. Identification of a 60s Preribosomal Particle That Is Closely Linked to Nuclear Export. Mol. Cell 2001, 8, 517–529. 10.1016/S1097-2765(01)00342-2. [DOI] [PubMed] [Google Scholar]
- Genuth N. R.; Barna M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 2018, 71, 364–374. 10.1016/j.molcel.2018.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilag L. L.; Videler H.; Mckay A. R.; Sobott F.; Fucini P.; Nierhaus K. H.; Robinson C. V. Heptameric (L12)6/L10 Rather Than Canonical Pentameric Complexes Are Found by Tandem MS of Intact Ribosomes from Thermophilic Bacteria. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 8192–8197. 10.1073/pnas.0502193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostom A. A.; Fucini P.; Benjamin D. R.; Juenemann R.; Nierhaus K. H.; Hartl F. U.; Dobson C. M.; Robinson C. V. Detection and Selective Dissociation of Intact Ribosomes in a Mass Spectrometer. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5185–5190. 10.1073/pnas.97.10.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay A. R.; Ruotolo B. T.; Ilag L. L.; Robinson C. V. Mass Measurements of Increased Accuracy Resolve Heterogeneous Populations of Intact Ribosomes. J. Am. Chem. Soc. 2006, 128, 11433–11442. 10.1021/ja061468q. [DOI] [PubMed] [Google Scholar]
- Abdillahi A. M.; Lee K. W.; McLuckey S. A. Mass Analysis of Macro-molecular Analytes via Multiply-Charged Ion Attachment. Anal. Chem. 2020, 92, 16301–16306. 10.1021/acs.analchem.0c04335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson J. L.; McLuckey S. A. Simplification of Product Ion Spectra Derived from Multiply Charged Parent Ions via Ion/Ion Chemistry. Anal. Chem. 1998, 70, 3533–3544. 10.1021/ac9802832. [DOI] [PubMed] [Google Scholar]
- Unverdorben P.; Beck F.; Sledz P.; Schweitzer A.; Pfeifer G.; Plitzko J. M.; Baumeister W.; Forster F. Deep Classification of a Large Cryo-Em Dataset Defines the Conformational Landscape of the 26s Proteasome. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 5544–5549. 10.1073/pnas.1403409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Luan B.; Wu J.; Shi Y. An Atomic Structure of the Human 26s Proteasome. Nat. Struct. Mol. Biol. 2016, 23, 778–785. 10.1038/nsmb.3273. [DOI] [PubMed] [Google Scholar]
- Bard J. A. M.; Goodall E. A.; Greene E. R.; Jonsson E.; Dong K. C.; Martin A. Structure and Function of the 26s Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. 10.1146/annurev-biochem-062917-011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo J. A.; Berhane B.; Kaddis C. S.; Wooding K. M.; Xie Y.; Kaufman S. L.; Chernushevich I. V. Electrospray Ionization Mass Spectrometry and Ion Mobility Analysis of the 20S Proteasome Complex. J. Am. Soc. Mass Spectrom. 2005, 16, 998–1008. 10.1016/j.jasms.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Sharon M.; Witt S.; Felderer K.; Rockel B.; Baumeister W.; Robinson C. V. 20S Proteasomes Have the Potential to Keep Substrates in Store for Continual Degradation. J. Biol. Chem. 2006, 281, 9569–9575. 10.1074/jbc.M511951200. [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G.; Vimer S.; Tarnavsky M.; Sharon M. Structural Mass Spectrometry Approaches to Study the 20S Proteasome. Methods Enzymol. 2019, 619, 179–223. 10.1016/bs.mie.2018.12.029. [DOI] [PubMed] [Google Scholar]
- Sharon M.; Taverner T.; Ambroggio X. I.; Deshaies R. J.; Robinson C. V. Structural Organization of the 19s Proteasome Lid: Insights from MS of Intact Complexes. PLoS Biol. 2006, 4, e267 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonn-Segev A.; Belacic K.; Bodrug T.; Young G.; VanderLinden R. T.; Schulman B. A.; Schimpf J.; Friedrich T.; Dip P. V.; Schwartz T. U.; et al. Quantifying the Heterogeneity of Macromolecular Machines by Mass Photometry. Nat. Commun. 2020, 11, 1772. 10.1038/s41467-020-15642-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothner B.; Siuzdak G. Electrospray Ionization of a Whole Virus: Analyzing Mass, Structure, and Viability. ChemBioChem 2004, 5, 258–260. 10.1002/cbic.200300754. [DOI] [PubMed] [Google Scholar]
- Fuerstenau S. D.; Benner W. H.; Thomas J. J.; Brugidou C.; Bothner B.; Siuzdak G. Mass Spectrometry of an Intact Virus. Angew. Chem., Int. Ed. 2001, 40, 541–544. . [DOI] [PubMed] [Google Scholar]
- Thomas J. J.; Bothner B.; Traina J.; Benner W. H.; Siuzdak G. Electrospray Ion Mobility Spectrometry of Intact Viruses. Spectroscopy 2004, 18, 31–36. 10.1155/2004/376572. [DOI] [Google Scholar]
- Kaddis C. S.; Lomeli S. H.; Yin S.; Berhane B.; Apostol M. I.; Kickhoefer V. A.; Rome L. H.; Loo J. A. Sizing Large Proteins and Protein Complexes by Electrospray Ionization Mass Spectrometry and Ion Mobility. J. Am. Soc. Mass Spectrom. 2007, 18, 1206–1216. 10.1016/j.jasms.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss V. U.; Pogan R.; Zoratto S.; Bond K. M.; Boulanger P.; Jarrold M. F.; Lyktey N.; Pahl D.; Puffler N.; Schelhaas M.; et al. Virus-Like Particle Size and Molecular Weight/Mass Determination Applying Gas-Phase Electrophoresis (Native Nes Gemma). Anal. Bioanal. Chem. 2019, 411, 5951–5962. 10.1007/s00216-019-01998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C.; Versluis C.; Watts N. R.; Wingfield P. T.; Steven A. C.; Heck A. J. Stability and Shape of Hepatitis B Virus Capsids in Vacuo. Angew. Chem., Int. Ed. 2008, 47, 6247–6251. 10.1002/anie.200802410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C.; Heck A. J. Modern Biomolecular Mass Spectrometry and Its Role in Studying Virus Structure, Dynamics, and Assembly. Angew. Chem., Int. Ed. 2011, 50, 8248–8262. 10.1002/anie.201008120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft A. E. Mass Spectrometry-Based Studies of Virus Assembly. Curr. Opin. Virol. 2019, 36, 17–24. 10.1016/j.coviro.2019.02.006. [DOI] [PubMed] [Google Scholar]
- Wörner T. P.; Shamorkina T. M.; Snijder J.; Heck A. J. R. Mass Spectrometry-Based Structural Virology. Anal. Chem. 2021, 93, 620–640. 10.1021/acs.analchem.0c04339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulfer J.; Kadek A.; Kopicki J. D.; Krichel B.; Uetrecht C. Structural Mass Spectrometry Goes Viral. Adv. Virus Res. 2019, 105, 189–238. 10.1016/bs.aivir.2019.07.003. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Barbu I. M.; Shoemaker G. K.; van Duijn E.; Heck A. J. Interrogating Viral Capsid Assembly with Ion Mobility-Mass Spectrometry. Nat. Chem. 2011, 3, 126–132. 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- Poliakov A.; van Duijn E.; Lander G.; Fu C. Y.; Johnson J. E.; Prevelige P. E. Jr.; Heck A. J. Macromolecular Mass Spectrometry and Electron Microscopy as Complementary Tools for Investigation of the Heterogeneity of Bacteriophage Portal Assemblies. J. Struct. Biol. 2007, 157, 371–383. 10.1016/j.jsb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Veesler D.; Khayat R.; Krishnamurthy S.; Snijder J.; Huang R. K.; Heck A. J.; Anand G. S.; Johnson J. E. Architecture of a Dsdna Viral Capsid in Complex with Its Maturation Protease. Structure 2014, 22, 230–237. 10.1016/j.str.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wörner T. P.; Bennett A.; Habka S.; Snijder J.; Friese O.; Powers T.; Agbandje-McKenna M.; Heck A. J. R. Adeno-Associated Virus Capsid Assembly Is Divergent and Stochastic. Nat. Commun. 2021, 12, 1642. 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J.; Uetrecht C.; Rose R. J.; Sanchez-Eugenia R.; Marti G. A.; Agirre J.; Guérin D. M. A.; Wuite G. J. L.; Heck A. J. R.; Roos W. H. Probing the Biophysical Interplay between a Viral Genome and Its Capsid. Nat. Chem. 2013, 5, 502–509. 10.1038/nchem.1627. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Snijder J.; Tsvetkova I. B.; Dragnea B. G.; Cornelissen J. J.; Heck A. J. Examining the Heterogeneous Genome Content of Multipartite Viruses Bmv and Ccmv by Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1000–1009. 10.1007/s13361-016-1348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch M.; de la Escosura A.; Ma Y.; Uetrecht C.; Heck A. J.; Torres T.; Cornelissen J. J. Encapsulation of Phthalocyanine Supramolecular Stacks into Virus-Like Particles. J. Am. Chem. Soc. 2011, 133, 6878–6881. 10.1021/ja110752u. [DOI] [PubMed] [Google Scholar]
- Sutter M.; Boehringer D.; Gutmann S.; Gunther S.; Prangishvili D.; Loessner M. J.; Stetter K. O.; Weber-Ban E.; Ban N. Structural Basis of Enzyme Encapsulation into a Bacterial Nanocompartment. Nat. Struct. Mol. Biol. 2008, 15, 939–947. 10.1038/nsmb.1473. [DOI] [PubMed] [Google Scholar]
- Gabashvili A. N.; Chmelyuk N. S.; Efremova M. V.; Malinovskaya J. A.; Semkina A. S.; Abakumov M. A. Encapsulins-Bacterial Protein Nanocompartments: Structure, Properties, and Application. Biomolecules 2020, 10, 966. 10.3390/biom10060966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J.; Kononova O.; Barbu I. M.; Uetrecht C.; Rurup W. F.; Burnley R. J.; Koay M. S.; Cornelissen J. J.; Roos W. H.; Barsegov V.; et al. Assembly and Mechanical Properties of the Cargo-Free and Cargo-Loaded Bacterial Nanocompartment Encapsulin. Biomacromolecules 2016, 17, 2522–2529. 10.1021/acs.biomac.6b00469. [DOI] [PubMed] [Google Scholar]
- Corchero J. L.; Cedano J. Self-Assembling, Protein-Based Intracellular Bacterial Organelles: Emerging Vehicles for Encapsulating, Targeting and Delivering Therapeutical Cargoes. Microb. Cell Fact. 2011, 10, 92. 10.1186/1475-2859-10-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigmund F.; Massner C.; Erdmann P.; Stelzl A.; Rolbieski H.; Desai M.; Bricault S.; Worner T. P.; Snijder J.; Geerlof A.; et al. Bacterial Encapsulins as Orthogonal Compartments for Mammalian Cell Engineering. Nat. Commun. 2018, 9, 1990. 10.1038/s41467-018-04227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rurup W. F.; Snijder J.; Koay M. S. T.; Heck A. J. R.; Cornelissen J. J. L. M. Self-Sorting of Foreign Proteins in a Bacterial Nanocompartment. J. Am. Chem. Soc. 2014, 136, 3828–3832. 10.1021/ja410891c. [DOI] [PubMed] [Google Scholar]
- Seebeck F. P.; Woycechowsky K. J.; Zhuang W.; Rabe J. P.; Hilvert D. A Simple Tagging System for Protein Encapsulation. J. Am. Chem. Soc. 2006, 128, 4516–4517. 10.1021/ja058363s. [DOI] [PubMed] [Google Scholar]
- Azuma Y.; Edwardson T. G. W.; Hilvert D. Tailoring Lumazine Synthase Assemblies for Bionanotechnology. Chem. Soc. Rev. 2018, 47, 3543–3557. 10.1039/C8CS00154E. [DOI] [PubMed] [Google Scholar]
- Sasaki E.; Bohringer D.; van de Waterbeemd M.; Leibundgut M.; Zschoche R.; Heck A. J.; Ban N.; Hilvert D. Structure and Assembly of Scalable Porous Protein Cages. Nat. Commun. 2017, 8, 14663. 10.1038/ncomms14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N. P.; Lai Y. T. Practical Approaches to Designing Novel Protein Assemblies. Curr. Opin. Struct. Biol. 2013, 23, 632–638. 10.1016/j.sbi.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Kibler R. D.; Hunt A.; Busch F.; Pearl J.; Jia M.; VanAernum Z. L.; Wicky B. I. M.; Dods G.; Liao H.; et al. De Novo Design of Protein Logic Gates. Science 2020, 368, 78–84. 10.1126/science.aay2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Fiala B.; Schafer A.; Wrenn S.; Pham M. N.; Murphy M.; Tse L. V.; Shehata L.; O’Connor M. A.; Chen C.; et al. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 2020, 183, 1367–1382. 10.1016/j.cell.2020.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wargacki A. J.; Wörner T. P.; Van De Waterbeemd M.; Ellis D.; Heck A. J. R.; King N. P. Complete and Cooperative in Vitro Assembly of Computationally Designed Self-Assembling Protein Nanomaterials. Nat. Commun. 2021, 12, 883. 10.1038/s41467-021-21251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabuddhe A.; Hsia Y.; Busch F.; Sheffler W.; King N. P.; Baker D.; Wysocki V. H. Confirmation of Intersubunit Connectivity and Topology of Designed Protein Complexes by Native MS. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 1268–1273. 10.1073/pnas.1713646115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyken S. E.; Benhaim M. A.; Busch F.; Jia M.; Bick M. J.; Choi H.; Klima J. C.; Chen Z.; Walkey C.; Mileant A.; et al. De Novo Design of Tunable, Ph-Driven Conformational Changes. Science 2019, 364, 658–664. 10.1126/science.aav7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer D. Z.; Motwani T.; Teschke C. M.; Jarrold M. F. Acquiring Structural Information on Virus Particles with Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1028–1036. 10.1007/s13361-016-1362-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G.; Degn S. E.; Thiel S.; Andersen G. R. Complement Activation, Regulation, and Molecular Basis for Complement-Related Diseases. EMBO J. 2015, 34, 2735–2757. 10.15252/embj.201591881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp T. H.; Boyle A. L.; Diebolder C. A.; Kros A.; Koster A. J.; Gros P. Insights into IgM-Mediated Complement Activation Based on in Situ Structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 11900–11905. 10.1073/pnas.1901841116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebolder C. A.; Beurskens F. J.; de Jong R. N.; Koning R. I.; Strumane K.; Lindorfer M. A.; Voorhorst M.; Ugurlar D.; Rosati S.; Heck A. J. R.; et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science 2014, 343, 1260–1263. 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong R. N.; Beurskens F. J.; Verploegen S.; Strumane K.; van Kampen M. D.; Voorhorst M.; Horstman W.; Engelberts P. J.; Oostindie S. C.; Wang G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of Igg Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; de Jong R. N.; van den Bremer E. T. J.; Beurskens F. J.; Labrijn A. F.; Ugurlar D.; Gros P.; Schuurman J.; Parren P. W. H. I.; Heck A. J. R. Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen. Mol. Cell 2016, 63, 135–145. 10.1016/j.molcel.2016.05.016. [DOI] [PubMed] [Google Scholar]
- Strasser J.; de Jong R. N.; Beurskens F. J.; Wang G.; Heck A. J. R.; Schuurman J.; Parren P. W. H. I.; Hinterdorfer P.; Preiner J. Unraveling the Macromolecular Pathways of IgG Oligomerization and Complement Activation on Antigenic Surfaces. Nano Lett. 2019, 19, 4787–4796. 10.1021/acs.nanolett.9b02220. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; Fleming G. R.; Olaya-Castro A.; Van Grondelle R. Lessons from Nature About Solar Light Harvesting. Nat. Chem. 2011, 3, 763–774. 10.1038/nchem.1145. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Goodson C.; Blankenship R. E.; Gross M. L. Primary and Higher Order Structure of the Reaction Center from the Purple Phototrophic Bacteriumblastochloris Viridis: A Test for Native Mass Spectrometry. J. Proteome Res. 2018, 17, 1615–1623. 10.1021/acs.jproteome.7b00897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Harrington L. B.; Lu Y.; Prado M.; Saer R.; Rempel D.; Blankenship R. E.; Gross M. L. Native Mass Spectrometry Characterizes the Photosynthetic Reaction Center Complex from the Purple Bacterium Rhodobacter Sphaeroides. J. Am. Soc. Mass Spectrom. 2017, 28, 87–95. 10.1007/s13361-016-1451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Liu H.; Saer R. G.; Zhang H.; Meyer C. M.; Li V. L.; Shi L.; King J. D.; Gross M. L.; Blankenship R. E. Native Mass Spectrometry Analysis of Oligomerization States of Fluorescence Recovery Protein and Orange Carotenoid Protein: Two Proteins Involved in the Cyanobacterial Photoprotection Cycle. Biochemistry 2017, 56, 160–166. 10.1021/acs.biochem.6b01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Liu H.; Saer R.; Li V. L.; Zhang H.; Shi L.; Goodson C.; Gross M. L.; Blankenship R. E. A Molecular Mechanism for Nonphotochemical Quenching in Cyanobacteria. Biochemistry 2017, 56, 2812–2823. 10.1021/acs.biochem.7b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Liu H.; Lu Y.; Wolf N. R.; Gross M. L.; Blankenship R. E. Native Mass Spectrometry and Ion Mobility Characterize the Orange Carotenoid Protein Functional Domains. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 734–739. 10.1016/j.bbabio.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Zhang H.; Lu X.; Lu Y.; Cuneo M. J.; O’Neill H. M.; Urban V.; Lo C. S.; Blankenship R. E. Oligomerization State and Pigment Binding Strength of the Peridinin-Chla-Protein. FEBS Lett. 2015, 589, 2713–2719. 10.1016/j.febslet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Liu H.; Niedzwiedzki D. M.; Prado M.; Jiang J.; Gross M. L.; Blankenship R. E. Molecular Mechanism of Photoactivation and Structural Location of the Cyanobacterial Orange Carotenoid Protein. Biochemistry 2014, 53, 13–19. 10.1021/bi401539w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J.; Zhang H.; Gross M. L.; Blankenship R. E. Native Electrospray Mass Spectrometry Reveals the Nature and Stoichiometry of Pigments in the Fmo Photosynthetic Antenna Protein. Biochemistry 2011, 50, 3502–3511. 10.1021/bi200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronrud D. E.; Schmid M. F.; Matthews B. W. Structure and X-ray Amino Acid Sequence of a Bacteriochlorophyll a Protein from Prosthecochloris Aestuarii Refined at 1.9 Å Resolution. J. Mol. Biol. 1986, 188, 443–454. 10.1016/0022-2836(86)90167-1. [DOI] [PubMed] [Google Scholar]
- Fenna R. E.; Matthews B. W. Chlorophyll Arrangement in a Bacteriochlorophyll Protein from Chlorobium Limicola. Nature 1975, 258, 573–577. 10.1038/258573a0. [DOI] [Google Scholar]
- Ma J.; You X.; Sun S.; Wang X.; Qin S.; Sui S.-F. Structural Basis of Energy Transfer in Porphyridium Purpureum Phycobilisome. Nature 2020, 579, 146–151. 10.1038/s41586-020-2020-7. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Ma J.; Liu D.; Qin S.; Sun S.; Zhao J.; Sui S.-F. Structure of Phycobilisome from the Red Alga Griffithsia Pacifica. Nature 2017, 551, 57–63. 10.1038/nature24278. [DOI] [PubMed] [Google Scholar]
- Leney A. C.; Tschanz A.; Heck A. J. R. Connecting Color with Assembly in the Fluorescent B-Phycoerythrin Protein Complex. FEBS J. 2018, 285, 178–187. 10.1111/febs.14331. [DOI] [PubMed] [Google Scholar]
- Saluri M.; Kaldmäe M.; Rospu M.; Sirkel H.; Paalme T.; Landreh M.; Tuvikene R. Spatial Variation and Structural Characteristics of Phycobiliproteins from the Red Algae Furcellaria Lumbricalis and Coccotylus Truncatus. Algal Res. 2020, 52, 102058. 10.1016/j.algal.2020.102058. [DOI] [Google Scholar]
- Tamara S.; Hoek M.; Scheltema R. A.; Leney A. C.; Heck A. J. R. A Colorful Pallet of B-Phycoerythrin Proteoforms Exposed by a Multimodal Mass Spectrometry Approach. Chem. 2019, 5, 1302–1317. 10.1016/j.chempr.2019.03.006. [DOI] [Google Scholar]
- Leney A. C. Subunit Pi Can. Influence Protein Complex Dissociation Characteristics. J. Am. Soc. Mass Spectrom. 2019, 30, 1389–1395. 10.1007/s13361-019-02198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldmäe M.; Sahin C.; Saluri M.; Marklund E. G.; Landreh M. A Strategy for the Identification of Protein Architectures Directly from Ion Mobility Mass Spectrometry Data Reveals Stabilizing Subunit Interactions in Light Harvesting Complexes. Protein Sci. 2019, 28, 1024–1030. 10.1002/pro.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho I. S.; Theisen A.; Johannissen L. O.; Díaz-Ramos L. A.; Christie J. M.; Jenkins G. I.; Bellina B.; Barran P.; Jones A. R. Native Mass Spectrometry Reveals the Conformational Diversity of the UVR8 Photoreceptor. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 1116–1125. 10.1073/pnas.1813254116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese P.; Tamara S.; Saracco G.; Scheltema R. A.; Pagliano C. How Paired PSII-LHCII Supercomplexes Mediate the Stacking of Plant Thylakoid Membranes Unveiled by Structural Mass-Spectrometry. Nat. Commun. 2020, 11, 1361. 10.1038/s41467-020-15184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konijnenberg A.; van Dyck J. F.; Kailing L. L.; Sobott F. Extending Native Mass Spectrometry Approaches to Integral Membrane Proteins. Biol. Chem. 2015, 396, 991–1002. 10.1515/hsz-2015-0136. [DOI] [PubMed] [Google Scholar]
- Bolla J. R.; Agasid M. T.; Mehmood S.; Robinson C. V. Membrane Protein-Lipid Interactions Probed Using Mass Spectrometry. Annu. Rev. Biochem. 2019, 88, 85–111. 10.1146/annurev-biochem-013118-111508. [DOI] [PubMed] [Google Scholar]
- Hopper J. T. S.; Yu Y. T.-C.; Li D.; Raymond A.; Bostock M.; Liko I.; Mikhailov V.; Laganowsky A.; Benesch J. L. P.; Caffrey M.; et al. Detergent-Free Mass Spectrometry of Membrane Protein Complexes. Nat. Methods 2013, 10, 1206–1208. 10.1038/nmeth.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty M. T.; Hoi K. K.; Gault J.; Robinson C. V. Probing the Lipid Annular Belt by Gas-Phase Dissociation of Membrane Proteins in Nanodiscs. Angew. Chem., Int. Ed. 2016, 55, 550–554. 10.1002/anie.201508289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid D. J.; Keener J. E.; Wheeler A. P.; Zambrano D. E.; Diesing J. M.; Reinhardt-Szyba M.; Makarov A.; Marty M. T. Engineering Nanodisc Scaffold Proteins for Native Mass Spectrometry. Anal. Chem. 2017, 89, 11189–11192. 10.1021/acs.analchem.7b03569. [DOI] [PubMed] [Google Scholar]
- Keener J. E.; Zambrano D. E.; Zhang G.; Zak C. K.; Reid D. J.; Deodhar B. S.; Pemberton J. E.; Prell J. S.; Marty M. T. Chemical Additives Enable Native Mass Spectrometry Measurement of Membrane Protein Oligomeric State within Intact Nanodiscs. J. Am. Chem. Soc. 2019, 141, 1054–1061. 10.1021/jacs.8b11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. R.; Marzluff E. M.; Townsend J. A.; Resager W. C.; Marty M. T. Native Mass Spectrometry of Antimicrobial Peptides in Lipid Nanodiscs Elucidates Complex Assembly. Anal. Chem. 2019, 91, 9284–9291. 10.1021/acs.analchem.9b02261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. R.; Marty M. T. Revealing the Specificity of a Range of Antimicrobial Peptides in Lipid Nanodiscs by Native Mass Spectrometry. Biochemistry 2020, 59, 2135–2142. 10.1021/acs.biochem.0c00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorev D. S.; Baker L. A.; Wu D.; Beilsten-Edmands V.; Rouse S. L.; Zeev-Ben-Mordehai T.; Jiko C.; Samsudin F.; Gerle C.; Khalid S.; et al. Protein Assemblies Ejected Directly from Native Membranes Yield Complexes for Mass Spectrometry. Science 2018, 362, 829–834. 10.1126/science.aau0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorev D. S.; Tang H.; Rouse S. L.; Bolla J. R.; von Kügelgen A.; Baker L. A.; Wu D.; Gault J.; Grünewald K.; Bharat T. A. M.; et al. The Use of Sonicated Lipid Vesicles for Mass Spectrometry of Membrane Protein Complexes. Nat. Protoc. 2020, 15, 1690–1706. 10.1038/s41596-020-0303-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J.; Kunji E. R. S.; Walker J. E. Comment on “Protein Assemblies Ejected Directly from Native Membranes Yield Complexes for Mass Spectrometry. Science 2019, 366, eaaw9830 10.1126/science.aaw9830. [DOI] [PubMed] [Google Scholar]
- Chorev D. S.; Robinson C. V. Response to Comment on “Protein Assemblies Ejected Directly from Native Membranes Yield Complexes for Mass Spectrometry. Science 2019, 366, eaax3102 10.1126/science.aax3102. [DOI] [PubMed] [Google Scholar]
- Alley W. R. Jr.; Mann B. F.; Novotny M. V. High-Sensitivity Analytical Approaches for the Structural Characterization of Glycoproteins. Chem. Rev. 2013, 113, 2668–2732. 10.1021/cr3003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak L. R.; Xu G.; Li Q.; Goonatilleke E.; Lebrilla C. B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118, 7886–7930. 10.1021/acs.chemrev.7b00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhrer M.; Catalina M. I.; Deelder A. M.; Hokke C. H. Glycoproteomics Based on Tandem Mass Spectrometry of Glycopeptides. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 849, 115–128. 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Čaval T.; Heck A. J. R.; Reiding K. R. Meta-Heterogeneity: Evaluating and Describing the Diversity in Glycosylation between Sites on the Same Glycoprotein. Mol. Cell. Proteomics 2021, 20, 100010. 10.1074/mcp.R120.002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Wang G.; Song T.; Lebrilla C. B.; Heck A. J. R. Resolving the Micro-Heterogeneity and Structural Integrity of Monoclonal Antibodies by Hybrid Mass Spectrometric Approaches. MAbs 2017, 9, 638–645. 10.1080/19420862.2017.1290033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin K. L.; Welply J. K.; Huang E.; Henion J. D. Characterization of N-Linked Oligosaccharides by Electrospray and Tandem Mass Spectrometry. Anal. Chem. 1992, 64, 1440–1448. 10.1021/ac00037a024. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Barendregt A.; Kamerling J. P.; Heck A. J. Analyzing Protein Micro-Heterogeneity in Chicken Ovalbumin by High-Resolution Native Mass Spectrometry Exposes Qualitatively and Semi-Quantitatively 59 Proteoforms. Anal. Chem. 2013, 85, 12037–12045. 10.1021/ac403057y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füssl F.; Criscuolo A.; Cook K.; Scheffler K.; Bones J. Cracking Proteoform Complexity of Ovalbumin with Anion-Exchange Chromatography–High-Resolution Mass Spectrometry under Native Conditions. J. Proteome Res. 2019, 18, 3689–3702. 10.1021/acs.jproteome.9b00375. [DOI] [PubMed] [Google Scholar]
- Hayase T.; Rice K. G.; Dziegielewska K. M.; Kuhlenschmidt M.; Reilly T.; Lee Y. C. Comparison of N-Glycosides of Fetuins from Different Species and Human Alpha 2-Hs-Glycoprotein. Biochemistry 1992, 31, 4915–4921. 10.1021/bi00135a024. [DOI] [PubMed] [Google Scholar]
- Windwarder M.; Altmann F. Site-Specific Analysis of the O-Glycosylation of Bovine Fetuin by Electron-Transfer Dissociation Mass Spectrometry. J. Proteomics 2014, 108, 258–268. 10.1016/j.jprot.2014.05.022. [DOI] [PubMed] [Google Scholar]
- Lin Y. H.; Franc V.; Heck A. J. R. Similar Albeit Not the Same: In-Depth Analysis of Proteoforms of Human Serum, Bovine Serum, and Recombinant Human Fetuin. J. Proteome Res. 2018, 17, 2861–2869. 10.1021/acs.jproteome.8b00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science 2020, 369, 330–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; McLellan J. S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struwe W. B.; Stuckmann A.; Behrens A. J.; Pagel K.; Crispin M. Global N-Glycan Site Occupancy of Hiv-1 Gp120 by Metabolic Engineering and High-Resolution Intact Mass Spectrometry. ACS Chem. Biol. 2017, 12, 357–361. 10.1021/acschembio.6b00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical Benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. 10.1038/nbt.4305. [DOI] [PubMed] [Google Scholar]
- Matsumiya S.; Yamaguchi Y.; Saito J.; Nagano M.; Sasakawa H.; Otaki S.; Satoh M.; Shitara K.; Kato K. Structural Comparison of Fucosylated and Nonfucosylated Fc Fragments of Human Immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. 10.1016/j.jmb.2007.02.034. [DOI] [PubMed] [Google Scholar]
- Hodoniczky J.; Zheng Y. Z.; James D. C. Control of Recombinant Monoclonal Antibody Effector Functions by Fc N-Glycan Remodeling in Vitro. Biotechnol. Prog. 2005, 21, 1644–1652. 10.1021/bp050228w. [DOI] [PubMed] [Google Scholar]
- Alter G.; Ottenhoff T. H. M.; Joosten S. A. Antibody Glycosylation in Inflammation, Disease and Vaccination. Semin. Immunol. 2018, 39, 102–110. 10.1016/j.smim.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajba L.; Szekrenyes A.; Borza B.; Guttman A. On the Glycosylation Aspects of Biosimilarity. Drug Discovery Today 2018, 23, 616–625. 10.1016/j.drudis.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Schellekens H.; Moors E. Biosimilars or Semi-Similars?. Nat. Biotechnol. 2015, 33, 19–20. 10.1038/nbt.3083. [DOI] [PubMed] [Google Scholar]
- Beck A.; Sanglier-Cianferani S.; Van Dorsselaer A. Biosimilar, Biobetter, and Next Generation Antibody Characterization by Mass Spectrometry. Anal. Chem. 2012, 84, 4637–4646. 10.1021/ac3002885. [DOI] [PubMed] [Google Scholar]
- Thompson N. J.; Rosati S.; Heck A. J. R. Performing Native Mass Spectrometry Analysis on Therapeutic Antibodies. Methods 2014, 65, 11–17. 10.1016/j.ymeth.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Tassi M.; De Vos J.; Chatterjee S.; Sobott F.; Bones J.; Eeltink S. Advances in Native High-Performance Liquid Chromatography and Intact Mass Spectrometry for the Characterization of Biopharmaceutical Products. J. Sep. Sci. 2018, 41, 125–144. 10.1002/jssc.201700988. [DOI] [PubMed] [Google Scholar]
- Struwe W. B.; Robinson C. V. Relating Glycoprotein Structural Heterogeneity to Function - Insights from Native Mass Spectrometry. Curr. Opin. Struct. Biol. 2019, 58, 241–248. 10.1016/j.sbi.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati S.; Yang Y.; Barendregt A.; Heck A. J. R. Detailed Mass Analysis of Structural Heterogeneity in Monoclonal Antibodies Using Native Mass Spectrometry. Nat. Protoc. 2014, 9, 967–976. 10.1038/nprot.2014.057. [DOI] [PubMed] [Google Scholar]
- Parsons T. B.; Struwe W. B.; Gault J.; Yamamoto K.; Taylor T. A.; Raj R.; Wals K.; Mohammed S.; Robinson C. V.; Benesch J. L. P.; et al. Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed. 2016, 55, 2361–2367. 10.1002/anie.201508723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schoot J. M. S.; Fennemann F. L.; Valente M.; Dolen Y.; Hagemans I. M.; Becker A. M. D.; Le Gall C. M.; van Dalen D.; Cevirgel A.; van Bruggen J. A. C.; et al. Functional Diversification of Hybridoma-Produced Antibodies by CRISPR/HDR Genomic Engineering. Sci. Adv. 2019, 5, eaaw1822 10.1126/sciadv.aaw1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson N. J.; Hendriks L. J. A.; de Kruif J.; Throsby M.; Heck A. J. R. Complex Mixtures of Antibodies Generated from a Single Production Qualitatively and Quantitatively Evaluated by Native Orbitrap Mass Spectrometry. MAbs 2014, 6, 197–203. 10.4161/mabs.27126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimer S.; Ben-Nissan G.; Sharon M. Direct Characterization of Overproduced Proteins by Native Mass Spectrometry. Nat. Protoc. 2020, 15, 236–265. 10.1038/s41596-019-0233-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser-Skala W.; Wohlschlager T.; Regl C.; Huber C. G. A Simple Strategy to Eliminate Hexosylation Bias in the Relative Quantification of N-Glycosylation in Biopharmaceuticals. Angew. Chem., Int. Ed. 2020, 59, 16225–16232. 10.1002/anie.202002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skala W.; Wohlschlager T.; Senn S.; Huber G. E.; Huber C. G. MoFi: A Software Tool for Annotating Glycoprotein Mass Spectra by Integrating Hybrid Data from the Intact Protein and Glycopeptide Level. Anal. Chem. 2018, 90, 5728–5736. 10.1021/acs.analchem.8b00019. [DOI] [PubMed] [Google Scholar]
- Brücher D.; Franc V.; Smith S. N.; Heck A. J. R.; Plückthun A. Malignant Tissues Produce Divergent Antibody Glycosylation of Relevance for Cancer Gene Therapy Effectiveness. MAbs 2020, 12, 1792084. 10.1080/19420862.2020.1792084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandra K.; Vandenheede I.; Sandra P. Modern Chromatographic and Mass Spectrometric Techniques for Protein Biopharmaceutical Characterization. J. Chromatogr. A 2014, 1335, 81–103. 10.1016/j.chroma.2013.11.057. [DOI] [PubMed] [Google Scholar]
- Bailey A. O.; Han G.; Phung W.; Gazis P.; Sutton J.; Josephs J. L.; Sandoval W. Charge Variant Native Mass Spectrometry Benefits Mass Precision and Dynamic Range of Monoclonal Antibody Intact Mass Analysis. MAbs 2018, 10, 1214–1225. 10.1080/19420862.2018.1521131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phung W.; Han G.; Polderdijk S. G. I.; Dillon M.; Shatz W.; Liu P.; Wei B.; Suresh P.; Fischer D.; Spiess C.; et al. Characterization of Bispecific and Mispaired IgGs by Native Charge-Variant Mass Spectrometry. Int. J. Mass Spectrom. 2019, 446, 116229. 10.1016/j.ijms.2019.116229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füssl F.; Cook K.; Scheffler K.; Farrell A.; Mittermayr S.; Bones J. Charge Variant Analysis of Monoclonal Antibodies Using Direct Coupled pH Gradient Cation Exchange Chromatography to High-Resolution Native Mass Spectrometry. Anal. Chem. 2018, 90, 4669–4676. 10.1021/acs.analchem.7b05241. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Liu A. P.; Wang S.; Daly T. J.; Li N. Ultrasensitive Characterization of Charge Heterogeneity of Therapeutic Monoclonal Antibodies Using Strong Cation Exchange Chromatography Coupled to Native Mass Spectrometry. Anal. Chem. 2018, 90, 13013–13020. 10.1021/acs.analchem.8b03773. [DOI] [PubMed] [Google Scholar]
- Ma F.; Raoufi F.; Bailly M. A.; Fayadat-Dilman L.; Tomazela D. Hyphenation of Strong Cation Exchange Chromatography to Native Mass Spectrometry for High Throughput Online Characterization of Charge Heterogeneity of Therapeutic Monoclonal Antibodies. mAbs 2020, 12, 1763762. 10.1080/19420862.2020.1763762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Xing T.; Wang S.; Daly T. J.; Li N. Online Coupling of Analytical Hydrophobic Interaction Chromatography with Native Mass Spectrometry for the Characterization of Monoclonal Antibodies and Related Products. J. Pharm. Biomed. Anal. 2020, 186, 113313. 10.1016/j.jpba.2020.113313. [DOI] [PubMed] [Google Scholar]
- Schachner L.; Han G.; Dillon M.; Zhou J.; McCarty L.; Ellerman D.; Yin Y.; Spiess C.; Lill J. R.; Carter P. J.; et al. Characterization of Chain Pairing Variants of Bispecific IgG Expressed in a Single Host Cell by High-Resolution Native and Denaturing Mass Spectrometry. Anal. Chem. 2016, 88, 12122–12127. 10.1021/acs.analchem.6b02866. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Xing T.; Wang S.; Daly T. J.; Li N. Coupling Mixed-Mode Size Exclusion Chromatography with Native Mass Spectrometry for Sensitive Detection and Quantitation of Homodimer Impurities in Bispecific IgG. Anal. Chem. 2019, 91, 11417–11424. 10.1021/acs.analchem.9b02793. [DOI] [PubMed] [Google Scholar]
- Gomes F. P.; Yates J. R. III Recent Trends of Capillary Electrophoresis-Mass Spectrometry in Proteomics Research. Mass Spectrom. Rev. 2019, 38, 445–460. 10.1002/mas.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov A. M.; Viner R.; Santos M. R.; Horn D. M.; Bern M.; Karger B. L.; Ivanov A. R. Analysis of Proteins, Protein Complexes, and Organellar Proteomes Using Sheathless Capillary Zone Electrophoresis - Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 2614–2634. 10.1007/s13361-017-1781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov A. M.; Zang L.; Sebastiano R.; Santos M. R.; Bush D. R.; Karger B. L.; Ivanov A. R. Complementary Middle-Down and Intact Monoclonal Antibody Proteoform Characterization by Capillary Zone Electrophoresis - Mass Spectrometry. Electrophoresis 2018, 39, 2069–2082. 10.1002/elps.201800067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füssl F.; Trappe A.; Carillo S.; Jakes C.; Bones J. Comparative Elucidation of Cetuximab Heterogeneity on the Intact Protein Level by Cation Exchange Chromatography and Capillary Electrophoresis Coupled to Mass Spectrometry. Anal. Chem. 2020, 92, 5431–5438. 10.1021/acs.analchem.0c00185. [DOI] [PubMed] [Google Scholar]
- Carillo S.; Jakes C.; Bones J. In-Depth Analysis of Monoclonal Antibodies Using Microfluidic Capillary Electrophoresis and Native Mass Spectrometry. J. Pharm. Biomed. Anal. 2020, 185, 113218. 10.1016/j.jpba.2020.113218. [DOI] [PubMed] [Google Scholar]
- de Goeij B. E.; Lambert J. M. New Developments for Antibody-Drug Conjugate-Based Therapeutic Approaches. Curr. Opin. Immunol. 2016, 40, 14–23. 10.1016/j.coi.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Valliere-Douglass J. F.; McFee W. A.; Salas-Solano O. Native Intact Mass Determination of Antibodies Conjugated with Monomethyl Auristatin E and F at Interchain Cysteine Residues. Anal. Chem. 2012, 84, 2843–2849. 10.1021/ac203346c. [DOI] [PubMed] [Google Scholar]
- Jones J.; Pack L.; Hunter J. H.; Valliere-Douglass J. F. Native Size-Exclusion Chromatography-Mass Spectrometry: Suitability for Antibody-Drug Conjugate Drug-to-Antibody Ratio Quantitation across a Range of Chemotypes and Drug-Loading Levels. MAbs 2020, 12, 1682895. 10.1080/19420862.2019.1682895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debaene F.; Boeuf A.; Wagner-Rousset E.; Colas O.; Ayoub D.; Corvaia N.; Van Dorsselaer A.; Beck A.; Cianferani S. Innovative Native MS Methodologies for Antibody Drug Conjugate Characterization: High Resolution Native MS and IM-MS for Average DAR and DAR Distribution Assessment. Anal. Chem. 2014, 86, 10674–10683. 10.1021/ac502593n. [DOI] [PubMed] [Google Scholar]
- Gautier V.; Boumeester A. J.; Lössl P.; Heck A. J. Lysine Conjugation Properties in Human IgGs Studied by Integrating High-Resolution Native Mass Spectrometry and Bottom-up Proteomics. Proteomics 2015, 15, 2756–2765. 10.1002/pmic.201400462. [DOI] [PubMed] [Google Scholar]
- Botzanowski T.; Erb S.; Hernandez-Alba O.; Ehkirch A.; Colas O.; Wagner-Rousset E.; Rabuka D.; Beck A.; Drake P. M.; Cianferani S. Insights from Native Mass Spectrometry Approaches for Top- and Middle- Level Characterization of Site-Specific Antibody-Drug Conjugates. MAbs 2017, 9, 801–811. 10.1080/19420862.2017.1316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. N.; Sasaki H.; Lopez L.; Fukuda M. Survival of Recombinant Erythropoietin in the Circulation: The Role of Carbohydrates. Blood 1989, 73, 84–89. 10.1182/blood.V73.1.84.84. [DOI] [PubMed] [Google Scholar]
- Erbayraktar S.; Grasso G.; Sfacteria A.; Xie Q. W.; Coleman T.; Kreilgaard M.; Torup L.; Sager T.; Erbayraktar Z.; Gokmen N.; et al. Asialoerythropoietin Is a Nonerythropoietic Cytokine with Broad Neuroprotective Activity in Vivo. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6741–6746. 10.1073/pnas.1031753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet F.; Paubel P.; Fusier I.; Cordonnier A. L.; Sinegre M.; Le Pen C. Biosimilar Versus Patented Erythropoietins: Learning from 5 Years of European and Japanese Experience. Appl. Health. Econ. Health Policy 2015, 13, 47–59. 10.1007/s40258-014-0125-6. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Liu F.; Franc V.; Halim L. A.; Schellekens H.; Heck A. J. Hybrid Mass Spectrometry Approaches in Glycoprotein Analysis and Their Usage in Scoring Biosimilarity. Nat. Commun. 2016, 7, 13397. 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čaval T.; Tian W.; Yang Z.; Clausen H.; Heck A. J. R. Direct Quality Control of Glycoengineered Erythropoietin Variants. Nat. Commun. 2018, 9, 3342. 10.1038/s41467-018-05536-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Wang S.; Halim A.; Schulz M. A.; Frodin M.; Rahman S. H.; Vester-Christensen M. B.; Behrens C.; Kristensen C.; Vakhrushev S. Y.; et al. Engineered Cho Cells for Production of Diverse, Homogeneous Glycoproteins. Nat. Biotechnol. 2015, 33, 842–844. 10.1038/nbt.3280. [DOI] [PubMed] [Google Scholar]
- Wohlschlager T.; Scheffler K.; Forstenlehner I. C.; Skala W.; Senn S.; Damoc E.; Holzmann J.; Huber C. G. Native Mass Spectrometry Combined with Enzymatic Dissection Unravels Glycoform Heterogeneity of Biopharmaceuticals. Nat. Commun. 2018, 9, 1713. 10.1038/s41467-018-04061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebede M.; Di Marco F.; Esser-Skala W.; Hennig R.; Wohlschlager T.; Huber C. G. Exploring the Chemical Space of Protein Glycosylation in Noncovalent Protein Complexes: An Expedition Along Different Structural Levels of Human Chorionic Gonadotropin Employing Mass Spectrometry. Anal. Chem. 2021, 93, 10424–10434. 10.1021/acs.analchem.1c02199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Rudnick P. A.; Martinez M. Y.; Cheek K. L.; Stein S. E.; Slebos R. J.; Liebler D. C. Depletion of Abundant Plasma Proteins and Limitations of Plasma Proteomics. J. Proteome Res. 2010, 9, 4982–4991. 10.1021/pr100646w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer P. E.; Kulak N. A.; Pichler G.; Holdt L. M.; Teupser D.; Mann M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016, 2, 185–195. 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- Toby T. K.; Fornelli L.; Srzentic K.; DeHart C. J.; Levitsky J.; Friedewald J.; Kelleher N. L. A Comprehensive Pipeline for Translational Top-Down Proteomics from a Single Blood Draw. Nat. Protoc. 2019, 14, 119–152. 10.1038/s41596-018-0085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. L. The Clinical Plasma Proteome: A Survey of Clinical Assays for Proteins in Plasma and Serum. Clin. Chem. 2010, 56, 177–185. 10.1373/clinchem.2009.126706. [DOI] [PubMed] [Google Scholar]
- Doherty M.; Theodoratou E.; Walsh I.; Adamczyk B.; Stockmann H.; Agakov F.; Timofeeva M.; Trbojevic-Akmacic I.; Vuckovic F.; Duffy F.; et al. Plasma N-Glycans in Colorectal Cancer Risk. Sci. Rep. 2018, 8, 8655. 10.1038/s41598-018-26805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Patwa T. H.; Xu L.; Shedden K.; Misek D. E.; Tuck M.; Jin G.; Ruffin M. T.; Turgeon D. K.; Synal S.; et al. Plasma Glycoprotein Profiling for Colorectal Cancer Biomarker Identification by Lectin Glycoarray and Lectin Blot. J. Proteome Res. 2008, 7, 1693–1703. 10.1021/pr700706s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavic T.; Dilber D.; Kifer D.; Selak N.; Keser T.; Ljubicic D.; Vukic Dugac A.; Lauc G.; Rumora L.; Gornik O. N-Glycosylation Patterns of Plasma Proteins and Immunoglobulin G in Chronic Obstructive Pulmonary Disease. J. Transl. Med. 2018, 16, 323. 10.1186/s12967-018-1695-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc V.; Yang Y.; Heck A. J. Proteoform Profile Mapping of the Human Serum Complement Component C9 Revealing Unexpected New Features of N-, O-, and C-Glycosylation. Anal. Chem. 2017, 89, 3483–3491. 10.1021/acs.analchem.6b04527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc V.; Zhu J.; Heck A. J. R. Comprehensive Proteoform Characterization of Plasma Complement Component C8αβγ by Hybrid Mass Spectrometry Approaches. J. Am. Soc. Mass Spectrom. 2018, 29, 1099–1110. 10.1007/s13361-018-1901-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Struwe W. B.; Harvey D. J.; Ferguson M. A. J.; Robinson C. V. N-Glycan Microheterogeneity Regulates Interactions of Plasma Proteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 8763–8768. 10.1073/pnas.1807439115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Li J.; Struwe W. B.; Robinson C. V. Probing N-Glycoprotein Microheterogeneity by Lectin Affinity Purification-Mass Spectrometry Analysis. Chem. Sci. 2019, 10, 5146–5155. 10.1039/C9SC00360F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-H.; Zhu J.; Meijer S.; Franc V.; Heck A. J. R. Glycoproteogenomics: A Frequent Gene Polymorphism Affects the Glycosylation Pattern of the Human Serum Fetuin/Alpha-2-Hs-Glycoprotein. Mol. Cell. Proteomics 2019, 18, 1479–1490. 10.1074/mcp.RA119.001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čaval T.; Lin Y.-H.; Varkila M.; Reiding K. R.; Bonten M. J. M.; Cremer O. L.; Franc V.; Heck A. J. R. Glycoproteoform Profiles of Individual Patients’ Plasma Alpha-1-Antichymotrypsin Are Unique and Extensively Remodeled Following a Septic Episode. Front. Immunol. 2021, 11, 608466. 10.3389/fimmu.2020.608466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye B.; Cramer D. W.; Skates S. J.; Gygi S. P.; Pratomo V.; Fu L.; Horick N. K.; Licklider L. J.; Schorge J. O.; Berkowitz R. S.; et al. Haptoglobin-Alpha Subunit as Potential Serum Biomarker in Ovarian Cancer: Identification and Characterization Using Proteomic Profiling and Mass Spectrometry. Clin. Cancer Res. 2003, 9, 2904–2911. [PubMed] [Google Scholar]
- Kruger A. J.; Yang C.; Tam S. W.; Hinerfeld D.; Evans J. E.; Green K. M.; Leszyk J.; Yang K.; Guberski D. L.; Mordes J. P.; et al. Haptoglobin as an Early Serum Biomarker of Virus-Induced Autoimmune Type 1 Diabetes in Biobreeding Diabetes Resistant and Lew1. Wr1 Rats. Exp. Biol. Med. (London, U. K.) 2010, 235, 1328–1337. 10.1258/ebm.2010.010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Chen Z.; Zhang J.; An M.; Wu J.; Yu Q.; Skilton S. J.; Bern M.; Ilker Sen K.; Li L.; et al. Differential Quantitative Determination of Site-Specific Intact N-Glycopeptides in Serum Haptoglobin between Hepatocellular Carcinoma and Cirrhosis Using LC-EThcD-MS/MS. J. Proteome Res. 2018, 18, 359–371. 10.1021/acs.jproteome.8b00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Shang S.; Li W.; Qin X.; Liu Y. Insights on N-Glycosylation of Human Haptoglobin and Its Association with Cancers. Glycobiology 2016, 26, 684–692. 10.1093/glycob/cww016. [DOI] [PubMed] [Google Scholar]
- Tamara S.; Franc V.; Heck A. J. R. A Wealth of Genotype-Specific Proteoforms Fine-Tunes Hemoglobin Scavenging by Haptoglobin. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 15554–15564. 10.1073/pnas.2002483117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler P. W.; Humar R.; Schaer D. J. Haptoglobin Therapeutics and Compartmentalization of Cell-Free Hemoglobin Toxicity. Trends Mol. Med. 2020, 26, 683–697. 10.1016/j.molmed.2020.02.004. [DOI] [PubMed] [Google Scholar]
- Kleppe R.; Rosati S.; Jorge-Finnigan A.; Alvira S.; Ghorbani S.; Haavik J.; Valpuesta J. M.; Heck A. J.; Martinez A. Phosphorylation Dependence and Stoichiometry of the Complex Formed by Tyrosine Hydroxylase and 14-3-3γ. Mol. Cell. Proteomics 2014, 13, 2017–2030. 10.1074/mcp.M113.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Lössl P.; Gautier V.; Marino F.; Yamashita M.; Conti E.; Scholten A.; Heck A. J. Simultaneous Assessment of Kinetic, Site-Specific, and Structural Aspects of Enzymatic Protein Phosphorylation. Angew. Chem., Int. Ed. 2014, 53, 9660–9664. 10.1002/anie.201404637. [DOI] [PubMed] [Google Scholar]
- Lössl P.; Brunner A. M.; Liu F.; Leney A. C.; Yamashita M.; Scheltema R. A.; Heck A. J. Deciphering the Interplay among Multisite Phosphorylation, Interaction Dynamics, and Conformational Transitions in a Tripartite Protein System. ACS Cent. Sci. 2016, 2, 445–455. 10.1021/acscentsci.6b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul Azeez K. R.; Chatterjee S.; Yu C.; Golub T. R.; Sobott F.; Elkins J. M. Structural Mechanism of Synergistic Activation of Aurora Kinase B/C by Phosphorylated INCENP. Nat. Commun. 2019, 10, 3166. 10.1038/s41467-019-11085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potel C. M.; Fasci D.; Heck A. J. R. Mix and Match of the Tumor Metastasis Suppressor Nm23 Protein Isoforms in Vitro and in Vivo. FEBS J. 2018, 285, 2856–2868. 10.1111/febs.14525. [DOI] [PubMed] [Google Scholar]
- Nguyen H. H.; Park J.; Kang S.; Kim M. Surface Plasmon Resonance: A Versatile Technique for Biosensor Applications. Sensors 2015, 15, 10481–10510. 10.3390/s150510481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puiu M.; Bala C. SPR and SPR Imaging: Recent Trends in Developing Nanodevices for Detection and Real-Time Monitoring of Biomolecular Events. Sensors 2016, 16, 870. 10.3390/s16060870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield N. J. Using Circular Dichroism Collected as a Function of Temperature to Determine the Thermodynamics of Protein Unfolding and Binding Interactions. Nat. Protoc. 2006, 1, 2527–2535. 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y. Applications of Isothermal Titration Calorimetry in Protein Science. Acta Biochim. Biophys. Sin. 2008, 40, 565–576. 10.1111/j.1745-7270.2008.00437.x. [DOI] [PubMed] [Google Scholar]
- Ganem B.; Li Y. T.; Henion J. D. Detection of Noncovalent Receptor-Ligand Complexes by Mass Spectrometry. J. Am. Chem. Soc. 1991, 113, 6294–6296. 10.1021/ja00016a069. [DOI] [Google Scholar]
- Loo R. R. O.; Goodlett D. R.; Smith R. D.; Loo J. A. Observation of a Noncovalent Ribonuclease S-Protein/S-Peptide Complex by Electrospray Ionization Mass Spectrometry. J. Am. Chem. Soc. 1993, 115, 4391–4392. 10.1021/ja00063a079. [DOI] [Google Scholar]
- Jørgensen T. J. D.; Roepstorff P.; Heck A. J. R. Direct Determination of Solution Binding Constants for Noncovalent Complexes between Bacterial Cell Wall Peptide Analogues and Vancomycin Group Antibiotics by Electrospray Ionization Mass Spectrometry. Anal. Chem. 1998, 70, 4427–4432. 10.1021/ac980563h. [DOI] [Google Scholar]
- Jørgensen T. J. D.; Staroske T.; Roepstorff P.; Williams D. H.; Heck A. J. R. Subtle Differences in Molecular Recognition between Modified Glycopeptide Antibiotics and Bacterial Receptor Peptides Identified by Electrospray Ionization Mass Spectrometry. J. Chem. Soc., Perkin Trans. 2 1999, 1859–1863. 10.1039/a904504j. [DOI] [Google Scholar]
- Cheng X.; Chen R.; Bruce J. E.; Schwartz B. L.; Anderson G. A.; Hofstadler S. A.; Gale D. C.; Smith R. D.; Gao J.; Sigal G. B.; et al. Using Electrospray Ionization Fticr Mass Spectrometry to Study Competitive Binding of Inhibitors to Carbonic Anhydrase. J. Am. Chem. Soc. 1995, 117, 8859–8860. 10.1021/ja00139a023. [DOI] [Google Scholar]
- Feng X.; Liu B.-F.; Li J.; Liu X. Advances in Coupling Microfluidic Chips to Mass Spectrometry. Mass Spectrom. Rev. 2015, 34, 535–557. 10.1002/mas.21417. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Van Pelt C. K.; Wilson D. B. Quantitative Determination of Noncovalent Binding Interactions Using Automated Nanoelectrospray Mass Spectrometry. Anal. Chem. 2003, 75, 3010–3018. 10.1021/ac034089d. [DOI] [PubMed] [Google Scholar]
- Daniel J. M.; Friess S. D.; Rajagopalan S.; Wendt S.; Zenobi R. Quantitative Determination of Noncovalent Binding Interactions Using Soft Ionization Mass Spectrometry. Int. J. Mass Spectrom. 2002, 216, 1–27. 10.1016/S1387-3806(02)00585-7. [DOI] [Google Scholar]
- Peschke M.; Verkerk U. H.; Kebarle P. Features of the Esi Mechanism That Affect the Observation of Multiply Charged Noncovalent Protein Complexes and the Determination of the Association Constant by the Titration Method. J. Am. Soc. Mass Spectrom. 2004, 15, 1424–1434. 10.1016/j.jasms.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Schermann S. M.; Simmons D. A.; Konermann L. Mass Spectrometry-Based Approaches to Protein-Ligand Interactions. Expert Rev. Proteomics 2005, 2, 475–485. 10.1586/14789450.2.4.475. [DOI] [PubMed] [Google Scholar]
- Van Berkel G. J.; Asano K. G.; Schnier P. D. Electrochemical Processes in a Wire-in-a-Capillary Bulk-Loaded, Nano-Electrospray Emitter. J. Am. Soc. Mass Spectrom. 2001, 12, 853–862. 10.1016/S1044-0305(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Van Berkel G. J.; Kertesz V. Using the Electrochemistry of the Electrospray Ion Source. Anal. Chem. 2007, 79, 5510–5520. 10.1021/ac071944a. [DOI] [PubMed] [Google Scholar]
- Zhou S.; Prebyl B. S.; Cook K. D. Profiling Ph Changes in the Electrospray Plume. Anal. Chem. 2002, 74, 4885–4888. 10.1021/ac025960d. [DOI] [PubMed] [Google Scholar]
- Girod M.; Dagany X.; Antoine R.; Dugourd P. Relation between Charge State Distributions of Peptide Anions and Ph Changes in the Electrospray Plume. A Mass Spectrometry and Optical Spectroscopy Investigation. Int. J. Mass Spectrom. 2011, 308, 41–48. 10.1016/j.ijms.2011.07.020. [DOI] [Google Scholar]
- Konermann L. Addressing a Common Misconception: Ammonium Acetate as Neutral Ph “Buffer” for Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1827–1835. 10.1007/s13361-017-1739-3. [DOI] [PubMed] [Google Scholar]
- Cech N. B.; Enke C. G. Relating Electrospray Ionization Response to Nonpolar Character of Small Peptides. Anal. Chem. 2000, 72, 2717–2723. 10.1021/ac9914869. [DOI] [PubMed] [Google Scholar]
- Robinson C. V.; Chung E. W.; Kragelund B. B.; Knudsen J.; Aplin R. T.; Poulsen F. M.; Dobson C. M. Probing the Nature of Noncovalent Interactions by Mass Spectrometry. A Study of Protein-Coa Ligand Binding and Assembly. J. Am. Chem. Soc. 1996, 118, 8646–8653. 10.1021/ja960211x. [DOI] [Google Scholar]
- Wigger M.; Eyler J. R.; Benner S. A.; Li W.; Marshall A. G. Fourier Transform-Ion Cyclotron Resonance Mass Spectrometric Resolution, Identification, and Screening of Non-Covalent Complexes of Hck Src Homology 2 Domain Receptor and Ligands from a 324-Member Peptide Combinatorial Library. J. Am. Soc. Mass Spectrom. 2002, 13, 1162–1169. 10.1016/S1044-0305(02)00439-7. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Kaltashov I. A.; Eyles S. J. Indirect Assessment of Small Hydrophobic Ligand Binding to a Model Protein Using a Combination of ESI MS and HDX/ESI MS. J. Am. Soc. Mass Spectrom. 2003, 14, 506–515. 10.1016/S1044-0305(03)00135-1. [DOI] [PubMed] [Google Scholar]
- Gabelica V.; Galic N.; Rosu F.; Houssier C.; De Pauw E. Influence of Response Factors on Determining Equilibrium Association Constants of Non-Covalent Complexes by Electrospray Ionization Mass Spectrometry. J. Mass Spectrom. 2003, 38, 491–501. 10.1002/jms.459. [DOI] [PubMed] [Google Scholar]
- Tjernberg A.; Carnö S.; Oliv F.; Benkestock K.; Edlund P.-O.; Griffiths W. J.; Hallén D. Determination of Dissociation Constants for Protein-Ligand Complexes by Electrospray Ionization Mass Spectrometry. Anal. Chem. 2004, 76, 4325–4331. 10.1021/ac0497914. [DOI] [PubMed] [Google Scholar]
- Kempen E. C.; Brodbelt J. S. A Method for the Determination of Binding Constants by Electrospray Ionization Mass Spectrometry. Anal. Chem. 2000, 72, 5411–5416. 10.1021/ac000540e. [DOI] [PubMed] [Google Scholar]
- Nguyen G. T. H.; Tran T. N.; Podgorski M. N.; Bell S. G.; Supuran C. T.; Donald W. A. Nanoscale Ion Emitters in Native Mass Spectrometry for Measuring Ligand-Protein Binding Affinities. ACS Cent. Sci. 2019, 5, 308–318. 10.1021/acscentsci.8b00787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susa A. C.; Xia Z.; Williams E. R. Native Mass Spectrometry from Common Buffers with Salts That Mimic the Extracellular Environment. Angew. Chem., Int. Ed. 2017, 56, 7912–7915. 10.1002/anie.201702330. [DOI] [PubMed] [Google Scholar]
- Hu J.; Guan Q.-Y.; Wang J.; Jiang X.-X.; Wu Z.-Q.; Xia X.-H.; Xu J.-J.; Chen H.-Y. Effect of Nanoemitters on Suppressing the Formation of Metal Adduct Ions in Electrospray Ionization Mass Spectrometry. Anal. Chem. 2017, 89, 1838–1845. 10.1021/acs.analchem.6b04218. [DOI] [PubMed] [Google Scholar]
- Wortmann A.; Jecklin M. C.; Touboul D.; Badertscher M.; Zenobi R. Binding Constant Determination of High-Affinity Protein-Ligand Complexes by Electrospray Ionization Mass Spectrometry and Ligand Competition. J. Mass Spectrom. 2008, 43, 600–608. 10.1002/jms.1355. [DOI] [PubMed] [Google Scholar]
- Wang W.; Kitova E. N.; Klassen J. S. Influence of Solution and Gas Phase Processes on Protein-Carbohydrate Binding Affinities Determined by Nanoelectrospray Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2003, 75, 4945–4955. 10.1021/ac034300l. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kirkup C. E.; Pi N.; Leary J. A. Characterization of Noncovalent Protein-Ligand Complexes and Associated Enzyme Intermediates of Glcnac-6-O-Sulfotransferase by Electrospray Ionization Ft-Icr Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1400–1407. 10.1016/j.jasms.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Mathur S.; Badertscher M.; Scott M.; Zenobi R. Critical Evaluation of Mass Spectrometric Measurement of Dissociation Constants: Accuracy and Cross-Validation against Surface Plasmon Resonance and Circular Dichroism for the Calmodulin-Melittin System. Phys. Chem. Chem. Phys. 2007, 9, 6187–6198. 10.1039/b707946j. [DOI] [PubMed] [Google Scholar]
- Shoemaker G. K.; Soya N.; Palcic M. M.; Klassen J. S. Temperature-Dependent Cooperativity in Donor-Acceptor Substrate Binding to the Human Blood Group Glycosyltransferases. Glycobiology 2008, 18, 587–592. 10.1093/glycob/cwn043. [DOI] [PubMed] [Google Scholar]
- Soya N.; Shoemaker G. K.; Palcic M. M.; Klassen J. S. Comparative Study of Substrate and Product Binding to the Human Abo(H) Blood Group Glycosyltransferases. Glycobiology 2009, 19, 1224–1234. 10.1093/glycob/cwp114. [DOI] [PubMed] [Google Scholar]
- Jecklin M. C.; Schauer S.; Dumelin C. E.; Zenobi R. Label-Free Determination of Protein-Ligand Binding Constants Using Mass Spectrometry and Validation Using Surface Plasmon Resonance and Isothermal Titration Calorimetry. J. Mol. Recognit. 2009, 22, 319–329. 10.1002/jmr.951. [DOI] [PubMed] [Google Scholar]
- Liu L.; Kitova E. N.; Klassen J. S. Quantifying Protein-Fatty Acid Interactions Using Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 310–318. 10.1007/s13361-010-0032-5. [DOI] [PubMed] [Google Scholar]
- Hofstadler S. A.; Sannes-Lowery K. A. Applications of ESI-MS in Drug Discovery: Interrogation of Noncovalent Complexes. Nat. Rev. Drug Discovery 2006, 5, 585–595. 10.1038/nrd2083. [DOI] [PubMed] [Google Scholar]
- Deng G.; Sanyal G. Applications of Mass Spectrometry in Early Stages of Target Based Drug Discovery. J. Pharm. Biomed. Anal. 2006, 40, 528–538. 10.1016/j.jpba.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Vivat Hannah V.; Atmanene C; Zeyer D; Van Dorsselaer A; Sanglier-Cianférani S. Native MS: An ‘ESI’ Way to Support Structure- and Fragment-Based Drug Discovery. Future Med. Chem. 2010, 2, 35–50. 10.4155/fmc.09.141. [DOI] [PubMed] [Google Scholar]
- Pacholarz K. J.; Garlish R. A.; Taylor R. J.; Barran P. E. Mass Spectrometry Based Tools to Investigate Protein-Ligand Interactions for Drug Discovery. Chem. Soc. Rev. 2012, 41, 4335–4355. 10.1039/c2cs35035a. [DOI] [PubMed] [Google Scholar]
- Pedro L.; Quinn R. J. Native Mass Spectrometry in Fragment-Based Drug Discovery. Molecules 2016, 21, 984. 10.3390/molecules21080984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschweiler J. D.; Kerr R.; Rabuck-Gibbons J.; Ruotolo B. T. Sizing up Protein-Ligand Complexes: The Rise of Structural Mass Spectrometry Approaches in the Pharmaceutical Sciences. Annu. Rev. Anal. Chem. 2017, 10, 25–44. 10.1146/annurev-anchem-061516-045414. [DOI] [PubMed] [Google Scholar]
- Swayze E. E.; Jefferson E. A.; Sannes-Lowery K. A.; Blyn L. B.; Risen L. M.; Arakawa S.; Osgood S. A.; Hofstadler S. A.; Griffey R. H. SAR by MS: A Ligand Based Technique for Drug Lead Discovery against Structured RNA Targets. J. Med. Chem. 2002, 45, 3816–3819. 10.1021/jm0255466. [DOI] [PubMed] [Google Scholar]
- Maple H. J.; Garlish R. A.; Rigau-Roca L.; Porter J.; Whitcombe I.; Prosser C. E.; Kennedy J.; Henry A. J.; Taylor R. J.; Crump M. P.; et al. Automated Protein-Ligand Interaction Screening by Mass Spectrometry. J. Med. Chem. 2012, 55, 837–851. 10.1021/jm201347k. [DOI] [PubMed] [Google Scholar]
- Woods L. A.; Dolezal O.; Ren B.; Ryan J. H.; Peat T. S.; Poulsen S.-A. Native State Mass Spectrometry, Surface Plasmon Resonance, and X-ray Crystallography Correlate Strongly as a Fragment Screening Combination. J. Med. Chem. 2016, 59, 2192–2204. 10.1021/acs.jmedchem.5b01940. [DOI] [PubMed] [Google Scholar]
- Gavriilidou A. F. M.; Holding F. P.; Coyle J. E.; Zenobi R. Application of Native ESI-MS to Characterize Interactions between Compounds Derived from Fragment-Based Discovery Campaigns and Two Pharmaceutically Relevant Proteins. SLAS Discovery 2018, 23, 951–959. 10.1177/2472555218775921. [DOI] [PubMed] [Google Scholar]
- Ren C.; Bailey A. O.; VanderPorten E.; Oh A.; Phung W.; Mulvihill M. M.; Harris S. F.; Liu Y.; Han G.; Sandoval W. Quantitative Determination of Protein-Ligand Affinity by Size Exclusion Chromatography Directly Coupled to High-Resolution Native Mass Spectrometry. Anal. Chem. 2019, 91, 903–911. 10.1021/acs.analchem.8b03829. [DOI] [PubMed] [Google Scholar]
- Bovet C.; Wortmann A.; Eiler S.; Granger F.; Ruff M.; Gerrits B.; Moras D.; Zenobi R. Estrogen Receptor-Ligand Complexes Measured by Chip-Based Nanoelectrospray Mass Spectrometry: An Approach for the Screening of Endocrine Disruptors. Protein Sci. 2007, 16, 938–946. 10.1110/ps.062664107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jecklin M. C.; Touboul D.; Jain R.; Toole E. N.; Tallarico J.; Drueckes P.; Ramage P.; Zenobi R. Affinity Classification of Kinase Inhibitors by Mass Spectrometric Methods and Validation Using Standard IC50 Measurements. Anal. Chem. 2009, 81, 408–419. 10.1021/ac801782c. [DOI] [PubMed] [Google Scholar]
- Maple H. J.; Scheibner O.; Baumert M.; Allen M.; Taylor R. J.; Garlish R. A.; Bromirski M.; Burnley R. J. Application of the Exactive Plus EMR for Automated Protein-Ligand Screening by Non-Covalent Mass Spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 1561–1568. 10.1002/rcm.6925. [DOI] [PubMed] [Google Scholar]
- Mehmood S.; Marcoux J.; Gault J.; Quigley A.; Michaelis S.; Young S. G.; Carpenter E. P.; Robinson C. V. Mass Spectrometry Captures Off-Target Drug Binding and Provides Mechanistic Insights into the Human Metalloprotease Zmpste24. Nat. Chem. 2016, 8, 1152–1158. 10.1038/nchem.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavriilidou A. F. M.; Holding F. P.; Mayer D.; Coyle J. E.; Veprintsev D. B.; Zenobi R. Native Mass Spectrometry Gives Insight into the Allosteric Binding Mechanism of M2 Pyruvate Kinase to Fructose-1,6-Bisphosphate. Biochemistry 2018, 57, 1685–1689. 10.1021/acs.biochem.7b01270. [DOI] [PubMed] [Google Scholar]
- Root K.; Barylyuk K.; Schwab A.; Thelemann J.; Illarionov B.; Geist J. G.; Gräwert T.; Bacher A.; Fischer M.; Diederich F.; et al. Aryl Bis-Sulfonamides Bind to the Active Site of a Homotrimeric Isoprenoid Biosynthesis Enzyme IspF and Extract the Essential Divalent Metal Cation Cofactor. Chem. Sci. 2018, 9, 5976–5986. 10.1039/C8SC00814K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Baba T. J.; Lutomski C. A.; Kantsadi A. L.; Malla T. R.; John T.; Mikhailov V.; Bolla J. R.; Schofield C. J.; Zitzmann N.; Vakonakis I.; et al. Allosteric Inhibition of the SARS-CoV-2 Main Protease: Insights from Mass Spectrometry Based Assays. Angew. Chem., Int. Ed. 2020, 59, 23544–23548. 10.1002/anie.202010316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gülbakan B.; Barylyuk K.; Schneider P.; Pillong M.; Schneider G.; Zenobi R. Native Electrospray Ionization Mass Spectrometry Reveals Multiple Facets of Aptamer-Ligand Interactions: From Mechanism to Binding Constants. J. Am. Chem. Soc. 2018, 140, 7486–7497. 10.1021/jacs.7b13044. [DOI] [PubMed] [Google Scholar]
- Nguyen G. T. H.; Leung W. Y.; Tran T. N.; Wang H.; Murray V.; Donald W. A. Mechanism for the Binding of Netropsin to Hairpin DNA Revealed Using Nanoscale Ion Emitters in Native Mass Spectrometry. Anal. Chem. 2020, 92, 1130–1137. 10.1021/acs.analchem.9b04209. [DOI] [PubMed] [Google Scholar]
- Gülbakan B.; Barylyuk K.; Zenobi R. Determination of Thermodynamic and Kinetic Properties of Biomolecules by Mass Spectrometry. Curr. Opin. Biotechnol. 2015, 31, 65–72. 10.1016/j.copbio.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Daneshfar R.; Kitova E. N.; Klassen J. S. Determination of Protein-Ligand Association Thermochemistry Using Variable-Temperature Nanoelectrospray Mass Spectrometry. J. Am. Chem. Soc. 2004, 126, 4786–4787. 10.1021/ja0316972. [DOI] [PubMed] [Google Scholar]
- Cong X.; Liu Y.; Liu W.; Liang X.; Russell D. H.; Laganowsky A. Determining Membrane Protein-Lipid Binding Thermodynamics Using Native Mass Spectrometry. J. Am. Chem. Soc. 2016, 138, 4346–4349. 10.1021/jacs.6b01771. [DOI] [PubMed] [Google Scholar]
- Moghadamchargari Z.; Huddleston J.; Shirzadeh M.; Zheng X.; Clemmer D. E.; Raushel F. M.; Russell D. H.; Laganowsky A. Intrinsic Gtpase Activity of K-RAS Monitored by Native Mass Spectrometry. Biochemistry 2019, 58, 3396–3405. 10.1021/acs.biochem.9b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand A.; Rosu F.; Zenobi R.; Gabelica V. Thermal Denaturation of DNA G-Quadruplexes and Their Complexes with Ligands: Thermodynamic Analysis of the Multiple States Revealed by Mass Spectrometry. J. Am. Chem. Soc. 2018, 140, 12553–12565. 10.1021/jacs.8b07302. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Zhang J.; Yin S.; Loo J. A. Top-Down ESI-ECD-FT-ICR Mass Spectrometry Localizes Noncovalent Protein-Ligand Binding Sites. J. Am. Chem. Soc. 2006, 128, 14432–14433. 10.1021/ja063197p. [DOI] [PubMed] [Google Scholar]
- Yin S.; Loo J. A. Elucidating the Site of Protein-ATP Binding by Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 899–907. 10.1016/j.jasms.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Yin S.; Loo J. A. Top-Down Mass Spectrometry of Supercharged Native Protein-Ligand Complexes. Int. J. Mass Spectrom. 2011, 300, 118–122. 10.1016/j.ijms.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke D. J.; Murray E.; Hupp T.; Mackay C. L.; Langridge-Smith P. R. Mapping a Noncovalent Protein-Peptide Interface by Top-Down FTICR Mass Spectrometry Using Electron Capture Dissociation. J. Am. Soc. Mass Spectrom. 2011, 22, 1432–1440. 10.1007/s13361-011-0155-3. [DOI] [PubMed] [Google Scholar]
- Cammarata M. B.; Thyer R.; Rosenberg J.; Ellington A.; Brodbelt J. S. Structural Characterization of Dihydrofolate Reductase Complexes by Top-Down Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2015, 137, 9128–9135. 10.1021/jacs.5b04628. [DOI] [PubMed] [Google Scholar]
- Cammarata M.; Thyer R.; Lombardo M.; Anderson A.; Wright D.; Ellington A.; Brodbelt J. S. Characterization of Trimethoprim Resistant E. coli Dihydrofolate Reductase Mutants by Mass Spectrometry and Inhibition by Propargyl-Linked Antifolates. Chem. Sci. 2017, 8, 4062–4072. 10.1039/C6SC05235E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn J. B. Electrospray Wings for Molecular Elephants (Nobel Lecture). Angew. Chem., Int. Ed. 2003, 42, 3871–3894. 10.1002/anie.200300605. [DOI] [PubMed] [Google Scholar]
- Vimer S.; Ben-Nissan G.; Sharon M. Mass Spectrometry Analysis of Intact Proteins from Crude Samples. Anal. Chem. 2020, 92, 12741–12749. 10.1021/acs.analchem.0c02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; Wisniewski J. R.; Geiger T.; Cox J.; Kircher M.; Kelso J.; Pääbo S.; Mann M. Deep Proteome and Transcriptome Mapping of a Human Cancer Cell Line. Mol. Syst. Biol. 2011, 7, 548. 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The smaller the better. Nat. Methods 2008, 5, 457. 10.1038/nmeth0608-457. [DOI] [PubMed] [Google Scholar]
- Albrecht T. Single-Molecule Analysis with Solid-State Nanopores. Annu. Rev. Anal. Chem. 2019, 12, 371–387. 10.1146/annurev-anchem-061417-125903. [DOI] [PubMed] [Google Scholar]
- Moerner W. E. Single-Molecule Spectroscopy, Imaging, and Photocontrol: Foundations for Super-Resolution Microscopy (Nobel Lecture). Angew. Chem., Int. Ed. 2015, 54, 8067–8093. 10.1002/anie.201501949. [DOI] [PubMed] [Google Scholar]
- Sage E.; Brenac A.; Alava T.; Morel R.; Dupre C.; Hanay M. S.; Roukes M. L.; Duraffourg L.; Masselon C.; Hentz S. Neutral Particle Mass Spectrometry with Nanomechanical Systems. Nat. Commun. 2015, 6, 6482. 10.1038/ncomms7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip K. M.; Fischer N.; Paknia E.; Chari A.; Stark H. Atomic-Resolution Protein Structure Determination by Cryo-EM. Nature 2020, 587, 157–161. 10.1038/s41586-020-2833-4. [DOI] [PubMed] [Google Scholar]
- Henderson R. From Electron Crystallography to Single Particle Cryo-EM (Nobel Lecture). Angew. Chem., Int. Ed. 2018, 57, 10804–10825. 10.1002/anie.201802731. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Schuller J. M.; Wiegard A.; Lössl P.; Schmelling N.; Axmann I. M.; Plitzko J. M.; Forster F.; Heck A. J. Structures of the Cyanobacterial Circadian Oscillator Frozen in a Fully Assembled State. Science 2017, 355, 1181–1184. 10.1126/science.aag3218. [DOI] [PubMed] [Google Scholar]
- Lau A. M.; Politis A. Integrative Mass Spectrometry-Based Approaches for Modeling Macromolecular Assemblies. Methods Mol. Biol. 2021, 2247, 221–241. 10.1007/978-1-0716-1126-5_12. [DOI] [PubMed] [Google Scholar]
- Bond K.; Tsvetkova I. B.; Wang J. C.; Jarrold M. F.; Dragnea B. Virus Assembly Pathways: Straying Away but Not Too Far. Small 2020, 16, 2004475 10.1002/smll.202004475. [DOI] [PubMed] [Google Scholar]
- Olinares P. D. B.; Kang J. Y.; Llewellyn E.; Chiu C.; Chen J.; Malone B.; Saecker R. M.; Campbell E. A.; Darst S. A.; Chait B. T. Native Mass Spectrometry-Based Screening for Optimal Sample Preparation in Single-Particle Cryo-EM. Structure 2021, 29, 186–195. 10.1016/j.str.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]