

Abstract
Targeted protein degradation (TPD) holds immense promise for drug discovery, but mechanisms of acquired resistance to degraders remain to be fully identified. Here, we used clustered regularly interspaced short palindromic repeats (CRISPR)-suppressor scanning to identify mechanistic classes of drug resistance mutations to molecular glue degraders in GSPT1 and RBM39, neosubstrates targeted by E3 ligase substrate receptors cereblon and DCAF15, respectively. While many mutations directly alter the ternary complex heterodimerization surface, distal resistance sites were also identified. Several distal mutations in RBM39 led to modest decreases in degradation, yet can enable cell survival, underscoring how small differences in degradation can lead to resistance. Integrative analysis of resistance sites across GSPT1 and RBM39 revealed varying levels of sequence conservation and mutational constraint that control the emergence of different resistance mechanisms, highlighting that many regions co-opted by TPD are nonessential. Altogether, our study identifies common resistance mechanisms for molecular glue degraders and outlines a general approach to survey neosubstrate requirements necessary for effective degradation.
Short abstract
Using clustered regularly interspaced short palindromic repeats (CRISPR)-suppressor scanning, we identified thematic classes of resistance mutations to molecular glue degraders and their drivers across two distinct neosubstrates for targeted protein degradation.
Introduction
In recent years, the discovery of molecular glue degraders has converged with the development of proteolysis targeting chimeras (PROTACs), revealing the remarkable ability of small molecules to co-opt the ubiquitin-proteasome system (UPS) and degrade protein targets.1−4 Molecular glue degraders chemically remodel E3 ligase substrate receptors, creating a small molecule–protein composite surface capable of de novo complexation with complementary yet otherwise unrelated protein substrates. These neosubstrates are then subsequently polyubiquitinated and proteolytically degraded via the UPS.5
Immunomodulatory drugs (IMiDs), including thalidomide and its analogues lenalidomide and pomalidomide, bind to cereblon (CRBN), a substrate receptor for the CUL4-RING (CRL4) E3 ubiquitin ligase, and induce its complexation with various neosubstrates that are subsequently degraded.6−9 Mechanistic studies of IMiDs revealed that the selectivity of the CRBN-IMiD recognition surface and their targeted neosubstrates could be broadly modulated through even subtle chemical changes to the IMiD structure.10,11 As a leading example, CC-885 (Figure 1a), an analogue of lenalidomide, was shown to gain the ability to induce degradation of GSPT1 (also known as eRF3A), a translation termination factor essential for acute myeloid leukemia (AML) cell proliferation.10 Structural studies on the CC-885-CRBN-GSPT111,12 ternary complex were critical in determining a β-hairpin structural degron, a unifying motif across the diverse array of IMiD-targeted neosubstrates necessary for CRL4CRBN-mediated degradation.10,12,13 New IMiD derivatives tailored to degrade novel neosubstrates, including GSPT1 and IKZF2, have entered clinical trials for oncology applications.
Figure 1.

CRISPR-suppressor scanning identifies regions of GSPT1 and RBM39 that mediate targeted protein degradation by molecular glue degraders. (a) Chemical structures of degraders used in this study. (b) Schematic showing the CRISPR-suppressor scanning workflow applied to molecular glue degraders. (c) Scatter plot showing resistance scores (y axis) in MOLM-13 under CC-885 (left) or ZXH-1-161 (right) treatment at four weeks. Resistance scores were calculated as the log2(fold-change sgRNA enrichment under drug treatment) normalized to the mean of the negative control sgRNAs (n = 22). The GSPT1-targeting sgRNAs (n = 239) are arrayed by amino acid position in the GSPT1 CDS on the x axis corresponding to the position of the predicted cut site. When the sgRNA cut site falls between two amino acids, both amino acids are denoted. Data points represent mean values across three replicate treatments. Protein domains and the structural degron site are demarcated by the colored panels. (d) Scatter plots showing resistance scores (y axis) in MOLM-13 under E7820 (left) or indisulam (right) treatment at four weeks. Resistance scores were calculated as the log2(fold-change sgRNA enrichment under drug treatment) normalized to the mean of the negative control sgRNAs (n = 77). The RBM39-targeting sgRNAs (n = 129) are arrayed by the amino acid position in the RBM39 CDS on the x axis corresponding to the position of the predicted cut site. Data points represent the mean values across three replicate treatments. Protein domains and the structural degron site are demarcated by the colored panels. (e) Scatter plot showing GSPT1-targeting sgRNA resistance scores under CC-885 (y axis) or ZHX-1-161 (x axis) treatment at 4 weeks. Dotted lines represent two s.d. above the mean of the negative control sgRNAs. Pearson’s r and two-sided P values are shown. (f) Scatter plot showing RBM39-targeting sgRNA resistance scores under E7820 (y axis) or indisulam (x axis) treatment at four weeks. Dotted lines represent two s.d. above the mean of the negative control sgRNAs. Pearson’s r and two-sided P values are shown. (g) Structural view of the CC-885-CRBN-GSPT1 ternary complex showing the location of top-enriched sgRNAs (red) (Protein Data Bank (PDB: 5HXB)). (h) Structural view of the E7820-DCAF15-RBM39(RRM2) ternary complex showing the location of top-enriched sgRNAs (red) (PDB: 6UE5).
Aside from IMiDs, the anticancer sulfonamides, including E7820 and indisulam (Figure 1a, Figure S1a), were discovered to also operate as molecular glue degraders, highlighting the structural diversity of small molecules, neosubstrates, and E3 ligases that can be involved in TPD.14−19 These sulfonamides induce ternary complex formation between the CRL4 E3 substrate receptor DCAF15 and the splicing factors RBM39 (also known as CAPERα) and RBM23, which share a common α-helical structural degron.14−19 Notably, CRBN-IMiD and DCAF15-sulfonamide complexes engage their respective targets through completely distinct structural degrons and kinetic pathways,17−19 highlighting their unique modes of action despite thematic similarities. Taken together, the ability to co-opt the UPS and diverse E3 substrate receptors to induce degradation of a wide repertoire of unrelated protein targets—spanning transcription factors, kinases, translation regulators, and RNA-binding proteins—underscores TPD as a transformative approach for developing therapeutics against targets previously considered undruggable.1,2,4
As an emerging therapeutic modality, molecular glue degraders may encounter mechanisms of acquired resistance that differ substantially from canonical occupancy-driven inhibitors, potentially exploiting the unique molecular requirements necessary to catalyze proteolytic degradation.2,4 For instance, loss-of-function (LOF) and missense mutations in the IMiD-binding domain of CRBN confer resistance to IMiDs, which have been observed in multiple myeloma patients refractory to lenalidomide and pomalidomide.4,20−22 Additionally, multiple studies have shown that loss of other UPS components or chaperones can interfere with TPD.23−29 By contrast, systematic characterization of resistance mutations arising in the targeted neosubstrate has been more limited.13,14 Profiling the landscape of neosubstrate resistance mutations could delineate thematic classes of resistance mechanisms available to cancer cells and identify the structural and functional constraints that modulate their accessibility. More broadly, these mutational landscapes could illuminate molecular requirements and structural features—both within and beyond the structural degron—necessary for effective TPD.13 Motivated by these considerations, here we conducted CRISPR-suppressor scanning to systematically identify mutations across GSPT1 and RBM39 that confer resistance to molecular glue degraders, with the aim of investigating potentially unifying principles across distinct E3 substrate receptors and neosubstrates.
Results
CRISPR-suppressor Scanning of GSPT1 and RBM39
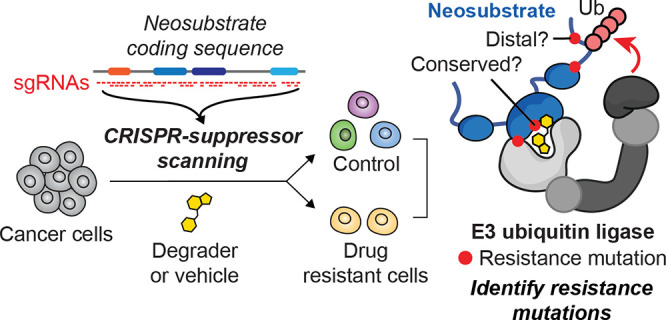
To identify candidate drug resistance mechanisms to molecular glue degraders, we performed CRISPR-suppressor scanning across two different TPD targets, GSPT1 and RBM39, which are recognized by distinct CRL4 substrate receptors. GSPT1 and RBM39 are both essential for the proliferation of AML cells and are clinical targets of interest for the treatment of AML.30,31 Consequently, we conducted CRISPR-mutagenesis of both GSPT1 and RBM39 in MOLM-13 cells, an MLL-rearranged AML cell line, to allow more facile comparisons across the two systems. In CRISPR-suppressor scanning,32 pools of single guide RNAs (sgRNAs) spanning a target protein coding sequence – in this case, GSPT1 or RBM39 – and control sgRNAs are transduced along with Streptococcus pyogenes Cas9 (SpCas9) into cells (Figure 1b). DNA double-strand breaks introduced by Cas9 can lead to the formation of diverse insertion/deletion (indel) mutations at positions proximal to the sgRNA cut site. Cells containing lethal LOF mutations in either GSPT1 or RBM39 drop out, leaving pools of cells containing viable in-frame variants that are then split and treated with either vehicle or the appropriate molecular glue degraders to select for candidate drug resistance-conferring mutations (Figure 1a, Figure S1a).
For GSPT1 CRISPR-suppressor scanning, the GSPT1 degraders CC-885 and ZXH-1-16133 were dosed in gradual escalation due to their acute cytotoxicity, starting at the approximate GI50 values and then gradually escalating to above the GI90 dose over four weeks (Figure S1a). In the case of RBM39 CRISPR-suppressor scanning, E7820 (1 μM) and indisulam (1 μM) were dosed at the approximate GI90 concentrations for four weeks (Figure S1b). After vehicle or degrader treatment, genomic DNA was isolated from surviving cells and sequenced to deconvolute sgRNA identities enriched in each condition, allowing us to calculate “resistance scores” for each sgRNA that correspond to their enrichment in degrader-treated cells and hence their propensity to generate drug resistance-conferring mutations (Figure 1c,d). Enriched sgRNAs were asymmetrically distributed across the GSPT1 and RBM39 coding sequences in the degrader-treatment conditions, consistent with the expansion of drug-selected populations. For both neosubstrate targets, sgRNAs enriched in either set of drug treatments (CC-885 and ZXH-1-161 or E7820 and indisulam) were strongly correlated (Figure 1e,f), reflecting the structural similarity between the degraders employed and the overall assay robustness.
The top-enriched sgRNAs across both screens targeted the structural degron of each respective TPD substrate (Figure 1c,d). For the GSPT1 screen, sgRNAs highly enriched in degrader treatment (i.e., sgC568, sgK573, sgQ611) clustered near the key β-hairpin structural degron and the CRBN-GSPT1 interface (Figure 1g).10 Likewise, top-enriched sgRNAs in the RBM39 screen, sgL266 and sgL266/R267, target the α-helical structural degron in the RRM2 domain helix 1 that mediates the ternary DCAF15-RBM39-sulfonamide interaction (Figure 1h).17−19 These highly enriched sgRNAs presumably lead to mutations that disrupt ternary complex formation. However, several sgRNAs enriched in RBM39, and to a lesser extent, GSPT1, targeted positions distal to the structural degron that have not been previously implicated in degradation (i.e., RBM39 sgD151/A152, sgE286, sgE343). Altogether, these data demonstrate that CRISPR-suppressor scanning can identify key binding sites in neosubstrate targets previously established to be critical for ternary complex formation and, by extension, TPD.
Identification of Resistance Mutations in the Primary Structural Degrons
To investigate resistance mechanisms at a molecular level, we performed targeted amplicon deep sequencing directly from the pooled CRISPR-suppressor scan, focusing on the sgRNA cut sites corresponding to the highest enriched sgRNAs. This included sgRNAs targeting (1) the region proximal to the β-hairpin structural degron of GSPT1 (Figure 1g, sgC568, sgK573, sgQ611), (2) the RRM2 helix 1 structural degron of RBM39 (Figure 1h, sgL266, sgL266/R267), and (3) the region N-terminal to the RRM1 domain of RBM39 (sgD151/A152). We genotyped GSPT1 and RBM39 variants and quantified their mutational allele frequency. Overall, variants across both GSPT1 and RBM39 exhibited considerable variation in enrichment and sequence diversity (Figure 2a, Figure 3a). This analysis revealed strong enrichment of diverse in-frame indel mutations within the GSPT1 structural degron in CC-885- and ZXH-1-161-treated conditions (Figure 2a). These mutations were predominantly centered around the sgK573 cut site, consistent with sgK573 being the most highly enriched sgRNA across both compound treatments, and frequently altered up to seven amino acids spanning D571 to K577 with distinct amino acid deletions and insertions (Figure 2a, bottom panel). Consideration of the CC-885-CRBN-GSPT1 structure suggested that many of these mutations may impede ternary complex formation by modifying the conformation of the β-hairpin structural degron and consequently disrupting contacts between GSPT1, CC-885, and CRBN (Figure 2b).10 For instance, GSPT1 S574del removes a key serine residue that, together with D571, forms an ST-turn that stabilizes the β-hairpin (Figure S2a). In addition, several mutants, such as GSPT1 S574_K577del, altogether remove the critical G575 residue—the only residue within the β-hairpin degron conserved across all reported CRBN-IMiD neosubstrates.10,13 By contrast, mutations surrounding C568 are predicted to indirectly perturb the position of the β-hairpin by altering the upstream N-terminal sequences (C568_L569delinsQ) or by disrupting the ASX-motif involving D571 (C568_D571del) (Figure S2b-c). Lastly, mutations surrounding Q611 could not be detected, consistent with the lower resistance score of sgQ611 in comparison to sgC568 and sgK573. Corroborating these predictions, GSPT1 mutants in the presence of CC-885 failed to (1) degrade as assessed by a HiBiT lytic bioluminescence assay34 (Figure 2c) and (2) undergo coimmunoprecipitation (co-IP) with CRBN in comparison to wild-type (wt) GSPT1 (Figure 2d). The diversity of resistance mutations that alter the sequence or position of the β-hairpin structural degron (vide infra) suggests that this region of GSPT1 can tolerate substantial sequence variation without compromising protein function essential for cell survival and hence can serve as a hotspot for resistance mutations.
Figure 2.

CC-885 resistance mutations alter the GSPT1 β-hairpin structural degron and impair GSPT1 degradation. (a) Left: Schematic shows the coding variants of the most abundant in-frame mutations enriched in the β-hairpin structural degron of GSPT1 (>1% frequency in any condition). Right: Bar plot showing frequency (%, x axis) of each variant. Bars represent the mean across three replicate treatments, and dots show the individual replicate values. Bottom: Heat map showing normalized mutational frequency (y axis, %) by sequence position (x axis). Mutational frequency was normalized as a percentage of the total frequency of the displayed variants. (b) Structural view of the CC-885-CRBN-GSPT1 ternary complex, with key residues in CRBN (gray) and GSPT1 (green) highlighted. Carbon atoms of CC-885 are depicted in yellow (PDB: 5HXB). (c) Dose–response curves for wt and mutant HiBiT-GSPT1-HA cellular protein levels, as indicated by vehicle-normalized luminescence (y axis, %), in HEK293T cells treated with CC-885 for 6 h. Data represent mean ± s.e.m. across three technical replicates. One of two independent experiments is shown. (d) Immunoblots showing co-IP of GSPT1-HA wt and mutant variants with CRBN after vehicle or CC-885 treatment (10 μM, 2 h) in transiently transfected HEK293T cells. All cells were first pretreated with MLN-4924 (1 μM, 3 h) prior to vehicle or CC-885 treatment. Co-IP was performed using anti-HA antibody. One of two independent replicates is shown.
Figure 3.

E7820 resistance mutations in different domains of RBM39 operate via distinct mechanisms. (a) Top: Schematic of the RBM39 coding sequence. Left: Schematic shows the coding variants of the most abundant in-frame mutations enriched in the RRM2 helix 1 structural degron of RBM39 (>1% frequency in any condition). Right: Bar plot showing frequency (%, x axis) of each variant. Bars represent the mean across three replicate treatments, and dots show the individual replicate values. Bottom: Heat map showing normalized mutational frequency (y axis, %) by sequence position (x axis). Mutational frequency was normalized as a percentage of the total frequency of the displayed variants. (b) Left: Schematic shows the coding variants of the most abundant in-frame mutations enriched in the RRM1 N-terminal extension of RBM39 (>1% frequency in any condition). Right: Bar plot showing frequency (%, x axis) of each variant. Bars represent the mean across three replicate treatments, and dots show the individual replicate values. Bottom: Heat map showing normalized mutational frequency (y axis, %) by sequence position (x axis). Mutation frequency was normalized as a percentage of the total frequency of the displayed variants. (c) Structural view of the E7820-DCAF15-RBM39(RRM2) ternary complex, with key residues of RBM39 highlighted in blue. RBM39 G268 and a water molecule are highlighted in orange and red, respectively. Carbon atoms of E7820 are depicted in yellow (PDB: 6UE5). (d) Structural view of the RBM39 RRM1 domain (light blue), with key residues corresponding to the RDA deletion highlighted in orange (PDB: 4YUD). RNA molecule from a CUGBP1 structure (PDB: 3NMR) is shown in yellow, overlaid and visualized by structural alignment. (e) Dose–response curves for wt and mutant HiBiT-RBM39-HA cellular protein levels, as indicated by vehicle-normalized luminescence (y axis, %), in HEK293T cells treated with E7820 for 24 h. Data represent mean ± s.e.m. across three technical replicates. The P value was calculated using a two-sided Student’s t-test. One of two independent experiments is shown. (f) Immunoblots showing co-IP of RBM39-HA wt and mutant variants with FLAG-DCAF15 after vehicle or E7820 treatment (1 μM, 4 h) in transiently transfected HEK293T cells. All cells were first pretreated with MLN-4924 (1 μM, 2 h) prior to vehicle or E7820 treatment. Co-IP was performed using an anti-FLAG antibody. One of two independent replicates is shown. (g) Top: Schematic of the fluorescent EGFP-IRES-mCherry degradation reporter vector. Bottom: Dose–response curves for wt and mutant RBM39 cellular protein levels, as indicated by vehicle-normalized EGFP to mCherry ratio (y axis, %), in MOLM-13 (left) or K562 (right) cells treated with E7820 for 24 h. Data represent mean ± s.e.m. across three technical replicates. The Dmax ± s.e.m. and P values (two-sided Student’s t-test) are shown below. One of two independent experiments is shown for MOLM-13 cells, while one independent experiment was conducted for K562 cells.
Likewise, for RBM39, we observed significant enrichment of in-frame mutations in the α-helical structural degron in RRM2 helix 1 across both E7820- and indisulam-treated conditions (Figure 3a). In contrast to GSPT1, the most prevalent mutations were double missense mutations primarily affecting R267 and G268 (Figure 3a, bottom panel), a previously identified hotspot for resistance.14,16 The tight packing of G268 against DCAF15 has been demonstrated in structural studies to preclude larger residues at this position without abrogating ternary complex formation (Figure 3c).17,19 Supporting this notion, RBM39 R267Q/G268R failed to degrade in the presence of E7820 in comparison to wt RBM39 (Figure 3e). Whereas the diversity of complex indels observed in the GSPT1 degron region suggests substantial mutational tolerance, the preponderance of double amino acid substitutions arising from extensive point mutations suggest stricter structural and/or functional constraints on the RBM39 degron. Altogether, these data spanning GSPT1 and RBM39 characterize the landscape of resistance mutations—comprising point substitutions and complex indels—that directly alter the structural degron while maintaining essential protein function.
E7820 Resistance Mutations in Different Domains of RBM39 Operate via Distinct Mechanisms
While the enrichment of sgRNAs targeting the structural degrons corroborate past findings, the enrichment of particular sgRNAs targeting regions outside of the RBM39 RRM2 helix 1 was unanticipated, as the binding affinity of RBM39 to DCAF15-sulfonamide predominantly depends on the RRM2 domain.15,17,19 Notably, sgD151/A152 is the second-most highly enriched sgRNA in the RBM39 CRISPR-suppressor scan for both sulfonamide treatments, suggesting that the resultant mutation(s) represents a major, competitive resistance mechanism on par with perturbing the structural degron (Figure 1d). Sequencing the amplicon surrounding sgD151/A152 revealed strong enrichment of RBM39 R150_A152del (abbreviated from here on as RDAdel) in the degrader- versus vehicle-treated pools (Figure 3b). The RDAdel mutation truncates an N-terminal α-helical extension that lies just outside of the annotated RRM1 domain (Figure 3d). Because of its striking enrichment, we conducted a deeper investigation of RBM39 RDAdel. In contrast to the RRM2 helix 1 R267Q/G268R mutant, RBM39 RDAdel led to modest differences in RBM39 degradation versus wt RBM39 in a HiBiT assay conducted in HEK293T cells (Figure 3e). These differences in RBM39 degradation were predominantly characterized by a decreased level of maximal degradation (Dmax) at higher E7820 doses versus a change in half-maximal degradation concentration (DC50) (wt RBM39: DC50 = 17 nM, Dmax = 46%; RBM39 RDAdel DC50 = 17 nM, Dmax = 29%) (Figure 3e, Figure S3a). Notably, the differential Dmax between wt and RDAdel RBM39 was dependent on expression levels of DCAF15, as overexpression of DCAF15 caused Dmax to converge between the variants (Figure S3b). Co-IP experiments demonstrated that RBM39 RDAdel formed a ternary complex with DCAF15 and E7820 at comparable propensity as wt RBM39, while RBM39 R267Q/G268R completely failed to do so (Figure 3f). These results demonstrate that the RDA deletion does not fully abrogate E7820-DCAF15-RBM39 ternary complex formation and subsequent RBM39 degradation, suggesting that it may operate through a more intricate mechanism.
To corroborate the partial effects on Dmax, we evaluated RBM39 degradation using a fluorescent reporter system, in which wt or mutant RBM39 is fused in-frame with EGFP followed by an internal ribosome entry site (IRES) and mCherry (Figure 3g).13,27 Using this reporter, levels of RBM39 are directly correlated with EGFP fluorescence, which can be normalized to mCherry fluorescence to account for differences in reporter integrations and transcript expression levels. After lentiviral transduction of the reporter into MOLM-13 cells, levels of EGFP and mCherry fluorescence were assessed by flow cytometry after treatment with vehicle or E7820. As anticipated, cells expressing wt RBM39-EGFP exhibited a dose-dependent decrease in EGFP to mCherry ratio upon treatment with E7820 (DC50 = 9 nM, Dmax = 81%) (Figure 3g, Figure S4a). By contrast, cells expressing RBM39-EGFP R267Q/G268R exhibited no decrease in the EGFP to mCherry ratio even at the highest E7820 dose, as expected due to this mutant’s inability to form the DCAF15-RBM39 ternary complex (Figure 3f). However, cells expressing RBM39-EGFP RDAdel recapitulated a partial but significant rescue in degradation in comparison to wt RBM39-EGFP (RBM39-EGFP RDAdel: DC50 = 15 nM, Dmax = 73%). Analogous effects were also observed in K562 and HEK293T cells (Figure 3g, Figure S4b), confirming our findings in multiple cell lines and across different degradation assays.
As ectopic expression of RBM39 RDAdel revealed modest differences in Dmax, we sought to characterize the RDA deletion in an endogenous context by generating clonal cell lines. We lentivirally transduced MOLM-13 cells with SpCas9 and sgD151/A152 and treated them with E7820 (1 μM) for four weeks, after which surviving cells were sorted, expanded, and genotyped. We identified clonal cell lines harboring homozygous RDAdel alleles, which we refer to as MOLM-13RDAdel (Figure S5a). We confirmed that MOLM-13RDAdel cells were resistant to treatment with E7820 and express RBM39 at levels comparable to wt MOLM-13 cells (Figure 4a, Figure S5b). Furthermore, immunoblotting after 24 h of E7820 treatment revealed elevated levels of RBM39 in MOLM-13RDAdel versus wt MOLM-13 cells at higher doses of E7820 tested (Figure 4b), consistent with the decreased Dmax observed in ectopic expression experiments.
Figure 4.

Mutations distal to the RBM39 RRM2 helix 1 structural degron alter maximum levels of RBM39 degradation to abrogate E7820 cytotoxicity. (a) Dose–response curves for wt MOLM-13 and MOLM-13RDAdel cell proliferation relative to vehicle-treated cells (y axis, % control) after E7820 treatment for 72 h. Data represent mean ± s.e.m. across three technical replicates. One of two independent experiments is shown. (b) Immunoblots showing levels of RBM39 and GAPDH after vehicle or E7820 treatment for 24 h. One of two independent replicates is shown. (c) Line graphs showing cell proliferation (y axis) over a time course (x axis) following lentiviral transduction of SpCas9 and sgRNAs targeting luciferase (sgLuc) or RBM39 (sgL266/R267) into wt MOLM-13 and MOLM-13RDAdel cells. Data represent mean ± s.e.m. across three technical replicates. One of two independent experiments is shown. (d) Bar graphs showing fraction of GFP-positive cells (y axis) in a competition growth assay with nontransduced cells at day 0 and day 10 after treatment with either vehicle or 1 μM E7820 following lentiviral transduction of plasmid overexpressing DCAF15 and GFP in wt MOLM-13 and MOLM-13RDAdel. One of three independent replicates is shown. (e) Schematic showing the coding variants of the most abundant in-frame RBM39 mutations enriched in E7820 treatment (1 μM) by each sgRNA tested. Variant frequencies in vehicle- and E7820-treatment conditions are indicated. (f) Bar plots showing wt and mutant RBM39 cellular protein levels, as indicated by vehicle-normalized EGFP to mCherry ratio (y axis, %), in MOLM-13 cells treated with E7820 for 24 h. Data represent mean ± s.e.m. across three technical replicates. Dotted gray line indicates the mean signal of wt MOLM-13 treated with 10 μM E7820. Values for Dmax ± s.e.m. are shown (right) with significance levels from a two-sided Student’s t-test comparing to wt RBM39 Dmax indicated in parentheses (P < 10–3: ***; P < 10–4: ****; ns: not significant; nd: not determined). One of two independent experiments is shown. Full dose–response curves are shown in Figure S5d.
On the basis of these results, we considered whether the RBM39 RDA deletion confers resistance by preventing depletion of RBM39 below a threshold level necessary to induce significant growth inhibition. Lowering RBM39 RDAdel levels by genetic depletion with CRISPR-Cas9 in MOLM-13RDAdel led to growth inhibition, supporting the idea that substantial depletion of RBM39 RDAdel remains antiproliferative in the MOLM-13RDAdel cell line (Figure 4c). As expression of DCAF15 is correlated to Dmax (Figure S3b), we considered whether ectopic overexpression of DCAF15 might resensitize MOLM-13RDAdel to E7820. Indeed, both wt MOLM-13 and MOLM-13RDAdel cells overexpressing DCAF15 exhibited growth inhibition upon E7820 treatment (Figure 4d). Taken together, our data support the possibility that even modest differences in target degradation can confer robust resistance to degraders and that mutations outside the ternary complex interface may be sufficient to achieve this.
Several Mutations Distal to the RBM39(RRM2) Structural Degron Decrease Dmax
Aside from sgD151/A152, we next considered whether other enriched sgRNAs targeting regions outside the RBM39 RRM2 helix 1 structural degron may generate resistance mutations that behave like the RDA deletion by partially decreasing Dmax and impeding maximal RBM39 degradation. As we were unable to detect mutations at these distal sites by directly sequencing the pooled cells derived from the CRISPR-suppressor scan, we individually transduced selected enriched sgRNAs (RBM39 sgS127, sgE286, sgE343, sgD350) along with SpCas9 into MOLM-13 cells to identify their corresponding mutations. sgL266/R267 and sgD151/A152 were also transduced individually as comparators. Transduced cells were subsequently split and treated with E7820 (1 μM) or vehicle for four weeks, and surviving cells were genotyped by targeted amplicon sequencing around the corresponding sgRNA cut sites.
With transduction of sgD151/A152 and sgL266/R267, we observed significant enrichment of in-frame mutations and concomitant depletion of the wt allele in the presence of E7820 versus vehicle control (Figure 4e). For sgL266/R267, in-frame variants constituted <0.5% of detected alleles under vehicle treatment, whereas the wt allele was highly prevalent at >50%. Under E7820 treatment, however, in-frame mutations and the wt allele represented >50% and <0.5% of detected alleles, respectively. These results suggest that RRM2 helix 1 variants may confer a significant fitness advantage to E7820 but may otherwise be rare and/or potentially deleterious in its absence (vide infra). By contrast, RDAdel was the predominant variant in cells transduced with sgD151/A152, comprising >90% and 17.5% of alleles in E7820- and vehicle-treatment, respectively. The prevalence of RDAdel in the control condition likely reflects its high predicted frequency as an editing outcome and limited effects on protein fitness and cell viability (Figure S5c).
In comparison to sgD151/A152, E7820 treatment led to more modest enrichment of in-frame alleles generated by sgS127, sgE286, and sgD350, consistent with these sgRNAs having lower resistance scores in the CRISPR-suppressor scan (Figure 4e). Mutations were not observed with sgE343 in this experiment. To assess possible effects on RBM39 degradation, we selected top enriched in-frame mutants generated by each sgRNA to evaluate with the RBM39-EGFP-IRES-mCherry reporter (Figure 4f, Figure S5d). As anticipated, complete rescue from E7820-induced degradation was observed with L266_E271del, which substantially alters the RRM2 helix 1 structural degron. By contrast, apart from I349_T353del, the remaining distal RBM39 mutants conferred partial resistance to E7820-induced degradation at levels similar to RDAdel (Figure 4f, Figure S5d). Notably, E286_T287del alters a β-hairpin within the RBM39 RRM2 domain formed by residues D284-R289 that may compromise a peripheral protein–protein interaction with DCAF15 (Figure S5e).17−19 Like RDAdel, S127_K128delinsR lies outside the RRM2 domain and is not structurally resolved.
We next considered if the distal mutations’ effects on Dmax might be dependent or additive. RBM39 constructs containing two or three of these distal mutations exhibited significant cumulative decreases in Dmax values of up to 25% (Figures 4f and S5d), showing that these mutations have additive effects and might operate independently of one another. Collectively, these findings show that several sites distal to the RBM39 structural degron, and in some cases distal to the known ternary complex interface altogether, can modulate Dmax and the efficacy of target degradation. Furthermore, the observation that many distal site mutations can decrease Dmax supports the notion that modest rescue of RBM39 levels is sufficient to confer resistance to E7820.
Resistance Mutation Sites across TPD Targets Exhibit Varying Levels of Sequence Conservation and Mutational Constraint
Evolutionary conservation of protein sequences is a strong indicator of function. Consequently, protein sequence conservation can influence the accessible landscape of drug resistance-conferring mutations, as highly conserved sites (e.g., enzyme active sites) are typically more constrained by their functional importance and hence more difficult to mutate than less conserved sites. As a result, small molecules that bind or mechanistically involve less conserved sites may be more susceptible to the emergence of resistance mutations. Unlike orthosteric inhibitors, which typically modulate target activity and exploit the conserved structural features of active sites, molecular glue degraders are not necessarily dependent on neosubstrate function for efficacy. Thus, the mechanism of TPD may co-opt regions of the neosubstrate that are otherwise nonfunctional and hence may exhibit varying levels of mutational constraint. These considerations raise questions as to what factors shape the accessibility of neosubstrate resistance mutations.
Taking advantage of our CRISPR-suppressor scanning data spanning GSPT1 and RBM39, we considered how sequence conservation and mutational constraint may influence the emergence and diversity of resistance mutations. To do so, we first calculated the conservation score of each residue in GSPT1 and RBM39 using ConSurf (Figure 5a), which estimates the relative conservation of each amino acid position (see Methods). As sequence conservation can vary substantially between adjacent residues and Cas9 generally mutates multiple amino acids around the cut site, we applied a LOESS regression to estimate per-residue conservation scores with respect to neighboring residues in the local region. As anticipated, this analysis highlighted the greater relative conservation of the well-defined protein domains versus the unstructured N-terminus and interdomain linkers of each respective protein, where more negative ConSurf scores indicate higher levels of evolutionary conservation. In support of these calculations, our CRISPR-suppressor scanning data under vehicle-treatment showed preferential depletion of sgRNAs targeting more conserved regions (Figure S6a,b). The depletion of sgRNAs targeting functional protein regions has been previously demonstrated to indicate their essentiality,35−37 and consequently we refer to a sgRNA’s depletion in the vehicle condition as the “fitness score,” with lower scores corresponding to higher levels of essentiality.
Figure 5.

Resistance mutation sites across TPD targets exhibit low levels of sequence conservation. (a) ConSurf conservation scores (y axis) of amino acid residues in GSPT1 (top panel) and RBM39 (bottom panel) shown as dots with the LOESS regression line in blue. Amino acids corresponding to enriched sgRNA cut site positions from the CRISPR-suppressor scanning are highlighted in red and key residues are labeled. (b) Box plots with jitter showing fitness scores and ConSurf LOESS scores for nonenriched (gray, n = 230 for GSPT1 and 119 for RBM39) or enriched (red, n = 9 for GSPT1 and 10 for RBM39) sgRNAs. Fitness scores were calculated as the log2(fold-change sgRNA enrichment at week 4 under vehicle treatment versus the plasmid library) normalized to the mean of the negative control sgRNAs. sgRNAs were assigned ConSurf LOESS scores based on the amino acid corresponding to their predicted cut site positions; sgRNAs cutting between amino acids were assigned the mean of the flanking amino acids’ scores. Dots represent the fitness scores or corresponding amino acid ConSurf LOESS scores for individual sgRNAs. Two-sided P values were calculated with the Mann–Whitney test (ns: not significant). The box shows the median, 25th, and 75th percentiles with whiskers denoting 1.5 × the interquartile range. (c) Structural view of GSPT1(I440-P634) (left) and RBM39(RRM2) (right), with residues colored according to ConSurf conservation scores. The top three most conserved bins of ConSurf scores are colored in red, orange, and yellow, respectively, and the bottom six bins are colored in gray. sgRNAs enriched in the CRISPR-suppressor scan are depicted as spheres. Sequences corresponding to the approximate region around the structural degrons are shown below and colored according to ConSurf scores. (d) Stacked bar plot showing the frequency distribution of variant types (y axis, % of total reads) after transduction of the indicated sgRNAs targeting GSPT1 and RBM39 and treatment with vehicle or drug molecules (see Methods) for four weeks. (e) Bar plots showing variant frequencies (x axis, % of total reads) for the top 50 variants (y axis) generated by the indicated sgRNAs after treatment with vehicle (gray bars, left) or drug molecules (red bars, right) for four weeks. Variants are rank-ordered on the y axis by decreasing frequency in vehicle treatment for each sgRNA. (f) Cumulative plot showing the normalized variant frequency (y axis) for the 100 most abundant in-frame edited variants (x axis) for each indicated sgRNA after drug treatment for four weeks. Variants are rank-ordered on the x axis by decreasing normalized frequency for each respective sgRNA condition. Variant frequency was normalized to the total frequency of all in-frame edited variants.
We next assessed the sgRNA fitness scores and conservation of positions in GSPT1 and RBM39 implicated in mediating resistance, focusing on enriched sgRNAs that had resistance scores ≥2 s.d. above the mean of the negative controls in either degrader condition. In RBM39 and GSPT1, top enriched sgRNAs by resistance score had fitness scores similar to nonenriched sgRNAs and were close to 0, the mean of the negative controls, indicating that they are functionally neutral and not likely targeting highly essential positions (Figure 5b). By ConSurf, these positions exhibited comparable or slightly greater conservation than those targeted by nonenriched sgRNAs. Altogether, these data suggest that resistance mutations to degraders can occur at sites that are not highly conserved relative to other residues.
Beyond evolutionary conservation, we next sought to directly assess the permissible mutational landscape across resistance sites validated in our study, including those affecting the (1) GSPT1 β-hairpin structural degron, (2) RBM39 RRM2 helix 1 structural degron, and (3) RBM39 distal positions. Although the neosubstrate structural degrons are both relatively conserved (Figure 5c), degrader treatments more strongly jackpot sgRNAs targeting the GSPT1 β-hairpin (sgC568, sgK573) than sgRNAs targeting the RBM39 RRM2 helix 1 (sgL266, sgL266/R267) within their respective CRISPR-suppressor scans, as the RBM39 distal mutations can presumably compete effectively with the RRM2 helix 1 mutations despite their partial rescue phenotype (Figure 4e,f). We therefore reasoned that the GSPT1 β-hairpin and RBM39 distal positions may tolerate more mutational variation than the highly structured RBM39 RRM2 helix 1, permitting more diversity of mutations in these regions that do not abrogate essential functions. To explore this notion further, we first individually transduced GSPT1 sgC568 and sgK573–sgRNAs that target the β-hairpin–along with SpCas9 into MOLM-13 cells, treated with either vehicle or CC-885 for 4 weeks, and then genotyped the surviving cellular pools by targeted amplicon sequencing. We then compared these allele frequency data for GSPT1 with those acquired previously for RBM39 sgL266/R267, sgS127, sgD151/A152, and sgE286–sgRNAs that target the RRM2 helix 1 and distal positions, respectively (Figure 4e).
We first considered alleles identified under the vehicle conditions. While frequencies of in-frame edited alleles typically ranged from 10% to 30% across both RBM39 and GSPT1, the total percentage of in-frame edited alleles generated by RBM39 sgL266/R267 was significantly lower (<1%) (Figure 5d), suggesting that the RRM2 helix 1 cannot tolerate mutational variation. It is unlikely that this difference in in-frame edited alleles is due to major discrepancies in sgRNA cutting efficiencies, as the fraction of total edited alleles for sgL266/R267 is second highest among the sgRNAs evaluated (Figure 5d, Figure S5c). We next scrutinized the distribution of the in-frame variants under vehicle conditions (Figure 5e, Figure S5c). sgRNAs targeting the GSPT1 β-hairpin—in particular sgK573—led to a wider spread of in-frame variant distributions than sgRNAs targeting RBM39. Altogether, these results suggest that resistance sites to degraders exhibit a wide range of mutational constraint under normal growth conditions and that the GSPT1 β-hairpin can tolerate mutational variation to a higher extent than positions in RBM39 despite its sequence conservation.
We next evaluated allele frequencies identified in the degrader-treated conditions. Across all sgRNAs evaluated, in-frame mutant alleles were enriched by degrader treatment (Figure 5d), and as anticipated, this enrichment was greatest for top-scoring sgRNAs in the CRISPR-suppressor scans (i.e., GSPT1 sgC568, GSPT1 sgK573, RBM39 sgD151/A152, RBM39 sgL266/R267). We considered how the distribution of in-frame variants may change between vehicle- and degrader-treated conditions. Under degrader treatment, the distributions of in-frame mutations generally become more skewed, and rarer variants can be highly selected for (Figure 5e,f, Figure S5c), consistent with not all mutations robustly conferring resistance. However, the level of skewing is highly variable. In particular, in-frame variant distributions generated by introduction of RBM39 sgL266/R267 and sgD151/A152 are dominated by 1–2 mutants each under E7820 treatment. For RBM39 sgL266/R267, this jackpotting supports the idea that RBM39 RRM2 helix 1 mutations are highly selected for and constrained. By contrast, the jackpotting observed with RBM39 sgD151/A152 likely reflects the prevalence of RDAdel as a favorable editing outcome that is both well-tolerated and selected (Figure 4e). Other RBM39 distal position mutations were also highly selected for by E7820, albeit to a lesser extent than those generated by sgL266/R267 and sgD151/A152 (Figure 5e,f, Figure S5c).
Strikingly, mutagenesis of the GSPT1 β-hairpin by sgK573 and, to a lesser extent, gC568 led to wider spread distributions of in-frame variants in comparison to mutagenesis of RBM39 (Figure 5e,f). Altogether, these data suggest that the GSPT1 β-hairpin can accommodate many mutations, a large fraction of which can robustly confer resistance (Figure S5c). Moreover, many of these in-frame mutations identified in the GSPT1 β-hairpin involve complex indel mutations altering variable stretches of multiple amino acids (Figure 2a) in contrast to the predominance of point mutations or smaller deletions observed in RBM39 upon E7820-treatment (Figure 3a, Figure 4e). As a result, despite its high sequence conservation, the GSPT1 degron can tolerate substantial mutational variation, in contrast to the highly conserved and mutationally constrained degron of RBM39. This mutational constraint imposed on the RBM39 degron, in tandem with the modest rescue effect required to restore growth, likely enabled the emergence of diverse resistance mutations across distal positions of RBM39 (i.e., S127_K128delinsR, RDAdel, E286_T287del). Altogether, our analysis highlights how various factors can constrain or enable the accessibility of resistance mutations to degraders and cooperate to ultimately shape neosubstrate-specific mutational landscapes.
Discussion
Whereas resistance mutations to occupancy-driven inhibitors are well-studied and fall broadly into several archetypal classes (e.g., drug-binding disrupting, enzyme activating), the analogous mutational landscape for molecular glue degraders remains poorly defined owing to their unique mode of action.2,4 To address these challenges, here we systematically profiled the landscape of resistance mutations afforded by CRISPR-mutagenesis across two distinct TPD neosubstrates, GSPT1 and RBM39. We demonstrate that CRISPR-suppressor scanning can rapidly identify mutations that confer resistance to molecular glue degraders and that most of these mutations disrupt the structural degron. Such mutations are consistent with structural data and reinforce the notion that high-grade resistance mutations may disrupt ternary complex formation by perturbing either small molecule-protein interactions, protein–protein interactions, or both. On the basis of these observations, we expect that CRISPR-suppressor scanning will be a powerful approach to study potential neosubstrate surfaces and their interactions involved in ternary complex formation, especially in the absence of structural information or where multiple binding modes may possibly occur (e.g., hetero-bifunctional degraders).
Because of their dominance in various screens and precedence from natural genetic variation,7,14,38,39 we speculate that resistance mutations that disrupt ternary complex formation may be encountered in the clinic, especially when degradation of a single neosubstrate drives therapeutic response within cancer cells, the structural degron is not mutationally constrained, or the E3 ubiquitin ligase is an essential gene. This might especially be the case for GSPT1 degraders, as our data demonstrate that the GSPT1 β-hairpin is not mutationally constrained. However, alteration of the E3 ubiquitin ligase—especially when it is nonessential (i.e., CRBN)—will likely be the predominant clinical resistance mechanism, as a wide spectrum of LOF mutations within the ligase or even its downregulation is sufficient to abrogate target degradation. Supporting this notion, several studies have reported LOF mutations or reduced expression of E3 ligase substrate receptors as a major pathway of resistance to degraders, which is consistent with the positive correlation of CRBN expression levels with response to lenalidomide and pomalidomide in multiple myeloma patients.4,20−27
While mutations at the molecular glue interface can readily disrupt ternary complex formation and TPD, we also identified unexpected resistance mutations in RBM39 that are distal to the structural degron. Through closer investigation, we show that some distal mutations decrease the depth of maximal protein degradation (i.e., Dmax), thereby preventing sufficient target depletion that is necessary for growth inhibition to occur. Moreover, these impacts on Dmax can be additive, showing how multiple, independent distal structural alterations may be sufficient to significantly alter target degradation. Interestingly, increased DCAF15 expression diminishes the impact of RDAdel on Dmax, suggesting that mutations that modestly decrease Dmax might have stronger effects in cells with lower E3 ligase expression. Of note, the threshold of target degradation necessary for phenotypic response may be highly context-dependent, which may further influence the potential of these “depth-altering” resistance pathways. For example, studies investigating BCL6-degraders suggest that very high levels of BCL6 degradation are necessary to achieve tumor regression,40 and, in these instances, mutations that only modestly influence Dmax may be sufficient to confer resistance. These observations highlight the advantage of investigating resistance mutations within their endogenous protein contexts, as dosage effects may be prevalent. Altogether, our findings suggest that distal mutations can be sufficient to decrease Dmax and degrader efficacy, raising the possibility that distal post-translational modifications, alternative isoforms, or even binding partners may have similar effects as well.
Lastly, by integrating CRISPR-suppressor scanning data spanning GSPT1 and RBM39, we demonstrate that mutational tolerance of the structural degron is a primary driver of the overall landscape of degrader resistance mutations within neosubstrates. Neosubstrates like GSPT1, where the β-hairpin structural degron is not mutationally constrained, may permit the formation of mutational “hotspots,” with an array of diverse mutations concentrated within a small region of the protein. On the other hand, neosubstrates like RBM39, where the α-helical structural degron is highly constrained, can lead to the emergence of degrader resistance mutations across numerous sites both within and distal to the structural degron, despite their weaker effects on Dmax. While these mutations are largely context-specific and generated by CRISPR-Cas9, which favors formation of indels, these results highlight the utility in profiling mutational constraint directly, which may complement evolutionary conservation analysis. More broadly, our analysis highlights how the interplay of relative mutational constraint across putative sites of resistance can shape divergent outcomes in the overall landscape of degrader resistance mutations.
In conclusion, systematic identification of drug resistance-conferring alleles through approaches like CRISPR-suppressor scanning can illuminate neosubstrate requirements that are necessary for chemically induced dimerization and for TPD to drive effective phenotypic responses. Notably, many secondary neosubstrate features beyond the structural degron are not well understood and typically involve flexible yet potentially functional regions that are not structurally resolved, highlighting the utility of this approach. Deeper investigation of the mutants identified in these types of studies might uncover additional resistance mechanisms. We anticipate that the strategy developed here will also be broadly applicable for the study of TPD across different types of degraders (e.g., PROTACs, autophagy-targeting chimeras), neosubstrates, and E3 ligase systems and will be informative for the design and optimization of degraders.
Methods
Please see the Supporting Information for detailed experimental protocols.
Safety Statement
No unexpected or unusually high safety hazards were encountered in this study.
Acknowledgments
The authors acknowledge the Gray lab for providing ZXH-1-161. The authors acknowledge Ceejay Lee for assistance in designing sgRNA libraries and members of the Liau lab for comments on the manuscript. The authors acknowledge Dr. Ralph Mazitschek and Connor Payne for experimental support and discussion. This research was supported by startup funds from Harvard University and the Ono Pharma Breakthrough Science Initiative Award. K.C.N. and N.Z.L. acknowledge NSF predoctoral fellowships (DGE1745303) for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c01603.
Author Contributions
# P.M.G. and K.C.N. contributed equally. P.M.G. and B.B.L. conceived the project. P.M.G., K.C.N., and B.B.L. designed the experiments. P.M.G. performed the CRISPR-suppressor screens and single guide experiments for RBM39 and GSPT1. K.C.N. performed computational analysis. P.M.G. and C.S. cloned and performed the coimmunoprecipitation studies. P.M.G. characterized single-cell clones and performed cell growth assays and degradation assays using Western blot. P.M.G. cloned HiBiT-tagged constructs and performed the HiBiT degradation assay. M.J.R.Y. and P.M.G. cloned and performed the protein degradation artichoke reporter assay in K562 and MOLM-13. K.C.N. performed the protein degradation artichoke reporter assay in HEK293T. J.L. synthesized select compounds used in the study. N.Z.L. performed computational protein modeling. S.M.H. performed the initial literature review. B.B.L., P.M.G., and K.C.N. wrote the manuscript. B.B.L. supervised the study and held overall responsibility of the study.
The authors declare the following competing financial interest(s): Brian Liau is on the scientific advisory board of H3 Biomedicine.
Supplementary Material
References
- Chamberlain P. P.; Hamann L. G. Development of Targeted Protein Degradation Therapeutics. Nat. Chem. Biol. 2019, 15 (10), 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov 2019, 18 (12), 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Wu T.; Yoon H.; Xiong Y.; Dixon-Clarke S. E.; Nowak R. P.; Fischer E. S. Targeted Protein Degradation as a Powerful Research Tool in Basic Biology and Drug Target Discovery. Nat. Struct Mol. Biol. 2020, 27 (7), 605–614. 10.1038/s41594-020-0438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M.; Sperling A. S.; Ebert B. L. Cancer Therapies Based on Targeted Protein Degradation — Lessons Learned with Lenalidomide. Nat. Rev. Clin Oncol 2021, 18, 401–417. 10.1038/s41571-021-00479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L. The Rise of Molecular Glues. Cell 2021, 184 (1), 3–9. 10.1016/j.cell.2020.12.020. [DOI] [PubMed] [Google Scholar]
- Ito T.; Ando H.; Suzuki T.; Ogura T.; Hotta K.; Imamura Y.; Yamaguchi Y.; Handa H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327 (5971), 1345–1350. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Krönke J.; Udeshi N. D.; Narla A.; Grauman P.; Hurst S. N.; McConkey M.; Svinkina T.; Heckl D.; Comer E.; Li X.; Ciarlo C.; Hartman E.; Munshi N.; Schenone M.; Schreiber S. L.; Carr S. A.; Ebert B. L. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343 (6168), 301–305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Middleton R. E.; Sun H.; Naniong M.; Ott C. J.; Mitsiades C. S.; Wong K.-K.; Bradner J. E.; Kaelin W. G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343 (6168), 305–309. 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi A. K.; Kang J.; Havens C. G.; Conklin T.; Ning Y.; Wu L.; Ito T.; Ando H.; Waldman M. F.; Thakurta A.; Klippel A.; Handa H.; Daniel T. O.; Schafer P. H.; Chopra R. Immunomodulatory Agents Lenalidomide and Pomalidomide Co-stimulate T Cells by Inducing Degradation of T Cell Repressors Ikaros and Aiolos via Modulation of the E3 Ubiquitin Ligase Complex CRL4CRBN. Br. J. Hamaetol. 2014, 164 (6), 811–821. 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Lu G.; Ito T.; Pagarigan B.; Lu C.-C.; Miller K.; Fang W.; Wang N.-Y.; Nguyen D.; Houston J.; Carmel G.; Tran T.; Riley M.; Nosaka L.; Lander G. C.; Gaidarova S.; Xu S.; Ruchelman A. L.; Handa H.; Carmichael J.; Daniel T. O.; Cathers B. E.; Lopez-Girona A.; Chamberlain P. P. A Novel Cereblon Modulator Recruits GSPT1 to the CRL4CRBN Ubiquitin Ligase. Nature 2016, 535 (7611), 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- Krönke J.; Fink E. C.; Hollenbach P. W.; MacBeth K. J.; Hurst S. N.; Udeshi N. D.; Chamberlain P. P.; Mani D. R.; Man H. W.; Gandhi A. K.; Svinkina T.; Schneider R. K.; McConkey M.; Järås M.; Griffiths E.; Wetzler M.; Bullinger L.; Cathers B. E.; Carr S. A.; Chopra R.; Ebert B. L. Lenalidomide Induces Ubiquitination and Degradation of CK1α in Del(5q) MDS. Nature 2015, 523 (7559), 183–188. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thomä N. H. Structural Basis of Lenalidomide-Induced CK1α Degradation by the CRL4CRBN Ubiquitin Ligase. Nature 2016, 532 (7597), 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Sievers Q. L.; Petzold G.; Bunker R. D.; Renneville A.; Słabicki M.; Liddicoat B. J.; Abdulrahman W.; Mikkelsen T.; Ebert B. L.; Thomä N. H. Defining the Human C2H2 Zinc Finger Degrome Targeted by Thalidomide Analogs through CRBN. Science 2018, 362 (6414), eaat0572. 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T.; Goralski M.; Gaskill N.; Capota E.; Kim J.; Ting T. C.; Xie Y.; Williams N. S.; Nijhawan D. Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 2017, 356 (6336), eaal3755. 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Ting T. C.; Goralski M.; Klein K.; Wang B.; Kim J.; Xie Y.; Nijhawan D. Aryl Sulfonamides Degrade RBM39 and RBM23 by Recruitment to CRL4-DCAF15. Cell Reports 2019, 29 (6), 1499–1510. 10.1016/j.celrep.2019.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T.; Minoshima Y.; Sagane K.; Sugi N. H.; Mitsuhashi K. O.; Yamamoto N.; Kamiyama H.; Takahashi K.; Kotake Y.; Uesugi M.; Yokoi A.; Inoue A.; Yoshida T.; Mabuchi M.; Tanaka A.; Owa T. Selective Degradation of Splicing Factor CAPERα by Anticancer Sulfonamides. Nat. Chem. Biol. 2017, 13 (6), 675–680. 10.1038/nchembio.2363. [DOI] [PubMed] [Google Scholar]
- Bussiere D. E.; Xie L.; Srinivas H.; Shu W.; Burke A.; Be C.; Zhao J.; Godbole A.; King D.; Karki R. G.; Hornak V.; Xu F.; Cobb J.; Carte N.; Frank A. O.; Frommlet A.; Graff P.; Knapp M.; Fazal A.; Okram B.; Jiang S.; Michellys P.-Y.; Beckwith R.; Voshol H.; Wiesmann C.; Solomon J. M.; Paulk J. Structural Basis of Indisulam-Mediated RBM39 Recruitment to DCAF15 E3 Ligase Complex. Nat. Chem. Biol. 2020, 16 (1), 15–23. 10.1038/s41589-019-0411-6. [DOI] [PubMed] [Google Scholar]
- Du X.; Volkov O. A.; Czerwinski R. M.; Tan H.; Huerta C.; Morton E. R.; Rizzi J. P.; Wehn P. M.; Xu R.; Nijhawan D.; Wallace E. M. Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 2019, 27 (11), 1625–1633. 10.1016/j.str.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Faust T. B.; Yoon H.; Nowak R. P.; Donovan K. A.; Li Z.; Cai Q.; Eleuteri N. A.; Zhang T.; Gray N. S.; Fischer E. S. Structural Complementarity Facilitates E7820-Mediated Degradation of RBM39 by DCAF15. Nat. Chem. Biol. 2020, 16 (1), 7–14. 10.1038/s41589-019-0378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio S.; Munawar U.; Zhu Y. X.; Giesen N.; Shi C.-X.; Viá M. D.; Sanchez R.; Bruins L.; Demler T.; Müller N.; Haertle L.; Garitano A.; Steinbrunn T.; Danhof S.; Cuenca I.; Barrio-Garcia C.; Braggio E.; Rosenwald A.; Martinez-Lopez J.; Rasche L.; Raab M. S.; Stewart A. K.; Einsele H.; Stühmer T.; Kortüm K. M. IKZF1/3 and CRL4-CRBN E3 Ubiquitin Ligase Mutations and IMiD Resistance in Multiple Myeloma. Haematologica 2020, 105 (5), e237–e241. 10.3324/haematol.2019.217943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortüm K. M.; Mai E. K.; Hanafiah N. H.; Shi C.-X.; Zhu Y.-X.; Bruins L.; Barrio S.; Jedlowski P.; Merz M.; Xu J.; Stewart R. A.; Andrulis M.; Jauch A.; Hillengass J.; Goldschmidt H.; Bergsagel P. L.; Braggio E.; Stewart A. K.; Raab M. S. Targeted Sequencing of Refractory Myeloma Reveals a High Incidence of Mutations in CRBN and Ras Pathway Genes. Blood 2016, 128 (9), 1226–1233. 10.1182/blood-2016-02-698092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding S.; Ansari-Pour N.; Towfic F.; Estévez M. O.; Chamberlain P. P.; Tsai K.-T.; Flynt E.; Hirst M.; Rozelle D.; Dhiman P.; Neri P.; Ramasamy K.; Bahlis N.; Vyas P.; Thakurta A. Multiple Cereblon Genetic Changes Are Associated with Acquired Resistance to Lenalidomide or Pomalidomide in Multiple Myeloma. Blood 2021, 137 (2), 232–237. 10.1182/blood.2020007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Weng S.; Matyskiela M.; Zheng X.; Fang W.; Wood S.; Surka C.; Mizukoshi R.; Lu C.-C.; Mendy D.; Jang I. S.; Wang K.; Marella M.; Couto S.; Cathers B.; Carmichael J.; Chamberlain P.; Rolfe M. UBE2G1 Governs the Destruction of Cereblon Neomorphic Substrates. Elife 2018, 7, e40958. 10.7554/eLife.40958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor-Ruiz C.; Jaeger M. G.; Bauer S.; Brand M.; Sin C.; Hanzl A.; Mueller A. C.; Menche J.; Winter G. E. Plasticity of the Cullin-RING Ligase Repertoire Shapes Sensitivity to Ligand-Induced Protein Degradation. Mol. Cell 2019, 75 (4), 849–858. 10.1016/j.molcel.2019.07.013. [DOI] [PubMed] [Google Scholar]
- Shirasaki R.; Matthews G. M.; Gandolfi S.; de Matos Simoes R.; Buckley D. L.; Raja Vora J.; Sievers Q. L.; Bruggenthies J. B.; Dashevsky O.; Poarch H.; Tang H.; Bariteau M. A.; Sheffer M.; Hu Y.; Downey-Kopyscinski S. L.; Hengeveld P. J.; Glassner B. J.; Dhimolea E.; Ott C. J.; Zhang T.; Kwiatkowski N. P.; Laubach J. P.; Schlossman R. L.; Richardson P. G.; Culhane A. C.; Groen R. W.J.; Fischer E. S.; Vazquez F.; Tsherniak A.; Hahn W. C.; Levy J.; Auclair D.; Licht J. D.; Keats J. J.; Boise L. H.; Ebert B. L.; Bradner J. E.; Gray N. S.; Mitsiades C. S. Functional Genomics Identify Distinct and Overlapping Genes Mediating Resistance to Different Classes of Heterobifunctional Degraders of Oncoproteins. Cell Reports 2021, 34 (1), 108532. 10.1016/j.celrep.2020.108532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottis P.; Palladino C.; Thienger P.; Britschgi A.; Heichinger C.; Berrera M.; Julien-Laferriere A.; Roudnicky F.; Kam-Thong T.; Bischoff J. R.; Martoglio B.; Pettazzoni P. Cellular Resistance Mechanisms to Targeted Protein Degradation Converge Toward Impairment of the Engaged Ubiquitin Transfer Pathway. ACS Chem. Biol. 2019, 14 (10), 2215–2223. 10.1021/acschembio.9b00525. [DOI] [PubMed] [Google Scholar]
- Sievers Q. L.; Gasser J. A.; Cowley G. S.; Fischer E. S.; Ebert B. L. Genome-Wide Screen Identifies Cullin-RING Ligase Machinery Required for Lenalidomide-Dependent CRL4CRBN Activity. Blood 2018, 132 (12), 1293–1303. 10.1182/blood-2018-01-821769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. V.; Li J.; Lu C.-C. J.; Mamrosh J. L.; Lu G.; Cathers B. E.; Deshaies R. J. P97/VCP Promotes Degradation of CRBN Substrate Glutamine Synthetase and Neosubstrates. Proc. Natl. Acad. Sci. 2017, 114 (14), 3565–3571. 10.1073/pnas.1700949114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan K. A.; Ferguson F. M.; Bushman J. W.; Eleuteri N. A.; Bhunia D.; Ryu S.; Tan L.; Shi K.; Yue H.; Liu X.; Dobrovolsky D.; Jiang B.; Wang J.; Hao M.; You I.; Teng M.; Liang Y.; Hatcher J.; Li Z.; Manz T. D.; Groendyke B.; Hu W.; Nam Y.; Sengupta S.; Cho H.; Shin I.; Agius M. P.; Ghobrial I. M.; Ma M. W.; Che J.; Buhrlage S. J.; Sim T.; Gray N. S.; Fischer E. S. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 2020, 183 (6), 1714–1731. 10.1016/j.cell.2020.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uy G. L.; Minden M. D.; Montesinos P.; DeAngelo D. J.; Altman J. K.; Koprivnikar J.; Vyas P.; Fløisand Y.; Vidriales M. B.; Gjertsen B. T.; Esteve J.; Buchholz T. J.; Couto S.; Fan J.; Hanna B.; Li L.; Pierce D. W.; Hege K.; Pourdehnad M.; Zeidan A. M. Clinical Activity of CC-90009, a Cereblon E3 Ligase Modulator and First-in-Class GSPT1 Degrader, As a Single Agent in Patients with Relapsed or Refractory Acute Myeloid Leukemia (R/R AML): First Results from a Phase I Dose-Finding Study. Blood 2019, 134 (Supplement_1), 232–232. 10.1182/blood-2019-123966. [DOI] [Google Scholar]
- Wang E.; Lu S. X.; Pastore A.; Chen X.; Imig J.; Lee S. C.-W.; Hockemeyer K.; Ghebrechristos Y. E.; Yoshimi A.; Inoue D.; Ki M.; Cho H.; Bitner L.; Kloetgen A.; Lin K.-T.; Uehara T.; Owa T.; Tibes R.; Krainer A. R.; Abdel-Wahab O.; Aifantis I. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35 (3), 369–384.e7. 10.1016/j.ccell.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinyard M. E.; Su C.; Siegenfeld A. P.; Waterbury A. L.; Freedy A. M.; Gosavi P. M.; Park Y.; Kwan E. E.; Senzer B. D.; Doench J. G.; Bauer D. E.; Pinello L.; Liau B. B. CRISPR-Suppressor Scanning Reveals a Nonenzymatic Role of LSD1 in AML. Nat. Chem. Biol. 2019, 15 (5), 529. 10.1038/s41589-019-0263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C. E.; Du G.; Che J.; He Z.; Donovan K. A.; Yue H.; Wang E. S.; Nowak R. P.; Zhang T.; Fischer E. S.; Gray N. S. Selective Degradation of GSPT1 by Cereblon Modulators Identified via a Focused Combinatorial Library. ACS Chem. Biol. 2020, 15 (10), 2722–2730. 10.1021/acschembio.0c00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinn M. K.; Machleidt T.; Zimmerman K.; Eggers C. T.; Dixon A. S.; Hurst R.; Hall M. P.; Encell L. P.; Binkowski B. F.; Wood K. V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13 (2), 467–474. 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- Shi J.; Wang E.; Milazzo J. P.; Wang Z.; Kinney J. B.; Vakoc C. R. Discovery of Cancer Drug Targets by CRISPR-Cas9 Screening of Protein Domains. Nat. Biotechnol. 2015, 33 (6), 661–667. 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher F.; Hossain M.; Seruggia D.; Schoonenberg V. A. C.; Yao Q.; Cifani P.; Dassama L. M. K.; Cole M. A.; Ren C.; Vinjamur D. S.; Macias-Trevino C.; Luk K.; McGuckin C.; Schupp P. G.; Canver M. C.; Kurita R.; Nakamura Y.; Fujiwara Y.; Wolfe S. A.; Pinello L.; Maeda T.; Kentsis A.; Orkin S. H.; Bauer D. E. Rational Targeting of a NuRD Subcomplex Guided by Comprehensive in Situ Mutagenesis. Nat. Genet. 2019, 51 (7), 1149–1159. 10.1038/s41588-019-0453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonenberg V. A. C.; Cole M. A.; Yao Q.; Macias-Treviño C.; Sher F.; Schupp P. G.; Canver M. C.; Maeda T.; Pinello L.; Bauer D. E. CRISPRO: Identification of Functional Protein Coding Sequences Based on Genome Editing Dense Mutagenesis. Genome Biol. 2018, 19 (1), 169. 10.1186/s13059-018-1563-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan K. A.; An J.; Nowak R. P.; Yuan J. C.; Fink E. C.; Berry B. C.; Ebert B. L.; Fischer E. S. Thalidomide Promotes Degradation of SALL4, a Transcription Factor Implicated in Duane Radial Ray Syndrome. Elife 2018, 7, e38430. 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B.; Gao Y.; Che J.; Lu W.; Kaltheuner I. H.; Dries R.; Kalocsay M.; Berberich M. J.; Jiang J.; You I.; Kwiatkowski N.; Riching K. M.; Daniels D. L.; Sorger P. K.; Geyer M.; Zhang T.; Gray N. S. Discovery and Resistance Mechanism of a Selective CDK12 Degrader. Nat. Chem. Biol. 2021, 17 (6), 675–683. 10.1038/s41589-021-00765-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoull W.; Cheung T.; Anderson E.; Barton P.; Burgess J.; Byth K.; Cao Q.; Castaldi M. P.; Chen H.; Chiarparin E.; Carbajo R. J.; Code E.; Cowan S.; Davey P. R.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gao N.; Hargreaves D.; Howard M. R.; Hu J.; Kawatkar A.; Kemmitt P. D.; Leo E.; Molina D. M.; O’Connell N.; Petteruti P.; Rasmusson T.; Raubo P.; Rawlins P. B.; Ricchiuto P.; Robb G. R.; Schenone M.; Waring M. J.; Zinda M.; Fawell S.; Wilson D. M. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13 (11), 3131–3141. 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.