

Abstract
PPP2R5D-related neurodevelopmental disorder is characterized by a range of neurodevelopmental and behavioral manifestations. We report the association of early-onset parkinsonism with the PPP2R5D p.E200K mutation. Clinical characterization and exome sequencing were performed on three patients, with postmortem neuropathologic examination for one patient. All patients had mild developmental delay and developed levodopa-responsive parkinsonism between the ages of 25 and 40 years. The PPP2R5D c.598G>A (p.E200K) mutation was identified in all patients. Neuropathologic examination demonstrated uneven, focally severe neuronal loss and gliosis in the substantia nigra pars compacta, without Lewy bodies. Our findings suggest the PPP2R5D p.E200K mutation to be a possible new cause of early-onset parkinsonism.
Introduction
PPP2R5D, primarily expressed in brain, encodes the regulatory subunit B566 of PP2A, a serine–threonine phosphatase playing a crucial regulatory role in multiple cellular processes, including proliferation, apoptosis, and signal transduction.1 Recurring de novo missense mutations in PPP2R5D have been associated with neurodevelopmental delay, intellectual disability, macrocephaly, motor and coordination deficits, epilepsy, visual impairment, and autism spectrum disorders (Mendelian Inheritance in Man (MIM) number 616355).2,5 Mast of these mutations, including p. E200K, occur in a hotspot coding for a conserved acidic ltxjp enabling B56δ to bind the scaffolding and catalytic PP2A subunits.6 p.E200K might impair this binding, perturbing the holoenzyme formation and hindering dephosphorylation of the normal PP2A substrates through a dominant-negative effect3 The associated phenotype of p. E200K appears to be milder, with greater functional capacity than other mutations, but with limited certainty given the condition’s rarity. Additionally, given that most confirmed genetic diagnoses of PPP2R5D-related disorder are in children, its natural history after 20 years of age remains largely unknown. We report clinical courses for three adults carrying the PPP2R5D p.E200K mutation with mild intellectual disability who developed early-onset parkinsonism. We report postmortem neuropathologic examination for one patient.
Patients and Methods
Cases 1 and 2 were identified through Movement Disorders clinics. Case 3 was identified through an overgrowth study.4 Genomic DNA was extracted from whole blood. Whole exome sequencing (WES) and Sanger confirmations 4‘7,8 and neuropathologic analysis9 were performed as previously described.
All participation was voluntary. Signed informed consent was obtained from each patient or representative before genetic testing. Patients were enrolled in institutional research studies, approved by the Columbia University Institutional Review Board, Strasbourg Ethics Committee,10 and London Multicentre Ethics Committee, respectively.
Results
Clinical Summaries
Case 1: The patient had mild developmental motor and language delays. At 40 years of age, he developed gait difficulty and bradykinesia. Brain magnetic resonance imaging (MRI) demonstrated T2 white matter hyperintensities (Fig 1A). He had co-morbid hypertension and diabetes mellitus. Motor symptoms improved with dopaminergic therapy, gradually complicated by motor and nonmotor fluctuations. Treatment with dopamine agonists was limited by impulse control disorders. At 57 years of age, he underwent implantation for deep brain stimulation, with unclear benefit. He died at 61 years of age from aspiration pneumonia.
FIGURE 1:
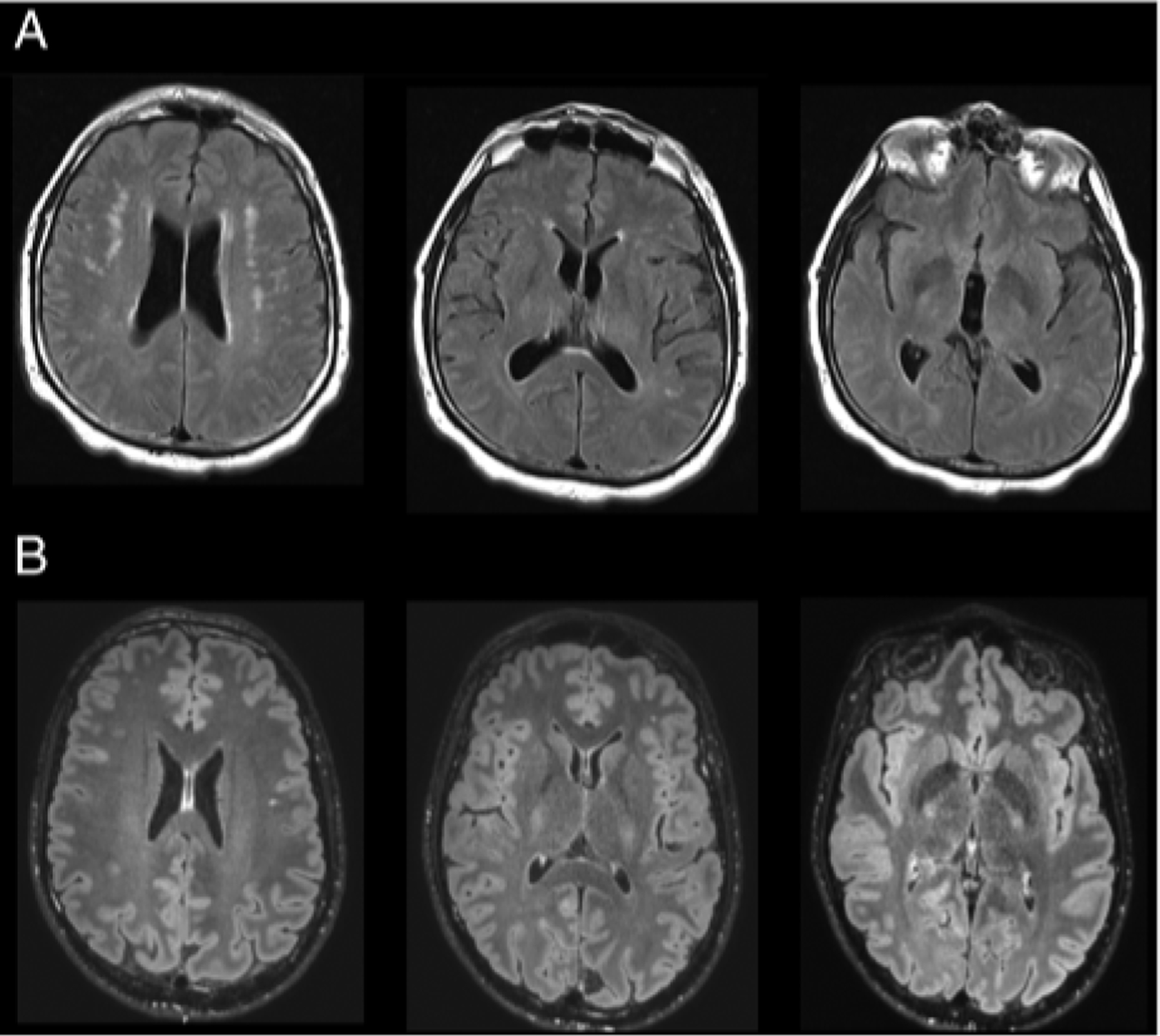
Neuroimaging: Cases 1 and 2. (A) Case 1: magnetic resonance imaging (MRI) brain T2 fluid-attenuated inversion recovery (FLAIR) images demonstrating hyperintensities in subcortical white matter bilaterally. (B) Case 2: MRI brain T2 FLAIR images demonstrating relative paucity of white matter signal abnormality.
Case 2: The patient had no motor delay but had intellectual disability. At 27 years of age, he developed rest tremor and myoclonus, evolving to akinetic rigid parkinsonism. Brain MRI was normal (Fig 1B). Motor symptoms improved with levodopa, limited by motor and nonmotor fluctuations, controlled with carbidopa/levodopa intestinal gel. Treatment with dopamine agonists was complicated by impulse control disorders. Early nonmotor features of parkinsonism were noted.
Case 3: The patient was previously reported (case COG0328).4 She was suspected to have Sotos syndrome based on overgrowth features; however, no mutations in NSDJ or NFIX were identified. She had delays in motor development and language acquisition, with mild intellectual disability. By 22 years of age, she developed rest tremor, incoordination, and parkinsonism with good levodopa responsiveness, ultimately complicated by motor fluctuations including dyskinesias.
Birth history was unremarkable in all cases. No family members were affected. Details of clinical courses are provided in Table and as Supplemental Material (Video SI examination).
TABLE.
Parkinsonism in PPP2R5D (NM_006245.3) p.E200K Mutation Carriers: Clinical Features
| Characteristic | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Ancestry | European | European | European |
| Sex | Male | Male | Female |
| Age at last visit | 61 years | 34 years | 44 years |
| Genetic test | Exome sequencing | Exome sequencing | Exome sequencing |
| Developmental history | Term birth. Motor delay: walked at 2–3 years. Language delayed. Mild intellectual disability: special education classes; graduated from vocational high school | Term birth: possible amniotic fluid aspiration. No motor delay. Language delayed. Severe learning disability: stopped schooling at 15 years | Term birth. Motor delay: sat at 9–10 months; cruised at 2 years. I Iypotonia. Language delayed. Mild intellectual disability |
| Seizures | No | No | No |
| Past medical history | Diabetes mellitus type 2, hypertension, sensorimotor neuropathy | None | Overgrowth (height 97th percentile by 2.5 years; macrocephaly) |
| Parkinsonism: age at onset | 40 years | 27 years | 22 years |
| Motor features | Onset of gait difficulty, generalized hradykinesia; subsequent freezing of gait. Atrcmulous | Onset of right arm rest tremor; subsequently, predominantly akinetic–rigid. Postural instability | Onset of asymmetric rest tremor; subsequent freezing of gait, postural instability, cervical dystonia |
| Nonmotor features | Autonomic: non motor off symptoms (sweats, urinary urgency). No cognitive decline | Autonomic: constipation, urinary urgency. Rapid eye movement sleep behavior disorder. Depression, anxiety. Early cognitive decline | Autonomic: none. Sleep disturbance: none. Visual hallucinations |
| Atypical features | None | Myoclonus; oculomotor abnormalities | None |
| Rate of progression | Fluctuations: 9 years after onset. Freezing of gait: 10 years after onset. Wheelchair bound: 17 years after onset | Fluctuations: 3 years after onset. Postural instability: 5 years after onset. Remains ambulatory without aids | Slow progression since onset (neady 20 years) |
| Diagnostic studies | Magnetic resonance imaging of brain without contrast: subcortical white matter T2 hyperintensities(Fig 1A) | Dopamine transporter imaging scan: abnormal. Magnetic resonance imaging of brain without contrast: normal (Fig 1B). Electromyography/Nerve conduction study: normal | None |
| Levodopa and other treatment response | Levodopa responsive. Deep brain stimulation (subthalamic nuclei, 17 years after onset): unclear benefit | Levodopa responsive | Levodopa responsive |
| Complications of therapy | Motor and nonmotor off symptoms. Impulse control disorders (gambling, overeating) with dopamine agonist | Motor and nonmotor off symptoms. Impulse control disorders (gambling, hypersexuality) with dopamine agonist | Fluctuations: motor off symptoms (marked rigidity); dopamine-induced dyskinesias |
| Agc/causc of death | 61 years/aspiration pneumonia | Not applicable | Not applicable |
Video S1.
Genetic Findings
In Case 1, the variant PPP2R5D c.598 G>A;p.E200K (NM_006245.3) was identified by proband-only WES,confirmed with Sanger sequencing as present in the patient and absent from his unaffected older sister. This variant was reported as pathogenic by multiple institutions in ClinVar (https://www.ncbi.nlm.nih.gOv/clinvar/variation/217456/). Assessment for likely gene-disrupting or deleterious missense variants in known Parkinson’s disease (PD) genes was negative. A heterozygous variant in PARK7 c.70delG; p.Asp24fs (NM_007262) was identified in the prohand and his unaffected sister.
The same PPP2R5D variant, c.598G>A;p.E200K, was identified through WES in both Cases 2 and 3. The mutation was confirmed by Sanger sequencing in both cases and was absent from both parents in Case 2. Although the parents were unavailable for testing in Case 3, the mutation was presumed de novo because they were reported to be clinically unaffected. No other likely pathogenic variants in genes associated with PD or early-onset parkinsonism10 were identified in Case 2. Known PD genes were not assessed specifically in Case 3.
Neuropathology (Case 1)
The external brain surface (l,792.2g) was normal except for the presence of bilateral frontal electrodes and atheromatous plaques involving the large arteries of the base (maximum 50% luminal stenosis). Bilateral electrode tracks extended from the cortex to the zona incerta. Transverse slices from the brainstem revealed marked depigmentation of the substantia nigra bilaterally, contrasting with the well-pigmented nucleus coeruleus. Microscopic assessment of the substantia nigra was performed using two levels: one near the red nucleus, the other near the decussation of the superior cerebellar peduncle (Fig 2). Bilateral, uneven loss of pigmented neurons, reactive gliosis, and the presence of scant, small macrophage clusters with pigmented cytoplasmic debris were notable. The density of pigmented neurons was either apparendy normal or mildly to moderately decreased in patches, flanked by areas showing severe loss of pigmented neurons with loose, glioric parenchyma Sections from the rostral level were less involved than the caudal. In the left pars compacta, loss of pigmented neurons prevailed medially. In contrast, on the right, loss was severe laterally but moderate medially. The pars reticulata was unremarkable. The dorsal nucleus of the vagus was normal bilaterally. Neither Lewy body-containing neurons nor Lewy neuritis was detected throughout the myelencephalon, mesencephalon, diencephalon, basal forebrain, or cerebral cortex. The nucleus coeruleus showed no abnormality. Marked criblures involved the polar subcortical white matter and lenticular nuclei; athero-arteriolosderosis was likewise marked.
FIGURE 2:
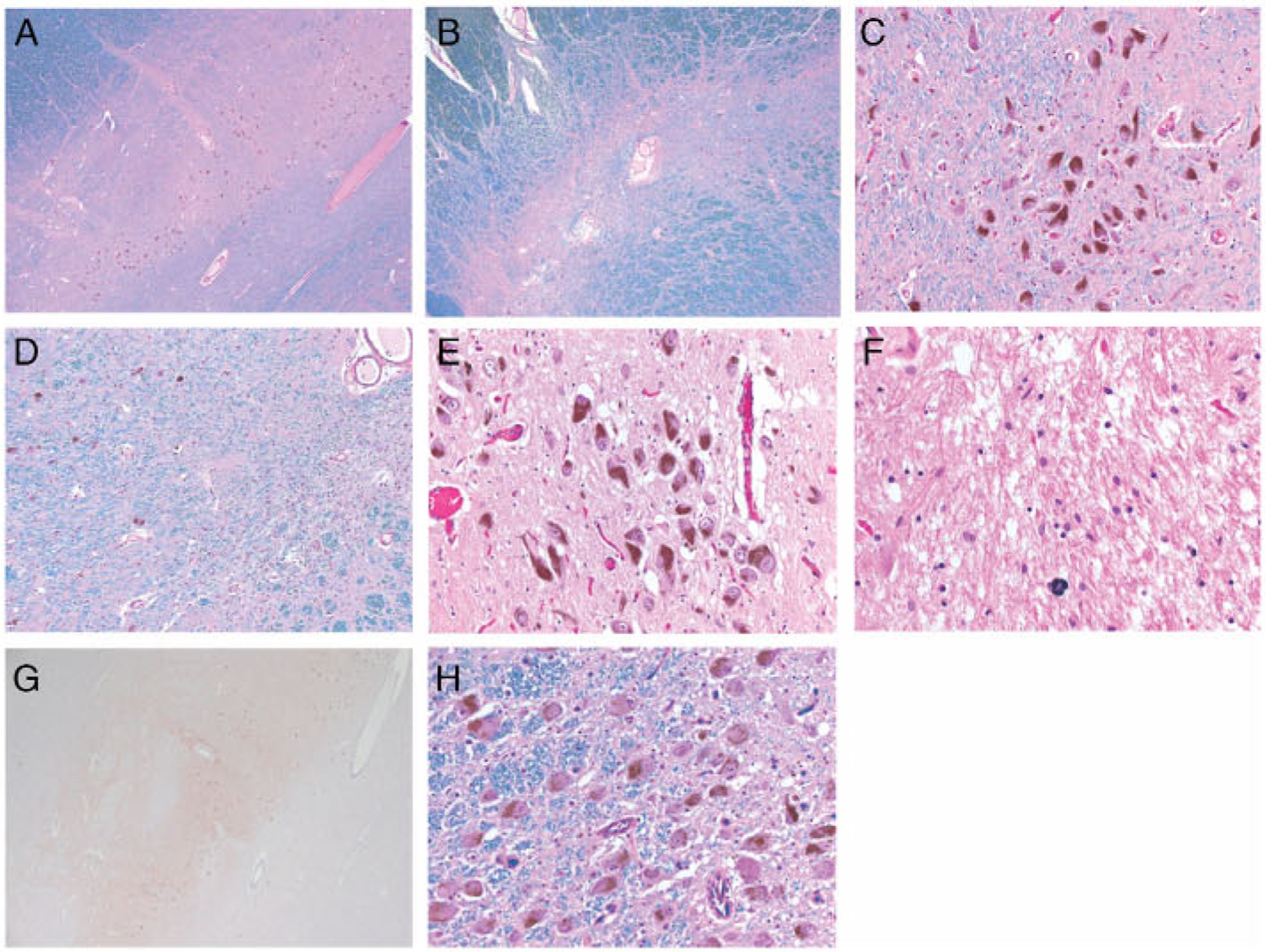
(A–D) Photomicrographs of the left substantia nigra: rostral, medial levels in A and C; caudal in B; and caudal, medial levels in D. The loss of pigmented neurons is uneven and prevails at the caudal levels. Relatively sharp demarcation between foci without or with resilient neurons in D. (E–H) Photomicrographs of the caudal levels of the right substantia nigra in E–G and of the nucleus coeruleus in H. (E) The density of pigmented neurons is apparently normal within the medial third. (F) The loss is subtotal within the lateral third. (G) Neither Lewy body-containing neurons nor Lewy neurites were detected. (H) The neuronal density of the nucleus coeruleus is normal. Staining: Luxol Fast Blue counterstained with Hematoxylin and Eosin in A–D and H; Hematoxylin and Eosin in E and F; and a-synudein in G. Original magnification: ×25 in A, B, and G; ×100 in D; ×200 in C, E, and H; and ×400 in F.
Discussion
The association of parkinsonism with a unique recurring de novo mutation, PPP2R5D p.E200K, is noteworthy. Although the association between the p.E200K mutation and impaired neurodevelopment is well established, our report is the first linking it to neurodegeneration, which represents an important advance in understanding of the natural history of this condition. The clinical courses of all three patients are notable for mild intellectual disability and early-onset parkinsonism, with onset from ages 25 to 40 years and variable rates of progression. This phenotype of stable intellectual disability during childhood with subsequent adult-onset parkinsonism has been described with mutations in only a few genes previously.8,11 All PPP2R5D p.E200K cases demonstrated levodopa responsiveness with motor fluctuations; two were complicated by impulse control disorders.
Incidence of the PPP2R5D neurodevelopmental disorder is estimated at 2.32 to 2.87 per 100,000 births.12 The mutation frequency in early-onset PD is unknown. We explored public PD genetic databases for the presence of the p.E200K mutation and did not identify any PD or control carriers, suggesting the variant to be extremely rare in the general population (JJ Kim; MB Makarious; S Bandres Ciga; JR Gibbs; J Ding; D Hernandez; J Brooks; F Grenn; H Iwaki; A Singleton; MA Nalls; G Blauwendraat; and International Parkinson’s Disease Genomics Gonsortium, https://pdgenetics.shinyapps.io/VariantBrowser/, manuscript in preparation). Notably, current databases do not distinguish between early-onset, familial, and other PD. We searched for PPP2R5D in a University of Strasbourg cohort of 122 patients with parkinsonism, including 60 early-onset and 35 familial cases; we did not identify any pathogenic or likely pathogenic variants. Further clarification will require study of a larger early-onset cohort.
The association of other PPP2R5D mutations with neurodegeneration is unknown. Parkinsonism has not been reported in association with other mutations to our knowledge. Increased screening (eg, inclusion of PPP2R5D in a genetic panel for early-onset parkinsonism) and longitudinal follow-up of PPP2R5D mutation carriers would allow better delineation of phenotype–genotype correlation. Biochemical/functional studies are reported in another manuscript currendy under review and suggest alteration in targets of phosphorylation with the mutation. Identification of the downstream targets of different mutations should help to elucidate the molecular mechanisms underlying neurological manifestations.
The neuropathologic findings represent the first such characterization in a PPP2R5D mutation carrier, to our knowledge. Focally severe substantia nigra atrophy and the absence of Lewy body pathology are notable. The absence of α-synuclein pathology is reminiscent of Parkin-associated PD13 or l-methyl-4-phenyl-l,2,3,6-tetrahydropyridine-related parkinsonism,14 both of which are thought to be primarily attributable to mitochondrial impairment.14,15 In contrast to most idiopathic PD cases, the nucleus coeruleus was unaffected here, the sparing of which is also reported in most Parkin-associated PD.13 PP2A-B56δ plays an important role in regulating phosphatidylinositol 3-kinase/protein kinase B and glycogen synthase kinase-3 beta-mediated growth control and tau phosphorylation, but tau deposits were not induced in a PPP2R5D knockout mouse model.16,17 Thus, although neurofibrillary tangles were not noted, we cannot exclude the possibility that altered dephosphorylation of tau might have contributed to the observed neurodegeneration.
We suspect that the noted vascular changes involving the subcortical white matter reflect incidental microvascular disease secondary to the patient’s vascular risk factors. Notably, brain MRI in Gase 2 was reassuring for significant white matter abnormalities (Fig 1B). Gase 1 is notable for a heterozygous DJ-1 frameshift variant, very likely to be incidental, given that his asymptomatic sister is also a carrier, only recessive mutations in DJ-1 have been linked to PD, and all DJ-1 PD cases with published neuropathology have demonstrated Lewy body pathology,18,19 absent here.
Rare neurogenetic conditions associated with intellectual disabilities, autism, and epilepsy are rapidly being identified, meriting further natural history data in adults. Although many will be static, identification of those associated with neurodegeneration or other systemic phenotypes is important. Understanding the precise molecular genetic basis for neurological conditions will allow individualized prognostication and, ultimately, treatment.
Acknowledgments
This research was supported in part by the Intramural Research Program of the US National Institutes of Health, National Institute on Aging and by grants from Jordan’s Guardian Angels, SFARI/Simons Foundation, the France Parkinson organization, the Revue Neurologique, and the JPB Foundation. The Columbia Parkinsonism Brain Bank is funded by the Parkinson’s Foundation (PF-SPE-1936 and PF-SPE-2032; RNA).
We thank the patients and their families for their generous contributions. We thank P. Greene for his clinical care and K. Tanji for her expert opinion of the neuropathologic findings for Case 1. We thank B. Jost for his support. We thank E. Ollivier and P. Nistchke for the bioinformatic treatment of the exome data from Case 2. We thank the Childhood Overgrowth Study, based at the Institute of Cancer Research, London, UK.
Footnotes
Potential Conflict of Interest
The authors report no conflicts of interest.
References
- 1.Seshacharyulu P, Pandey P, Datta K, Batra SK. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett 2013;335:9–18. 10.1016/j-canlet2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015;519:223–228. 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houge G, Haesen D, Vissers LE, et al. B56delta-related protein phosphatase 2A dysfunction identified in patients with intellectual disability. J Clin Invest 2015;125:3051–3062. 10.1172/jci79860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loveday C, Tatton-Brown K, Clarke M, et al. Mutations in the PP2A regulatory subunit B family genes PPP2R5B, PPP2R5C and PPP2R5D cause human overgrowth. Hum Mol Genet 2015;24:4775–4779. 10.1093/hmg/ddv182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shang L, Henderson LB, Cho MT, et al. De novo missense variants in PPP2R5D are associated with intellectual disability, macrocephaly, hypotonia, and autism. Neurogenetics 2016;17:43–49. 10.1007/s10048-015-0466-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007;445:53–57. 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka AJ, Cho MT, Millan F, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 2015;97:457–464. 10.1016/j-ajhg.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wirth T, Mariani LL, Bergant G, et al. Loss-of-function mutations in NR4A2 cause dopa-responsive dystonia parkinsonism. Mov Dis 2020;35:880–885. 10.1002/mds.27982. [DOI] [PubMed] [Google Scholar]
- 9.Vonsattel JP, Del Amaya MP, Keller CE. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol 2008;115:509–532. 10.1007/s00401-007-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montaut S, Tranchant C, Drouot N, et al. Assessment of a targeted gene panel for identification of genes associated with movement disorders. J AMA Neurol 2018;75:1234–1245. 10.1001/jamaneurol.2018.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nature Genet 2013;45: 445–449, 9e1. 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Rivera JA, Perez-Palma E, Symonds J, et al. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain 2020;143:1099–1105. 10.1093/brain/awaa051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord 2017;32:1504–1523. 10.1002/mds.27193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983;219:979–980. 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 15.Palacino JJ, Sagi D, Goldberg MS, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 2004; 279:18614–18622. 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 16.Louis JV, Martens E, Borghgraef P, et al. Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc Natl Acad Sci U S A 2011;108:6957–6962. 10.1073/pnas.1018777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu UY, Yoo BC, Ahn JH. Regulatory B subunits of protein phosphatase 2A are involved in site-specific regulation of tau protein phosphorylation. Kor J Physiol Pharmacol 2014;18:155–161. 10.4196/kjpp.2014.18.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taipa R, Pereira C, Reis I, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016;139:1680–1687. 10.1093/brain/aww080. [DOI] [PubMed] [Google Scholar]
- 19.Narendra DP, Isonaka R, Nguyen D, et al. Peripheral synucleinopathy in a DJ1 patient with Parkinson disease, cataracts, and hearing loss. Neurology 2019;92:1113–1115. 10.1212/wnl.0000000000007614. [DOI] [PMC free article] [PubMed] [Google Scholar]