

Abstract

Cells encode information in the sequence of biopolymers, such as nucleic acids, proteins, and glycans. Although glycans are essential to all living organisms, surprisingly little is known about the “sugar code” and the biological roles of these molecules. The reason glycobiology lags behind its counterparts dealing with nucleic acids and proteins lies in the complexity of carbohydrate structures, which renders their analysis extremely challenging. Building blocks that may differ only in the configuration of a single stereocenter, combined with the vast possibilities to connect monosaccharide units, lead to an immense variety of isomers, which poses a formidable challenge to conventional mass spectrometry. In recent years, however, a combination of innovative ion activation methods, commercialization of ion mobility–mass spectrometry, progress in gas-phase ion spectroscopy, and advances in computational chemistry have led to a revolution in mass spectrometry-based glycan analysis. The present review focuses on the above techniques that expanded the traditional glycomics toolkit and provided spectacular insight into the structure of these fascinating biomolecules. To emphasize the specific challenges associated with them, major classes of mammalian glycans are discussed in separate sections. By doing so, we aim to put the spotlight on the most important element of glycobiology: the glycans themselves.
1. Introduction
Carbohydrates—often referred to as glycans—are the most abundant organic polymers found on Earth. They are essential to all known living organisms and regulate a variety of vital functions.1 Structurally, glycans are composed of monosaccharide building blocks linked together via glycosidic bonds. The certainly most prominent form are polysaccharides: mostly linear polymers with a regular structure and a high degree of polymerization. They are highly abundant and omnipresent in our daily life, for example, in the form of starch during cooking or as cellulose in plant tissues.2 In biology, however, smaller but structurally more diverse structures are often found to play an important role. Glycosylation is the most abundant and most complex post-translational modification (PTM) found in proteins.3,4 Here, highly branched glycan structures consisting of 10 to 20 monosaccharide building blocks are covalently attached to certain residues in the protein chain. The involved glycans are usually exposed to the exterior of the protein, where they promote protein folding5,6 and regulate a variety of often remarkably specific functions, such as immune response7,8 and fertilization.9−11 Similarly complex structures can also occur as free oligosaccharides, for example in milk, where they serve as crucial constituents to develop and retain a healthy microbiota.12 Also the extracellular matrix (ECM) is crowded with sugars, in particular glycosaminoglycans (GAGs), that are attached to proteins as proteoglycans.13,14 Even though GAGs are usually linear assemblies of up to 250 repeating disaccharide units, they are diversely sulfated at different sites in the molecule, making them exceptionally complex and heterogeneous.
Example structures of abundant glycan classes are shown in Figure 1A. Unlike the biosynthesis of DNA (replication), RNA (transcription), or proteins (translation), glycan biosynthesis is not a template-driven process. Instead, glycans are assembled in a complex cascade of en bloc attachment and multiple consecutive de- and reglycosylation steps.15 The structural diversity in the resulting oligosaccharides is therefore vast and determined by multiple parameters (Figure 1B). The monosaccharide composition (I) describes the types of building blocks that are connected. Monosaccharides can be depicted either by chemical structures or by employing the simplified symbol nomenclature for glycans (SNFG, Figure 1C). In vertebrates, the number of monosaccharides is limited to d-glucose (Glu), d-galactose (Gal), d-mannose (Man), N-acetyl-d-glucosamine (GlcNAc), N-acetyl-d-galactosamine (GalNAc), d-glucuronic acid (GlcA), l-iduronic acid (IdoA), d-xylose (Xyl), l-fucose (Fuc), and N-acetyl-d-neuraminic acid (Neu5Ac). In invertebrates, plants, fungi, protists, bacteria, or archaea, on the other hand, this number is considerably larger, which tremendously increases structural complexity. The second important parameter is the connectivity (II), i.e., the regiochemistry of the glycosidic bond that connects the individual building blocks. In contrast to proteins and oligonucleotides, monosaccharides bear multiple glycosidic OH groups which all serve as possible linkage sites. Upon condensation of two monosaccharides, a new stereocenter emerges at the linkage site with two possible anomeric configurations (III). Finally, glycans are not necessarily assembled in a strictly linear fashion but may also exhibit one or more branching sites (IV).
Figure 1.
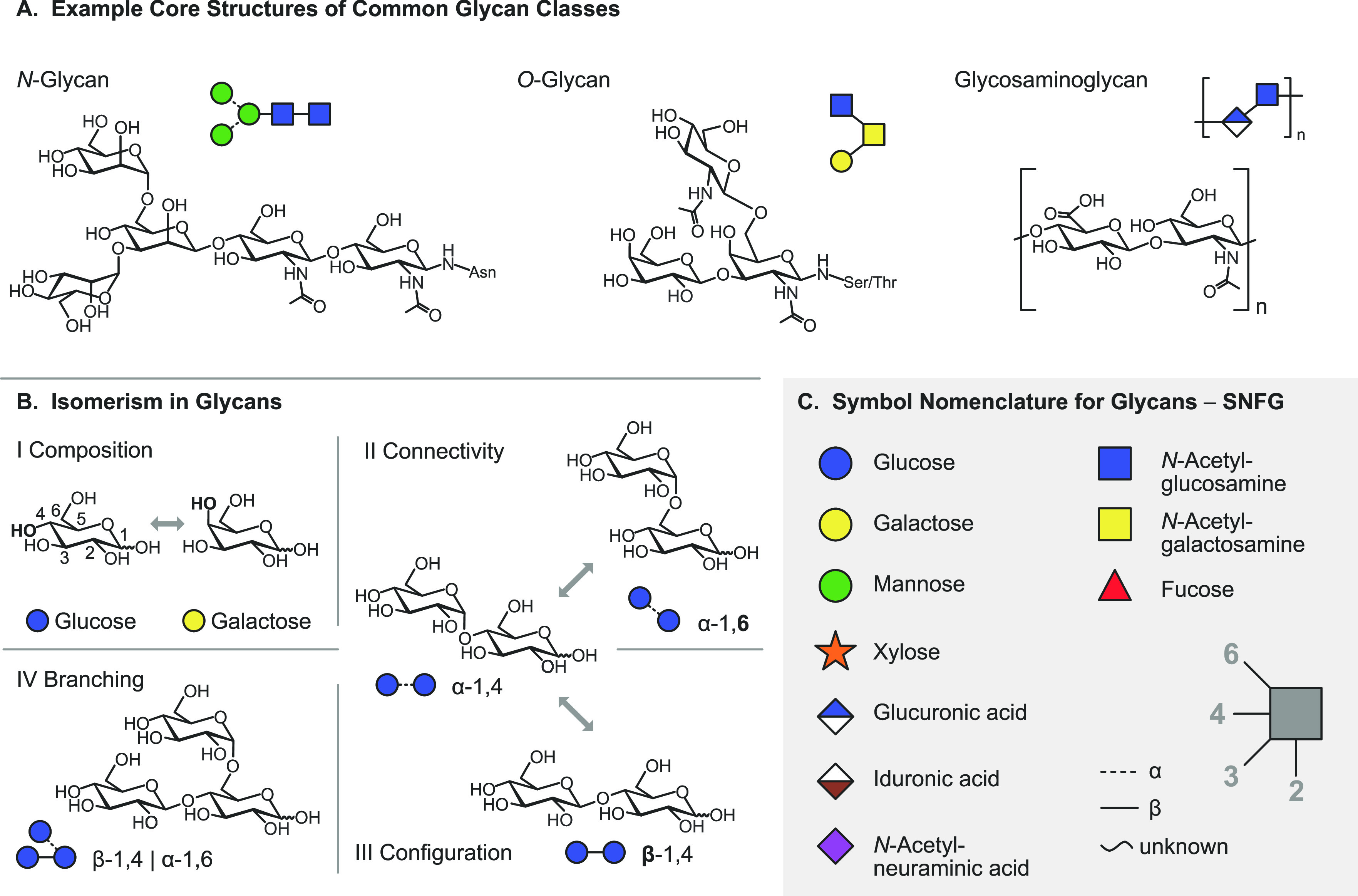
Representation of glycan structures and frequently occurring isomers. (A) Example core structures of N-glycans, O-glycans, and glycosaminoglycans are represented using chemical structures and equivalent symbol nomenclature. (B) Isomerism in glycans occurs on different levels: glycan composition, connectivity, configuration, and branching. (C) The symbol nomenclature for glycans offers simplified but unambiguous representations of complex glycan structures. Symbols are shown for the most abundant monosaccharides found in vertebrates.
All the above-mentioned structural parameters can lead to isomers—molecules that exhibit an identical atomic composition but differ in their structure. An analysis based on mass spectrometry (MS) therefore has inherent limitations to resolve and unambiguously identify the underlying molecular structure. Due to its enormous prevalence in proteomics16−20 and its exceptional sensitivity, however, MS is still a widely used and valuable tool for the analysis of glycans.21−24 The isomer problem is often addressed by coupling MS with orthogonal techniques.25−28 The most obvious of these combinations is certainly a separation of the analytes prior to MS analysis using liquid chromatography (LC)—again an approach that is extremely prevalent in other omics fields. However, even though of great use for certain applications, LC-MS-based approaches often struggle with the high polarity of glycans or the amphiphilic nature of glycoconjugates.
In recent years, a series of promising novel techniques directed at the specific needs of glycan analysis emerged.29 Many of these techniques are directly implemented within the mass spectrometer, enabling an improved duty cycle while still providing sufficient diagnostic potential. High energy activation methods such as electron-based dissociation (ExD)30,31 or ultraviolet photodissociation (UVPD)32 help to disentangle complex oligosaccharide structures based on more informative fragmentation patterns. Approaches such as ion mobility spectrometry (IMS) and gas-phase spectroscopy, on the other hand, are directly sensitive to the structure of the investigated molecule.33−36 Here we discuss recent developments in the structural analysis of glycans and glycoconjugates using MS-based techniques. As the present review is focused on the above-mentioned emerging techniques, traditional slow-heating methods such as collision-induced dissociation (CID) are addressed only briefly. The selection of references also reflects the growing importance of electrospray ionization (ESI) in the field, opposed to matrix-assisted laser desorption/ionization (MALDI) that dominated MS-based glycan analysis in the 1990s and early 2000s.
In the first part of the review (Section 2), general concepts and the fundamentals of each of the techniques will be described briefly, while the second part (Sections 3 to 8) addresses specific aspects of individual glycan and glycoconjugate classes. Sections dedicated to the different molecule classes may be read independently of each other, and the reader is encouraged to freely select those in their interest in any desired order.
2. Techniques
2.1. Mass Spectrometry
Natural glycans often occur in complex matrices and thus require isolation and purification prior to MS analysis. N- and O-glycans, for example, need to be released from glycoproteins by enzymatic or chemical methods for subsequent glycan analysis. Because glycan enrichment and purification strategies constitute a crucial prerequisite for the quality of MS analyses, they are briefly mentioned here to refer the reader to relevant literature. Commonly employed methods for glycan purification and enrichment include solid-phase extraction, liquid-phase extraction, chromatography, and electrophoresis.37,38 Chromatographic and electrophoretic methods routinely employed for glycan separation are reversed-phase high-performance liquid chromatography (RP-HPLC), hydrophilic interaction liquid chromatography (HILIC), porous graphitized carbon (PGC) chromatography, strong anion exchange (SAX) chromatography, and capillary electrophoresis (CE), which are discussed in more detail in excellent recent reviews.28,39−43 Solid-phase isolation can be combined with reversible or irreversible chemical coupling strategies, such as boronic acid capturing and hydrazide capturing, or exploits specific noncovalent interactions in lectin-capturing approaches.44 Following the extraction from natural sources, glycans are often derivatized to enhance ionization, detectability, and separation. Derivatization strategies for glycans are comprehensively discussed in the related literature.45−47
The present review focuses on developments accomplished in the past 15 years in the field of electrospray ionization–mass spectrometry (ESI-MS) based techniques for the structural characterization of glycans. Owing to the development of novel ion activation methods, the commercialization of ion mobility–mass spectrometry (IM-MS), and the introduction of gas-phase ion spectroscopy to the field of glycomics, the amount and specificity of structural information obtainable by MS-based methods has significantly increased in this period. The ESI process and various ion activation methods are discussed herein, followed by a brief description of IM-MS and the emerging gas-phase spectroscopic techniques in the infrared (IR) and ultraviolet (UV) region, which gain increasing importance for the structural analysis of glycans.
2.1.1. Ionization
Mass spectrometry became widely applicable to the analysis of biomolecules with the advent of soft ionization techniques in the late 1980s. Today, the main ionization techniques employed for the transfer of intact glycans into the gas phase are matrix-assisted laser desorption/ionization (MALDI)48 and ESI.49,50 While around the turn of the millennium MALDI was regarded as the method of choice to generate gas-phase carbohydrate ions, in the past decade ESI has become the dominant ion source in MS-based glycan analysis, which is also reflected by the works selected herein. For the latest developments in glycan analysis with specific focus on MALDI, the reader is referred to excellent recent reviews dedicated to the subject.51,52 MALDI is often used to ionize glycans directly from tissue samples, whereas ESI enables ionization of glycans from solution and is thus compatible with liquid chromatography (LC) online coupling. Upon application of a high voltage between the emitter containing the sample solution and the entrance of the mass spectrometer, charge separation leads to the formation of a Taylor cone at the tip of the emitter, from which droplets are released. Evaporation of solvent and charge repulsion lead to a Coulomb explosion, which generates multiple smaller droplets from the original droplet. Finally, the ion is either released from the shrinking droplet (ion evaporation model), or the bare ion is obtained after complete evaporation of the solvent (charged residue model).53 Ion evaporation occurs for low molecular weight analytes, whereas the release of large analytes such as native (glyco-)proteins is more accurately described by the charged residue model.
In general, ESI yields even-electron molecular ions, which are typically protonated or metal adducts in positive ion mode. In negative ion mode, deprotonated or anion-adducted species are observed. The occurrence of multiple charge states is common for ESI, especially in large molecules. This can be beneficial for the detection of large molecules in a restricted m/z range. The charge state depends on the ESI conditions such as pH and the presence of salts or detergents. Glycans usually do not yield abundant ion signals due to their hydrophilicity and the absence of basic sites that can be protonated. The ion yield can be increased by derivatization such as permethylation, peracetylation, or labeling at the reducing end.46 Permethylation not only enhances the ionization efficiency but also stabilizes labile moieties such as sialic acid.
Once in the gas phase, glycans can undergo specific fragmentation upon activation. In principle, two fundamentally different types of cleavage can occur in oligo- and polysaccharides: glycosidic cleavage and cross-ring cleavage. The resulting fragments are commonly designated using the Domon–Costello nomenclature (Figure 2).54 The letters A, B, and C designate fragments containing the nonreducing end, whereas X-, Y-, and Z-ions are the complementary counterparts that retain the reducing end or the aglycone. A- and X-fragments result from cross-ring cleavage within the sugar ring of a monosaccharide, while the other ions are produced by glycosidic cleavage between two monosaccharides on either side of the glycosidic oxygen. The position of cleavage within the glycan chain is expressed by subscript numerals, whereas superscript numerals on cross-ring fragments indicate the bonds that are broken within the respective monosaccharide unit. The subscript numerals indicating the number of the glycosidic bond being cleaved depend on whether the fragments contain the nonreducing or the reducing end: in A-, B-, and C-type fragments, the glycosidic bonds are counted from the nonreducing end starting with 1, whereas the position of cleavage leading to X-, Y-, and Z-type fragments is counted from the reducing end or aglycone. The glycosidic bond between the glycan portion and the aglycone in glycoconjugates is by convention numbered 0. Glycosidic cleavage yields information about glycan sequence, whereas cross-ring cleavage is important to deduce information about linkage and branching. Glycosidic B-fragments are also particularly interesting for chemical synthesis of glycans as they are believed to occur as intermediates of SN1 reactions in solution, and their reactivity can be predicted by gas-phase studies.55,56 Glycosidic B- and C-fragments showed in some cases a memory of the stereoinformation on the glycosidic linkage in unprotected glycosides57−61 but not in protected glycosides.62
Figure 2.

Domon–Costello nomenclature of carbohydrate fragmentation. A- and X-fragments result from cross-ring cleavages, whereas B- and C-fragments and their Y and Z counterparts originate from glycosidic cleavage between two monosaccharide units. The numbering of bonds in the sugar ring, which is indicated as superscript numerals in cross-ring fragments, is exemplarily shown for one monosaccharide. Figure is based on ref (54).
Apart from the intended formation of fragments yielding sequence and linkage information, unexpected rearrangement reactions of protonated glycans have been observed in the gas phase, which can lead to erroneous structural assignment. The process is often compared to peptide scrambling, a well-studied rearrangement reaction in proteomics.63 Misleading fragments that do not represent the original glycan structure are formed by so-called internal residue loss, during which a glycan fragment is lost from an internal position of the chain.64,65 At the same time, the monosaccharide at the tip of the chain migrates to adjacent or remote positions, which were not occupied in the original structure. This phenomenon of migrating hexoses was mainly observed for the deoxy sugar fucose. Typically, an asterisk in combination with the Domon–Costello nomenclature is used to denote the rearrangement products. Hexose migration must be considered for the structural investigation of protonated glycans and ammonium adducts, whereas metal adducts and negatively charged glycans do not undergo internal residue loss.66 Glycan rearrangement reactions will be discussed in more detail in Section 5.6.
2.1.2. Ion Activation Methods
With the advent of soft ionization techniques, which transfer intact ions into the gas phase with little or no fragmentation, a number of ion activation methods have been developed to allow for structural investigation of molecular ions by targeted fragmentation within the mass spectrometer.67−69 Ion activation methods differ in the total amount of energy deposited into the precursor ion, as well as in the activation time, i.e., the time scale of this energy deposition (Figure 3). Consequently, different methods lead to substantially different, often complementary, fragmentation mechanisms. In general, dissociation of isolated precursor ions in tandem MS can occur either via collision-, photon-, or electron-mediated activation. The most important ion activation methods with respect to the structural analysis of glycans and glycoconjugates will be briefly presented and compared among each other in the following.
Figure 3.

Time scale and specificity of different ion activation methods. Slow heating methods involve multiple activation events, leading to the cleavage of labile chemical bonds. Fast energy deposition occurs in a single activation step and yields complementary, site-specific fragments. UVPD = ultraviolet photodissociation; ExD = electron-mediated dissociation (including ECD, ETD, EDD, and EID/EED); HCD = higher-energy collisional dissociation; EThcD = electron-transfer/higher-energy collision dissociation; a-EPD = activated electron photodetachment; IRMPD = infrared multiple photon dissociation; CID = collision-induced dissociation.
Collision-mediated activation was the first activation method to be employed and is still the most widely used method in modern tandem MS instruments. Precursor ions are accelerated by an electric field and undergo inelastic collisions with a heavy neutral gas, which leads to partial conversion of the kinetic energy into internal energy. Collision-induced dissociation (CID)70 employs collision energies in the eV range and usually allows for long activation times during which multiple collisions of the precursor ion and a target gas occur. CID is thus a slow heating method71 with sequential increase of internal energy until the weakest bonds break. Those are usually glycosidic bonds in unmodified glycans or labile glycan modifications such as sulfates (Figure 4A–C). Higher-energy collisional dissociation (HCD)72 employing collision energies in the keV range results in single or few collisions and fast ion activation. The technique is less established but can yield a multitude of fragments from direct bond cleavage that are inaccessible by CID, such as abundant fragmentation of both the glycan and peptide backbone in glycopeptides.73
Figure 4.
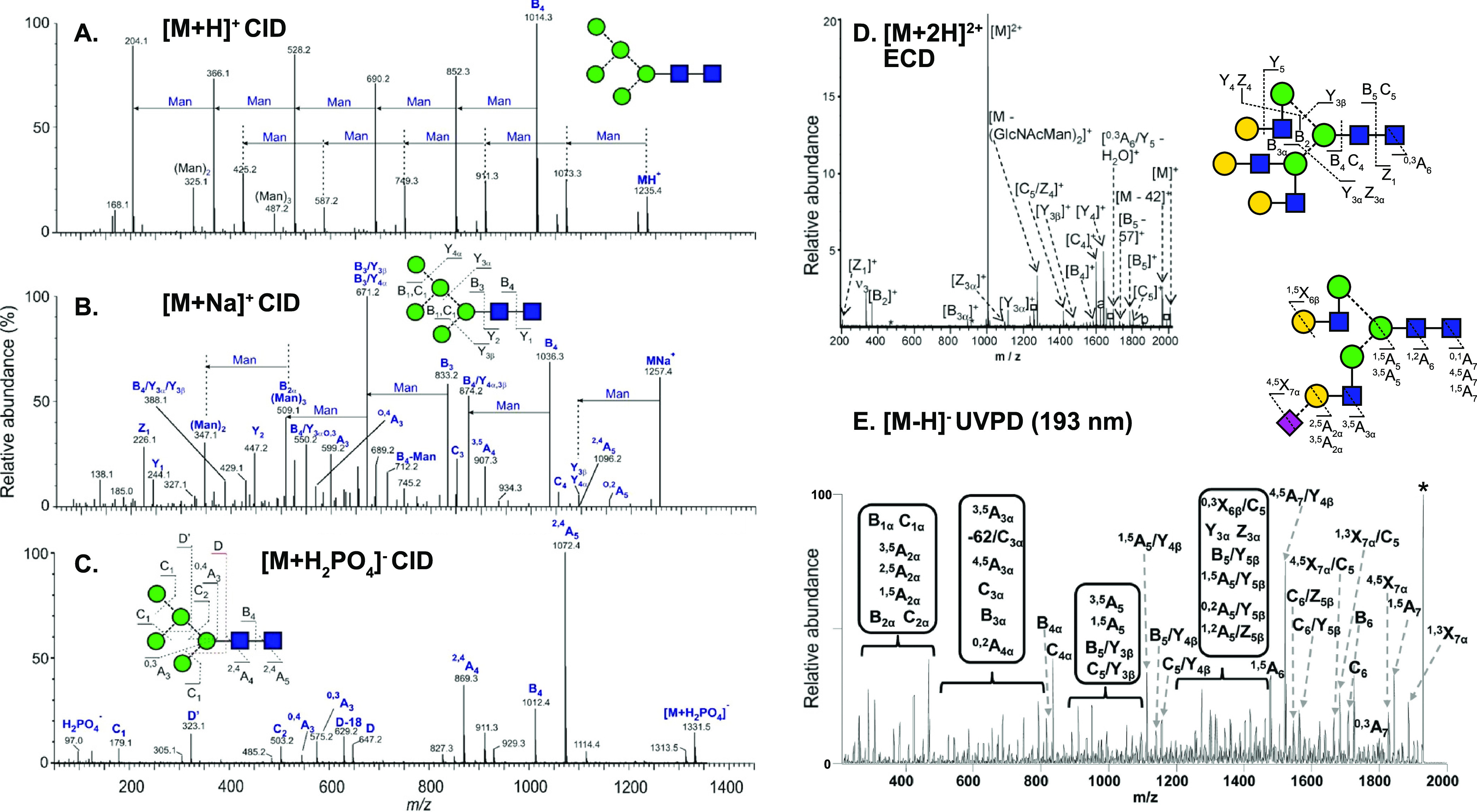
Influence of ion polarity and the ion activation method on the fragmentation pattern of N-glycans. (A) CID in positive ion mode mainly yields B-fragments from glycosidic cleavage. The abundance of cross-ring fragments can be increased by the coordination of metal cations (B) or by employing CID in negative ion mode (C). Figure reprinted with permission from ref (95). 2020 Copyright Wiley-VCH. (D) ECD yields both glycosidic and cross-ring cleavage. Figure adapted with permission from ref (85). Copyright 2007 American Chemical Society. (E) UVPD of N-glycans yields a wealth of fragments, which are informative but challenging to interpret. Only cross-ring fragments are shown in the accompanying structure for clarity. Figure adapted with permission from ref (76). Copyright 2011 American Chemical Society.
Photon-mediated fragmentation induces ion activation by irradiation with photons from different regions of the electromagnetic spectrum. Infrared multiple photon dissociation (IRMPD)74 is a slow heating method based on the sequential absorption of IR photons, which are usually provided by a CO2 laser (λ = 10.6 μm, 117 meV). After each photon absorption event, the increase in internal energy in the precursor ion is redistributed over all vibrational modes by intramolecular vibrational redistribution (IVR). After multiple absorption–IVR cycles, the dissociation threshold is reached, and the weakest bonds are cleaved as in CID.71 Using a tunable light source instead of a fixed wavelength, IRMPD can be employed to record gas-phase IR spectra, as discussed in more detail in Section 2.3. Ultraviolet photodissociation (UVPD)75 employs UV photons of wavelengths between 10 and 400 nm, which are sufficiently energetic for single-photon dissociation. UVPD is thus a fast activation method occurring via electronic excitation of the precursor ion. It yields abundant fragmentation including A- and X-fragments resulting from cross-ring cleavage and site-specific cleavage on sialic acids (Figure 4E).32,76 A prerequisite for UVPD is a suitable UV-absorbing chromophore on the precursor ion. UVPD of multiply charged anions can trigger a radical fragmentation mechanism by electron detachment (activated electron photodetachment; a-EPD).77
Electron-mediated ion activation methods (ExDs) are classified as fast activation methods inducing specific bond cleavage and are particularly important for structure analysis of glycoconjugates but also free glycans.78 They include electron capture dissociation (ECD),79 electron transfer dissociation (ETD),80 electron detachment dissociation (EDD),81 electron-induced dissociation (EID),82 and electronic excitation dissociation (EED). Except for EID and EED, these methods are only applicable to multiply charged cations (ECD and ETD) or anions (EDD), as they induce charge state reduction by adding or detaching an electron, respectively. ECD and ETD induce very similar fragmentation mechanisms but differ by the technical implementation of the initial electron transfer and thus require different instrumentation. ECD employs a low-energy electron source and requires the magnetic fields of an FTICR cell for trapping electrons. ETD, on the other hand, is also compatible with linear and 3D quadrupole ion traps because trapped electrons are not employed for ion activation. Instead, electron transfer is mediated by stable odd-electron anions generated in a separate ion source. Both ECD and ETD transform multiply charged cations into radical cations, which subsequently fragment. Importantly, the fragmentation mechanism depends on the location of the captured electron rather than dissociation barriers. As a result, weak bonds and even noncovalent interactions that are preferentially cleaved in CID or IRMPD can remain intact. Therefore, ECD and ETD are extensively harnessed for the investigation of labile post-translational modifications in peptides and proteins, such as O-glycosylation,83 but also for sequence analysis of oligosaccharides, which can yield informative cross-ring fragments upon electron capture (Figure 4D).84,85 EDD is the negative ion analogue to ECD, transforming multiply charged anions into radical anions by electron detachment, employing electrons of moderate kinetic energy. The method is particularly useful for the analysis of sulfated oligosaccharides such as GAGs, which yield informative fragments from both glycosidic and cross-ring cleavages that locate sites of sulfation.86 However, EED, as well as ECD, rely on FTICR instrumentation because electrons and ions must be trapped simultaneously. Contrary to the ExD methods described before, EID and EED can be applied to singly charged positive or negative ions and are thus suited for the investigation of small glycoconjugates such as glycopeptides and -lipids that are only observable as singly charged species.87 EED results in extensive cross-ring cleavage and therefore holds much potential for linkage analysis.88
A drawback of many ExD methods is their low fragmentation efficiency, making long reaction/interaction periods necessary to achieve sufficiently intense fragment ion peaks. Since a single successful ion–electron encounter usually deposits enough energy to induce fragmentation, ExD activation times are short. However, such successful ion–electron encounters are of low probability due to the small interaction cross sections, leading to inefficient fragmentation. Ion–electron interaction periods ranging from hundreds of milliseconds to seconds—significantly longer than those employed in CID—have hindered the straightforward online coupling of many ExD methods to chromatographic and electrophoretic separations. As electrostatic repulsion between polyanions and electrons renders EDD particularly inefficient, the development of LC-ExD-MS/MS workflows for the analysis of acidic glycans as negative ions proved to be especially challenging. However, the introduction of NETD, a technique relying on more efficient anion–cation interactions and requiring shorter interaction periods, opened the way recently for such multidimensional workflows.89,90
Several mass spectrometer designs allow for multiple fragmentation of ions in time or in space. Multistage MS or MSn in time is provided by mass spectrometers based on ion trapping, which can repeatedly isolate and fragment ions. This approach is useful for detailed structure elucidation of fragments91 and can reveal information about glycan branching patterns.92 Due to considerable ion losses in each round of isolation and fragmentation, a sufficient precursor ion intensity is required. Different ion activation methods can also be combined in space to yield complementary information in a single experiment.93
In summary, ion activation methods can be classified either by the source of activation (collision, photon, electron) or by the activation time, which is long for multiple discrete activation events or short if the activation occurs quickly relative to the unimolecular dissociation. For example, IRMPD and UVPD are both classified as photon-mediated activation methods but are located at opposite sites on the activation time and specificity scale. In general, long activation times lead to fragmentation according to bond dissociation energies, whereas short activation times induce direct bond cleavage before the energy is redistributed over the whole molecule. Each ion activation method has its own area of application, and their combination can yield a more comprehensive picture of complex glycan and glycoconjugate structures.
2.1.3. Ion Polarity
The nature of fragments generated in tandem MS is not only dependent on the choice of ion activation method but also crucially influenced by ion polarity and the type of coordinating cations or anions.94,95 In positive ion mode, CID of protonated oligosaccharides mainly yields B-fragments resulting from glycosidic bond cleavage (Figure 4A). Coordination of monovalent and divalent metal cations was shown to increase the abundance of diagnostic cross-ring cleavage in positive ion mode in numerous studies (Figure 4B).69 Contrary to protons, metal cations can coordinate to multiple oxygen atoms simultaneously, and their site of coordination influences the type of fragment ions. Metal cations coordinating to the ring oxygen were suggested to prevent glycosidic cleavage by localizing the oxygen’s electrons while not impeding cross-ring fragmentation.96 Negative ion mode CID provides details on linkage by yielding abundant A-fragments from cross-ring cleavage (Figure 4C).
In order to obtain more comprehensive linkage information, orthogonal techniques can be coupled to MS to allow for in-depth structural investigation of glycans.35 IMS and gas-phase action spectroscopy coupled to MS are increasingly employed for glycan analysis and will be detailed in the following.
2.2. Ion Mobility Spectrometry
Ion mobility spectrometry (IMS) is a gas-phase electrophoretic separation technique, widely employed as a stand-alone method for the detection of drugs, explosives, and chemical warfare agents.97−99 It has a long history as part of ion mobility–mass spectrometry (IM-MS) couplings, with applications in fields as diverse as molecular physics and structural biology.100−114 Since the commercialization of the first integrated instruments, IM-MS has gained remarkable popularity in bioanalytical chemistry115−126 and become a key element of the glycomics toolbox.23,29,34,35,127−130 Glycan isomers often exhibit identical fragment ion spectra, impeding their tandem MS-based distinction, while the presence of multiple isomers in a mixture calls for efficient separations. IMS proved to be extremely useful for the distinction, postionization separation, and relative quantification of glycan isomers, helping to overcome key challenges associated with MS-based glycan analysis.131−134 In addition, as comprehensive IMS analyses take place generally on the millisecond time scale, they fit perfectly between condensed-phase separations and fast MS experiments, further increasing the peak capacity of multidimensional workflows.135
In general, IMS separates ions based on differences in their gas-phase mobilities K, a transport property related to the ions’ mass, charge, size, and shape. K is also influenced by the nature and number density of the buffer gas and by the effective temperature at high reduced fields, as will be discussed later in relation to field asymmetric IMS. As they traverse a suitable gas-filled cell under the influence of an electric field, ions undergo binary collisions with the neutral gas particles. Larger, extended species collide more frequently, reaching lower velocities upon their electrophoretic motion than more compact ions. This simplified picture reveals the aptitude of IMS to separate isomers and conformers, species having identical m/z ratios but often differing in size and shape.
Within the low-field limit, ion–neutral collisions are essentially thermal, and K is basically independent of the electric field strength E:
| 1 |
Equation 1 is the fundamental low-field ion mobility equation, also called the Mason–Schamp equation.136−138 Here, μ is the reduced mass of the ion–neutral collision complex, kB the Boltzmann constant, T the buffer gas temperature, z the ionic charge state, e the elementary charge, n the buffer gas number density, and CCS the rotationally averaged collision integral, often referred to as the collision cross section. CCSs are related to the size and shape of the collision partners but also depend on interaction potentials.101,139,140 They serve as effective areas, generally expressed in units of Å2, reflecting momentum transfer between the colliding particles.141 The larger the CCS of an ion–neutral pair, the more efficient the momentum transfer between the ion and the gas particles in IMS. CCSs are independent of gas pressure or number density and vary generally less with temperature than mobilities, making them suitable molecular descriptors. They are comparable across different IMS platforms and can be readily stored in databases, facilitating the identification of analytes. Besides experimental determination in suitable IMS experiments, CCSs may also be calculated by computational methods, addressed in more detail at the end of this section.
Various IMS techniques have been developed and commercialized in recent years that efficiently harness electric forces for separating ions in gases.142−144Figure 5 provides an overview of the methods applied most successfully in glycan analysis, accompanied by a brief description of each technique below.
Figure 5.
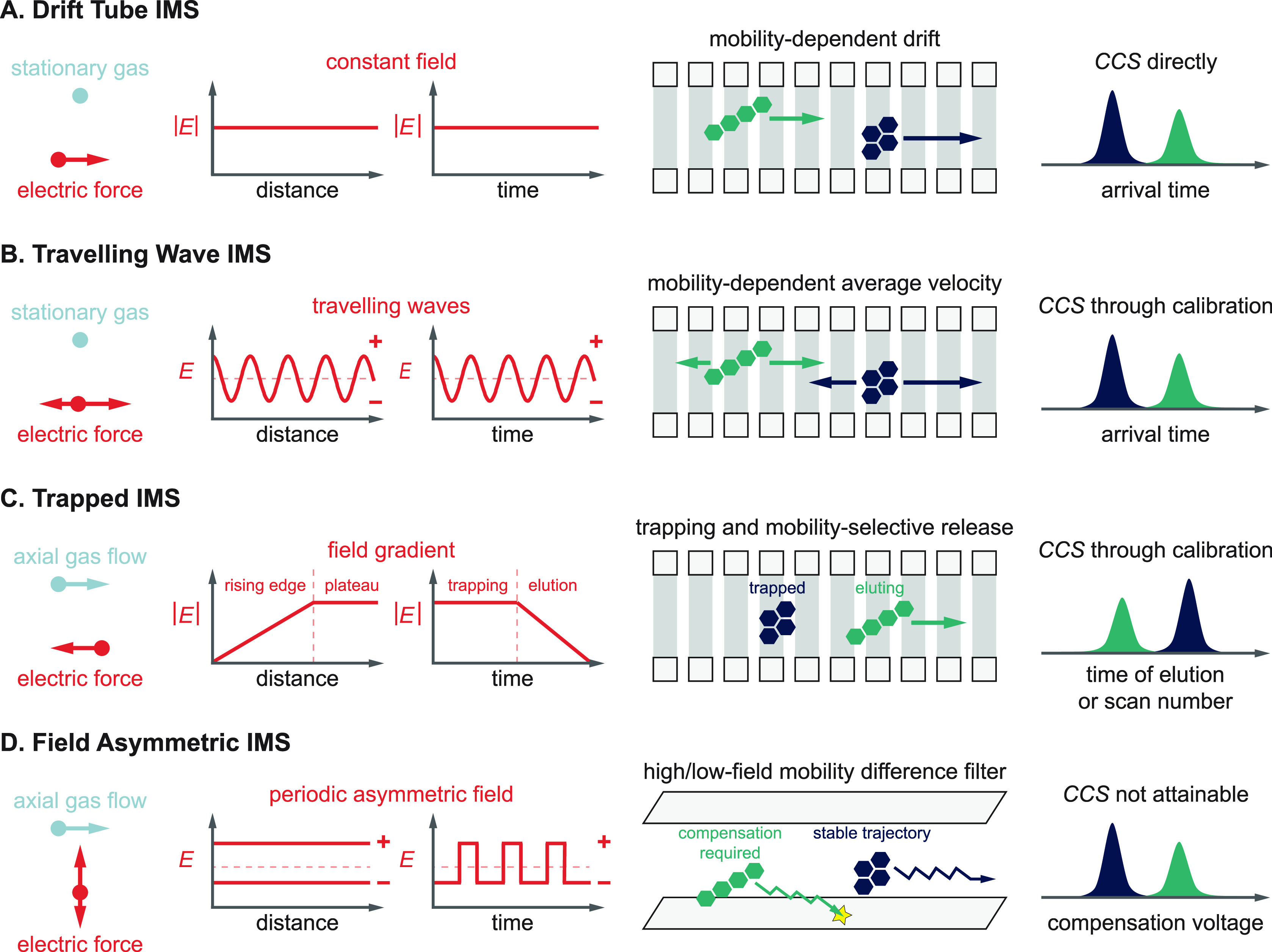
Overview and basic principles of ion mobility spectrometry techniques commonly applied in glycan analysis.
Drift Tube Ion Mobility Spectrometry (DTIMS)
Ions are propelled through a stationary buffer gas by a constant electric field, invariable in both space and time. Ions with higher mobilities traverse the drift cell faster and reach the detector at shorter arrival times, making DTIMS a time-dispersive method.145 In the constant electric fields applied, drift velocities are directly proportional to mobilities, enabling determination of the latter directly from first principles. When experiments are performed within the low-field regime, obtaining CCSs from mobilities using eq 1 is straightforward. To date, the stepped-field method using DTIMS/IM-MS represents the most accurate way of determining K and CCS values experimentally, with expanded uncertainties reported as low as 0.5%.146 Although these features make the technique very attractive, enhancing resolving power requires increasingly long pathways and short injection pulses combined with high DC voltages, which may become impractical or difficult to achieve beyond a certain limit.144
Traveling Wave Ion Mobility Spectrometry (TWIMS)147−149
TWIMS is a time-dispersive method where ions are driven through a gas-filled cell by the eponymous traveling potential waves. In brief, the approximately sinusoidal waves traverse the mobility cell with velocities generally between 100 and 500 m s–1, while the ions follow their movement in a mobility-dependent manner. Analytes with higher mobilities are able to “surf” longer on each wave, experiencing fewer discrete roll-over events and reaching higher average velocities. The electric field itself changes sign between the leading and trailing edges of each electric potential wave, causing the ion velocity vectors to fluctuate. This results in a characteristic “n steps forward, one step back” motion of the analytes. Despite remarkable improvements in TWIMS theory,150,151 to date no complete and general analytical model has been developed that would allow for inferring mobilities directly from transit times under a broad range of practical operating conditions. Thus, obtaining CCSs from TWIMS measurements generally requires calibrant ions whose CCS values had previously been determined in low-field DTIMS experiments. An advantage of TWIMS technology over DTIMS is the flexibility it enables in instrument geometry: cyclic arrangements or structures for lossless ion manipulations (SLIMs) with serpentine routes provide extremely long separation pathways within compact architectures.152,153 Such instruments are capable of achieving resolving power values currently unattainable in linear drift tubes, allowing for the separation of glycan anomers in the gas phase.154,155
Trapped Ion Mobility Spectrometry (TIMS)156−158
TIMS represents an alternative concept to time-dispersive methods for separating ions according to their gas-phase mobilities. Here, ions are held stationary along a linear electric field gradient by two counteracting forces: a fast axial gas flow (>100 m s–1) drags the ions toward the exit, while the electric field propels them in the opposite direction. The position along the axial field gradient where the two forces exactly balance each other, focusing the ions into steady-state zones, depends on the ions’ mobility. Ions with high K are trapped close to the entrance (lower fields), while species with low K occupy the region closer to the exit (higher fields). Having reached their mobility-dependent stationary positions, the ions are eluted selectively by gradually decreasing the magnitude of the electric field. The higher their mobility, the later the ions elute, causing the species to reach the exit in an order of increasing mobility. Due to the high linear gas velocities in the tunnel, the effective path length covered by ions during their elution can be orders of magnitude higher than the physical dimensions of the TIMS tunnel, enabling outstanding resolving powers (RP > 250) to be reached in a compact device.144 Although TIMS in principle enables the direct determination of K and CCS values, measuring the gas velocity and pressure with sufficient accuracy is extremely difficult. Thus, establishing calibration curves using ions of known CCS is preferred in practice.159 Finally, as radial focusing is achieved using radiofrequency fields in the TIMS tunnel, ion heating during long trapping times is worth being considered when analyzing labile species.
Field Asymmetric Ion Mobility Spectrometry (FAIMS)160−162
In all three techniques above, ion transport and separation are dictated by the absolute value of mainly low-field mobilities. In stark contrast, FAIMS devices filter ions based on the shift in their mobility between low and high fields. The dependence of an ion’s mobility on the reduced field strength E/n is considered negligible below the low-field limit but becomes significant once this limit is exceeded. To harness this dependence, FAIMS employs a periodic, highly asymmetric field perpendicular to a gas flow that propels ions uniformly toward the exit of the planar device. The field changes between positive and negative values, generating high-field conditions for a shorter time and low-field conditions at the opposite polarity for a proportionally longer time (the time integrals of the two fields are equal). This causes the ions to oscillate along the field axis, experiencing deflection toward the top or bottom electrode if the low- and high-field mobilities are different. To force ions on stable trajectories, a compensation voltage is applied between the electrodes. Scanning this DC voltage enables ions with different mobility shifts to traverse the cell without hitting the electrodes and pass through the exit. Being essentially a mobility shift filter, FAIMS does not allow for comprehensive analysis in a single run. Instead, it can provide a continuous beam of a selected species at 100% duty cycle, making it highly compatible with slower mass analyzers, such as FTICR-MS instruments.163 Although FAIMS does not allow for inferring absolute K or CCS values from the experiments, it is superior to the above three IMS techniques in terms of orthogonality to MS. Shifts in mobilities upon moving from low to high fields are difficult to predict, and they show generally weaker correlation with m/z ratios than low-field mobilities.
Although experimentally determined CCSs are highly useful to distinguish and identify analytes, they ultimately reduce molecular structures to a single value: an effective area. These areas are linked to the overall shape of ions but do not carry direct, atomic level information about the underlying molecular structure. Without prior knowledge and based merely on experimentally determined CCSs, it is extremely difficult—if not impossible—to tell unambiguously whether two separated isomers differ in their conformation, configuration, or constitution. To obtain atomic level structural details with the help of CCSs, complementing theoretical approaches are indispensable. The strategy, in principle, is simple: experimentally determined CCSs are compared to those calculated for structural candidates generated by computational methods, and the model structures are evaluated on the basis of CCS agreement. This approach proved to be very helpful for the structural analysis of charged clusters164−166 and has been successfully adopted for biomolecular ions.167,168
To calculate CCSs, the first, computationally less expensive methods treated the colliding partners as hard spheres, ignoring details of the interaction potentials.164,169 The development of the trajectory method (TM) applying realistic interaction potentials put CCS calculations on a more solid physical basis, albeit at the expense of significant computational cost.139 Since then, continuous improvements in theoretical methods and computational tools have manifested in increasingly fast and accurate CCS calculations in atomic and molecular gases.170−179 Today, the main bottleneck for glycans is not the calculation of CCSs for given model structures with satisfying accuracy (at least in common buffer gases) but the generation of reliable structural candidates. It requires advanced quantum chemical methods, most prominently density functional theory calculations.35 As isolated carbohydrates represent an immense challenge in theoretical chemistry, Section 2.5 is dedicated entirely to the subject, focusing on electronic structure calculations and their merging with methods to explore the vast conformational space of glycans.
2.3. Gas-Phase Infrared Spectroscopy of Mass-Selected Ions
Infrared (IR) spectroscopy is a powerful tool used to identify unknown molecules and deduce information on their structure, such as functional groups, intra- and intermolecular interactions, as well as molecular conformations. Electromagnetic radiation in the IR range can excite molecules if the frequency of the incident radiation is in resonance with IR active vibrational transitions. IR radiation expands from the edge of the visible spectrum to the microwave range and can be further divided into the higher energy, near IR (>4000 cm–1), mid-IR (4000–400 cm–1) and lower energy, far IR (400–10 cm–1) regime. Fundamental vibrations are typically found in the mid-IR range. Classical IR spectroscopy techniques are based on direct absorption spectroscopy, which measures the attenuation of light after passing through a solid, liquid, or gaseous sample. The absorbance as a function of the frequency is derived from the Lambert–Beer law. The concept of direct absorption spectroscopy is depicted in Figure 6A. For m/z-selected ions in the high vacuum of a mass spectrometer, the sample density is usually limited to 106 singly charged ions per cubic centimeter due to Coulomb repulsion and the resulting space-charge limit.180 The attenuation of light after passing through the low-density sample is so low that it is difficult or even impossible to measure. Therefore, vibrational spectroscopy of ions in the gas phase has evolved and is today typically performed as action spectroscopy, as addressed in recent reviews.181,182 Here, the absorption of photons is measured indirectly by following an action, e.g., the dissociation of the intact ion or electron detachment (see Section 2.4). A schematic concept of action spectroscopy can be found in Figure 6B. The number of unaffected ions n at a specific frequency ν can be expressed as a function of the number of precursor ion n0, the absorption cross section σ(ν), and the photon fluence F(ν) in the following equation:181
| 2 |
The challenge is that IR light is usually much lower in energy than the threshold to dissociation; therefore, the analytes must have a low barrier to dissociate, or the laser system must be intense enough to enable multiple photon absorption. With the advance of tunable and powerful laser systems such as IR free-electron lasers (FELs)183−185 and benchtop laser systems such as optical parametric oscillators and optical parametric amplifiers (OPO/OPA) in the early 2000s, the interest in gas-phase IR spectroscopy of m/z-selected ions for their structural analyses started growing. Early works by the Simons group186,187 investigated small, neutral glycans in a UV-IR double-resonance experiment from a free jet expansion. Limited to the presence of UV chromophores and the harsh ionization method, the method was only applicable to small glycans. Combined with ESI sources, the field of IR spectroscopy finally opened up for the investigation of larger glycan ions.188−190
Figure 6.

Direct absorption vs action spectroscopy. (A) Concept of direct absorption spectroscopy. The Lambert–Beer law relates the intensity of transmitted light I at a specific frequency ν to the intensity of the incident light I0, the absorption cross section σ, the path length l, and the particle density N. (B) Concept of action spectroscopy (upper panel). The equation is derived from the Lambert–Beer law and relates the number of unaffected ions n at a specific frequency ν to the number of precursor ions n0, the absorption cross section σ(ν), and the photon fluence F(ν). IRMPD time-of-flight mass spectra of a m/z-selected highly sulfated pentasaccharide without (middle panel) and with (lower panel) resonant IR irradiation. Two fragments with a sequential loss of neutral SO3 are observed upon multiple photon dissociation at 1264 cm–1.
Infrared Multiple Photon Dissociation (IRMPD) Spectroscopy
One of the most widely used types of IR action spectroscopy is IRMPD spectroscopy, which induces fragmentation of weakly bound clusters or ions with energetically low fragmentation barriers upon irradiation.181,182 An IR spectrum is recorded by measuring the fragmentation yield as a function of the wavelength. The IRMPD mechanism is a nonlinear process involving the sequential absorption of a large number of IR photons (typically tens to hundreds) in which the energy of a single photon is distributed throughout the ion via IVR; i.e., the original vibrational mode relaxes via anharmonic coupling to vibrational background states. The sequential absorption of single photons, hence, takes place via the same fundamental vibrational level; yet, the internal energy of the ion rises with vibrational excitation until the dissociation threshold is reached and the ion fragments. With the IVR-coupled excitation process, the vibrational excitation is randomized throughout the ion so that the fragmentation occurs statistically, and usually the weakest bonds dissociate. For the above reason, IRMPD fragmentation patterns closely resemble those obtained by CID. Inherent to the process, red-shifting of absorption bands and spectral congestion can leave an imprint on the IRMPD spectrum. Additionally, glycans typically populate a large number of coexisting conformers at room temperature due to their conformational flexibility, which further increases spectral congestion and limits IRMPD to mostly mono- and disaccharides.188,191−193
Various designs of IRMPD instruments are published, yet in the most basic approach an interaction region with electrodynamic ion optics is sufficient to radially define the ion cloud and achieve an efficient overlap of the ions with the laser. Ion storage proves beneficial for slow dissociation processes so that the ions can be irradiated for a longer time. Online coupling of IRMPD with LC-MS workflows is challenging because the time required to record an IRMPD spectrum usually exceeds 10 min, which is not compatible with the time scale of LC peaks. However, efforts are underway to generate IRMPD spectra from chromatographically separated glycans.194
Messenger-Tagging Spectroscopy
Another approach that does not require multiple photon excitation steps is messenger-tagging spectroscopy which follows the detachment of a weakly bound, noninteracting messenger atom or tag upon resonant irradiation. Typical tags for the investigation of glycans are helium and nitrogen, and for other (bio)molecules the use of argon and hydrogen has been explored. Contrary to IRMPD spectroscopy, the IVR-mediated detachment mechanism of the tag in messenger-tagging spectroscopy is a linear single-photon process overcoming red-shifting and reducing spectral congestion. Furthermore, the power of the tunable IR laser required for messenger-tagging spectroscopy is significantly lower and thus renders the approach more compatible with benchtop lasers. Prerequisites for the attachment of a tag are temperatures close to the boiling point of the tag. Coincidently, this limits the numbers of glycan conformers present, therewith further reducing spectral broadening. Several IR spectroscopic studies on glycans, small building blocks up to oligosaccharides,189 using messenger-tagging spectroscopy are reviewed in the respective chapter of their glycan classes.
Already in the 1980s, messenger-tagging spectroscopy was developed as a concept using a supersonic jet expansion followed by electron ionization or corona discharge. With the low temperatures in the supersonic jet, ions or clusters formed weakly bound complexes with coexpanded hydrogen. The detachment of the hydrogen tag upon irradiation was detected in a quadrupole mass analyzer (Figure 7). Since the mid-1990s, the development of cryogenic ion traps allowed the combination of this technique with soft ionization sources.195,196 The ions are transferred to the cryogenic ion trap, cooled by collisions, and eventually tagged with buffer gas at temperatures between 3 and 70 K. Various designs for cryogenic ion traps serve different purposes, such as temperatures below 3 K, long storage times, spatial spread of the ion cloud, or space focusing of ions with the ejection from the trap. As the first designs, multipole ion traps were developed,197−199 followed by planar multipole ion traps200−202 and ring ion guide traps.199 It was recently published203 that a wire quadrupole ion trap, with a linear quadrupole geometry where each of the rods is approximated by six copper wires, reaches temperatures below 3 K and is attached to a commercial mass spectrometer. In all recently published designs, the interaction region with the laser is a cryogenic ion trap.
Figure 7.
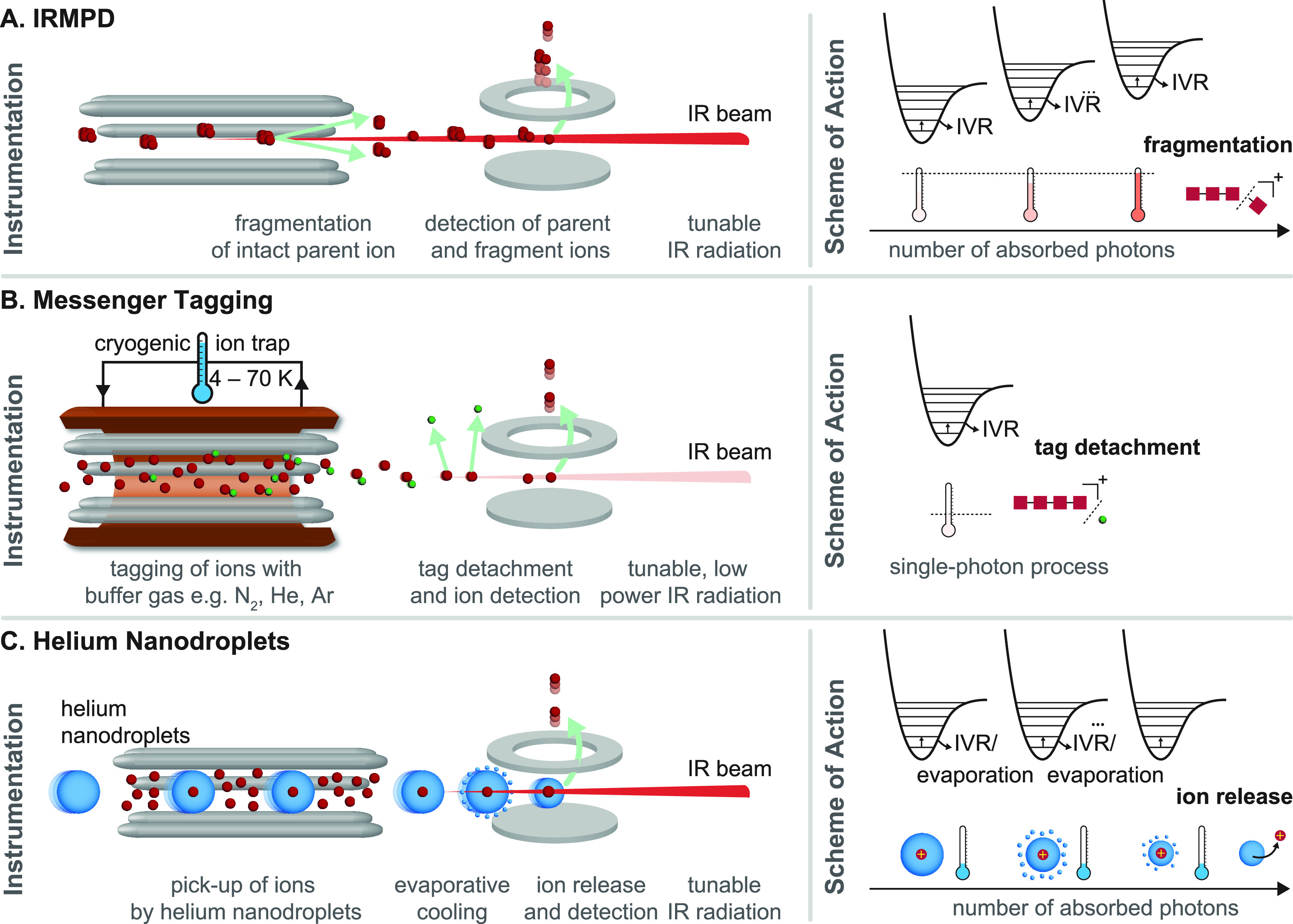
Comparison of types of IR action spectroscopy. (A) Instrumentation and scheme of action for infrared multiple photon dissociation (IRMPD) spectroscopy. With resonant irradiation, multiple IR photons excite the intact parent ion until the fragmentation threshold is reached. Fragment and parent ions are detected. (B) Instrumentation and scheme of action for messenger (tagging) spectroscopy. Ions are tagged with buffer gas atoms or molecules, e.g., N2, He, or Ar, in a cryogenic ion trap. With resonant irradiation typically with a single photon, the tag is detached, and the bare ion is detected. (C) Instrumentation and scheme of action for cryogenic spectroscopy in helium nanodroplets. The ions are picked up by helium nanodroplets in an ion trap and cooled to 0.4 K. With resonant irradiation, the ion is excited and immediately cooled again by evaporative cooling. After several iterations, the ion is released from the nanodroplet and detected.
IR Action Spectroscopy in Helium Nanodroplets
IR action spectroscopy in helium nanodroplets is currently applied in basic research only, yet the spectral quality which is achievable for glycan ions sets new benchmarks in the field.190 The superfluid helium nanodroplets are produced from a precooled reservoir of helium with the opening of a valve into the mass spectrometer, subsequent evaporative cooling, and formation of nanodroplets of a defined size (typically 105 helium atoms). Before irradiation, the trapped ions are picked up by the traversing helium nanodroplets and cooled to the equilibrium temperature of the droplet of 0.37 K. Upon irradiation with a resonant photon, the ion is vibrationally excited and immediately cooled again to its ground state by evaporation of helium from the shell of the droplet. After several iterations, the ion is eventually released from the droplet and detected background-free. With this approach, a unique resolving power and distinct spectral fingerprints have been recorded for various classes of glycans up to pentasaccharides.55,204,205
2.4. Gas-Phase Ultraviolet Spectroscopy of Mass-Selected Ions
Besides IR spectroscopy that probes vibrational transitions, molecular ions may also be studied using UV spectroscopy inside a mass spectrometer.206 UV spectra reflect the electronic structure of analytes. They are sensitive to the local environment of chromophores and thereby to the constitution, configuration, and conformation of molecules. When performing UV spectroscopy on isolated ions, one needs to rely on action spectroscopic approaches, similarly to gas-phase IR spectroscopy discussed above. On one hand, the attenuation of UV radiation by a low-density ion cloud of absorbing species is extremely weak, which makes it unfeasible to record spectra by measuring changes in light intensity. On the other hand, absorption of energetic UV photons leads to electronic excitation and initiates a variety of traceable processes in isolated ions. By monitoring such a photoinduced process, serving as the action, one can readily detect photon absorption. Following electronic excitation, the ions can undergo various relaxation pathways, often leading to dissociation or also to electron photodetachment (EPD)207 in polyanions. Because both UVPD and EPD are accompanied by changes in the m/z ratio, they can be readily monitored by mass spectrometers in action spectroscopy schemes. Plotting the UVPD and/or EPD yield as a function of irradiation wavelength yields UV spectra for m/z-selected ions.
UVPD and EPD are usually single-photon processes, enabling experiments to be performed on cold ions using cryogenic ion traps. Cooling analytes can greatly reduce thermal broadening, leading to less congested electronic spectra, often with vibrational resolution.208 UV ion spectroscopy requires bright, tunable UV sources, with optical parametric oscillators (OPOs) and dye lasers being commonly applied. UV optical spectra of biomolecular ions are usually recorded in ranges between 200 and 400 nm (3.1–6.2 eV or 25 000–50 000 cm–1). For analytical purposes, simpler one-color experiments are more suited, requiring only a single laser and less sophisticated optical layouts.
In contrast to slow-heating IRMPD, fragmentation patterns in UVPD may depend markedly on the irradiation wavelength. Dissociation pathways are affected by the initial site of excitation, i.e., the location of electrons involved in the electronic transition induced by photon absorption. This fundamental aspect of UVPD is utilized for analytical purposes in 2D UV-MS, an alternative way of handling and representing UV ion spectroscopic data.208,209 Instead of plotting the overall fragmentation yield as a function of photon energy, 2D UV-MS deals with 2D data arrays depicting entire fragment ion spectra as a function of irradiation wavelength. Figure 8 provides a graphical overview of gas-phase UV ion spectroscopy, its methodology, and the different ways of representing spectroscopic information.
Figure 8.
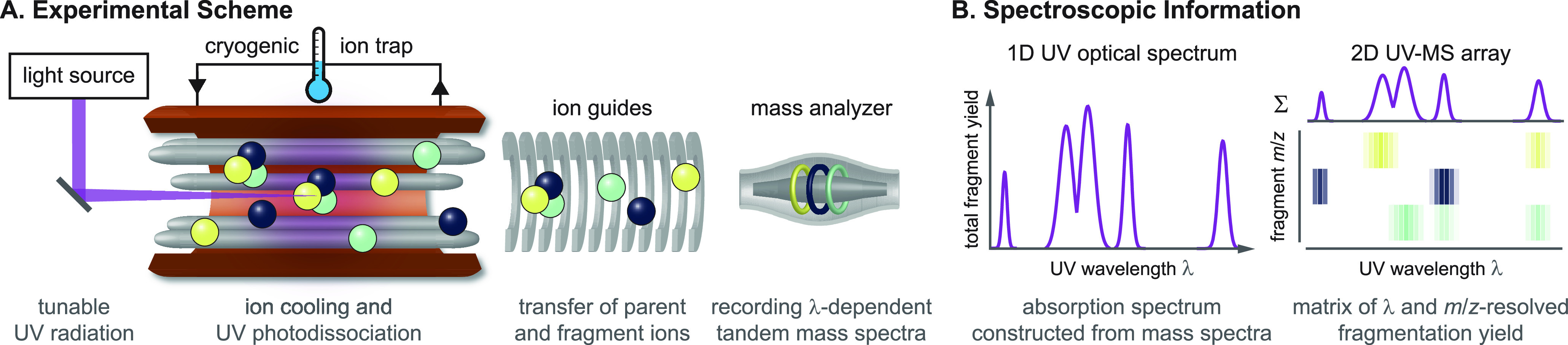
Schematic overview of cold-ion UV spectroscopy. (A) Simplified representation of an experimental scheme for recording UV spectra of cold ions. m/z-Selected parent ions, represented by three joint spheres, are cooled in a cryogenic ion trap and probed by UV radiation from a tunable laser, inducing photodissociation and/or photodetachment (latter not shown). The parent species and its photofragments (lone spheres) are extracted from the trap and transferred to a simultaneous mass analyzer, such as a ToF or FTMS device. (B) Plotting the overall fragmentation yield as a function of photon energy yields the UV optical spectrum of the parent ion. 2D UV-MS fingerprints correlate UV optical spectroscopic information and MS data. The fingerprints display entire fragment ion spectra as the function of excitation wavelength, condensing more analytical information into UV-MS matrices.
Various biomolecular ions have been studied by UV action spectroscopy, including amino acids,198 peptides,210−212 and nucleic acids.207 Although still in its infancy, recording UV spectra of m/z-selected glycan ions has gained momentum in recent years. Glycans generally lack strong chromophores, which makes their UV spectroscopic analysis challenging. Species containing, for example, C=C double bonds or −COOH/–COO– groups (with corresponding π → π* or n → π* transitions) may be analyzed directly,213,214 but many carbohydrates require alternative strategies. Boyarkin and co-workers introduced a method where glycans (analytes) were attached to protonated aromatics (chromophore reporters) through noncovalent interactions.215 The complexes formed spontaneously in the ESI process and allowed for the UV-spectroscopy-based distinction of carbohydrate isomers with weak or no inherent UV absorption.
Recently, UV ion spectroscopy has been performed using circularly polarized light, resulting in mass-resolved electronic circular dichroism spectroscopy of oligonucleotides (monitoring EPD)216 and amino acids (tracking UVPD).217 Although not yet applied for oligosaccharides, the method holds potential for MS-based glycan analysis in the future.
2.5. Theoretical Methods to Study Glycan Structure in Vacuo
Using gas-phase spectroscopy and IM-MS discussed above, detailed information may be obtained on the structure of isolated glycan ions, including the overall shape of the analytes, their functional groups, and the spatial orientation thereof. With computational chemistry, glycan structures can be modeled and then correlated with the experimentally obtained IR spectra or collision cross sections (CCSs). Subsequently, experimental CCSs and absorption bands can be matched with their computed counterparts, leading to an assignment of the structure to the probed glycan ion and their vibrations. The challenges lie in finding the correct structure by modeling the glycans’ conformational space and then extracting the computational IR spectra and CCSs out of that structure with the most efficient method.
The computational chemist’s toolbox usually comprises three different methods to tackle modeling of glycans: empirical force field and semiempirical and first-principles methods. Commonly, accuracy increases with computational expense that in turn is rising with the size of the system. Thus, the size of the system is the limiting factor for the accuracy, and each calculation is a trade-off between accuracy and computational expense.
A widespread approach to sample isolated carbohydrate ions is to employ empirical force fields.218−220 While promising force fields have been parametrized for glycans, they were commonly not developed for sampling isolated charged molecules in the gas phase and are therefore often limited to the analysis of neutral carbohydrates in the condensed phase.35 Contrary to first-principles methods, force field methods do not have their origin in quantum mechanics but in Newtonian physics. Therefore, force field methods are outperformed by first-principles methods that consider all electrons of the system or its electron density without an empirical bias.221 From all first-principles methods, mainly density functional theory (DFT) methods are used for carbohydrates. Promising results in terms of accuracy and reasonable computational expense have been obtained with hybrid DFT functionals221 such as PBE0222,223 or B3LYP,224 including dispersion correction225,226 and triple-ζ basis sets. Usage of basis sets from the Ahlrichs227 family is recommended, although accurate results can also be obtained with Pople228 or Dunning229 basis sets. However, the high accuracy of DFT comes at the cost of computational expense. Here, semiempirical methods that are based on DFT could fill the gap. Some integrals are replaced by empirical parameters leading to a significant increase in computation speed. Although some of these methods performed underwhelmingly for carbohydrates,221 several new methods are more promising.230
Each method can be used for sampling of glycans to generate a high number of structures, from which the most promising candidates are subsequently used for reoptimization at a higher level of theory (usually DFT). Commonly used methods for generating conformers from a reasonable starting structure are (replica exchange) molecular dynamics, genetic algorithms, or Monte Carlo simulations. It has been suggested that ideally the conformational search should start with DFT from the very beginning as the usage of a force field with a strong bias might lead to the exclusion of the most stable conformers, which cannot be recovered at a later stage of the process.35 As electrons are not considered in force field calculations, the initial connectivity of a molecule is rigid, and bond-breaking/-formation processes are not considered. It is a major limitation of methods relying on empirical force fields since rapid charge migration may occur in isolated carbohydrate ions. First-principles methods, in turn, can correctly determine structural features, such as ring puckering, glycosidic bond geometry, charge migration processes, or hydrogen bonding. For sampling, using a GGA-DFT functional with a small basis set is reasonable. The genetic algorithm FAFOOM231 can be interfaced with various software packages for DFT optimizations and yielded promising results for carbohydrate sampling.55,56,221,232,233 Other DFT-based approaches using molecular dynamics234 or Monte Carlo simulations235−237 have been implemented to sample carbohydrates. Tools based on semiempirical methods, such as CREST,238 were successfully used for modeling the conformational space of glycans.239,240
To get a reliable computed IR spectrum or CCS from a sampled structure, the structure needs to be reoptimized at a high DFT level of theory (such as PBE0+D3/def2-TZVP). The structure can subsequently be used to compute harmonic frequencies and Merz–Singh–Kollman (MK) charges. After scaling, harmonic frequencies often agree well with the experimental spectra. However, in various cases, certain absorption bands are strongly anharmonic. The issue can be circumvented by calculating anharmonic frequencies, albeit at an increased computational expense. The MK charges, on the other hand, are very reliable for calculating theoretical CCSs.
To get coordinates of a structure to theoretical CCSs, no quantum chemical calculation needs to be performed. Thus, computing CCSs is comparably fast, even for larger structures. Historically, CCSs were computed with the projection approximation164 (PA) or the exact hard-sphere scattering model241,242 (EHSS). For both methods, a mere structure is sufficient to compute the corresponding CCS. The PA differentiates only between hits and misses of the ion and the drift gas. In EHSS, also scattering is considered.141 The most reliable method is the trajectory method243 (TM) that has been implemented in the MobCal179 and hpccs178 packages. Here, partial charges ideally derived from a quantum chemical calculation are used for the calculation to predict accurate trajectories of the gas, leading to more reliable computed CCS values. The MK charges are recommended among all charge schemes.179
In summary, the available computational methods are fully capable of reliably calculating reasonable structures of isolated carbohydrate ions in the gas phase, and the presented workflow can also be applied to other classes of biomolecules, such as lipids244 or nucleotides.245 In most cases, it is recommended to use DFT methods not only for computing the energetics, spectra, and charges of the promising structural candidates but also for conformational sampling. Accuracy remains a question of computational expense, and for most mammalian glycans, not exceeding more than eight monosaccharide units, DFT approaches are feasible. In the long run, computational resources will increase, and sampling of larger carbohydrate ions at a higher accuracy will become possible. In the meantime, promising new semiempirical methods can fill the gap.
3. Human Milk Oligosaccharides
3.1. Structure and Analytical Challenges
The facts that breast-fed infants enjoy a variety of health benefits compared to their bottle-fed peers and that infant physiology is strongly influenced by intestinal bacteria have been known for over a century. After a connection between breast feeding and the composition of infant gut microbiota had been established, intense research led to the discovery of human milk oligosaccharides (HMOs) as the milk fraction responsible for promoting the growth of certain desired bacteria.12 HMOs are unconjugated glycans that represent the third largest fraction in colostrum and human milk (5–25 g L–1), following lactose and lipids.12,246 In contrast to lactose that serves as a caloric nutrient for infants, HMOs are digested and absorbed minimally, their functions being more diverse and complex.247−249 Besides helping to establish a healthy gut microbiota as prebiotics, HMOs also serve as antiadhesive antimicrobials. By resembling cell surface glycan epitopes, they act as soluble decoy receptors for enteric pathogens relying on glycan-mediated attachment to mucosal surfaces for infection. In addition, HMOs also perform regulatory roles, modulating intestinal epithelial cell and immune responses in infants.250,251
Members of the HMO family can be derived from five monosaccharide building blocks: d-glucose (Glu), d-galactose (Gal), N-acetyl-d-glucosamine (GlcNAc), l-fucose (Fuc), and N-acetyl-d-neuraminic acid (Neu5Ac), the only sialic acid identified in human milk to date. The structure of HMOs follows a strict general blueprint, highlighted in Figure 9.12,252−254 During assembly of the core, a lactose (Lac) moiety—found at the reducing end of all HMOs—can be extended by N-acetyllactosamine (LacNAc, type 2) and lacto-N-biose (LNB, type 1) disaccharide units. While further disaccharide units may be attached to type 2 LacNAc residues, addition of a type 1 LNB unit terminates the respective antenna. Branching points in the core are introduced through β1,6-glycosidic linkages, leading to iso-HMOs with multiple antennae. The absence of such branching points results in para-HMOs with unbranched core structures, containing exclusively β1,3-linkages between the disaccharide units. Terminal and internal residues of the HMO core may be fucosylated via α1,2-, α1,3-, or α1,6-linkages. In addition, Neu5Ac residues may be attached to the core via α2,3- or α2,6-glycosidic bonds, leading to acidic oligosaccharides. The majority of HMOs is fucosylated, while 5–20% carry one or more sialic acids.12,246 As a result of these modifications, HMOs often display a diverse array of Lewis and blood group epitopes, frequently observed across various classes of glycans.
Figure 9.

Overview of the structure of human milk oligosaccharides. (A) The five monosaccharide building blocks and basic structural blueprint of human milk oligosaccharides (HMOs), shown on the example of a hypothetical triantennary glycan. (B) Typical examples of neutral HMOs, highlighting both unmodified and fucosylated structures. (C) Representative acidic HMOs carrying one or more sialic acid residues.
Despite the limited number of building blocks and the strict logic of their assembly, HMOs show remarkable structural diversity, with well over 100 different structures identified to date.246,255 Many of these oligosaccharides are isomers, differing only in their branching, fucosylation, or sialylation patterns. Although the anomeric configuration of Gal, GlcNAc, Fuc, and Neu5Ac residues is invariable in HMOs and determining the composition is straightforward owing to mass differences between the building blocks, elucidating branching and connectivity are major challenges in traditional MS-based HMO analysis. Thus, the present section focuses on recent developments in the field that facilitate isomer distinction and provide information on linkage positions, a crucial element of HMO structure.
3.2. Electron-Based Dissociation Methods in HMO Analysis
B-, C-, Y-, and Z-type fragment ions, resulting from glycosidic bond cleavages, carry essential information on the sequence and composition of oligosaccharides. A- and X-type cross-ring fragments, on the other hand, are indispensable for the assignment of linkage positions. As branching and connectivity are key aspects of HMO structure, considering both the core architecture and the attachment of modifications, dissociation methods capable of generating diverse and abundant cross-ring fragments have been at the center of MS-based HMO research. Low-energy CID and IRMPD are based on the gradual heating of ions and lead primarily to cleavage of the most labile bonds. This manifests in extensive glycosidic cleavages, while cross-ring fragments are generally scarce, especially in the case of protonated species.256,257 In addition, Fuc and Neu5Ac residues may be lost upon vibrational excitation of the analytes, impeding structural assignment.23 Through removing mobile protons that facilitate glycosidic bond cleavage, permethylation and adduction of metal ions may enhance the formation of cross-ring fragments in slow-heating methods.258−260 Despite these advances, information provided by CID and IRMPD on oligosaccharides is rather limited, inspiring scientists to explore gas-phase ion chemistry and alternative ion activation methods better suited for the specific needs of HMO research.
Electron-based or electron-mediated dissociation (ExD) methods, adopted from MS-based peptide and protein analysis,79,81,261−265 have proven to be extremely useful for the structural characterization of HMOs. Briefly, ExD methods are based on ion–electron interactions in the gas phase, inducing either rapid electronic excitation of analytes, the formation of charge-reduced radical ions, or both. Fragmentation pathways involving such radicals and excited electronic states lead to markedly different product ion spectra than those observed upon CID or IRMPD. In principle, ExD methods accompanied by charge reduction require multiply charged precursors: ECD and ETD are applied to polycations, while EDD and negative (N)ETD are suited for polyanionic species. In contrast, EED and EID do not involve electron transfer and are generally performed on singly charged precursors, irrespective of ion polarity.88,266,267 Although ETD and NETD utilize gas-phase ion–ion reactions instead of interactions between ions and cotrapped electrons, they are generally included into the broader family of ExD methods owing to similarities of the resulting fragmentation patterns and will be discussed as such herein.
The first ECD MS experiments on a milk oligosaccharide were performed by Adamson and Håkansson in 2007, employing an FTICR-MS platform and the unbranched para-lacto-N-hexaose (pLNH, see Figure 9) as the model compound.85 To facilitate the formation of doubly charged precursors and systematically investigate the influence of metal ion adduction on ECD, native pLNH was cationized with divalent alkaline earth and transition metal ions, such as Mg2+, Ca2+, Ba2+, Mn2+, Co2+, and Zn2+. In general, ECD provided complementary information to IRMPD, generating several cross-ring fragments not observed upon slow-heating photodissociation of pLNH dications. Subtle differences observed in the ECD patterns of the various pLNH adducts were attributed to the impact of metal ion coordination site on fragmentation and to that of the second ionization energy influencing the electron capture of analytes.
Following the introduction of ECD to the analysis of neutral HMOs in positive ion mode, the same authors employed its negative ion counterpart, EDD, for the structural characterization of sialylated structures.268 Disialyllacto-N-tetraose (DSLNT) and the sialyllacto-N-tetraose isomers LST a and LST b (see Figure 9) were studied as doubly deprotonated ions using FTICR-MS. Electron detachment, induced by electrons of moderate (20–30 eV) kinetic energy, led to charge-reduced anions that readily underwent radical-driven dissociation pathways, producing singly charged fragments. For all three model compounds, EDD led to a more diverse set of cross-ring fragments than CID or IRMPD of the respective [M – H]− and [M – 2H]2– species. In the case of LST b, as an example, 0,4A2 and 3,5A2 ions confirmed the 2,6-linkage of Neu5Ac, while the 1,3X2 fragment indicated 1,3-linkage of the terminal Gal residue, in accordance with the lacto-N-tetraose (LNT) core of the analyte. In a subsequent study employing the same underivatized model compounds, EDD of acidic HMOs was extended to their chloride adducts with the general formula [M – H + Cl]2–. In general, EDD spectra of chloride-adducted HMOs were dominated by singly charged product ions and contained a more diverse array of glycosidic and cross-ring fragments than that previously observed for doubly deprotonated species. While generating DSLNT chloride adducts by ESI proved to be challenging, LST isomers readily formed singly deprotonated chloride adducts with sufficient abundance. Figure 10 shows the EDD spectrum of [LST b – H + Cl]2–. Linkage positions of the Gal and Neu5Ac residues at the nonreducing termini may be unambiguously determined, owing to diagnostic fragments arising from cross-ring cleavages of the GlcNAc pyranoside.
Figure 10.

Electron detachment dissociation (EDD) of human milk oligosaccharide dianions. (A) EDD tandem mass spectrum of LST b as [M – H + Cl]2–, along with (B) the corresponding fragmentation pattern. Product ions depicted in bold appeared uniquely for the chloride-adducted species and were not observed upon EDD of the doubly deprotonated analogue. Fragments resulting from multiple cleavage sites are designated with a slash. Reprinted with permission from ref (269). Copyright 2012 American Society for Mass Spectrometry.
In 2011, Han and Costello employed ETD for the first time for the MS-based structural characterization of glycans.257 ETD is based on electron transfer from a suitable radical anion, generated usually from fluoranthene, to polycationic analytes. As ETD exploits ion–ion chemistry and does not require trapping electrons like most other ExD methods, it is readily compatible with linear and 3D quadrupole ion traps. The neutral tetrasaccharide LNT, its linkage isomer lacto-N-neotetraose (LNnT, see Figure 9), and three monosialylated LST isomers with either a LNT or LNnT core were studied as reduced, permethylated species. Systematic analysis of Na+, K+, Mg2+, and Ca2+ adducts revealed a strong dependence of ETD patterns and fragmentation efficiencies on the nature of the metal ion chosen. Alkali metal adducts of HMOs gave rise to a very limited number of fragments, proving unsuitable for ETD experiments. In contrast, Ca2+ and especially Mg2+ adducts yielded highly informative product ion spectra: numerous cross-ring fragments and extensive glycosidic cleavages enabled distinction of isomeric HMOs and assignment of the underlying structures. Figure 11 shows the ETD spectrum of reduced, permethylated LST c as a Mg2+ adduct, along with the assignment of product ions, among which both odd- and even-electron species are present. The 2,6-linkage of Neu5Ac in LST c could be confirmed based on 3,5X3 and 0,4X3 fragments, while the ions 3,5A3, 0,3A3, and 2,4A3 indicated a 1,4-linkage between the Gal and GlcNAc residues, verifying the LNnT core of the monosialylated analyte. In cases when MS2 with ETD did not allow for an unambiguous structural assignment, the authors utilized the MSn capabilities of the quadrupole ion trap instrument to combine ETD and CID in a sequential manner. While the linkage position of the nonreducing end Gal in LNT and LNnT could not be determined by single stage ETD, subsequent CID of a singly charged internal ETD fragment provided the information required to decipher connectivity in each isomer, demonstrating the potential of MSn strategies to combine complementary ion activation methods.
Figure 11.

Electron transfer dissociation (ETD) of metal-ion-adducted human milk oligosaccharides. (A) ETD fragmentation pattern of reduced, permethylated LST c as [M + Mg]2+ (m/z 652.3). Note the diagnostic cross-ring fragments enabling the assignment of the LNnT core and the Neu5Ac linkage position. (B) Corresponding ETD tandem mass spectrum. Fragments between m/z 1142.2 and 1216.4 are attributed to cleavages within the sialic acid residue. Reprinted with permission from ref (257). Copyright 2011 American Society for Mass Spectrometry.
The latest ExD method introduced to MS-based HMO analysis is EED. EID and EED refer to closely related processes and are sometimes used interchangeably in the literature, with the former acronym applied as a more general term. To avoid confusion, we always use the term favored by the authors in the respective studies. Unlike the techniques discussed so far in the present section, EED is not accompanied by charge reduction and may be readily performed on singly charged species. Taking advantage of this feature and building on the work of Gao et al.,270,271 Lin and co-workers performed EDD on five LNFP isomers derivatized with the N-methylated form of a sequestered proton reagent for acid-catalyzed glycan sequencing (Me-PRAGS).272 Me-PRAGS is a quaternary ammonium compound with a fixed positive charge that can be readily attached to the reducing end of glycans. EDD of Me-PRAGS-derivatized LNFP cations in an FTICR cell generated primarily fragments retaining the charge at the reducing end. Upon irradiating the analytes with 12 eV electrons, complete sets of sequence-informative 1,5X-, Y-, and Z-type ions were observed. Employing electrons with 16 eV kinetic energy led to more efficient fragmentation, to the formation of doubly charged product ions, as well as to a higher number of linkage-informative cross-ring and secondary fragments. The study reveals the capabilities of EED MS to sequence neutral, unmethylated glycans following charge tagging and to distinguish isomers based on differences in their product ion spectra. We hope the selected works above not only reflect the utility of ExD methods in HMO analysis but also demonstrate the importance of basic research to develop techniques that provide information previously unattainable by conventional MS-based methods.
3.3. Ultraviolet Photodissociation Mass Spectrometry of HMOs
Another ion activation method that has been successfully employed to gain information on branching and connectivity in HMOs is UVPD. The rapid increase of internal energy, electronic excitation, and occasional EPD accompanying the absorption of energetic UV photons open up diverse fragmentation pathways in glycan ions, manifesting in abundant cross-ring and internal fragments.32 Unlike charge-reducing ExD methods, such as (N)ETD or ECD/EDD, photodissociation may be readily performed on singly charged species. In addition, UVPD is compatible with a variety of MS platforms, provided suitable optical access is established for a sufficiently bright UV source, most commonly an excimer or solid-state Nd:YAG laser.
In general, native HMOs lack strong UV chromophores, exhibiting relatively low absorption cross sections at lower UV photon energies. Thus, the first UVPD MS study on HMOs was performed using 157 nm vacuum ultraviolet (VUV) radiation, produced by a nanosecond F2 excimer laser.273 Following these initial experiments on native and reducing end-modified oligosaccharides, Reilly and co-workers systematically studied the VUV photofragmentation of acidic HMOs as positive ions in a linear ion trap.274 The analytes—permethylated to avoid the loss of labile Neu5Ac residues—showed extensive fragmentation upon interaction with 7.9 eV VUV photons, resulting in numerous cross-ring fragments. Isomeric 3′- and 6′-sialyllactose (3′- and 6′-SL, see Figure 9) could be unambiguously distinguished based on their markedly different product ion spectra. In addition to mere distinction, the sialic acid linkage position could be confidently assigned in both species, owing to diagnostic cross-ring fragments whose formation involves the cleavage of the C3–C4 or C4–C5 bond in the Gal residues. Following the assignment of the sialic acid linkage position in the simplest acidic HMOs, the authors demonstrated the capabilities of 157 nm UVPD MS for the structural characterization of larger sialylated species. In the monosialylated biantennary lacto-N-neohexaose SLNnH II (see Figure 9), the combination of 0,4A4, 1,5X3β, 1,5X3β, and 2,4A4/1,5X3α fragments confirmed the location of Neu5Ac being on the β3 antenna of the heptasaccharide.
Ko and Brodbelt analyzed acidic HMOs (dp3–dp7, dp being the degree of polymerization) in their native form by UVPD MS in negative ion mode.76 Deprotonated ions were irradiated in a linear ion trap using a 193 nm (6.4 eV photon energy) ArF excimer laser, and the resulting fragmentation patterns were compared to those obtained by CID on the same MS instrument. The ArF laser with 8 mJ pulse energy provided efficient photofragmentation, allowing for short UVPD interaction periods on the low millisecond time scale. In general, UVPD provided not only better sequence coverage than CID but also more extensive cross-ring cleavages, as highlighted in Figure 12, on the example of LST b. Abundant A- and X-type photofragments carry information on connectivity, e.g., on the linkage position of the terminal Gal in LST b, that could not be assigned based on CID experiments. Interestingly, UVPD of dianions resulted mainly in singly charged fragments, while CID mass spectra were rich in doubly charged product ions. This phenomenon could be attributed to structural differences between photo- and CID fragments, retaining in general one vs two sialic acids, respectively. Charge-reduced EPD products among the photofragments of multiply charged anions were of relatively low abundance.
Figure 12.

193 nm ultraviolet photodissociation (UVPD) of deprotonated human milk oligosaccharides. (A) Product ion spectra of singly deprotonated LST b (m/z 997) employing (A) collision-induced dissociation (CID) and (B) 193 nm UVPD. The corresponding (C) CID and (D) UVPD fragmentation patterns and assignments. The precursor ion is labeled with an asterisk. Reproduced with permission from ref (76). Copyright 2011 American Chemical Society.
An inventive approach to make HMOs more suitable for UVPD MS at longer wavelengths utilizes covalent attachment of aromatic chromophores to the glycans. In a proof-of-concept study, four isomeric lacto-N-fucopentaoses (LNFP I, II, III, and V) and three lacto-N-difucohexaose isomers (LNDFH Ia, Ib, and II) were derivatized with various fluorophores via reductive amination.275 The labels, such as 6-aminoquinoline (6-AQ) and 7-aminomethylcoumarin (AMC), are commonly applied in LC-MS glycomics workflows to facilitate fluorescent detection. Here, the tags served to increase the absorption cross section of the analytes at 355 nm (3.5 eV), corresponding to the third harmonic of a Nd:YAG laser. While CID of singly sodiated species generated abundant Y-type ions retaining the fluorophore at the reducing end, photodissociation at 355 nm led mainly to A- and C-type fragments. In general, photodissociation pathways were largely unaffected by the nature of the fluorophore, whereas UVPD efficiencies appeared to be strongly influenced by the label, with 6-AQ providing the most efficient fragmentation. Although UVPD in general provided superior isomer differentiation compared to CID, LNFP II and III—both carrying a Fuc on their subterminal GlcNAc residue, only at different linkage positions—could not be distinguished based on their photofragments. In addition to direct UVPD of singly sodiated species, the authors performed electron photodetachment dissociation—also termed activated (a-)EPD—on LNDFH II anions. HMOs derivatized with 7-amino-1,3-naphthalenedisulfonic acid (AGA), a fluorophore with high solution- and gas-phase acidity, readily form multiply deprotonated species when electrosprayed in negative ion mode. Upon photon absorption at 355 nm, part of the [(LNDFH II + AGA) – 2H]2– ion population underwent EPD, resulting in charge-reduced radical anions. CID of the singly charged radical species and of its even-electron analogue led to remarkably different fragmentation patterns, the former displaying a more diverse array of product ions. Thus, a-EPD has the potential to complement more widespread ion activation techniques and become a powerful hybrid dissociation method in MS-based glycan analysis.276
In the Julian lab, an alternative strategy was developed to enable efficient UVPD MS analysis of HMOs at lower photon energies.277 The method utilizes radical chemistry and relies on labeling glycans with the aromatic radical precursor 2-iodoaniline at their reducing end. Homolytic cleavage of the carbon–iodine bond is induced upon absorption of 4.7 eV photons, provided by a Nd:YAG laser (266 nm). The photocleavage generates hydrogen-deficient oligosaccharide radical ions, which readily undergo radical migration and dissociation upon collisional activation. This radical-directed dissociation (RDD) approach enabled unambiguous distinction of two pairs of fucosylated HMO isomers. The extensive set of glycosidic and cross-ring fragments generated from singly protonated parent ions by RDD is shown in Figure 13, on the example of LNDFH I and II. Although the most abundant, 1,5X-type cross-ring fragments do not reveal the underlying branching and connectivity, several diagnostic signals can be observed that appear uniquely for only one species, demonstrating the utility of RDD for the distinction of isomeric HMOs.
Figure 13.

Radical-directed dissociation (RDD) of protonated human milk oligosaccharides at 266 nm. (a) RDD tandem mass spectrum and product ion assignment of singly protonated LNDFH I labeled with 4-iodoaniline. (b) RDD of the singly protonated LNDFH II isomer labeled with the same radical precursor. Reprinted with permission from ref (277). Copyright 2014 Elsevier B.V.
3.4. Ion Mobility–Mass Spectrometry in HMO Analysis
With the advent of commercial IM-MS instruments, the separation of isomeric HMOs based on their gas-phase mobilities has gained significant momentum. In general, the CCS and mobility of molecular ions strongly depend on the three-dimensional structures they adopt in the gas phase. As differences in composition, configuration, connectivity, or branching often manifest in different size and shape, isomeric glycans may be efficiently distinguished, separated, identified, and quantified by IM-MS.128,129
In a comparative study, Williams et al. employed a custom-built DTIM-MS and a commercial TWIM-MS device to study pairs of LNFP and LNDFH isomers as singly charged cations.278 Each HMO Na+ adduct displayed a sole, symmetric arrival time distribution (ATD) on both IM-MS platforms. This observation was in accordance with molecular modeling calculations, revealing a single family of low-energy conformers for all four sodiated glycans ions. Experimental DTCCSHe values were generally in good agreement with those obtained by TM calculations, further verifying the low-energy structural candidates. The rather small difference in the mobilities of isomeric HMOs, however, limited the efficiency of separations, which underlines the importance of resolving power and selectivity in IMS experiments.
In comparison to chromatography or condensed-phase electrophoretic techniques, the possibilities to improve selectivity in IMS through altering the separation medium are rather restricted. Although changing the buffer gas composition may lead to better separation in certain cases, the effects are not comparable to those resulting from tuning a myriad of experimental parameters over a broad range, as possible in HPLC. Therefore, various strategies were developed in IMS to influence the conformation of analytes, with the aim of increasing the relative CCS difference (ΔCCS/CCSavg) of isomers and ultimately improving the resolution between critical peaks.
Fenn and McLean applied an in-house developed DTIM-MS instrument to study an extensive set of carbohydrate standards and recorded over 300 DTCCSHe values for intact and fragment ions generated though in-source dissociation.132 Among the 31 model compounds, four LNFP and two LNDFH isomers represented HMOs. In accordance with previous works on smaller carbohydrates,131 metal ions had a significant impact on the measured mobilities and CCSs of larger glycans. These findings highlight the role metal ions play in determining the gas-phase structure of HMOs, an effect successfully harnessed in various studies to improve isomer separations by IMS.
In a series of closely related experiments, Huang and Dodds investigated group I–II metal ion adducts of fucosylated HMOs, employing a commercial TWIM-MS device with N2 as the drift gas. First, two LNFP and two LNDFH isomers were analyzed as singly charged cations formed with Li+, Na+, K+, Rb+, and Cs+ ions.279 To convert the measured arrival times to TWCCSN2→He values, protonated polyalanine ions with known DTCCSHe were used as calibrants (the notation to designate the buffer gas and IMS technique of choice as super- and subscript follows widely accepted recommendations138). Despite the buffer gas mismatch, the TWCCSN2→He values of the alkali-metal-adducted glycans formed a consistent set. CCSs of LNDFH I and II metal adducts varied in parallel, first increasing with increasing metal ion radius and then stagnating at around 224 and 220 Å2, respectively. In stark contrast, the CCSs of LNFP V ions increased steadily with increasing metal ion size, while the measured CCS values of LNFP I adducts decreased in the following order: [M + Cs]+ > [M + Na]+ > [M + K]+ > [M + Rb]+ > [M + Li]+. Owing to the remarkably large CCS of [LNFP I + Na]+, the best separation for LNFP isomers was achieved using Na+ adduction. The results clearly demonstrate the possibility to improve selectivity in IM separations of HMOs through the careful choice of adduct-forming metal ions. Building on these findings, the authors combined gas-phase nondissociative electron transfer (ETnoD) with subsequent TWIM separation of Ca2+- and Ba2+-adducted HMOs.280 Employing the same four model glycans as previously and the 1,4-dicyanobenzene radical anion as the electron donor, the separation of isomers with and without prior electron transfer to the analytes was compared. Figure 14 shows that charge-reduced [M + Ca]+• radical ETnoD products show better separation than their even-electron counterparts. In contrast, no significant improvement could be achieved for Ba2+ adducts with this strategy, due to comparable shifts in the CCS of the isomers upon ETnoD. Although the aforementioned electron-transfer-induced CCS shifts are rather unpredictable, they appear to be both isomer-specific and metal-ion-dependent, revealing the clear analytical potential of the method. The ETnoD-TWIM-MS approach was successfully extended to additional fucosylated compounds, in combination with the full set of nonradioactive alkaline earth metals.281 Complete rationalization of these experimental observations would require high-level theory. Although the sheer size and complexity of the analytes make such attempts extremely challenging, state-of-the-art theoretical methods may reveal the underlying atomic level structure of oligosaccharide metal ion adducts in the foreseeable future.
Figure 14.

Nondissociative electron transfer (ETnoD) combined with traveling wave ion mobility (TWIM) separations for the analysis of fucosylated milk oligosaccharides. (a) Collision cross section (CCS) distributions of LNFP I and V isomers as calcium and barium ion adducts, along with (b) the CCS distributions of the respective singly charged radical ETnoD products. (c) CCS distributions of LNDFH I and II isomers as calcium and barium ion adducts and (d) that of the respective ETnoD products. Reproduced with permission from ref (280). Copyright 2015 American Chemical Society.
The utility of metal ion adduction to improve HMO isomer separations was further demonstrated by Baker and co-workers, employing DTIM-MS with N2 as a buffer gas.282 While singly sodiated LNT and LNnT were inseparable due to nearly identical mobilities, K+ and singly deprotonated Zn2+ adducts of the same ions showed increasingly better separation, enabling their distinction. Interestingly, [LNT + 2Na]2+ and [LNnT + 2Na]2+ were baseline resolved, with the isomers exhibiting reverse migration order compared to K+ and Zn2+ adducts. Another important finding of the study concerns the effect of ion polarity. While small glycan isomers displayed in general better separation as Na+ adducts, dp4–dp6 oligosaccharide isomers exhibited larger differences in their mobilities when analyzed as deprotonated species. As an example, lacto-N-hexaose (LNH) and lacto-N-neohexaose (LNnH, see Figure 9) showed only partial separation as sodiated ions, but the same compounds could be baseline resolved when analyzed as singly deprotonated species, in accordance with previous findings.283
Inspired by the efficient IM separation of deprotonated carbohydrate isomers,133 Struwe et al. studied the influence of ion polarity and adduct formation on the gas-phase structure of glycans, combining TWIM-MS experiments with DFT and ab initio molecular dynamics (MD).283 LNH and LNnH showed significantly improved separation in N2 as deprotonated species compared to their chlorinated, sodiated, and protonated counterparts. To rationalize the observations, experimental TWCCSN2→He values were compared to those calculated for DFT-optimized candidates. In general, experiment and theory showed a good match for sodiated ions; [LNT + Na]+ and [LNnT + Na]+ adopt similar, compact gas-phase structures, governed mainly by charge solvation. Interestingly, TWCCSN2→He of the respective deprotonated ions matched those calculated for neutral species. The ab initio MD simulations performed on exemplary conformations of [LNH – H]− revealed rapid charge migration on the picosecond time scale, blurring the negative charge over the molecule during the millisecond-long IM separation (Figure 15). This charge delocalization provides a rationale for the above-mentioned unusual agreement: the experimental CCS of the deprotonated species represents an average of several, rapidly interconverting deprotonation site isomers, thereby approaching the CCS of the respective neutral species.
Figure 15.

Ab initio molecular dynamics reveals rapid charge migration in a deprotonated milk oligosaccharide. (a) Snapshots of singly deprotonated LNH at different simulation times. Colored spheres indicate the OH groups involved in charge migration. The position of the charge within each structure is highlighted by a black circle. (b) Chemical structure of LNH; the colored spheres correspond to the OH groups in the upper panel that are deprotonated over the 20 ps simulation. Reprinted from ref (283). Published by The Royal Society of Chemistry. Copyright 2016 Struwe et al. (Creative Commons Attribution 3.0 Unported License).
Although selectivity is a key aspect of IM separations, resolving power is equally important in determining the extent to which two peaks are separated. To demonstrate the role of resolving power, Pu et al. combined TIMS with EED on an FTICR-MS platform to separate and identify the linkage isomers LNT and LNnT as permethylated, singly sodiated ions.284 The method, termed selected accumulation (SA-)TIMS,285 enables the hyphenation of millisecond-long IM separations to slow mass analyzers and ExD techniques requiring comparably long interaction periods. Briefly, mobility-selected ions were accumulated on the electric field plateau of the TIMS tunnel, in front of a small potential barrier near the exit funnel. After reaching a sufficiently large population of mobility-selected ions, the barrier may be lowered to extract ions for subsequent fragmentation and mass analysis. The tetrasaccharides were baseline resolved by TIMS, and the linkage position of the terminal Gal could be determined in each isomer owing to diagnostic cross-ring fragments previously not observed in CID, ECD, or ETD spectra. Thus, TIMS-EED-MS/MS enabled both rapid distinction and structural assignment of HMO linkage isomers. TIMSCCSN2 values obtained through calibration agreed with those determined by a commercial DTIM-MS device, providing further structural information on the analytes.
Another platform that enabled a significant increase in the resolving power of IM separations is termed structures for lossless ion manipulations (SLIMs), developed by Smith and co-workers.286−289 SLIM technology utilizes printed circuit boards (PCBs) to pattern electrodes on planar surfaces, and ion manipulation is achieved in a suitable buffer gas by a combination of RF and DC fields. SLIM-based devices have been successfully applied for TWIM separations at low pressures, offering the possibility to create extremely long separation pathways through serpentine routes and multipass devices. Employing a 13 m long serpentine SLIM module with N2 buffer gas, singly sodiated LNFP I and II ions could be fully resolved within 550 ms.290 In a SLIM device enabling multiple passes through its 13.5 m long separation route, [LNH + H + K]2+ and [LNnH + H + K]2+ were baseline resolved already after a single pass.152 After accomplishing nine rounds and thereby covering a total of 121.5 m, an additional feature appeared in the ATD of [LNnH + H + K]2+. This feature had remained unnoticed in previous IM-MS experiments but appeared here as a distinct peak owing to the extremely high resolving power provided by the multipass SLIM technology. Building on these intriguing findings, the authors set out to unravel hidden features in the ATDs of various glycan ions.134 Upon high-resolution SLIM-based TWIM-MS analysis, four distinct peaks could be observed in the ATD of singly protonated LNT and two in that of its isomer LNnT. Similarly, several well-resolved features appeared in the ATD of LNFP I, II, and III ions, analyzed as [M + H + K]2+. These features, highlighted in Figure 16 may correspond to α/β-anomers, protomers, or conformers that do not interconvert on the subsecond time scale of the separations. Gaining more information on the nature of these species would require the hyphenation of SLIM-based IM-MS to orthogonal analytical methods, such as IR ion spectroscopy, which is addressed in Section 3.5.
Figure 16.
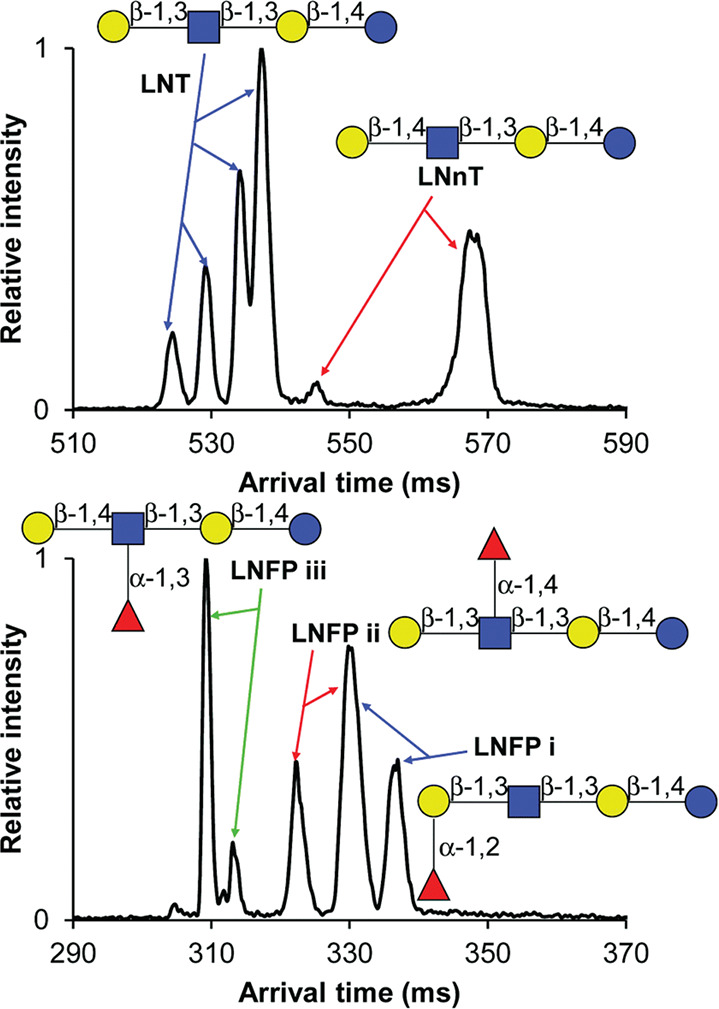
Ion mobility separation of human milk oligosaccharides in structures for lossless ion manipulations. The upper panel shows the arrival time distributions (ATDs) of singly protonated LNT and LNnT, resulting from a 31.5 m separation. The lower panel depicts the ATDs of three LNFP isomers as the outcome of a 45 m separation. LNFP isomers were measured as doubly charged [M + H + K]2+ species. Reprinted with permission from ref (134). Copyright 2018 The Royal Society of Chemistry.
At last, an interesting study by Li and co-workers—dealing with the integration of IMS into a multidimensional glycomics workflow—needs to be mentioned.291 The authors coupled capillary zone electrophoresis (CZE) to TWIM-MS and successfully analyzed three LST isomers labeled with a carbonyl-reactive aminoxy tandem mass tag (aminoxyTMT) for quantitation. Although LST b and LST c comigrated in CZE, they could be readily separated by TWIMS as [M + H + Na]2+ ions. The orthogonality observed between the two electrophoretic techniques demonstrates that “size-to-charge ratios” of glycans are governed by markedly different effects in solution and in the gas phase.
An important aspect of MS-based glycan analysis is fucose migration, which also affects the structural characterization of HMOs. Owing to their importance and relevance to a variety of glycan classes, rearrangement reactions of gas-phase oligosaccharide ions will be addressed separately in Section 5.6.
3.5. HMO Analysis by Gas-Phase Ion Spectroscopy
Gas-phase ion spectroscopy in the UV and IR region for the characterization of HMOs has been the subject of recent research and shows great potential.33,36,208 In a more general approach across different glycan classes, the first IR spectrum using messenger-tagging spectroscopy in the OH-stretching region (here 3200 to 3700 cm–1) of N-acteyllactosamine, a core termination in HMOs, as a sodium adduct was published.189 The IR spectrum is well-resolved and distinguishable from the IR spectra of five isomeric disaccharides. The approach was challenged with the identification of HMOs up to hexasaccharides which yielded highly resolved IR spectra that are shown in Figure 17.292 A high-resolution IMS stage prior to messenger-tagging serves as additional separation in the case where multiple conformers and anomers are present.201,293
Figure 17.
Gas-phase IR spectroscopy of human milk oligosaccharides. Arrival time distributions and vibrational spectra of sodiated (a) LNDFH I and (b) LNDFH II, each tagged with one molecule of N2. The drift times and peak widths are (a) 9.21 ms, fwhm = 0.13 ms; and (b) 9.32 ms, fwhm = 0.13 ms. Reprinted with permission from ref (292). Copyright 2018 The Royal Society of Chemistry.
Conformer-selective IR spectroscopy can, furthermore, be performed in IR-IR double resonance spectroscopy in which the first pump laser is fixed to a certain wavelength which is specific to one conformer only, and the second probe laser is scanned through the wavenumber range. For N-acetyllactosamine, the IR signature is identical to the IR signature using high-resolution IMS prior to IR spectroscopy,294 yet both approaches require advanced instrumental setups. Recently, the first cryogenic UV spectra of HMOs were recorded as 2D UV-MS spectra which is an alternative way of representing the spectroscopic data in 2D data arrays of fragment ions, as described in Section 2.4.295 The lack of a chromophore in HMOs is circumvented by mixing the glycans in solution with aromatic molecules which ionize as noncovalent protonated complexes. Gas-phase ion spectroscopy in combination with rich databases is capable of identifying glycans in cases in which LC, tandem MS, and IMS reach their limits.
4. N-Linked Glycans
4.1. Structure and Analytical Challenges
Glycosylation is the most important post-translational modification of proteins.26N-Glycans are branched oligosaccharides that are bound, most commonly, via GlcNAc to an Asn residue of the protein backbone. Protein glycosylation generally occurs only to Asn-Xxx-Ser/Thr sequons, where Xxx can be any amino acid except for proline.26,296 In rare cases, the third amino acid of the sequon can also be cysteine.297N-Glycans can be found in all living beings and viruses and exhibit various important physiological roles. N-Glycoslyation influences several properties of glycoproteins, such as, for example, “their conformation, solubility, antigenicity, activity, and recognition by glycan-binding proteins”.298 Furthermore, several human diseases, such as arthritis, can be linked to N-glycosylation.299,300
Here, we will focus on N-glycans that can be found on mammalian proteins. Commonly, N-glycans are enzymatically cleaved from proteins or peptides using peptide N-glycosidases (PNGase) prior to analysis. It is also possible to directly analyze glycopeptides, which will be discussed later. A common motif of all N-glycans is the chitobiose core (Figure 18B), composed of three mannose and two GlcNAc moieties, which is commonly attached to the protein backbone via GlcNAc. The mannose residue is branched and connected via α1,3- and α1,6-glycosidic linkages to the two other mannose building blocks. Based on the chitobiose core, there are three types of N-glycans: high mannose, complex, and hybrid (Figure 18A). In high-mannose N-glycans, the mannose antennae are extended by further mannose building blocks, whereas in complex N-glycans the mannose antennae are extended by GlcNAc and subsequently other monosaccharide units (e.g., Gal). In hybrid N-glycans, the C6-antenna is extended by mannose building blocks and the C3-antenna similarly to complex antennae. The core can also be extended by a bisecting GlcNAc (β1,4) at the intermediary mannose, and its GlcNAc residues can be α1,6-fucosylated (Figure 18B). Commonly, N-glycans have two, three, or four antennae (Figure 18C). In complex N-glycans, the mannose residues can be extended by a GlcNAc residue by a β1,2-, β1,4-, or α1,6-glycosidic linkage. The GlcNAc residues are in turn commonly substituted by β1,4-linked Gal that can subsequently be substituted with N-acetylneuraminic acid via an α2,6- or α2,3-glycosidic linkage. The antennary Gal and GlcNAc residues can be α1,2- or α1,3-fucosylated, respectively. Further modifications of the antennae are possible, such as, for example, the substitution with multiple LacNAc (PolyLacNAc) moieties that can in turn also be branched. Furthermore, other fucosylation and sialylation patterns, substitution with blood group epitopes, as well as sulfations are possible.301
Figure 18.

Most common types and modifications of N-glycans found in mammals. (A) Exemplary structures for three types of N-glycans: high-mannose (sometimes called oligomannose), complex, and hybrid. (B) Common core modifications of mammalian N-glycans and chitobiose core, a subunit common to all N-glycans. (C) Common antennary modifications.
The analysis of N-glycans by means of chromatography often coupled to mass spectrometry is possible and established in many laboratories. Experimental retention times can be matched to values stored in databases and structures assigned.302 Although this technique has proven to be reliable, it does not always yield sufficient structural information about the analytes. In the past decades, the usage of mass spectrometry-based techniques for structural characterization of N-glycans has become a valuable tool both coupled to chromatography and as a stand-alone technique. Although it is possible to ionize the N-glycopeptide analogues of N-glycans by fast-atom bombardment (FAB) mass spectrometry,54 it was not feasible to ionize underivatized N-glycans efficiently using this technique. This changed when ESI and MALDI were introduced, which exhibit an increased sensitivity toward both native and derivatized carbohydrates.303−305
Mass spectrometry yields mass-to-charge ratios of ionized N-glycans. From the resulting masses, it is commonly possible to estimate the number of monosaccharide units that constitute the N-glycan. However, monosaccharide units such as Man and Gal or GlcNAc and GalNAc exhibit identical masses, and discrimination purely based on mass spectrometry is therefore usually not possible. Furthermore, information about branching and the configuration of glycosidic linkages cannot be obtained. By combining MS and various fragmentation techniques, IMS, and spectroscopic techniques, multidimensional information can be obtained to resolve the aforementioned issues. Here, we will give a brief overview of recent works employing CID, MSn, ExD, UVPD, IM-MS, and IR spectroscopy for in-depth structural analysis of N-glycans.
4.2. Collision-Induced Dissociation of N-Glycans
CID is the most common fragmentation method. It is a slow activation method, where the weakest bonds are cleaved first. Based on the formed fragments, information about the connectivity of the monosaccharide units can be gained. Depending on whether cross-ring fragments (A and X) are formed, additional information about linkage and branching can be obtained.306 However, the fragmentation patterns are strongly dependent on whether the spectra are recorded in positive or negative ion mode. Moreover, the information content obtained from CID of N-glycans depends on the type of adducts that are fragmented (protonated ions, metal adducts, or phosphate adducts) and whether the N-glycan is derivatized or labeled. It must be noted that activation by IRMPD essentially yields the same fragments as CID.307
An early report shows that CID of sodiated isomeric and permethylated high-mannose N-glycans leads to distinct fragmentation patterns upon which some of the isomers can be discriminated.305 It was shown that cross-ring fragments can be formed, and increased branching of N-glycans leads to wealthier fragmentation patterns. However, in complex or hybrid N-glycans, diagnostic cross-ring fragments are formed at a much lower abundance. Harvey studied CID of derivatized and underivatized N-glycans as protonated ions and sodium adducts generated by ESI-MS. CID of [M + H]+ mainly yields glycosidic cleavage fragments, whereas for [M + Na]+ additional diagnostic cross-ring fragments can be detected. Also, the degree of linkage information by cross-ring fragments is higher in mass spectra of high mannose than of complex N-glycans. Complex N-glycans preferentially fragment at the glycosidic linkage of GlcNAc residues instead of yielding cross-ring fragments.94,308,309 CID of silver-adducted underivatized N-glycans, formed during ESI-MS, promotes the cleavage of glycosidic bonds. However, contrary to [M + Na]+ adducts, no diagnostic cross-ring fragments are formed.310
In positive ion mode CID, cross-ring fragments (A/X) that are diagnostic for the structure can only be obtained for certain metal adducts, usually at low abundance. The issue can be circumvented in negative ion mode CID. Harvey showed that CID of derivatized N-glycosidic [M – H]− ions leads to diagnostic A-, Y-, and Z-fragments, which enable unambiguous identification of the type of N-glycan and certain structural motifs. The abundance of cross-ring fragments in these spectra is rationalized by competitive loss of a hydrogen from a hydroxy group.311 The fragmentation behavior in negative and positive ion modes was confirmed for derivatized N-glycans using MALDI-MS by Wuhrer et al.312 In contrast to other anionic adducts, [M + NO3]− adducts, formed by ESI, tremendously increased the sensitivity, and CID yielded diagnostic C- and A-fragments, contrary to the less diagnostic B- and Y-fragments that are commonly observed in the positive ion mode.313−315 A similar fragmentation behavior has been determined for [M + H2PO4]− adducts.313,316 Domann et al. found that ionization of N-glycans in the negative mode by MALDI does not readily occur. By using a 2,4,6-trihydroxyacetophenone (THAP) matrix, they were able to efficiently form [M – H]− ions, and subsequently LIFT-CID yielded comparable spectra to ESI/CID.317
It is not straightforward to ionize acidic N-glycans (e.g., sialylated or sulfated) in positive ion mode. Generally, sialic acids (in mammals only neuraminic acid) are lost during ionization in positive ion mode, which can, however, be circumvented by prior esterification.318 Clean MS and MS2 spectra of sialylated species in positive ion mode were obtained by MALDI-MS.319 Reiding et al. showed that α2,3- and α2,6-sialylated complex N-glycans can be distinguished by MALDI in positive ion mode after esterification.320,321 Ionization of these species is facilitated in negative ion mode, but does it help to elucidate N-glycan structures? Wheeler and Harvey were able to discriminate two isomeric complex disialylated N-glycans based on the MS2 (and MS3) CID fragmentation patterns of the [M – 2H]2– ions and their C4 fragments generated via ESI-MS.322 Seymour et al. emphasized the difference in CID spectra obtained from sialylated and asialylated N-glycans in negative ion mode ESI-MS. Due to charge localization, the MS2 spectra of sialylated N-glycans are much less comprehensive than their asialylated counterparts.323 Using a similar approach based on PGC-LC-ESI-MS/MS and CID combined with the Skyline software,324 Ashwood et al. were able to identify the linkage and branching positions of sialic acids on N-glycans ionized as [M – H]−.325 Sulfated N-glycans exhibit similar fragmentation patterns as sialylated compounds, due to the localized charge in their [M – H]− ions.315,326
Wuhrer et al. determined that Fuc and several other hexoses in N-glycans can migrate under tandem MS conditions in both ESI- and MALDI-MS, which may lead to potentially erroneous structure assignments. The phenomenon only occurs for protonated ions and ammonium adducts in positive ion mode.327,328 Fuc and bisecting GlcNAc moieties can be reliably determined by the fragmentation pattern observed in negative ion mode CID of [M + NO3]− and [M + H2PO4]− adducts.315,329 Sulfated and fucosylated complex N-glycans were characterized in positive and negative ion modes via MALDI-MS/CID.330 Zhou et al. analyzed permethylated N-glycans as [M + H]+ using PGC-LC-ESI-MS/MS. Based on the collisional fragmentation patterns, a diagnostic ion for core fucosylation is reported, and β1,3- and β1,4-linked Gal could be discriminated.331
CID of N-glycans in the positive and the negative ion mode studied by ESI- and MALDI-MS has been extensively reviewed.95,332,333
4.3. Characterization of N-Glycans by MSn Methods
MSn essentially yields the same information as CID, but controlled sequential collisional fragmentation provides more details about the parent ion. Sheeley et al. and Weiskopf et al. showed that branching patterns and linkage data can be obtained by MSn of high-mannose and complex sodiated and permethylated N-glycans generated by ESI-MS.334,335 AP-MALDI experiments led to sodiated N-glycans [M + Na]+ and showed that multistage experiments are possible without permethylation or other derivatizations.336 Harvey et al. showed that high-mannose, complex, and hybrid [M + Na]+N-glycans can be generated by MALDI. Here, MSn helps the understanding of the mechanisms leading to fragmentation, facilitating interpretation of tandem mass spectra.337 Lapadula et al. presented an algorithm for processing MSn spectra of oligosaccharides, as shown for permethylated N-glycans, that helps to reconstruct glycan structures based on the MSn spectra, without biosynthetic constraints or comparing against previously reported structures.338−340 Ashline et al. showed that MSn of permethylated sodium-adducted N-glycans leads to the same fragments as chemically synthesized epitope precursors.341 Fucosylation positions and fragments indicative for polylactosamine could be identified by MSn.342 In conclusion, MSn leads to a higher degree of information than simple fragmentation via CID. Reliable structural assignment based on algorithms interpreting MSn data is possible. However, recording MSn spectra comes at the cost of higher sample consumption and is therefore not always a viable option.
4.4. Electron-Based Dissociation Methods in N-Glycan Analysis
ExD often delivers complementary information to CID or IRMPD for carbohydrates. While in slow activation methods (e.g., CID and IRMPD) usually the weakest bonds are cleaved first, ExD methods are fast activation methods, where a bond in close proximity to the site of electronic excitation is broken. Commonly, the techniques involve multiply charged species that still carry a charge after an electron is attached to or detached from the precursor ion.
Adamson et al. compared the IRMPD and ECD fragmentation spectra of a complex N-glycan. In ECD, electrons from a low-energy electron source are captured by a multiply charged cation, leading to fragmentation. For N-glycans, the fragmentation efficiency was observed to be very weak so that vibrational activation by IRMPD was necessary before ECD fragmentation. It is suggested that the low fragmentation efficiency is connected to the secondary structure of the N-glycan, as ECD fragmentation was not an issue in linear glycans. Because most of the fragments obtained by activated ECD are identical to those obtained by IRMPD, the authors expected them to be formed rather by vibrational excitation. Metal-adducted N-glycans, e.g., [M + Co]2+, exhibited richer fragmentation patterns than their protonated counterparts.85 Zhou et al. compared ECD and IRMPD spectra obtained from Ca2+-, Co2+-, and Mg2+-adducted sulfated hybrid N-glycans. IRMPD generally leads to cleavage of the labile sulfate group, while it is retained after ECD, which allows us to determine the sites of sulfation.343 Zhao et al. probed permethylated high-mannose and complex N-glycans (sialylated and asialylated) with hot ECD, where the electrons are higher in energy than in common ECD. Triply and doubly sodiated adducts mainly yielded C- and Z-ions but also A- and X-fragments, which helped to determine branching and sialylation patterns.344 Yu et al. compared ECD (1.5 eV), hot ECD (9 eV), and EED (14 eV) for a high-mannose N-glycan as [M + 2Li]2+ adducts. In EED, the cations are irradiated with electrons (>9 eV), leading to much richer fragmentations than ECD. In the study, most cross-ring fragments were obtained from EED, so that it was possible to find the positions of five out of six glycosidic linkages.345 Wei et al. used a PGC-LC-EED MS/MS workflow for the de novo analysis of glycan structures from isomeric high-mannose N-glycans with a software called GlycoDeNovo.346 The method can also be used for quantification.
Another promising technique for N-glycan analysis is EDD of multiply charged anions. Similar to cations in ECD, the anions are irradiated by low-energy electrons, leading to electron detachment and subsequent fragmentation. Besides the commonly obtained B/C/Y/Z-fragments, this technique also yields more diagnostic A- and X-fragments, such as 1,5A, 3,5A, 1,5X, and 3,5X (contrary to 0,2A and 2,4A observed in negative ion CID/IRMPD), in high abundance. Both asialylated and sialylated complex N-glycans were probed as doubly charged deprotomers. Based on the fragments, core fucosylation could be reliably determined.268 In acidic-labeled sialylated complex N-glycans, the EDD spectra are less rich in fragments, probably due to the location of the charge, altered by the label.347 EDD of a chloride-adducted, deprotonated asialylated complex N-glycan [M – H + Cl]2– leads to extensive fragmentation compared to CID (Figure 19).269
Figure 19.

Fourier transform ion cyclotron resonance tandem mass spectra of a deprotonated and chlorinated asialylated complex biantennary N-glycan, [M – H + Cl]2–. Mass spectrum employing (top) EDD and (middle) CID on the precursor ion [M – H + Cl]2–. Product ions shown in bold are unique to fragmentation of [M – H + Cl]2– compared to the doubly deprotonated species [M – 2H]2–. An asterisk indicates doubly charged product ions, whereas squares indicate water loss from adjacent product ions. Product ions bearing a chloride anion are highlighted with superscripted Cl. (bottom) observed fragmentation pattern after EDD of [M – H + Cl]2–. Reprinted with permission from reference.269 Copyright 2012 American Society for Mass Spectrometry.
4.5. Ultraviolet Photodissociation Mass Spectrometry of N-Glycans
In UVPD, the probed ions are electronically excited by UV photons (10–400 nm), leading to fast activation with subsequent fragmentation. Devakumar et al. analyzed permethylated high-mannose and (asialylated and core fucosylated) complex N-glycans as [M + Na]+ ions (Table 1). Here, 157 nm UVPD leads to a considerable amount of A- and X-fragments by which connectivity of the monosaccharide units can be determined. However, also B-, C-, Y-, and Z-fragments can be observed.348 In a second study, the same authors showed that 157 nm UVPD in combination with ion trap MSn is able to analyze isomeric N-glycans released from ovalbumin due to extensive cross-ring fragments.349 Ko et al. analyzed mono- and disialylated complex N-glycans as [M – H]− and [M – 2H]2–, respectively, with CID and 193 nm UVPD. In contrast to their CID counterparts, the obtained UVPD fragmentation patterns are generally very rich and contain a lot of signals for cross-ring A- and X-fragments, on the basis of which sialylation patterns can be analyzed.76
Table 1. Fragments and Informational Content Delivered by Applying Diverse Fragmentation Techniques in MS to N-Glycan Ions.
| technique (ion mode) | species | fragments | diagnostic information | references |
|---|---|---|---|---|
| CID (+) | [M + H]+ | B/Y | composition, sequence | b,308; a,94 |
| CID (+) | [M + Na]+ | (A)/B/Y | composition, sequence, (connectivity, branching) | b,308,309; a,94 |
| CID (+) | [M + Ag]+ | B/Y | composition, sequence | a,310 |
| CID (−) | [M – H]− | A/Y/Z | composition, sequence, connectivity, branching | b,311 |
| CID (−) | [M + NO3]− | A/C | composition, sequence, connectivity, branching | a,313,315; b,314 |
| CID (−) | [M + H2PO4]− | A/C | composition, sequence, connectivity, branching | a,313,316 |
| IRMPD (+) | [M + 2H]2+ | B/C/Y/Z | composition, sequence | a,85 |
| IRMPD (+) | [M + Co]2+ | A/B/C/Y/Z | composition, sequence, connectivity, branching | a,85,343 |
| IRMPD (−) | [M – 2H]2– | A/B/C/Y/Z | composition, sequence, connectivity, branching | a,268; b,347 |
| ECD (+) | [M + 2H]2+ | (A)/B/C/Y/Z | composition, sequence, (connectivity, branching) | a,85 |
| ECD (+) | [M + Co]2+ | A/B/C/X/Y/Z | composition, sequence, connectivity, branching | a,85,343 |
| hot ECD (+) | [M + 2Na]2+/[M + 3Na]3+ | A/B/C/X/Y/Z | composition, sequence, connectivity, branching | c,344 |
| ECD (+) | [M + 2Li]2+ | A/B/C/Y/Z | composition, sequence, connectivity, branching | c,345 |
| EED (+) | [M + 2Li]2+ | A/B/C/X/Y/Z | composition, sequence, connectivity, branching | c,345 |
| EDD (−) | [M – 2H]2– | A/B/C/X/Y/Z | composition, sequence, connectivity, branching | a,268; b,347 |
| UVPD (+) | [M + Na]+ | A/(B/C)/X/(Y/Z) | composition, sequence, connectivity, branching | c,348 |
| UVPD (−) | [M – H]− | A/B/C/X/Y/Z | composition, sequence, connectivity, branching | a,76 |
Not derivatized.
Labeled
Permethylated.
All fragmentation techniques yield valuable information on the structure of N-glycans. However, there are some limitations. Analysis of mass spectra of isomeric N-glycan mixtures is still difficult, which is the reason why separation by chromatography prior to MS is still necessary. Although some isomers yield distinct fragmentation patterns, databases and/or expert knowledge are furthermore required for structural assignments.350 Separation by chromatography can, however, be replaced with a separation technique in the gas phase, namely, IMS. The technique can be easily coupled to various ionization sources and MS instrumentation.
4.6. Ion Mobility–Mass Spectrometry in N-Glycan Analysis
Preliminary studies showed that compositional, configurational, and linkage isomers of glycans can be distinguished by IM-MS.133,351−353 The advantage of IM-MS over MS is that ions are separated not only by their mass and charge but also by their size and shape. How can these results be transferred to N-glycans? Isailovic et al. provided the first evidence that it is possible to separate permethylated high-mannose and complex N-glycan isomers as [M + Na]+ ions using DTIM-MS.354 Plasencia et al. were able to resolve and assign structural isomers of permethylated high-mannose and hybrid N-glycans from ovalbumin ionized as [M + Na]+ and [M + 2Na]2+ adducts using DTIM-MS.355 Similar approaches were used by others to separate labeled and nonlabeled N-glycans released from biological samples with356 or without357−361 prior chromatographic separation. Here, the IM separation made it possible to measure peptides and glycans from the same sample by “cleaning up” the mass spectra. Thus, IM-MS can reliably separate different groups of molecules with similar m/z. Williams et al. characterized [M + Na]+ and [M + H2PO4]− adducts of high-mannose and complex N-glycans using DTIM-MS and TWIM-MS. It is shown that for the same N-glycan released from different proteins the same drift times are obtained. Complex isomeric N-glycans were separated and subsequently analyzed via MS/MS (see Figure 20). Thus, depending on which antennae the GlcNAc residue is located, different mobilities are obtained.278
Figure 20.

Ion mobility separation with subsequent collision-induced dissociation (CID) of a mixture of complex N-glycans as [M + H2PO4]− ions. (a) TWIM-MS ATD for GlcNAc1Man3GlcNAc2 released from chicken albumin. CID tandem mass spectra for the features (b) b, (c) c, and (d) d in the ATD. Reprinted with permission from ref (278). Copyright 2009 Elsevier B.V.
Harvey et al. used TWIM-MS to yield glycan profiles of underivatized N-glycans (high-mannose, hybrid, complex, bisecting GlcNAc, core fucosylated, and sialylated) released from various proteins and determined that isobaric N-glycans can be separated based on their drift times in positive ion mode as [M + Na]+ ions and in negative ion mode as [M + Cl]−, [M + NO3]−, and [M + H2PO4]− ions followed by CID.362 The technique can also be applied to differentiate between molecular ions and in-source fragments. However, they conclude that the resolution of IM-MS needs to be increased to unfold its full potential as an analytical technique for separating isomers.363 Yamaguchi et al. were able to separate isomeric labeled N-glycans by HILIC-TWIM-MS. They determined that the best separation is observed for [M + 2H]2+ ions, contrary to [M + H]+, [M + H + Na]2+, and [M + H + K]2+. Although fucosylated samples were also part of the analysis, fucose migration was not taken into consideration.364 Zhu et al. analyzed permethylated isomeric high-mannose (Man7) N-glycans from ribonuclease B as [M + 2Na]2+ adducts. The DTIM-MS arrival time distribution (ATD) of the selected m/z value showed four features corresponding to each isomer, respectively.365
Pagel and Harvey showed that the ATDs of sodiated N-glycans [M + Na]+ recorded on a second-generation TWIM-MS instrument are superior to the previous generation. They showed that the collision cross sections (CCSs), an instrument-independent parameter of the ion derived from the ATDs, are clearly differentiable for isomeric N-glycans. They emphasize using the obtained CCSs as storable parameters in databases for complex carbohydrate analysis.366,367 Another IM-MS study from Harvey et al. showed that conformer resolution of N-glycans released with endoH or endoS (no terminal GlcNAc) is smaller than that of N-glycans released with PNGase F; however, the molecular ions have a similar fragmentation behavior.368 Struwe et al. determined the CCSs of high-mannose N-glycans measured as [M + H]+, [M + Na]+, [M + K]+, [M – H]−, [M + Cl]−, and [M + H2PO4]− adducts using a combination of TWIM-MS and DTIM-MS. [M – H]− ions exhibited an unusually broad and multimodal ATD, which provided evidence for the coexistence of different conformers.369 Reduction of the reducing end to alditol, however, later showed that the broad ATD results from reducing end anomers rather than the presence of multiple conformers.370
Zhu et al. measured the CCSs (DTIMS-MS) of doubly charged metalated, permethylated high-mannose N-glycans [M + X]2+, where X = Mn, Fe, Co, Ni, Cu, Mg, Ca, and Ba. The ATDs suggest the presence of multiple conformers, arising from the metal binding to different sites of the N-glycan. Using MS/MS of ATD-selected conformers, the location of the binding site was assessed, leading mainly to B- and Y-fragments. Interestingly, larger cations do not necessarily lead to higher CCSs, as determined for Ba2+ and Ca2+. The former presumably binds to multiple sites, leading to an overall tighter conformation.371 Harvey and Abrahams measured the arrival times (TWIM-MS) of a large set of reduced high-mannose, complex, and hybrid N-glycans in the negative ion mode as [M + H2PO4]− adducts. Compared to nonreduced glycans, two main differences were observed: (1) fragmentation of the chitobiose core was less abundant and (2) the observed ATDs were more symmetric than for nonreduced compounds. The authors therefore concluded that the asymmetry was due to anomer separation.370 Harvey et al. determined that isomeric separation (TWIM-MS) of hybrid and complex N-glycans is possible in positive and negative ion mode. Although the separation seems to be marginally better in positive ion mode, the tandem mass spectra give more informative in negative ion mode.372 In another study, the authors show that the separation of high-mannose N-glycans by the same method is less distinct. This issue was resolved using an instrument with higher resolution.373 Hofmann et al. analyzed pentafucosylated biantennary complex N-glycan in positive ion mode as sodium adducts with IM-MS/MS. Based on the CCSs of the obtained fragments, it was possible to determine that the antennae are substituted with the Ley epitope (Figure 21).374 Harvey and Struwe reported that TWIM-MS/MS of complex and hybrid fucosylated N-glycans measured as [M + H2PO4]− can reveal the presence or absence of bisecting GlcNAcs, branching patterns of antennae, and the location of fucose.375,376 Similar findings were reported by Harvey et al. for [M + H2PO4]− anions of high-mannose N-glycans that produce fragments with unique CCSs under CID conditions that can be used for assignment of isomers.377 Re et al. and Toraño et al. correlated the CCSs of complex N-glycans as [M + H]+ and [M – H]− ions. In the negative ion mode, the sialylation pattern of these ion can be readily distinguished by their unique CCS. Furthermore, they show that N-glycans can be sampled via force field molecular dynamics to obtain theoretical CCS values of N-glycans and predict their conformational distribution in the gas phase.219,378 Pallister et al. present an LC-TWIM-MS/MS approach for high-throughput analysis of labeled complex N-glycans in the positive ion mode ([M + Na + H]2+). Based on the CCSs of N-glycans and their fragments, isomeric N-glycans can be comprehensively differentiated.379 Wei et al. utilized a TIMS instrument to study a permethylated complex and hybrid N-glycans measured as [M + 2Na]2+. From the data, it can be concluded that many mobility features of N-glycans are due to distinct conformers and not structural isomerism. EED of the conformer-selected N-glycans provided many characteristic fragments, showing that the fragmentation mechanism is similar for different conformers of the same molecule.380
Figure 21.

TWIM-MS/MS of sodiated complex N-glycan from a human parotid gland. (A) ATDs of diagnostic fragments with TWCCSN2. (B) Tandem mass spectrum of [M + Na]+, including the representation of the precursor structure and fragment assignments. Glycan structures are represented using the Oxford system (notable differences to SNFG nomenclature: yellow diamond = Gal, red diamond = Fuc). Reproduced with permission from ref (374). Copyright 2017 American Chemical Society.
4.7. Gas-Phase Ion Spectroscopy of N-Glycans
While there are many studies on IM-MS of N-glycans, gas-phase spectroscopic studies have remained scarce. UV spectra of mass-selected N-glycans have not been reported to date, and their IR action spectroscopic investigation is still in its infancy. Using this method, the vibrational modes of the ions can be excited by IR photons, leading to a characteristic spectrum unique to each ion. Depraz Depland et al. measured gas-phase IR spectra using IRMPD of isomeric sialylated epitopes that can be commonly found on the antennae of N-glycans. Infrared spectra were recorded in the 3 μm range of protonated ions and sodium adducts based on which α2,3- and α2,6-sialylated isomers can be clearly distinguished.381 Likewise, Mucha et al. used IR spectroscopy in helium nanodroplets in the 5–10 μm range to study fucosylated trisaccharide epitopes, showing that fucose migration in protonated glycans is a universal phenomenon in mass spectrometry. By probing the sodium adducts of these epitopes, it was, however, possible to clearly distinguish epitopes with distinct fucosylation patterns.205 Dyukova et al. used messenger-tagging spectroscopy with N2 for recording IR spectra of complex N-glycans before and after enzymatic cleavage and compared the IR signatures to those obtained from synthetic glycan standards in the 3 μm range. With increasing size of the glycan, the spectra become more complex. However, based on the spectra it can be clearly seen that the signatures of the synthetic standards and the enzymatically cleaved sugars are identical (Figure 22).382 Yalovenko et al. combined SLIM-IM-MS and messenger-tagging spectroscopy with N2 to study protonated complex (and fucosylated) N-glycans in the 3 μm range. With the presented approach, IR signatures with a spectral window of 300 cm–1 can be obtained within 55 s, which allows incorporation of this method into analytical workflows.202 Based on the findings, the authors emphasize the ultimate goal to compile a database of IR signatures of intact N-glycans and their fragments. In a recent study, Dyukova et al. employed the same setup to distinguish complex and fucosylated N-glycan positional isomers cleaved from monoclonal antibodies as sodium adducts.383
Figure 22.

Comparison of IR spectra of protonated complex N-glycans. IR spectrum of the GlcNAc2Man3GlcNAc2 reference (purple) and of GlcNAc2Man3GlcNAc2 after enzymatic cleavage from Gal2GlcNAc2Man3GlcNAc2 (gray). Figure adapted with permission from ref (382). Copyright 2020 American Chemical Society.
5. Mucin-Type O-Glycans
5.1. Structure and Analytical Challenges
O-Glycans are highly abundant in extracellular proteins. The largest number of O-glycans occurs on mucins: large proteins that often carry hundreds of O-glycosylation sites and which are found throughout the body. Therefore, the involved oligosaccharide structures are often termed mucin-type O-glycans. However, O-glycosylation is not exclusive to mucins and is also common among other glycoproteins such as immunoglobulins. Mucins are characterized by a large number of tandem repeat domains that are rich in Ser, Thr, and Pro residues, giving rise to a vast number of O-glycosylation sites. Extensive O-glycosylation in mucins can make up 50 to 80% of the overall mass of the glycoprotein.384 This often has a tremendous impact on the overall properties of the molecule. In comparison to the protein backbone, oligosaccharides are generally more hydrophilic and, depending on their terminal modifications, often negatively charged. As a result, O-glycans are usually extensively solvated by water and salt ions, which induce a high viscosity and gel-like structure of the mucus. The crucial impact of glycans on the physiochemical properties becomes especially obvious when O-glycosylation is altered, for example, in diseases. The certainly most prominent example for such a disease is cystic fibrosis (CF). Here sulfation and sialylation of sputum mucins are significantly increased, while a decreased sialylation/increased fucosylation is observed in membrane-embedded mucins of airway cells.385 This drastically increases the viscosity of the mucus in the lungs and leads to obstruction and malfunction of the mucus as a pathogen barrier, which in turn can result in infections with bacteria such as Pseudomonas aeruginosa.
O-Glycosylation occurs at hydroxyl groups of Ser and Thr residues. It is initiated by enzymatic transfer of an N-acetylgalactosamine (GalNAc) residue in the Golgi apparatus. There is a relatively special modification with a single N-acetylglucosamine, often simply termed O-GlcNAcylation, which will not be covered here as there are no oligosaccharides involved.386 In contrast to N-glycosylation, no defined sequons are required for O-glycosylation. In addition, biosynthesis occurs stepwise and does not include trimming, i.e., cleavage of certain residues by glycosidases during the final processing steps. As a result, O-glycans are typically smaller in size compared to N-glycans; however, the underlying structural space is vast with multiple sources of isomerism.
Generally, O-glycans are extended following four major core structures (Figure 23A, first row). Starting from the GalNAc linked to Ser or Thr residues, attachment of β1,3Gal leads to core 1; further attachment of a β1,6-linked GlcNAc to core 1 gives rise to core 2. Both are by far the most common core structures in O-glycosylation and are found throughout the body. Cores 3 and 4 are formed by subsequent attachment of a β1,3GlcNAc and a β1,6-linked GlcNAc and are more exclusive to gastric and lung glycoproteins, in particular mucins. In addition, a further four rare core structures have been observed (Figure 23A, second row). However, their underlying biosynthesis and further processing are still poorly understood, and it cannot be excluded that they are arising from enzyme side reactions.387 The exceptional structural diversity of O-linked glycans is generated by further extension of the core structures, for example, with β1,3- and β1,6-linked N-acetyllactosamine (LacNAc) repeats, sulfation, sialylation, and/or fucosylation (Figure 23B). The resulting glycans are highly complex and often contain antigenic structures at their nonreducing termini such as the ABO and Lewis blood group determinants (Figure 23C).
Figure 23.

Typical O-glycan structures. (A) The most abundant O-glycan core structures 1–4 and the four less common cores 5–8 shown using the symbol nomenclature for glycans (SNFG). (B) Typical extensions of glycan cores are the attachment of N-acetyllactosamine units, sialylation, and fucosylation. (C) Examples for fully processed O-glycan structures based on core 1 (upper structure) and core 3 (lower structure) including their linkage site to the glycoprotein. At the nonreducing end, O-glycans often contain antigenic structures such as the ABO and Lewis blood group determinants, which are highlighted by gray boxes.
The analysis of O-glycosylation is generally more challenging than that of N-glycosylation. As mentioned above, O-glycosylation does not follow defined sequons. Statistically, prolines are often found adjacent to O-glycosylation sites; however, this is a mere statistical effect and is of little analytical use. O-Glycosylation positions within a protein are therefore considerably more challenging to identify than N-linkages. To make matters worse, there are no specific enzymes, such as PNGaseF in the case of N-glycans, that specifically and selectively hydrolyze the glycosidic bond to the protein. Instead, rather harsh chemical procedures are usually employed. The linkage between GalNAc and Ser/Thr residues is labile under basic conditions, such that β-elimination can be used for their release. Usually, this is combined with a reductive workup with NaBH4 to reduce side reactions often referred to as “peeling”,388 yielding stable oligosaccharides with N-acetylgalactosaminitol at the reducing end. As a downside, treatment with NaBH4 can lead to partial degradation of the peptide backbone, and the resulting alditols are chemically inactive at the reducing end. In order to reduce the complexity of the resulting mixture, the O-glycans can furthermore be trimmed prior to release, for example, by removal of the terminal sialic acid using sialidases. For more details on O-glycan release reactions and nonreductive procedures, the reader is referred to recent review articles.388−390 Analytical approaches to study intact O-linked glycopeptides are briefly discussed in the glycopeptide chapter of this review.
Following the release, O-glycans are usually purified and/or separated using chromatography. Reversed-phase (RP) chromatography only shows a poor separation of O-glycans due to their high polarity and often negative charge. Hydrophilic interaction liquid chromatography (HILIC)391 and porous graphitized carbon liquid chromatography (PGC-LC)392 on the other hand are much better suited and often show a remarkable separation even for isomeric structures.393 However, all chromatographic techniques have to cope with a fundamental limitation: due to the reductive workup, the released products are alditols, which cannot be labeled with UV and fluorescent-active tags at the reducing end. Nonreductive workup on the other hand shows reproducibility issues and often suffers from unpredictable peeling.390 As a result, the workflows established for N-glycans are only of limited use for the analysis of O-glycosides—one of the major reasons that O-linked glycomics is substantially lagging behind the advances made in N-linked glycomics. Instead, reductively released and chromatographically separated O-glycans are usually analyzed directly as alditols or after permethylation using ESI-MS. The molecular identity of the underlying structures is usually obtained using a tedious but often highly informative fragment analysis.394 In the following chapter, we will summarize the recent developments in O-glycan tandem MS and IM-MS analysis.
5.2. Collision-Induced Dissociation and Higher-Energy Collisional Dissociation of O-Glycans
CID and higher-energy collisional dissociation (HCD) are the most widespread and therefore most frequently used dissociation techniques in tandem MS. In this regard, O-glycan analysis is no exception. Similarly to N-glycans, tandem MS experiments are usually preceded by a chromatographic separation, which is directly hyphenated to the mass spectrometer. In contrast to N-glycans, however, even the very early MS studies on released O-glycans were mostly focused on ions with negative polarity.392 On one hand, this is a result of the higher abundance of negatively charged sialic acid and sulfate moieties in O-glycans, which generally lead to considerably increased ionization efficiency for negative ions. Especially in nano ESI, also neutral released O-glycans tend to ionize well when derived from a reductive workup, i.e., as alditols.395 On the other hand—and similarly to N-glycans—negatively charged O-glycans tend to form C-type fragments, which in turn can rearrange in a retro-aldol reaction into highly diagnostic A-ions. Albeit less pronounced in negatively charged O-glycans, such cross-ring cleavages significantly increase the informational content of the resulting fragmentation spectra and facilitate more confident structural assignments.394 As a downside, tandem mass spectra of negatively charged O-glycans are often very complex, and their interpretation usually requires expert knowledge—an aspect which so far limits applications to a few specialized laboratories.
Mucins and other hydrogel glycoproteins are usually densely O-glycosylated. As a result, they exhibit a characteristic structure in which the glycans are sticking out like the brittles of a bottle brush. These brittles shield the protein backbone from the exterior and prevent proteolysis from enzymes such as trypsin. Generating glycopeptides from fully glycosylated mucins is therefore highly challenging, if not impossible. The majority of MS-based glycomics analyses is therefore limited to a profiling of structural features rather than a full, site-specific structural identification. The probed motifs can include core structures, ABO and Lewis blood group motifs, sialylation, and even sulfation.396 Often, O-glycan profiling is combined with exoglycosidase digestion prior to analysis to remove certain terminal residues such as sialic acids.
Early works on eggs from Xenopus frogs showed that characteristic O-glycosylation features can be identified based on the CID mass spectra of glycans released from egg jelly and digested with exoglycosidases.397 Using tetradeuterioborate instead of tetrahydroborate during the release and workup procedure was furthermore used for comparison and relative quantification of O-glycans from different sources.398 Later works showed that similar approaches can also be utilized for the profiling of O-glycans on isolated and purified glycoproteins.399 The underlying workflows are built on the sequential release of N- and O-glycans from the glycoprotein as well as exoglycosidase digestion. When combined with glycopeptide analysis,400 a detailed and site-specific glycosylation profile can be obtained. Furthermore, an approach for the combined release and labeling of O-glycans with 1-phenyl-3-methyl-5-pyrazolone (PMP) has been reported.401 This is particularly useful for the analysis of released oligosaccharides in positive ion mode. A very recent work showed the exceptional utility and robustness of O-glycan profiling for the analysis of gastric mucin samples from patients.396 Extensive O-glycosylation features of ten individuals were determined. The results revealed that each individual carries more than 100 glycan structures, with up to 14 of them being unique to the particular patient. Even though the number of probed individuals was low, a consistent increase in the level of sialylation and sulfation on gastric O-glycans was observed for cancerous tissue.
Very few works deal with the unique mechanistic aspects of O-glycan CID fragmentation. A comparison between O-glycan alditols and aldoses revealed distinct fragmentation pathways, some with and some without participation of the charge carrier.402 This study furthermore revealed that O-linked aldoses are present as α/β-anomers—an aspect that was later also observed in IM-MS for other oligosaccharides.154 Further works revealed that HCD fragmentation is mechanistically not that different from CID but can clearly help to identify diagnostic fragments that are not accessible by CID, in particular at lower masses. This, for example, enabled the identification of O-glycans from fungi that are exclusively composed of hexose and hexuronic acid.403 Likewise, low-mass HCD fragments were recently shown to be diagnostic to the regiochemistry of sulfate modification (3 vs 6) in mucin O-glycans.404
5.3. Characterization of O-Glycans by MSn Methods
O-Glycans are highly branched oligosaccharides. An identification of the branched site is not always straightforward using conventional CID experiments, as fragments from branched and linear chains are often isomeric. A strategy to solve this problem is MSn of permethylated glycans using ion trap instruments.350,405 Here, all accessible OH and N-acetylated amide groups are derivatized using reactive methylating agents such as methyl iodide.406 Subsequent sequential fragmentation of fragments reveals the number of methylated sites per monosaccharide and with that the relative position of the building block within the glycan network. As a downside, the time and sample consumption in MSn experiments is usually high, and a clear annotation of the spectra is elaborate. MSn experiments on permethylated O-linked glycans are usually performed on sodium adducts and can yield exceptional structural insights, including the identification of isomers.341,342,407 Furthermore, MSn experiments on nonderivatized glycans were used to unravel the details of O-glycan fragmentation and identify more complex structures.408,409 As in conventional O-glycan CID, exoglycosidases were used to reduce the sample complexity, and ions of negative polarity were investigated.
5.4. Ion Mobility–Mass Spectrometry of O-Glycans
In contrast to other glycoconjugates, O-glycans are only sparsely studied by IM-MS and related methods. This is likely a result of the tedious release procedure and the lack of well-characterized standards, and not least because of their exceptional structural complexity. A series of synthetic O-glycan-like structures were investigated as part of a very extensive study on the IM-MS behavior of oligosaccharide isomers.282 Here, the impact of ion polarity and adduct formation on the separation of isomeric structures was focused. Very similar to other glycoconjugates, the investigated O-glycans did not show clear trends, which would enable a prediction of the best separation conditions. An optimization of the probed ionic species as well as the experimental conditions are therefore required for each individual separation problem. Moreover, the potential of a combination of IM-MS and UVPD was recently tested.410 Two synthetic tetrasaccharides were mobility-separated in a DTIMS cell and subsequently subjected to UV radiation from a 193 nm ArF excimer laser. As metal adducts, both isomers are clearly distinguishable by IMS alone. However, UVPD adds an additional dimension in the form of rich tandem mass spectra. This gain in informational content becomes especially apparent, when the obtained UVPD mass spectra are compared with their counterparts from CID.
Very recently, IM-MS was extensively used to test the utility of the method in established PGC negative ion LC-MS workflows—the current gold-standard technology for the separation of isomeric O-glycans (Figure 24).411 In particular, O-glycans released from porcine gastric and human salivary mucins were systematically investigated and compared using both methods. The experiments reveal that IM-MS is a promising tool for O-glycan structural analysis that is capable of resolving the structural complexity that LC cannot always resolve. The combination of IMS-selected precursor ions and their negative ion MS fragmentation spectra was found to be particularly useful. Moreover, this study presents one of the few examples in which the utility of IMS as a separation technique is directly compared to that of LC. A couple of years ago, this comparison would have been a clear overestimation; however, the resolution of state-of-the-art IMS systems is increasing at a rapid pace, and LC-like separations in IMS are therefore not out of reach any longer.144
Figure 24.
Ion-mobility-based comparison of two sets of O-glycan isomers as [M – H]– and [M + H2PO4]– ions. (A) Arrival time distributions (ATDs) of two isomers composed of Hex1HexNAc2 measured as deprotonated (m/z 587, blue) or phosphate adduct (m/z 685, black) ions. (B) ATDs of two Hex1HexNAc 2dHex1 isomers as deprotonated (m/z 733, blue) or phosphate adduct (m/z 831, black) ions. Structures are depicted using the symbol nomenclature for glycans (SNFGs): yellow square = GalNAc, yellow circle = Gal, blue square = GlcNAc, red triangle = Fuc. Figure adapted and modified with permission from ref (411). Copyright 2019 American Chemical Society.
5.5. Bioinformatics Tools
One of the fundamental outcomes of the above-mentioned tandem-MS studies is that the analysis and annotation of glycan fragment spectra are often very tedious and require expert knowledge. A promising approach to tackle this problem is spectral libraries that can be used as references to annotate complex spectra. Due to the universal nature of glycan fragments, this approach is generally not limited to O-glycans but rather universal for all glycoconjugate structures that exhibit a comparable CID fragmentation behavior and fragment structure.
The general feasibility of a database approach was exemplified in the LC-ESI-MS/MS analysis over 200 N- and O-glycans from human saliva glycoproteins.412 A library of diagnostic fragment ions was compiled and combined with other specific structural features from cross-ring and glycosidic cleavages. These reference values were subsequently used to characterize and differentiate potential isomers. The success of this study led to the release of UniCarb-DB, a curated open access database containing comprehensive LC MS/MS data of synthetic standards as well as N- and O-linked glycans released from glycoproteins.413 The deposited spectra have been evaluated by independent laboratories and were annotated with glycosidic and cross-ring fragmentation ions, retention times, and associated experimental metadata descriptions. Also IM-MS-derived CCS values of glycoconjugates and their fragments were implemented with UniCarb under the name GlycoMob.414 A similar, albeit much less extensive, library of LC and MS/MS data of human milk oligosaccharides was recently released within the NIST framework (https://chemdata.nist.gov/glycan/spectra).415
Another important aspect toward routine use is the automation of a reliable spectral assignment and structural identification, ideally with minimal human intervention. The recently reported software tool Glycoforest 1.0 is a first step toward this goal. Glycoforest uses a de novo algorithm that is fed with manually annotated tandem mass spectra, either prepared in-house or obtained externally for example from UniCarb-DB. Unknown structures are identified based on scores that reflect a matching and connection of the fragment spectra. Recently, the freely available software Skyline416 was furthermore used to identify fragments that are diagnostic for the discrimination of N- and O-glycan isomers.
Regardless whether used as a reference in publicly available databases or for structural identifications, the quality of the underlying raw data is absolutely crucial. This includes not only obvious measures in MS such as m/z resolution, mass accuracy, etc., but also less obvious metadata about data acquisition, processing, and storage. In order to ensure common quality standards in databases and publications, the MIRAGE (Minimum Information Required for a Glycomics Experiment) project, a community initiative coordinated by the Beilstein-Institute, was formed in 2011.417 Since then, a variety of specific, method-oriented guidelines—covering MS,418 LC,419 and bioinformatics420—have been established.
5.6. Fucose Migration
A reoccurring challenge in tandem MS of fucosylated glycans is fucose migration, an intramolecular rearrangement reaction, which can lead to erroneous sequence assignments (Figure 25).421 Owing to its importance in Lewis and blood group epitopes, fucose migration is discussed in the present section dedicated to O-glycans. However, this rearrangement reaction has been observed in various other glycan classes as well and may be regarded as a universal phenomenon in the MS-based analysis of fucosylated oligosaccharides. During migration, a fucose (6-deoxy-l-galactose) residue is transferred from the nonreducing end of a glycan to an adjacent or remote site within the same molecule. In fragmentation experiments using CID, fucose migration may be accompanied by internal residue loss (IRL). Unexpected m/z fragments indirectly indicate the occurrence of the reaction. The topic is of relevance not only for O-glycans but also for all classes of glycans that potentially contain fucose monosaccharides. Besides fucose, rearrangement reactions have been observed for xylose,422,423 rhamnose,423,424 and glucuronic acid,423 with distinct masses, but also mannose328 is able to migrate and more challenging to detect.
Figure 25.
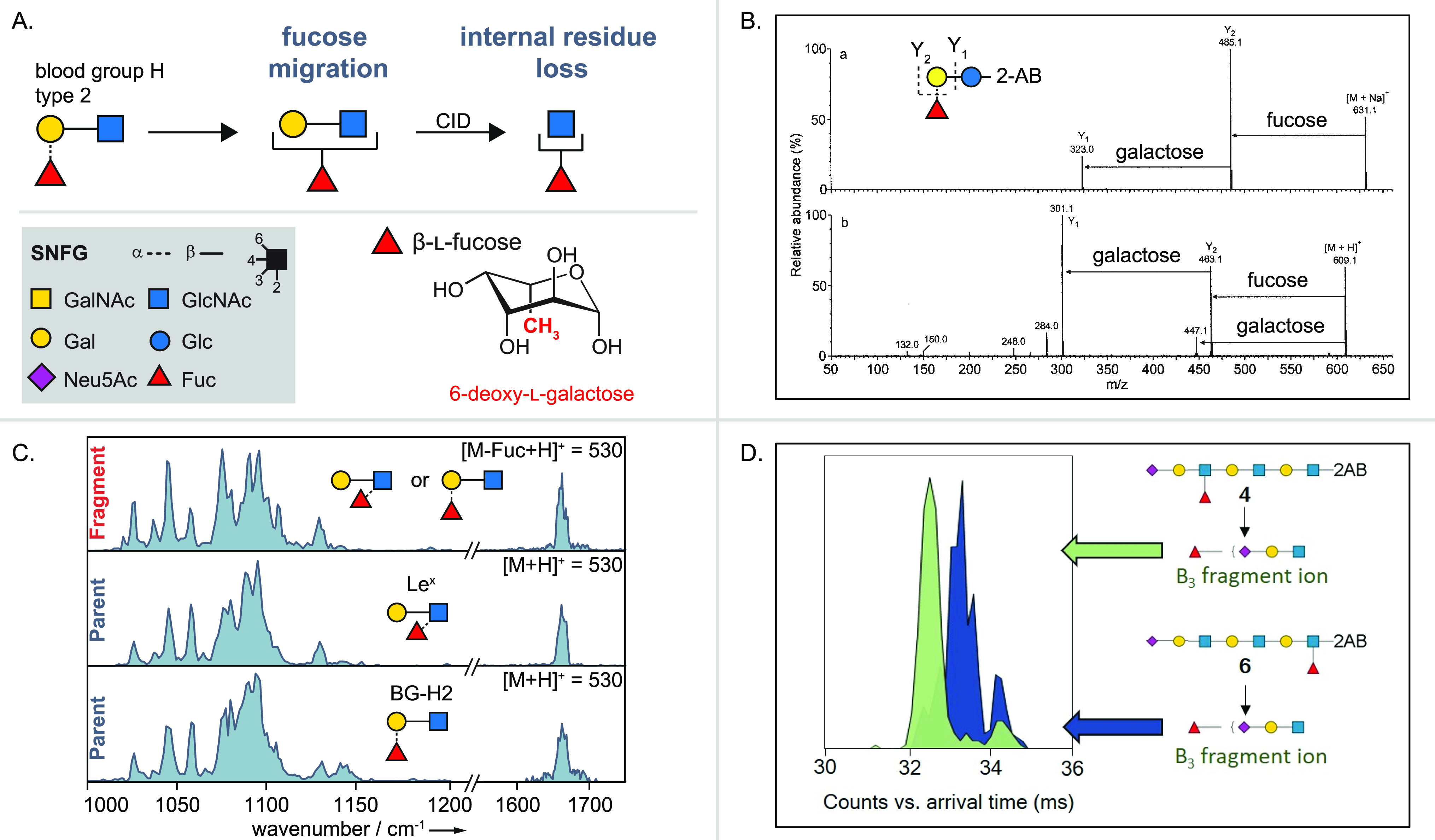
From early tandem MS experiments to IR spectroscopy and ion mobility experiments investigating fucose migration and internal residue loss. (A) Schematic example of fucose migration and internal residue loss for a trisaccharide (upper panel) in the symbol nomenclature for glycans (SNFG) (lower panel, left) and chemical representation of β-l-fucose, a 6-deoxy-l-galactose (lower panel, right). (B) Tandem MS experiments showing unexpected m/z-fragments from internal residue loss. Figure adapted with permission from ref (66). Copyright 2002 American Chemical Society. (C) IR spectroscopy experiments showing fucose migration in the trisaccharides Lex and BG-H2 as in-source fragmentation products of a tetrasaccharide (upper panel) and as intact parent ions (middle and lower panel). Figure reprinted with permission from ref (205). Copyright 2018 Wiley-VCH. (D) IMS experiments distinguishing two fragments with identical m/z ratio but at least one with a rearranged fucose monosaccharide from their ATD. Figure reprinted with permission from ref (425). Copyright 2019 Wiley-VCH.
Several studies based on tandem MS, IMS,425 and IR spectroscopy205,426 have been published investigating the reaction mechanism and driving forces, the kinetics and energetics, and, of utmost concern, the destination of the migrating monosaccharide. The reaction is charge-induced, and therefore mobile protons or at least poor charge fixation is a necessity. The free hydroxyl groups within the oligosaccharides display a hydrogen bonding network of vicinal and syn-diaxial hydrogen bonds in which proton or charge transfer can take place.426 Both short-66 and long-range427 migration reactions are possible. For both cases, the close proximity in space of the migrating residue and the destination within the chain are important yet difficult to predict. The rearrangement reaction has been observed for 1,2-, 1,3-, 1,4-, and 1,6-linked migrating monosaccharides. Considering the time scale of the reaction, experiments using MALDI-TOF/TOF-MS show that the reaction for the investigated ions is faster than microseconds.327 The abundance of ion signals resulting from IRL is collision-energy-dependent. When plotting the relative abundance of an ion against the collision energy, the ion from a simple loss of a terminal fucose and the ion from IRL with migration of fucose show the same curve shape, indicating a similar mechanism.66
The exact mechanism of fucose migration remains unresolved to date, yet in the following, different approaches are reviewed. In oligosaccharides with a reducing-end modification of 2-aminobenzamide, the attack from the nitrogen in the linker with a transfer of the migrating residue and a subsequent glycosidic bond cleavage of the terminal residue has been proposed.66 The mechanism combines a migration reaction, which is independent of an internal loss, with a subsequent fragmentation. A similar reaction pathway but with migration to a remote hydroxyl group within the oligosaccharide has been suggested.427 Generally, functional groups other than the amine linker are plausible for the destination of migration since internal residue loss is observable in oligosaccharides with a methylated amine linker. In glycans containing sialic acids, it has been proposed428 that the oxygen of the amide group of a sialic acid attacks the anomeric center of fucose, and a new bond forms, leading to an imine group with the proton located at the reducing end of the chain.
In the case of rhamnose migration, a 6-deoxy-l-mannose, the following detailed mechanism has been suggested.424 The ring oxygen atom is protonated with subsequent cleavage of the adjacent C-1–O bond, resulting in a carbenium ion at the anomeric center of the migrating group. In 6-deoxy monosaccharides, the ring oxygen has a slightly higher proton affinity than in other monosaccharides. Then, the oxygen of the flavonoide residue linked to the reducing end of the diglycoside attacks the carbenium ion at the anomeric center, and a semirigid internal residue is eliminated from the oligosaccharide.
A very effective way to prevent the rearrangement reaction in mass spectrometry experiments is to measure metal adducts or deprotonated ions.429 Furthermore, reducing-end modifications such as procainamide labeling430 or a modification with a free radical precursor and a fixed charge on a pyridine moiety271 successfully suppress the reaction. All approaches aim at either demobilizing the proton at locations of high proton affinity or generally avoiding the presence of protons. Permethylation and peracetylation, on the other hand, do not in all cases prevent the rearrangement.64,427,431
6. Glycosaminoglycans
6.1. Structure and Analytical Challenges
Glycosaminoglycans (GAGs) are unbranched, highly acidic polysaccharides (kDa to MDa range), expressed by essentially all animal cells.432 Although also present intracellularly, GAGs are much more prominent on cell surfaces and in the extracellular matrix (ECM). They interact with a diverse set of soluble and membrane proteins, as well as ECM components: cytokines and chemokines, growth factors and their receptors, morphogens, blood coagulation factors, lipoproteins, integrins, and collagens.433−437 In addition to these endogenous binding partners, GAGs may also interact with viral and microbial proteins.438,439 Thus, they participate in various physio- and pathophysiological processes, such as embryonic and neural development, angiogenesis, hemostasis, inflammation, cancer progression, and infection.13,14,440−444
Albeit a structurally heterogeneous class of complex carbohydrates, some common, distinct features of GAGs render them a unique, well-recognizable group within the glycome. All GAGs possess a linear sequence formed by repeating disaccharide units, where an (occasionally deacetylated) N-acetylhexosamine alternates with hexuronic acids or galactose. Based on the structure of these disaccharides, four families of GAGs are distinguished traditionally: hyaluronan, chondroitin sulfate jointly with dermatan sulfate, heparan sulfate together with heparin, and keratan sulfate, as portrayed in Figure 26.432,445
Figure 26.
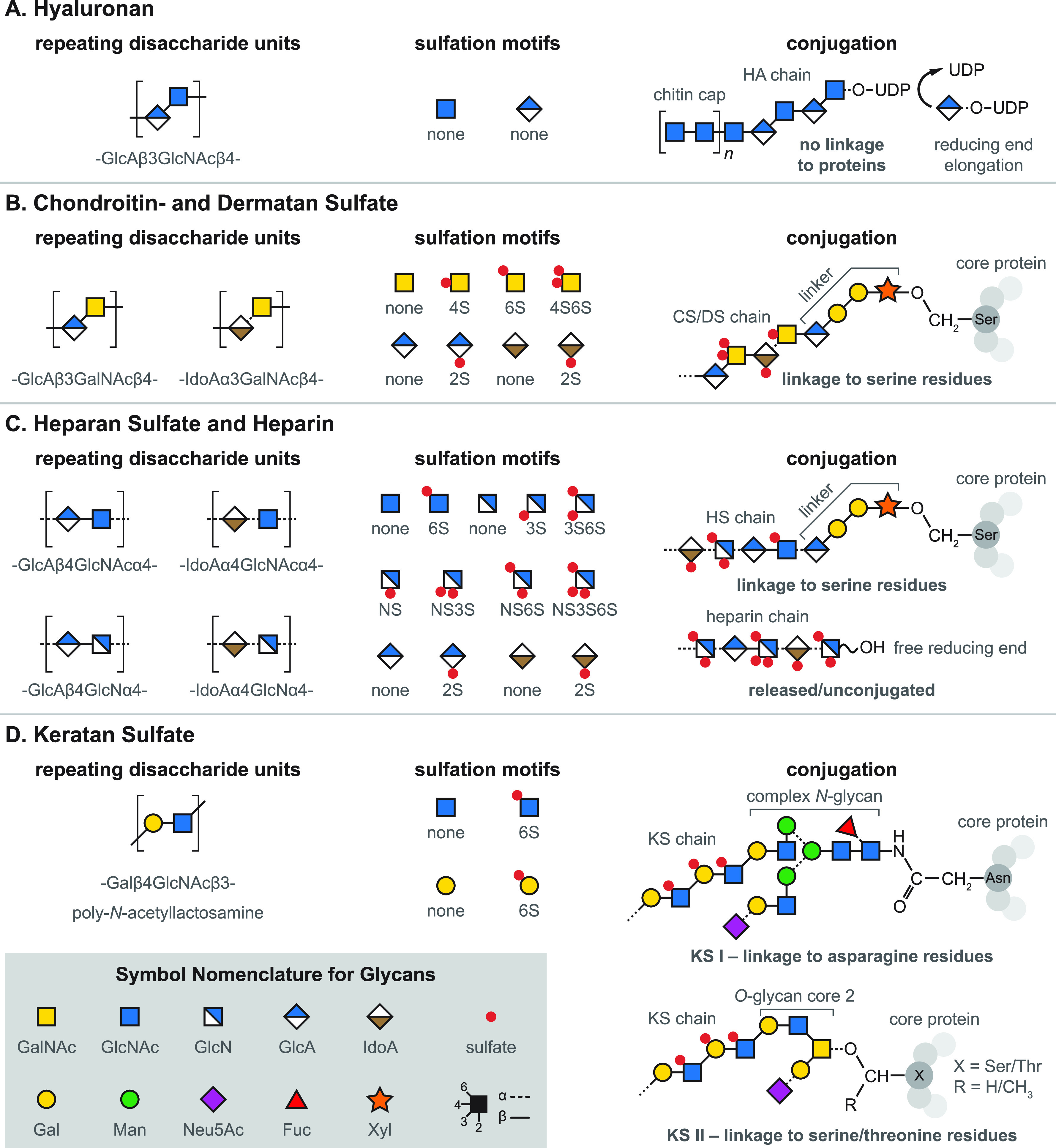
General structure of glycosaminoglycans. Overview of the characteristic disaccharide units, sulfation motifs, and potential protein linkages found in the four main glycosaminoglycan families. (A) Repetitive hyaluronan chains are not modified further by sulfation or epimerization. Uniquely, biosynthesis starts with the formation of a chitin cap and proceeds toward the reducing end. (B) Chondroitin and dermatan sulfate display a variety of sulfation motifs. The chains are linked to serine residues of proteoglycan core proteins through a specific tetrasaccharide linker. (C) Heparan sulfate and heparin represent the most diverse family of glycosaminoglycans. The heparin chain depicted corresponds to the antithrombin III binding sequence, mimicked by the synthetic anticoagulant fondaparinux. Discovery of additional sulfation motifs in the future cannot be ruled out. (D) Keratan sulfate contains galactose instead of hexuronic acid. The chains may be linked to both asparagine and serine/threonine residues of core proteins.
Hyaluronan or hyaluronic acid (HA) is an extremely high molecular weight polysaccharide that consists of alternating N-acetyl-d-glucosamine (GlcNAc) and d-glucuronic acid (GlcA) residues, forming a poly(GlcAβ3-GlcNAcβ4) chain of several thousand disaccharide units (Figure 26A).446 HA emerged relatively late in the evolution of animals and appears to be an exception among GAGs in many aspects. It stands out as the only GAG where the repetitive copolymeric chain is not modified further by sulfation or epimerization. In addition, HA is not linked covalently to proteins, in stark contrast to other GAGs that form specific glycoproteins, so-called proteoglycans, whose functions are determined principally by their GAG constituents.
Chondroitin sulfate (CS) and dermatan sulfate (DS) are closely related galactosaminoglycans, often referred to as a single family and discussed in conjunction, owing to substantial similarities in structure, biosynthesis, and function (Figure 26B).447 The main difference between the two concerns their hexuronic acid residues. CS contains exclusively GlcA, alternating with N-acetyl-d-galactosamine (GalNAc) to build poly(GlcAβ3-GalNAcβ4) chains. In contrast, DS contains not only GlcA but also its C5 epimer, l-iduronic acid (IdoA), to varying extents. It leads to a more complex polymer backbone where two basic disaccharide building blocks vary across the sequence: (GlcAβ3-GalNAcβ4) and (IdoAα3-GalNAcβ4). It is, however, also popular to distinguish GlcA-containing CS disaccharides and IdoA-containing DS disaccharides categorically while referring to longer sequences containing both kinds of hexuronic acid as hybrid CS/DS chains. CS and DS chains may contain over 100 disaccharide units, and unlike HA, they are extensively sulfated. GlcA and IdoA may carry 2-O-sulfation, while GalNAc can be sulfated at the 4-O and 6-O positions. The resulting sulfation motifs give rise to several isomeric building blocks and a complex sulfation pattern across the chains that influences molecular recognition and may encode functional information.448 Typical GAG epitopes found within longer chains range from tetra- to decasaccharides (dp4–dp10). Elucidating the “sulfation code” of such bioactive protein-binding sequences is a major challenge in the analysis of all sulfated GAGs.
Heparan sulfate (HS) and heparin form the structurally most complex GAG family (Figure 26C). During their biosynthesis, the initial poly(GlcAβ4-GlcNAcα4) chain undergoes extensive modifications: epimerization of GlcA to IdoA, 2-O-sulfation of the hexuronic acids, N-deacetylation/N-sulfation, and 6-O- and the rare 3-O-sulfation of GlcNAc may all occur, affecting a varying number of residues.449−451 The result is a heterogeneous copolymer of enormous complexity, with extremely high density of ionizable functional groups.452,453 Like CS and DS, HS is linked to serine residues of specific core proteins via a xylose-containing tetrasaccharide. The sulfation and epimerization pattern of GAG chains in these proteoglycans show temporal and spatial variation across tissues. In general, the composition of HS chains found on a certain kind of proteoglycan, but in different cell types, shows higher variability than the chains found on different proteoglycans within the same cell. Although sharing the same set of disaccharide building blocks and basic sulfation motifs, important differences exist between HS and heparin.434,454 HS is expressed by virtually all animal cells, whereas heparin is produced by only a few cell types, most prominently connective tissue mastocytes. HS is attached to core proteins localized on cell surfaces and in the ECM. Heparin, on the other hand, is stored intracellularly in secretory granules, attached to its cytoplasmic core protein, serglycin. HS chains typically consist of 50–250 disaccharide units, while heparin chains are significantly shorter with an average molecular weight of 12–15 kDa. In HS, regions showing extensive sulfation and epimerization are clustered along the chain (NS domains), separated by largely unmodified regions (NA domains). Heparin lacks such domain structure, serving as a single extended NS region: most of its GlcA residues undergo epimerization to IdoA, and the chains are more heavily sulfated (around 2.5 sulfates per disaccharide) than in HS (roughly one sulfate per disaccharide on average). Heparin is the largest biopharmaceutical in production and widely used as an anticoagulant in unconjugated form.
Keratan sulfate (KS) is unique among GAGs as the chains lack hexuronic acid, containing instead d-galactose (Gal).455,456 In KS chains, up to 50 repeating (GalAβ4–GlcNAcβ3) disaccharide units form the linear poly-N-acetyllactosamine backbone, whose strong acidic character stems from sulfation. Sulfate groups can be installed at the 6-O-position of both Gal and GlcNAc residues, with sulfated Gal occurring mainly adjacent to sulfated GlcNAc. In addition to O-mannose (KS III, not shown) and O-GalNAc (KS II) linkages, the chains may also be linked to asparagine residues (KS I) of core proteins in KS proteoglycans. Although the complex-type N-glycan linker may possess multiple antennae, KS chains themselves are not branched (Figure 26D).
In general, sulfated GAGs represent an immense structural complexity and are among the most challenging biopolymers to characterize. Obtaining information on the sequence of even the simplest full-length chains is a formidable task.457−459 Complexity and the associated challenges stem from four chief aspects of GAG structure: high degree of polymerization combined with size polydispersity, sequence microheterogeneity, high negative charge density, and the potentially isomeric building blocks. Being highly polydisperse, the length of GAG chains found on a certain proteoglycan at a given position is not uniform. Because of their microheterogeneity, GAGs cannot be characterized by a single, well-defined sequence, in contrast to biopolymers with template-driven biosynthesis, such as proteins or coding DNA. The dense sulfation of GAGs complicates their MS analysis due to Coulomb repulsion, sulfate loss, and the formation of multiple adducts. Finally, epimerization and sulfation at various positions lead to a large number of isomeric building blocks, difficult to distinguish by MS-based methods relying ultimately on the measurement of m/z ratios.
Due to the sheer size of full-length GAG polysaccharides, (partial) enzymatic or chemical depolymerization of the chains (Figure 27) is crucial for obtaining smaller oligosaccharides tractable by state-of-the-art MS methods.453,460−462 Thus, strategies to characterize and sequence GAGs tend to follow a bottom-up approach. The complexity of the mixture resulting from depolymerization, along with the inherent polydispersity and sequence microheterogeneity, imply that extensive multistep separations are indispensable in GAG analysis, as addressed in excellent reviews.453,463−465 Depolymerization, combined with chromatographic and electrophoretic separations, provides the link between the full-length GAG chains and the shorter oligosaccharides (dp < 12) compatible with MS analysis. Instead of focusing on sample preparation, condensed-phase separations, or disaccharide profiling, herein we review developments in the methodology and instrumentation of MS-based techniques in the context of oligosaccharide analysis and sequencing. These developments include novel ion activation methods, the hyphenation of IMS to MS, and action spectroscopy of mass-selected GAG ions. In the past 15 years, the above inventions significantly increased the amount and specificity of structural information obtainable on GAGs at the oligosaccharide level. They successfully tackle challenges arising from dense sulfation and isomerism, two aspects of GAGs that have impeded their analysis using traditional MS techniques.
Figure 27.

Common glycosaminoglycan depolymerization strategies shown through the example of heparan sulfate/heparin. (A, left) Enzymatic depolymerization of GAG chains may be performed using glycosidases, resulting in hydrolytic cleavage that preserves the hexuronic acid stereochemistry. To obtain oligosaccharide fragments covering the full sequence, enzymes with endolytic activity are necessary. Heparanases are endo-β-glucuronidases cleaving at the reducing end of GlcA residues in moderately sulfated HS/heparin chains. (A, right) Prokaryotic lyases, such as heparinase I–III, act via a β-eliminative mechanism, leading to Δ4,5-unsaturated uronic acid residues at the new nonreducing end. Consequently, stereochemical information is lost in the process. (B, left) Benzyl esterification with alkaline β-elimination may be applied for the depolymerization of GAGs, mimicking lyase activity. (B, right) Deaminative cleavage preserves hexuronic acid stereochemical information at the cleavage site but alters the structure of the glucosamine through the formation of 2,5-anhydromannose. The reaction is blocked in the presence of N-acetyl groups on glucosamines, making prior deacetylation necessary.
6.2. Electron-Based Dissociation Methods in GAG Analysis
Owing to their acidity, GAGs have mainly been analyzed in negative ion mode by ESI-MS. They exhibit high ionization efficiencies, with sulfates and—to a lesser extent—carboxylates carrying the negative charges.465 Mass spectra of GAGs are highly complex, as even a single, well-defined oligosaccharide can give rise to a multitude of ions: various charge states may be simultaneously present, each with a distribution of adducts due to H+/metal ion exchange. A major challenge in the MS analysis of highly sulfated species is the undesirable decomposition of sulfate modifications, appearing in the form of SO3 neutral loss (79.96 Da).466 Sulfate loss hampers compositional analysis and the localization of sulfate modifications. This unimolecular reaction has a low activation barrier in the gas phase and affects protonated sulfate groups. Removal of the proton from sulfates successfully inhibits the process and the resulting loss of information, as depicted in Figure 28. Exchanging the neutralizing proton to an aprotic tetraalkylammonium/metal cation is a common strategy to reduce intramolecular Coulomb repulsion between charged sulfate groups, thereby facilitating deprotonation in densely sulfated species. Although ESI is among the softest ionization methods, sulfate loss products often occur as in-source fragments, even without additional ion activation. Therefore, applying the softest possible source conditions is essential when analyzing highly sulfated compounds. As slow-heating methods favor dissociation channels with the lowest barriers, sulfate loss products dominate CID and IRMPD product ion spectra. Both of these traditional dissociation methods yield abundant glycosidic bond cleavages, while cross-ring fragments—often crucial for the precise localization of sulfate modifications—are scarce.465 Various attempts were made to overcome these shortcomings, facilitate cross-ring cleavages, and reduce sulfate losses in CID mass spectra. Although metal ion adduction,467−469 derivatization,470−472 or positive ion mode analysis473 proved to be successful in many aspects, inherent characteristics of slow-heating methods limit their utility for GAG analysis.
Figure 28.

Sulfate equivalent loss of glycosaminoglycans in the gas phase. (A) Sulfate loss in the form of neutral SO3 upon ion heating. Protonated sites are more prone to undergo decomposition. (B) Deprotonation of sulfate groups inhibits the undesirable reaction. Quaternary ammonium or metal ions (M+) may neutralize the charge of deprotonated sulfate groups, reducing intramolecular Coulomb repulsion and facilitating the removal of protons.
In a landmark study, Wolff et al. employed EDD for the first time for GAG analysis, comparing the EDD fragmentation pattern of four modestly sulfated HS tetrasaccharide dianions to those obtained by CID and IRMPD.86 EDD may be regarded as the negative ion counterpart of ECD, a nonergodic fragmentation process developed for polycations.79 In EDD, multiply charged anions are irradiated by electrons of moderate kinetic energy (15–20 eV), causing electronic excitation and electron detachment, initiating radical-driven dissociation pathways. EDD product ion spectra of the HS tetrasaccharides were informative, highly reproducible, and rich in A- and X-type cross-ring fragments; yielded a full set of glycosidic cleavages for all analytes; and exhibited much more favorable sulfate retention than CID or IRMPD (see Figure 29). Thus, this fragmentation technique overcame many challenges associated with the tandem MS analysis of GAG oligosaccharides. A drawback of EDD is the dependency on expensive FTICR-MS platforms, as trapping molecular ions and electrons simultaneously in the presence of radiofrequency (RF) fields, such as in linear ion traps and Paul traps, is not straightforward.474−479 In addition, electrostatic repulsion between polyanions and electrons of moderate kinetic energy makes the process rather inefficient, with consequently long interaction periods required.
Figure 29.
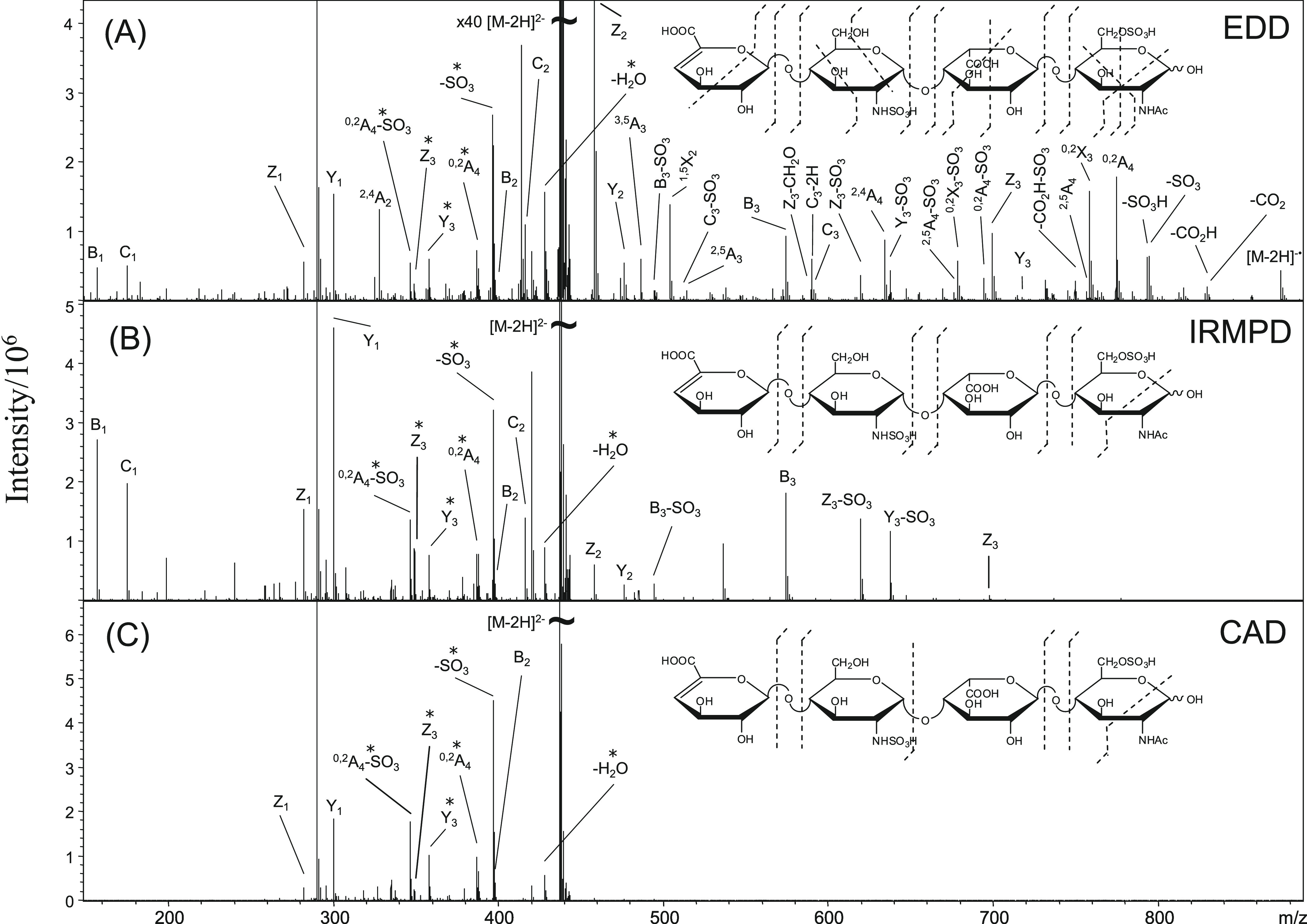
Electron detachment dissociation (EDD) vs slow-heating fragmentation of glycosaminoglycans. (A) EDD tandem mass spectrum and corresponding fragmentation pattern of a synthetic heparan sulfate tetrasaccharide as [M – 2H]2–. Tandem mass spectra and fragmentation patterns resulting from (B) infrared multiple photon dissociation (IRMPD) and (C) collision-induced dissociation (CID) of the same precursor ion. Doubly charged product ions are indicated with an asterisk. Figure reprinted with permission from ref (86). Copyright 2007 American Society for Mass Spectrometry.
The capabilities of EDD were further explored for the analysis of more heavily sulfated HS species480 and for larger DS oligosaccharides (dp4–dp10).481 A systematic analysis concerning the influence of charge state and cation adduction on the EDD fragmentation pattern of DS oligosaccharides revealed that electron detachment from carboxylates is preferred over sulfates for thermodynamic reasons, provided the carboxylates are not protonated.482 Deprotonation of carboxyl groups increases sulfate retention upon EDD but requires high precursor charge states or cation adduction due to the higher proton affinity of carboxylates over sulfates.
Another fruitful application of EDD concerns the stereochemistry of hexuronic acid residues. Distinguishing GlcA from its C5 epimer IdoA in HS/heparin or hybrid CS/DS chains is an important task in GAG research.483 However, it is extremely difficult to accomplish by MS methods that ultimately reduce structural information to m/z ratios. In a study by the Amster group, pairs of diastereomeric HS-related tetrasaccharides were used as model compounds: each species carried GlcA or IdoA in the second position and a Δ4,5 unsaturated uronic acid residue at the nonreducing end. EDD provided diagnostic fragments, enabling the distinction of GlcA-containing oligosaccharides from their counterparts carrying IdoA. According to the proposed hypothesis, the formation of these fragments involves hydrogen atom transfer to the carboxyl radical from neighboring carbons or hydroxyl groups. The site of hydrogen abstraction, which will determine subsequent reaction steps and the nature of fragment ions formed, depends on key interatomic distances and thereby on the configuration of C5. In less unambiguous cases, when no diagnostic fragments could be identified, EDD was combined with principal component analysis (PCA). Multivariate statistical analysis facilitated both the distinction of diastereomeric HS and CS/DS tetrasaccharides and the stereochemical assignment of certain hexuronic acid residues.484−487 The possibilities of assigning the configuration of hexuronic acid units farther away from the reducing end, or determining the stereochemistry of multiple residues simultaneously in longer chains, are intriguing questions that may inspire future research.
Following the introduction of EDD to the field, the family of ExD methods applied for GAG oligosaccharides has been extended by EID266 and NETD.89 EID is performed by irradiating singly charged anions with electrons of moderate kinetic energy (6–20 eV), inducing electronic excitation. Electron detachment from monoanions leads to neutral species, invisible by MS. Similarly to EDD, EID produces both even- and odd-electron fragment ions and cross-ring cleavages that affect primarily hexuronic acid residues.266 However, it seems to be less sensitive to hexuronic acid stereochemistry and, lacking clear advantages over EDD, has not gained widespread popularity in the field. NETD, in contrast, earned popularity in GAG research, owing to its compatibility with a variety of MS platforms, the rich fragmentation patterns, and the short interaction periods it requires for efficient fragmentation.488 NETD represents the negative ion mode counterpart of ETD: instead of transferring electrons to positive analytes with radical anions as in the latter, NETD utilizes radical cations to abstract electrons from polyanions, initiating radical-driven dissociation pathways.489 Common sources of the reactive radical cations are fluoranthene and Xe, but other gases may also be applied.465 Although NETD is based on ion–ion reactions and not on direct ion–electron interactions, the resulting dissociation pathways and fragmentation patterns closely resemble those observed in other ExD techniques, justifying the inclusion of NETD into this family of ion activation methods.
Huang and Yu et al. compared NETD and EDD on an FTICR-MS platform, using highly sulfated HS/heparin model oligosaccharides up to dp6.490 Despite remarkable advances in the chemical,491−499 enzymatic,500,501 and chemoenzymatic502−508 synthesis of GAGs, the accessibility of standards with well-defined structures is not comparable to that found in the fields of peptide and nucleic acid research. One of the few commercially available standards is fondaparinux (Arixtra), a fully synthetic anticoagulant mimicking the antithrombin III binding sequence of heparin. This pentasaccharide, carrying 8 sulfate modifications, showed comparable fragmentation patterns in EDD and NETD, with a full set of glycosidic cleavages and plentiful cross-ring fragments, enabling MS-based sequence assignment. In general, NETD proved to be a more efficient dissociation process for highly charged species, while EDD was better suited for the analysis of dianions. Interestingly, NETD led to fewer sulfate loss products, both in number and in abundance. This phenomenon may be related to the absence of direct electronic excitation when using cation radicals instead of ∼20 eV electrons.
Zaia and co-workers systematically studied the capabilities of NETD to distinguish the rare 3-O-sulfation from the common 6-O-sulfate modification in HS/heparin oligosaccharides.509 Previously, NETD did not allow for unambiguous localization of sulfate modifications (4-O- vs 6-O-sulfation) in a CS oligosaccharide, due to the lack of diagnostic product ions, e.g., cross-ring fragments with a bond cleavage between C4 and C5 of GalNAc residues.510 In contrast, diagnostic fragments were generated by NETD for 3-O-sulfated HS species, enabling the assignment of 3-O- vs 6-O-sulfation position in GlcNS units, as highlighted in Figure 30.
Figure 30.

Negative electron transfer dissociation (NETD) fragmentation patterns of synthetic heparin/heparan sulfate hexasaccharides. (a) GlcA-GlcNS6S-IdoA-GlcNS3S-GlcAGlcNS6S; (b) GlcA-GlcNS6S-IdoA-GlcNS6S-GlcA-GlcNS6S; and (c) GlcA-GlcNS6S-IdoA-GlcNS3S6S-GlcA-GlcNS6S. The highly sulfated glycans were measured as [M – H]5– polyanions. Note the diagnostic cross-ring fragments enabling the localization of sulfate groups within the glucosamine residues. Reprinted with permission from ref (509). Copyright 2018 American Society for Mass Spectrometry.
Taking advantage of its short interaction periods, MS with NETD has been successfully coupled in an online fashion to HILIC by Wu and co-workers.90 A library of 15 HS oligosaccharide standards, containing di- and tetrasaccharides with varying degrees of sulfation, including many isomeric species, was used to demonstrate the potential of the workflow for sequencing. Although NETD fragment ion spectra alone did not enable the assignment of hexuronic acid stereochemistry, the diastereomers could be successfully separated using HILIC. Strong structure–retention relationships were revealed, with IdoA-containing chains eluting earlier than their GlcA-containing counterparts. In accordance with previous results, 3-O- and 6-O-sulfation motifs could be unambiguously assigned by NETD MS/MS on the chromatographic time scale. Similarly to the HILIC coupling, hyphenation of NETD MS/MS to capillary zone electrophoresis (CZE) has been recently accomplished.511 The 50–100 ms interaction periods required for efficient NETD of GAGs enabled sufficiently frequent sampling of the narrow (30 s) CZE peaks. Although hexuronic acid isomers could be separated by CZE, their identification based on the NETD fragmentation patterns remains an unresolved challenge.
Finally, in relation to ExD techniques, it is worth mentioning charge-transfer dissociation (CTD), an activation method recently demonstrated to yield informative fragment ion spectra for HS, CS, and DS oligosaccharides.512 In CTD, ions are collided with high-energy (several keV) He+ ions. As He has an exceptionally high ionization energy (24.6 eV), He+ ions abstract electrons from the analytes. Electron transfer is accompanied by rapid excitation of the oxidized radical product ion, promoting radical-driven dissociation pathways. Like NETD, CTD is based on ion–ion reactions and may be readily implemented into various MS platforms. Although providing abundant cross-ring cleavages in general, CTD did not deliver diagnostic fragments, enabling the unambiguous assignment of sulfate positions (4-O vs 6-O) in GalNAc residues of CS/DS oligosaccharides. This intriguing aspect of GAG structural diversity represents an important challenge in the field, likely to inspire future research and developments.
6.3. Ultraviolet Photodissociation Mass Spectrometry of GAGs
Although not as widespread in GAG analysis as the various ExD methods, UVPD MS has become increasingly popular in recent years and is expected to further expand in the near future, owing to the commercialization of the technique. An advantage of UVPD is its compatibility with linear and 3D quadrupole ion traps,32 opposed to EDD that is difficult to implement in the above-mentioned platforms and is mainly performed using expensive FTICR-MS (Penning traps). To date, UVPD on GAGs has been performed exclusively in negative ion mode. In general, UVPD fragmentation patterns of GAG anions are characterized by (1) retention of sulfate modifications to an extent comparable to that observed in ExD methods, (2) extensive glycosidic bond cleavages, resulting in good sequence coverage, and (3) abundant cross-ring fragments that help verify backbone linkage positions, localize sulfate modifications, and distinguish isomers. Many of these favorable characteristics stem from the fact that absorption of energetic (>3 eV) UV photons leads to fast energy deposition and electronic excitation, in contrast to the gradual vibrational excitation in slow-heating CID and IRMPD. Direct dissociation from excited electronic states is not the only fragmentation form associated with the UV photoexcitation of isolated GAG anions. EPD from multiply charged ions enables radical-driven dissociation pathways, while internal conversion in combination with IVR leads to thermal fragments, similar to those observed upon CID. The capabilities and potential of UVPD MS in GAG analysis are demonstrated through selected works below.
A series of pioneering experimental and theoretical works on UVPD MS of GAGs were carried out by Racaud et al. In a proof-of-principle study, UVPD was performed in a linear ion trap using a tunable optical parametric oscillator (OPO) laser between 220 and 290 nm (4.3 to 5.6 eV).513 By plotting the fragmentation yield as a function of the irradiation wavelength, the authors recorded gas-phase UV action spectra of oligosaccharide ions for the first time. One nonsulfated and two singly sulfated HS/heparin-derived disaccharides served as model compounds, investigated as singly deprotonated ions. Fragmentation yield increased linearly with the laser power, indicating that UVPD of the investigated species is a single-photon process. UVPD fragmentation patterns were recorded at 240 nm, the absorption maximum of the sulfated species, and compared to those obtained by CID. In general, UVPD led to more abundant cross-ring cleavages that helped localize sulfate modifications. Some of these X- and A-type fragments were specific to UVPD and likely result from direct dissociation from excited electronic states. The unspecific UVPD fragments observed also upon CID, on the other hand, were probably generated following internal conversion and the redistribution of vibrational energy. As the ions carried at maximum one deprotonated sulfate group, SO3 loss channels were not dominant.
In a follow-up publication, the strategy of inducing fragmentation of HS/heparin-derived anions by UV photoexcitation was further expanded.514 At 220 nm irradiation wavelength, two competing processes were observed for di- and tetrasaccharide dianions: UVPD and EPD. Electron detachment led to charge-reduced, oxidized radical species that were further subjected to CID. The resulting hybrid dissociation method that merges UV irradiation with collisional activation was previously termed activated (a-)EPD by the authors. By opening up new, radical-driven dissociation pathways in GAG anions, a-EPD gave rise to informative fragments distinct from those observed upon UVPD. In addition, the relative contributions of UVPD and EPD could be systematically tuned by Na+ adduction. While EPD was suppressed when all carboxyl groups were protonated, it became more dominant when the proton on one or more carboxyls was exchanged to Na+, leaving the precursor ion charge state unaltered. Thus, EPD in GAG ions is strongly linked to the presence of deprotonated carboxylates.
In a joint study by the Amster and Brodbelt groups, the fragmentation of various GAG oligosaccharide anions was investigated in UVPD experiments, employing a 193 nm ArF excimer laser on a Fourier transform mass spectrometry (FTMS) platform.515 UVPD was compared to high-energy collisional dissociation (HCD), EDD, and NETD. The model compounds included moderately sulfated HS and DS oligosaccharides (dp4 and dp10) and the highly sulfated pentasaccharide fondaparinux. HCD generated mainly glycosidic fragments and abundant sulfate loss products. In contrast, EDD, NETD, and UVPD led to more informative product ion spectra with abundant sulfate-retaining fragments, including cross-ring cleavage products. Besides providing a complete set of glycosidic cleavages and thus full sequence coverage for all model species, UVPD exhibited the highest abundance of cross-ring fragments. Many of these informative fragments were observed exclusively upon UVPD, including diagnostic 2,4X- and 1,4A-type fragments enabling unambiguous assignment of 4-O-sulfation in a DS tetrasaccharide, highlighted in Figure 31. To the best of our knowledge, such an achievement was not reported previously for ExD methods. UVPD experiments also revealed charge-reduced EPD products and the dependence of fragmentation patterns on precursor ion charge state and cation adduction. Moreover, the data indicated occasional sulfate transfer between monosaccharide units, a form of mass spectrometric rearrangement worthy of further investigation. Finally, the authors assessed the capabilities of UVPD using a more user-friendly solid-state Nd:YAG laser at 213 nm. The fragmentation pattern of the model DS tetrasaccharide at 213 nm was similar to that observed employing the 193 nm excimer laser and the same MS instrument, with discrepancies likely stemming from the difference in photon energies (5.8 vs 6.4 eV). However, an order of magnitude longer irradiation periods were needed at 213 nm, due to decreased fragmentation efficiency caused by the significantly lower pulse energy of the solid-state laser. This observation underlines the importance of bright UV sources, necessary to achieve sufficiently intense fragment ion peaks in UVPD MS within an interaction period comparable to that in NETD.
Figure 31.
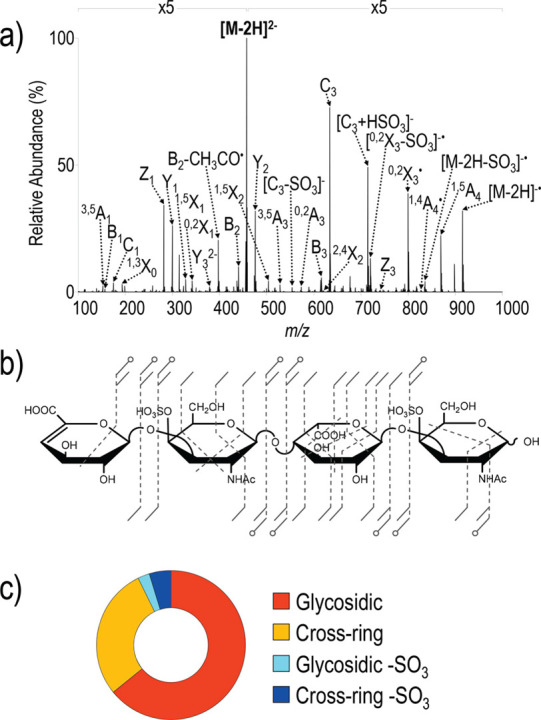
Ultraviolet photodissociation (UVPD) tandem MS of a dermatan sulfate tetrasaccharide at 193 nm. (a) Product ion spectrum of the doubly deprotonated oligosaccharide (m/z 458). (b) The corresponding UVPD fragmentation pattern and structural assignment. (c) Donut plot depicting the distribution of photofragments based on summed abundances of fragment types. Reproduced with permission from ref (515). Copyright 2019 American Chemical Society.
Building on the above findings, the authors systematically studied the influence of key experimental parameters on the fragmentation of CS and DS tetrasaccharides upon UVPD.516 Importantly, the ionization state of the precursor anions appeared to have a greater impact on the fragmentation patterns than the photon energy or the number of laser pulses employed. Upon choosing the suitable precursor ions, where the number of deprotonated groups either equaled the number of sulfates or exceeded it by one, UVPD at both 193 and 213 nm led to significant sulfate retention and enabled the assignment of 4-O-sulfation by generating diagnostic 1,4A-, 2,4A-, and 2,4X-type fragments.
Recently, UVPD MS was performed on fondaparinux ions in negative ion mode, using a commercial FTMS setup equipped with a 213 nm solid-state Nd:YAG laser.517 In the product ion spectrum of the doubly deprotonated fondaparinux precursor, a Y3/C3 internal glycosidic fragment was observed at m/z 417.94. This [GlcNS3S6S – H]− ion corresponds to the monosaccharide residue located in the middle of the pentasaccharide sequence, carrying the rare 3-O-sulfate modification that is found in some specific protein binding sequences. The characteristic triply sulfated monosaccharide residue is essential for antithrombin III binding and the resulting anticoagulant effect of heparin and related pharmaceuticals, but little is known about its other biological functions. Searching for exotic structural elements in the hope of finding specific protein binding sites along the glycan chain is an important aspect of GAG research.518 Based on their results, the authors envisaged an LC-MS/MS screening method using UVPD to detect HS/heparin sequences containing the rare GlcNS3S6S motif. Although such a method would have far-reaching applications in biomedical research and drug discovery, it is yet to be clarified whether the [GlcNS3S6S – H]− fragment ion is specific to the doubly deprotonated fondaparinux or appears more generally among the UVPD fragments of HS/heparin oligosaccharides carrying the GlcNS3S6S motif, thereby enabling the proposed screening strategy.
6.4. Ion Mobility–Mass Spectrometry in GAG Analysis
Since the commercialization of the first IM-MS instruments, gas-phase electrophoretic separations have contributed significantly to GAG sequencing approaches. They proved to be especially useful for separating GlcA- vs IdoA-containing stereoisomers, complementing tandem MS approaches. Employing a TWIM-MS instrument, Leary and co-workers demonstrated the separation of two HS hexasaccharide epimers as dianions.519 Although CCS values were not reported, the separation of stereoisomers differing merely in the configuration of a single carbon clearly showed the potential of IMS in GAG analysis. In a related systematic study, six synthetic HS octasaccharide stereoisomers—containing GlcA/IdoA residues in different ratios and positions—were investigated as multiply deprotonated species by TWIM-MS using He buffer gas.520TWCCSHe values were determined using a calibration curve based on a set of oligonucleotide standards. Oligosaccharides containing IdoA at the reducing end had systematically higher CCS values than their GlcA-containing counterparts. Although extraordinary care must be taken when comparing solution and gas-phase glycan conformations, it is reasonable to assume that IdoA and GlcA residues adopt different ring puckers not only in solution but also in the gas phase, which provides a rationale for the observed sequence–CCS correlation.
Multiple studies demonstrated the separation of sulfation positional isomers for HS521−523 and CS/DS species.524 Interestingly, the separation of the isomeric CS/DS disaccharides ΔUA-GalNAc4S and ΔUA-GalNAc6S in N2 buffer gas was more efficient when analyzing them as triply sodiated, singly charged cations. As the mobilities of the isomers were very close to each other, baseline resolution required outstanding resolving power, provided by an atmospheric pressure DTIM-MS instrument.
An important aspect of coupling IMS to MS, especially to tandem MS, is the compatibility of time scales. FTICR-MS is a relatively slow mass analyzer, while ExD methods highly suited for GAG analysis often require interaction periods on the order of 100 ms, rendering comprehensive, nested IM-MS2 experiments unfeasible. Strategies to overcome this difficulty include the application of filtering IMS devices, such as FAIMS, or the storage of mobility-selected ions for longer time periods before fragmentation and mass analysis. Kailemia et al. coupled FAIMS to FTICR-MS, which enabled the postionization separation of epimeric HS tetrasaccharides in combination with their selective MS/MS analysis employing EDD.163 FAIMS also separated members of a CS dp4–dp10 homologue series, where the species differed in both mass and charge but yielded overlapping peaks on the m/z scale. This charge state separation underlines the advantage of IMS as an additional, orthogonal separation step that reduces stress on subsequent MS analysis, resulting in less congested mass spectra.
Lin and co-workers combined TIMS with NETD MS/MS on an FTICR-MS instrument, analyzing a set of highly sulfated synthetic HS/heparin oligosaccharides (dp4–dp6).525 As TIM separations and NETD MS/MS take place on comparable time scales, their combination requires decoupling fragmentation and m/z analysis from the preceding mobility separation. This was achieved using a gated-TIMS strategy.526 Mobility- and m/z-selected ions emerging from the TIMS funnel and the downstream quadrupole mass filter were first trapped in a hexapole storage cell, which enabled the accumulation of ions from multiple TIMS cycles. Then, following multiple rounds of storage cell filling and potential NETD in the cell, the ions were released into the high-resolution mass analyzer. FTMS analysis and ion accumulation in the hexapole cell can be performed in parallel, increasing the duty cycle of the workflow. In contrast to prolonged residence times in TIMS funnels that may lead to significant rf heating, extensive storage times in the hexapole cell were not accompanied by notable ion activation: sulfate losses were negligible even after several hundred filling cycles. Sulfation position isomers and epimers differing only in one hexuronic acid residue could be successfully separated in N2 buffer gas, and TIMSCCSN2 values were obtained using perfluoroalkyl phosphazine ions to establish the calibration curves. Baseline resolution of two hexasaccharide epimers by TIMS allowed for their relative quantification (Figure 32), while NETD in combination with FTICR-MS provided diagnostic cross-ring fragments and excellent mass resolution, enabling the localization of sulfate modifications. Finally, an interesting aspect of the TIMS NETD MS/MS coupling concerns selection of the optimal charge state. While NETD works better for highly charged ions in general, isomer separations in TIMS were often more efficient when selecting lower charge states, making the choice of precursor ions all the more important.
Figure 32.
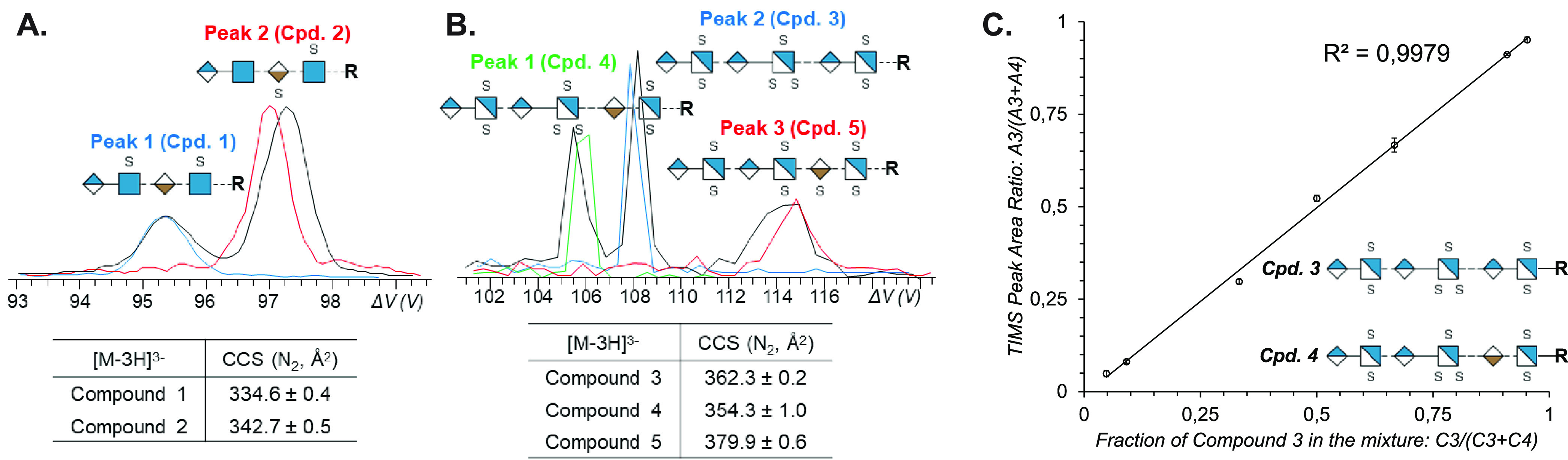
Gated trapped ion mobility separation of heparin/heparan sulfate oligosaccharides. (A) Arrival time distributions (ATDs) of two isomeric tetrasaccharides (blue and red traces) and that of their mixture (black trace). (B) ATDs of three highly sulfated hexasaccharide isomers (blue, green, and red traces) and that of their mixture (black trace). Collision cross sections of each compound (measured as triply deprotonated species) are listed below. R stands for an aminopentyl linker. (C) Relative quantification of the two hexasaccharide stereoisomers (compounds 3 and 4) enabled by trapped ion mobility spectrometry. The peak area ratio, A3/(A3 + A4), was averaged over three technical replicates and plotted against the ratio of the concentration of compound 3 over the total concentration, C3/(C3 + C4). Error bars represent the standard deviations of three measurements. Figure adapted with permission from ref (525). Copyright 2019 American Chemical Society.
Recently, Miller et al. introduced a fragment-based shotgun IM-MS sequencing (SIMMS2) approach, utilizing the unique pre-IMS fragmentation capabilities of a Q-DTIMS-ToF platform.527 First, a library of DTCCSHe values was created using the stepped-field method and 36 HS oligosaccharide standards (dp2–dp10) of well-defined structure, including species carrying 3-O-sulfation. CCS values were determined for both intact and CID fragment ions generated from each of the 36 standards, resulting in a library containing sequence–CCS–m/z trios in numbers far exceeding the number of standard compounds used. This strategy also circumvents certain issues arising from potential reducing-end modification mismatches, by utilizing internal and A-, B-, and C-type fragments. To demonstrate the speed and potential of SIMMS2, an unknown hexasaccharide was rapidly sequenced by comparing the CCS values of its fragments to CCSs in the library of known structures. The known sequences, in this case, stemmed from two dp4 standards sharing common structural elements with the unknown hexasaccharide. SIMMS2 is currently limited mainly by IMS resolving power, the uncertainty of CCS measurements, and the availability of standards. Improvements are expected in all three aspects in the future, decreasing potential degeneracy in the library (i.e., one CCS value corresponding to multiple structures) and thereby reducing the possibility of structural misassignments. As the method is based on an ever-expanding library of CCS values, it may help circumvent the meticulous analysis of fragment ion spectra that relies heavily on expertise and could represent a step toward high-throughput GAG sequencing. A graphical overview of the SIMMS2 workflow is shown in Figure 33.
Figure 33.

Graphical overview of the SIMMS2 strategy for de novo glycosaminoglycan sequencing. (a) Characteristic domain structure of heparan sulfate (HS) chains and a matching set of shorter HS oligosaccharide standards. (b) MS and tandem MS provide m/z values for intact and fragment ions, while ion mobility spectrometry allows for the determination of collision cross sections (CCSs). (c) Through comparison of unknowns with database elements, the library containing m/z values and CCSs of ions generated from known standards enables the determination of unknown HS sequences. (d) Illustration of the iterative loop process for expanding the CCS data set, enabling continuous development of the library-based SIMMS2 strategy. Figure reprinted from ref (527). Copyright 2020 Miller et al. (Creative Commons Attribution 4.0 International License).
Because the present review is focused on glycan sequencing and structure elucidation, many excellent studies dealing with GAG–protein interactions were not addressed above.528−532 This decision reflects only the scope of the review and not the importance of these interactions or that of the respective studies.
6.5. GAG Analysis by Gas-Phase Ion Spectroscopy
As a relatively new field augmenting the GAG sequencing toolbox, GAG analysis by gas-phase ion spectroscopy in the UV and IR range emerged only throughout the past decade.34,533 As already mentioned in Section 6.3, the Dugourd group published the first experimental, gas-phase UV spectrum of sulfated disaccharide anions from 220 to 290 nm.513 The exact gas-phase molecular structure of the set was further investigated using density functional theory (DFT). The experimental spectra were found to be in reasonable agreement with the calculated spectra and revealed that UV spectroscopy is sensitive to the modes of the sulfate groups present.213
The first gas-phase IR spectrum from 3400 to 3700 cm–1, using IRMPD spectroscopy in an FT-ICR cell of d-glucuronic and l-iduronic acid monosaccharides, was published by Polfer and co-workers.534 They used rubidium to ionize the monosaccharides as positively charged ions and showed that the method is sensitive to the epimerization at C5. Similar results were obtained for the same monosaccharides investigated as anions.535
Compagnon and co-workers188 used IRMPD spectroscopy from 3200 to 3700 cm–1 to record the spectrum of a sulfated monosaccharide, i.e., glucosamine 6-sulfate, as a protonated cation. In their publication, they describe the potential of gas-phase spectroscopy coupled to MS as a tool to differentiate sulfated from phosphated saccharides based on their vibrational modes. Harmonic and anharmonic (VPT2 and finite temperature molecular dynamics) frequency simulations were used to predict the wavenumber of certain OH stretches in the sulfate and phosphate functional groups. Glucosamine 6-sulfate was later used as a model to test the accuracy of a large number of frequency simulation methods.536 Methods which are satisfying at higher wavenumbers are shown to fail from 500 and 1700 cm–1 as the anharmonic nature of the sulfates challenges the calculations. The best match between experimental and theoretical spectra from 500 to 1700 cm–1 was obtained using the computationally most demanding method. DFT simulations were further improved for anionic monosaccharide ions and compared to the experimental spectra.537
Disaccharides are the smallest building blocks in GAGs, and consequently, the use of IR spectroscopy advanced most recently to characterize larger fragments. Rizzo and co-workers538 published IR spectra from 3200 to 3700 cm–1 using messenger-tagging spectroscopy coupled to IM separation of isomeric CS and HS disaccharides as positively charged sodium adducts. At cryogenic temperatures, the resolving power is sufficient to differentiate the five isomers only based on their IR fingerprints. Four isomeric CS and HS disaccharides were investigated from 2700 to 3700 cm–1 and two of the set also from 550 to 1850 cm–1 using IRMPD spectroscopy (see Figure 34).537 The authors explored the use of different charge states, species, and ionization modes on exemplary ions to gain resolving power in IRMPD spectroscopy. In another study, MSn in a 3D ion trap analyzer was combined with IRMPD spectroscopy from 2800 to 3700 cm–1 for Y- and B-type fragment anions of CS isomers with O-sulfation at the C4 and C6 position.539 The two isomers cannot be differentiated by MS alone and are challenging in IMS. The resolving power of IRMPD reaches its limitations, yet the isomers can be distinguished by their spectroscopic fingerprints.
Figure 34.

IRMPD spectra of heparan sulfate (HS) and chondroitin sulfate (CS) disaccharides. Scheme: Δ4,5 unsaturated disaccharides derived from HS (left) and CS (right). (A, B) IRMPD spectra of deprotonated Hp II-A (red: [Hp II-A – H]−) and Hp III-A (black: [Hp III-A – H]−) in the 550–1850 and 2700–3700 cm–1 spectral ranges. Right panels: IRMPD spectra of CS-A (red) and CS-C (blue) in different charge states. (C) Singly deprotonated [CS-A – H]− and [CS-C – H]−. (D) Doubly deprotonated [CS-A – 2H]2– and [CS-C – 2H]2–. (E) NH4+ cationic complexes [CS-A + NH4]+ and [CS-C – NH4]+. Figure reproduced with permission from ref (537). Copyright 2017 American Chemical Society.
Going larger, IR spectroscopy from 1000 to 1800 cm–1 was used to investigate an intact, highly sulfated pentasaccharide, i.e., the synthetic anticoagulant fondaparinux or Arixtra, as a protonated ion and sodium adduct.204 The pentasaccharide challenges most MS fragmentation methods due to the fragile nature of the eight sulfate groups. The IR spectra were recorded using both IRMPD spectroscopy and cryogenic IR spectroscopy in helium nanodroplets. The cryogenic temperatures are essential for the efficient IR spectroscopic study of larger GAGs. For a set of synthetic, sulfated HA derivatives, it has been shown that, even for tetrasaccharides, the IR signature of the sulfate groups exhibits a distinct pattern. The charge state, with respect to the number of equivalent, acidic functional groups, plays an important role in IR spectroscopy of anionic GAGs to prevent charge migration and further increase the spectral resolution. Using this high resolving power, the distinct IR signatures of four HS tetrasaccharide anions with varying configurations at C5 revealed a clear spectra–structure relationship, which was further rationalized by quantum chemical calculations.233 Structural motifs in the hydrogen bonding network could be deduced for either d-glucuronic or l-iduronic acid. In another study, the IR spectra in combination with quantum chemical calculations of CS/DS disaccharides from bacterial chondroitinase digestion (exclusively as Δ4,5 unsaturated uronic acids, SNFG symbol: white diamond) with all known motifs of sulfation revealed that the charge state at the sulfates, which are exclusively deprotonated, defines the size of the gas-phase conformational landscape and therewith the spectral signature (see Figure 35).540 In higher sulfated and charged CS/DS disaccharides, as in the triply sulfated disaccharide shown in Figure 35, less low-energy conformers are present than in lower charged CS/DS disaccharides. Quantum chemical calculations aid the understanding of the IR spectra of GAGs to evolve the method and its ability within the analytical toolkit.
Figure 35.
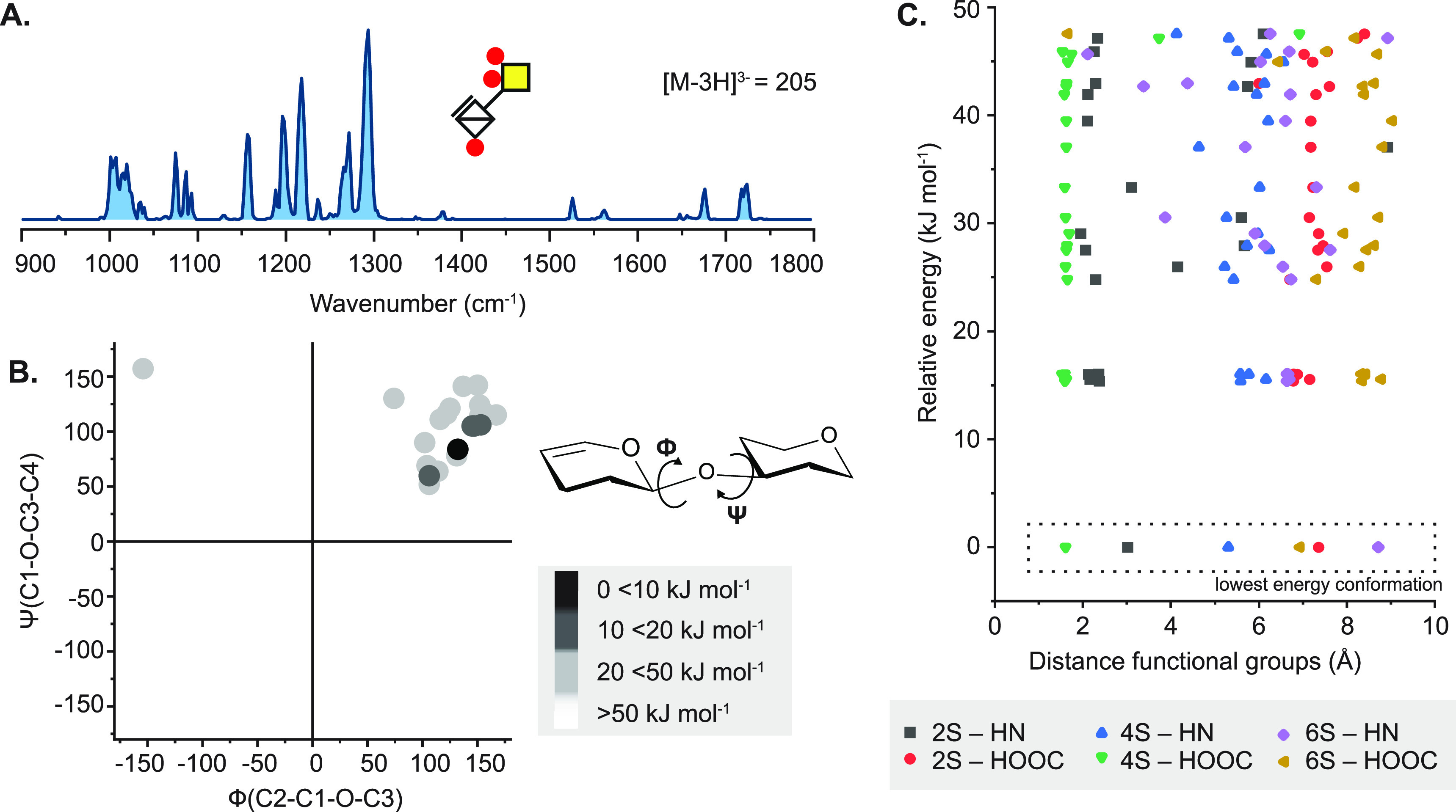
Conformation of chondroitin sulfate disaccharides in the gas phase revealed by IR spectroscopy and quantum chemical calculations. (A) Cryogenic IR spectra of a triply sulfated disaccharide investigated as a [M – 3H]3– anion with m/z of 205. (B) Calculation of the dihedral angles at the glycosidic bond with respect to the relative energies of the conformers presented in a Ramachandran-type plot for the glycosidic linkage. (C) Calculation of the intramolecular distance of charged sulfates to carboxyl and amide groups in Å in conformers with relative energies ΔEPBE < 50 kJ mol–1. In the case of the triply sulfated and triply charged disaccharide, the low-energy conformers present in the gas phase are similar to each other. The empty diamond symbol represents a Δ4,5-unsaturated hexuronic acid residue. Figure reproduced from ref (540). Copyright 2021 Lettow et al. (Creative Commons Attribution 4.0 International License).
7. Glycopeptides
The field of glycomics experienced a rapid growth in recent years, and many fundamental insights into structural properties of all glycan classes, including N- and O-glycans, GAGs, and glycolipids were gained. Especially MS-based N- and O-glycan analysis found its way as a routine measurement in clinical context due to the fact that alterations in their glycan profile are directly associated with various diseases such as rheumatoid arthritis,541,542 diabetes,1,543 and cancer.544,545 However, most of the information gained on N- and O-glycans emerged from experiments on the released glycoforms, while very little information is raised from glycoproteins directly. The traditional approach for glycan analysis is mostly separated from peptide/protein analysis to make the process simpler but comes at the cost of losing glycan site information.
This has drastic consequences for our understanding of the biological function of glycans. We lose the ability to study the prevalence for specific glycosylation sites on proteins and the influence of glycan location on biological processes.546 The assignment of specific glycan structures in the intact proteome could have a huge impact on our current understanding of structure–function relationships and will likely lead to a boost in finding potential disease biomarkers and therapeutic targets.547 Therefore, the field of glycoproteomics aims to merge glycomics and proteomics to obtain a comprehensive picture of both glycan and peptide identity, for any given glycoprotein in a cell or tissue. MS-based analytical techniques play an essential role in this process as they provide the necessary resolution to enable global glycoprotein analysis. MS often provides more detailed information compared to solution-based approaches, e.g., immunological assays, because it is possible to characterize and identify species on a molecular level.
7.1. Top-Down Analysis of Glycoproteins
The MS methods used in glycoproteomics are very similar to classical “bottom-up” and “top-down” proteomic approaches. While top-down glycoproteomics focuses on the direct analysis of intact glycoproteins via LC-MS/MS, bottom-up glycoproteomics first utilizes proteolytic enzymes to digest the glycoproteins and subsequently characterizes the glycopeptides by LC-MS/MS.
The advantage of top-down glycoproteomics is the information gained on the complete amino acid sequence, which includes all post-translational modifications. The charge state and the isotopic distribution of each precursor embody a very detailed but extremely complex set of information on the glycoprotein structure with minimal sample preparation. This information richness also has its downsides: low charge states (which often represent the native conformation of the analyte) result in very high glycoprotein precursor m/z and require suitable high-mass instruments with extraordinary resolution and/or complex detection systems548 to unravel the data.
On the other hand, high charge states (which are often the result of denaturing, non-native conditions) lead to low precursor m/z with multiple charge states that are readily detectable by most mass spectrometers. The wide distribution of charge states, however, can significantly dilute signal sensitivity and leads to overlapping of different glycoprotein precursors, resulting in problems with identification. This situation is aggravated due to the macro- and microheterogeneity of glycosylation sites. Their inherent structural diversity leads to a diverse mixture of differently glycosylated proteins, which represents a major challenge for the comprehensive analysis of glycoproteins via intact glycoproteomics.549 Therefore, the complex application of top-down analysis still hampers its routine use in therapeutic and diagnostic approaches despite the potential advantages in gained information.
7.2. Bottom-Up Analysis of Glycoproteins
7.2.1. Sample Preparation and Condensed-Phase Separations
Currently, it is more straightforward to digest glycoproteins with proteolytic enzymes into smaller glycopeptides to reduce the complexity and simplify the analysis by MS. Although the data set for glycopeptides needs to be combined and backtracked to obtain information on the intact glycoprotein, modern strategies facilitate this process and allow a high coverage of both a glycan and peptide moiety. This led to the rise of bottom-up approaches as a prominent technique for the analysis of glycosylated proteins already today. Bottom-up experiments follow a typical workflow that can be divided into sample preparation, enrichment/separation strategies, and MS analysis (Figure 36).
Figure 36.
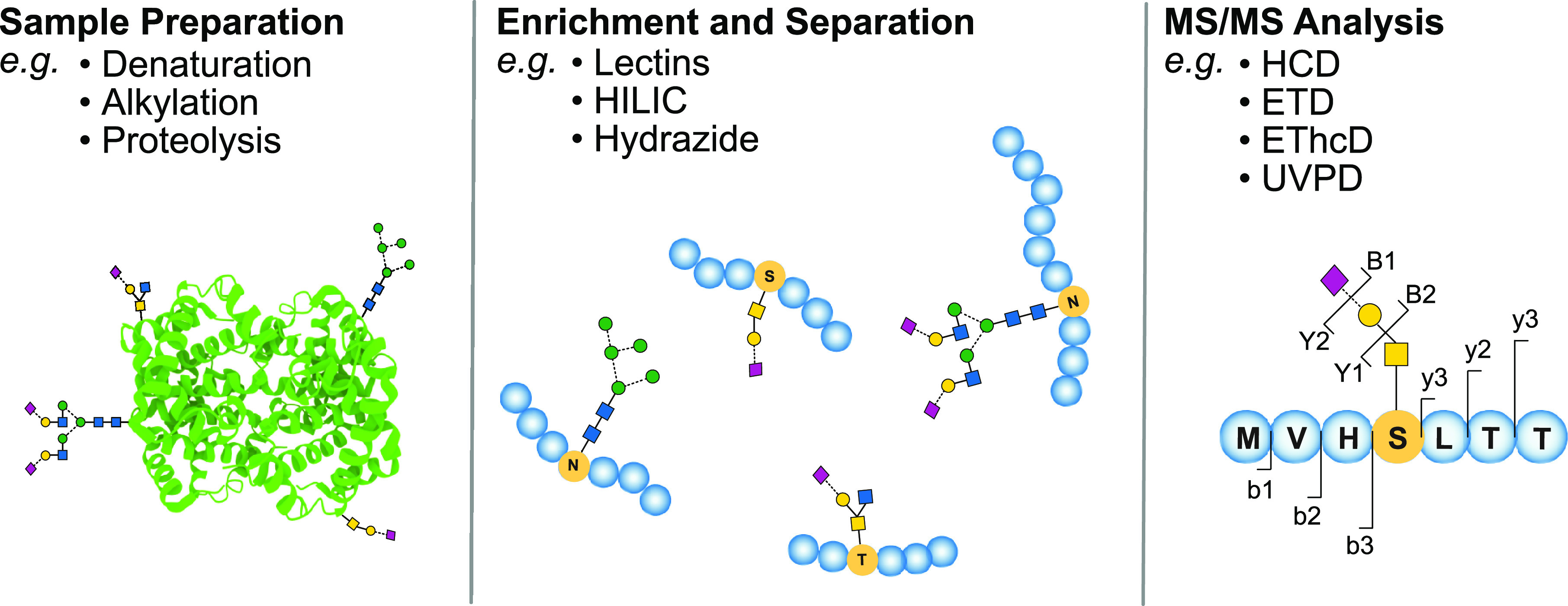
General scheme of bottom-up glycoproteomics workflows. Glycoproteins are extracted and enriched from biological samples. The glycoproteins are digested by proteolytic enzymes, and the resulting glycopeptides are enriched from the digestion mixture. The concentrated glycopeptides are separated via HPLC or CE to facilitate isomeric identification and subsequently analyzed via MS/MS. Complete glycopeptide characterization requires three elements: the sequencing of the peptide backbone, the sequencing of the glycan moiety, and the localization of the glycosylation site within the amino acid sequence.
Sample preparation generally includes an optional enrichment of the glycoprotein from its complex matrix and the subsequent proteolytic digestion with a suitable enzyme. The proteolytic digestion mixture can be directly analyzed by MS, but the great variety of glycans attached to each glycan site (microheterogeneity) and their heterogeneous site occupancy (macroheterogeneity) results in low abundancies for each glycopeptide. Furthermore, glycopeptides generally show lower ionization efficiencies compared to nonglycosylated species; therefore, it can be necessary to separate the glycopeptides from their complex matrix before MS analysis. There are several enrichment strategies available. The most prominent strategies include for example lectin-based approaches, HILIC SPE workflows, metabolic labeling, or covalent conjugation to hydrazide resins.4
Although enrichment strategies can concentrate the glycopeptides from the digestion mixture, they cannot completely compensate for the low abundance of glycosylated species. Commonly used data-dependent acquisition (DDA) approaches for MS analysis are often struggling with these low abundancies as MS/MS acquisition is generally triggered by intensity thresholds. Targeted and semitargeted approaches are alternative approaches to facilitate data acquisition, but including all possible glycan structures in inclusion lists is a great challenge due to their large heterogeneity.550 A promising approach to overcome this problem was developed by Woo et al. in 2015, which combines metabolic labeling with an analysis of the isotope pattern. By introducing a bromide-containing azide or alkyne function into the glycan residue via metabolic labeling, the enrichment and proteolytic digestion could be combined into one coherent workflow called IsoTag.551 Furthermore, the subsequent analysis of the characteristic bromide isotopic pattern via MS enables a pattern-searching algorithm for both detection and validation of isotopically encoded species. This approach gained increasing attention as it enables us to identify glycopeptides in complex mixtures in MS experiments even at low concentrations.547,552
Due to the large variety of glycan structures and the possibility of multiple glycosylation sites on the glycopeptides, enrichment of the glycopeptides is often accompanied by an additional separation step before MS characterization. This allows the separation of isomeric structures and simplifies the identification of glycopeptides by MS. Traditionally, reversed-phase methods with C8 and C18 stationary phases are applied to peptides due to the hydrophobic character of the peptide backbone; however, the hydrophilic glycan moiety is not well retained, and therefore only small or no isomeric resolution of glycopeptides is achieved. This led to the application of more specific chromatographic methods such as PGC553 and HILIC,554 which have unique properties to resolve both peptides and glycopeptides, on an isomeric level. Furthermore, capillary electrophoresis showed its potential as a promising separation technique as it allows the separation of isomeric glycopeptides in very short time frames.555,556 Further details on sample preparation and separation strategies are summarized in recent review articles.4,557,558
7.2.2. Tandem Mass Spectrometry for Glycopeptide Analysis
Complete characterization of glycoproteins requires not only sequencing of the protein backbone and glycan chains but also the localization of glycosylation sites within the amino acid sequence. This is especially difficult for O-glycans because, in contrast to N-glycosylation, there is no defined consensus peptide sequence for the various types of O-glycosylation. Furthermore, as the intact glycopeptide mass is generally not sufficient to characterize the components,559 it is necessary to fragment it via MS/MS. One of the major analytical challenges in the analysis of glycopeptides is the generation of informative fragments to fully identify a peptide and glycan moiety at the same time. In this regard, it is important to mention that peptide560 and glycan fragments54 are denoted using very similar nomenclatures. In order to avoid confusion, it is therefore common in glycoproteomics to denote peptide fragments using small letters (e.g., b/y) and glycan fragments using capital letters (e.g., B/Y).
Traditional slow-heating techniques such as CID are commonly used ion dissociation methods in glycoproteomics as they are built in many mass spectrometers due to their simple implementation. Low-energy CID predominantly leads to the dissociation of the weakest bonds, resulting mainly in glycosidic bond cleavages (B- and Y-fragments), and the loss of the labile sugar moieties from the peptide backbone. As such, it provides little information on the primary structure of peptide constituents, and the information regarding the sites of glycosylation is lost. High-energy CID further generates some peptide backbone fragments (b/y). The combination of low- and high-energy CID is mostly applied for sequencing the glycan and peptide moieties separately561 and represents a robust sequencing method that is well suited for a broad range of N- and O-glycosylated peptides.562 However, as it hardly can generate Y1 fragments, it is more commonly used for determining N-glycosylation, as the prediction of N-glycosylation sites is more straightforward due to the defined attachment points.
For a more detailed analysis of glycan attachment sites, higher-energy CID (HCD), which is implemented into modern Orbitrap instruments, is a promising alternative. In contrast to standard CID, typical HCD ion activation mainly results in peptide backbone fragmentation as well as fewer oxonium ions resulting from glycan fragmentation. Furthermore, b/y type fragments of the peptide backbone that still contain an N-acetylglucosamine (GlcNAc) moiety (Y1) are often generated, which provide clues to the location of glycosylation.563 Recent studies showed that, similar to standard CID, higher HCD collision energies predominantly generate b/y-fragments, while lower collision energies yield B/Y-fragments which are preferable for glycan characterization. The combination of multiple collision energies in one MS/MS run, termed stepped-energy HCD, is used for high-throughput application and is very popular in the studies of N-glycopeptides.73,564 In contrast to the defined amino acid sequence for N-glycosylation, O-glycosides generally have multiple serine or threonine residues that can serve as potential attachment points. The exact determination of O-glycosylation sites therefore often requires orthogonal approaches in addition to the popular HCD methods. The O-glycosidic bond connecting the peptide to the glycan is more labile than the amide bond of N-glycans and readily dissociates during vibrational activation in the mass spectrometer.
ExD methods such as ETD and ECD are more likely to generate the preferential dissociation of the peptide backbone before glycosidic cleavage.565 They produce predominantly c/z-fragments, whereas the glycosylation site remains intact. This allows for the identification of the peptide sequence and the localization of the modification site, which is especially helpful in O-glycoproteomics.566,567 Modern MS instruments can combine electron-based and vibrational activation (EThcD,568 ETD/CID569) into a hybrid approach that allows efficient glycopeptide characterization within a single MS/MS experiment. In principle, the hybrid fragmentation can be applied in a sequential or combinatorial manner. Typical combinatorial approaches perform ETD and apply CID on the undissociated charge-reduced species. Low-energy CID results in glycan fragmentation, while ETD allows cleavage of the peptide moiety. The information on both can be combined into a comprehensive data set.570 This method is relatively straightforward to apply and produces orthogonal tandem mass spectra to fully identify the glycopeptide (Figure 37). A relatively new method performs ETD and applies HCD on all ions, resulting from this process in a sequential way (EthcD). The hybrid fragmentation method generates considerably more fragment signals, which can be used for glycopeptide analysis and database matching. The requirements for a comprehensive and rapid characterization significantly differ for N- and O-glycopeptides and were recently summarized.571 Stepped-energy HCD and EthcD replaced CID in terms of popularity and are widely used for glycopeptide characterization. However, a general drawback of all ETD hybrid methods is the relatively low signal sensitivity for larger glycopeptides because they are commonly observed as lowly charged ions with high m/z values which typically produce less fragment ions with ETD.
Figure 37.

Ion-mobility-resolved parallel fragmentation of a triantennary, fully sialylated complex N-glycopeptide structure via collision-induced dissociation (CID) and electron transfer dissociation (ETD) in positive ion mode. (a) 2D plot of glycopeptide precursor intensity against m/z and drift time. Precursor activation via ETD results in charge reduction which allows separation of activated (dt2, green) and nonactivated precursor (dt1, blue) on the IMS level. (b) Tandem mass spectrum of the glycosylated species at dt1 after CID activation. (c) Tandem mass spectrum of the glycosylated species at dt2 after ETD activation. (d) Visual representation of the glycopeptide precursor and the resulting fragmentation pattern after CID (blue) and ETD (green) fragmentation. Figure reprinted with permission from ref (569). Copyright 2017 The Royal Society of Chemistry.
Complementary alternatives to collisional and electron-based techniques are photofragmentation approaches by IRMPD or UVPD. IRMPD is not very common in glycoproteomics as it is technically demanding and typically only used in ion trap instruments, for example, as a fragmentation method in FTICR. Although IRMPD yields very similar fragmentation spectra to traditional CID, it has several advantages. One major advantage of IRMPD over traditional CID is that no collision gas is required for ion activation. Therefore, there is no degradation of the vacuum inside the instrument which speeds up the analysis and minimizes ion losses during measurement.572 Furthermore, the efficiency of CID fragmentation can be highly dependent on the precursor mass, while IRMPD can be easily applied for larger precursor ions and even intact proteins.573 IRMPD was successfully applied for the analysis of N- and O-glycopeptides574,575 but is currently limited by the complex instrumental demands.
Another dissociation method employing ion–photon interaction, UVPD, recently gained attention for the analysis of glycans and glycoconjugates.32 Here, the rapid absorption of energetic photons by molecular ions leads to the population of excited electronic states, opening up new dissociation pathways that lead to highly informative fragmentation patterns. Many of the fragment ions formed are usually not accessible by conventional ion activation methods, which enables a much more detailed structural assignment. UVPD efficiently generates fragment ions from both the peptide and glycan components, enabling the simultaneous analysis of the two constituents by tandem MS. Photofragments resulting solely from peptide bond cleavages (a/x-fragments) that still carry the sugar moieties also allow for the localization of the glycosylated sites. In addition, less abundant B-, Y-, C-, and Z-ions and cross-ring cleavages provided information on the connectivity of sugar moieties.576 UVPD was successfully applied on N-glycopeptides,577 but it could be even more helpful for the characterization of O-glycosylation. O-Glycopeptides are often very acidic and generally hard to ionize in positive ionization mode, which is traditionally used for glycopeptide analysis. UVPD generates extensive fragmentation for deprotonated species and showed very good sequence coverage for both the glycan and peptide moiety, in recent studies.578 Although photodissociation via UVPD was introduced more than 30 years ago, its application was limited by the low signal-to-noise ratio of mass spectra and difficult light sources.32 However, recent technical advances will facilitate the analysis of biomolecules via UVPD, and it is expected to take an important role in the future glycoproteomics field.
7.2.3. Ion Mobility–Mass Spectrometry of Glycopeptides
In many cases glycans and glycopeptides have multiple coexisting isomers that exhibit an identical atomic composition and, as a result, an identical mass. Even with sophisticated MS/MS- and software-based approaches, it is often simply not possible to clearly assign a particular molecular structure by MS alone. A powerful technique that has the ability to compensate this shortcoming is IMS. The combination of IM-MS is a promising orthogonal tool for the analysis of biomolecules and glycoconjugates in particular.579−581 It showed great potential as a prefilter before MS detection and allows us to separate glycopeptides from nonglycosylated peptides in the gas phase. In this function it complements the enrichment/separation step to prevent overlapping of precursors and to increase identification confidence. This is especially important for O-glycosylated species as they are traditionally harder to enrich via conventional methods.582
Furthermore, IMS can be used to distinguish isomeric glycopeptides with identical peptide sequence but different glycosylation sites583 or different glycoforms584 before ion activation with ETD or CID.585,586 This combination can increase peptide sequencing coverage587 and glycan composition information.582 In addition to the orthogonal separation dimension produced by the combination of IMS and MS, the resulting drift times can be used to obtain CCS values, which can provide structural information on the glycopeptides as they represent the rotationally averaged surface area of the ions. CCS values can be used as additional identification parameters to complement retention time and mass information to increase identification confidence. Various databases for CCSs of glycopeptides have been created,414,588 and it could be feasible to predict the glycopeptide identity solely based on their CCSs in the near future, comparable with an approach for nonglycosylated peptides.589
For very similar isomers, the differences in CCS decrease with increasing glycopeptide size, and therefore IM separation becomes less efficient for larger glycoconjugates. This often complicates the differentiation of intact ions. The fragmentation of intact precursors, however, can lead to diagnostic fragments with distinct CCSs, from which information on the structure of the precursor can be deduced. Based on mono- and disaccharide fragments cleaved from glycopeptides, it is possible to identify glycan motifs independent of the peptide sequence.353 Larger fragments can also be used to identify the branching from complex N-glycopeptides which, as a result, can be used to deduce larger oligosaccharide structures.379
However, fragment CCS values not only are useful to reconstruct the structure of larger oligosaccharides but also have a great potential to be used for the rapid screening of common structural features. In many diagnostic applications, a full structural elucidation of the glycans and glycopeptides is not necessary, and it is often sufficient to identify specific glycan epitopes. Such reoccurring patterns in N- and O-glycosylation are often found in fucosylation and sialylation. Under low-energy CID conditions, glycan-only fragments are usually released from intact, cationized glycopeptides. This aspect is usually regarded as a major drawback of CID in glycoproteomics, as it precludes glycosylation site analysis; however, it turned out to be exceptionally useful for the IM-MS analysis of sialylation patterns (Figure 38). Upon CID, diagnostic antennary trisaccharide fragments, which showed characteristic drift times depending on the regiochemistry of the sialic acid linkage, were obtained. This enabled the rapid and reliable identification of the sialic acid connectivity based on fragment CCSs.590,591 A similar fragment-based approach was used for the analysis of fucosylated glycans via IM-MS; however, until now it was only applied for released glycans, and it still needs to be verified for fucosylated glycopeptide structures.374
Figure 38.
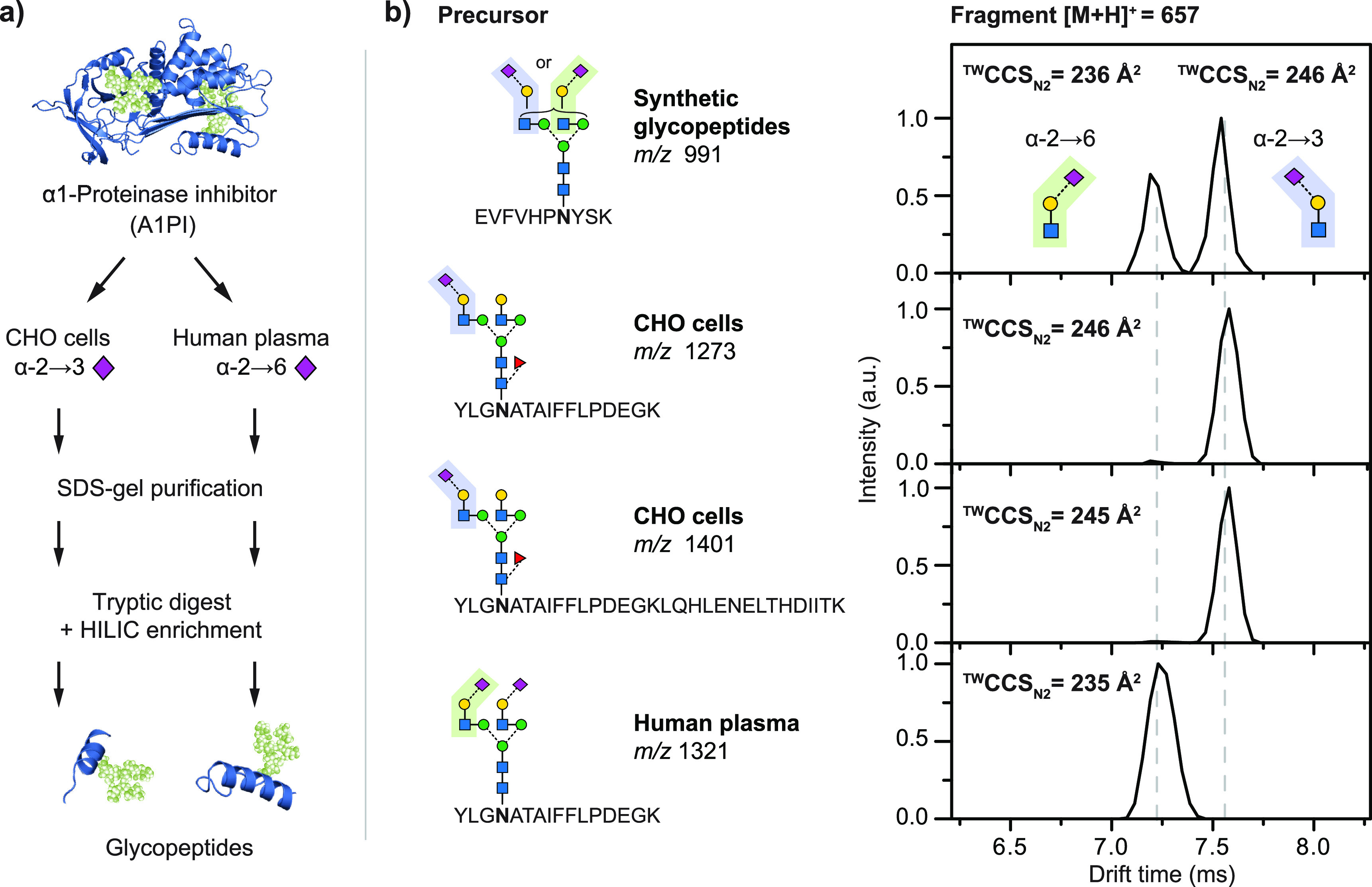
Ion mobility–mass spectrometry (IM-MS) workflow for regiochemistry analysis of N-acetylneuraminic acid (Neu5Ac) linkages in a1-proteinase inhibitor (A1PI). (A) A1PI isolated from human plasma and recombinantly expressed in Chinese hamster ovary (CHO) cells was purified and digested with trypsin, and the glycopeptides were HILIC-enriched. (B) Fragmentation of the obtained glycopeptides and subsequent IM-MS analysis of the characteristic B3-trisaccharide fragments (m/z 657) enabled the differentiation of α2,3- from α2,6-linked Neu5Ac. The observed fragment drift times and TWCCSN2 are independent of the underlying precursor sequence. Reprinted from ref (590). Published by The Royal Society of Chemistry. Copyright 2016 Hinneburg et al. (Creative Commons Attribution 3.0 Unported License).
7.2.4. Spectroscopic Approaches for Glycopeptides
In recent years, IR-based spectroscopic approaches have been introduced for the analysis for many glycan classes. In contrast to MS and IMS, which produce mass- and surface-related information on a given species, IR ion spectroscopy is able to generate a picture of internal interactions and molecular conformations. While IRMPD is regularly used as slow-heating ion activation in various tandem MS approaches for glycopeptides, IR spectroscopy of glycopeptides is often hindered by the intense signals generated by the peptide backbone. IR spectra of peptide-related species are generally dominated by the amide I, II, and III bands. The shape and intensity of the amide I/II bands are characteristic for the globular folding of peptides/proteins and can be used to deduce information on their secondary structure.592,593 The intensity of all three absorption bands is usually very high due to the large number of aligned amide oscillators inside the peptide backbone. N-Acetylated glycans, e.g., GlcNAc and GalNAc structures, are overshadowed in these regions.574 Furthermore, also other distinct differences in the vibrational signature of glycans such as connectivity and configurational isomers usually appear above 1300 cm–1.35 The most pronounced spectral difference of oligosaccharides and peptides is found in the IR fingerprint region around 1100 cm–1574 and the hydrogen stretching region (3400–3750 cm–1). The comprehensive characterization of a glycan moiety on an intact glycopeptide is highly impeded as most of the informative frequency range is overshadowed by the peptide backbone. At that moment, the only feasible approach is the chemical/enzymatic release of the glycans and subsequent IR spectroscopy.202 Similar to IMS workflows, however, a potential solution to this problem could lie in fragmentation-based approaches. Prior fragmentation via slow-heating ion activation methods such as CID could cleave the glycan residue of the peptide backbone in the gas phase. IR ion spectroscopy could then be applied on the glycan component to deduce structural and conformational information.35
Another promising alternative could be ion spectroscopy in the UV range. Peptides and proteins can be readily excited due to the presence of aromatic side chains like phenylalanine, tryptophan, or tyrosine and can be used to generate fingerprints of the electronic structures of these chromophores. Neutral, deprotonated, and radical aromatic amino acids display strong bathochromic shifts in the UV spectra which can be associated with the reactivity and biological function of the respective peptide.594,595 The application on the structural characterization of glycopeptides, however, has yet to be implemented.
8. Glycolipids
8.1. Structure and Analytical Challenges
Glycolipids are important glycoconjugates and essential components of biological membranes, originating from the glycosylation of lipids.596 They occur in living organisms ranging from bacteria to humans and fulfill numerous vital functions, which make glycolipids indispensable for the development and differentiation of multicellular organisms.597−599 The amphiphilic molecules can easily insert their hydrophobic lipid tail into cell membranes, while the glycan headgroup points outward into the extracellular medium. This configuration enables glycolipids to mediate cellular interaction and signaling processes600,601 and to modulate the function of membrane proteins.602 Furthermore, glycolipids are required to ensure the permeability barrier function of skin.603 The impairment of β-Glc ceramide catabolism in Gaucher disease results in an increased skin permeability and a concomitant risk of dehydration. Gaucher disease604 and other lysosomal storage disorders605,606 also frequently affect neural functions. This finding illustrates the importance of glycolipids for the functioning of the nerve system, e.g., for the myelination of axons.607 Furthermore, several glycolipids bearing α-glycosidic bonds were found to play an important role in innate immunity.608,609 In this context of antibody–antigen interactions, seemingly small structural details, such as the stereochemistry of the glycosidic bond, can affect the antigenic activity of glycolipids tremendously.
Glycolipids are divided into three categories according to the lipid class (Figure 39A): glycosphingolipids (GSLs), glycoglycerolipids, and glycosylphosphatidylinositols. However, the overwhelming majority of glycolipids in mammalian cells is constituted by GSLs.596 In GSLs, the glycan headgroup is attached to an amino alcohol, a so-called sphingoid base, which is mainly sphingosine in mammalian GSLs.610 In shorthand nomenclature, sphingosine is denoted as d18:1, indicating the number of hydroxyl groups (m = mono, d = di, t = tri), the total length of the hydrocarbon chain (18), and the number of C=C double bonds (1), separated by a colon. Other common sphingoid bases are phytosphingosine (t18:0) and sphinganine (d18:0).611,612 Before the attachment of a glycan to the sphingoid base, the latter is N-acetylated by a fatty acid to form a ceramide (Cer).613 Both lipid chains of the ceramide can vary in length, degree, and position of unsaturation and hydroxylation.611 Beyond the structural diversity of the individual building blocks, the structural richness of GSLs also results from the combinatorial, modular assembly of the two lipid chains and the glycan headgroup (Figure 39B).
Figure 39.

Structural diversity of glycolipids. (A) Glycolipids are classified according to the lipid core structure into three categories. (B) The most common glycolipids in mammals are glycosphingolipids, which are assembled in a modular fashion from a sphingoid base, a fatty acid, and a glycan headgroup. (C) The first monosaccharide attached to ceramides in mammals is either β-Gal or β-Glc. The latter can be elongated to yield common glycan core structures. (D) Bacterial glycoglycerolipids often contain building blocks that are uncommon for mammalian glycolipids, including α-linked monosaccharides and lipid chain branching or cyclopropane rings. The glycoglycerolipid shown here was isolated from Lactobacillus plantarum.
Even though the number of imaginable glycan structures is countless, only few glycan core structures were identified in mammalian GSLs. Typically, either β-Glc or -Gal is attached as the first monosaccharide to ceramide in mammals. Gal can be sulfated to yield so-called sulfatides but is usually not extended by further monosaccharides.596 Glc, on the other hand, is often extended to yield typical glycan structures classified into the lacto, neolacto, globo, isoglobo, and ganglio series (Figure 39C).610 Based on their acidity, GSLs are further divided into neutral and acidic GSLs. The latter are either sulfatides or bear at least one sialic acid residue. Acidic glycolipids with sialic acids are historically designated as gangliosides and are mostly expressed in the central nerve system and, in particular, the brain.614 Gangliosides are labeled according to the historical nomenclature of Svennerholm,615 which is still employed today and therefore explained in the following. Notations such as GM3 or GD1 start by the letter G (=ganglioside), followed by M, D, or T (=mono, di, or tri), designating the number of sialic acid residues, and a number referring to the relative position of the ganglioside after migration on a TLC plate.
GSLs also occur in plants, fungi, and bacteria, which often exhibit uncommon sphingolipids with chain branching, hydroxylation, and cyclopropane rings.612 Glycoglycerolipids are also much more common in plants and microbes.596 In addition to lipid modifications, bacterial glycolipids can exhibit abundant α-linked glycans, as shown for a glycoglycerolipid extracted from a Lactobacillus species (Figure 39D).616 Bacteria also present another class of macromolecules composed of a glycan and lipid part, the so-called lipopolysaccharides. However, those are not classified as glycolipids but saccharolipids617 and will not be discussed herein. Glycosylphosphatidylinositols that serve as anchors for membrane proteins and are structurally very different from GSLs and glycoglycerolipids will not be addressed either. In view of the huge diversity of glycolipids found in flora and fauna, the following sections will be restricted to the analysis of mammalian GSLs.
The structural analysis of glycolipids poses several challenges arising from (1) the amphiphilicity, i.e., opposed properties of the glycan and lipid part, (2) isomerism in the glycan headgroup, further complicated by modifications such as sulfation, and (3) heterogeneity in the lipid chains, induced by varying chain lengths, unsaturation, and degree of hydroxylation. Recent progress in glycolipid analysis can be ascribed mainly to improvements in separation, innovation of ion activation methods, chemical derivatization strategies, and novel spectroscopic techniques.
Accurate profiling of complex GSLs usually requires the application of separation techniques prior to MS analysis.618 The amphiphilic character of GSLs challenges the choice of ideal conditions for chromatographic separation and has resulted in two different modes of operation in liquid chromatography.619 Separation on polar stationary phases resolves glycan headgroups, while different ceramide moieties coelute. Reversely, ceramide moieties can be separated on reversed-phase HPLC columns to the detriment of glycan headgroup separation. Recently, an LC-MS/MS method employing a chiral LC column and multichannel–multireaction monitoring was introduced to analyze both the glycan and ceramide.620 This experimental setup allows for the simultaneous analysis of neutral GSLs, gangliosides, and sulfatides with different ceramide moieties. The chiral column is also adapted to separate diastereoisomers such as Glc- and GalCer (d18:1/18:0), which were separated by more than half a minute. Chip-based chromatographic methods also hold much potential for glycolipid profiling. Gangliosides and sulfatides were identified and quantified using MS combined with a nano-HPLC chip, which provided reproducible retention times and allowed for efficient separation of isomeric glycolipids.621 Another approach for separate glycan profiling consists of enzymatic release of glycans from glycolipids and chromatographic analysis of released glycans, e.g., by porous graphitized carbon (PGC) chromatography.622,623
8.2. Ion Activation Methods for Glycolipids
Historically, glycolipids have been analyzed by CID MS/MS with or without prior chromatographic separation.624 CID of even-electron glycolipid ions does, however, not result in diagnostic fragments that allow for the identification of structural details. Typical fragments of neutral GSLs in positive ion mode result from the loss of water, neutral loss of the fatty acid, and neutral loss of the glycan headgroup due to cleavage of the relatively weak glycosidic bond.625 A universal fragment of sphingosine-containing GSLs searched for in precursor ion scans is doubly dehydrated sphingosine at m/z 264.609 CID does not induce intrachain fragmentation and therefore yields information neither on C=C bond positions in the lipid chain nor on the stereochemistry in the glycan headgroup (Figure 41A). However, relative intensities of fragments at different collision energies can differ between isomers, even though the fragments are not unique. Such an approach thus requires calibration with standards, as was shown for α- and β-GlcCer.626 Multistage MS can reveal further information that is not available by single CID MS/MS. MSn of deprotonated ceramides in a linear ion trap allowed identification of different sphingoid bases, hydroxylation, and different isomeric structures.91
Figure 41.

Comparison of the performance of several MS-based techniques in distinguishing between the diastereoisomeric glycolipids Glc- and Gal-sphingosine (So). (A) Classical CID induces cleavage of the glycosidic bond and concomitant loss of the stereochemical information. (B/C) RDD and charge inversion induce alternative fragmentation mechanisms and yield different relative intensities of fragments. (D) SLIM IM-MS can partially separate sodiated Glc and Gal sphingosine after four passes (ca. 60 m). (E) Gas-phase IR spectroscopy is sensitive toward stereochemical changes and yields diagnostic spectroscopic fingerprints for Glc and Gal epimers.
Alternative activation methods besides CID have been applied to glycolipids even though their usage has remained restricted. For example, UVPD of glycolipids was shown to yield more diagnostic fragments than CID and HCD by inducing cross-ring cleavages in the glycan headgroup in addition to several C–N and C–C cleavages in the sphingoid base and fatty acid (Figure 40).627 Isobaric gangliosides were thus differentiated in an Orbitrap mass spectrometer by irradiation with 193 nm UV light. ExD methods including ECD, EDD, and EID were also tested for glycolipid analysis, and ECD was found to induce extensive fragmentation of the GM1 ganglioside, which enabled characterization of the glycan sequence and both lipid chains of the ceramide.628 In this respect, ECD largely outperformed other dissociation techniques typically applied to FTICR-MS, including IRMPD and EDD. EID was established as a useful technique for the analysis of small glycoconjugates including glycolipids and yielded informative fragments of the gangliosides GD1a and GD1b, contrary to CID.87 EID alone revealed only little information about the lipid tails in the ceramide but is to date the only tandem MS method allowing for the distinction of cis/trans double bond isomers in lipid chains.629
Figure 40.
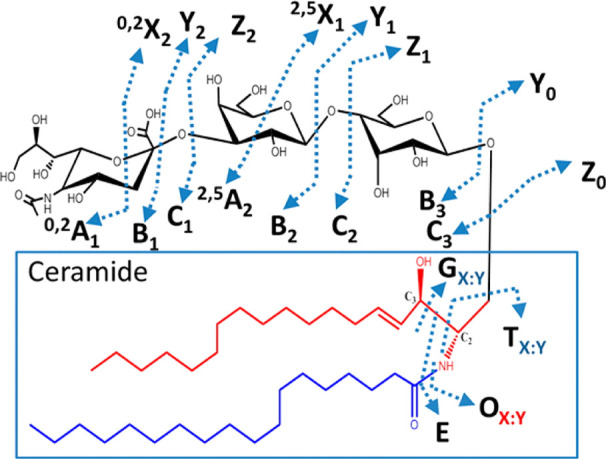
Illustration of fragment ions produced by ultraviolet photodissociation (UVPD) of GSLs. Contrary to collision-induced dissociation, UVPD induces cross-ring cleavage (A/X-ions) in addition to glycosidic bond cleavage. Furthermore, irradiation with 193 nm UV light causes several unique cleavages within the sphingoid base and the fatty acid of the ceramide tail. Figure reproduced with permission from ref (627). Copyright 2013 American Chemical Society.
8.3. Derivatization Strategies
Besides different ion activation methods, chemical derivatization strategies can help to characterize glycolipid structures in combination with classical CID fragmentation. Glycan sequences have been investigated by permethylation since the implementation of the derivatization strategy 50 years ago.630 Permethylation was recently adapted to achieve relative quantification of glycolipids by differential isotope labeling and RPLC-MS/MS. The protocol is based on a comparison of 12C-permethylated samples with 13C-permethylated internal standards.631 The need for chromatographic separation to resolve glycolipid isomers can be circumvented by other, more specialized derivatization strategies. For example, lipid and glycolipid isomers form informative fragments upon radical-directed dissociation (RDD).632−634 RDD is based on covalent or noncovalent modification of the analyte by bifunctional molecules that contain both a radical initiator and a functional group that can be attached to the analyte. Upon irradiation of the lipid ion–molecule complex, a radical is formed, which is subsequently activated by CID to induce fragmentation within the lipid moiety. The fragmentation pattern reveals the position of C=C bonds, branching, and hydroxylation in the hydrocarbon chain.632,634 RDD was furthermore employed to quantitatively distinguish between Glc and Gal headgroups by noncovalent attachment of a modified crown ether bearing a photocleavable C–I bond (Figure 41B).633 The approach is only applicable to lyso-GSLs, which lack the fatty acid and therefore exhibit a primary amine. Glc and Gal ceramides do not interact with the crown ether but can alternatively be distinguished by their reactivity toward boronic acid.633 A different approach used to distinguish between Glc and Gal ceramides is based on gas-phase ion chemistry and MS/MS.635 Deprotonated GSLs form charge-inverted complexes with terpyridine–magnesium complex dications in a linear ion trap, which are subsequently fragmented by CID. The fragment spectra allow for distinction and relative quantification not only of Glc and Gal epimers (Figure 41C) but also of alpha versus beta anomeric linkages and provide further information on double bond positions in the fatty acyl chains.636 Complementary information about the lipid residues can be obtained by ozone-induced dissociation (OzID),637 which is based on the 1,3-dipolar cycloaddition of ozone to C=C bonds, followed by cleavage of the latter. OzID was applied to pinpoint C=C bonds in both the fatty acid and the sphingoid base of unsaturated GSLs based on their differential reactivity toward ozone.638,639 A similar approach is followed by the Paternò–Büchi reaction, which is a photochemical [2 + 2] cycloaddition between a C=C bond and a carbonyl group and leads to cleavage of C=C bonds upon activation by CID.640 The technique was applied to distinguish the C=C Δ15 and Δ17 regioisomers in the fatty acid of GalCer (d18:1/24:1) directly from tissue samples by MALDI-MS/MS.641 A comprehensive summary of advanced tandem mass spectrometry strategies for full structural characterization of lipids is provided in an excellent recent review.642
8.4. Ion Mobility–Mass Spectrometry of Glycolipids
The use of IMS in glycosphingolipidomics has been limited to very few studies in the past 15 years and remained largely restricted to the separation of gangliosides. MALDI-IM-MS was first used to separate all major brain gangliosides based on the number and position of sialic acids in the glycan headgroup in a drift tube setup.643 Even the structural isomers GD1a and GD1b, which differ by the position of a single sialic acid residue, were partially resolved in a mixture. Only recently was IMS coupled to desorption (D)ESI for MS imaging of gangliosides under ambient conditions using a commercial traveling wave instrument.644 DESI-IM-MS yielded complementary information to MALDI-IM-MS and offered clear trendlines enabling the separation of gangliosides from other lipid classes. The first workflow for the analysis of gangliosides by ESI-IM-MS(/MS) was implemented at the same time for the investigation of human brain gangliosides.645 The number of gangliosides detected in human hippocampi was increased up to 10-fold compared with MS analyses without prior IM separation,646,647 and the results confirmed a correlation between the degree of ganglioside sialylation and brain development. Atmospheric pressure DTIMS in combination with an optimized drift gas composition recently allowed for an enhanced separation of GD1a and GD1b isomers compared with previous DTIMS studies.648 In some cases, isomer resolution can be increased by the addition of coordinating cations. For instance, the separation of t18:0 GSL stereoisomers was shown to increase significantly by the addition of silver(I) ions in ESI-DTIMS.239 IM separation by traveling waves in SLIM649 lifted IMS resolution of isomeric lipids and glycolipids to new levels.650,651 The isomeric gangliosides GD1a and GD1b, which were only partially resolved by DTIMS, were baseline-separated in a mixture after a path length of only 1.25 m. Neutral GSLs could, however, not be separated to a sufficient degree. Sodiated Glc and Gal sphingosine were only partially separated after 60 m path length (Figure 41D), whereas no separation was achieved for the sodiated ceramide analogues Glc and GalCer. Recently, IMS was integrated into a workflow for automated GSL glycan identification.652 The HILIC–IM–MS workflow combines glucose units, CCS, and m/z information for automated structural assignment of glycans from procainamide-labeled GSLs and allowed differentiation between breast cancer subtypes based on the GSL profiles. The integration of IMS into workflows for GSL identification, for example, as potential tumor markers,653 is expected to increase in the future.
8.5. Gas-Phase Infrared Spectroscopy of Glycolipids
Recently, the first investigation of glycolipids by cryogenic gas-phase IR spectroscopy based on an MS detection scheme demonstrated that isomeric glycan headgroups can be distinguished by unique spectroscopic fingerprints in the mid-IR regime.239 The IR spectra not only are informative of Glc/Gal epimers (Figure 41E) but also reveal the anomeric configuration, which plays a crucial role in antibody–antigen interactions during natural killer T cell activation.609 The lipid residue, too, was found to influence the spectral signature and leads to a clear distinction between GSLs and glycoglycerolipids. Most importantly, biological mixtures of naturally coexisting GSL isomers were analyzed with the help of reference spectra obtained from synthetic standards (Figure 42). Spectral deconvolution by non-negative matrix factorization provided evidence of an increase in α-GlcCer in α-glucosidase-deficient mice. Regarding the power of high-resolution gas-phase IR spectroscopy to identify a range of different isomers and determine their relative abundance in isomeric mixtures, the development of commercial benchtop systems is highly desirable for future glycolipid analysis. First applications of UVPD spectroscopy are equally to be expected in the future.
Figure 42.

Spectral deconvolution of biological glycolipid mixtures into single isomers. IR spectra of biological mixtures from α-galactosidase (1) and α-glucosidase (2) knockout mice were deconvoluted to identify contributing isomers. IR spectra of the synthetic isomers α- and β-Glc and GalCer are required for isomer assignment. The main isomer in both biological samples is β-GlcCer, but α-glucosidase deficiency also leads to a measurable increase in α-GlcCer. Figure reprinted from ref (239). Copyright 2021 Kirschbaum et al. (Creative Commons Attribution 4.0 International License).
9. Outlook
Even though no automated de novo glycan-sequencing method exists to date, significant progress has been made in the growing field of glycomics in the past decade. As outlined in this review, the analysis of mammalian glycans has profited enormously from recent developments of advanced MS-based techniques. Novel ion activation techniques exploiting ion–photon, ion–electron, and ion–ion interactions have given rise to previously inaccessible glycan fragments. Innovative derivatization strategies have not only improved detectability in MS experiments but also often led to more informative fragmentation patterns when combined with suitable dissociation methods. Hyphenation of ion mobility spectrometry to MS has enabled the postionization separation of isomeric species, and collision cross sections have been efficiently used to facilitate the identification and structural assignment of various glycans. Finally, gas-phase ion spectroscopy in the UV and IR range has provided information on the electronic and vibrational structure of mass-selected carbohydrate ions. When combined with advanced quantum chemical calculations, these analytical techniques reveal invaluable details on the underlying structure of the analytes, such as charge migration, conformational constraints, or intramolecular interactions in glycans in the gas phase.
The implementation of advanced MS-based techniques into multidimensional glycomics workflows holds great promise for the future. Democratization of MS-based technologies within the coming years is to be expected, which will render recently developed techniques accessible to the broader glycobiology community. The next decade will likely witness an increased implementation of high-energy ion activation techniques into LC-MS workflows. Advantages of novel fragmentation methods tailored specifically for the unique challenges posed by glycans or benefits stemming from increased resolution of ion mobility separations are evident. The commercialization of action spectroscopy would open a vast and largely unexplored field to nonexperts. Similarly to the situation of UVPD a couple of years ago, the key to this will lie in the commercial availability of affordable, easy-to-use light sources. Tunable benchtop laser systems are currently developing at a rapid pace, and significant progress can be expected in the near future. In addition, ion soft landing in combination with single-molecule surface analysis techniques has just demonstrated its immense potential for glycan analysis and awaits further exploration.654,655 Although several challenges are still waiting to be tackled, such as the integration of action spectroscopy into fast LC-MS methods, the importance of basic research to provide innovative solutions has become clearly apparent. In particular, the targeted development of techniques that consider the structure of carbohydrates and the unique challenges arising from them—instead of adopting methods from the MS-based proteomics toolbox—needs to be promoted.
Acknowledgments
The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (SFB 1340 and FOR 2177), the Fonds der Chemischen Industrie, the Fonds National de la Recherche, Luxembourg (GlycoCat, 13549747), the European Union’s Horizon 2020 Research and Innovation Program (HS-SEQ, 899687), and the European Research Council (ERC-2019-CoG-863934-GlycoSpec). We would also like to thank Prof. Gert von Helden and Prof. Gerard Meijer for the long-standing fruitful collaboration and their continuous support.
Glossary
ABBREVIATIONS
- MS
mass spectrometry
- IMS
ion mobility spectrometry
- DTIMS
drift tube ion mobility spectrometry
- FAIMS
field asymmetric ion mobility spectrometry
- TIMS
trapped ion mobility spectrometry
- TWIMS
traveling wave ion mobility spectrometry
- IM-MS
ion mobility–mass spectrometry
- CID
collision-induced dissociation
- HCD
higher-energy collisional dissociation
- ExD
electron-based or electron-mediated dissociation
- ECD
electron capture dissociation
- EDD
electron detachment dissociation
- EED
electronic excitation dissociation
- EID
electron-induced dissociation
- ETD
electron transfer dissociation
- ETnoD
electron transfer with no dissociation
- NETD
negative electron transfer dissociation
- CTD
charge-transfer dissociation
- RDD
radical-directed dissociation
- EPD
electron photodetachment
- a-EPD
activated electron photodetachment
- IRMPD
infrared multiple photon dissociation
- UVPD
ultraviolet photodissociation
- DFT
density functional theory
- MD
molecular dynamics
Biographies
Márkó Grabarics is a Ph.D. student at the Freie Universität Berlin and guest researcher at the Fritz Haber Institute of the Max Planck Society. He completed his M.Sc. at Semmelweis University in Budapest, studying human milk oligosaccharides under the supervision of Dr. Szabolcs Béni. Following a brief encounter with inductively coupled plasma mass spectrometry and metal-based anticancer agents in the lab of Prof. Gunda Köllensperger, Márkó joined the group of Prof. Kevin Pagel in 2017. His research is focused on the application of ion mobility–mass spectrometry and gas-phase ion spectroscopy for the structural characterization of glycosaminoglycans, as well as on developing theoretical models to address resolution and peak capacity in ion mobility separations.
Maike Lettow is a Ph.D. candidate in the department of Molecular Physics at the Fritz Haber Institute in Berlin. From 2013 to 2018, she studied chemistry at Freie Universität in Berlin. During her bachelor thesis at the Max Planck Institute of Colloids and Interfaces in Potsdam, she worked on the synthesis of glycans, and for her master thesis, she used this synthetic chemical background to analyze the gas-phase structures of glycans in the laboratories of Prof. Kevin Pagel and Prof. Gert von Helden. Her research interest centers around the application of different types of infrared action spectroscopy and ion mobility spectrometry. She showed that fucosylated glycan ions can undergo a gas-phase rearrangement reaction, termed fucose migration, as intact ions.
Carla Kirschbaum is a Ph.D. student at Freie Universität Berlin and guest scientist at the Fritz Haber Institute of the Max Planck Society. From 2014 to 2019, she studied chemistry at Freie Universität Berlin and École Normale Supérieure de Paris, supported by the German Academic Scholarship Foundation. In October 2019, Carla started her Ph.D. thesis in the Pagel group, funded by the German chemical industry association VCI. Her research is focused on lipid and glycolipid analysis using innovative mass spectrometry-based techniques such as ion mobility–mass spectrometry and cryogenic gas-phase infrared spectroscopy.
Kim Greis studied chemistry at Humboldt-Universität zu Berlin and University of Melbourne. Since November 2019, he has been a Ph.D. candidate, funded by the Luxembourg National Research Fund, at Freie Universität Berlin and Fritz Haber Institute in the Pagel group. He is interested in characterizing reactive intermediates, such as the glycosyl cation, using cryogenic gas-phase infrared spectroscopy in helium nanodroplets and computational chemistry. As a Fulbright Scholar, he will join the group of Prof. Mark Johnson at Yale University in 2022 to deepen his knowledge about mass spectrometry-based techniques.
Christian Manz is a Ph.D. candidate at Freie Universität Berlin and a guest scientist at the Fritz Haber Institute of the Max Planck Society. He studied chemistry at the Technical University of Darmstadt and Freie Universität Berlin before starting his Ph.D. thesis in 2017 in the Pagel group. He is interested in the analysis of complex N-glycans via liquid chromatography and mass spectrometry. In particular, he focuses on the identification of important structural motifs, such as sialylation and fucosylation patterns, by implementing ion mobility spectrometry into the standard workflow. For this purpose, he also develops novel tools to facilitate LC and IMS calibration to speed up data acquisition and data analysis.
Kevin Pagel is Full Professor for Bioorganic Chemistry at the Institute of Chemistry and Biochemistry of the Freie Universität Berlin and guest researcher at the Fritz Haber Institute of the Max Planck Society. He earned a diploma in Organic Chemistry from the University of Leipzig in 2003 and a Ph.D. from the Freie Universität Berlin in 2007. From 2008 to 2011 he pursued postdoctoral research with Prof. Dame Carol V. Robinson at the University of Cambridge and later the University of Oxford. Kevin and his group received several recognitions including the Mattauch Herzog Award of the German Mass Spectrometry Society (2016), the Ron Hites Award of the American Society for Mass Spectrometry (2016), the Award of the Division of Analytical Chemistry of the German Chemical Society (2017), and a Consolidator Grant from the European Research Council (2019). Research in the Pagel group is focused on the structural analysis of biological macromolecules, and in particular glycans, using ion mobility–mass spectrometry and gas-phase IR spectroscopy techniques.
Open access funded by Max Planck Society.
The authors declare no competing financial interest.
References
- Varki A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm D.; Heublein B.; Fink H.-P.; Bohn A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem., Int. Ed. 2005, 44, 3358–3393. 10.1002/anie.200460587. [DOI] [PubMed] [Google Scholar]
- Hart G. W.; Copeland R. J. Glycomics Hits the Big Time. Cell 2010, 143, 672–676. 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniappan K. K.; Bertozzi C. R. Chemical Glycoproteomics. Chem. Rev. 2016, 116, 14277–14306. 10.1021/acs.chemrev.6b00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrescu A.-J.; Wormald M. R.; Dwek R. A. Structural Aspects of Glycomes with a Focus on N-Glycosylation and Glycoprotein Folding. Curr. Opin. Struct. Biol. 2006, 16, 600–607. 10.1016/j.sbi.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Shental-Bechor D.; Levy Y. Folding of Glycoproteins: Toward Understanding the Biophysics of the Glycosylation Code. Curr. Opin. Struct. Biol. 2009, 19, 524–533. 10.1016/j.sbi.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Rudd P. M.; Elliott T.; Cresswell P.; Wilson I. A.; Dwek R. A. Glycosylation and the Immune System. Science 2001, 291, 2370. 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- Marth J. D.; Grewal P. K. Mammalian Glycosylation in Immunity. Nat. Rev. Immunol. 2008, 8, 874–887. 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell W. J.; White J. M. The Molecules of Mammalian Fertilization. Cell 1996, 85, 629–637. 10.1016/S0092-8674(00)81230-1. [DOI] [PubMed] [Google Scholar]
- Tulsiani D. R. P.; Yoshida-Komiya H.; Araki Y. Mammalian Fertilization: A Carbohydrate-Mediated Event. Biol. Reprod. 1997, 57, 487–494. 10.1095/biolreprod57.3.487. [DOI] [PubMed] [Google Scholar]
- Wassarman P. M.; Jovine L.; Litscher E. S. A Profile of Fertilization in Mammals. Nat. Cell Biol. 2001, 3, E59–E64. 10.1038/35055178. [DOI] [PubMed] [Google Scholar]
- Bode L. Human Milk Oligosaccharides: Every Baby Needs a Sugar Mama. Glycobiology 2012, 22, 1147–1162. 10.1093/glycob/cws074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop J. R.; Schuksz M.; Esko J. D. Heparan Sulphate Proteoglycans Fine-Tune Mammalian Physiology. Nature 2007, 446, 1030–1037. 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Karamanos N. K.; Piperigkou Z.; Theocharis A. D.; Watanabe H.; Franchi M.; Baud S.; Brézillon S.; Götte M.; Passi A.; Vigetti D.; et al. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem. Rev. 2018, 118, 9152–9232. 10.1021/acs.chemrev.8b00354. [DOI] [PubMed] [Google Scholar]
- Ajit Varki R. D. C.; Esko J. D.; Stanley P.; Hart G. W; Aebi M.; Darvill A. G.; Kinoshita T.; Packer N. H.; Prestegard J. H.; Schnaar R. L.; Seeberger P. H.. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press, 2017. [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass Spectrometry-Based Proteomics. Nature 2003, 422, 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Benesch J. L. P.; Ruotolo B. T.; Simmons D. A.; Robinson C. V. Protein Complexes in the Gas Phase: Technology for Structural Genomics and Proteomics. Chem. Rev. 2007, 107, 3544–3567. 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- Bensimon A.; Heck A. J. R.; Aebersold R. Mass Spectrometry-Based Proteomics and Network Biology. Annu. Rev. Biochem. 2012, 81, 379–405. 10.1146/annurev-biochem-072909-100424. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M.-C.; Yates J. R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass-Spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537, 347–355. 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Zaia J. Mass Spectrometry and the Emerging Field of Glycomics. Chem. Biol. 2008, 15, 881–892. 10.1016/j.chembiol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelle W.; Michalski J.-C. Analysis of Protein Glycosylation by Mass Spectrometry. Nat. Protoc. 2007, 2, 1585–1602. 10.1038/nprot.2007.227. [DOI] [PubMed] [Google Scholar]
- Ruhaak L. R.; Xu G.; Li Q.; Goonatilleke E.; Lebrilla C. B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118, 7886–7930. 10.1021/acs.chemrev.7b00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan N.; Yang S.; Cipollo J.; Wuhrer M. Glycomics Studies Using Sialic Acid Derivatization and Mass Spectrometry. Nat. Rev. Chem. 2020, 4, 229–242. 10.1038/s41570-020-0174-3. [DOI] [PubMed] [Google Scholar]
- Wuhrer M.; de Boer A. R.; Deelder A. M. Structural Glycomics Using Hydrophilic Interaction Chromatography (Hilic) with Mass Spectrometry. Mass Spectrom. Rev. 2009, 28, 192–206. 10.1002/mas.20195. [DOI] [PubMed] [Google Scholar]
- Mariño K.; Bones J.; Kattla J. J.; Rudd P. M. A Systematic Approach to Protein Glycosylation Analysis: A Path through the Maze. Nat. Chem. Biol. 2010, 6, 713–723. 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- Alley W. R.; Novotny M. V. Structural Glycomic Analyses at High Sensitivity: A Decade of Progress. Annu. Rev. Anal. Chem. 2013, 6, 237–265. 10.1146/annurev-anchem-062012-092609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Crihfield C. L.; Gattu S.; Veltri L. M.; Holland L. A. Capillary Electrophoresis Separations of Glycans. Chem. Rev. 2018, 118, 7867–7885. 10.1021/acs.chemrev.7b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray C. J.; Migas L. G.; Barran P. E.; Pagel K.; Seeberger P. H.; Eyers C. E.; Boons G.-J.; Pohl N. L. B.; Compagnon I.; Widmalm G.; et al. Advancing Solutions to the Carbohydrate Sequencing Challenge. J. Am. Chem. Soc. 2019, 141, 14463–14479. 10.1021/jacs.9b06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley W. R.; Mann B. F.; Novotny M. V. High-Sensitivity Analytical Approaches for the Structural Characterization of Glycoproteins. Chem. Rev. 2013, 113, 2668–2732. 10.1021/cr3003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia M. J.; Ruhaak L. R.; Lebrilla C. B.; Amster I. J. Oligosaccharide Analysis by Mass Spectrometry: A Review of Recent Developments. Anal. Chem. 2014, 86, 196–212. 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S.; Morrison L. J.; Santos I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120, 3328–3380. 10.1021/acs.chemrev.9b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamrath M. Z.; Rizzo T. R. Combining Ion Mobility and Cryogenic Spectroscopy for Structural and Analytical Studies of Biomolecular Ions. Acc. Chem. Res. 2018, 51, 1487–1495. 10.1021/acs.accounts.8b00133. [DOI] [PubMed] [Google Scholar]
- Manz C.; Pagel K. Glycan Analysis by Ion Mobility-Mass Spectrometry and Gas-Phase Spectroscopy. Curr. Opin. Chem. Biol. 2018, 42, 16–24. 10.1016/j.cbpa.2017.10.021. [DOI] [PubMed] [Google Scholar]
- Mucha E.; Stuckmann A.; Marianski M.; Struwe W. B.; Meijer G.; Pagel K. In-Depth Structural Analysis of Glycans in the Gas Phase. Chemical Science 2019, 10, 1272–1284. 10.1039/C8SC05426F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray C. J.; Compagnon I.; Flitsch S. L. Mass Spectrometry Hybridized with Gas-Phase Infrared Spectroscopy for Glycan Sequencing. Curr. Opin. Struct. Biol. 2020, 62, 121–131. 10.1016/j.sbi.2019.12.014. [DOI] [PubMed] [Google Scholar]
- Ahn Y. H.; Kim J. Y.; Yoo J. S. Quantitative Mass Spectrometric Analysis of Glycoproteins Combined with Enrichment Methods. Mass Spectrom. Rev. 2015, 34, 148–165. 10.1002/mas.21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Bertozzi C. R.; Pitteri S. J. A Pragmatic Guide to Enrichment Strategies for Mass Spectrometry-Based Glycoproteomics. Molecular & Cellular Proteomics 2021, 20, 100029. 10.1074/mcp.R120.002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M.; Novotny M. V.; Mechref Y. Sequential Enrichment of Sulfated Glycans by Strong Anion-Exchange Chromatography Prior to Mass Spectrometric Measurements. J. Am. Soc. Mass Spectrom. 2010, 21, 348–357. 10.1016/j.jasms.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Zauner G.; Deelder A. M.; Wuhrer M. Recent Advances in Hydrophilic Interaction Liquid Chromatography (Hilic) for Structural Glycomics. Electrophoresis 2011, 32, 3456–3466. 10.1002/elps.201100247. [DOI] [PubMed] [Google Scholar]
- Pabst M.; Altmann F. Glycan Analysis by Modern Instrumental Methods. Proteomics 2011, 11, 631–643. 10.1002/pmic.201000517. [DOI] [PubMed] [Google Scholar]
- Veillon L.; Huang Y.; Peng W.; Dong X.; Cho B. G.; Mechref Y. Characterization of Isomeric Glycan Structures by Lc-Ms/Ms. Electrophoresis 2017, 38, 2100–2114. 10.1002/elps.201700042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreeker G. C.; Wuhrer M. Reversed-Phase Separation Methods for Glycan Analysis. Anal. Bioanal. Chem. 2017, 409, 359–378. 10.1007/s00216-016-0073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Zhang H. Solid-Phase Glycan Isolation for Glycomics Analysis. Proteomics: Clin. Appl. 2012, 6, 596–608. 10.1002/prca.201200045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak L. R.; Zauner G.; Huhn C.; Bruggink C.; Deelder A. M.; Wuhrer M. Glycan Labeling Strategies and Their Use in Identification and Quantification. Anal. Bioanal. Chem. 2010, 397, 3457–3481. 10.1007/s00216-010-3532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.; Veillon L.; Dong X.; Huang Y.; Mechref Y. Direct Comparison of Derivatization Strategies for Lc-Ms/Ms Analysis of N-Glycans. Analyst 2017, 142, 4446–4455. 10.1039/C7AN01262D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J. Derivatization of Carbohydrates for Analysis by Chromatography; Electrophoresis and Mass Spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2011, 879, 1196–1225. 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Karas M.; Hillenkamp F. Laser Desorption Ionization of Proteins with Molecular Masses Exceeding 10,000 Da. Anal. Chem. 1988, 60, 2299–2301. 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- Fenn J.; Mann M.; Meng C.; Wong S.; Whitehouse C. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246, 64–71. 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Fenn J. B.; Mann M.; Meng C. K.; Wong S. F.; Whitehouse C. M. Electrospray Ionization-Principles and Practice. Mass Spectrom. Rev. 1990, 9, 37–70. 10.1002/mas.1280090103. [DOI] [Google Scholar]
- Harvey D. J. Analysis of Carbohydrates and Glycoconjugates by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry: An Update for 2013–2014. Mass Spectrom. Rev. 2018, 37, 353–491. 10.1002/mas.21530. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Analysis of Carbohydrates and Glycoconjugates by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry: An Update for 2015–2016. Mass Spectrom. Rev. 2021, 40, 408–565. 10.1002/mas.21651. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Ahadi E.; Rodriguez A. D.; Vahidi S. Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85, 2–9. 10.1021/ac302789c. [DOI] [PubMed] [Google Scholar]
- Domon B.; Costello C. E. A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms/Ms Spectra of Glycoconjugates. Glycoconjugate J. 1988, 5, 397–409. 10.1007/BF01049915. [DOI] [Google Scholar]
- Mucha E.; Marianski M.; Xu F.-F.; Thomas D. A.; Meijer G.; von Helden G.; Seeberger P. H.; Pagel K. Unravelling the Structure of Glycosyl Cations Via Cold-Ion Infrared Spectroscopy. Nat. Commun. 2018, 9, 4174. 10.1038/s41467-018-06764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marianski M.; Mucha E.; Greis K.; Moon S.; Pardo A.; Kirschbaum C.; Thomas D. A.; Meijer G.; von Helden G.; Gilmore K.; et al. Remote Participation During Glycosylation Reactions of Galactose Building Blocks: Direct Evidence from Cryogenic Vibrational Spectroscopy. Angew. Chem., Int. Ed. 2020, 59, 6166–6171. 10.1002/anie.201916245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler B.; Barnes L.; Renois G.; Gray C.; Chambert S.; Fort S.; Flitsch S.; Loison C.; Allouche A. R.; Compagnon I. Anomeric Memory of the Glycosidic Bond Upon Fragmentation and Its Consequences for Carbohydrate Sequencing. Nat. Commun. 2017, 8, 973. 10.1038/s41467-017-01179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray C. J.; Schindler B.; Migas L. G.; Picmanova M.; Allouche A. R.; Green A. P.; Mandal S.; Motawia M. S.; Sanchez-Perez R.; Bjarnholt N.; et al. Bottom-up Elucidation of Glycosidic Bond Stereochemistry. Anal. Chem. 2017, 89, 4540–4549. 10.1021/acs.analchem.6b04998. [DOI] [PubMed] [Google Scholar]
- Pellegrinelli R. P.; Yue L.; Carrascosa E.; Warnke S.; Ben Faleh A.; Rizzo T. R. How General Is Anomeric Retention During Collision-Induced Dissociation of Glycans?. J. Am. Chem. Soc. 2020, 142, 5948–5951. 10.1021/jacs.0c00264. [DOI] [PubMed] [Google Scholar]
- Mookherjee A.; Uppal S. S.; Murphree T. A.; Guttman M. Linkage Memory in Underivatized Protonated Carbohydrates. J. Am. Soc. Mass Spectrom. 2021, 32, 581–589. 10.1021/jasms.0c00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollivier S.; Tarquis L.; Fanuel M.; Li A.; Durand J.; Laville E.; Potocki-Veronese G.; Ropartz D.; Rogniaux H. Anomeric Retention of Carbohydrates in Multistage Cyclic Ion Mobility (Imsn): De Novo Structural Elucidation of Enzymatically Produced Mannosides. Anal. Chem. 2021, 93, 6254. 10.1021/acs.analchem.1c00673. [DOI] [PubMed] [Google Scholar]
- Greis K.; Mucha E.; Lettow M.; Thomas D. A.; Kirschbaum C.; Moon S.; Pardo-Vargas A.; von Helden G.; Meijer G.; Gilmore K.; et al. The Impact of Leaving Group Anomericity on the Structure of Glycosyl Cations of Protected Galactosides. ChemPhysChem 2020, 21, 1905–1907. 10.1002/cphc.202000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachet R. W.; Bishop B. M.; Erickson B. W.; Glish G. L. Novel Peptide Dissociation: Gas-Phase Intramolecular Rearrangement of Internal Amino Acid Residues. J. Am. Chem. Soc. 1997, 119, 5481–5488. 10.1021/ja9640758. [DOI] [Google Scholar]
- McNeil M. Elimination of Internal Glycosyl Residues During Chemical Ionization-Mass Spectrometry of Per-O-Alkylated Oligosaccharide-Alditols. Carbohydr. Res. 1983, 123, 31–40. 10.1016/0008-6215(83)88378-5. [DOI] [Google Scholar]
- Wuhrer M.; Deelder A. M.; van der Burgt Y. E.M. Mass Spectrometric Glycan Rearrangements. Mass Spectrom. Rev. 2011, 30, 664–680. 10.1002/mas.20337. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Mattu T. S.; Wormald M. R.; Royle L.; Dwek R. A.; Rudd P. M. Internal Residue Loss“: Rearrangements Occurring During the Fragmentation of Carbohydrates Derivatized at the Reducing Terminus. Anal. Chem. 2002, 74, 734–740. 10.1021/ac0109321. [DOI] [PubMed] [Google Scholar]
- Sleno L.; Volmer D. A. Ion Activation Methods for Tandem Mass Spectrometry. J. Mass Spectrom. 2004, 39, 1091–1112. 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- Bayat P.; Lesage D.; Cole R. B. Tutorial: Ion Activation in Tandem Mass Spectrometry Using Ultra-High Resolution Instrumentation. Mass Spectrom. Rev. 2020, 39, 680–702. 10.1002/mas.21623. [DOI] [PubMed] [Google Scholar]
- Han L.; Costello C. E. Mass Spectrometry of Glycans. Biochemistry (Moscow) 2013, 78, 710–720. 10.1134/S0006297913070031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey S. A. Principles of Collisional Activation in Analytical Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1992, 3, 599–614. 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- McLuckey S. A.; Goeringer D. E. Special Feature:Tutorial Slow Heating Methods in Tandem Mass Spectrometry. J. Mass Spectrom. 1997, 32, 461–474. . [DOI] [Google Scholar]
- Olsen J. V.; Macek B.; Lange O.; Makarov A.; Horning S.; Mann M. Higher-Energy C-Trap Dissociation for Peptide Modification Analysis. Nat. Methods 2007, 4, 709–712. 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- Cao L.; Tolic N.; Qu Y.; Meng D.; Zhao R.; Zhang Q.; Moore R. J.; Zink E. M.; Lipton M. S.; Pasa-Tolic L.; et al. Characterization of Intact N- and O-Linked Glycopeptides Using Higher Energy Collisional Dissociation. Anal. Biochem. 2014, 452, 96–102. 10.1016/j.ab.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodin R. L.; Bomse D. S.; Beauchamp J. L. Multiphoton Dissociation of Molecules with Low Power Continuous Wave Infrared Laser Radiation. J. Am. Chem. Soc. 1978, 100, 3248–3250. 10.1021/ja00478a065. [DOI] [Google Scholar]
- Ly T.; Julian R. R. Ultraviolet Photodissociation: Developments Towards Applications for Mass-Spectrometry-Based Proteomics. Angew. Chem., Int. Ed. 2009, 48, 7130–7137. 10.1002/anie.200900613. [DOI] [PubMed] [Google Scholar]
- Ko B. J.; Brodbelt J. S. 193 Nm Ultraviolet Photodissociation of Deprotonated Sialylated Oligosaccharides. Anal. Chem. 2011, 83, 8192–8200. 10.1021/ac201751u. [DOI] [PubMed] [Google Scholar]
- Antoine R.; Lemoine J.; Dugourd P. Electron Photodetachment Dissociation for Structural Characterization of Synthetic and Bio-Polymer Anions. Mass Spectrom. Rev. 2014, 33, 501–522. 10.1002/mas.21402. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Volmer D. A. Electron-Based Fragmentation Methods in Mass Spectrometry: An Overview. Mass Spectrom. Rev. 2017, 36, 4–15. 10.1002/mas.21482. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc. 1998, 120, 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Syka J. E.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik B. A.; Haselmann K. F.; Zubarev R. A. Electron Detachment Dissociation of Peptide Di-Anions: An Electron-Hole Recombination Phenomenon. Chem. Phys. Lett. 2001, 342, 299–302. 10.1016/S0009-2614(01)00501-2. [DOI] [Google Scholar]
- Cody R. B.; Freiser B. S. Electron Impact Excitation of Ions from Organics: An Alternative to Collision Induced Dissociation. Anal. Chem. 1979, 51, 547–551. 10.1021/ac50040a022. [DOI] [Google Scholar]
- Mirgorodskaya E.; Roepstorff P.; Zubarev R. A. Localization of O-Glycosylation Sites in Peptides by Electron Capture Dissociation in a Fourier Transform Mass Spectrometer. Anal. Chem. 1999, 71, 4431–4436. 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- Han L.; Costello C. E. Electron Transfer Dissociation of Milk Oligosaccharides. J. Am. Soc. Mass Spectrom. 2011, 22, 997–1013. 10.1007/s13361-011-0117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson J. T.; Håkansson K. Electron Capture Dissociation of Oligosaccharides Ionized with Alkali, Alkaline Earth, and Transition Metals. Anal. Chem. 2007, 79, 2901–2910. 10.1021/ac0621423. [DOI] [PubMed] [Google Scholar]
- Wolff J. J.; Amster I. J.; Chi L.; Linhardt R. J. Electron Detachment Dissociation of Glycosaminoglycan Tetrasaccharides. J. Am. Soc. Mass Spectrom. 2007, 18, 234–244. 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Y. L. E.; Chen X.; Wu R.; Hung Y. L. W.; Chan T. D. Structural Characterization of Intact Glycoconjugates by Tandem Mass Spectrometry Using Electron-Induced Dissociation. Anal. Chem. 2017, 89, 10111–10117. 10.1021/acs.analchem.7b03128. [DOI] [PubMed] [Google Scholar]
- Yu X.; Jiang Y.; Chen Y.; Huang Y.; Costello C. E.; Lin C. Detailed Glycan Structural Characterization by Electronic Excitation Dissociation. Anal. Chem. 2013, 85, 10017–10021. 10.1021/ac402886q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J. J.; Leach F. E.; Laremore T. N.; Kaplan D. A.; Easterling M. L.; Linhardt R. J.; Amster I. J. Negative Electron Transfer Dissociation of Glycosaminoglycans. Anal. Chem. 2010, 82, 3460–3466. 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Wei J.; Chopra P.; Boons G.-J.; Lin C.; Zaia J. Sequencing Heparan Sulfate Using Hilic Lc-Netd-Ms/Ms. Anal. Chem. 2019, 91, 11738–11746. 10.1021/acs.analchem.9b02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F. F. Complete Structural Characterization of Ceramides as [M-H](−) Ions by Multiple-Stage Linear Ion Trap Mass Spectrometry. Biochimie 2016, 130, 63–75. 10.1016/j.biochi.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Zhang J.; Dong J.; Hou M.; Pan W.; Bu D.; Zhou J.; Zhang Q.; Wang Y.; Zhao K.; et al. Identification of Glycan Branching Patterns Using Multistage Mass Spectrometry with Spectra Tree Analysis. J. Proteomics 2020, 217, 103649. 10.1016/j.jprot.2020.103649. [DOI] [PubMed] [Google Scholar]
- Klein D. R.; Holden D. D.; Brodbelt J. S. Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2016, 88, 1044–1051. 10.1021/acs.analchem.5b04218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J. Collision-Induced Fragmentation of Underivatized N-Linked Carbohydrates Ionized by Electrospray. J. Mass Spectrom. 2000, 35, 1178–1190. . [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Negative Ion Mass Spectrometry for the Analysis of N-Linked Glycans. Mass Spectrom. Rev. 2020, 39, 586–679. 10.1002/mas.21622. [DOI] [PubMed] [Google Scholar]
- Cancilla M. T.; Penn S. G.; Carroll J. A.; Lebrilla C. B. Coordination of Alkali Metals to Oligosaccharides Dictates Fragmentation Behavior in Matrix Assisted Laser Desorption Ionization/Fourier Transform Mass Spectrometry. J. Am. Chem. Soc. 1996, 118, 6736–6745. 10.1021/ja9603766. [DOI] [Google Scholar]
- Ewing R. G.; Atkinson D. A.; Eiceman G. A.; Ewing G. J. A Critical Review of Ion Mobility Spectrometry for the Detection of Explosives and Explosive Related Compounds. Talanta 2001, 54, 515–529. 10.1016/S0039-9140(00)00565-8. [DOI] [PubMed] [Google Scholar]
- Borsdorf H.; Eiceman G. A. Ion Mobility Spectrometry: Principles and Applications. Appl. Spectrosc. Rev. 2006, 41, 323–375. 10.1080/05704920600663469. [DOI] [Google Scholar]
- Eiceman G. A.; Karpas Z.; Hill H. H. J.. Ion Mobility Spectrometry, 3rd ed.; CRC Press, 2013. [Google Scholar]
- McDaniel E. W.; Martin D. W.; Barnes W. S. Drift Tube-Mass Spectrometer for Studies of Low-Energy Ion–Molecule Reactions. Rev. Sci. Instrum. 1962, 33, 2–7. 10.1063/1.1717656. [DOI] [Google Scholar]
- Mason E. A.; McDaniel E. W.. Transport Properties of Ions in Gases, 1st ed.; Wiley: New York, 1988. [Google Scholar]
- von Helden G.; Hsu M. T.; Kemper P. R.; Bowers M. T. Structures of Carbon Cluster Ions from 3 to 60 Atoms: Linears to Rings to Fullerenes. J. Chem. Phys. 1991, 95, 3835–3837. 10.1063/1.460783. [DOI] [Google Scholar]
- Bowers M. T.; Kemper P. R.; von Helden G.; van Koppen P. A. M. Gas-Phase Ion Chromatography: Transition Metal State Selection and Carbon Cluster Formation. Science 1993, 260, 1446–1451. 10.1126/science.260.5113.1446. [DOI] [PubMed] [Google Scholar]
- Clemmer D. E.; Hudgins R. R.; Jarrold M. F. Naked Protein Conformations: Cytochrome C in the Gas Phase. J. Am. Chem. Soc. 1995, 117, 10141–10142. 10.1021/ja00145a037. [DOI] [Google Scholar]
- Jarrold M. F. Peptides and Proteins in the Vapor Phase. Annu. Rev. Phys. Chem. 2000, 51, 179–207. 10.1146/annurev.physchem.51.1.179. [DOI] [PubMed] [Google Scholar]
- Ruotolo B. T.; Giles K.; Campuzano I.; Sandercock A. M.; Bateman R. H.; Robinson C. V. Evidence for Macromolecular Protein Rings in the Absence of Bulk Water. Science 2005, 310, 1658–1661. 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- Ruotolo B. T.; Benesch J. L. P.; Sandercock A. M.; Hyung S.-J.; Robinson C. V. Ion Mobility-Mass Spectrometry Analysis of Large Protein Complexes. Nat. Protoc. 2008, 3, 1139. 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- Bleiholder C.; Dupuis N. F.; Wyttenbach T.; Bowers M. T. Ion Mobility-Mass Spectrometry Reveals a Conformational Conversion from Random Assembly to B-Sheet in Amyloid Fibril Formation. Nat. Chem. 2011, 3, 172–177. 10.1038/nchem.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C.; Barbu I. M.; Shoemaker G. K.; van Duijn E.; Heck A. J. R. Interrogating Viral Capsid Assembly with Ion Mobility-Mass Spectrometry. Nat. Chem. 2011, 3, 126–132. 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- Warnke S.; von Helden G.; Pagel K. Protein Structure in the Gas Phase: The Influence of Side-Chain Microsolvation. J. Am. Chem. Soc. 2013, 135, 1177–1180. 10.1021/ja308528d. [DOI] [PubMed] [Google Scholar]
- Abi-Ghanem J.; Gabelica V. Nucleic Acid Ion Structures in the Gas Phase. Phys. Chem. Chem. Phys. 2014, 16, 21204–21218. 10.1039/C4CP02362E. [DOI] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Allison T. M.; Ulmschneider M. B.; Degiacomi M. T.; Baldwin A. J.; Robinson C. V. Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510, 172. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servage K. A.; Silveira J. A.; Fort K. L.; Russell D. H. Cryogenic Ion Mobility-Mass Spectrometry: Tracking Ion Structure from Solution to the Gas Phase. Acc. Chem. Res. 2016, 49, 1421–1428. 10.1021/acs.accounts.6b00177. [DOI] [PubMed] [Google Scholar]
- Hochberg G. K. A.; Shepherd D. A.; Marklund E. G.; Santhanagoplan I.; Degiacomi M. T.; Laganowsky A.; Allison T. M.; Basha E.; Marty M. T.; Galpin M. R.; et al. Structural Principles That Enable Oligomeric Small Heat-Shock Protein Paralogs to Evolve Distinct Functions. Science 2018, 359, 930. 10.1126/science.aam7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedair M.; Sumner L. W. Current and Emerging Mass-Spectrometry Technologies for Metabolomics. TrAC, Trends Anal. Chem. 2008, 27, 238–250. 10.1016/j.trac.2008.01.006. [DOI] [Google Scholar]
- Helm D.; Vissers J. P. C.; Hughes C. J.; Hahne H.; Ruprecht B.; Pachl F.; Grzyb A.; Richardson K.; Wildgoose J.; Maier S. K.; et al. Ion Mobility Tandem Mass Spectrometry Enhances Performance of Bottom-up Proteomics. Molecular & Cellular Proteomics 2014, 13, 3709–3715. 10.1074/mcp.M114.041038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May J. C.; Goodwin C. R.; McLean J. A. Ion Mobility-Mass Spectrometry Strategies for Untargeted Systems, Synthetic, and Chemical Biology. Curr. Opin. Biotechnol. 2015, 31, 117–121. 10.1016/j.copbio.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglia G.; Angel P.; Williams J. P.; Richardson K.; Olivos H. J.; Thompson J. W.; Menikarachchi L.; Lai S.; Walsh C.; Moseley A.; et al. Ion Mobility-Derived Collision Cross Section as an Additional Measure for Lipid Fingerprinting and Identification. Anal. Chem. 2015, 87, 1137–1144. 10.1021/ac503715v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle J. E.; Zhang X.; Weitz K. K.; Monroe M. E.; Ibrahim Y. M.; Moore R. J.; Cha J.; Sun X.; Lovelace E. S.; Wagoner J.; et al. Uncovering Biologically Significant Lipid Isomers with Liquid Chromatography, Ion Mobility Spectrometry and Mass Spectrometry. Analyst 2016, 141, 1649–1659. 10.1039/C5AN02062J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglia G.; Astarita G. Metabolomics and Lipidomics Using Traveling-Wave Ion Mobility Mass Spectrometry. Nat. Protoc. 2017, 12, 797. 10.1038/nprot.2017.013. [DOI] [PubMed] [Google Scholar]
- Hinz C.; Liggi S.; Griffin J. L. The Potential of Ion Mobility Mass Spectrometry for High-Throughput and High-Resolution Lipidomics. Curr. Opin. Chem. Biol. 2018, 42, 42–50. 10.1016/j.cbpa.2017.10.018. [DOI] [PubMed] [Google Scholar]
- Mairinger T.; Causon T. J.; Hann S. The Potential of Ion Mobility-Mass Spectrometry for Non-Targeted Metabolomics. Curr. Opin. Chem. Biol. 2018, 42, 9–15. 10.1016/j.cbpa.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Burnum-Johnson K. E.; Zheng X.; Dodds J. N.; Ash J.; Fourches D.; Nicora C. D.; Wendler J. P.; Metz T. O.; Waters K. M.; Jansson J. K.; et al. Ion Mobility Spectrometry and the Omics: Distinguishing Isomers, Molecular Classes and Contaminant Ions in Complex Samples. TrAC, Trends Anal. Chem. 2019, 116, 292–299. 10.1016/j.trac.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaptrot K. L.; May J. C.; Dodds J. N.; McLean J. A. Ion Mobility Conformational Lipid Atlas for High Confidence Lipidomics. Nat. Commun. 2019, 10, 985. 10.1038/s41467-019-08897-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilopoulou C. G.; Sulek K.; Brunner A. D.; Meitei N. S.; Schweiger-Hufnagel U.; Meyer S. W.; Barsch A.; Mann M.; Meier F. Trapped Ion Mobility Spectrometry and Pasef Enable in-Depth Lipidomics from Minimal Sample Amounts. Nat. Commun. 2020, 11, 331. 10.1038/s41467-019-14044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglia G.; Smith A. J.; Astarita G. Ion Mobility Mass Spectrometry in the Omics Era: Challenges and Opportunities for Metabolomics and Lipidomics. Mass Spectrom. Rev. 2021, 10.1002/mas.21686. [DOI] [PubMed] [Google Scholar]
- Gray C. J.; Thomas B.; Upton R.; Migas L. G.; Eyers C. E.; Barran P. E.; Flitsch S. L. Applications of Ion Mobility Mass Spectrometry for High Throughput, High Resolution Glycan Analysis. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860, 1688–1709. 10.1016/j.bbagen.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Hofmann J.; Pagel K. Glycan Analysis by Ion Mobility-Mass Spectrometry. Angew. Chem., Int. Ed. 2017, 56, 8342–8349. 10.1002/anie.201701309. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Glover M. S.; Li L. Recent Advances in Ion Mobility-Mass Spectrometry for Improved Structural Characterization of Glycans and Glycoconjugates. Curr. Opin. Chem. Biol. 2018, 42, 1–8. 10.1016/j.cbpa.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookherjee A.; Guttman M. Bridging the Structural Gap of Glycoproteomics with Ion Mobility Spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 86–92. 10.1016/j.cbpa.2017.11.012. [DOI] [PubMed] [Google Scholar]
- Dwivedi P.; Bendiak B.; Clowers B. H.; Hill H. H. Rapid Resolution of Carbohydrate Isomers by Electrospray Ionization Ambient Pressure Ion Mobility Spectrometry-Time-of-Flight Mass Spectrometry (Esi-Apims-Tofms). J. Am. Soc. Mass Spectrom. 2007, 18, 1163–1175. 10.1016/j.jasms.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Fenn L. S.; McLean J. A. Structural Resolution of Carbohydrate Positional and Structural Isomers Based on Gas-Phase Ion Mobility-Mass Spectrometry. Phys. Chem. Chem. Phys. 2011, 13, 2196–2205. 10.1039/C0CP01414A. [DOI] [PubMed] [Google Scholar]
- Hofmann J.; Hahm H. S.; Seeberger P. H.; Pagel K. Identification of Carbohydrate Anomers Using Ion Mobility-Mass Spectrometry. Nature 2015, 526, 241. 10.1038/nature15388. [DOI] [PubMed] [Google Scholar]
- Nagy G.; Attah I. K.; Garimella S. V. B.; Tang K.; Ibrahim Y. M.; Baker E. S.; Smith R. D. Unraveling the Isomeric Heterogeneity of Glycans: Ion Mobility Separations in Structures for Lossless Ion Manipulations. Chem. Commun. 2018, 54, 11701–11704. 10.1039/C8CC06966B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causon T. J.; Hann S. Theoretical Evaluation of Peak Capacity Improvements by Use of Liquid Chromatography Combined with Drift Tube Ion Mobility-Mass Spectrometry. J. Chromatogr. A 2015, 1416, 47–56. 10.1016/j.chroma.2015.09.009. [DOI] [PubMed] [Google Scholar]
- Mason E. A.; Schamp H. W. Mobility of Gaseous Ions in Weak Electric Fields. Ann. Phys. 1958, 4, 233–270. 10.1016/0003-4916(58)90049-6. [DOI] [Google Scholar]
- Viehland L. A.Gaseous Ion Mobility, Diffusion, and Reaction, 1st ed.; Springer, 2018. [Google Scholar]
- Gabelica V.; Shvartsburg A. A.; Afonso C.; Barran P.; Benesch J. L. P.; Bleiholder C.; Bowers M. T.; Bilbao A.; Bush M. F.; Campbell J. L.; et al. Recommendations for Reporting Ion Mobility Mass Spectrometry Measurements. Mass Spectrom. Rev. 2019, 38, 291–320. 10.1002/mas.21585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesleh M. F.; Hunter J. M.; Shvartsburg A. A.; Schatz G. C.; Jarrold M. F. Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential. J. Phys. Chem. 1996, 100, 16082–16086. 10.1021/jp961623v. [DOI] [Google Scholar]
- Wyttenbach T.; von Helden G.; Batka J. J.; Carlat D.; Bowers M. T. Effect of the Long-Range Potential on Ion Mobility Measurements. J. Am. Soc. Mass Spectrom. 1997, 8, 275–282. 10.1016/S1044-0305(96)00236-X. [DOI] [Google Scholar]
- Gabelica V.; Marklund E. Fundamentals of Ion Mobility Spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 51–59. 10.1016/j.cbpa.2017.10.022. [DOI] [PubMed] [Google Scholar]
- D’Atri V.; Causon T.; Hernandez-Alba O.; Mutabazi A.; Veuthey J.-L.; Cianferani S.; Guillarme D. Adding a New Separation Dimension to Ms and Lc-Ms: What Is the Utility of Ion Mobility Spectrometry?. J. Sep. Sci. 2018, 41, 20–67. 10.1002/jssc.201700919. [DOI] [PubMed] [Google Scholar]
- Dodds J. N.; Baker E. S. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30, 2185–2195. 10.1007/s13361-019-02288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk A. T.; Bohnhorst A.; Raddatz C.-R.; Allers M.; Zimmermann S. Ultra-High-Resolution Ion Mobility Spectrometry—Current Instrumentation, Limitations, and Future Developments. Anal. Bioanal. Chem. 2019, 411, 6229–6246. 10.1007/s00216-019-01807-0. [DOI] [PubMed] [Google Scholar]
- May J. C.; McLean J. A. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem. 2015, 87, 1422–1436. 10.1021/ac504720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stow S. M.; Causon T. J.; Zheng X.; Kurulugama R. T.; Mairinger T.; May J. C.; Rennie E. E.; Baker E. S.; Smith R. D.; McLean J. A.; et al. An Interlaboratory Evaluation of Drift Tube Ion Mobility-Mass Spectrometry Collision Cross Section Measurements. Anal. Chem. 2017, 89, 9048–9055. 10.1021/acs.analchem.7b01729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K.; Pringle S. D.; Worthington K. R.; Little D.; Wildgoose J. L.; Bateman R. H. Applications of a Travelling Wave-Based Radio-Frequency-Only Stacked Ring Ion Guide. Rapid Commun. Mass Spectrom. 2004, 18, 2401–2414. 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- Giles K.; Williams J. P.; Campuzano I. Enhancements in Travelling Wave Ion Mobility Resolution. Rapid Commun. Mass Spectrom. 2011, 25, 1559–1566. 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- Campuzano I. D. G.; Giles K. Historical, Current and Future Developments of Travelling Wave Ion Mobility Mass Spectrometry: A Personal Perspective. TrAC, Trends Anal. Chem. 2019, 120, 115620. 10.1016/j.trac.2019.115620. [DOI] [Google Scholar]
- Shvartsburg A. A.; Smith R. D. Fundamentals of Traveling Wave Ion Mobility Spectrometry. Anal. Chem. 2008, 80, 9689–9699. 10.1021/ac8016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson K.; Langridge D.; Giles K. Fundamentals of Travelling Wave Ion Mobility Revisited: I. Smoothly Moving Waves. Int. J. Mass Spectrom. 2018, 428, 71–80. 10.1016/j.ijms.2018.03.007. [DOI] [Google Scholar]
- Deng L.; Webb I. K.; Garimella S. V. B.; Hamid A. M.; Zheng X.; Norheim R. V.; Prost S. A.; Anderson G. A.; Sandoval J. A.; Baker E. S.; et al. Serpentine Ultralong Path with Extended Routing (Super) High Resolution Traveling Wave Ion Mobility-Ms Using Structures for Lossless Ion Manipulations. Anal. Chem. 2017, 89, 4628–4634. 10.1021/acs.analchem.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K.; Ujma J.; Wildgoose J.; Pringle S.; Richardson K.; Langridge D.; Green M. A Cyclic Ion Mobility-Mass Spectrometry System. Anal. Chem. 2019, 91, 8564–8573. 10.1021/acs.analchem.9b01838. [DOI] [PubMed] [Google Scholar]
- Ujma J.; Ropartz D.; Giles K.; Richardson K.; Langridge D.; Wildgoose J.; Green M.; Pringle S. Cyclic Ion Mobility Mass Spectrometry Distinguishes Anomers and Open-Ring Forms of Pentasaccharides. J. Am. Soc. Mass Spectrom. 2019, 30, 1028–1037. 10.1007/s13361-019-02168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnke S.; Ben Faleh A.; Scutelnic V.; Rizzo T. R. Separation and Identification of Glycan Anomers Using Ultrahigh-Resolution Ion-Mobility Spectrometry and Cryogenic Ion Spectroscopy. J. Am. Soc. Mass Spectrom. 2019, 30, 2204–2211. 10.1007/s13361-019-02333-0. [DOI] [PubMed] [Google Scholar]
- Hernandez D. R.; DeBord J. D.; Ridgeway M. E.; Kaplan D. A.; Park M. A.; Fernandez-Lima F. Ion Dynamics in a Trapped Ion Mobility Spectrometer. Analyst 2014, 139, 1913–1921. 10.1039/C3AN02174B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelmann K.; Silveira J. A.; Ridgeway M. E.; Park M. A. Fundamentals of Trapped Ion Mobility Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 14–24. 10.1007/s13361-014-0999-4. [DOI] [PubMed] [Google Scholar]
- Silveira J. A.; Michelmann K.; Ridgeway M. E.; Park M. A. Fundamentals of Trapped Ion Mobility Spectrometry Part Ii: Fluid Dynamics. J. Am. Soc. Mass Spectrom. 2016, 27, 585–595. 10.1007/s13361-015-1310-z. [DOI] [PubMed] [Google Scholar]
- Ridgeway M. E.; Lubeck M.; Jordens J.; Mann M.; Park M. A. Trapped Ion Mobility Spectrometry: A Short Review. Int. J. Mass Spectrom. 2018, 425, 22–35. 10.1016/j.ijms.2018.01.006. [DOI] [Google Scholar]
- Buryakov I. A.; Krylov E. V.; Nazarov E. G.; Rasulev U. K. A New Method of Separation of Multi-Atomic Ions by Mobility at Atmospheric Pressure Using a High-Frequency Amplitude-Asymmetric Strong Electric Field. Int. J. Mass Spectrom. Ion Processes 1993, 128, 143–148. 10.1016/0168-1176(93)87062-W. [DOI] [Google Scholar]
- Purves R. W.; Guevremont R.; Day S.; Pipich C. W.; Matyjaszczyk M. S. Mass Spectrometric Characterization of a High-Field Asymmetric Waveform Ion Mobility Spectrometer. Rev. Sci. Instrum. 1998, 69, 4094–4105. 10.1063/1.1149255. [DOI] [Google Scholar]
- Shvartsburg A. A.; Li F.; Tang K.; Smith R. D. High-Resolution Field Asymmetric Waveform Ion Mobility Spectrometry Using New Planar Geometry Analyzers. Anal. Chem. 2006, 78, 3706–3714. 10.1021/ac052020v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia M. J.; Park M.; Kaplan D. A.; Venot A.; Boons G.-J.; Li L.; Linhardt R. J.; Amster I. J. High-Field Asymmetric-Waveform Ion Mobility Spectrometry and Electron Detachment Dissociation of Isobaric Mixtures of Glycosaminoglycans. J. Am. Soc. Mass Spectrom. 2014, 25, 258–268. 10.1007/s13361-013-0771-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Helden G.; Hsu M. T.; Gotts N.; Bowers M. T. Carbon Cluster Cations with up to 84 Atoms: Structures, Formation Mechanism, and Reactivity. J. Phys. Chem. 1993, 97, 8182–8192. 10.1021/j100133a011. [DOI] [Google Scholar]
- Ho K.-M.; Shvartsburg A. A.; Pan B.; Lu Z.-Y.; Wang C.-Z.; Wacker J. G.; Fye J. L.; Jarrold M. F. Structures of Medium-Sized Silicon Clusters. Nature 1998, 392, 582–585. 10.1038/33369. [DOI] [Google Scholar]
- Furche F.; Ahlrichs R.; Weis P.; Jacob C.; Gilb S.; Bierweiler T.; Kappes M. M. The Structures of Small Gold Cluster Anions as Determined by a Combination of Ion Mobility Measurements and Density Functional Calculations. J. Chem. Phys. 2002, 117, 6982–6990. 10.1063/1.1507582. [DOI] [Google Scholar]
- D’Atri V.; Porrini M.; Rosu F.; Gabelica V. Linking Molecular Models with Ion Mobility Experiments. Illustration with a Rigid Nucleic Acid Structure. J. Mass Spectrom. 2015, 50, 711–726. 10.1002/jms.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund E. G.; Degiacomi M. T.; Robinson C. V.; Baldwin A. J.; Benesch J. L.P. Collision Cross Sections for Structural Proteomics. Structure 2015, 23, 791–799. 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Jarrold M. F. An Exact Hard-Spheres Scattering Model for the Mobilities of Polyatomic Ions. Chem. Phys. Lett. 1996, 261, 86–91. 10.1016/0009-2614(96)00941-4. [DOI] [Google Scholar]
- Viehland L. A.; Chang Y. Transport Cross Sections for Collisions between Particles. Comput. Phys. Commun. 2010, 181, 1687–1696. 10.1016/j.cpc.2010.06.008. [DOI] [Google Scholar]
- Bleiholder C.; Wyttenbach T.; Bowers M. T. A Novel Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (I). Int. J. Mass Spectrom. 2011, 308, 1–10. 10.1016/j.ijms.2011.06.014. [DOI] [Google Scholar]
- Larriba C.; Hogan C. J. Ion Mobilities in Diatomic Gases: Measurement Versus Prediction with Non-Specular Scattering Models. J. Phys. Chem. A 2013, 117, 3887–3901. 10.1021/jp312432z. [DOI] [PubMed] [Google Scholar]
- Larriba C.; Hogan C. J. Free Molecular Collision Cross Section Calculation Methods for Nanoparticles and Complex Ions with Energy Accommodation. J. Comput. Phys. 2013, 251, 344–363. 10.1016/j.jcp.2013.05.038. [DOI] [Google Scholar]
- Larriba-Andaluz C.; Hogan C. J. Collision Cross Section Calculations for Polyatomic Ions Considering Rotating Diatomic/Linear Gas Molecules. J. Chem. Phys. 2014, 141, 194107. 10.1063/1.4901890. [DOI] [PubMed] [Google Scholar]
- Bleiholder C. A Local Collision Probability Approximation for Predicting Momentum Transfer Cross Sections. Analyst 2015, 140, 6804–6813. 10.1039/C5AN00712G. [DOI] [PubMed] [Google Scholar]
- Ewing S. A.; Donor M. T.; Wilson J. W.; Prell J. S. Collidoscope: An Improved Tool for Computing Collisional Cross-Sections with the Trajectory Method. J. Am. Soc. Mass Spectrom. 2017, 28, 587–596. 10.1007/s13361-017-1594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav V.; Nahin M.; Hogan C. J.; Larriba-Andaluz C. Benchmark Comparison for a Multi-Processing Ion Mobility Calculator in the Free Molecular Regime. J. Am. Soc. Mass Spectrom. 2017, 28, 1540–1551. 10.1007/s13361-017-1661-8. [DOI] [PubMed] [Google Scholar]
- Zanotto L.; Heerdt G.; Souza P. C. T.; Araujo G.; Skaf M. S. High Performance Collision Cross Section Calculation—Hpccs. J. Comput. Chem. 2018, 39, 1675–1681. 10.1002/jcc.25199. [DOI] [PubMed] [Google Scholar]
- Ieritano C.; Crouse J.; Campbell J. L.; Hopkins W. S. A Parallelized Molecular Collision Cross Section Package with Optimized Accuracy and Efficiency. Analyst 2019, 144, 1660–1670. 10.1039/C8AN02150C. [DOI] [PubMed] [Google Scholar]
- Oomens J.; Sartakov B. G.; Meijer G.; von Helden G. Gas-Phase Infrared Multiple Photon Dissociation Spectroscopy of Mass-Selected Molecular Ions. Int. J. Mass Spectrom. 2006, 254, 1–19. 10.1016/j.ijms.2006.05.009. [DOI] [Google Scholar]
- Rijs A. M.; Oomens J. In Gas-Phase Ir Spectroscopy and Structure of Biological Molecules; Rijs A. M., Oomens J., Eds.; Springer International Publishing: Cham, 2015; 10.1007/128_2014_621. [DOI] [Google Scholar]
- Polfer N. C.; Oomens J. Vibrational Spectroscopy of Bare and Solvated Ionic Complexes of Biological Relevance. Mass Spectrom. Rev. 2009, 28, 468–494. 10.1002/mas.20215. [DOI] [PubMed] [Google Scholar]
- Schöllkopf W.; Gewinner S.; Junkes H.; Paarmann A.; von Helden G.; Bluem H.; Todd A. M. M. In Proc. Of Spie - Advances in X-Ray Free-Electron Lasers Instrumentation Iii; Biedron S. G., Ed.; Prague, Czech Republic, 2015; Vol. 9512. [Google Scholar]
- Lucas B.; Grégoire G.; Lemaire J.; Maître P.; Glotin F.; Schermann J. P.; Desfrançois C. Infrared Multiphoton Dissociation Spectroscopy of Protonated N-Acetyl-Alanine and Alanyl-Histidine. Int. J. Mass Spectrom. 2005, 243, 105–113. 10.1016/j.ijms.2005.01.002. [DOI] [Google Scholar]
- Oomens J.; van Roij A. J. A.; Meijer G.; von Helden G. Gas-Phase Infrared Photodissociation Spectroscopy of Cationic Polyaromatic Hydrocarbons. Astrophys. J. 2000, 542, 404–410. 10.1086/309545. [DOI] [Google Scholar]
- Talbot F. O.; Simons J. P. Sugars in the Gas Phase: The Spectroscopy and Structure of Jet-Cooled Phenyl B-D-Glucopyranoside. Phys. Chem. Chem. Phys. 2002, 4, 3562–3565. 10.1039/b204132d. [DOI] [Google Scholar]
- Jockusch R. A.; Kroemer R. T.; Talbot F. O.; Snoek L. C.; Carcabal P.; Simons J. P.; Havenith M.; Bakker J. M.; Compagnon I.; Meijer G.; et al. Probing the Glycosidic Linkage: Uv and Ir Ion-Dip Spectroscopy of a Lactoside. J. Am. Chem. Soc. 2004, 126, 5709–5714. 10.1021/ja031679k. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Joshi J.; Allouche A. R.; Simon D.; Chambert S.; Brites V.; Gaigeot M. P.; Compagnon I. Distinguishing Isobaric Phosphated and Sulfated Carbohydrates by Coupling of Mass Spectrometry with Gas Phase Vibrational Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 22131–22138. 10.1039/C4CP02898H. [DOI] [PubMed] [Google Scholar]
- Masellis C.; Khanal N.; Kamrath M. Z.; Clemmer D. E.; Rizzo T. R. Cryogenic Vibrational Spectroscopy Provides Unique Fingerprints for Glycan Identification. J. Am. Soc. Mass Spectrom. 2017, 28, 2217–2222. 10.1007/s13361-017-1728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha E.; Gonzalez Florez A. I.; Marianski M.; Thomas D. A.; Hoffmann W.; Struwe W. B.; Hahm H. S.; Gewinner S.; Schollkopf W.; Seeberger P. H.; et al. Glycan Fingerprinting Via Cold-Ion Infrared Spectroscopy. Angew. Chem., Int. Ed. 2017, 56, 11248–11251. 10.1002/anie.201702896. [DOI] [PubMed] [Google Scholar]
- Barnes L.; Schindler B.; Chambert S.; Allouche A.-R.; Compagnon I. Conformational Preferences of Protonated N-Acetylated Hexosamines Probed by Infrared Multiple Photon Dissociation (Irmpd) Spectroscopy and Ab Initio Calculations. Int. J. Mass Spectrom. 2017, 421, 116–123. 10.1016/j.ijms.2017.05.005. [DOI] [Google Scholar]
- Tan Y.; Polfer N. C. Linkage and Anomeric Differentiation in Trisaccharides by Sequential Fragmentation and Variable-Wavelength Infrared Photodissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 359–368. 10.1007/s13361-014-1025-6. [DOI] [PubMed] [Google Scholar]
- Tan Y.; Zhao N.; Liu J.; Li P.; Stedwell C. N.; Yu L.; Polfer N. C. Vibrational Signatures of Isomeric Lithiated N-Acetyl-D-Hexosamines by Gas-Phase Infrared Multiple-Photon Dissociation (Irmpd) Spectroscopy. J. Am. Soc. Mass Spectrom. 2017, 28, 539–550. 10.1007/s13361-016-1575-x. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Laloy-Borgna G.; Barnes L.; Allouche A. R.; Bouju E.; Dugas V.; Demesmay C.; Compagnon I. Online Separation and Identification of Isomers Using Infrared Multiple Photon Dissociation Ion Spectroscopy Coupled to Liquid Chromatography: Application to the Analysis of Disaccharides Regio-Isomers and Monosaccharide Anomers. Anal. Chem. 2018, 90, 11741–11745. 10.1021/acs.analchem.8b02801. [DOI] [PubMed] [Google Scholar]
- Gerlich D. Adv. Chem. Phys. 2007, 1. 10.1002/9780470141397.ch1. [DOI] [Google Scholar]
- Gerlich D. Ion-Neutral Collisions in a 22-Pole Trap at Very Low Energies. Phys. Scr. 1995, T59, 256–263. 10.1088/0031-8949/1995/T59/035. [DOI] [Google Scholar]
- Wang Y. S.; Tsai C. H.; Lee Y. T.; Chang H. C.; Jiang J. C.; Asvany O.; Schlemmer S.; Gerlich D. Investigations of Protonated and Deprotonated Water Clusters Using a Low-Temperature 22-Pole Ion Trap. J. Phys. Chem. A 2003, 107, 4217–4225. 10.1021/jp022156m. [DOI] [Google Scholar]
- Boyarkin O. V.; Mercier S. R.; Kamariotis A.; Rizzo T. R. Electronic Spectroscopy of Cold, Protonated Tryptophan and Tyrosine. J. Am. Chem. Soc. 2006, 128, 2816–2817. 10.1021/ja058383u. [DOI] [PubMed] [Google Scholar]
- Asmis K. R.; Brümmer M.; Kaposta C.; Santambrogio G.; von Helden G.; Meijer G.; Rademann K.; Wöste L. Mass-Selected Infrared Photodissociation Spectroscopy of V4o10+. Phys. Chem. Chem. Phys. 2002, 4, 1101–1104. 10.1039/b111056j. [DOI] [Google Scholar]
- Lorenz U. J.; Rizzo T. R. Planar Multipole Ion Trap/Time-of-Flight Mass Spectrometer. Anal. Chem. 2011, 83, 7895–7901. 10.1021/ac201790d. [DOI] [PubMed] [Google Scholar]
- Bansal P.; Yatsyna V.; AbiKhodr A. H.; Warnke S.; Ben Faleh A.; Yalovenko N.; Wysocki V. H.; Rizzo T. R. Using Slim-Based Ims-Ims Together with Cryogenic Infrared Spectroscopy for Glycan Analysis. Anal. Chem. 2020, 92, 9079–9085. 10.1021/acs.analchem.0c01265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalovenko N.; Yatsyna V.; Bansal P.; AbiKhodr A. H.; Rizzo T. R. Analyzing Glycans Cleaved from a Biotherapeutic Protein Using Ultrahigh-Resolution Ion Mobility Spectrometry Together with Cryogenic Ion Spectroscopy. Analyst 2020, 145, 6493–6499. 10.1039/D0AN01206H. [DOI] [PubMed] [Google Scholar]
- Roithova J.; Gray A.; Andris E.; Jasik J.; Gerlich D. Helium Tagging Infrared Photodissociation Spectroscopy of Reactive Ions. Acc. Chem. Res. 2016, 49, 223–230. 10.1021/acs.accounts.5b00489. [DOI] [PubMed] [Google Scholar]
- Lettow M.; Grabarics M.; Mucha E.; Thomas D. A.; Polewski L.; Freyse J.; Rademann J.; Meijer G.; von Helden G.; Pagel K. Ir Action Spectroscopy of Glycosaminoglycan Oligosaccharides. Anal. Bioanal. Chem. 2020, 412, 533–537. 10.1007/s00216-019-02327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha E.; Lettow M.; Marianski M.; Thomas D. A.; Struwe W. B.; Harvey D. J.; Meijer G.; Seeberger P. H.; von Helden G.; Pagel K. Fucose Migration in Intact Protonated Glycan Ions: A Universal Phenomenon in Mass Spectrometry. Angew. Chem., Int. Ed. 2018, 57, 7440–7443. 10.1002/anie.201801418. [DOI] [PubMed] [Google Scholar]
- Rizzo T. R.; Stearns J. A.; Boyarkin O. V. Spectroscopic Studies of Cold, Gas-Phase Biomolecular Ions. Int. Rev. Phys. Chem. 2009, 28, 481–515. 10.1080/01442350903069931. [DOI] [Google Scholar]
- Daly S.; Porrini M.; Rosu F.; Gabelica V. Electronic Spectroscopy of Isolated DNA Polyanions. Faraday Discuss. 2019, 217, 361–382. 10.1039/C8FD00207J. [DOI] [PubMed] [Google Scholar]
- Boyarkin O. V. Cold Ion Spectroscopy for Structural Identifications of Biomolecules. Int. Rev. Phys. Chem. 2018, 37, 559–606. 10.1080/0144235X.2018.1547453. [DOI] [Google Scholar]
- Kopysov V.; Makarov A.; Boyarkin O. V. Colors for Molecular Masses: Fusion of Spectroscopy and Mass Spectrometry for Identification of Biomolecules. Anal. Chem. 2015, 87, 4607–4611. 10.1021/acs.analchem.5b00822. [DOI] [PubMed] [Google Scholar]
- Nagornova N. S.; Rizzo T. R.; Boyarkin O. V. Interplay of Intra- and Intermolecular H-Bonding in a Progressively Solvated Macrocyclic Peptide. Science 2012, 336, 320–323. 10.1126/science.1218709. [DOI] [PubMed] [Google Scholar]
- Nguyen H. T. H.; Shaffer C. J.; Pepin R.; Tureček F. Uv Action Spectroscopy of Gas-Phase Peptide Radicals. J. Phys. Chem. Lett. 2015, 6, 4722–4727. 10.1021/acs.jpclett.5b02354. [DOI] [PubMed] [Google Scholar]
- Kwon J. H.; Lee M. J.; Song G.; Tsuruta K.; Ishiuchi S.-i.; Fujii M.; Kang H. Cryogenic Ion Spectroscopy of a Singly Protonated Peptide Dyyvvr: Locating Phosphorylation Sites of a Kinase Domain. J. Phys. Chem. Lett. 2020, 11, 7103–7108. 10.1021/acs.jpclett.0c01802. [DOI] [PubMed] [Google Scholar]
- Racaud A.; Allouche A. R.; Antoine R.; Lemoine J.; Dugourd P. Uv Electronic Excitations in Acidic Sugars. J. Mol. Struct.: THEOCHEM 2010, 960, 51–56. 10.1016/j.theochem.2010.08.024. [DOI] [Google Scholar]
- Ortiz D.; Enjalbert Q.; MacAleese L.; Dugourd P.; Salpin J. Y. Effects of Calcium Complexation on Heparin-Like Disaccharides. A Combined Theoretical, Tandem Mass Spectrometry and Ultraviolet Experiment. Rapid Commun. Mass Spectrom. 2015, 29, 1135–1144. 10.1002/rcm.7204. [DOI] [PubMed] [Google Scholar]
- Saparbaev E.; Kopysov V.; Yamaletdinov R.; Pereverzev A. Y.; Boyarkin O. V. Interplay of H-Bonds with Aromatics in Isolated Complexes Identifies Isomeric Carbohydrates. Angew. Chem., Int. Ed. 2019, 58, 7346–7350. 10.1002/anie.201902377. [DOI] [PubMed] [Google Scholar]
- Daly S.; Rosu F.; Gabelica V. Mass-Resolved Electronic Circular Dichroism Ion Spectroscopy. Science 2020, 368, 1465–1468. 10.1126/science.abb1822. [DOI] [PubMed] [Google Scholar]
- Eun H. J.; Min A.; Jeon C. W.; Yoo I. T.; Heo J.; Kim N. J. Chiral and Isomeric Discrimination of Chiral Molecular Ions by Cold Ion Circular Dichroism Spectroscopy. J. Phys. Chem. Lett. 2020, 11, 4367–4371. 10.1021/acs.jpclett.0c01081. [DOI] [PubMed] [Google Scholar]
- Jin L.; Barran P. E.; Deakin J. A.; Lyon M.; Uhrin D. Conformation of Glycosaminoglycans by Ion Mobility Mass Spectrometry and Molecular Modelling. Phys. Chem. Chem. Phys. 2005, 7, 3464–3471. 10.1039/b508644b. [DOI] [PubMed] [Google Scholar]
- Sastre Torano J.; Aizpurua-Olaizola O.; Wei N.; Li T.; Unione L.; Jiménez-Osés G.; Corzana F.; Somovilla V. J.; Falcon-Perez J. M.; Boons G.-J. Identification of Isomeric n-Glycans by Conformer Distribution Fingerprinting Using Ion Mobility-Mass Spectrometry. Chem. - Eur. J. 2021, 27, 2149–2154. 10.1002/chem.202004522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.; Elferink H.; van Hengst J. M. A.; Houthuijs K. J.; Remmerswaal W. A.; Kromm A.; Berden G.; van der Vorm S.; Rijs A. M.; Overkleeft H. S.; et al. Characterization of Glycosyl Dioxolenium Ions and Their Role in Glycosylation Reactions. Nat. Commun. 2020, 11, 2664. 10.1038/s41467-020-16362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marianski M.; Supady A.; Ingram T.; Schneider M.; Baldauf C. Assessing the Accuracy of across-the-Scale Methods for Predicting Carbohydrate Conformational Energies for the Examples of Glucose and Alpha-Maltose. J. Chem. Theory Comput. 2016, 12, 6157–6168. 10.1021/acs.jctc.6b00876. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Barone V. Toward Reliable Density Functional Methods without Adjustable Parameters: The Pbe0Model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. Iii. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (Dft-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Tkatchenko A.; Scheffler M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. Self—Consistent Molecular Orbital Methods. Xii. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Dunning T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Bannwarth C.; Ehlert S.; Grimme S. Gfn2-Xtb-an Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]
- Supady A.; Blum V.; Baldauf C. First-Principles Molecular Structure Search with a Genetic Algorithm. J. Chem. Inf. Model. 2015, 55, 2338–2348. 10.1021/acs.jcim.5b00243. [DOI] [PubMed] [Google Scholar]
- Greis K.; Kirschbaum C.; Leichnitz S.; Gewinner S.; Schöllkopf W.; von Helden G.; Meijer G.; Seeberger P. H.; Pagel K. Direct Experimental Characterization of the Ferrier Glycosyl Cation in the Gas Phase. Org. Lett. 2020, 22, 8916–8919. 10.1021/acs.orglett.0c03301. [DOI] [PubMed] [Google Scholar]
- Lettow M.; Grabarics M.; Greis K.; Mucha E.; Thomas D. A.; Chopra P.; Boons G.-J.; Karlsson R.; Turnbull J. E.; Meijer G.; et al. Cryogenic Infrared Spectroscopy Reveals Structural Modularity in the Vibrational Fingerprints of Heparan Sulfate Diastereomers. Anal. Chem. 2020, 92, 10228–10232. 10.1021/acs.analchem.0c02048. [DOI] [PubMed] [Google Scholar]
- Struwe W. B.; Baldauf C.; Hofmann J.; Rudd P. M.; Pagel K. Ion Mobility Separation of Deprotonated Oligosaccharide Isomers - Evidence for Gas-Phase Charge Migration. Chem. Commun. 2016, 52, 12353–12356. 10.1039/C6CC06247D. [DOI] [PubMed] [Google Scholar]
- Hansen T.; Lebedel L.; Remmerswaal W. A.; van der Vorm S.; Wander D. P. A.; Somers M.; Overkleeft H. S.; Filippov D. V.; Desire J.; Mingot A.; et al. Defining the Sn1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Cent. Sci. 2019, 5, 781–788. 10.1021/acscentsci.9b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.; Ofman T. P.; Vlaming J. G. C.; Gagarinov I. A.; van Beek J.; Gote T. A.; Tichem J. M.; Ruijgrok G.; Overkleeft H. S.; Filippov D. V.; et al. Reactivity-Stereoselectivity Mapping for the Assembly of Mycobacterium Marinum Lipooligosaccharides. Angew. Chem., Int. Ed. 2021, 60, 937–945. 10.1002/anie.202010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vorm S.; Hansen T.; van Rijssel E. R.; Dekkers R.; Madern J. M.; Overkleeft H. S.; Filippov D. V.; van der Marel G. A.; Codee J. D. C. Furanosyl Oxocarbenium Ion Conformational Energy Landscape Maps as a Tool to Study the Glycosylation Stereoselectivity of 2-Azidofuranoses, 2-Fluorofuranoses and Methyl Furanosyl Uronates. Chem. - Eur. J. 2019, 25, 7149–7157. 10.1002/chem.201900651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracht P.; Bohle F.; Grimme S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. 10.1039/C9CP06869D. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C.; Greis K.; Mucha E.; Kain L.; Deng S.; Zappe A.; Gewinner S.; Schöllkopf W.; von Helden G.; Meijer G.; et al. Unravelling the Structural Complexity of Glycolipids with Cryogenic Infrared Spectroscopy. Nat. Commun. 2021, 12, 1201. 10.1038/s41467-021-21480-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray C. J.; Schindler B.; Migas L. G.; Pičmanová M.; Allouche A. R.; Green A. P.; Mandal S.; Motawia M. S.; Sánchez-Pérez R.; Bjarnholt N.; et al. Bottom-up Elucidation of Glycosidic Bond Stereochemistry. Anal. Chem. 2017, 89, 4540–4549. 10.1021/acs.analchem.6b04998. [DOI] [PubMed] [Google Scholar]
- Singh U. C.; Kollman P. A. An Approach to Computing Electrostatic Charges for Molecules. J. Comput. Chem. 1984, 5, 129–145. 10.1002/jcc.540050204. [DOI] [Google Scholar]
- Shvartsburg A. A.; Jarrold M. F. An Exact Hard-Spheres Scattering Model for the Mobilities of Polyatomic Ions. Chem. Phys. Lett. 1996, 261, 86–91. 10.1016/0009-2614(96)00941-4. [DOI] [Google Scholar]
- Mesleh M. F.; Hunter J. M.; Shvartsburg A. A.; Schatz G. C.; Jarrold M. F. Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential. J. Phys. Chem. 1996, 100, 16082–16086. 10.1021/jp961623v. [DOI] [Google Scholar]
- Kirschbaum C.; Saied E. M.; Greis K.; Mucha E.; Gewinner S.; Schöllkopf W.; Meijer G.; von Helden G.; Poad B. L. J.; Blanksby S. J.; et al. Resolving Sphingolipid Isomers Using Cryogenic Infrared Spectroscopy. Angew. Chem., Int. Ed. 2020, 59, 13638–13642. 10.1002/anie.202002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. A.; Chang R.; Mucha E.; Lettow M.; Greis K.; Gewinner S.; Schöllkopf W.; Meijer G.; von Helden G. Probing the Conformational Landscape and Thermochemistry of DNA Dinucleotide Anions Via Helium Nanodroplet Infrared Action Spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 18400–18413. 10.1039/D0CP02482A. [DOI] [PubMed] [Google Scholar]
- Zivkovic A. M.; German J. B.; Lebrilla C. B.; Mills D. A. Human Milk Glycobiome and Its Impact on the Infant Gastrointestinal Microbiota. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 4653–4658. 10.1073/pnas.1000083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnoth M. J.; Kunz C.; Kinne-Saffran E.; Rudloff S. Human Milk Oligosaccharides Are Minimally Digested in Vitro. J. Nutr. 2000, 130, 3014–3020. 10.1093/jn/130.12.3014. [DOI] [PubMed] [Google Scholar]
- Bode L. Human Milk Oligosaccharides: Prebiotics and Beyond. Nutr. Rev. 2009, 67, S183–S191. 10.1111/j.1753-4887.2009.00239.x. [DOI] [PubMed] [Google Scholar]
- Charbonneau M. R.; Blanton L. V.; DiGiulio D. B.; Relman D. A.; Lebrilla C. B.; Mills D. A.; Gordon J. I. A Microbial Perspective of Human Developmental Biology. Nature 2016, 535, 48–55. 10.1038/nature18845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza-Díaz J.; Fontana L.; Gil A. Human Milk Oligosaccharides and Immune System Development. Nutrients 2018, 10, 1038. 10.3390/nu10081038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantis V.; Bode L.; van Neerven R. J. J. Immunological Effects of Human Milk Oligosaccharides. Frontiers in Pediatrics 2018, 6, 190. 10.3389/fped.2018.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz C.; Rudloff S.; Baier W.; Klein N.; Strobel S. Oligosaccharides in Human Milk: Structural, Functional, and Metabolic Aspects. Annu. Rev. Nutr. 2000, 20, 699–722. 10.1146/annurev.nutr.20.1.699. [DOI] [PubMed] [Google Scholar]
- Grabarics M.; Csernak O.; Balogh R.; Beni S. Analytical Characterization of Human Milk Oligosaccharides - Potential Applications in Pharmaceutical Analysis. J. Pharm. Biomed. Anal. 2017, 146, 168–178. 10.1016/j.jpba.2017.08.039. [DOI] [PubMed] [Google Scholar]
- Prudden A. R.; Liu L.; Capicciotti C. J.; Wolfert M. A.; Wang S.; Gao Z.; Meng L.; Moremen K. W.; Boons G.-J. Synthesis of Asymmetrical Multiantennary Human Milk Oligosaccharides. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 6954. 10.1073/pnas.1701785114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode L. Recent Advances on Structure, Metabolism, and Function of Human Milk Oligosaccharides. J. Nutr. 2006, 136, 2127–2130. 10.1093/jn/136.8.2127. [DOI] [PubMed] [Google Scholar]
- Pikulski M.; Hargrove A.; Shabbir S. H.; Anslyn E. V.; Brodbelt J. S. Sequencing and Characterization of Oligosaccharides Using Infrared Multiphoton Dissociation and Boronic Acid Derivatization in a Quadrupole Ion Trap. J. Am. Soc. Mass Spectrom. 2007, 18, 2094–2106. 10.1016/j.jasms.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Han L.; Costello C. E. Electron Transfer Dissociation of Milk Oligosaccharides. J. Am. Soc. Mass Spectrom. 2011, 22, 997–1013. 10.1007/s13361-011-0117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeister G. E.; Zhou Z.; Leary J. A. Linkage Position Determination in Lithium-Cationized Disaccharides: Tandem Mass Spectrometry and Semiempirical Calculations. J. Am. Chem. Soc. 1991, 113, 5964–5970. 10.1021/ja00016a007. [DOI] [Google Scholar]
- Harvey D. J. Ionization and Collision-Induced Fragmentation of N-Linked and Related Carbohydrates Using Divalent Cations. J. Am. Soc. Mass Spectrom. 2001, 12, 926–937. 10.1016/S1044-0305(01)00268-9. [DOI] [PubMed] [Google Scholar]
- Zaia J. Mass Spectrometry of Oligosaccharides. Mass Spectrom. Rev. 2004, 23, 161–227. 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A. Reactions of Polypeptide Ions with Electrons in the Gas Phase. Mass Spectrom. Rev. 2003, 22, 57–77. 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon J. J.; Shabanowitz J.; Hunt D. F.; Syka J. E. P. Electron Transfer Dissociation of Peptide Anions. J. Am. Soc. Mass Spectrom. 2005, 16, 880–882. 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Cooper H. J.; Håkansson K.; Marshall A. G. The Role of Electron Capture Dissociation in Biomolecular Analysis. Mass Spectrom. Rev. 2005, 24, 201–222. 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- Lioe H.; O’Hair R. A. J. Comparison of Collision-Induced Dissociation and Electron-Induced Dissociation of Singly Protonated Aromatic Amino Acids, Cystine and Related Simple Peptides Using a Hybrid Linear Ion Trap-Ft-Icr Mass Spectrometer. Anal. Bioanal. Chem. 2007, 389, 1429–1437. 10.1007/s00216-007-1535-1. [DOI] [PubMed] [Google Scholar]
- Wolff J. J.; Laremore T. N.; Aslam H.; Linhardt R. J.; Amster I. J. Electron-Induced Dissociation of Glycosaminoglycan Tetrasaccharides. J. Am. Soc. Mass Spectrom. 2008, 19, 1449–1458. 10.1016/j.jasms.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Pu Y.; Yu X.; Costello C. E.; Lin C. Mechanistic Study on Electronic Excitation Dissociation of the Cellobiose-Na+ Complex. J. Am. Soc. Mass Spectrom. 2016, 27, 319–328. 10.1007/s13361-015-1277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson J. T.; Håkansson K. Electron Detachment Dissociation of Neutral and Sialylated Oligosaccharides. J. Am. Soc. Mass Spectrom. 2007, 18, 2162–2172. 10.1016/j.jasms.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kornacki J. R.; Adamson J. T.; Håkansson K. Electron Detachment Dissociation of Underivatized Chloride-Adducted Oligosaccharides. J. Am. Soc. Mass Spectrom. 2012, 23, 2031–2042. 10.1007/s13361-012-0459-y. [DOI] [PubMed] [Google Scholar]
- Gao J.; Thomas D. A.; Sohn C. H.; Beauchamp J. L. Biomimetic Reagents for the Selective Free Radical and Acid-Base Chemistry of Glycans: Application to Glycan Structure Determination by Mass Spectrometry. J. Am. Chem. Soc. 2013, 135, 10684–10692. 10.1021/ja402810t. [DOI] [PubMed] [Google Scholar]
- Desai N.; Thomas D. A.; Lee J.; Gao J.; Beauchamp J. L. Eradicating Mass Spectrometric Glycan Rearrangement by Utilizing Free Radicals. Chemical Science 2016, 7, 5390–5397. 10.1039/C6SC01371F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Pu Y.; Gao J.; Hong P.; Costello C. E.; Lin C. De Novo Glycan Sequencing by Electronic Excitation Dissociation and Fixed-Charge Derivatization. Anal. Chem. 2018, 90, 3793–3801. 10.1021/acs.analchem.7b04077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devakumar A.; Thompson M. S.; Reilly J. P. Fragmentation of Oligosaccharide Ions with 157 Nm Vacuum Ultraviolet Light. Rapid Commun. Mass Spectrom. 2005, 19, 2313–2320. 10.1002/rcm.2058. [DOI] [PubMed] [Google Scholar]
- Devakumar A.; Mechref Y.; Kang P.; Novotny M. V.; Reilly J. P. Laser-Induced Photofragmentation of Neutral and Acidic Glycans inside an Ion-Trap Mass Spectrometer. Rapid Commun. Mass Spectrom. 2007, 21, 1452–1460. 10.1002/rcm.2981. [DOI] [PubMed] [Google Scholar]
- Wilson J. J.; Brodbelt J. S. Ultraviolet Photodissociation at 355 Nm of Fluorescently Labeled Oligosaccharides. Anal. Chem. 2008, 80, 5186–5196. 10.1021/ac800315k. [DOI] [PubMed] [Google Scholar]
- Reiding K. R.; Bondt A.; Franc V.; Heck A. J. R. The Benefits of Hybrid Fragmentation Methods for Glycoproteomics. TrAC, Trends Anal. Chem. 2018, 108, 260–268. 10.1016/j.trac.2018.09.007. [DOI] [Google Scholar]
- Zhang X.; Julian R. R. Radical Mediated Dissection of Oligosaccharides. Int. J. Mass Spectrom. 2014, 372, 22–28. 10.1016/j.ijms.2014.07.045. [DOI] [Google Scholar]
- Williams J. P.; Grabenauer M.; Holland R. J.; Carpenter C. J.; Wormald M. R.; Giles K.; Harvey D. J.; Bateman R. H.; Scrivens J. H.; Bowers M. T. Characterization of Simple Isomeric Oligosaccharides and the Rapid Separation of Glycan Mixtures by Ion Mobility Mass Spectrometry. Int. J. Mass Spectrom. 2010, 298, 119–127. 10.1016/j.ijms.2009.08.006. [DOI] [Google Scholar]
- Huang Y.; Dodds E. D. Ion Mobility Studies of Carbohydrates as Group I Adducts: Isomer Specific Collisional Cross Section Dependence on Metal Ion Radius. Anal. Chem. 2013, 85, 9728–9735. 10.1021/ac402133f. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Dodds E. D. Discrimination of Isomeric Carbohydrates as the Electron Transfer Products of Group Ii Cation Adducts by Ion Mobility Spectrometry and Tandem Mass Spectrometry. Anal. Chem. 2015, 87, 5664–5668. 10.1021/acs.analchem.5b00759. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Dodds E. D. Ion-Neutral Collisional Cross Sections of Carbohydrate Isomers as Divalent Cation Adducts and Their Electron Transfer Products. Analyst 2015, 140, 6912–6921. 10.1039/C5AN01093D. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Zhang X.; Schocker N. S.; Renslow R. S.; Orton D. J.; Khamsi J.; Ashmus R. A.; Almeida I. C.; Tang K.; Costello C. E.; et al. Enhancing Glycan Isomer Separations with Metal Ions and Positive and Negative Polarity Ion Mobility Spectrometry-Mass Spectrometry Analyses. Anal. Bioanal. Chem. 2017, 409, 467–476. 10.1007/s00216-016-9866-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struwe W. B.; Baldauf C.; Hofmann J.; Rudd P. M.; Pagel K. Ion Mobility Separation of Deprotonated Oligosaccharide Isomers - Evidence for Gas-Phase Charge Migration. Chem. Commun. 2016, 52, 12353–12356. 10.1039/C6CC06247D. [DOI] [PubMed] [Google Scholar]
- Pu Y.; Ridgeway M. E.; Glaskin R. S.; Park M. A.; Costello C. E.; Lin C. Separation and Identification of Isomeric Glycans by Selected Accumulation-Trapped Ion Mobility Spectrometry-Electron Activated Dissociation Tandem Mass Spectrometry. Anal. Chem. 2016, 88, 3440–3443. 10.1021/acs.analchem.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benigni P.; Thompson C. J.; Ridgeway M. E.; Park M. A.; Fernandez-Lima F. Targeted High-Resolution Ion Mobility Separation Coupled to Ultrahigh-Resolution Mass Spectrometry of Endocrine Disruptors in Complex Mixtures. Anal. Chem. 2015, 87, 4321–4325. 10.1021/ac504866v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmachev A. V.; Webb I. K.; Ibrahim Y. M.; Garimella S. V. B.; Zhang X.; Anderson G. A.; Smith R. D. Characterization of Ion Dynamics in Structures for Lossless Ion Manipulations. Anal. Chem. 2014, 86, 9162–9168. 10.1021/ac502054p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb I. K.; Garimella S. V. B.; Tolmachev A. V.; Chen T.-C.; Zhang X.; Norheim R. V.; Prost S. A.; LaMarche B.; Anderson G. A.; Ibrahim Y. M.; et al. Experimental Evaluation and Optimization of Structures for Lossless Ion Manipulations for Ion Mobility Spectrometry with Time-of-Flight Mass Spectrometry. Anal. Chem. 2014, 86, 9169–9176. 10.1021/ac502055e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid A. M.; Ibrahim Y. M.; Garimella S. V. B.; Webb I. K.; Deng L.; Chen T.-C.; Anderson G. A.; Prost S. A.; Norheim R. V.; Tolmachev A. V.; et al. Characterization of Traveling Wave Ion Mobility Separations in Structures for Lossless Ion Manipulations. Anal. Chem. 2015, 87, 11301–11308. 10.1021/acs.analchem.5b02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim Y. M.; Hamid A. M.; Deng L.; Garimella S. V. B.; Webb I. K.; Baker E. S.; Smith R. D. New Frontiers for Mass Spectrometry Based Upon Structures for Lossless Ion Manipulations. Analyst 2017, 142, 1010–1021. 10.1039/C7AN00031F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Ibrahim Y. M.; Hamid A. M.; Garimella S. V. B.; Webb I. K.; Zheng X.; Prost S. A.; Sandoval J. A.; Norheim R. V.; Anderson G. A.; et al. Ultra-High Resolution Ion Mobility Separations Utilizing Traveling Waves in a 13 M Serpentine Path Length Structures for Lossless Ion Manipulations Module. Anal. Chem. 2016, 88, 8957–8964. 10.1021/acs.analchem.6b01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X.; Chen Z.; Snovida S.; Liu Y.; Rogers J. C.; Li L. Capillary Electrophoresis-Electrospray Ionization-Mass Spectrometry for Quantitative Analysis of Glycans Labeled with Multiplex Carbonyl-Reactive Tandem Mass Tags. Anal. Chem. 2015, 87, 6527–6534. 10.1021/acs.analchem.5b01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanal N.; Masellis C.; Kamrath M. Z.; Clemmer D. E.; Rizzo T. R. Cryogenic Ir Spectroscopy Combined with Ion Mobility Spectrometry for the Analysis of Human Milk Oligosaccharides. Analyst 2018, 143, 1846–1852. 10.1039/C8AN00230D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Faleh A.; Warnke S.; Rizzo T. R. Combining Ultrahigh-Resolution Ion-Mobility Spectrometry with Cryogenic Infrared Spectroscopy for the Analysis of Glycan Mixtures. Anal. Chem. 2019, 91, 4876–4882. 10.1021/acs.analchem.9b00659. [DOI] [PubMed] [Google Scholar]
- Warnke S.; Ben Faleh A.; Pellegrinelli R. P.; Yalovenko N.; Rizzo T. R. Combining Ultra-High Resolution Ion Mobility Spectrometry with Cryogenic Ir Spectroscopy for the Study of Biomolecular Ions. Faraday Discuss. 2019, 217, 114–125. 10.1039/C8FD00180D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saparbaev E.; Kopysov V.; Aladinskaia V.; Ferrieres V.; Legentil L.; Boyarkin O. V. Identification and Quantification of Any Isoforms of Carbohydrates by 2d Uv-Ms Fingerprinting of Cold Ions. Anal. Chem. 2020, 92, 14624–14632. 10.1021/acs.analchem.0c03122. [DOI] [PubMed] [Google Scholar]
- Kornfeld R.; Kornfeld S. Assembly of Asparagine-Linked Oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Dell A.; Galadari A.; Sastre F.; Hitchen P. Similarities and Differences in the Glycosylation Mechanisms in Prokaryotes and Eukaryotes. Int. J. Microbiol. 2010, 2010, 148178. 10.1155/2010/148178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P.; Taniguchi N.; Aebi M. In Essentials of Glycobiology, 3rd ed.; Varki A., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2017, 10.1101/glycobiology.3e.009. [DOI] [Google Scholar]
- Ohtsubo K.; Marth J. D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Freeze H. H.; Aebi M. Altered Glycan Structures: The Molecular Basis of Congenital Disorders of Glycosylation. Curr. Opin. Struct. Biol. 2005, 15, 490–498. 10.1016/j.sbi.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Stanley P.; Cummings R. D. In Essentials of Glycobiology, 3rd ed.; Varki A., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2017, 10.1101/glycobiology.3e.014. [DOI] [PubMed] [Google Scholar]
- Campbell M. P.; Royle L.; Radcliffe C. M.; Dwek R. A.; Rudd P. M. Glycobase and Autogu: Tools for Hplc-Based Glycan Analysis. Bioinformatics 2008, 24, 1214–1216. 10.1093/bioinformatics/btn090. [DOI] [PubMed] [Google Scholar]
- Dwek R. A.; Edge C. J.; Harvey D. J.; Wormald M. R.; Parekh R. B. Analysis of Glycoprotein-Associated Oligosaccharides. Annu. Rev. Biochem. 1993, 62, 65–100. 10.1146/annurev.bi.62.070193.000433. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Rudd P. M.; Bateman R. H.; Bordoli R. S.; Howes K.; Hoyes J. B.; Vickers R. G. Examination of Complex Oligosaccharides by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry on Time-of-Flight and Magnetic Sector Instruments. Org. Mass Spectrom. 1994, 29, 753–766. 10.1002/oms.1210291208. [DOI] [Google Scholar]
- Reinhold V. N.; Reinhold B. B.; Costello C. E. Carbohydrate Molecular Weight Profiling, Sequence, Linkage, and Branching Data: Es-Ms and Cid. Anal. Chem. 1995, 67, 1772–1784. 10.1021/ac00107a005. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Naven T. J. P.; Küster B.; Bateman R. H.; Green M. R.; Critchley G. Comparison of Fragmentation Modes for the Structural Determination of Complex Oligosaccharides Ionized by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 1995, 9, 1556–1561. 10.1002/rcm.1290091517. [DOI] [PubMed] [Google Scholar]
- Lancaster K. S.; An H. J.; Li B.; Lebrilla C. B. Interrogation of N-Linked Oligosaccharides Using Infrared Multiphoton Dissociation in Ft-Icr Mass Spectrometry. Anal. Chem. 2006, 78, 4990–4997. 10.1021/ac0600656. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Electrospray Mass Spectrometry and Fragmentation of N-Linked Carbohydrates Derivatized at the Reducing Terminus. J. Am. Soc. Mass Spectrom. 2000, 11, 900–915. 10.1016/S1044-0305(00)00156-2. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Electrospray Mass Spectrometry and Collision-Induced Fragmentation of 2-Aminobenzamide-Labelled Neutral N-Linked Glycans. Analyst 2000, 125, 609–617. 10.1039/a908997g. [DOI] [Google Scholar]
- Harvey D. J. Ionization and Fragmentation of N-Linked Glycans as Silver Adducts by Electrospray Mass Spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 484–492. 10.1002/rcm.1815. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Collision-Induced Fragmentation of Negative Ions from N-Linked Glycans Derivatized with 2-Aminobenzoic Acid. J. Mass Spectrom. 2005, 40, 642–653. 10.1002/jms.836. [DOI] [PubMed] [Google Scholar]
- Wuhrer M.; Deelder A. M. Negative-Mode Maldi-Tof/Tof-Ms of Oligosaccharides Labeled with 2-Aminobenzamide. Anal. Chem. 2005, 77, 6954–6959. 10.1021/ac051117e. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Fragmentation of Negative Ions from Carbohydrates: Part 1. Use of Nitrate and Other Anionic Adducts for the Production of Negative Ion Electrospray Spectra from N-Linked Carbohydrates. J. Am. Soc. Mass Spectrom. 2005, 16, 622–630. 10.1016/j.jasms.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Fragmentation of Negative Ions from Carbohydrates: Part 2. Fragmentation of High-Mannose N-Linked Glycans. J. Am. Soc. Mass Spectrom. 2005, 16, 631–646. 10.1016/j.jasms.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Fragmentation of Negative Ions from Carbohydrates: Part 3. Fragmentation of Hybrid and Complex N-Linked Glycans. J. Am. Soc. Mass Spectrom. 2005, 16, 647–659. 10.1016/j.jasms.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Royle L.; Radcliffe C. M.; Rudd P. M.; Dwek R. A. Structural and Quantitative Analysis of N-Linked Glycans by Matrix-Assisted Laser Desorption Ionization and Negative Ion Nanospray Mass Spectrometry. Anal. Biochem. 2008, 376, 44–60. 10.1016/j.ab.2008.01.025. [DOI] [PubMed] [Google Scholar]
- Domann P.; Spencer D. I.; Harvey D. J. Production and Fragmentation of Negative Ions from Neutral N-Linked Carbohydrates Ionized by Matrix-Assisted Laser Desorption/Ionization. Rapid Commun. Mass Spectrom. 2012, 26, 469–479. 10.1002/rcm.5322. [DOI] [PubMed] [Google Scholar]
- Powell A. K.; Harvey D. J. Stabilization of Sialic Acids in N-Linked Oligosaccharides and Gangliosides for Analysis by Positive Ion Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1027–1032. . [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Bateman R. H.; Bordoli R. S.; Tyldesley R. Ionisation and Fragmentation of Complex Glycans with a Quadrupole Time-of-Flight Mass Spectrometer Fitted with a Matrix-Assisted Laser Desorption/Ionisation Ion Source. Rapid Commun. Mass Spectrom. 2000, 14, 2135–2142. . [DOI] [PubMed] [Google Scholar]
- Reiding K. R.; Blank D.; Kuijper D. M.; Deelder A. M.; Wuhrer M. High-Throughput Profiling of Protein N-Glycosylation by Maldi-Tof-Ms Employing Linkage-Specific Sialic Acid Esterification. Anal. Chem. 2014, 86, 5784–5793. 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
- Bladergroen M. R.; Reiding K. R.; Hipgrave Ederveen A. L.; Vreeker G. C.; Clerc F.; Holst S.; Bondt A.; Wuhrer M.; van der Burgt Y. E. Automation of High-Throughput Mass Spectrometry-Based Plasma N-Glycome Analysis with Linkage-Specific Sialic Acid Esterification. J. Proteome Res. 2015, 14, 4080–4086. 10.1021/acs.jproteome.5b00538. [DOI] [PubMed] [Google Scholar]
- Wheeler S. F.; Harvey D. J. Negative Ion Mass Spectrometry of Sialylated Carbohydrates: Discrimination of N-Acetylneuraminic Acid Linkages by Maldi-Tof and Esi-Tof Mass Spectrometry. Anal. Chem. 2000, 72, 5027–5039. 10.1021/ac000436x. [DOI] [PubMed] [Google Scholar]
- Seymour J. L.; Costello C. E.; Zaia J. The Influence of Sialylation on Glycan Negative Ion Dissociation and Energetics. J. Am. Soc. Mass Spectrom. 2006, 17, 844–854. 10.1016/j.jasms.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood C.; Lin C. H.; Thaysen-Andersen M.; Packer N. H. Discrimination of Isomers of Released N- and O-Glycans Using Diagnostic Product Ions in Negative Ion Pgc-Lc-Esi-Ms/Ms. J. Am. Soc. Mass Spectrom. 2018, 29, 1194–1209. 10.1007/s13361-018-1932-z. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Rudd P. M. Fragmentation of Negative Ions from N-Linked Carbohydrates. Part 5: Anionic N-Linked Glycans. Int. J. Mass Spectrom. 2011, 305, 120–130. 10.1016/j.ijms.2010.10.016. [DOI] [Google Scholar]
- Wuhrer M.; Koeleman C. A. M.; Hokke C. H.; Deelder A. M. Mass Spectrometry of Proton Adducts of Fucosylated N-Glycans: Fucose Transfer between Antennae Gives Rise to Misleading Fragments. Rapid Commun. Mass Spectrom. 2006, 20, 1747–1754. 10.1002/rcm.2509. [DOI] [PubMed] [Google Scholar]
- Wuhrer M.; Koeleman C. A. M.; Deelder A. M. Hexose Rearrangements Upon Fragmentation of N-Glycopeptides and Reductively Aminated N-Glycans. Anal. Chem. 2009, 81, 4422–4432. 10.1021/ac900278q. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Baruah K.; Scanlan C. N. Application of Negative Ion Ms/Ms to the Identification of N-Glycans Released from Carcinoembryonic Antigen Cell Adhesion Molecule 1 (Ceacam1). J. Mass Spectrom. 2009, 44, 50–60. 10.1002/jms.1470. [DOI] [PubMed] [Google Scholar]
- Wheeler S. F.; Harvey D. J. Extension of the in-Gel Release Method for Structural Analysis of Neutral and Sialylated N-Linked Glycans to the Analysis of Sulfated Glycans: Application to the Glycans from Bovine Thyroid-Stimulating Hormone. Anal. Biochem. 2001, 296, 92–100. 10.1006/abio.2001.5199. [DOI] [PubMed] [Google Scholar]
- Zhou S.; Dong X.; Veillon L.; Huang Y.; Mechref Y. Lc-Ms/Ms Analysis of Permethylated N-Glycans Facilitating Isomeric Characterization. Anal. Bioanal. Chem. 2017, 409, 453–466. 10.1007/s00216-016-9996-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J. Structural Determination of N-Linked Glycans by Matrix-Assisted Laser Desorption/Ionization and Electrospray Ionization Mass Spectrometry. Proteomics 2005, 5, 1774–1786. 10.1002/pmic.200401248. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Dwek R. A.; Rudd P. M. Determining the Structure of Glycan Moieties by Mass Spectrometry. Curr. Protoc. Protein Sci. 2006, 10.1002/0471140864.ps1207s43. [DOI] [PubMed] [Google Scholar]
- Sheeley D. M.; Reinhold V. N. Structural Characterization of Carbohydrate Sequence, Linkage, and Branching in a Quadrupole Ion Trap Mass Spectrometer: Neutral Oligosaccharides and N-Linked Glycans. Anal. Chem. 1998, 70, 3053–3059. 10.1021/ac9713058. [DOI] [PubMed] [Google Scholar]
- Weiskopf A. S.; Vouros P.; Harvey D. J. Electrospray Ionization-Ion Trap Mass Spectrometry for Structural Analysis of Complex N-Linked Glycoprotein Oligosaccharides. Anal. Chem. 1998, 70, 4441–4447. 10.1021/ac980289r. [DOI] [PubMed] [Google Scholar]
- Creaser C. S.; Reynolds J. C.; Harvey D. J. Structural Analysis of Oligosaccharides by Atmospheric Pressure Matrix-Assisted Laser Desorption/Ionisation Quadrupole Ion Trap Mass Spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 176–184. 10.1002/rcm.563. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Martin R. L.; Jackson K. A.; Sutton C. W. Fragmentation of N-Linked Glycans with a Matrix-Assisted Laser Desorption/Ionization Ion Trap Time-of-Flight Mass Spectrometer. Rapid Commun. Mass Spectrom. 2004, 18, 2997–3007. 10.1002/rcm.1709. [DOI] [PubMed] [Google Scholar]
- Lapadula A. J.; Hatcher P. J.; Hanneman A. J.; Ashline D. J.; Zhang H.; Reinhold V. N. Congruent Strategies for Carbohydrate Sequencing. 3. Oscar: An Algorithm for Assigning Oligosaccharide Topology from Msn Data. Anal. Chem. 2005, 77, 6271–6279. 10.1021/ac050726j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashline D. J.; Lapadula A. J.; Liu Y. H.; Lin M.; Grace M.; Pramanik B.; Reinhold V. N. Carbohydrate Structural Isomers Analyzed by Sequential Mass Spectrometry. Anal. Chem. 2007, 79, 3830–3842. 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold V.; Zhang H.; Hanneman A.; Ashline D. Toward a Platform for Comprehensive Glycan Sequencing. Mol. Cell. Proteomics 2013, 12, 866–873. 10.1074/mcp.R112.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashline D. J.; Hanneman A. J. S.; Zhang H.; Reinhold V. N. Structural Documentation of Glycan Epitopes: Sequential Mass Spectrometry and Spectral Matching. J. Am. Soc. Mass Spectrom. 2014, 25, 444–453. 10.1007/s13361-013-0776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashline D. J.; Zhang H.; Reinhold V. N. Isomeric Complexity of Glycosylation Documented by Msn. Anal. Bioanal. Chem. 2017, 409, 439–451. 10.1007/s00216-016-0018-7. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Hakansson K. Electron Capture Dissociation of Divalent Metal-Adducted Sulfated N-Glycans Released from Bovine Thyroid Stimulating Hormone. J. Am. Soc. Mass Spectrom. 2013, 24, 1798–1806. 10.1007/s13361-013-0700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Xie B.; Chan S. Y.; Costello C. E.; O’Connor P. B. Collisionally Activated Dissociation and Electron Capture Dissociation Provide Complementary Structural Information for Branched Permethylated Oligosaccharides. J. Am. Soc. Mass Spectrom. 2008, 19, 138–150. 10.1016/j.jasms.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Yu X.; Huang Y.; Lin C.; Costello C. E. Energy-Dependent Electron Activated Dissociation of Metal-Adducted Permethylated Oligosaccharides. Anal. Chem. 2012, 84, 7487–7494. 10.1021/ac301589z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Tang Y.; Bai Y.; Zaia J.; Costello C. E.; Hong P.; Lin C. Toward Automatic and Comprehensive Glycan Characterization by Online Pgc-Lc-Eed Ms/Ms. Anal. Chem. 2020, 92, 782–791. 10.1021/acs.analchem.9b03183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Håkansson K. Electron Detachment Dissociation of Fluorescently Labeled Sialylated Oligosaccharides. Electrophoresis 2011, 32, 3526–3535. 10.1002/elps.201100327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devakumar A.; Mechref Y.; Kang P.; Novotny M. V.; Reilly J. P. Laser-Induced Photofragmentation of Neutral and Acidic Glycans inside an Ion-Trap Mass Spectrometer. Rapid Commun. Mass Spectrom. 2007, 21, 1452–1460. 10.1002/rcm.2981. [DOI] [PubMed] [Google Scholar]
- Devakumar A.; Mechref Y.; Kang P.; Novotny M.; Reilly J. Identification of Isomeric N-Glycan Structures by Mass Spectrometry with 157 Nm Laser-Induced Photofragmentation. J. Am. Soc. Mass Spectrom. 2008, 19, 1027–1040. 10.1016/j.jasms.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prien J. M.; Ashline D. J.; Lapadula A. J.; Zhang H.; Reinhold V. N. The High Mannose Glycans from Bovine Ribonuclease B Isomer Characterization by Ion Trap Ms. J. Am. Soc. Mass Spectrom. 2009, 20, 539–556. 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabryelski W.; Froese K. L. Rapid and Sensitive Differentiation of Anomers, Linkage, and Position Isomers of Disaccharides Using High-Field Asymmetric Waveform Ion Mobility Spectrometry (Faims). J. Am. Soc. Mass Spectrom. 2003, 14, 265–277. 10.1016/S1044-0305(03)00002-3. [DOI] [PubMed] [Google Scholar]
- Clowers B. H.; Dwivedi P.; Steiner W. E.; Hill H. H.; Bendiak B. Separation of Sodiated Isobaric Disaccharides and Trisaccharides Using Electrospray Ionization-Atmospheric Pressure Ion Mobility-Time of Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2005, 16, 660–669. 10.1016/j.jasms.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Both P.; Green A. P.; Gray C. J.; Šardzík R.; Voglmeir J.; Fontana C.; Austeri M.; Rejzek M.; Richardson D.; Field R. A.; et al. Discrimination of Epimeric Glycans and Glycopeptides Using Im-Ms and Its Potential for Carbohydrate Sequencing. Nat. Chem. 2014, 6, 65–74. 10.1038/nchem.1817. [DOI] [PubMed] [Google Scholar]
- Isailovic D.; Kurulugama R. T.; Plasencia M. D.; Stokes S. T.; Kyselova Z.; Goldman R.; Mechref Y.; Novotny M. V.; Clemmer D. E. Profiling of Human Serum Glycans Associated with Liver Cancer and Cirrhosis by Ims-Ms. J. Proteome Res. 2008, 7, 1109–1117. 10.1021/pr700702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasencia M. D.; Isailovic D.; Merenbloom S. I.; Mechref Y.; Clemmer D. E. Resolving and Assigning N-Linked Glycan Structural Isomers from Ovalbumin by Ims-Ms. J. Am. Soc. Mass Spectrom. 2008, 19, 1706–1715. 10.1016/j.jasms.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen C. W. N.; Chen W.; Chakraborty A. B.; Oosterhout M.; Mazzeo J. R.; Gebler J. C.; Schellens J. H. M.; Rosing H.; Beijnen J. H. Electrospray Ionization Quadrupole Ion-Mobility Time-of-Flight Mass Spectrometry as a Tool to Distinguish the Lot-to-Lot Heterogeneity in N-Glycosylation Profile of the Therapeutic Monoclonal Antibody Trastuzumab. J. Am. Soc. Mass Spectrom. 2009, 20, 2021–2033. 10.1016/j.jasms.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Fenn L. S.; Mclean J. A. Simultaneous Glycoproteomics on the Basis of Structure Using Ion Mobility-Mass Spectrometry. Mol. BioSyst. 2009, 5, 1298. 10.1039/b909745g. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Sobott F.; Crispin M.; Wrobel A.; Bonomelli C.; Vasiljevic S.; Scanlan C. N.; Scarff C. A.; Thalassinos K.; Scrivens J. H. Ion Mobility Mass Spectrometry for Extracting Spectra of N-Glycans Directly from Incubation Mixtures Following Glycan Release: Application to Glycans from Engineered Glycoforms of Intact, Folded Hiv Gp120. J. Am. Soc. Mass Spectrom. 2011, 22, 568–581. 10.1007/s13361-010-0053-0. [DOI] [PubMed] [Google Scholar]
- Gaye M. M.; Valentine S. J.; Hu Y.; Mirjankar N.; Hammoud Z. T.; Mechref Y.; Lavine B. K.; Clemmer D. E. Ion Mobility-Mass Spectrometry Analysis of Serum N-Linked Glycans from Esophageal Adenocarcinoma Phenotypes. J. Proteome Res. 2012, 11, 6102–6110. 10.1021/pr300756e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isailovic D.; Plasencia M. D.; Gaye M. M.; Stokes S. T.; Kurulugama R. T.; Pungpapong V.; Zhang M.; Kyselova Z.; Goldman R.; Mechref Y.; et al. Delineating Diseases by Ims-Ms Profiling of Serum N-Linked Glycans. J. Proteome Res. 2012, 11, 576–585. 10.1021/pr200777u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Crispin M.; Bonomelli C.; Scrivens J. H. Ion Mobility Mass Spectrometry for Ion Recovery and Clean-up of Ms and Ms/Ms Spectra Obtained from Low Abundance Viral Samples. J. Am. Soc. Mass Spectrom. 2015, 26, 1754–1767. 10.1007/s13361-015-1163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Scarff C. A.; Crispin M.; Scanlan C. N.; Bonomelli C.; Scrivens J. H. Maldi-Ms/Ms with Traveling Wave Ion Mobility for the Structural Analysis of N-Linked Glycans. J. Am. Soc. Mass Spectrom. 2012, 23, 1955–1966. 10.1007/s13361-012-0425-8. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Scarff C. A.; Edgeworth M.; Crispin M.; Scanlan C. N.; Sobott F.; Allman S.; Baruah K.; Pritchard L.; Scrivens J. H. Travelling Wave Ion Mobility and Negative Ion Fragmentation for the Structural Determination of N-Linked Glycans. Electrophoresis 2013, 34, 2368–2378. 10.1002/elps.201200669. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y.; Nishima W.; Re S.; Sugita Y. Confident Identification of Isomeric N-Glycan Structures by Combined Ion Mobility Mass Spectrometry and Hydrophilic Interaction Liquid Chromatography. Rapid Commun. Mass Spectrom. 2012, 26, 2877–2884. 10.1002/rcm.6412. [DOI] [PubMed] [Google Scholar]
- Zhu F.; Lee S.; Valentine S. J.; Reilly J. P.; Clemmer D. E. Mannose7 Glycan Isomer Characterization by Ims-Ms/Ms Analysis. J. Am. Soc. Mass Spectrom. 2012, 23, 2158–2166. 10.1007/s13361-012-0491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel K.; Harvey D. J. Ion Mobility-Mass Spectrometry of Complex Carbohydrates: Collision Cross Sections of Sodiated N-Linked Glycans. Anal. Chem. 2013, 85, 5138–5145. 10.1021/ac400403d. [DOI] [PubMed] [Google Scholar]
- Hofmann J.; Struwe W. B.; Scarff C. A.; Scrivens J. H.; Harvey D. J.; Pagel K. Estimating Collision Cross Sections of Negatively Charged N-Glycans Using Traveling Wave Ion Mobility-Mass Spectrometry. Anal. Chem. 2014, 86, 10789–10795. 10.1021/ac5028353. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Edgeworth M.; Krishna B. A.; Bonomelli C.; Allman S. A.; Crispin M.; Scrivens J. H. Fragmentation of Negative Ions from N-Linked Carbohydrates: Part 6. Glycans Containing One N-Acetylglucosamine in the Core. Rapid Commun. Mass Spectrom. 2014, 28, 2008–2018. 10.1002/rcm.6980. [DOI] [PubMed] [Google Scholar]
- Struwe W. B.; Benesch J. L.; Harvey D. J.; Pagel K. Collision Cross Sections of High-Mannose N-Glycans in Commonly Observed Adduct States--Identification of Gas-Phase Conformers Unique to [M-H](−) Ions. Analyst 2015, 140, 6799–6803. 10.1039/C5AN01092F. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Abrahams J. L. Fragmentation and Ion Mobility Properties of Negative Ions from N-Linked Carbohydrates: Part 7. Reduced Glycans. Rapid Commun. Mass Spectrom. 2016, 30, 627–634. 10.1002/rcm.7467. [DOI] [PubMed] [Google Scholar]
- Zhu F.; Glover M. S.; Shi H.; Trinidad J. C.; Clemmer D. E. Populations of Metal-Glycan Structures Influence Ms Fragmentation Patterns. J. Am. Soc. Mass Spectrom. 2015, 26, 25–35. 10.1007/s13361-014-1000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Scarff C. A.; Edgeworth M.; Pagel K.; Thalassinos K.; Struwe W. B.; Crispin M.; Scrivens J. H. Travelling-Wave Ion Mobility Mass Spectrometry and Negative Ion Fragmentation of Hybrid and Complex N-Glycans. J. Mass Spectrom. 2016, 51, 1064–1079. 10.1002/jms.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Scarff C. A.; Edgeworth M.; Struwe W. B.; Pagel K.; Thalassinos K.; Crispin M.; Scrivens J. Travelling-Wave Ion Mobility and Negative Ion Fragmentation of High-Mannose N-Glycans. J. Mass Spectrom. 2016, 51, 219–235. 10.1002/jms.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J.; Stuckmann A.; Crispin M.; Harvey D. J.; Pagel K.; Struwe W. B. Identification of Lewis and Blood Group Carbohydrate Epitopes by Ion Mobility-Tandem-Mass Spectrometry Fingerprinting. Anal. Chem. 2017, 89, 2318–2325. 10.1021/acs.analchem.6b03853. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Struwe W. B. Structural Studies of Fucosylated N-Glycans by Ion Mobility Mass Spectrometry and Collision-Induced Fragmentation of Negative Ions. J. Am. Soc. Mass Spectrom. 2018, 29, 1179–1193. 10.1007/s13361-018-1950-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Watanabe Y.; Allen J. D.; Rudd P.; Pagel K.; Crispin M.; Struwe W. B. Collision Cross Sections and Ion Mobility Separation of Fragment Ions from Complex N-Glycans. J. Am. Soc. Mass Spectrom. 2018, 29, 1250–1261. 10.1007/s13361-018-1930-1. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Seabright G. E.; Vasiljevic S.; Crispin M.; Struwe W. B. Isomer Information from Ion Mobility Separation of High-Mannose Glycan Fragments. J. Am. Soc. Mass Spectrom. 2018, 29, 972–988. 10.1007/s13361-018-1890-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re S.; Watabe S.; Nishima W.; Muneyuki E.; Yamaguchi Y.; Mackerell A. D.; Sugita Y. Characterization of Conformational Ensembles of Protonated N-Glycans in the Gas-Phase. Sci. Rep. 2018, 10.1038/s41598-018-20012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallister E. G.; Choo M. S. F.; Walsh I.; Tai J. N.; Tay S. J.; Yang Y. S.; Ng S. K.; Rudd P. M.; Flitsch S. L.; Nguyen-Khuong T. Utility of Ion-Mobility Spectrometry for Deducing Branching of Multiply Charged Glycans and Glycopeptides in a High-Throughput Positive Ion Lc-Flr-Ims-Ms Workflow. Anal. Chem. 2020, 92, 15323–15335. 10.1021/acs.analchem.0c01954. [DOI] [PubMed] [Google Scholar]
- Wei J.; Tang Y.; Ridgeway M. E.; Park M. A.; Costello C. E.; Lin C. Accurate Identification of Isomeric Glycans by Trapped Ion Mobility Spectrometry-Electronic Excitation Dissociation Tandem Mass Spectrometry. Anal. Chem. 2020, 92, 13211–13220. 10.1021/acs.analchem.0c02374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depraz Depland A.; Renois-Predelus G.; Schindler B.; Compagnon I. Identification of Sialic Acid Linkage Isomers in Glycans Using Coupled Infrared Multiple Photon Dissociation (Irmpd) Spectroscopy and Mass Spectrometry. Int. J. Mass Spectrom. 2018, 434, 65–69. 10.1016/j.ijms.2018.09.005. [DOI] [Google Scholar]
- Dyukova I.; Carrascosa E.; Pellegrinelli R. P.; Rizzo T. R. Combining Cryogenic Infrared Spectroscopy with Selective Enzymatic Cleavage for Determining Glycan Primary Structure. Anal. Chem. 2020, 92, 1658–1662. 10.1021/acs.analchem.9b04776. [DOI] [PubMed] [Google Scholar]
- Dyukova I.; Ben Faleh A.; Warnke S.; Yalovenko N.; Yatsyna V.; Bansal P.; Rizzo T. R. A New Approach for Identifying Positional Isomers of Glycans Cleaved from Monoclonal Antibodies. Analyst 2021, 146, 4789–4795. 10.1039/D1AN00780G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhausen I.; Stanley P. In Essentials of Glycobiology, 3rd ed.; Ajit Varki R. D. C., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Ed.; Cold Spring Harbor Laboratory Press, 2017. [PubMed] [Google Scholar]
- Venkatakrishnan V.; Packer N. H.; Thaysen-Andersen M. Host Mucin Glycosylation Plays a Role in Bacterial Adhesion in Lungs of Individuals with Cystic Fibrosis. Expert Rev. Respir. Med. 2013, 7, 553–576. 10.1586/17476348.2013.837752. [DOI] [PubMed] [Google Scholar]
- Zachara N.; Akimoto Y.; Hart G. W. In Essentials of Glycobiology, 3rd ed.; Ajit Varki R. D. C., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita Taroh, Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Ed.; Cold Spring Harbor Laboratory Press, 2017. [PubMed] [Google Scholar]
- Brockhausen I. In Comprehensive Glycoscience; Kamerling H., Ed.; Elsevier: Oxford, 2007, 10.1016/B978-044451967-2/00035-0. [DOI] [Google Scholar]
- Wilkinson H.; Saldova R. Current Methods for the Characterization of O-Glycans. J. Proteome Res. 2020, 19, 3890–3905. 10.1021/acs.jproteome.0c00435. [DOI] [PubMed] [Google Scholar]
- Zauner G.; Kozak R. P.; Gardner R. A.; Fernandes D. L.; Deelder A. M.; Wuhrer M. Protein O-Glycosylation Analysis. Biol. Chem. 2012, 393, 687–708. 10.1515/hsz-2012-0144. [DOI] [PubMed] [Google Scholar]
- Karlsson N. G.; Jin C.; Rojas-Macias M. A.; Adamczyk B. Next Generation O-Linked Glycomics. Trends Glycosci. Glycotechnol. 2017, 29, E35–E46. 10.4052/tigg.1602.1E. [DOI] [Google Scholar]
- Buszewski B.; Noga S. Hydrophilic Interaction Liquid Chromatography (Hilic)—a Powerful Separation Technique. Anal. Bioanal. Chem. 2012, 402, 231–247. 10.1007/s00216-011-5308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsson K. A.; Karlsson N. G.; Hansson G. C. Liquid Chromatography-Electrospray Mass Spectrometry as a Tool for the Analysis of Sulfated Oligosaccharides from Mucin Glycoproteins. J. Chromatogr. A 1999, 854, 131–139. 10.1016/S0021-9673(99)00625-1. [DOI] [PubMed] [Google Scholar]
- Melmer M.; Stangler T.; Premstaller A.; Lindner W. Comparison of Hydrophilic-Interaction, Reversed-Phase and Porous Graphitic Carbon Chromatography for Glycan Analysis. J. Chromatogr. A 2011, 1218, 118–123. 10.1016/j.chroma.2010.10.122. [DOI] [PubMed] [Google Scholar]
- Leymarie N.; Zaia J. Effective Use of Mass Spectrometry for Glycan and Glycopeptide Structural Analysis. Anal. Chem. 2012, 84, 3040–3048. 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia J. Mass Spectrometry and Glycomics. OMICS 2010, 14, 401–418. 10.1089/omi.2009.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C.; Kenny D. T.; Skoog E. C.; Padra M.; Adamczyk B.; Vitizeva V.; Thorell A.; Venkatakrishnan V.; Lindén S. K.; Karlsson N. G. Structural Diversity of Human Gastric Mucin Glycans*. Molecular & Cellular Proteomics 2017, 16, 743–758. 10.1074/mcp.M117.067983. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Lindsay L. L.; Hedrick J. L.; Lebrilla C. B. Strategy for Profiling and Structure Elucidation of Mucin-Type Oligosaccharides by Mass Spectrometry. Anal. Chem. 2004, 76, 5990–6001. 10.1021/ac049666s. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Liu J.; Zhang J.; Hedrick J. L.; Lebrilla C. B. Method for the Comparative Glycomic Analyses of O-Linked, Mucin-Type Oligosaccharides. Anal. Chem. 2004, 76, 5186–5197. 10.1021/ac0496953. [DOI] [PubMed] [Google Scholar]
- Jensen P. H.; Karlsson N. G.; Kolarich D.; Packer N. H. Structural Analysis of N- and O-Glycans Released from Glycoproteins. Nat. Protoc. 2012, 7, 1299–1310. 10.1038/nprot.2012.063. [DOI] [PubMed] [Google Scholar]
- Kolarich D.; Jensen P. H.; Altmann F.; Packer N. H. Determination of Site-Specific Glycan Heterogeneity on Glycoproteins. Nat. Protoc. 2012, 7, 1285–1298. 10.1038/nprot.2012.062. [DOI] [PubMed] [Google Scholar]
- Zauner G.; Koeleman C. A. M.; Deelder A. M.; Wuhrer M. Mass Spectrometric O-Glycan Analysis after Combined O-Glycan Release by Beta-Elimination and 1-Phenyl-3-Methyl-5-Pyrazolone Labeling. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1420–1428. 10.1016/j.bbagen.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Doohan R. A.; Hayes C. A.; Harhen B.; Karlsson N. G. Negative Ion Cid Fragmentation of O-Linked Oligosaccharide Aldoses—Charge Induced and Charge Remote Fragmentation. J. Am. Soc. Mass Spectrom. 2011, 22, 1052. 10.1007/s13361-011-0102-3. [DOI] [PubMed] [Google Scholar]
- Ashwood C.; Abrahams J. L.; Nevalainen H.; Packer N. H. Enhancing Structural Characterisation of Glucuronidated O-Linked Glycans Using Negative Mode Ion Trap Higher Energy Collision-Induced Dissociation Mass Spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 851–858. 10.1002/rcm.7851. [DOI] [PubMed] [Google Scholar]
- Issa S. M. A.; Vitiazeva V.; Hayes C. A.; Karlsson N. G. Higher Energy Collisional Dissociation Mass Spectrometry of Sulfated O-Linked Oligosaccharides. J. Proteome Res. 2018, 17, 3259–3267. 10.1021/acs.jproteome.8b00376. [DOI] [PubMed] [Google Scholar]
- Jiao J.; Zhang H.; Reinhold V. N. High Performance It-Msn Sequencing of Glycans: Spatial Resolution of Ovalbumin Isomers. Int. J. Mass Spectrom. 2011, 303, 109–117. 10.1016/j.ijms.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciucanu I.; Kerek F. A Simple and Rapid Method for the Permethylation of Carbohydrates. Carbohydr. Res. 1984, 131, 209–217. 10.1016/0008-6215(84)85242-8. [DOI] [Google Scholar]
- Li Z.; Zhang Q.; Ashline D.; Zhu Y.; Lasanajak Y.; Chernova T.; Reinhold V.; Cummings R. D.; Wang P. G.; Ju T.; et al. Amplification and Preparation of Cellular O-Glycomes for Functional Glycomics. Anal. Chem. 2020, 92, 10390–10401. 10.1021/acs.analchem.0c00632. [DOI] [PubMed] [Google Scholar]
- Karlsson N. G.; Schulz B. L.; Packer N. H. Structural Determination of Neutral O-Linked Oligosaccharide Alditols by Negative Ion Lc-Electrospray-Msn. J. Am. Soc. Mass Spectrom. 2004, 15, 659–672. 10.1016/j.jasms.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Ali L.; Kenny D. T.; Hayes C. A.; Karlsson N. G. Structural Identification of O-Linked Oligosaccharides Using Exoglycosidases and Msn Together with Unicarb-Db Fragment Spectra Comparison. Metabolites 2012, 2, 648. 10.3390/metabo2040648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison K. A.; Clowers B. H. Differential Fragmentation of Mobility-Selected Glycans Via Ultraviolet Photodissociation and Ion Mobility-Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1236–1241. 10.1007/s13361-017-1621-3. [DOI] [PubMed] [Google Scholar]
- Jin C.; Harvey D. J.; Struwe W. B.; Karlsson N. G. Separation of Isomeric O-Glycans by Ion Mobility and Liquid Chromatography-Mass Spectrometry. Anal. Chem. 2019, 91, 10604–10613. 10.1021/acs.analchem.9b01772. [DOI] [PubMed] [Google Scholar]
- Everest-Dass A. V.; Abrahams J. L.; Kolarich D.; Packer N. H.; Campbell M. P. Structural Feature Ions for Distinguishing N- and O-Linked Glycan Isomers by Lc-Esi-It Ms/Ms. J. Am. Soc. Mass Spectrom. 2013, 24, 895–906. 10.1007/s13361-013-0610-4. [DOI] [PubMed] [Google Scholar]
- Campbell M. P.; Nguyen-Khuong T.; Hayes C. A.; Flowers S. A.; Alagesan K.; Kolarich D.; Packer N. H.; Karlsson N. G. Validation of the Curation Pipeline of Unicarb-Db: Building a Global Glycan Reference Ms/Ms Repository. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 108–116. 10.1016/j.bbapap.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Struwe W. B.; Pagel K.; Benesch J. L. P.; Harvey D. J.; Campbell M. P. Glycomob: An Ion Mobility-Mass Spectrometry Collision Cross Section Database for Glycomics. Glycoconjugate J. 2016, 33, 399–404. 10.1007/s10719-015-9613-7. [DOI] [PubMed] [Google Scholar]
- Remoroza C. A.; Mak T. D.; De Leoz M. L. A.; Mirokhin Y. A.; Stein S. E. Creating a Mass Spectral Reference Library for Oligosaccharides in Human Milk. Anal. Chem. 2018, 90, 8977–8988. 10.1021/acs.analchem.8b01176. [DOI] [PubMed] [Google Scholar]
- Pino L. K.; Searle B. C.; Bollinger J. G.; Nunn B.; MacLean B.; MacCoss M. J. The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York W. S.; Agravat S.; Aoki-Kinoshita K. F.; McBride R.; Campbell M. P.; Costello C. E.; Dell A.; Feizi T.; Haslam S. M.; Karlsson N.; et al. Mirage: The Minimum Information Required for a Glycomics Experiment. Glycobiology 2014, 24, 402–406. 10.1093/glycob/cwu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarich D.; Rapp E.; Struwe W. B.; Haslam S. M.; Zaia J.; McBride R.; Agravat S.; Campbell M. P.; Kato M.; Ranzinger R.; et al. The Minimum Information Required for a Glycomics Experiment (Mirage) Project: Improving the Standards for Reporting Mass-Spectrometry-Based Glycoanalytic Data. Molecular & Cellular Proteomics 2013, 12, 991–995. 10.1074/mcp.O112.026492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M. P.; Abrahams J. L.; Rapp E.; Struwe W. B.; Costello C. E.; Novotny M.; Ranzinger R.; York W. S.; Kolarich D.; Rudd P. M.; et al. The Minimum Information Required for a Glycomics Experiment (Mirage) Project: Lc Guidelines. Glycobiology 2019, 29, 349–354. 10.1093/glycob/cwz009. [DOI] [PubMed] [Google Scholar]
- Rojas-Macias M. A.; Mariethoz J.; Andersson P.; Jin C.; Venkatakrishnan V.; Aoki N. P.; Shinmachi D.; Ashwood C.; Madunic K.; Zhang T.; et al. Towards a Standardized Bioinformatics Infrastructure for N- and O-Glycomics. Nat. Commun. 2019, 10, 3275. 10.1038/s41467-019-11131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhrer M.; Deelder A. M.; van der Burgt Y. E. Mass Spectrometric Glycan Rearrangements. Mass Spectrom. Rev. 2011, 30, 664–680. 10.1002/mas.20337. [DOI] [PubMed] [Google Scholar]
- Hecht E. S.; Loziuk P. L.; Muddiman D. C. Xylose Migration During Tandem Mass Spectrometry of N-Linked Glycans. J. Am. Soc. Mass Spectrom. 2017, 28, 729–732. 10.1007/s13361-016-1588-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kováčik V.; Hirsch J.; Kováč P.; Heerma W.; Thomas-Oates J.; Haverkamp J. Oligosaccharide Characterization Using Collision-Induced Dissociation Fast Atom Bombardment Mass Spectrometry: Evidence for Internal Monosaccharide Residue Loss. J. Mass Spectrom. 1995, 30, 949–958. 10.1002/jms.1190300704. [DOI] [Google Scholar]
- Ma Y.-L.; Vedernikova I.; Heuvel H.; Claeys M. Internal Glucose Residue Loss in Protonated O-Diglycosyl Flavonoids Upon Low-Energy Collision-Induced Dissociation. J. Am. Soc. Mass Spectrom. 2000, 11, 136–144. 10.1016/S1044-0305(99)00133-6. [DOI] [PubMed] [Google Scholar]
- Sastre Torano J.; Gagarinov I. A.; Vos G. M.; Broszeit F.; Srivastava A. D.; Palmer M.; Langridge J. I.; Aizpurua-Olaizola O.; Somovilla V. J.; Boons G. J. Ion-Mobility Spectrometry Can Assign Exact Fucosyl Positions in Glycans and Prevent Misinterpretation of Mass-Spectrometry Data after Gas-Phase Rearrangement. Angew. Chem., Int. Ed. 2019, 58, 17616–17620. 10.1002/anie.201909623. [DOI] [PubMed] [Google Scholar]
- Lettow M.; Mucha E.; Manz C.; Thomas D. A.; Marianski M.; Meijer G.; von Helden G.; Pagel K. The Role of the Mobile Proton in Fucose Migration. Anal. Bioanal. Chem. 2019, 411, 4637–4645. 10.1007/s00216-019-01657-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A. H.; Lebrilla C. B. Evidence for Long-Range Glycosyl Transfer Reactions in the Gas Phase. J. Am. Soc. Mass Spectrom. 2002, 13, 325–337. 10.1016/S1044-0305(02)00343-4. [DOI] [PubMed] [Google Scholar]
- Ernst B.; Müller D. R.; Richter W. J. False Sugar Sequence Ions in Electrospray Tandem Mass Spectrometry of Underivatized Sialyl-Lewis-Type Oligosaccharides. Int. J. Mass Spectrom. Ion Processes 1997, 160, 283–290. 10.1016/S0168-1176(96)04487-4. [DOI] [Google Scholar]
- Brüll L. P.; Kovácik V.; Thomas-Oates J. E.; Heerma W.; Haverkamp J. Sodium-Cationized Oligosaccharides Do Not Appear to Undergo ‘Internal Residue Loss’ Rearrangement Processes on Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 1998, 12, 1520–1532. . [DOI] [PubMed] [Google Scholar]
- Nwosu C.; Yau H. K.; Becht S. Assignment of Core Versus Antenna Fucosylation Types in Protein N-Glycosylation Via Procainamide Labeling and Tandem Mass Spectrometry. Anal. Chem. 2015, 87, 5905–5913. 10.1021/ac5040743. [DOI] [PubMed] [Google Scholar]
- Brüll L. P.; Heerma W.; Thomas-Oates J.; Haverkamp J.; Kovácik V.; Kovác P. Loss of Internal 1 - 6 Substituted Monosaccharide Residues from Underivatized and Per-O-Methylated Trisaccharides. J. Am. Soc. Mass Spectrom. 1997, 8, 43–49. 10.1016/S1044-0305(96)00134-1. [DOI] [Google Scholar]
- Lindahl U.; Couchman J.; Kimata K.; Esko J. D. In Essentials of Glycobiology, 3rd ed.; Ajit Varki R. D. C., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Ed.; Cold Spring Harbor Laboratory Press, 2017. [PubMed] [Google Scholar]
- Capila I.; Linhardt R. J. Heparin-Protein Interactions. Angew. Chem., Int. Ed. 2002, 41, 390–412. . [DOI] [PubMed] [Google Scholar]
- Xu D.; Esko J. D. Demystifying Heparan Sulfate-Protein Interactions. Annu. Rev. Biochem. 2014, 83, 129–157. 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumoto S.; Yamada S.; Sugahara K. Molecular Interactions between Chondroitin-Dermatan Sulfate and Growth Factors/Receptors/Matrix Proteins. Curr. Opin. Struct. Biol. 2015, 34, 35–42. 10.1016/j.sbi.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Kjellén L.; Lindahl U. Specificity of Glycosaminoglycan-Protein Interactions. Curr. Opin. Struct. Biol. 2018, 50, 101–108. 10.1016/j.sbi.2017.12.011. [DOI] [PubMed] [Google Scholar]
- Vallet S. D.; Clerc O.; Ricard-Blum S. Glycosaminoglycan-Protein Interactions: The First Draft of the Glycosaminoglycan Interactome. J. Histochem. Cytochem. 2021, 69, 93–104. 10.1369/0022155420946403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laremore T. N.; Zhang F.; Dordick J. S.; Liu J.; Linhardt R. J. Recent Progress and Applications in Glycosaminoglycan and Heparin Research. Curr. Opin. Chem. Biol. 2009, 13, 633–640. 10.1016/j.cbpa.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S.; Lamanna W. C.; Esko J. D. Heparan Sulfate Proteoglycans. Cold Spring Harbor Perspect. Biol. 2011, 3, a004952. 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasisekharan R.; Shriver Z.; Venkataraman G.; Narayanasami U. Roles of Heparan-Sulphate Glycosaminoglycans in Cancer. Nat. Rev. Cancer 2002, 2, 521. 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- Toole B. P. Hyaluronan: From Extracellular Glue to Pericellular Cue. Nat. Rev. Cancer 2004, 4, 528–539. 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Parish C. R. The Role of Heparan Sulphate in Inflammation. Nat. Rev. Immunol. 2006, 6, 633–643. 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- Mikami T.; Kitagawa H. Biosynthesis and Function of Chondroitin Sulfate. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 4719–4733. 10.1016/j.bbagen.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Lu H.; Peixoto R. T.; Pines M. K.; Ge Y.; Oku S.; Siddiqui T. J.; Xie Y.; Wu W.; Archer-Hartmann S.; et al. Heparan Sulfate Organizes Neuronal Synapses through Neurexin Partnerships. Cell 2018, 174, 1450–1464. 10.1016/j.cell.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasisekharan R.; Raman R.; Prabhakar V. Glycomics Approach to Structure-Function Relationships of Glycosaminoglycans. Annu. Rev. Biomed. Eng. 2006, 8, 181–231. 10.1146/annurev.bioeng.8.061505.095745. [DOI] [PubMed] [Google Scholar]
- Hascall V.; Esko J. D. In Essentials of Glycobiology, 3rd ed.; Ajit Varki R. D. C., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Ed.; Cold Spring Harbor Laboratory Press, 2017. [PubMed] [Google Scholar]
- Sugahara K.; Mikami T.; Uyama T.; Mizuguchi S.; Nomura K.; Kitagawa H. Recent Advances in the Structural Biology of Chondroitin Sulfate and Dermatan Sulfate. Curr. Opin. Struct. Biol. 2003, 13, 612–620. 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Gama C. I.; Tully S. E.; Sotogaku N.; Clark P. M.; Rawat M.; Vaidehi N.; Goddard III W. A.; Nishi A.; Hsieh-Wilson L. C. Sulfation Patterns of Glycosaminoglycans Encode Molecular Recognition and Activity. Nat. Chem. Biol. 2006, 2, 467. 10.1038/nchembio810. [DOI] [PubMed] [Google Scholar]
- Esko J. D.; Selleck S. B. Order out of Chaos: Assembly of Ligand Binding Sites in Heparan Sulfate. Annu. Rev. Biochem. 2002, 71, 435–471. 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Kreuger J.; Kjellén L. Heparan Sulfate Biosynthesis: Regulation and Variability. J. Histochem. Cytochem. 2012, 60, 898–907. 10.1369/0022155412464972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker B. E.; Xu D.; Lawrence R.; Esko J. D. Heparan Sulfate 3-O-Sulfation: A Rare Modification in Search of a Function. Matrix Biol. 2014, 35, 60–72. 10.1016/j.matbio.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko J. D.; Lindahl U. Molecular Diversity of Heparan Sulfate. J. Clin. Invest. 2001, 108, 169–173. 10.1172/JCI200113530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. J.; Beni S.; Limtiaco J. F. K.; Langeslay D. J.; Larive C. K. Heparin Characterization: Challenges and Solutions. Annu. Rev. Anal. Chem. 2011, 4, 439–465. 10.1146/annurev-anchem-061010-113911. [DOI] [PubMed] [Google Scholar]
- Linhardt R. J. 2003 Claude S. Hudson Award Address in Carbohydrate Chemistry. Heparin: Structure and Activity. J. Med. Chem. 2003, 46, 2551–2564. 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- Funderburgh J. L. Mini Review Keratan Sulfate: Structure, Biosynthesis, and Function. Glycobiology 2000, 10, 951–958. 10.1093/glycob/10.10.951. [DOI] [PubMed] [Google Scholar]
- Caterson B.; Melrose J. Keratan Sulfate, a Complex Glycosaminoglycan with Unique Functional Capability. Glycobiology 2018, 28, 182–206. 10.1093/glycob/cwy003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly M.; Leach III F. E.; Laremore T. N.; Toida T.; Amster I. J.; Linhardt R. J. The Proteoglycan Bikunin Has a Defined Sequence. Nat. Chem. Biol. 2011, 7, 827. 10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Duan J.; Leach F. E.; Toida T.; Higashi K.; Zhang H.; Zhang F.; Amster I. J.; Linhardt R. J. Sequencing the Dermatan Sulfate Chain of Decorin. J. Am. Chem. Soc. 2017, 139, 16986–16995. 10.1021/jacs.7b10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kuppevelt T. H.; Oosterhof A.; Versteeg E. M. M.; Podhumljak E.; van de Westerlo E. M. A.; Daamen W. F. Sequencing of Glycosaminoglycans with Potential to Interrogate Sequence-Specific Interactions. Sci. Rep. 2017, 7, 14785. 10.1038/s41598-017-15009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Ly M.; Linhardt R. J. Proteoglycan Sequence. Mol. BioSyst. 2012, 8, 1613–1625. 10.1039/c2mb25021g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlmann L.; Tredwell G. D.; Yu X.; Chang C.-W.; Haselhorst T.; Winger M.; Dyason J. C.; Thomson R. J.; Tiralongo J.; Beacham I. R.; et al. Functional and Structural Characterization of a Heparanase. Nat. Chem. Biol. 2015, 11, 955–957. 10.1038/nchembio.1956. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Williams A.; Zhang X.; Fu L.; Xia K.; Xu Y.; Zhang F.; Liu J.; Koffas M.; Linhardt R. J. Specificity and Action Pattern of Heparanase Bp, a B-Glucuronidase from Burkholderia Pseudomallei. Glycobiology 2019, 29, 572–581. 10.1093/glycob/cwz039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia J. On-Line Separations Combined with Ms for Analysis of Glycosaminoglycans. Mass Spectrom. Rev. 2009, 28, 254–272. 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Zhang F.; Linhardt R. J. Analysis of the Glycosaminoglycan Chains of Proteoglycans. J. Histochem. Cytochem. 2021, 69, 121–135. 10.1369/0022155420937154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepi L. E.; Sanderson P.; Stickney M.; Amster I. J. Developments in Mass Spectrometry for Glycosaminoglycan Analysis: A Review. Molecular & Cellular Proteomics 2021, 20, 100025. 10.1074/mcp.R120.002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia J. Glycosaminoglycan Glycomics Using Mass Spectrometry. Mol. Cell. Proteomics 2013, 12, 885–892. 10.1074/mcp.R112.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia M. J.; Li L.; Ly M.; Linhardt R. J.; Amster I. J. Complete Mass Spectral Characterization of a Synthetic Ultralow-Molecular-Weight Heparin Using Collision-Induced Dissociation. Anal. Chem. 2012, 84, 5475–5478. 10.1021/ac3015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J.; Noborn F.; Gomez Toledo A.; Nasir W.; Sihlbom C.; Larson G. Characterization of Glycan Structures of Chondroitin Sulfate-Glycopeptides Facilitated by Sodium Ion-Pairing and Positive Mode Lc-Ms/Ms. J. Am. Soc. Mass Spectrom. 2017, 28, 229–241. 10.1007/s13361-016-1539-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyer S.; Lopin-Bon C.; Jacquinet J.-C.; Salpin J.-Y.; Daniel R. Isomer Separation and Effect of the Degree of Polymerization on the Gas-Phase Structure of Chondroitin Sulfate Oligosaccharides Analyzed by Ion Mobility and Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 2003–2010. 10.1002/rcm.7987. [DOI] [PubMed] [Google Scholar]
- Shi X.; Huang Y.; Mao Y.; Naimy H.; Zaia J. Tandem Mass Spectrometry of Heparan Sulfate Negative Ions: Sulfate Loss Patterns and Chemical Modification Methods for Improvement of Product Ion Profiles. J. Am. Soc. Mass Spectrom. 2012, 23, 1498–1511. 10.1007/s13361-012-0429-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Liu J.; Sharp J. S. An Approach for Separation and Complete Structural Sequencing of Heparin/Heparan Sulfate-Like Oligosaccharides. Anal. Chem. 2013, 85, 5787–5795. 10.1021/ac400439a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Zong C.; Venot A.; Chiu Y.; Zhou D.; Boons G.-J.; Sharp J. S. De Novo Sequencing of Complex Mixtures of Heparan Sulfate Oligosaccharides. Anal. Chem. 2016, 88, 5299–5307. 10.1021/acs.analchem.6b00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmnitzer K.; Kohling S.; Freyse J.; Rademann J.; Schiller J. Characterization of Defined Sulfated Heparin-Like Oligosaccharides by Electrospray Ionization Ion Trap Mass Spectrometry. J. Mass Spectrom. 2021, 56, e4692 10.1002/jms.4692. [DOI] [PubMed] [Google Scholar]
- Baba T.; Hashimoto Y.; Hasegawa H.; Hirabayashi A.; Waki I. Electron Capture Dissociation in a Radio Frequency Ion Trap. Anal. Chem. 2004, 76, 4263–4266. 10.1021/ac049309h. [DOI] [PubMed] [Google Scholar]
- Silivra O. A.; Kjeldsen F.; Ivonin I. A.; Zubarev R. A. Electron Capture Dissociation of Polypeptides in a Three-Dimensional Quadrupole Ion Trap: Implementation and First Results. J. Am. Soc. Mass Spectrom. 2005, 16, 22–27. 10.1016/j.jasms.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Kjeldsen F.; Silivra O. A.; Ivonin I. A.; Haselmann K. F.; Gorshkov M.; Zubarev R. A. CαC Backbone Fragmentation Dominates in Electron Detachment Dissociation of Gas-Phase Polypeptide Polyanions. Chem. - Eur. J. 2005, 11, 1803–1812. 10.1002/chem.200400806. [DOI] [PubMed] [Google Scholar]
- Satake H.; Hasegawa H.; Hirabayashi A.; Hashimoto Y.; Baba T.; Masuda K. Fast Multiple Electron Capture Dissociation in a Linear Radio Frequency Quadrupole Ion Trap. Anal. Chem. 2007, 79, 8755–8761. 10.1021/ac071462z. [DOI] [PubMed] [Google Scholar]
- Voinov V. G.; Deinzer M. L.; Barofsky D. F. Electron Capture Dissociation in a Linear Radiofrequency-Free Magnetic Cell. Rapid Commun. Mass Spectrom. 2008, 22, 3087–3088. 10.1002/rcm.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T.; Campbell J. L.; Le Blanc J. C. Y.; Hager J. W.; Thomson B. A. Electron Capture Dissociation in a Branched Radio-Frequency Ion Trap. Anal. Chem. 2015, 87, 785–792. 10.1021/ac503773y. [DOI] [PubMed] [Google Scholar]
- Leach F. E.; Arungundram S.; Al-Mafraji K.; Venot A.; Boons G.-J.; Amster I. J. Electron Detachment Dissociation of Synthetic Heparan Sulfate Glycosaminoglycan Tetrasaccharides Varying in Degree of Sulfation and Hexuronic Acid Stereochemistry. Int. J. Mass Spectrom. 2012, 330–332, 152–159. 10.1016/j.ijms.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J. J.; Laremore T. N.; Busch A. M.; Linhardt R. J.; Amster I. J. Electron Detachment Dissociation of Dermatan Sulfate Oligosaccharides. J. Am. Soc. Mass Spectrom. 2008, 19, 294–304. 10.1016/j.jasms.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J. J.; Laremore T. N.; Busch A. M.; Linhardt R. J.; Amster I. J. Influence of Charge State and Sodium Cationization on the Electron Detachment Dissociation and Infrared Multiphoton Dissociation of Glycosaminoglycan Oligosaccharides. J. Am. Soc. Mass Spectrom. 2008, 19, 790–798. 10.1016/j.jasms.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia J.; Li X. Q.; Chan S. Y.; Costello C. E. Tandem Mass Spectrometric Strategies for Determination of Sulfation Positions and Uronic Acid Epimerization in Chondroitin Sulfate Oligosaccharides. J. Am. Soc. Mass Spectrom. 2003, 14, 1270–1281. 10.1016/S1044-0305(03)00541-5. [DOI] [PubMed] [Google Scholar]
- Oh H. B.; Leach F. E.; Arungundram S.; Al-Mafraji K.; Venot A.; Boons G.-J.; Amster I. J. Multivariate Analysis of Electron Detachment Dissociation and Infrared Multiphoton Dissociation Mass Spectra of Heparan Sulfate Tetrasaccharides Differing Only in Hexuronic Acid Stereochemistry. J. Am. Soc. Mass Spectrom. 2011, 22, 582–590. 10.1007/s13361-010-0047-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach F. E.; Ly M.; Laremore T. N.; Wolff J. J.; Perlow J.; Linhardt R. J.; Amster I. J. Hexuronic Acid Stereochemistry Determination in Chondroitin Sulfate Glycosaminoglycan Oligosaccharides by Electron Detachment Dissociation. J. Am. Soc. Mass Spectrom. 2012, 23, 1488–1497. 10.1007/s13361-012-0428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agyekum I.; Patel A. B.; Zong C.; Boons G.-J.; Amster I. J. Assignment of Hexuronic Acid Stereochemistry in Synthetic Heparan Sulfate Tetrasaccharides with 2-O-Sulfo Uronic Acids Using Electron Detachment Dissociation. Int. J. Mass Spectrom. 2015, 390, 163–169. 10.1016/j.ijms.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agyekum I.; Zong C.; Boons G.-J.; Amster I. J. Single Stage Tandem Mass Spectrometry Assignment of the C-5 Uronic Acid Stereochemistry in Heparan Sulfate Tetrasaccharides Using Electron Detachment Dissociation. J. Am. Soc. Mass Spectrom. 2017, 28, 1741–1750. 10.1007/s13361-017-1643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach F. E.; Riley N. M.; Westphall M. S.; Coon J. J.; Amster I. J. Negative Electron Transfer Dissociation Sequencing of Increasingly Sulfated Glycosaminoglycan Oligosaccharides on an Orbitrap Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2017, 28, 1844–1854. 10.1007/s13361-017-1709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron W. J.; Goeringer D. E.; McLuckey S. A. Gas-Phase Electron Transfer Reactions from Multiply-Charged Anions to Rare Gas Cations. J. Am. Chem. Soc. 1995, 117, 11555–11562. 10.1021/ja00151a021. [DOI] [Google Scholar]
- Huang Y.; Yu X.; Mao Y.; Costello C. E.; Zaia J.; Lin C. De Novo Sequencing of Heparan Sulfate Oligosaccharides by Electron-Activated Dissociation. Anal. Chem. 2013, 85, 11979–11986. 10.1021/ac402931j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgueira H. A.; Bartolozzi A.; Schell P.; Litjens R. E. J. N.; Palmacci E. R.; Seeberger P. H. Modular Synthesis of Heparin Oligosaccharides. Chem. - Eur. J. 2003, 9, 140–169. 10.1002/chem.200390009. [DOI] [PubMed] [Google Scholar]
- Prabhu A.; Venot A.; Boons G.-J. New Set of Orthogonal Protecting Groups for the Modular Synthesis of Heparan Sulfate Fragments. Org. Lett. 2003, 5, 4975–4978. 10.1021/ol0359261. [DOI] [PubMed] [Google Scholar]
- Arungundram S.; Al-Mafraji K.; Asong J.; Leach F. E.; Amster I. J.; Venot A.; Turnbull J. E.; Boons G.-J. Modular Synthesis of Heparan Sulfate Oligosaccharides for Structure-Activity Relationship Studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boltje T. J.; Buskas T.; Boons G.-J. Opportunities and Challenges in Synthetic Oligosaccharide and Glycoconjugate Research. Nat. Chem. 2009, 1, 611–622. 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller S.; Collot M.; Yin J.; Hahm H. S.; Seeberger P. H. Automated Solid-Phase Synthesis of Chondroitin Sulfate Glycosaminoglycans. Angew. Chem., Int. Ed. 2013, 52, 5858–5861. 10.1002/anie.201210132. [DOI] [PubMed] [Google Scholar]
- Schworer R.; Zubkova O. V.; Turnbull J. E.; Tyler P. C. Synthesis of a Targeted Library of Heparan Sulfate Hexa- to Dodecasaccharides as Inhibitors of Beta-Secretase: Potential Therapeutics for Alzheimer’s Disease. Chem. - Eur. J. 2013, 19, 6817–6823. 10.1002/chem.201204519. [DOI] [PubMed] [Google Scholar]
- Zong C.; Venot A.; Dhamale O.; Boons G.-J. Fluorous Supported Modular Synthesis of Heparan Sulfate Oligosaccharides. Org. Lett. 2013, 15, 342–345. 10.1021/ol303270v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C.; Venot A.; Li X.; Lu W.; Xiao W.; Wilkes J.-S. L.; Salanga C. L.; Handel T. M.; Wang L.; Wolfert M. A.; et al. Heparan Sulfate Microarray Reveals That Heparan Sulfate-Protein Binding Exhibits Different Ligand Requirements. J. Am. Chem. Soc. 2017, 139, 9534–9543. 10.1021/jacs.7b01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Chopra P.; Boons G.-J. Modular Synthesis of Heparan Sulfate Oligosaccharides Having N-Acetyl and N-Sulfate Moieties. J. Org. Chem. 2020, 85, 16082–16098. 10.1021/acs.joc.0c01881. [DOI] [PubMed] [Google Scholar]
- Kuberan B.; Lech M. Z.; Beeler D. L.; Wu Z. L.; Rosenberg R. D. Enzymatic Synthesis of Antithrombin Iii-Binding Heparan Sulfate Pentasaccharide. Nat. Biotechnol. 2003, 21, 1343–1346. 10.1038/nbt885. [DOI] [PubMed] [Google Scholar]
- Gottschalk J.; Elling L. Current State on the Enzymatic Synthesis of Glycosaminoglycans. Curr. Opin. Chem. Biol. 2021, 61, 71–80. 10.1016/j.cbpa.2020.09.008. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Masuko S.; Takieddin M.; Xu H.; Liu R.; Jing J.; Mousa S. A.; Linhardt R. J.; Liu J. Chemoenzymatic Synthesis of Homogeneous Ultralow Molecular Weight Heparins. Science 2011, 334, 498. 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Cai C.; Chandarajoti K.; Hsieh P.-H.; Li L.; Pham T. Q.; Sparkenbaugh E. M.; Sheng J.; Key N. S.; Pawlinski R.; et al. Homogeneous Low-Molecular-Weight Heparins with Reversible Anticoagulant Activity. Nat. Chem. Biol. 2014, 10, 248–250. 10.1038/nchembio.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Chandarajoti K.; Zhang X.; Pagadala V.; Dou W.; Hoppensteadt D. M.; Sparkenbaugh E. M.; Cooley B.; Daily S.; Key N. S.; et al. Synthetic Oligosaccharides Can Replace Animal-Sourced Low-Molecular Weight Heparins. Sci. Transl. Med. 2017, 9, eaan5954 10.1126/scitranslmed.aan5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling S.; Künze G.; Lemmnitzer K.; Bermudez M.; Wolber G.; Schiller J.; Huster D.; Rademann J. Chemoenzymatic Synthesis of Nonasulfated Tetrahyaluronan with a Paramagnetic Tag for Studying Its Complex with Interleukin-10. Chem. - Eur. J. 2016, 22, 5563–5574. 10.1002/chem.201504459. [DOI] [PubMed] [Google Scholar]
- Lu W.; Zong C.; Chopra P.; Pepi L. E.; Xu Y.; Amster I. J.; Liu J.; Boons G.-J. Controlled Chemoenzymatic Synthesis of Heparan Sulfate Oligosaccharides. Angew. Chem., Int. Ed. 2018, 57, 5340–5344. 10.1002/anie.201800387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cress B. F.; Bhaskar U.; Vaidyanathan D.; Williams A.; Cai C.; Liu X.; Fu L.; M-Chari V.; Zhang F.; Mousa S. A. Heavy Heparin: A Stable Isotope-Enriched, Chemoenzymatically-Synthesized, Poly-Component Drug. Angew. Chem., Int. Ed. 2019, 58, 5962. 10.1002/anie.201900768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Lin L.; Huang H.; Linhardt R. J. Chemoenzymatic Synthesis of Glycosaminoglycans. Acc. Chem. Res. 2020, 53, 335–346. 10.1021/acs.accounts.9b00420. [DOI] [PubMed] [Google Scholar]
- Wu J.; Wei J.; Hogan J. D.; Chopra P.; Joshi A.; Lu W.; Klein J.; Boons G.-J.; Lin C.; Zaia J. Negative Electron Transfer Dissociation Sequencing of 3-O-Sulfation-Containing Heparan Sulfate Oligosaccharides. J. Am. Soc. Mass Spectrom. 2018, 29, 1262–1272. 10.1007/s13361-018-1907-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach F. E.; Wolff J. J.; Xiao Z.; Ly M.; Laremore T. N.; Arungundram S.; Al-Mafraji K.; Venot A.; Boons G.-J.; Linhardt R. J.; et al. Negative Electron Transfer Dissociation Fourier Transform Mass Spectrometry of Glycosaminoglycan Carbohydrates. Eur. J. Mass Spectrom. 2011, 17, 167–176. 10.1255/ejms.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickney M.; Sanderson P.; Leach F. E.; Zhang F.; Linhardt R. J.; Amster I. J. Online Capillary Zone Electrophoresis Negative Electron Transfer Dissociation Tandem Mass Spectrometry of Glycosaminoglycan Mixtures. Int. J. Mass Spectrom. 2019, 445, 116209. 10.1016/j.ijms.2019.116209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepi L. E.; Sasiene Z. J.; Mendis P. M.; Jackson G. P.; Amster I. J. Structural Characterization of Sulfated Glycosaminoglycans Using Charge-Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2020, 31, 2143–2153. 10.1021/jasms.0c00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaud A.; Antoine R.; Joly L.; Mesplet N.; Dugourd P.; Lemoine J. Wavelength-Tunable Ultraviolet Photodissociation (Uvpd) of Heparin-Derived Disaccharides in a Linear Ion Trap. J. Am. Soc. Mass Spectrom. 2009, 20, 1645–1651. 10.1016/j.jasms.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Racaud A.; Antoine R.; Dugourd P.; Lemoine J. Photoinduced Dissociation of Heparin-Derived Oligosaccharides Controlled by Charge Location. J. Am. Soc. Mass Spectrom. 2010, 21, 2077–2084. 10.1016/j.jasms.2010.08.021. [DOI] [PubMed] [Google Scholar]
- Klein D. R.; Leach F. E.; Amster I. J.; Brodbelt J. S. Structural Characterization of Glycosaminoglycan Carbohydrates Using Ultraviolet Photodissociation. Anal. Chem. 2019, 91, 6019–6026. 10.1021/acs.analchem.9b00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepi L. E.; Leach F. E.; Klein D. R.; Brodbelt J. S.; Amster I. J. Investigation of the Experimental Parameters of Ultraviolet Photodissociation for the Structural Characterization of Chondroitin Sulfate Glycosaminoglycan Isomers. J. Am. Soc. Mass Spectrom. 2021, 32, 1759. 10.1021/jasms.1c00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkridge A. M.; Hackbusch S. Ultraviolet Photodissociation of Fondaparinux Generates Signature Antithrombin-Like 3-O-Sulfated -Glcns3s6s- Monosaccharide Fragment (Y3/C3). Anal. Bioanal. Chem. 2020, 412, 7925–7935. 10.1007/s00216-020-02925-w. [DOI] [PubMed] [Google Scholar]
- Chopra P.; Joshi A.; Wu J.; Lu W.; Yadavalli T.; Wolfert M. A.; Shukla D.; Zaia J.; Boons G.-J. The 3-O-Sulfation of Heparan Sulfate Modulates Protein Binding and Lyase Degradation. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2012935118 10.1073/pnas.2012935118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenauer M. R.; Meissen J. K.; Seo Y.; Ames J. B.; Leary J. A. Heparan Sulfate Separation, Sequencing, and Isomeric Differentiation: Ion Mobility Spectrometry Reveals Specific Iduronic and Glucuronic Acid-Containing Hexasaccharides. Anal. Chem. 2009, 81, 10179–10185. 10.1021/ac902186h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. L.; Wei W.; Schwörer R.; Zubkova O. V.; Tyler P. C.; Turnbull J. E.; Leary J. A. Composition, Sequencing and Ion Mobility Mass Spectrometry of Heparan Sulfate-Like Octasaccharide Isomers Differing in Glucuronic and Iduronic Acid Content. Eur. J. Mass Spectrom. 2015, 21, 245–254. 10.1255/ejms.1337. [DOI] [PubMed] [Google Scholar]
- Seo Y.; Andaya A.; Leary J. A. Preparation, Separation, and Conformational Analysis of Differentially Sulfated Heparin Octasaccharide Isomers Using Ion Mobility Mass Spectrometry. Anal. Chem. 2012, 84, 2416–2423. 10.1021/ac203190k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. L.; Guimond S. E.; Shivkumar M.; Blocksidge J.; Austin J. A.; Leary J. A.; Turnbull J. E. Heparin Isomeric Oligosaccharide Separation Using Volatile Salt Strong Anion Exchange Chromatography. Anal. Chem. 2016, 88, 11542–11550. 10.1021/acs.analchem.6b02801. [DOI] [PubMed] [Google Scholar]
- Miller R. L.; Dykstra A. B.; Wei W.; Holsclaw C.; Turnbull J. E.; Leary J. A. Enrichment of Two Isomeric Heparin Oligosaccharides Exhibiting Different Affinities toward Monocyte Chemoattractant Protein-1. Anal. Chem. 2016, 88, 11551–11558. 10.1021/acs.analchem.6b02803. [DOI] [PubMed] [Google Scholar]
- Lemmnitzer K.; Riemer T.; Groessl M.; Süβ R.; Knochenmuss R.; Schiller J. Comparison of Ion Mobility-Mass Spectrometry and Pulsed-Field Gradient Nuclear Magnetic Resonance Spectroscopy for the Differentiation of Chondroitin Sulfate Isomers. Anal. Methods 2016, 8, 8483–8491. 10.1039/C6AY02531E. [DOI] [Google Scholar]
- Wei J.; Wu J.; Tang Y.; Ridgeway M. E.; Park M. A.; Costello C. E.; Zaia J.; Lin C. Characterization and Quantification of Highly Sulfated Glycosaminoglycan Isomers by Gated-Trapped Ion Mobility Spectrometry Negative Electron Transfer Dissociation Ms/Ms. Anal. Chem. 2019, 91, 2994–3001. 10.1021/acs.analchem.8b05283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgeway M. E.; Wolff J. J.; Silveira J. A.; Lin C.; Costello C. E.; Park M. A. Gated Trapped Ion Mobility Spectrometry Coupled to Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Int. J. Ion Mobility Spectrom. 2016, 19, 77–85. 10.1007/s12127-016-0197-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. L.; Guimond S. E.; Schwörer R.; Zubkova O. V.; Tyler P. C.; Xu Y.; Liu J.; Chopra P.; Boons G.-J.; Grabarics M.; et al. Shotgun Ion Mobility Mass Spectrometry Sequencing of Heparan Sulfate Saccharides. Nat. Commun. 2020, 11, 1481. 10.1038/s41467-020-15284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y.; Andaya A.; Bleiholder C.; Leary J. A. Differentiation of Cc Vs Cxc Chemokine Dimers with Gag Octasaccharide Binding Partners: An Ion Mobility Mass Spectrometry Approach. J. Am. Chem. Soc. 2013, 135, 4325–4332. 10.1021/ja310915m. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Singh A.; Li L.; Linhardt R. J.; Xu Y.; Liu J.; Woods R. J.; Amster I. J. Investigating Changes in the Gas-Phase Conformation of Antithrombin Iii Upon Binding of Arixtra Using Traveling Wave Ion Mobility Spectrometry (Twims). Analyst 2015, 140, 6980–6989. 10.1039/C5AN00908A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Singh A.; Xu Y.; Zong C.; Zhang F.; Boons G.-J.; Liu J.; Linhardt R. J.; Woods R. J.; Amster I. J. Gas-Phase Analysis of the Complex of Fibroblast Growthfactor 1 with Heparan Sulfate: A Traveling Wave Ion Mobility Spectrometry (Twims) and Molecular Modeling Study. J. Am. Soc. Mass Spectrom. 2017, 28, 96–109. 10.1007/s13361-016-1496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Yang J. Y.; Thieker D. F.; Xu Y.; Zong C.; Boons G.-J.; Liu J.; Woods R. J.; Moremen K. W.; Amster I. J. A Traveling Wave Ion Mobility Spectrometry (Twims) Study of the Robo1-Heparan Sulfate Interaction. J. Am. Soc. Mass Spectrom. 2018, 29, 1153–1165. 10.1007/s13361-018-1903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Kaltashov I. A. Evaluation of Top-Down Mass Spectrometry and Ion-Mobility Spectroscopy as a Means of Mapping Protein-Binding Motifs within Heparin Chains. Analyst 2020, 145, 3090–3099. 10.1039/D0AN00097C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnon I.; Schindler B.; Renois-Predelus G.; Daniel R. Lasers and Ion Mobility: New Additions to the Glycosaminoglycanomics Toolkit. Curr. Opin. Struct. Biol. 2018, 50, 171–180. 10.1016/j.sbi.2018.06.006. [DOI] [PubMed] [Google Scholar]
- Cagmat E. B.; Szczepanski J.; Pearson W. L.; Powell D. H.; Eyler J. R.; Polfer N. C. Vibrational Signatures of Metal-Chelated Monosaccharide Epimers: Gas-Phase Infrared Spectroscopy of Rb+-Tagged Glucuronic and Iduronic Acid. Phys. Chem. Chem. Phys. 2010, 12, 3474–3479. 10.1039/b924027f. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Renois-Predelus G.; Bagdadi N.; Melizi S.; Barnes L.; Chambert S.; Allouche A. R.; Compagnon I. Ms/Ir, a New Ms-Based Hyphenated Method for Analysis of Hexuronic Acid Epimers in Glycosaminoglycans. Glycoconjugate J. 2017, 34, 421–425. 10.1007/s10719-016-9741-8. [DOI] [PubMed] [Google Scholar]
- Barnes L.; Schindler B.; Allouche A. R.; Simon D.; Chambert S.; Oomens J.; Compagnon I. Anharmonic Simulations of the Vibrational Spectrum of Sulfated Compounds: Application to the Glycosaminoglycan Fragment Glucosamine 6-Sulfate. Phys. Chem. Chem. Phys. 2015, 17, 25705–25713. 10.1039/C5CP02079D. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Barnes L.; Gray C. J.; Chambert S.; Flitsch S. L.; Oomens J.; Daniel R.; Allouche A. R.; Compagnon I. Irmpd Spectroscopy Sheds New (Infrared) Light on the Sulfate Pattern of Carbohydrates. J. Phys. Chem. A 2017, 121, 2114–2120. 10.1021/acs.jpca.6b11642. [DOI] [PubMed] [Google Scholar]
- Khanal N.; Masellis C.; Kamrath M. Z.; Clemmer D. E.; Rizzo T. R. Glycosaminoglycan Analysis by Cryogenic Messenger-Tagging Ir Spectroscopy Combined with Ims-Ms. Anal. Chem. 2017, 89, 7601–7606. 10.1021/acs.analchem.7b01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renois-Predelus G.; Schindler B.; Compagnon I. Analysis of Sulfate Patterns in Glycosaminoglycan Oligosaccharides by Ms(N) Coupled to Infrared Ion Spectroscopy: The Case of Galnac4s and Galnac6s. J. Am. Soc. Mass Spectrom. 2018, 29, 1242–1249. 10.1007/s13361-018-1955-5. [DOI] [PubMed] [Google Scholar]
- Lettow M.; Greis K.; Grabarics M.; Horlebein J.; Miller R. L.; Meijer G.; von Helden G.; Pagel K. Chondroitin Sulfate Disaccharides in the Gas Phase: Differentiation and Conformational Constraints. J. Phys. Chem. A 2021, 125, 4373–4379. 10.1021/acs.jpca.1c02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht S.; Unwin L.; Muniyappa M.; Rudd P. M. Glycosylation as a Marker for Inflammatory Arthritis. Cancer Biomarkers 2014, 14, 17–28. 10.3233/CBM-130373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C.; Stewart T. J.; Renfrow M. B.; Novak J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 346–366. 10.1038/s41581-019-0129-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K.; Chen M. Z.; Olefsky J. M.; Marth J. D. Pathway to Diabetes through Attenuation of Pancreatic Beta Cell Glycosylation and Glucose Transport. Nat. Med. 2011, 17, 1067–1075. 10.1038/nm.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A. F.; Campos D.; Reis C. A.; Gomes C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. 10.1016/j.trecan.2020.04.002. [DOI] [PubMed] [Google Scholar]
- Peixoto A.; Relvas-Santos M.; Azevedo R.; Santos L. L.; Ferreira J. A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. 10.3389/fonc.2019.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Westphall M. S.; Coon J. J. Capturing Site-Specific Heterogeneity with Large-Scale N-Glycoproteome Analysis. Nat. Commun. 2019, 10, 1311. 10.1038/s41467-019-09222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Felix A.; Byrd W. E.; Zuegel D. K.; Ishihara M.; Azadi P.; Iavarone A. T.; Pitteri S. J.; Bertozzi C. R. Development of Isotag, a Chemical Glycoproteomics Technique for Profiling Intact N- and O-Glycopeptides from Whole Cell Proteomes. J. Proteome Res. 2017, 16, 1706–1718. 10.1021/acs.jproteome.6b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worner T. P.; Shamorkina T. M.; Snijder J.; Heck A. J. R. Mass Spectrometry-Based Structural Virology. Anal. Chem. 2021, 93, 620–640. 10.1021/acs.analchem.0c04339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struwe W. B.; Stuckmann A.; Behrens A. J.; Pagel K.; Crispin M. Global N-Glycan Site Occupancy of Hiv-1 Gp120 by Metabolic Engineering and High-Resolution Intact Mass Spectrometry. ACS Chem. Biol. 2017, 12, 357–361. 10.1021/acschembio.6b00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Su X.; Zhu Z.; Go E. P.; Desaire H. Glycopep Masslist: Software to Generate Massive Inclusion Lists for Glycopeptide Analyses. Anal. Bioanal. Chem. 2017, 409, 561–570. 10.1007/s00216-016-9896-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Iavarone A. T.; Spiciarich D. R.; Palaniappan K. K.; Bertozzi C. R. Isotope-Targeted Glycoproteomics (Isotag): A Mass-Independent Platform for Intact N- and O-Glycopeptide Discovery and Analysis. Nat. Methods 2015, 12, 561–567. 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Felix A.; Zhang L.; Elias J. E.; Bertozzi C. R. Isotope-Targeted Glycoproteomics (Isotag) Analysis of Sialylated N- and O-Glycopeptides on an Orbitrap Fusion Tribrid Using Azido and Alkynyl Sugars. Anal. Bioanal. Chem. 2017, 409, 579–588. 10.1007/s00216-016-9934-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R.; Huang Y.; Zhao J.; Zhong J.; Mechref Y. Isomeric Separation of N-Glycopeptides Derived from Glycoproteins by Porous Graphitic Carbon (Pgc) Lc-Ms/Ms. Anal. Chem. 2020, 92, 9556–9565. 10.1021/acs.analchem.0c00668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Nie Y.; Boyes B.; Orlando R. Resolving Isomeric Glycopeptide Glycoforms with Hydrophilic Interaction Chromatography (Hilic). J. Biomol Tech 2016, 27, 98–104. 10.7171/jbt.16-2703-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera-Arteu M.; Lleshi N.; Sanz-Nebot V.; Gimenez E.; Benavente F. Analysis of Glycopeptide Biomarkers by on-Line Tio2 Solid-Phase Extraction Capillary Electrophoresis-Mass Spectrometry. Talanta 2020, 209, 120563. 10.1016/j.talanta.2019.120563. [DOI] [PubMed] [Google Scholar]
- Pont L.; Kuzyk V.; Benavente F.; Sanz-Nebot V.; Mayboroda O. A.; Wuhrer M.; Lageveen-Kammeijer G. S. M. Site-Specific N-Linked Glycosylation Analysis of Human Carcinoembryonic Antigen by Sheathless Capillary Electrophoresis-Tandem Mass Spectrometry. J. Proteome Res. 2021, 20, 1666–1675. 10.1021/acs.jproteome.0c00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H.; Sun F.; Suttapitugsakul S.; Wu R. Global and Site-Specific Analysis of Protein Glycosylation in Complex Biological Systems with Mass Spectrometry. Mass Spectrom. Rev. 2019, 38, 356–379. 10.1002/mas.21586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez Reyes C. D.; Jiang P.; Donohoo K.; Atashi M.; Mechref Y. S. Glycomics and Glycoproteomics: Approaches to Address Isomeric Separation of Glycans and Glycopeptides. J. Sep. Sci. 2021, 44, 403–425. 10.1002/jssc.202000878. [DOI] [PubMed] [Google Scholar]
- Desaire H.; Hua D. When Can Glycopeptides Be Assigned Based Solely on High-Resolution Mass Spectrometry Data?. Int. J. Mass Spectrom. 2009, 287, 21–26. 10.1016/j.ijms.2008.12.001. [DOI] [Google Scholar]
- Roepstorff P.; Fohlman J. Proposal for a Common Nomenclature for Sequence Ions in Mass Spectra of Peptides. Biomed. Mass Spectrom. 1984, 11, 601–601. 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- Kelly M. I.; Dodds E. D. Parallel Determination of Polypeptide and Oligosaccharide Connectivities by Energy-Resolved Collison-Induced Dissociation of Protonated O-Glycopeptides Derived from Nonspecific Proteolysis. J. Am. Soc. Mass Spectrom. 2020, 31, 624–632. 10.1021/jasms.9b00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinneburg H.; Stavenhagen K.; Schweiger-Hufnagel U.; Pengelley S.; Jabs W.; Seeberger P. H.; Silva D. V.; Wuhrer M.; Kolarich D. The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-Tof) Instruments for Glycopeptide-Based Glycoproteomics. J. Am. Soc. Mass Spectrom. 2016, 27, 507–519. 10.1007/s13361-015-1308-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segu Z. M.; Mechref Y. Characterizing Protein Glycosylation Sites through Higher-Energy C-Trap Dissociation. Rapid Commun. Mass Spectrom. 2010, 24, 1217–1225. 10.1002/rcm.4485. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yang C.; Sun T. Characterization of Glycopeptides Using a Stepped Higher-Energy C-Trap Dissociation Approach on a Hybrid Quadrupole Orbitrap. Rapid Commun. Mass Spectrom. 2018, 32, 1353–1362. 10.1002/rcm.8191. [DOI] [PubMed] [Google Scholar]
- Alagesan K.; Hinneburg H.; Seeberger P. H.; Silva D. V.; Kolarich D. Glycan Size and Attachment Site Location Affect Electron Transfer Dissociation (Etd) Fragmentation and Automated Glycopeptide Identification. Glycoconjugate J. 2019, 36, 487–493. 10.1007/s10719-019-09888-w. [DOI] [PubMed] [Google Scholar]
- Darula Z.; Pap A.; Medzihradszky K. F. Extended Sialylated O-Glycan Repertoire of Human Urinary Glycoproteins Discovered and Characterized Using Electron-Transfer/Higher-Energy Collision Dissociation. J. Proteome Res. 2018, 18, 280–291. 10.1021/acs.jproteome.8b00587. [DOI] [PubMed] [Google Scholar]
- Pap A.; Klement E.; Hunyadi-Gulyas E.; Darula Z.; Medzihradszky K. F. Status Report on the High-Throughput Characterization of Complex Intact O-Glycopeptide Mixtures. J. Am. Soc. Mass Spectrom. 2018, 29, 1210–1220. 10.1007/s13361-018-1945-7. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Wang B.; Chen Z.; Urabe G.; Glover M. S.; Shi X.; Guo L. W.; Kent K. C.; Li L. Electron-Transfer/Higher-Energy Collision Dissociation (Ethcd)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764. 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolli V.; Schumacher K. N.; Dodds E. D. Ion Mobility-Resolved Collision-Induced Dissociation and Electron Transfer Dissociation of N-Glycopeptides: Gathering Orthogonal Connectivity Information from a Single Mass-Selected Precursor Ion Population. Analyst 2017, 142, 4691–4702. 10.1039/C7AN01196B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechref Y. Use of Cid/Etd Mass Spectrometry to Analyze Glycopeptides. Curr. Protoc Protein Sci. 2012, 68, 11–11. 10.1002/0471140864.ps1211s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Malaker S. A.; Driessen M. D.; Bertozzi C. R. Optimal Dissociation Methods Differ for N- and O-Glycopeptides. J. Proteome Res. 2020, 19, 3286–3301. 10.1021/acs.jproteome.0c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H. J.; Lebrilla C. B. Structure Elucidation of Native N- and O-Linked Glycans by Tandem Mass Spectrometry (Tutorial). Mass Spectrom. Rev. 2011, 30, 560–578. 10.1002/mas.20283. [DOI] [PubMed] [Google Scholar]
- Jebanathirajah J. A.; Pittman J. L.; Thomson B. A.; Budnik B. A.; Kaur P.; Rape M.; Kirschner M.; Costello C. E.; O’Connor P. B. Characterization of a New Qqq-Fticr Mass Spectrometer for Post-Translational Modification Analysis and Top-Down Tandem Mass Spectrometry of Whole Proteins. J. Am. Soc. Mass Spectrom. 2005, 16, 1985–1999. 10.1016/j.jasms.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Fukui K.; Takahashi K. Infrared Multiple Photon Dissociation Spectroscopy and Computational Studies of O-Glycosylated Peptides. Anal. Chem. 2012, 84, 2188–2194. 10.1021/ac202379v. [DOI] [PubMed] [Google Scholar]
- Seipert R. R.; Dodds E. D.; Clowers B. H.; Beecroft S. M.; German J. B.; Lebrilla C. B. Factors That Influence Fragmentation Behavior of N-Linked Glycopeptide Ions. Anal. Chem. 2008, 80, 3684–3692. 10.1021/ac800067y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J. A.; Ko B. J.; Xu H.; Iwashkiw J. A.; Robotham S. A.; Shaw J. B.; Feldman M. F.; Brodbelt J. S. Concurrent Automated Sequencing of the Glycan and Peptide Portions of O-Linked Glycopeptide Anions by Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2013, 85, 9253–9261. 10.1021/ac4021177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Reilly J. P. Extracting Both Peptide Sequence and Glycan Structural Information by 157 Nm Photodissociation of N-Linked Glycopeptides. J. Proteome Res. 2009, 8, 734–742. 10.1021/pr800766f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko B. J.; Brodbelt J. S. Comparison of Glycopeptide Fragmentation by Collision Induced Dissociation and Ultraviolet Photodissociation. Int. J. Mass Spectrom. 2015, 377, 385–392. 10.1016/j.ijms.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglia G.; Kliman M.; Claude E.; Geromanos S.; Astarita G. Applications of Ion-Mobility Mass Spectrometry for Lipid Analysis. Anal. Bioanal. Chem. 2015, 407, 4995–5007. 10.1007/s00216-015-8664-8. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Hyung S. J.; Ruotolo B. T. Ion Mobility-Mass Spectrometry for Structural Proteomics. Expert Rev. Proteomics 2012, 9, 47–58. 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn L. S.; McLean J. A. Structural Separations by Ion Mobility-Ms for Glycomics and Glycoproteomics. Methods Mol. Biol. 2013, 951, 171–194. 10.1007/978-1-62703-146-2_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad Izaham A. R.; Ang C. S.; Nie S.; Bird L. E.; Williamson N. A.; Scott N. E. What Are We Missing by Using Hydrophilic Enrichment? Improving Bacterial Glycoproteome Coverage Using Total Proteome and Faims Analyses. J. Proteome Res. 2021, 20, 599–612. 10.1021/acs.jproteome.0c00565. [DOI] [PubMed] [Google Scholar]
- Creese A. J.; Cooper H. J. Separation and Identification of Isomeric Glycopeptides by High Field Asymmetric Waveform Ion Mobility Spectrometry. Anal. Chem. 2012, 84, 2597–2601. 10.1021/ac203321y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak P.; Baird M. A.; Shvartsburg A. A. High-Resolution Ion Mobility Separations of Isomeric Glycoforms with Variations on the Peptide and Glycan Levels. J. Am. Soc. Mass Spectrom. 2020, 31, 1603–1609. 10.1021/jasms.0c00183. [DOI] [PubMed] [Google Scholar]
- Campbell J. L.; Baba T.; Liu C.; Lane C. S.; Le Blanc J. C. Y.; Hager J. W. Analyzing Glycopeptide Isomers by Combining Differential Mobility Spectrometry with Electron- and Collision-Based Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1374–1381. 10.1007/s13361-017-1663-6. [DOI] [PubMed] [Google Scholar]
- Kolli V.; Schumacher K. N.; Dodds E. D. Ion Mobility-Resolved Collision-Induced Dissociation and Electron Transfer Dissociation of N-Glycopeptides: Gathering Orthogonal Connectivity Information from a Single Mass-Selected Precursor Ion Population. Analyst 2017, 142, 4691. 10.1039/C7AN01196B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese A. J.; Shimwell N. J.; Larkins K. P.; Heath J. K.; Cooper H. J. Probing the Complementarity of Faims and Strong Cation Exchange Chromatography in Shotgun Proteomics. J. Am. Soc. Mass Spectrom. 2013, 24, 431–443. 10.1007/s13361-012-0544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaskin R. S.; Khatri K.; Wang Q.; Zaia J.; Costello C. E. Construction of a Database of Collision Cross Section Values for Glycopeptides, Glycans, and Peptides Determined by Im-Ms. Anal. Chem. 2017, 89, 4452–4460. 10.1021/acs.analchem.6b04146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F.; Kohler N. D.; Brunner A. D.; Wanka J. H.; Voytik E.; Strauss M. T.; Theis F. J.; Mann M. Deep Learning the Collisional Cross Sections of the Peptide Universe from a Million Experimental Values. Nat. Commun. 2021, 12, 1185. 10.1038/s41467-021-21352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinneburg H.; Hofmann J.; Struwe W. B.; Thader A.; Altmann F.; Varon Silva D.; Seeberger P. H.; Pagel K.; Kolarich D. Distinguishing N-Acetylneuraminic Acid Linkage Isomers on Glycopeptides by Ion Mobility-Mass Spectrometry. Chem. Commun. 2016, 52, 4381–4384. 10.1039/C6CC01114D. [DOI] [PubMed] [Google Scholar]
- Guttman M.; Lee K. K. Site-Specific Mapping of Sialic Acid Linkage Isomers by Ion Mobility Spectrometry. Anal. Chem. 2016, 88, 5212–5217. 10.1021/acs.analchem.6b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J.; Hoffmann W.; Warnke S.; Huang X.; Gewinner S.; Schollkopf W.; Bowers M. T.; von Helden G.; Pagel K. An Infrared Spectroscopy Approach to Follow Beta-Sheet Formation in Peptide Amyloid Assemblies. Nat. Chem. 2017, 9, 39–44. 10.1038/nchem.2615. [DOI] [PubMed] [Google Scholar]
- Hoffmann W.; von Helden G.; Pagel K. Ion Mobility-Mass Spectrometry and Orthogonal Gas-Phase Techniques to Study Amyloid Formation and Inhibition. Curr. Opin. Struct. Biol. 2017, 46, 7–15. 10.1016/j.sbi.2017.03.002. [DOI] [PubMed] [Google Scholar]
- Antoine R.; Dugourd P. Visible and Ultraviolet Spectroscopy of Gas Phase Protein Ions. Phys. Chem. Chem. Phys. 2011, 13, 16494–16509. 10.1039/c1cp21531k. [DOI] [PubMed] [Google Scholar]
- Nguyen H. T.; Shaffer C. J.; Pepin R.; Turecek F. Uv Action Spectroscopy of Gas-Phase Peptide Radicals. J. Phys. Chem. Lett. 2015, 6, 4722–4727. 10.1021/acs.jpclett.5b02354. [DOI] [PubMed] [Google Scholar]
- Schnaar R. L.; Kinoshita T. In Essentials of Glycobiology; 3. ed.; Varki A.; Cummings R. D.; Esko J. D.; Stanley P.; Hart G. W.; Aebi M.; Darvill A. G.; Kinoshita T.; Packer N. H.; Prestegard J. H.. et al. , Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2015, 10.1101/glycobiology.3e.011. [DOI] [PubMed] [Google Scholar]
- Curatolo W. The Physical Properties of Glycolipids. Biochim. Biophys. Acta, Rev. Biomembr. 1987, 906, 111–136. 10.1016/0304-4157(87)90008-6. [DOI] [PubMed] [Google Scholar]
- Yamashita T.; Wada R.; Sasaki T.; Deng C.; Bierfreund U.; Sandhoff K.; Proia R. L. A Vital Role for Glycosphingolipid Synthesis During Development and Differentiation. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9142–9147. 10.1073/pnas.96.16.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degroote S.; Wolthoorn J.; van Meer G. The Cell Biology of Glycosphingolipids. Semin. Cell Dev. Biol. 2004, 15, 375–387. 10.1016/j.semcdb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Hakomori S. Glycosphingolipids in Cellular Interaction, Differentiation, and Oncogenesis. Annu. Rev. Biochem. 1981, 50, 733–764. 10.1146/annurev.bi.50.070181.003505. [DOI] [PubMed] [Google Scholar]
- Todeschini A. R.; Hakomori S. I. Functional Role of Glycosphingolipids and Gangliosides in Control of Cell Adhesion, Motility, and Growth, through Glycosynaptic Microdomains. Biochim. Biophys. Acta, Gen. Subj. 2008, 1780, 421–433. 10.1016/j.bbagen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S. J.; Nakayama K.; Hikita T.; Handa K.; Hakomori S. I. Epidermal Growth Factor Receptor Tyrosine Kinase Is Modulated by Gm3 Interaction with N-Linked Glcnac Termini of the Receptor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18987–18991. 10.1073/pnas.0609281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold K. R.; Elias P. M. Role of Lipids in the Formation and Maintenance of the Cutaneous Permeability Barrier. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 280–294. 10.1016/j.bbalip.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Aerts J.; Kuo C. L.; Lelieveld L. T.; Boer D. E. C.; van der Lienden M. J. C.; Overkleeft H. S.; Artola M. Glycosphingolipids and Lysosomal Storage Disorders as Illustrated by Gaucher Disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. 10.1016/j.cbpa.2019.10.006. [DOI] [PubMed] [Google Scholar]
- Platt F. M. Sphingolipid Lysosomal Storage Disorders. Nature 2014, 510, 68–75. 10.1038/nature13476. [DOI] [PubMed] [Google Scholar]
- Freeze H. H.; Kinoshita T.; Schnaar R. L. In Essentials of Glycobiology; Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H.. et al. , Eds.; Cold Spring Harbor (NY), 2015, 10.1101/glycobiology.3e.044. [DOI] [PubMed] [Google Scholar]
- Stoffel W.; Bosio A. Myelin Glycolipids and Their Functions. Curr. Opin. Neurobiol. 1997, 7, 654–661. 10.1016/S0959-4388(97)80085-2. [DOI] [PubMed] [Google Scholar]
- Kinjo Y.; Wu D.; Kim G.; Xing G. W.; Poles M. A.; Ho D. D.; Tsuji M.; Kawahara K.; Wong C. H.; Kronenberg M. Recognition of Bacterial Glycosphingolipids by Natural Killer T Cells. Nature 2005, 434, 520–525. 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- Kain L.; Webb B.; Anderson B. L.; Deng S.; Holt M.; Costanzo A.; Zhao M.; Self K.; Teyton A.; Everett C.; et al. The Identification of the Endogenous Ligands of Natural Killer T Cells Reveals the Presence of Mammalian Alpha-Linked Glycosylceramides. Immunity 2014, 41, 543–554. 10.1016/j.immuni.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. H. Jr. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. 10.1021/cr2002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Liu Y.; Sullards M. C.; Merrill A. H. An Introduction to Sphingolipid Metabolism and Analysis by New Technologies. NeuroMol. Med. 2010, 12, 306–319. 10.1007/s12017-010-8132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruett S. T.; Bushnev A.; Hagedorn K.; Adiga M.; Haynes C. A.; Sullards M. C.; Liotta D. C.; Merrill A. H. Jr. Biodiversity of Sphingoid Bases (“Sphingosines“) and Related Amino Alcohols. J. Lipid Res. 2008, 49, 1621–1639. 10.1194/jlr.R800012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Wang M. D.; Park M.; Sullards M. C. (Glyco)Sphingolipidology: An Amazing Challenge and Opportunity for Systems Biology. Trends Biochem. Sci. 2007, 32, 457–468. 10.1016/j.tibs.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Schnaar R. L. The Biology of Gangliosides. Adv. Carbohydr. Chem. Biochem. 2019, 76, 113–148. 10.1016/bs.accb.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Svennerholm L. The Gangliosides. J. Lipid Res. 1964, 5, 145–155. 10.1016/S0022-2275(20)40231-7. [DOI] [PubMed] [Google Scholar]
- Sauvageau J.; Ryan J.; Lagutin K.; Sims I. M.; Stocker B. L.; Timmer M. S. Isolation and Structural Characterisation of the Major Glycolipids from Lactobacillus Plantarum. Carbohydr. Res. 2012, 357, 151–156. 10.1016/j.carres.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Fahy E.; Subramaniam S.; Brown H. A.; Glass C. K.; Merrill A. H.; Murphy R. C.; Raetz C. R. H.; Russell D. W.; Seyama Y.; Shaw W.; et al. A Comprehensive Classification System for Lipids. Eur. J. Lipid Sci. Technol. 2005, 107, 337–364. 10.1002/ejlt.200405001. [DOI] [PubMed] [Google Scholar]
- Sarbu M.; Zamfir A. D. Modern Separation Techniques Coupled to High Performance Mass Spectrometry for Glycolipid Analysis. Electrophoresis 2018, 39, 1155–1170. 10.1002/elps.201700461. [DOI] [PubMed] [Google Scholar]
- Haynes C. A.; Allegood J. C.; Park H.; Sullards M. C. Sphingolipidomics: Methods for the Comprehensive Analysis of Sphingolipids. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 2696–2708. 10.1016/j.jchromb.2008.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y.; Hama K.; Yokoyama K. Mass Spectrometry in Combination with a Chiral Column and Multichannel-Mrm Allows Comprehensive Analysis of Glycosphingolipid Molecular Species from Mouse Brain. Carbohydr. Res. 2020, 490, 107959. 10.1016/j.carres.2020.107959. [DOI] [PubMed] [Google Scholar]
- Lee H.; Lerno L. A.; Choe Y.; Chu C. S.; Gillies L. A.; Grimm R.; Lebrilla C. B.; German J. B. Multiple Precursor Ion Scanning of Gangliosides and Sulfatides with a Reversed-Phase Microfluidic Chip and Quadrupole Time-of-Flight Mass Spectrometry. Anal. Chem. 2012, 84, 5905–5912. 10.1021/ac300254d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson H.; Halim A.; Teneberg S. Differentiation of Glycosphingolipid-Derived Glycan Structural Isomers by Liquid Chromatography/Mass Spectrometry. Glycobiology 2010, 20, 1103–1116. 10.1093/glycob/cwq070. [DOI] [PubMed] [Google Scholar]
- Anugraham M.; Everest-Dass A. V.; Jacob F.; Packer N. H. A Platform for the Structural Characterization of Glycans Enzymatically Released from Glycosphingolipids Extracted from Tissue and Cells. Rapid Commun. Mass Spectrom. 2015, 29, 545–561. 10.1002/rcm.7130. [DOI] [PubMed] [Google Scholar]
- Farwanah H.; Kolter T. Lipidomics of Glycosphingolipids. Metabolites 2012, 2, 134–164. 10.3390/metabo2010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. C.; Axelsen P. H. Mass Spectrometric Analysis of Long-Chain Lipids. Mass Spectrom. Rev. 2011, 30, 579–599. 10.1002/mas.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P. J.; Cheng T. Y.; Pellicci D. G.; Watts G. F. M.; Veerapen N.; Young D. C.; Rossjohn J.; Besra G. S.; Godfrey D. I.; Brenner M. B.; et al. Structural Determination of Lipid Antigens Captured at the Cd1d-T-Cell Receptor Interface. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8348–8353. 10.1073/pnas.1705882114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J. P.; Brodbelt J. S. Structural Characterization of Gangliosides and Glycolipids Via Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2013, 85, 10399–10407. 10.1021/ac402379y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland M. A.; Marshall A. G.; Hendrickson C. L.; Nilsson C. L.; Fredman P.; Mansson J. E. Structural Characterization of the Gm1 Ganglioside by Infrared Multiphoton Dissociation, Electron Capture Dissociation, and Electron Detachment Dissociation Electrospray Ionization Ft-Icr Ms/Ms. J. Am. Soc. Mass Spectrom. 2005, 16, 752–762. 10.1016/j.jasms.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Baba T.; Campbell J. L.; Le Blanc J. C. Y.; Baker P. R. S. Distinguishing Cis and Trans Isomers in Intact Complex Lipids Using Electron Impact Excitation of Ions from Organics Mass Spectrometry. Anal. Chem. 2017, 89, 7307–7315. 10.1021/acs.analchem.6b04734. [DOI] [PubMed] [Google Scholar]
- Ciucanu I. Per-O-Methylation Reaction for Structural Analysis of Carbohydrates by Mass Spectrometry. Anal. Chim. Acta 2006, 576, 147–155. 10.1016/j.aca.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Barrientos R. C.; Zhang Q. Differential Isotope Labeling by Permethylation and Reversed-Phase Liquid Chromatography-Mass Spectrometry for Relative Quantification of Intact Neutral Glycolipids in Mammalian Cells. Anal. Chem. 2019, 91, 9673–9681. 10.1021/acs.analchem.9b01206. [DOI] [PubMed] [Google Scholar]
- Pham H. T.; Ly T.; Trevitt A. J.; Mitchell T. W.; Blanksby S. J. Differentiation of Complex Lipid Isomers by Radical-Directed Dissociation Mass Spectrometry. Anal. Chem. 2012, 84, 7525–7532. 10.1021/ac301652a. [DOI] [PubMed] [Google Scholar]
- Pham H. T.; Julian R. R. Characterization of Glycosphingolipid Epimers by Radical-Directed Dissociation Mass Spectrometry. Analyst 2016, 141, 1273–1278. 10.1039/C5AN02383A. [DOI] [PubMed] [Google Scholar]
- Pham H. T.; Trevitt A. J.; Mitchell T. W.; Blanksby S. J. Rapid Differentiation of Isomeric Lipids by Photodissociation Mass Spectrometry of Fatty Acid Derivatives. Rapid Commun. Mass Spectrom. 2013, 27, 805–815. 10.1002/rcm.6503. [DOI] [PubMed] [Google Scholar]
- Chao H. C.; McLuckey S. A. Differentiation and Quantification of Diastereomeric Pairs of Glycosphingolipids Using Gas-Phase Ion Chemistry. Anal. Chem. 2020, 92, 13387–13395. 10.1021/acs.analchem.0c02755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H.-C.; McLuckey S. A. In-Depth Structural Characterization and Quantification of Cerebrosides and Glycosphingosines with Gas-Phase Ion Chemistry. Anal. Chem. 2021, 93, 7332–7340. 10.1021/acs.analchem.1c01021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M. C.; Mitchell T. W.; Harman D. G.; Deeley J. M.; Nealon J. R.; Blanksby S. J. Ozone-Induced Dissociation: Elucidation of Double Bond Position within Mass-Selected Lipid Ions. Anal. Chem. 2008, 80, 303–311. 10.1021/ac7017684. [DOI] [PubMed] [Google Scholar]
- Barrientos R. C.; Vu N.; Zhang Q. B. Structural Analysis of Unsaturated Glycosphingolipids Using Shotgun Ozone-Induced Dissociation Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 2330–2343. 10.1007/s13361-017-1772-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos R. C.; Zhang Q. Fragmentation Behavior and Gas-Phase Structures of Cationized Glycosphingolipids in Ozone-Induced Dissociation Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 1609–1620. 10.1007/s13361-019-02267-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.; Xia Y. Pinpointing Double Bonds in Lipids by Paterno-Buchi Reactions and Mass Spectrometry. Angew. Chem., Int. Ed. 2014, 53, 2592–2596. 10.1002/anie.201310699. [DOI] [PubMed] [Google Scholar]
- Bednarik A.; Bolsker S.; Soltwisch J.; Dreisewerd K. An on-Tissue Paterno-Buchi Reaction for Localization of Carbon-Carbon Double Bonds in Phospholipids and Glycolipids by Matrix-Assisted Laser-Desorption-Ionization Mass-Spectrometry Imaging. Angew. Chem., Int. Ed. 2018, 57, 12092–12096. 10.1002/anie.201806635. [DOI] [PubMed] [Google Scholar]
- Heiles S. Advanced Tandem Mass Spectrometry in Metabolomics and Lipidomics—Methods and Applications. Anal. Bioanal. Chem. 2021, 10.1007/s00216-021-03425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. N.; Colsch B.; Egan T.; Lewis E. K.; Schultz J. A.; Woods A. S. Gangliosides’ Analysis by Maldi-Ion Mobility Ms. Analyst 2011, 136, 463–466. 10.1039/C0AN00732C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skraskova K.; Claude E.; Jones E. A.; Towers M.; Ellis S. R.; Heeren R. M. Enhanced Capabilities for Imaging Gangliosides in Murine Brain with Matrix-Assisted Laser Desorption/Ionization and Desorption Electrospray Ionization Mass Spectrometry Coupled to Ion Mobility Separation. Methods 2016, 104, 69–78. 10.1016/j.ymeth.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Sarbu M.; Robu A. C.; Ghiulai R. M.; Vukelic Z.; Clemmer D. E.; Zamfir A. D. Electrospray Ionization Ion Mobility Mass Spectrometry of Human Brain Gangliosides. Anal. Chem. 2016, 88, 5166–5178. 10.1021/acs.analchem.6b00155. [DOI] [PubMed] [Google Scholar]
- Sarbu M.; Vukelic Z.; Clemmer D. E.; Zamfir A. D. Ion Mobility Mass Spectrometry Provides Novel Insights into the Expression and Structure of Gangliosides in the Normal Adult Human Hippocampus. Analyst 2018, 143, 5234–5246. 10.1039/C8AN01118D. [DOI] [PubMed] [Google Scholar]
- Sarbu M.; Vukelic Z.; Clemmer D. E.; Zamfir A. D. Electrospray Ionization Ion Mobility Mass Spectrometry Provides Novel Insights into the Pattern and Activity of Fetal Hippocampus Gangliosides. Biochimie 2017, 139, 81–94. 10.1016/j.biochi.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Kaszycki J. L.; La Rotta A.; Colsch B.; Fenaille F.; Dauly C.; Kamleh A.; Wu C. Separation of Biologically Relevant Isomers on an Orbitrap Mass Spectrometer Using High-Resolution Drift Tube Ion Mobility and Varied Drift Gas Mixtures. Rapid Commun. Mass Spectrom. 2019, 33, 3–10. 10.1002/rcm.8414. [DOI] [PubMed] [Google Scholar]
- Webb I. K.; Garimella S. V.; Tolmachev A. V.; Chen T. C.; Zhang X.; Norheim R. V.; Prost S. A.; LaMarche B.; Anderson G. A.; Ibrahim Y. M.; et al. Experimental Evaluation and Optimization of Structures for Lossless Ion Manipulations for Ion Mobility Spectrometry with Time-of-Flight Mass Spectrometry. Anal. Chem. 2014, 86, 9169–9176. 10.1021/ac502055e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik R.; Webb I. K.; Deng L.; Garimella S. V.; Prost S. A.; Ibrahim Y. M.; Baker E. S.; Smith R. D. Lipid and Glycolipid Isomer Analyses Using Ultra-High Resolution Ion Mobility Spectrometry Separations. Int. J. Mol. Sci. 2017, 18, 183. 10.3390/ijms18010183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May J. C.; Leaptrot K. L.; Rose B. S.; Moser K. L. W.; Deng L.; Maxon L.; DeBord D.; McLean J. A. Resolving Power and Collision Cross Section Measurement Accuracy of a Prototype High-Resolution Ion Mobility Platform Incorporating Structures for Lossless Ion Manipulation. J. Am. Soc. Mass Spectrom. 2021, 32, 1126–1137. 10.1021/jasms.1c00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongtrakul-Kish K.; Walsh I.; Sim L. C.; Mak A.; Liau B.; Ding V.; Hayati N.; Wang H.; Choo A.; Rudd P. M.; et al. Combining Glucose Units, M/Z, and Collision Cross Section Values: Multiattribute Data for Increased Accuracy in Automated Glycosphingolipid Glycan Identifications and Its Application in Triple Negative Breast Cancer. Anal. Chem. 2019, 91, 9078–9085. 10.1021/acs.analchem.9b01476. [DOI] [PubMed] [Google Scholar]
- Sarbu M.; Petrica L.; Clemmer D. E.; Vukelić Ž.; Zamfir A. D. Gangliosides of Human Glioblastoma Multiforme: A Comprehensive Mapping and Structural Analysis by Ion Mobility Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2021, 32, 1249. 10.1021/jasms.1c00088. [DOI] [PubMed] [Google Scholar]
- Abb S.; Tarrat N.; Cortés J.; Andriyevsky B.; Harnau L.; Schön J. C.; Rauschenbach S.; Kern K. Carbohydrate Self-Assembly at Surfaces: Stm Imaging of Sucrose Conformation and Ordering on Cu(100). Angew. Chem., Int. Ed. 2019, 58, 8336–8340. 10.1002/anie.201901340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Delbianco M.; Anggara K.; Michnowicz T.; Pardo-Vargas A.; Bharate P.; Sen S.; Pristl M.; Rauschenbach S.; Schlickum U.; et al. Imaging Single Glycans. Nature 2020, 582, 375–378. 10.1038/s41586-020-2362-1. [DOI] [PubMed] [Google Scholar]