

Although COVID-19 cases around the world have declined in recent months, that trend has been upended by the highly contagious BA.2 Omicron variant. Paxlovid, Pfizer’s oral COVID-19 treatment, could be a lifeline for those at high risk of serious disease. The antiviral showed tremendous promise in clinical trials: compared with a placebo, it reduced the risk of hospitalization or death from COVID-19 by 88% if given within 5 days of the onset of symptoms.
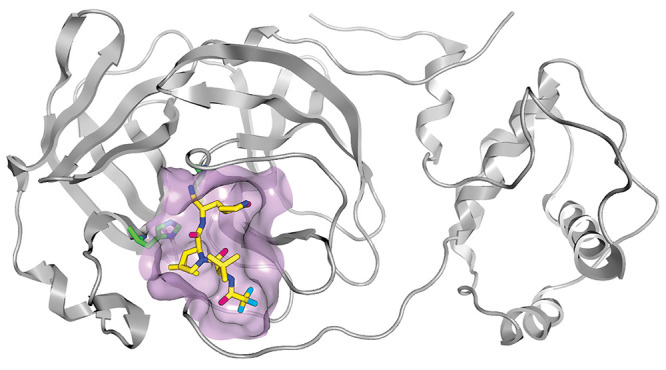
Nirmatrelvir (PF-07321332) bound to the main protease of SARS-CoV-2 (binding pocket is outlined in purple; C = yellow, N = dark blue, O = pink, and F = light blue; a histidine side chain is also shown in green and dark blue). Credit: Joy Yang/Pfizer.
Based on that data, the U.S. Food and Drug Administration gave Paxlovid emergency use authorization in December, and the federal government ordered enough of the antiviral to treat 20 million people through 2022. Pfizer announced that it plans to make 120 million courses of treatment this year and will seek full regulatory approval for the antiviral.
Paxlovid’s existence is remarkable, considering that its antiviral ingredient—originally called PF-07321332 and recently dubbed nirmatrelvir—was not even a sketch in a medicinal chemist’s lab notebook 2 years ago. The antiviral went from an idea to the first clinical test in a person in 12 months—a breathtaking pace at which to deliver a bespoke drug candidate.
The drug discovery story began Friday, March 13, 2020, when medicinal chemist Dafydd Owen was sent home from his job at Pfizer in Cambridge, Massachusetts. Like many employers at the time, the pharmaceutical giant was halting most on-site operations in the face of the national emergency over COVID-19. Owen’s bosses asked him to spend the weekend thinking about what resources he would need to lead a program to develop an oral drug to fight the emerging pandemic. It was an unusual choice in some ways: Owen had never worked on an antiviral and had never been a project leader.
Owen has spent his entire career at Pfizer. He started working at the company’s Sandwich, England, site in 1999 and moved to the U.S. in 2011. For the past decade, he says, his role has been to help pick targets for medicinal chemistry programs. He’s helped kick off campaigns in oncology, pain management, and cardiovascular disease. “I wanted to start the right programs, which I think is a critical part of a good decision-making at big pharma companies,” he says.
But Owen says he was happy to take on leadership of the oral antiviral program. His status as a rookie project leader may have given him an advantage. “We were all sent home on that Friday, and the world was completely different,” he says. There was a certain degree of urgency, and having never led a drug discovery project before, Owen did not feel bound by any preconceived notions of how such a program should be run.
Over the weekend, Owen brushed up on his antiviral chemistry. In this case, he did not have to pick the target. Pfizer scientists knew they wanted to make an antiviral that would go after the main protease (also known as the 3CL protease) of SARS-CoV-2—the coronavirus that causes COVID-19. Inhibiting a viral protease has been a successful strategy for developing drugs that fight HIV and hepatitis C. While there’s not a direct comparison between those viruses and SARS-CoV-2, Owen says it was a sound strategy.
What’s more, Pfizer already had a lead in hand. In 2003, researchers at the company developed an antiviral, known as PF-00835231, that could block the main protease of a coronavirus that emerged in 2002 and causes severe acute respiratory syndrome (SARS). But by the time they were ready to test it in patients, the SARS outbreak had been contained. PF-00835231 is structurally similar to a peptide that binds within SARS’s main protease. That binding site in SARS is identical to the one in SARS-CoV-2, so Pfizer researchers thought the molecule could work against the new virus. Tests showed they were right.
But with its peptide-like structure, PF-00835231 is rich in hydrogen-bond donors. It has five of them, and they give the molecule a polar surface area that traps it in the gut if swallowed. This means the molecule must be administered intravenously—something that can typically be done only in a hospital setting. Pfizer scientists wanted an antiviral pill that people could take at the first signs of infection, long before they had to go to the hospital.
The challenge Owen and his colleagues faced: find a molecule that can be given orally. “That’s a chemistry problem,” Owen says. After a weekend of mapping out strategies, he and his colleagues had a plan, and by Monday, March 16, 2020, their oral antiviral program was underway. Owen would not return to his office in Cambridge until April 2021. He would spend the next 13 months working in a makeshift office in his home, getting the occasional glimpse of wildlife on his deck while his children navigated remote schooling.
One key part of the chemists’ strategy was to eliminate some of the hydrogen-bond donors that keep PF-00835231 in the gut. Owen says the team took a systematic approach, interrogating each hydrogen bond. If a particular hydrogen bond is critical to making the compound bind to the protease, it had to stay in the molecule, Owen says. “We were looking for hydrogen bonds that we could get rid of without losing antiviral potency.”
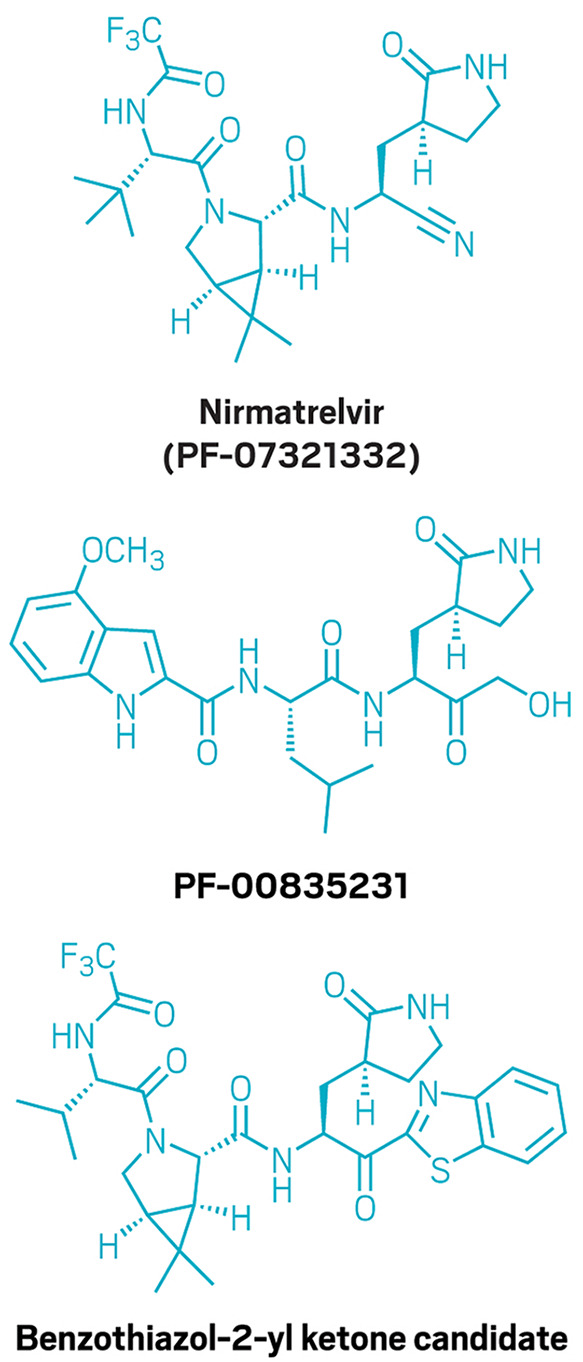
To create the oral COVID-19 antiviral nirmatrelvir (top), Pfizer scientists altered an antiviral compound called PF-00835231 (center) to make it orally bioavailable. They also pursued a benzothiazol-2-yl ketone candidate (bottom) before choosing nirmatrelvir for its better solubility and potential for scale-up. Credit: C&EN.
The first hydrogen-bond donor the team removed from PF-00835231 was the α-hydroxymethyl ketone. This site reacts covalently with a cysteine in the main protease, so the chemists reasoned they could replace it with a different reactive group that was not a hydrogen-bond donor. They chose to pursue two series of compounds: one with a benzothiazol-2-yl ketone as the reactive group and one with a nitrile reactive group. It would not be until near the end of the campaign that the chemists picked between the two.
Another hydrogen-bond donor the chemists realized they could eliminate was at PF-00835231’s leucine moiety. They decided to replace that portion with a cyclic amino acid that would eliminate an N–H bond. “Once you’ve made it cyclic, it still has to look like leucine, because the protease is expecting to see leucine at that position,” Owen says. To achieve leucine-like binding, the motif also features a fused cyclopropyl ring with two methyl groups. Computational studies suggested the structure would slot into the binding site. And there was precedent for the motif: Schering-Plough scientists had used it in the hepatitis C antiviral boceprevir.
“Making rings is kind of boom or bust in med chem,” Owen says. “You either win big or you lose big, because you’re only sampling one conformation when going to a cyclic molecule. So that conformation had better be right, or else you’re going to end up with an inactive molecule.” While the chemists saw a modest drop in potency when they introduced the cyclic element to their molecule, Owen says it was not enough to kill the compound’s activity—so the cyclic moiety became a critical feature.
But removing the hydrogen-bond donor also came a cost—the team lost a critical contact to a glutamine in the protease’s binding pocket. Seeking to restore that interaction with that amino acid, the chemists swapped in several different groups for PF-00835231’s indole moiety—trying out a methane sulfonamide, an acetamide, and a trifluoroacetamide. Those three molecules look similar, and you’d expect them to behave similarly. But that was not the case at all. The trifluoroacetamide stood out in its ability to permeate the gut barrier in assays.
That trifluoroacetamide would not be the first choice for most medicinal chemists, according to Jeremy Green, a veteran antiviral chemist and consultant who was not involved in the project. “But that seemed to really confer the permeability that they were looking for,” Green says.
The day they realized the effect of the trifluoroacetamide was an important one, Owen says. “We were then able to blend that in with other changes that we had underway, thinking that we may have a way to secure the oral bioavailability that was critical to delivering an oral agent.”
Finally, the chemists had to decide between their reactive groups—the benzothiazol-2-yl ketone and the nitrile. Both molecules looked promising, Owen says, but “when we had both in hand, we were able to draw a distinction between the two.” They chose the nitrile because of three key differences. First, the nitrile was more soluble than the benzothiazol-2-yl ketone. The more soluble a molecule, the easier it is to make high-concentration solutions of the molecule required for preclinical toxicology studies, Owen explains.
Another factor the chemists had to consider was that both reactive groups are next to a chiral hydrogen that can epimerize, and the benzothiazol-2-yl ketone was more prone to scrambling than the nitrile. Finally, Owen says, there was the matter of scaling up. “Frankly, the way to make that nitrile is a heck of a lot easier than the benzothiazol-2-yl ketone on scale,” he says. “There’s not much difference in a discovery-scale lab. But if you’re thinking manufacturing is in your future, it makes all the difference.”
On July 22, 2020, Pfizer chemists made PF-07321332—which would eventually be known as nirmatrelvir—for the first time. “That was just one of the 20 compounds we made that week,” Owen says. “We didn’t know what we had then.”
The molecule incorporated all the strategic tweaks the chemists had come up with to make it both a potent antiviral and a compound that could be taken as a pill. It was not until the morning of September 1, 2020—when the results of the pharmacokinetic study in rats came back—that the researchers knew they were on the right path.
A few weeks earlier, they had shown that PF-07321332 had good SARS-CoV-2 antiviral activity, and the rat study confirmed that the compound could be given orally. Owen says that there was still a ways to go before they would know if PF-07321332 was a viable drug candidate, but that early September morning was an exciting day for the team.
From there, Pfizer chemists quickly scaled up their synthesis of PF-07321332, delivering 1.4 kg of the compound for toxicological studies by early November 2020. Because the molecule mimics a peptide, Owen says, its synthesis essentially boils down to snapping together amide bonds. He notes that there are some complex starting materials in PF-07321332—the bicyclic structure and the lactam, which is the only portion of the molecule that remains from PF-00835231. Owen says the team had already established a good supply chain for the latter component.
To make their antiviral candidate Paxlovid, the team combined PF-07321332 with the HIV antiviral ritonavir. Owen says this approach was one the researchers thought they might have to take from the beginning of the project. Ritonavir has no activity against SARS-CoV-2 but does bind to metabolizing enzymes, thereby preventing PF-07321332 from being broken down before it has done its job mucking up the virus’s main protease.
Consultant Jeremy Green calls PF-07321332’s development impressive. “They’ve gone fast and far in a short amount of time,” he says. In the protease field, getting gut-permeable compounds can be challenging, Green says, particularly when the molecules look like peptides, as PF-07321332 does.
Owen says that, at this stage, thousands of people at Pfizer have helped bring Paxlovid to patients. As for the discovery of PF-07321332, he says, scientists in many disciplines brought the project to fruition. “It’s really great how big pharma can bring these things together, where people play their expert roles, brought their absolute best,” Owen says. “Happily, we have something to show for it.”
Bethany Halford is a senior correspondent at
Chemical & Engineering News, an independent news outlet of the American Chemical Society. A version of this story first appeared in C&EN.
In collaboration with C&EN.