

Abstract
Photosystem II (PSII) catalyzes light-driven water oxidization, releasing O2 into the atmosphere and transferring the electrons for the synthesis of biomass. However, despite decades of structural and functional studies, the water oxidation mechanism of PSII has remained puzzling and a major challenge for modern chemical research. Here, we show that PSII catalyzes redox-triggered proton transfer between its oxygen-evolving Mn4O5Ca cluster and a nearby cluster of conserved buried ion-pairs, which are connected to the bulk solvent via a proton pathway. By using multi-scale quantum and classical simulations, we find that oxidation of a redox-active Tyrz (Tyr161) lowers the reaction barrier for the water-mediated proton transfer from a Ca2+-bound water molecule (W3) to Asp61 via conformational changes in a nearby ion-pair (Asp61/Lys317). Deprotonation of this W3 substrate water triggers its migration toward Mn1 to a position identified in recent X-ray free-electron laser (XFEL) experiments [Ibrahim et al. Proc. Natl. Acad. Sci. USA 2020, 117, 12,624–12,635]. Further oxidation of the Mn4O5Ca cluster lowers the proton transfer barrier through the water ligand sphere of the Mn4O5Ca cluster to Asp61 via a similar ion-pair dissociation process, while the resulting Mn-bound oxo/oxyl species leads to O2 formation by a radical coupling mechanism. The proposed redox-coupled protonation mechanism shows a striking resemblance to functional motifs in other enzymes involved in biological energy conversion, with an interplay between hydration changes, ion-pair dynamics, and electric fields that modulate the catalytic barriers.
Introduction
Photosystem II (PSII) captures the energy of visible-light photons, powering water oxidization catalysis at the Mn4O5Ca cluster and reduction of plastoquinone at the QB site (Figure 1A,B).1−3 The photoexcitation of chlorophyll induces a charge separation (ChlD1•+/PheoD1•–) followed by electron transfer from PD1 to ChlD1•+, from PheoD1•– to QA, the first quinone acceptor, and then from QA•– to QB, the exchangeable quinone (Figure 1B). The resulting radical cation, PD1•+, is one of the strongest oxidants in biology, with a redox potential of ∼1.25 V that is capable of extracting electrons from a redox-active tyrosine, Tyr161 of the D1 subunit (termed Tyrz or Yz), forming a tyrosyl radical, Tyrz• (Yz•), in a reaction linked to proton transfer to the adjacent His190 (Figure 1C). Tyrz• further oxidizes the Mn4O5Ca cluster, the catalytic site for water oxidation (Figure 1B,C).4 Four sequential photochemical turnovers increase the oxidation state of the active site from S0Yz to the S4Yz state of the Kok cycle (Figure 1D).5 Upon formation of the S4Yz state, two water molecules (H2O) are oxidized into molecular oxygen (O2) while releasing the protons to the luminal side of the membrane.2,6 The release of the O2 product during the S4Yz to S0Yz transition occurs with kinetics that match the decay of the S3Yz• state; therefore, in kinetic terms, the S4Yz state is S3Yz•.
Figure 1.

Structure and function of PSII. (A) Structure of PSII from T. vulcanus (PDB ID: 3WU2), modeled in a lipid membrane, showing the setup employed in our molecular simulations. (B) Light-absorption at the ChlD1 chlorophyll triggers electron transfer along the D1 branch to QB. The arrow numbers show the order of the electron transfer events. Four photochemical turnovers couple to the sequential reduction of two quinones to quinol and the oxidation of two water molecules to oxygen, releasing the protons to the luminal side of the membrane. (C) Closeup of the oxygen-evolving Mn4O5Ca complex, the redox-active Tyrz, and its hydrogen-bonding histidine (His190), as well as the putative proton acceptor Asp61, forming an ion-pair with Lys317. (D) The catalytic cycle, where the chlorophyll-driven photo-oxidation, drives the stepwise oxidation and proton release from the Mn4O5Ca cluster. After the extraction of four electrons and four protons from two water molecules, molecular dioxygen is formed in the S4Yz → S0Yz transition.
When the full enzyme cycle is complete, each photochemical charge separation results on average in the release of one proton into the lumen from water oxidation (2H2O → O2 + 4e– + 4H+lumen) and the uptake of one proton from the stomal side coupled with QB reduction (2Q + 4e– + 4H+stroma→2QH2). Thus, a proton motive force (pmf) is generated across the thylakoid membranes, powering ATP synthesis, while the two electron-reduced and fully protonated QB is released as plastoquinol (QH2) into the membrane pool and is available to provide the electrons to cytochrome b6f and thence down-stream photosynthetic electron transfer, leading eventually to CO2 fixation and the formation of biomass.1
Despite recent structural advances,7−13 some molecular principles of the water oxidation process in PSII remain elusive, particularly the exact nature of the substrate water molecules involved in the O–O bond formation. To this end, most computational and experimental studies support the overall oxidation and spin states of the Mn4O5Ca cluster, although it has also been suggested that a low oxidation state model could explain some structural and spectroscopic features of the Mn4O5Ca cluster14 (but see ref (15) for thorough discussion on problems with this model). We have here modeled the catalytic cycle according to the strongly supported consensus high-valent redox scheme.
The Mn4O5Ca center binds four water ligands (W1–W4) and the five oxo/hydroxo bridges that connect the metal ions (Figure 1C). The O5 between Mn1 and Mn4 is considered to be a likely candidate for one of the substrate water molecules (Figure 1C and Figure S1A).7 Each light pulse triggers sequential oxidation and proton release from the Mn4O5Ca cluster, except in the S1Yz → S2Yz transition, which presumably accumulates the total charge of the cluster (Figure 1D).2,6 Electron paramagnetic resonance (EPR) spectroscopy, structural, and computational studies7,10,16−23 identified high spin (HS) and low spin (LS) states18−21,24−26 associated with the open and closed cubane conformations of the S2Yz state of the Mn4O5Ca cluster.7,10,22,23 Upon further oxidation of these states in the S2Yz → S3Yz transition, it was suggested that the Mn4-bound W1 or W2, or alternatively, the Ca2+-bound W3,25−27 could deprotonate and function as a substrate water molecule6,20,28−31 that together with O5 establishes the O–O bond.11 In this regard, Guidoni and co-workers32,33 observed a rapid deprotonation of W1/W2 to Asp61 during picosecond ab initio MD simulations, whereas Pantazis and co-workers34 found that the proton affinity for W1/W2 is lowered during the S2Yz → S3Yz transition. Ugur et al.(26) suggested that W3-deprotonation triggers the migration of this water molecule toward Mn1 (or to Mn4 in the closed cubane form), in a process that is favored by the oxidation of Tyrz, a finding that has recently gained indirect support from time-resolved X-ray free electron laser (XFEL) structures of PSII in the S2Yz → S3Yz transition23 (but cf. also refs (35) and (36)), Fourier transform infrared (FTIR) spectroscopic studies,37,38 time-resolved membrane inlet mass spectrometric (TR-MIMS) experiments,39 and other computational studies.40 Importantly, water-exchange experiments revealed that all substrate water molecules are already bound in the S2Yz state27 and that the (water/hydroxo/oxo) ligands observed in the refined X-ray structural models of this state7,9,11,23 are thus likely substrate water candidates. After deprotonation of the substrate water molecule(s), the O2 formation could take place by radical coupling with the O5 oxo-bridge,11,26,41−43 although nucleophilic attack-mechanisms have also been proposed44 (but cf. ref (45)).
Despite these recent advances, it still remains unclear how deprotonation of the substrate water molecules is achieved, including the exact pathway used for transferring the protons across the membrane.13 Moreover, the discrepancy between the proposed substrate water candidates still remains unsolved. Although the involvement of W3 in the O2 formation process26 has recently gained further support,9,11,22,39 it is currently unclear how Asp61, a residue that could be an important part in the proton pathway en route to the luminal aqueous phase,13,46 functions as a proton acceptor for W3, as this residue is located >10 Å away from W3 on the other side of the Mn4O5Ca cluster (Figure 1C). Interestingly, in this context, recent time-resolved XFEL experiments13 revealed conformational- and hydration changes along the Cl1 water/proton channel that is close to both Asp61 and Glu65 of subunit D1.
To probe the light-driven water oxidation mechanism in PSII, we develop here a multi-scale molecular simulation approach,47 where we combine quantum chemical density functional theory (DFT) and hybrid quantum/classical (QM/MM) calculations to study the energetics and dynamics of the bond-breaking/bond-formation processes, with classical atomistic molecular dynamics (MD) simulations, which allows us to sample the large-scale dynamics of PSII on microsecond timescales in a biological membrane-water environment (Figure 1 and Figure S1). To gain insight into the protonation dynamics, we focus here on the S2Yz → S3Yz and S3Yz → S4/S0Yz transitions that couple to the oxidation and deprotonation of the substrate water molecule and formation of the O–O bond.
Results
Redox-Driven Ion-Pair Dynamics Drives Substrate Water Deprotonation
In order to probe the redox-driven proton transfer energetics in the S2Yz → S3Yz transition, we performed quantum chemical DFT calculations of the Mn4O5Ca center and its nearby conserved environment, built from an experimental high-resolution X-ray structure of PSII (PDB ID: 3WU2)7. These rather large DFT-models comprised around 210 atoms with key first and second sphere ligands described quantum mechanically (Figure 2A,B and Figure S1B). The energetics is reported here at the B3LYP*-D3 level (with 15% exact exchange), as this functional has been found to accurately capture the energetics of the Mn4O5Ca cluster.31,41 However, we obtain similar energetics of the key transitions also with other density functionals (Figure S7), suggesting that the overall results are robust.
Figure 2.
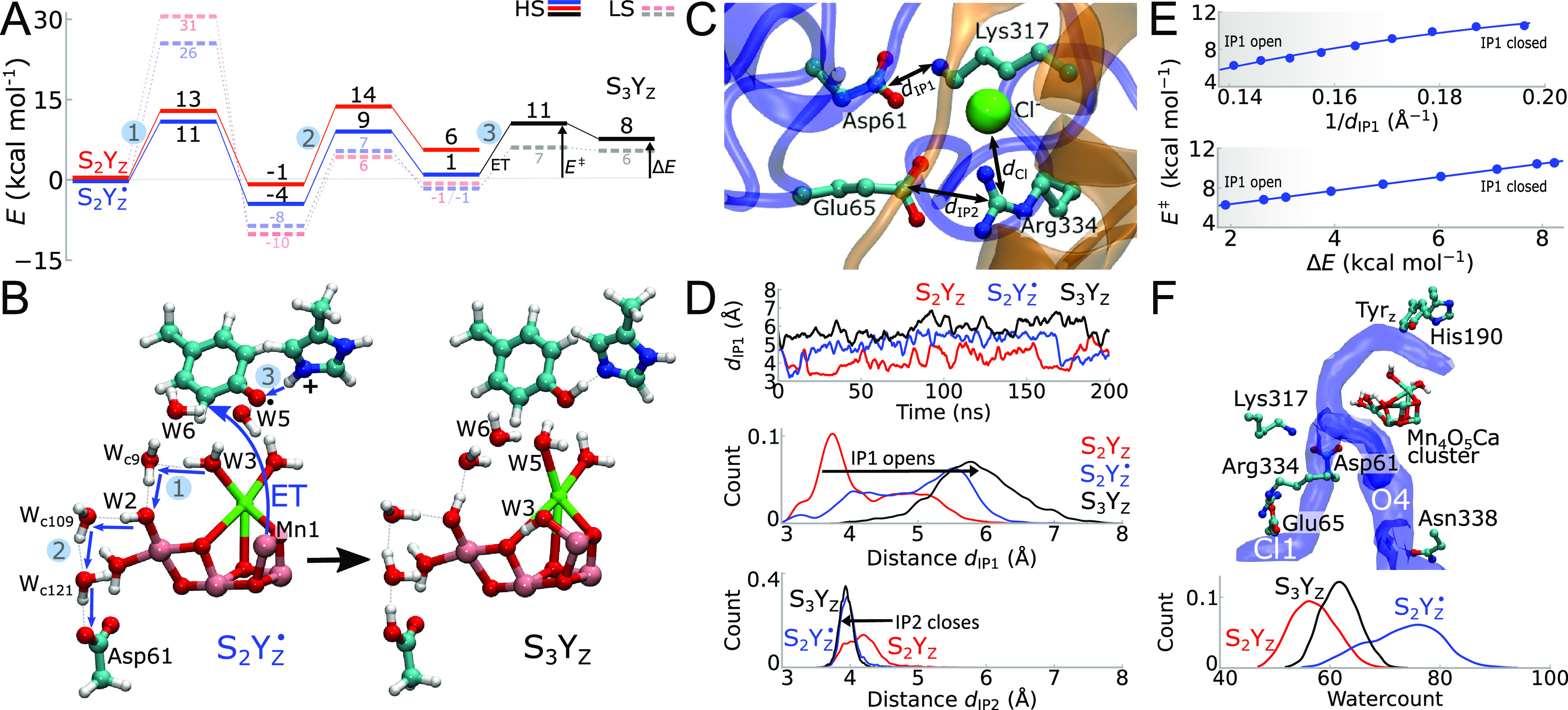
Redox-coupled protonation dynamics in the S2Yz → S3Yz transition. (A) Energetics of proton transfer from W3 to Asp61 in the open cubane/LS and closed cubane/HS states (see also Figure S2). The calculations were performed in the S2Yz (red), S2Yz• (blue), and S3Yz (black) states. All energies were obtained via reaction pathway optimizations using DFT models. The full reaction pathway for S2Yz• is shown in the Supporting Information, Movie S1 (for LS) and Movie S2 (fFor HS). See also SI Methods section for detailed description of the steps involved. (B) Optimized molecular structures of intermediate states along the S2Yz → S3Yz transition using DFT models. Only residues that participate in the direct reaction are shown (see Figure S1 and Table S1 for the complete model with 212 atoms). The proton is transferred (1) from W3 via Wc9 to W2 and further along (2) W2 via Wc109-Wc121 to Asp61. (3) Protonation of Asp61 couples to re-reduction of Tyrz• and deprotonation of His190 (HisH+), forming the S3Yz state. (C) Snapshot from MD simulations of the ion-paired network next to the Mn4O5Ca cluster. (D) Redox-coupled dynamics from classical MD simulations of the Asp61/Lys317 (dIP1) and Glu65/Arg334 (dIP2) ion-pairs. See Figure S4 for Arg334-Cl– (dCl) distances. Formation of S2Yz• and S3Yz states is coupled to a conformational change in the ion-pairs. (E) Top: proton transfer barriers from W3 to Asp61, obtained from DFT models by varying the Asp61-Lys317 headgroup distances (dIP1). The barrier has a 1/dIP1 dependence, indicative of electrostatic tuning. Bottom: the proton transfer barrier has a linear dependence on the thermodynamic driving force of the reaction, with a Brønsted slope of α = 0.7 (E‡ = αΔE + β). (F) Top: formation of putative proton pathways (Cl1 and O4) during the MD simulations with a focus on the possible proton release tunnels. Bottom: the S2Yz• state leads to a significant increase in the hydration state of Cl1. See Figure S4F for open and closed IP1 and IP2 conformations.
We modeled the Mn4O5Ca cluster in both closed (short O5-Mn1 distance) and open (long O5-Mn1 distance) cubane structures in the respective high spin (HS) and low spin (LS) configurations (see Materials and Methods, Figure S1A), as both configurations are accessible in the S2Yz state.16,18−21,25,26,48 We also studied the dynamics of the S2Yz → S3Yz transition using hybrid quantum/classical molecular dynamics (QM/MM MD) simulations, where the active QM region was polarized by the surrounding PSII structure, described at the classical force field (MM) level (Figure S1C).
To study the energetics of the proton transfer reactions, we optimized minimum energy reaction pathways connecting the optimized states prior and after the proton transfer reactions. We find that the proton transfer from the Ca2+-bound W3 to Asp61 takes place via an array of five water molecules, with an overall reaction barrier of ∼14 kcal mol–1 in the S2Yz state (k = 1.1 ms–1 according to transition state theory (TST), assuming a standard pre-exponential factor) (Figure 2A,B), whereas upon formation of the S2Yz•, we observe a reduction of the overall proton transfer barrier to ∼11 kcal mol–1 (k = 9 μs–1), suggesting that the Tyrz oxidation/cationic His190 (HisH+) formation favors the proton transfer from W3 to Asp61 (Figure 2A and Figure S2). This notion is supported by removal of the His/Tyrz pair from the QM system that significantly increases the reaction barrier for the S2Yz• state but leaves the barrier for the S2Yz state unchanged (Figure S2F). The proton is transferred first from W3 to W2 via Wc9 and then via two crystallographic water molecules (Wc109, Wc121) to Asp61 (Figure 2B and Figure S2). During the initial deprotonation of W3, the W3-hydroxide ligand migrates to a location between Mn1 and O5 (in the open cubane structure), while the empty ligand vacancy at the Ca2+ is immediately filled by a nearby crystallographically resolved water molecule (W5) (Figure 2B and Figure S2A), consistent with previous calculations.26 This process couples to the re-orientation of another nearby water molecule (W6), forming hydrogen-bonded contacts between the Mn1-bound W3(OH–) and O5, as well as subtle conformational changes also in the surrounding residues Ala344 and Arg357 (Figure S2G). The resulting geometry of the open/LS form of S3Yz is in overall good agreement with the refined XFEL models (Figure S9 and Table S6).
We obtain overall similar reaction profiles and redox-state dependence also in the closed cubane/HS form, with the exception that W3 migrates upon deprotonation to a position between O5 and Mn4 (Figure S2A,D). However, while the proton transfer barriers are more favorable in the high spin (HS) state (Figure 2A and Figure S2B–E), our calculations show that the two spin configurations/conformational states become nearly isoenergetic in the S2Yz and S2Yz• states, while the S3Yz state strongly favors the low spin form (Figure 2A and Table S2; cf. also refs (24−26) and (48)). This indicates that the proton transfer reaction could additionally couple to a spin transition in S2Yz → S3Yz (Figure S2A).18,19,26,49 Further calculations, where we restrained W3 to remain bound to Ca2+, reveal that the W3 migration strongly favors the proton transfer toward Asp61 (Figure S2B,C). During the final steps of this reaction, the ε-proton of His190 (HisH+) moves back to Tyrz in a process that couples to electron transfer from Mn1/Mn4 (open/closed cubane), resulting in the formation of the S3Yz state (Figure 2A,B and Figure S2).
The local dynamics of these states are also supported by our 8–10 ps QM/MM MD simulations (Figure S3): although we do not observe spontaneous complete proton transfer from W3 to Asp61 on the picosecond QM/MM simulation timescale, we find that oxidation of Tyrz and formation of HisH+ in the S2Yz• state strongly orients the water wire from W3 toward Asp61, which results in a transient protonation of Asp61 by W1 (LS/open, Figure S3C), consistent with previous studies.20 The picosecond QM/MM MD timescale allows for sampling of low barrier transitions but does not account for conformational changes linked to these transitions that take place on a longer timescale. However, to partially circumvent the limited timescale, we initiated simulations from different intermediate states along the S2Yz → S3Yz transition (see SI Methods and Models, and Figure S3). These local protonation steps (cf. also ref (20)) could comprise a part of the longer proton pathway from W3 to Asp61 (see above) and may unify in part the discrepancies between the W1/W2–20,21,32−34,50 and W3-mediated21,25,26 pathways.
In the simulations of the S2Yz• state (HS/closed), the deprotonated W3 spontaneously coordinates to Mn4 during the initial 3 ps (Figure S3B), supporting the re-organization and migration of the ligand water structure (see above), whereas in the LS QM/MM trajectory, we observe a spontaneous proton transfer between W1 and W3 (Figure S3E). Interestingly, upon the water migration, we observe an uptake of another water molecule from the nearby O1 channel (Figure S3F, see also ref (13)), whereas in the S3Yz state, we observe a rotation of the W1–W3 water/hydroxo ligands connected to Mn4 (Figure S3D), suggesting that these water molecules could interchange, as also proposed based on QM/MM geometry optimizations.51
Asp61, which functions as an acceptor in the proton transfer from W3, forms an ion-pair with Lys317 (here called IP1) and close contacts also with a Cl– ion and a second ion-pair (here IP2) (Glu65/Arg334, Figure 2C). These findings are consistent with previous simulations by Batista and co-workers,52 who found that the Cl– affects the conformation of the surrounding ion-pairs. To probe how the proton transfer reaction is linked to the conformational dynamics of this buried charged cluster, we performed atomistic molecular dynamics (MD) simulations of the S2Yz → S3Yz transition that allowed us to sample much longer (∼200 ns) timescales. Interestingly, in these simulations, the Asp61/Lys317 ion-pair samples a closed conformation in the S2Yz state (Figure 2C,D), while oxidation of Tyrz and the coupled protonation of His190(HisH+) in S2Yz• strongly favor opening of the ion-pair (dIP1 in Figure 2D). Protonation of Asp61 in the S3Yz state further stabilizes the open-ion pair conformations (Figure 2D, see also Figure S4F for open/closed ion-pair conformations).
Dissociation of this ion-pair reduces the proton transfer barrier with an electrostatic 1/distance-dependence of the Asp61-Lys317 headgroup separation (Figure 2E, top), which linearly correlates with an increase in the driving force of the reaction/proton affinity of Asp61 (Figure 2E, bottom). The open ion-pair conformation lowers the proton transfer barriers along the pathway from W3 to Asp61 (Figure 2E, top; Figure S2), while strongly increasing the thermodynamic driving force of the reaction (Figure 2E, bottom). Opening of the ion-pair thus lowers the proton transfer barrier in the S2Yz• state from ∼11 kcal mol–1 (Figure 2A) to ∼6 kcal mol–1 (Figure 2E).
Further analysis of the MD simulation trajectories show that this ion-pair opening correlates with the conformational state of the Glu65/Arg334 ion-pair (IP2), as well as the position of the Cl– ion relative to Arg334 (Figure 2C,D and Figure S4).52 These redox-triggered conformational changes are also supported by recent XFEL experiments.13 Although more challenging to experimentally validate, we suggest that the correlated dynamics establishes a gate that directs the protons toward the luminal aqueous phase, preventing back leakage reactions at high luminal proton concentrations.
As water molecules provide crucial conduits for protons, we next analyzed the hydration dynamics around the Mn4O5Ca cluster in the S2Yz → S3Yz transition. Our MD simulations predict that bulk water molecules flow in the vicinity of the Mn4O5Ca cluster (Figure 2F and Figure S4). We observe a bifurcated proton pathway that leads from the bulk solvent to Asp61 and further all the way to the Ca2+-bound W3. One of the branching points (the Cl1 tunnel13) passes the Asp61/Lys317 and Glu65/Arg334 ion-pairs, while the other pathway connects Asp61 via Asn338 to the bulk solvent (Figure 2F and Figure S4), resembling key observations in recent time-resolved XFEL experiments of the S2Yz → S3Yz transition.13
In MD simulations of the S2Yz• state, we observe a drastic increase of the hydration level relative to the S2Yz state (Figure 2F), an effect that is likely to arise from the electric field originating from the cationic His190 (HisH+). This S2Yz• state results in a strong oriented electric field (increase of E up to ∼0.8 VÅ–1 relative to S2Yz) along the pathway between His190 and Asp61, an effect that is further enhanced by opening of the Asp61-Lys317 ion pair (Figure S5). Interestingly, electrostatic interactions originating from the Yz• formation also influence the magnetic properties of the Mn4O5Ca cluster.48 These findings suggest that oxidation of the Tyrz induces local deprotonation of the substrate water molecule and also favors hydration and proton release to the bulk solvent.
Protonation Dynamics during the S3Yz → S4Yz Transition and Mechanism of O–O Bond Formation
To test whether the Mn1-W3(OH–) can undergo a second deprotonation involving the Asp61 route, we next performed DFT calculations along the S3Yz → S4Yz transition (Figure 3A,B). Despite the rather convoluted proton pathway from the Mn1-bound W3 to Asp61 (Figure 3B), we obtain a proton transfer barrier of 11 kcal mol–1 (in S3Yz•)/13 kcal mol–1 (in S3Yz) (k = 8.8 μs–1/0.2 ms–1) along the W3(OH–)-W6-O5-W2-Wc109-W1-Asp61 pathway (Figure 3A,B and Figure S6). This proton pathway is both kinetically and thermodynamically favored by the oxidation of Tyrz and formation of the cationic His190 (HisH+) in S3Yz•, as well as the dissociation of the Asp61/Lys317 ion-pair (Figure 3D). The optimized minimum energy proton transfer pathway takes place by the initial protonation of Asp61 by W1 followed by re-protonation of the resulting (W1) hydroxide by proton transfer from the Mn1-W3 (Figure 3B and Figure S6). However, the process involves multiple intermediate micro-states connected by rather low reaction barriers (Figure S6), suggesting that other competing reaction pathways could be favored and affected by temperature effects. Nevertheless, as found for the S2Yz• state, the cationic His190 formed by oxidation of Tyrz in S3Yz• induces dissociation of the Asp61/Lys317 ion-pair (Figure 3C), which in turn lowers the proton transfer barrier (Figure 3D).
Figure 3.
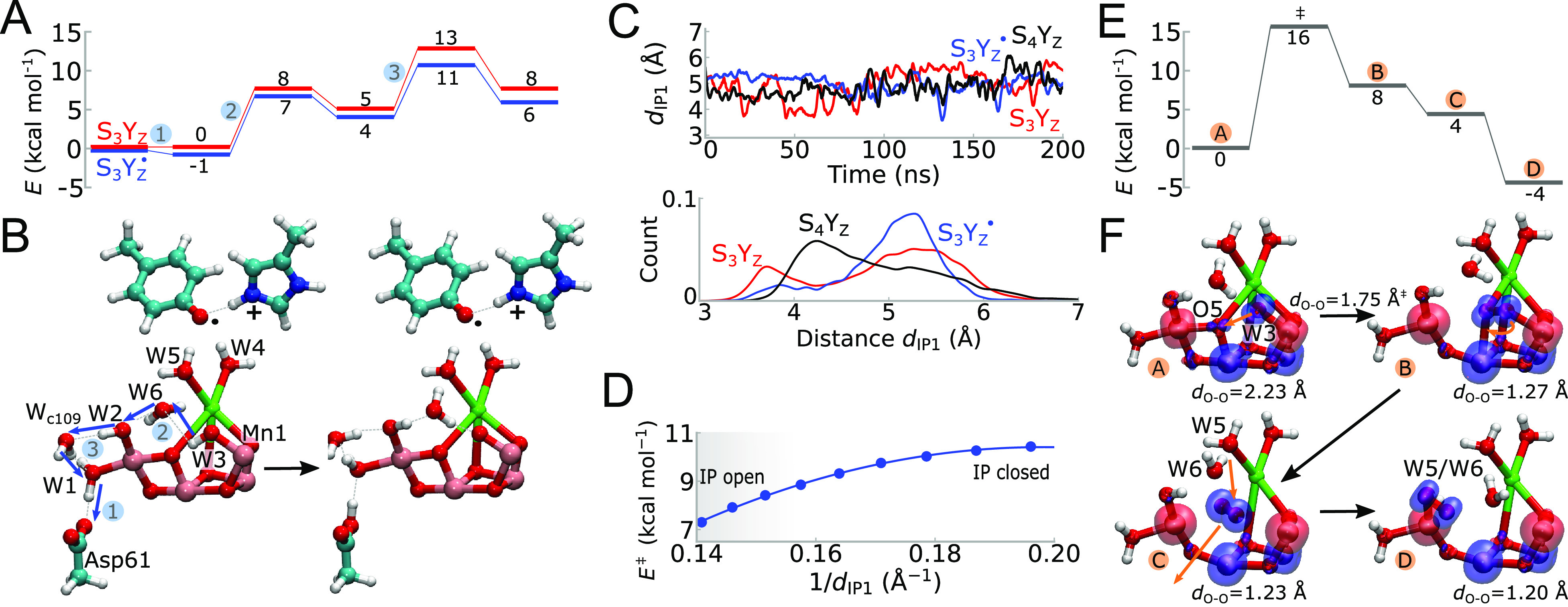
Redox-coupled protonation dynamics in the S3Yz → S4Yz transition. (A) Energetics of proton transfer from the Mn1-bound W3(OH–) to Asp61 in the open cubane (LS) configuration. The calculations were performed in the S3Yz (red) and S3Yz• states (blue). All energies were obtained via DFT reaction pathway optimizations. See SI Methods and Movie S3, for the visualization of the complete reaction pathway. (B) Optimized molecular structures of intermediate states occurring in the S3Yz → S4Yz transition. The steps involve proton transfer: 1) from W1 to Asp61, 2) from W3 via W6 to W2, and 3) from W2 via Wc109 to W1. (C) Dynamics of the Asp61-Lys317 ion-pair in the S3Yz, S3Yz•, and S4Yz states obtained from MD simulations. (D) The DFT models show that opening of the Asp61-Lys317 ion-pair lowers the proton transfer barrier from W3(OH–) to Asp61 similar as occurs in the S2Yz• state. (E), (F) Energetics of O-O bond formation and optimized molecular structures in the S4Yz → S0Yz transition obtained using DFT models, showing an oxyl radical, which precedes the formation of the O-O bond. Spin densities are shown at the threshold of ±0.02e/Å3 for α (in blue) and β (in red) spin.
Re-reduction of Tyrz by proton-coupled electron transfer from the hydroxy (W3) ligand on Mn1 results in a radical species (O•) in the S4Yz state (Figure 3E,F). Consistent with previous models,26,42,43 we find that this oxyl radical can form an O–O bond with O5, with a rather low energy barrier (∼16 kcal mol–1 with a d‡O–O ≈ 1.75 Å; Figure 3E). Dissociation of the oxygen molecule from the Mn4O5Ca cluster could lead to the further migration of the Ca2+-bound W5 or the nearby W6 into the gap between Mn1 and Mn4, forming a new O5 oxo-ligand upon deprotonation,26 which again, could take place via the Asp61/Lys317 gate (Figure 3F). A similar water insertion model was recently supported by QM/MM calculations.53
Discussion
We have studied here the redox-driven deprotonation steps in the S2Yz → S3Yz and S3Yz → S4/0Yz transitions of the Mn4O5Ca cluster of PSII. Both transitions involve a light-driven oxidation of Tyrz, creating a neutral tyrosyl radical (Yz•) and a cationic His190 (HisH+), which in turn induces dissociation of a nearby Asp61/Lys317 ion-pair and hydration of a putative proton pathway leading to the bulk solvent (Figures 2 and 3). These redox-coupled changes lower the reaction barrier and increase the thermodynamic driving force for the proton transfer from the Mn4O5Ca cluster to Asp61, resulting in proton release along a pathway recently supported by time-resolved XFEL experiments.13
We suggest that the origin of this redox-driven protonation dynamics is electrostatic: Tyrz oxidation and the coupled His190 protonation induce an oriented electric field along the proton pathway from the Ca2+-bound W3 to Asp61 (Figure 4). W3 deprotonation triggers a concerted re-organization of the water network and the migration of the hydroxide formed to Mn1, while the ligand sphere of the Ca2+ is filled by a nearby water molecule (W5 or W6). The S3Yz → S4/0Yz transition induces via similar proton-coupled electron transfer steps, the breaking of the Asp61/Lys317 ion-pair, which then drives the further deprotonation of the Mn1-bound OH–, again initiated by electric field effects from the Tyrz•/His190(HisH+) pair (Figure 4). A water-mediated deprotonation of the Mn1-bound hydroxide, but involving different water molecules, was also observed in QM/MM MD (PBE + U/MM) calculations in the 5/2 and 7/2 states of S3Yz.50 These transitions form an oxyl radical intermediate on Mn1, which can undergo radical coupling with the O5 oxo-ligand, forming a peroxo-intermediate with the reduction on Mn4 to MnIII, and then form molecular oxygen by the reduction of Mn1 and Mn3, consistent with previous findings.11,26,41−43 These terminal transitions of the PSII photocycle are thus suggested to involve ion-pair dissociation and electric field modulations that drive the proton transfer reaction via a coupled re-organization of the hydrogen-bonded water network.
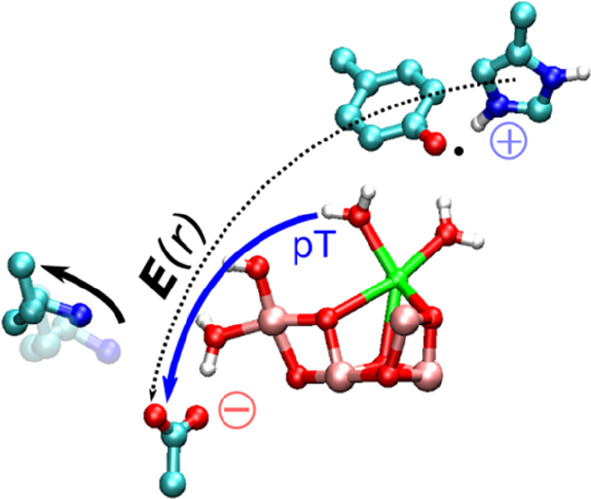
Figure 4.

Proposed redox-driven water splitting mechanism of PSII. Each light-flash leads to the formation of Tyrz• and a cationic His190 (HisH+) that opens up the Asp61/Lys317 ion-pair (solid black arrow), inducing an electric field toward Asp61. The electric field (E(r), black dotted line) drives the proton transfer reaction (solid blue arrow) from W3 and O5 via the Asp61/Lys317 gate. O2 formation between the Mn1-bound W3 and O5 leads to formation of a new O5 ligand from the Ca2+-bound W5 upon reorganization of the water structure. Deprotonation of the new O5 oxo-bridges initiates the next photocycle. Only open cubane forms of the cluster are shown for S3/S4/S0, whereas the S2Yz and S2Yz• states are shown in both open (LS; transparent) and closed (HS; solid) conformations, consistent with computational and experimental data.16−21,25,26,31−34,48,49,54−56,69
The catalytic steps studied here are likely to involve changes in both the conformational (open/closed) and spin states, as supported by previous computational and experimental studies,16−21,25,26,31−34,48,49,54−56 as well as our current findings. The S3Yz→S4Yz transition was modeled here in the low spin open conformation, which has been suggested to establish the major form of the S3Yz state.24,42 However, recent computational works suggest that the S3Yz state can also have a manifold of high spin states that could be relevant for the O–O bond formation process.55,56 We suggest that the Mn1/4-bound W3 is the prime substrate water candidate following its Tyrz oxidation-mediated deprotonation. However, we note that several previous computational studies found that the O–O bond formation can take place via W2 and O5.32−34,50,57 In addition to methodological differences, these studies modeled a Ca2+-bound W3 in the S3Yz state,25,26 and an additional water molecule (Wx) bound to Mn1/Mn4. Recent experiments have further supported W3 as a substrate water molecule,37−39 whereas previous experimental assignment supported W2.27−30 We also note that rotation of the Mn4 ligands, as observed in the present (Figure S3D) as well as previous QM/MM simulations,51 could exchange the identity of the water molecules.
In addition to PSII, other bioenergetically relevant protein complexes may also employ similar redox-driven protonation steps via conformational changes in conserved buried ion-pairs, electrostatically modulating the reaction barriers for the catalytic steps.47,58 Such conformational changes in ion-pairs could be responsible for long-range proton transport within the membrane domain of respiratory complex I,59−61 a gigantic redox-driven proton pump that initiates cell respiration in the mitochondria.58,62 Similar effects are also found in cytochrome c oxidase, where redox-driven dissociation of a propionate-arginine (Δ-propionate/Arg438 in bovine numbering) ion-pair modulates the proton transfer barriers across the membrane.63,64 Conformational changes in ion-pairs have also been suggested to trigger ion-transport and/or catalysis in bacterial light-driven Na+ pumps,65 molecular chaperones,66 and in artificial designer proteins,67 suggesting that the described key functional motifs are generally applied also in other proteins.
Conclusions
We have studied here the molecular principles of light-driven water oxidation in photosystem II by combining large-scale quantum and classical molecular simulation approaches. We found that Tyrz oxidation in the S2Yz → S3Yz and S3Yz → S4/0Yz transitions lowers the proton transfer barrier from a Mn4O5Ca cluster-bound water molecule (W3) to Asp61, a residue located in a proton pathway leading to the luminal bulk water phase. The barrier modulation takes place by electrostatic tuning,47,68 which leads to conformational changes in a network of buried ion-pairs and hydration changes in the proton pathway leading to the bulk solvent. Recent time-resolved X-ray free electron laser studies13 provide experimental support for some of the key features of the present model. We also find striking similarities between the redox-driven protonation dynamics in PSII and other bioenergetically relevant enzymes, suggesting that the proposed mechanistic principles may be broadly applicable to energy converting enzymes.
Materials and Methods
Molecular Dynamics Simulations
The crystal structure of cyanobacterial PSII from T. vulcanus (PDB ID: 3WU2)7 was embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane and solvated with TIP3P water molecules and 100 mM NaCl. The total system comprised ca. 535,000 atoms. Parameters for all cofactors were derived based on DFT (B3LYP-D3/def2-TZVP) calculations, and the remaining system was treated using the CHARMM36 force field.70 Fifteen independent molecular dynamics (MD) simulations, each 200 ns (3 μs in total), were carried out with the Mn4O5Ca cluster modeled in the S2Yz, S2Yz•, S3Yz (with both protonated and deprotonated Asp61), S3Yz•, and S4Yz states (see Table S4). The MD simulations were performed in an NPT ensemble at T = 300, 310, or 320 K and p = 1 atm and using a 1 fs integration timestep. The ion-pair opening is enhanced in simulations at higher temperatures. Long-range electrostatics was treated using the particle mesh Ewald (PME) method with a 1 Å grid separation. The system was gradually relaxed for 4 ns with harmonic restraints of 1 kcal mol–1 Å–1 followed by 20 ns equilibration without restraints and the 200 ns production runs. All classical MD simulations were performed using NAMD2.1371 and simulations were analyzed using VMD.72 All classical simulations are reported in Table S4.
DFT Models
DFT models were constructed to study the redox-driven protonation energetics of the Mn4O5Ca cluster. The models comprised the Mn4O5Ca cluster, its four coordinating water molecules (W1–W4), Asp61D1, Tyr161D1 (Tyrz), its neighboring His190D1, and additionally, 13 crystal water molecules found in close proximity to the cluster, and all amino acids directly coordinating to the cluster: His332D1, Asp342D1, Ala344D1, Glu333D1, Asp170D1, Asn181D1, Glu189D1, Glu354CP43, His337D1, Lys317D2, Arg357CP43, and a Cl– ion (Table S1). His337D1 was modeled in its protonated state (HisH+). All models were constructed based on the crystal structure of PSII (PDB ID: 3WU2)7. The Mn4O5Ca cluster was modeled in low-spin (LS, S = 0 or 1/2) and high spin (HS, S = 5/2 or 6/2) using the broken symmetry spin-flip DFT approach.73,74 The quantum chemical models comprised 210–212 atoms. Protein residues were cut between Cβ and Cα atoms, except for glutamate, lysine, and arginine residues, which were cut between Cγ and Cβ. For Ala344D1, the backbone was included in the QM region, with C and O atoms included from the neighboring Leu343D1 (Table S1). Terminal carbon atoms were saturated with hydrogen atoms, and kept fixed during geometry optimizations at the B3LYP-D3/def2-SVP/def2-TZVP (Mn,Ca,Cl) level of theory.75−78 The surroundings was modeled as a polarizable dielectric medium using the COSMO79 solvation model with ε set to 4. As the models included explicit functional groups of first and second solvation sphere residues, the energetics was found to be rather insensitive to the exact choice of the dielectric constant (Figure S7G), except when the charge moves close to the edge of the DFT model, when we observed a maximum effect of 4 kcal mol–1 between ε = 4 and ε = 80 (Figure S7G). Single point energy calculations were performed at the B3LYP*-D3 (with 15% exact exchange80)/def2-TZVP/ε = 4 level of theory. Based on our benchmarking calculations, the overall energetics was found to be similar with different hybrid functionals,75−78,81−84 suggesting that the overall results are robust (see Figure S7). All calculations were performed using TURBOMOLE v. 7.2–7.5,85 with VMD72 used for analysis. Reaction pathway optimizations were performed using the woelfling method,86 which is related to the zero-temperature string simulation approach. The reported barriers correspond to approximate transition states/saddle points and show an imaginary frequency along the proton transfer coordinate (Figure S8 and Table S5). See Table S1 for details of the DFT calculations.
QM/MM Models
Hybrid QM/MM calculations were performed based on the MD-relaxed structure of PSII (see above), with the same QM region and theory level as used in the DFT models and 62,767 MM atoms, described at a classical force field level (Figure S1C). Link atoms were introduced between Cα and Cβ atoms, except for Glu, Lys, and Arg with link atoms introduced between Cβ and Cγ atoms. The QM/MM systems were relaxed using the adopted basis Newton–Raphson algorithm, followed by QM/MM dynamics at T = 310 K with a 1 fs integration time step. A 12 Å sphere around the QM region was allowed to fully relax during the QM/MM simulations. QM/MM calculations were initiated from the S2Yz, S2Yz• (with both protonated and deprotonated W3; deprotonated/protonated Asp61), and S3Yz states, in both HS and LS forms. All QM/MM calculations were performed using the CHARMM/TURBOMOLE interface.87 See Table S3 for the list of QM/MM calculations performed.
Acknowledgments
This work was supported by the Knut and Alice Wallenberg Foundation (to V.R.I.K.), the Royal Society Wolfson Research Merit Award (to A.W.R.), and Biotechnology and Biological Sciences Research Council (BBSRC) grants BB/K002627/1 and BB/R00921X (to A.W.R.). Computational resources were provided by Leibniz Rechenzentrum (LRZ) and the Swedish National Infrastructure for Computing SNIC/PDC (SNIC 2021/6-143) at PDC Centre, partially funded by the Swedish Research Council through grant agreement no. 2016-07213. This work was also supported by the TU Munich-Imperial College Collaboration Fund (to V.R.I.K. and A.W.R). We thank the TUM Global Incentive Fund, funded under the Excellence Strategy of the Federal Government and the Länder. V.R.I.K. also acknowledges support from the German Research Foundation (DFG) via the Collaborative Research Centre (SFB1078) as Mercator Fellow.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c13041.
Elaborations on models, methods and mechanisms; figures showing detailed energetics and structures, electric field calculations, benchmarking, transition states, comparison to XFEL structures and computational details (PDF)
Optimized reaction pathways for the proton transfer in the S2Yz• state (MPG)
Optimized reaction pathways for the proton transfer in the S3Yz state (MPG)
Optimized reaction pathways for the proton transfer in the S3Yz• state (MPG)
The authors declare no competing financial interest.
Supplementary Material
References
- Nelson N.; Yocum C. F. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 2006, 57, 521–565. 10.1146/annurev.arplant.57.032905.105350. [DOI] [PubMed] [Google Scholar]
- Dau H.; Zaharieva I. Principles, Efficiency, and Blueprint Character of Solar-Energy Conversion in Photosynthetic Water Oxidation. Acc. Chem. Res. 2009, 42, 1861–1870. 10.1021/ar900225y. [DOI] [PubMed] [Google Scholar]
- Cox N.; Messinger J. Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1020–1030. 10.1016/j.bbabio.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Cardona T.; Sedoud A.; Cox N.; Rutherford A. W. Charge separation in Photosystem II: A comparative and evolutionary overview. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 26–43. 10.1016/j.bbabio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Kok B.; Forbush B.; Mcgloin M. Cooperation of charges in photosynthetic O2 evolution–1. A linear four-step mechanism. Photochem. Photobiol. 1970, 11, 457–475. 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- Cox N.; Messinger J. Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta, Bioenerg. 1827, 2013, 1020–1030. 10.1016/j.bbabio.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Hirata K.; Ueno G.; Murakami H.; Nakajima Y.; Shimizu T.; Yamashita K.; Yamamoto M.; Ago H.; Shen J.-R. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- Young I. D.; Ibrahim M.; Chatterjee R.; Gul S.; Fuller F. D.; Koroidov S.; Brewster A. S.; Tran R.; Alonso-Mori R.; Kroll T.; Michels-Clark T.; Laksmono H.; Sierra R. G.; Stan C. A.; Hussein R.; Zhang M.; Douthit L.; Kubin M.; De Lichtenberg C.; Vo Pham L.; Nilsson H.; Cheah M. H.; Shevela D.; Saracini C.; Bean M. A.; Seuffert I.; Sokaras D.; Weng T.-C.; Pastor E.; Weninger C.; Fransson T.; Lassalle L.; Bräuer P.; Aller P.; Docker P. T.; Andi B.; Orville A. M.; Glownia J. M.; Nelson S.; Sikorski M.; Zhu D.; Hunter M. S.; Lane T. J.; Aquila A.; Koglin J. E.; Robinson J.; Liang M.; Boutet S.; Lyubimov A. Y.; Uervirojnangkoorn M.; Moriarty N. W.; Liebschner D.; Afonine P. V.; Waterman D. G.; Evans G.; Wernet P.; Dobbek H.; Weis W. I.; Brunger A. T.; Zwart P. H.; Adams P. D.; Zouni A.; Messinger J.; Bergmann U.; Sauter N. K.; Kern J.; Yachandra V. K.; Yano J. Structure of photosystem II and substrate binding at room temperature. Nature 2016, 540, 453–457. 10.1038/nature20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Sugahara M.; Kubo M.; Nakajima Y.; Nakane T.; Yamashita K.; Umena Y.; Nakabayashi M.; Yamane T.; Nakano T.; Suzuki M.; Masuda T.; Inoue S.; Kimura T.; Nomura T.; Yonekura S.; Yu L.-J.; Sakamoto T.; Motomura T.; Chen J.-H.; Kato Y.; Noguchi T.; Tono K.; Joti Y.; Kameshima T.; Hatsui T.; Nango E.; Tanaka R.; Naitow H.; Matsuura Y.; Yamashita A.; Yamamoto M.; Nureki O.; Yabashi M.; Ishikawa T.; Iwata S.; Shen J.-R. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Yamashita K.; Nakajima Y.; Ueno G.; Li H.; Yamane T.; Hirata K.; Umena Y.; Yonekura S.; Yu L.-J.; Murakami H.; Nomura T.; Kimura T.; Kubo M.; Baba S.; Kumasaka T.; Tono K.; Yabashi M.; Isobe H.; Yamaguchi K.; Yamamoto M.; Ago H.; Shen J.-R. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 2019, 366, 334–338. 10.1126/science.aax6998. [DOI] [PubMed] [Google Scholar]
- Cox N.; Pantazis D. A.; Lubitz W. Current Understanding of the Mechanism of Water Oxidation in Photosystem II and Its Relation to XFEL Data. Annu. Rev. Biochem. 2020, 89, 795–820. 10.1146/annurev-biochem-011520-104801. [DOI] [PubMed] [Google Scholar]
- Hussein R.; Ibrahim M.; Bhowmick A.; Simon P. S.; Chatterjee R.; Lassalle L.; Doyle M.; Bogacz I.; Kim I.-S.; Cheah M. H.; Gul S.; De Lichtenberg C.; Chernev P.; Pham C. C.; Young I. D.; Carbajo S.; Fuller F. D.; Alonso-Mori R.; Batyuk A.; Sutherlin K. D.; Brewster A. S.; Bolotovsky R.; Mendez D.; Holton J. M.; Moriarty N. W.; Adams P. D.; Bergmann U.; Sauter N. K.; Dobbek H.; Messinger J.; Zouni A.; Kern J.; Yachandra V. K.; Yano J. Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat. Commun. 2021, 12, 6531. 10.1038/s41467-021-26781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie S.; Terrett R.; Stranger R.; Pace R. J. Rationalizing the Geometries of the Water Oxidising Complex in the Atomic Resolution, Nominal S3 State Crystal Structures of Photosystem II. ChemPhysChem 2020, 21, 785–801. 10.1002/cphc.201901106. [DOI] [PubMed] [Google Scholar]
- Pantazis D. A. Evaluation of new low-valent computational models for the oxygen-evolving complex of photosystem II. Chem. Phys. Lett. 2020, 753, 137629. 10.1016/j.cplett.2020.137629. [DOI] [Google Scholar]
- Boussac A.; Girerd J.-J.; Rutherford A. W. Conversion of the Spin State of the Manganese Complex in Photosystem II Induced by Near-Infrared Light. Biochemistry 1996, 35, 6984–6989. 10.1021/bi960636w. [DOI] [PubMed] [Google Scholar]
- Peloquin J. M.; Campbell K. A.; Randall D. W.; Evanchik M. A.; Pecoraro V. L.; Armstrong W. H.; Britt R. D. 55Mn ENDOR of the S2-State Multiline EPR Signal of Photosystem II: Implications on the Structure of the Tetranuclear Mn Cluster. J. Am. Chem. Soc. 2000, 122, 10926–10942. 10.1021/ja002104f. [DOI] [Google Scholar]
- Pantazis D. A.; Ames W.; Cox N.; Lubitz W.; Neese F. Two Interconvertible Structures that Explain the Spectroscopic Properties of the Oxygen-Evolving Complex of Photosystem II in the S2 State. Angew. Chem., Int. Ed. Engl. 2012, 51, 9935–9940. 10.1002/anie.201204705. [DOI] [PubMed] [Google Scholar]
- Bovi D.; Narzi D.; Guidoni L. The S2 State of the Oxygen-Evolving Complex of Photosystem II Explored by QM/MM Dynamics: Spin Surfaces and Metastable States Suggest a Reaction Path Towards the S3 State. Angew. Chem., Int. Ed. Engl. 2013, 52, 11744–11749. 10.1002/anie.201306667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzi D.; Bovi D.; Guidoni L. Pathway for Mn-cluster oxidation by tyrosine-Z in the S2 state of photosystem II. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 8723–8728. 10.1073/pnas.1401719111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussac A.; Ugur I.; Marion A.; Sugiura M.; Kaila V. R. I.; Rutherford A. W. The low spin - high spin equilibrium in the S2-state of the water oxidizing enzyme. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 342–356. 10.1016/j.bbabio.2018.02.010. [DOI] [PubMed] [Google Scholar]
- Kern J.; Chatterjee R.; Young I. D.; Fuller F. D.; Lassalle L.; Ibrahim M.; Gul S.; Fransson T.; Brewster A. S.; Alonso-Mori R.; Hussein R.; Zhang M.; Douthit L.; De Lichtenberg C.; Cheah M. H.; Shevela D.; Wersig J.; Seuffert I.; Sokaras D.; Pastor E.; Weninger C.; Kroll T.; Sierra R. G.; Aller P.; Butryn A.; Orville A. M.; Liang M.; Batyuk A.; Koglin J. E.; Carbajo S.; Boutet S.; Moriarty N. W.; Holton J. M.; Dobbek H.; Adams P. D.; Bergmann U.; Sauter N. K.; Zouni A.; Messinger J.; Yano J.; Yachandra V. K. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. 10.1038/s41586-018-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M.; Fransson T.; Chatterjee R.; Cheah M. H.; Hussein R.; Lassalle L.; Sutherlin K. D.; Young I. D.; Fuller F. D.; Gul S.; Kim I.-S.; Simon P. S.; De Lichtenberg C.; Chernev P.; Bogacz I.; Pham C. C.; Orville A. M.; Saichek N.; Northen T.; Batyuk A.; Carbajo S.; Alonso-Mori R.; Tono K.; Owada S.; Bhowmick A.; Bolotovsky R.; Mendez D.; Moriarty N. W.; Holton J. M.; Dobbek H.; Brewster A. S.; Adams P. D.; Sauter N. K.; Bergmann U.; Zouni A.; Messinger J.; Kern J.; Yachandra V. K.; Yano J. Untangling the sequence of events during the S2→ S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 12624–12635. 10.1073/pnas.2000529117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N.; Retegan M.; Neese F.; Pantazis D. A.; Boussac A.; Lubitz W. Electronic structure of the oxygen-evolving complex in photosystem II prior to O-O bond formation. Science 2014, 345, 804–808. 10.1126/science.1254910. [DOI] [PubMed] [Google Scholar]
- Shoji M.; Isobe H.; Yamaguchi K. QM/MM study of the S2 to S3 transition reaction in the oxygen-evolving complex of photosystem II. Chem. Phys. Lett. 2015, 636, 172–179. 10.1016/j.cplett.2015.07.039. [DOI] [PubMed] [Google Scholar]
- Ugur I.; Rutherford A. W.; Kaila V. R. I. Redox-coupled substrate water reorganization in the active site of Photosystem II—The role of calcium in substrate water delivery. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 740–748. 10.1016/j.bbabio.2016.01.015. [DOI] [PubMed] [Google Scholar]
- Nilsson H.; Krupnik T.; Kargul J.; Messinger J. Substrate water exchange in photosystem II core complexes of the extremophilic red alga Cyanidioschyzon merolae. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 1257–1262. 10.1016/j.bbabio.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Hillier W.; Wydrzynski T. Oxygen ligand exchange at metal sites – implications for the O2 evolving mechanism of photosystem II. Biochim. Biophys. Acta, Bioenerg. 2001, 1503, 197–209. 10.1016/S0005-2728(00)00225-5. [DOI] [PubMed] [Google Scholar]
- Hillier W.; Wydrzynski T. Substrate water interactions within the Photosystem II oxygen evolving complex. Phys. Chem. Chem. Phys. 2004, 6, 4882. 10.1039/b407269c. [DOI] [Google Scholar]
- Hillier W.; Wydrzynski T. 18O-Water exchange in photosystem II: Substrate binding and intermediates of the water splitting cycle. Coord. Chem. Rev. 2008, 252, 306–317. 10.1016/j.ccr.2007.09.004. [DOI] [Google Scholar]
- Siegbahn P. E. M. Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys. Chem. Chem. Phys. 2012, 14, 4849. 10.1039/c2cp00034b. [DOI] [PubMed] [Google Scholar]
- Capone M.; Bovi D.; Narzi D.; Guidoni L. Reorganization of Substrate Waters between the Closed and Open Cubane Conformers during the S2 to S3 Transition in the Oxygen Evolving Complex. Biochemistry 2015, 54, 6439–6442. 10.1021/acs.biochem.5b00782. [DOI] [PubMed] [Google Scholar]
- Capone M.; Narzi D.; Bovi D.; Guidoni L. Mechanism of Water Delivery to the Active Site of Photosystem II along the S2 to S3 Transition. J. Phys. Chem. Lett. 2016, 7, 592–596. 10.1021/acs.jpclett.5b02851. [DOI] [PubMed] [Google Scholar]
- Retegan M.; Krewald V.; Mamedov F.; Neese F.; Lubitz W.; Cox N.; Pantazis D. A. A five-coordinate Mn(iv) intermediate in biological water oxidation: spectroscopic signature and a pivot mechanism for water binding. Chem. Sci. 2016, 7, 72–84. 10.1039/C5SC03124A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Armstrong W. H.; Batista V. S. Do crystallographic XFEL data support binding of a water molecule to the oxygen-evolving complex of photosystem II exposed to two flashes of light?. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2023982118 10.1073/pnas.2023982118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M.; Moriarty N. W.; Kern J.; Holton J. M.; Brewster A. S.; Bhowmick A.; Bergmann U.; Zouni A.; Messinger J.; Yachandra V. K.; Yano J.; Dobbek H.; Sauter N. K.; Adams P. D. Reply to Wang et al.: Clear evidence of binding of Ox to the oxygen-evolving complex of photosystem II is best observed in the omit map. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2102342118 10.1073/pnas.2102342118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. J.; Debus R. J. Evidence from FTIR Difference Spectroscopy That a Substrate H2O Molecule for O2 Formation in Photosystem II Is Provided by the Ca Ion of the Catalytic Mn4CaO5 Cluster. Biochemistry 2017, 56, 2558–2570. 10.1021/acs.biochem.6b01278. [DOI] [PubMed] [Google Scholar]
- Kim C. J.; Debus R. J. One of the Substrate Waters for O2 Formation in Photosystem II Is Provided by the Water-Splitting Mn4CaO5 Cluster’s Ca2+ Ion. Biochemistry 2019, 58, 3185–3192. 10.1021/acs.biochem.9b00418. [DOI] [PubMed] [Google Scholar]
- De Lichtenberg C.; Kim C. J.; Chernev P.; Debus R. J.; Messinger J. The exchange of the fast substrate water in the S2 state of photosystem II is limited by diffusion of bulk water through channels – implications for the water oxidation mechanism. Chem. Sci. 2021, 12, 12763–12775. 10.1039/D1SC02265B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. M. The S2 to S3 transition for water oxidation in PSII (photosystem II), revisited. Phys. Chem. Chem. Phys. 2018, 20, 22926–22931. 10.1039/C8CP03720E. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M. O-O Bond Formation in the S4 State of the Oxygen-Evolving Complex in Photosystem II. Chem. – Eur. J. 2006, 12, 9217–9227. 10.1002/chem.200600774. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M. Water oxidation mechanism in photosystem II, including oxidations, proton release pathways, O—O bond formation and O2 release. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1003–1019. 10.1016/j.bbabio.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Krewald V.; Retegan M.; Cox N.; Messinger J.; Lubitz W.; Debeer S.; Neese F.; Pantazis D. A. Metal oxidation states in biological water splitting. Chem. Sci. 2015, 6, 1676–1695. 10.1039/C4SC03720K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinyard D. J.; Khan S.; Brudvig G. W. Photosynthetic water oxidation: binding and activation of substrate waters for O–O bond formation. Faraday Discuss. 2015, 185, 37–50. 10.1039/C5FD00087D. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M. Nucleophilic water attack is not a possible mechanism for O–O bond formation in photosystem II. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 4966–4968. 10.1073/pnas.1617843114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda H.; Kawashima K.; Ueda K.; Ikeda T.; Saito K.; Ninomiya R.; Hida C.; Takahashi Y.; Ishikita H. Proton transfer pathway from the oxygen-evolving complex in photosystem II substantiated by extensive mutagenesis. Biochim. Biophys. Acta, Bioenerg. 2021, 1862, 148329. 10.1016/j.bbabio.2020.148329. [DOI] [PubMed] [Google Scholar]
- Kaila V. R. I. Resolving Chemical Dynamics in Biological Energy Conversion: Long-Range Proton-Coupled Electron Transfer in Respiratory Complex I. Acc. Chem. Res. 2021, 54, 4462–4473. 10.1021/acs.accounts.1c00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retegan M.; Cox N.; Lubitz W.; Neese F.; Pantazis D. A. The first tyrosyl radical intermediate formed in the S2–S3 transition of photosystem II. Phys. Chem. Chem. Phys. 2014, 16, 11901. 10.1039/c4cp00696h. [DOI] [PubMed] [Google Scholar]
- Boussac A.; Rutherford A. W.; Sugiura M. Electron transfer pathways from the S2-states to the S3 -states either after a Ca2+ /Sr2+ or a Cl– /I– exchange in Photosystem II from Thermosynechococcus elongatus. Biochim. Biophys. Acta, Bioenerg. 1847, 2015, 576–586. 10.1016/j.bbabio.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Narzi D.; Capone M.; Bovi D.; Guidoni L. Evolution from S3 to S4 States of the Oxygen-Evolving Complex in Photosystem II Monitored by Quantum Mechanics/Molecular Mechanics (QM/MM) Dynamics. Chem. – Eur. J. 2018, 24, 10820–10828. 10.1002/chem.201801709. [DOI] [PubMed] [Google Scholar]
- Wang J.; Askerka M.; Brudvig G. W.; Batista V. S. Crystallographic Data Support the Carousel Mechanism of Water Supply to the Oxygen-Evolving Complex of Photosystem II. ACS Energy Lett. 2017, 2, 2299–2306. 10.1021/acsenergylett.7b00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivalta I.; Amin M.; Luber S.; Vassiliev S.; Pokhrel R.; Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N.; Bruce D.; Brudvig G. W.; Gunner M. R.; Batista V. S. Structural–Functional Role of Chloride in Photosystem II. Biochemistry 2011, 50, 6312–6315. 10.1021/bi200685w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone M.; Narzi D.; Guidoni L. Mechanism of Oxygen Evolution and Mn4CaO5 Cluster Restoration in the Natural Water-Oxidizing Catalyst. Biochemistry 2021, 60, 2341–2348. 10.1021/acs.biochem.1c00226. [DOI] [PubMed] [Google Scholar]
- Isobe H.; Shoji M.; Suzuki T.; Shen J.-R.; Yamaguchi K. Exploring reaction pathways for the structural rearrangements of the Mn cluster induced by water binding in the S3 state of the oxygen evolving complex of photosystem II. J. Photochem. Photobiol., A 2021, 405, 112905. 10.1016/j.jphotochem.2020.112905. [DOI] [Google Scholar]
- Isobe H.; Shoji M.; Suzuki T.; Shen J.-R.; Yamaguchi K. Spin, Valence, and Structural Isomerism in the S3 State of the Oxygen-Evolving Complex of Photosystem II as a Manifestation of Multimetallic Cooperativity. J. Chem. Theory Comput. 2019, 15, 2375–2391. 10.1021/acs.jctc.8b01055. [DOI] [PubMed] [Google Scholar]
- Zahariou G.; Ioannidis N.; Sanakis Y.; Pantazis D. A. Arrested Substrate Binding Resolves Catalytic Intermediates in Higher-Plant Water Oxidation. Angew. Chem., Int. Ed. Engl. 2021, 60, 3156–3162. 10.1002/anie.202012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Zhang B.; Kloo L.; Sun L. Necessity of structural rearrangements for O O bond formation between O5 and W2 in photosystem II. J. Energy Chem. 2021, 57, 436–442. 10.1016/j.jechem.2020.09.008. [DOI] [Google Scholar]
- Kaila V. R. I. Long-range proton-coupled electron transfer in biological energy conversion: towards mechanistic understanding of respiratory complex I. J. R. Soc., Interface 2018, 15, 20170916. 10.1098/rsif.2017.0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca A.; Gamiz-Hernandez A. P.; Kaila V. R. I. Symmetry-related proton transfer pathways in respiratory complex I. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6314–E6321. 10.1073/pnas.1706278114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlbauer M. E.; Saura P.; Nuber F.; Di Luca A.; Friedrich T.; Kaila V. R. I. Water-Gated Proton Transfer Dynamics in Respiratory Complex I. J. Am. Chem. Soc. 2020, 142, 13718–13728. 10.1021/jacs.0c02789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röpke M.; Saura P.; Riepl D.; Pöverlein M. C.; Kaila V. R. I. Functional Water Wires Catalyze Long-Range Proton Pumping in the Mammalian Respiratory Complex I. J. Am. Chem. Soc. 2020, 142, 21758–21766. 10.1021/jacs.0c09209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila V. R. I.; Wikström M. Architecture of bacterial respiratory chains. Nat. Rev. Microbiol. 2021, 19, 319–330. 10.1038/s41579-020-00486-4. [DOI] [PubMed] [Google Scholar]
- Kaila V. R. I.; Verkhovsky M. I.; Wikström M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev. 2010, 110, 7062–7081. 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- Supekar S.; Gamiz-Hernandez A. P.; Kaila V. R. I. A Protonated Water Cluster as a Transient Proton-Loading Site in Cytochrome c Oxidase. Angew. Chem., Int. Ed. Engl. 2016, 55, 11940–11944. 10.1002/anie.201603606. [DOI] [PubMed] [Google Scholar]
- Suomivuori C. M.; Gamiz-Hernandez A. P.; Sundholm D.; Kaila V. R. I. Energetics and dynamics of a light-driven sodium-pumping rhodopsin. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7043–7048. 10.1073/pnas.1703625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader S. L.; Lopez A.; Lawatscheck J.; Luo Q.; Rutz D. A.; Gamiz-Hernandez A. P.; Sattler M.; Buchner J.; Kaila V. R. I. Conformational dynamics modulate the catalytic activity of the molecular chaperone Hsp90. Nat. Commun. 2020, 11, 1410. 10.1038/s41467-020-15050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart M.; Röpke M.; Mühlbauer M. E.; Asami S.; Mader S. L.; Fredriksson K.; Groll M.; Gamiz-Hernandez A. P.; Kaila V. R. I. Design of buried charged networks in artificial proteins. Nat. Commun. 2021, 12, 1895. 10.1038/s41467-021-21909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura P.; Frey D. M.; Gamiz-Hernandez A. P.; Kaila V. R. I. Electric field modulated redox-driven protonation and hydration energetics in energy converting enzymes. Chem. Commun. 2019, 55, 6078–6081. 10.1039/C9CC01135H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe H.; Shoji M.; Yamanaka S.; Umena Y.; Kawakami K.; Kamiya N.; Shen J.-R.; Yamaguchi K. Theoretical illumination of water-inserted structures of the CaMn4O5 cluster in the S2 and S3 states of oxygen-evolving complex of photosystem II: full geometry optimizations by B3LYP hybrid density functional. Dalton Trans. 2012, 41, 13727. 10.1039/c2dt31420g. [DOI] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; De Groot B. L.; Grubmüller H.; Mackerell A. D. Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Noodleman L.; Davidson E. R. Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys. 1986, 109, 131–143. 10.1016/0301-0104(86)80192-6. [DOI] [Google Scholar]
- Soda T.; Kitagawa Y.; Onishi T.; Takano Y.; Shigeta Y.; Nagao H.; Yoshioka Y.; Yamaguchi K. Ab initio computations of effective exchange integrals for H–H, H–He–H and Mn2O2 complex: comparison of broken-symmetry approaches. Chem. Phys. Lett. 2000, 319, 223–230. 10.1016/S0009-2614(00)00166-4. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Reiher M.; Salomon O.; Artur Hess B. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. 10.1007/s00214-001-0300-3. [DOI] [Google Scholar]
- Staroverov V. N.; Scuseria G. E.; Tao J.; Perdew J. P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. 10.1063/1.1626543. [DOI] [Google Scholar]
- Becke A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Shao Y.; Mei Y.; Sundholm D.; Kaila V. R. I. Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores. J. Chem. Theory Comput. 2020, 16, 587–600. 10.1021/acs.jctc.9b00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Balasubramani S. G.; Chen G. P.; Coriani S.; Diedenhofen M.; Frank M. S.; Franzke Y. J.; Furche F.; Grotjahn R.; Harding M. E.; Hättig C.; Hellweg A.; Helmich-Paris B.; Holzer C.; Huniar U.; Kaupp M.; Marefat Khah A.; Karbalaei Khani S.; Müller T.; Mack F.; Nguyen B. D.; Parker S. M.; Perlt E.; Rappoport D.; Reiter K.; Roy S.; Rückert M.; Schmitz G.; Sierka M.; Tapavicza E.; Tew D. P.; Van Wüllen C.; Voora V. K.; Weigend F.; Wodyński A.; Yu J. M. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. 10.1063/5.0004635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessow P. Reaction Path Optimization without NEB Springs or Interpolation Algorithms. J. Chem. Theory Comput. 2013, 9, 1305–1310. 10.1021/ct300951j. [DOI] [PubMed] [Google Scholar]
- Riahi S.; Rowley C. N. The CHARMM-TURBOMOLE interface for efficient and accurate QM/MM molecular dynamics, free energies, and excited state properties. J. Comput. Chem. 2014, 35, 2076–2086. 10.1002/jcc.23716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.