

Abstract
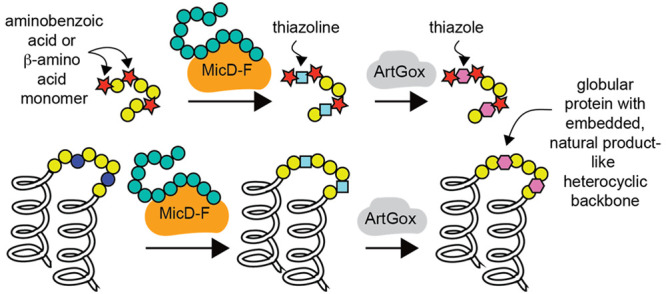
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are peptide-derived natural products with potent antibiotic, antiviral, and anticancer properties. RiPP enzymes known as cyclodehydratases and dehydrogenases work together to catalyze intramolecular, inter-residue condensation and dehydrogenation reactions that install oxazoline/oxazole and thiazoline/thiazole heterocycles within ribosomally produced polypeptide chains. Here, we show that the previously reported enzymes MicD-F and ArtGox accept backbone-modified monomers—including aminobenzoic acid derivatives and beta-amino acids—within leader-free polypeptides, even at positions immediately preceding or following the site of cyclization/dehydrogenation. The products are sequence-defined chemical polymers with multiple, diverse non-α-amino acid subunits. We show further that MicD-F and ArtGox can install heterocyclic backbones within protein loops and linkers without disrupting the native tertiary fold. Calculations reveal the extent to which these heterocycles restrict conformational space; they also eliminate a peptide bond—both features could improve the stability or add function to linker sequences now commonplace in emerging biotherapeutics. This work represents a general strategy to expand the chemical diversity of the proteome beyond and in synergy with what can now be accomplished by expanding the genetic code.
Short abstract
The ribosomally synthesized and post-translationally modified peptide (RiPP) enzymes MicD-F and ArtGox effectively embed thiazole and thiazoline heterocyclic backbones within folded proteins and diverse heteropolymers.
Introduction
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are peptide-derived natural products that include the FDA-approved analgesic ziconotide1,2 as well as compounds with potent antibiotic, antiviral, and anticancer properties.3 RIPP biosynthesis begins with a ribosomally synthesized polypeptide whose N-terminal leader sequence (∼20–110 aa) recruits one or more endogenous enzymes capable of diverse post-translational modification (PTM) of an adjacent C-terminal substrate sequence.3,4 Researchers have leveraged this leader-dependent mechanism to direct RiPP PTM enzymes to C-terminal substrate sequences containing diverse noncanonical α-amino acids (nc-α-AAs).5,6
Cyclodehydratases and dehydrogenases represent an exceptionally well-studied class of RiPP enzymes.3 These enzymes work together to catalyze intramolecular cyclization3,7 and subsequent aromatization reactions that install oxazoline/oxazole and thiazoline/thiazole heterocycles within polypeptide chains (Figure 1A,B). Previous work has shown that the cyclodehydratases PatD8 and TruD9 support leader sequence-dependent oxazoline/thiazoline formation within substrates containing nc-α-AAs adjacent to10 or at the cyclization site itself.11−13 In related work, it was shown that a chimeric leader peptide could direct the cyclodehydratase LynD9 and the dehydrogenase TbtE14 to install thiazol(in)es within substrates containing nc-α-AAs adjacent to the cyclization site.15 Finally, reconstituted lactazole biosynthesis,16 including the cyclodehydratase-dehydrogenase pair LazDE/LazF, was found to install oxazoles and thiazoles within polypeptide substrates containing α-hydroxy, N-methyl, cyclic α-, and β3-amino acids17 at sites distal from the site of heterocyclization (>4 residues away).
Figure 1.

Overview of cyclodehydratase/dehydrogenase chemistry. (A) Scheme illustrating the natural conversion of a serine, threonine, or cysteine-containing polypeptide into a oxazoline or thiazoline-containing product through the action of MicD21 and subsequent dehydrogenation into an oxazole or thiazole through the action of ArtGox.24 (B) Natural substrates for MicD34 and ArtGox35 consist of a core sequence that includes the reaction site and an extended upstream leader sequence. (C) Fusion of the leader sequence to the N-terminus of MicD generates a constitutively activated enzyme MicD-F that processes leaderless substrates.21 (D) This work: MicD-F and ArtGox accept leaderless polypeptide substrates containing diverse non-α-amino acid monomers, including aminobenzoic acid derivatives, at the +1 and −1 sites. MicD-F and ArtGox can also install thiazoline and thiazole linkages within leaderless globular proteins such as (E) mCherry and (F) Rop.
Previous work has also shown that certain cyclodehydratase enzymes can process leader sequence-free substrates when the leader peptide is provided in trans (Figure 1C).18,19 Building on this observation and earlier work on lantibiotic synthetases by van der Donk,20 Naismith and co-workers engineered a family of cyclodehydratases in which the leader peptide is fused to the N-terminus of the cyclodehydratase catalyst as opposed to the N-terminus of the substrate polypeptide. These constitutively activated enzymes, notably, LynD Fusion (LynD-F)19 and MicD Fusion (MicD-F),21 act in a leader peptide-independent manner to promote the cyclodehydration of polypeptides containing a C-terminal Ala-Tyr-Asp (AYD) recognition sequence.7,19,21−23 In complementary work, Schmidt and co-workers demonstrated that two dehydrogenases, ArtGox and ThcOx, also accept leaderless peptide substrates.24 Taken together, these enzymes represent a fully leader-free route toward polypeptides (and proteins, vide infra) containing mRNA-programmed thiazole and oxazole linkages—distinct from approaches based on chimeric leader peptides25 or leader peptide exchange.26 Indeed, some tolerance for noncanonical α-amino acid residues has been reported: LynD-F was shown to install a thiazoline in a peptide substrate containing 3-azido-l-alanine positioned four residues away from the site of cyclization,23 and the combination of LynD-F and ArtGox installed a thiazole in an AYD-containing peptide with a polyethylene glycol spacer two residues from the site of cyclization.22
Here, we report that MicD-F21,24 and ArtGox21,24 act together to process polypeptide substrates containing diverse translation-compatible27−32 aminobenzoic acid derivatives and β-amino acids, even at sites directly flanking the reaction site (Figure 1D). We show further that MicD-F21,24 and ArtGox21,24 process substrates even when the CAYD sequence is positioned at the C-terminus of mCherry, a large β-barrel protein, or embedded within the loop of the dimeric α-helical bundle protein Rop. The products are folded, globular proteins containing a conformationally restricted, fully unnatural, heterocyclic backbone. To the best of our knowledge, these studies represent the first example of leader-free azol(in)e biosynthesis within polypeptides containing diverse non-α-amino acid monomers flanking the site of cyclization and the first report of a cooperatively folded protein containing a post-translationally installed heterocyclic ring.33 The effects of the embedded heterocycle on local conformational flexibility are examined computationally, providing important insight into the backbone restrictions that could be leveraged to improve the physiochemical properties of therapeutic proteins. This work represents a general strategy to expand the chemical diversity of the proteome beyond and in synergy with what can now be accomplished by expanding the genetic code.
Results
MicD-F and ArtGox Accept Substrates with Diverse Structures at the +1 Site
We began by exploring the tolerance of MicD-F for sequences containing non-α-amino acid monomers at the +1 site (Figure 1A and Figure 2A,B). A series of nine potential substrates were prepared in which a non-α-amino acid preceded the reaction site (substrates 1(a-i)). Monomers evaluated included arenes, aminobenzoic acid derivatives, fluorophores, and linear and cyclic β-amino acids. All substrates contained a C-terminal AYD recognition sequence36 and were incubated with MicD-F (Supplementary Figure 1A,B) (5 mol %) under mild conditions (pH 8.0, 37 °C, 4 h) that resulted in complete conversion of ICAYDG, a substrate with the natural α-amino acid Ile at the +1 site (Supplementary Figure 2). Cyclization was analyzed initially via liquid chromatography–mass spectrometry (LC-MS), and the extent of product formation was estimated by integrating the extracted ion chromatogram (Figure 2B,C). Virtually every substrate examined underwent MicD-F-catalyzed cyclization to the corresponding thiazoline under these conditions. Substrates containing electron-withdrawing or donating aromatic rings, bulky multiring systems, and linear and cyclic β-amino acids were all cyclized efficiently by MicD-F, with yields between 91 and 99% (products 2(a-i)). UHPLC analysis of each reaction mixture confirmed that thiazolines 2a-i were the sole reaction products under these conditions (Supplementary Figure 3). It is notable that monomers with highly divergent structures are accepted almost equally by MicD-F, suggesting that the +1 residue interacts minimally if at all with the enzyme active site.
Figure 2.
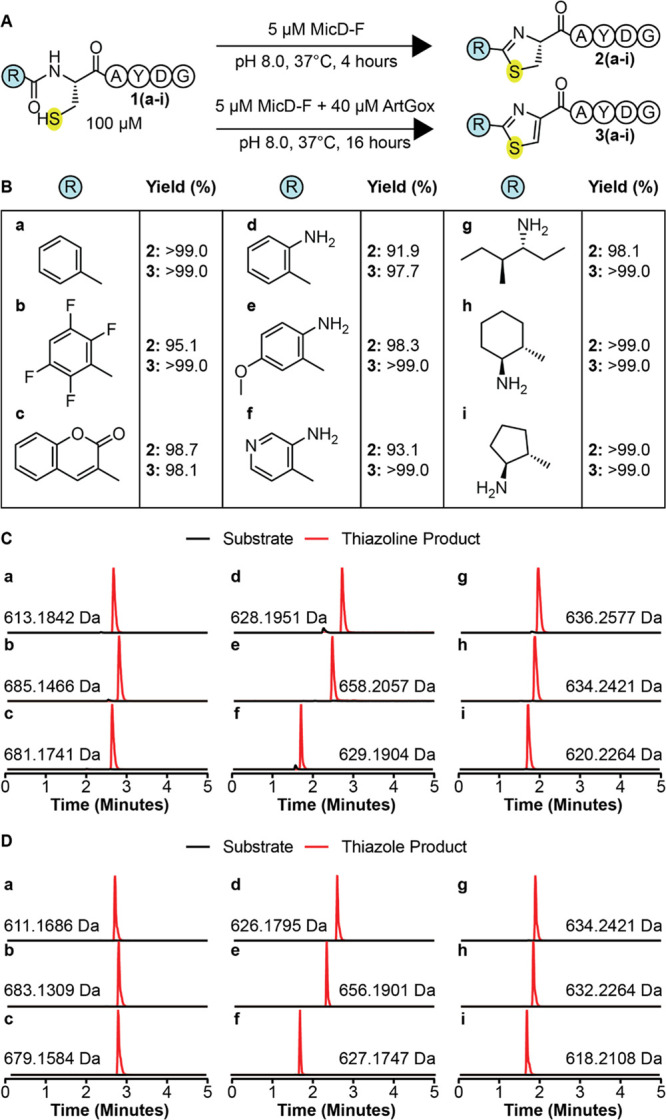
MicD-F and ArtGox tolerate diverse nonproteinogenic, non-α-amino acid monomers at the +1 site. (A) Scheme illustrating the conditions used for the reaction of ArtGox and/or MicD-F with substrates containing non-α-amino acid monomers N-terminal to the reaction site (+1 site). (B) Yields of thiazoline 2(a-i) and thiazole (3(a-i)) products obtained for substrates containing non-α-amino acid monomers at the +1 site. Extracted ion chromatograms illustrating the products of (C) MicD-F and (D) MicD-F + ArtGox-catalyzed reactions.
Next, we explored whether MicD-F and ArtGox could act in synergy to convert peptides containing non-α-amino acids at the +1 site directly into the corresponding thiazoles 3(a-i) (Figure 2A). Substrates 1(a-i) were incubated with MicD-F (5 mol %) and ArtGox (Supplementary Figure 1A,C) (40 mol %) under conditions (pH 8.0, 37 °C, 16 h) that resulted in complete two-step conversion of ICAYDG into the corresponding thiazole product (Supplementary Figure 4). ArtGox efficiently oxidized each thiazoline to the corresponding thiazole in yields that exceeded 97% over the two steps for every example (products 3(a-i)) (Figure 2B,D). UHPLC analysis of each reaction mixture confirmed that thiazoles 3a-e were the sole reaction product. Coelution with excess flavin mononucleotide precluded UHPLC analysis of thiazoles 3f-i (Supplementary Figure 5). These results indicate that MicD-F and ArtGox tolerate diverse nonproteinogenic, non-α-amino acid monomers at the +1 site. Many of these non-α-amino acid monomers have been installed at the N-termini of ribosomally translated peptides in vitro,30,31,37,38 suggesting a path toward proteins and polypeptides with highly unique N-terminal appendages.
MicD-F and ArtGox Accept Substrates with Diverse Structures at the −1 Site
Next, we explored whether MicD-F and ArtGox would accept leader-free polypeptide substrates containing non-α-amino acid monomers at the −1 site (Figure 3). We explored a diverse array of monomers – β3-amino acids, β2-amino acids, cyclic β2, β3-amino acids, as well as substituted and unsubstituted aminobenzoic acid derivatives. Notably, inserting a single α-amino acid (Ile) residue between the site of cyclization and the C-terminal AYD motif required higher concentrations (50 mol %) of MicD-F and up to 24 h reaction time to complete the cyclodehydration reaction (Supplementary Figure 6).
Figure 3.

MicD-F and ArtGox tolerate diverse nonproteinogenic, non-α-amino acid monomers at the −1 site. (A) MicD-F and ArtGox reactions of substrates with non-α-amino acid monomers immediately C-terminal to the site of cyclization. (B) Yields of thiazoline 5(a-i) and thiazole (6(a-i)) products obtained. (C) Extracted ion chromatograms illustrating the products of MicD-F and ArtGox-catalyzed reactions.
All −1 site substrates (substrates 4(a-g), Figure 3A,B) contained a C-terminal AYD recognition sequence36 and were incubated with MicD-F (50 mol %) and ArtGox (8 mol %) under conditions (pH 8.0, 37 °C, 24 h) that resulted in complete conversion of a substrate with a natural α-amino acid at the −1 site to the corresponding thiazole (Supplementary Figure 7). Reactions were analyzed as described above. Under these conditions, every peptide evaluated was a substrate for MicD-F, and a few were substrates for both MicD-F and ArtGox (Figure 3B–C). Substrates containing β3-amino acid-, β2-amino acid-, or cyclic β2,β3-amino acids at the −1 site (substrates 4(a-d)) were fully consumed under these conditions (<1% unmodified peptide). Those with β3-alkyl substituents (4a, c, and d) were converted cleanly into the corresponding thiazolines 5a, c, and d, with little (4a) or no (4c,d) thiazole formation. In contrast, substrate 4b, with geminal β2-methyl substituents, was converted into a 30/70 mixture of thiazoline 5b and thiazole 6b. Substrates 4e-g containing aminobenzoic acid derivatives at the −1 position reacted more slowly under these conditions, producing the analogous thiazoline products in 65–85% yield after 24 h reaction at pH 8 (Figure 3B,C). Surprisingly, while all substrates containing +1 site modifications were efficiently oxidized to the corresponding thiazole (Figure 2B,D), only the substrate containing a geminal β2-dimethyl substituent at the −1 site was efficiently oxidized by ArtGox (70%) (Figure 3B,C). With the exception of substrate 4c, increasing the pH to 9.0 promoted formation of the desired thiazole product (Supplementary Figure 8B,C). However, even under these conditions only substrate 4b (88%) yielded a greater than 41% thiazole product (Supplementary Figure 8B,C). These data indicate that MicD-F and ArtGox are both less tolerant of non-α-amino acid monomers at the −1 site than at the +1 site. ArtGox appears especially intolerant of substitution or sp2 hybridization at the β3-position of substrates at the −1 site.
MicD-F Is Sensitive to Amino Acid Identity at the Cyclization Site
To complete the exploration of the substrate tolerance of MicD-F and ArtGox, we synthesized a set of potential substrates containing a non-α-amino acid directly at the cyclization site. Each contained a C-terminal AYD sequence preceded by either l-β3- or d-β3-threonine (Supplementary Figure 9A). Incubation of these substrates with MicD-F (50 mol %) under conditions (pH 8.0, 37 °C, 24 h) that resulted in substantial cyclization of a substrate containing l-α-threonine at the cyclization site led to no detectable cyclization (<1%) (Supplementary Figure 9B). Even at pH 9.0, no cyclization occurred (Supplementary Figure 9C), indicating that MicD-F is highly sensitive to amino acid identity at the site of cyclization. This result is in line with previous work that demonstrated the cyclodehydratase PatD failed to react with substrates containing d-α-threonine at the cyclization site.12
Redirecting RiPP Biosynthetic Enzymes to Intact Folded Proteins
Thiazolines and thiazole are replete in natural products39−41 and synthetic drug-like small molecules,42,43 and calculations confirm the expected decrease in conformational freedom that derives from aromatic and/or sp2 character within the peptide backbone.44 This finding and the leader-independent nature of MicD-F and ArtGox-mediated thiazol(in)e biosynthesis inspired us to explore substrates in which the site of cyclodehydration/dehydrogenation is embedded within a stable protein fold (Figure 4). We first asked whether MicD-F and ArtGox could install thiazol(in)e linkages within loops and/or at the termini of mCherry. mCherry is a prototypic fluorescent beta-barrel protein derived from DsRed, isolated originally from Discosoma sea anemones.45 We cloned, expressed, and purified a set of mCherry variants in which the core sequence MCAYDG was appended to the mCherry C-terminus (mCherryC+) or inserted into a loop immediately downstream of residues D137 (mCherry137+), D174 (mCherry174+), V192 (mCherry192+), or E211 (mCherry211+) (Figure 4A, Supplementary Table 2, Supplementary Figure 10). Although mCherry137+ and mCherry211+ were partially/completely nonfluorescent or could not be purified, mCherryC+, mCherry174+ and mCherry192+ were soluble and fluorescent. In all three of these cases, mass spectrometry of the purified proteins showed the characteristic loss of 22 Da, indicating chromophore maturation (Supplementary Figure 11).
Figure 4.
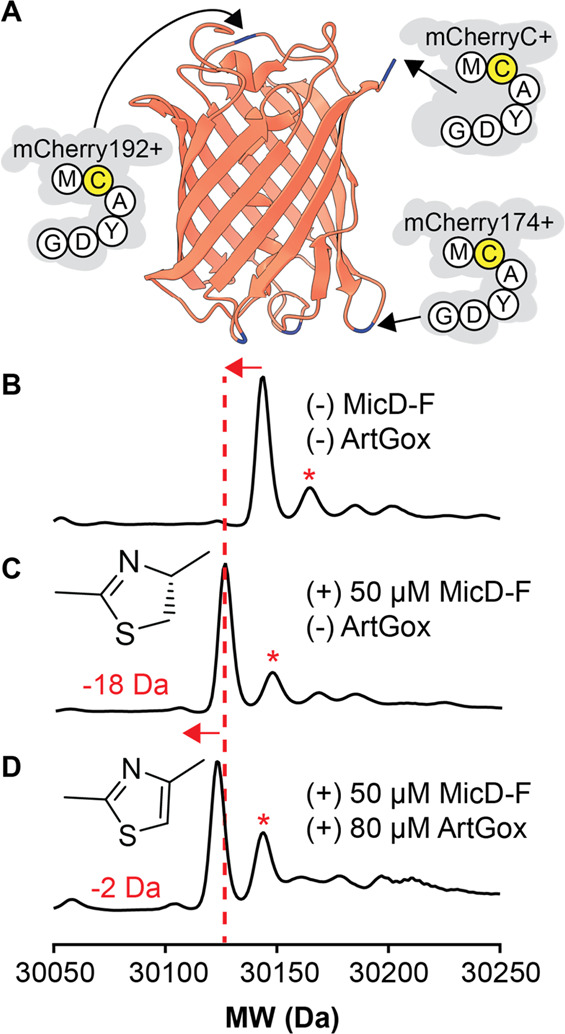
(A) mCherry variants evaluated as substrates for ArtGox and/or MicD-F. Each variant contains the sequence MCAYDG inserted following the residue shown. LC/MS analysis of mCherryC+ both (B) before and (C) after reaction with 50 mol % MicD-F or (D) 50 mol % MicD-F and 80 mol % ArtGox. Data reported are normalized counts from deconvoluted mass spectra. Asterisks indicate the molecular weight of the parent protein without a mature chromophore (+22 Da).
Treatment of mCherryC+ with 50 mol % MicD-F (pH 9.0, 24 h, 37 °C) led to virtually complete conversion to the thiazoline product as indicated by a loss of water in the deconvoluted mass spectrum (Figure 4B,C). No such mass change was observed in an analogous reaction containing mCherryC-, which carries the sequence MAAYDG in place of MCAYDG at the C-terminus, providing evidence that the observed cyclodehydration demanded a Cys residue immediately upstream of the AYD recognition sequence (Supplementary Figure 12B,C). Neither mCherry174+ nor mCherry192+ displayed the loss of water characteristic of successful cyclodehydration even after 24 h at 37 °C (Supplementary Figure 12D,E). Nevertheless, we explored the potential for MicD-F and ArtGox to act in tandem to install an aromatic thiazole backbone in mCherryC+. Simultaneous treatment of mCherryC+ for 24 h (pH 9.0, 37 °C) with MicD-F (50 mol %) and ArtGox (80 mol %) resulted in the expected −2 Da shift in the deconvoluted mass spectrum (Figure 4D) relative to that of mCherryC+ treated with only MicD-F (Figure 4C). This result indicates that the MicD-F/ArGox enzyme pair can post-translationally install an aromatic thiazole backbone within a structurally unconstrained region of a well-folded beta-barrel protein.
We hypothesized that the absence of cyclodehydration reactivity for mCherry174+ and mCherry192+ at 37 °C was due to neighboring structural elements that disfavor productive interaction with MicD-F and/or enzyme-promoted thiazoline formation. Therefore, we carried out a second set of cyclodehydration reactions at 42 °C, the highest temperature at which MicD-F remained stable in our hands. At this elevated temperature, mCherryC+ again displayed cysteine-specific loss of water characteristic of successful cyclodehydration (Supplementary Figure 13B,C). However, again neither mCherry174+ or mCherry192+ displayed the loss of water characteristic of successful cyclodehydration after 24 h at 42 °C (Supplementary Figure 13D,E). It has been reported46 that the apparent melting temperature of mCherry is upward of 90 °C. Taken together with our results, this finding suggests that there is an inherent mismatch between the temperature stability of MicD-F and the thermodynamic stabilities of the mCherry loop insertions evaluated here.
To test this hypothesis, we sought a folded, globular protein with a lower melting temperature than mCherry with the expectation that it would be more amenable to insertion of an internal thiazol(in)e linkage. Rop is a homodimeric four-helix bundle protein formed by the antiparallel association of two helix-turn-helix monomers.47 Regan and co-workers reported many years ago that the native two-residue turn in Rop could be replaced by up to 10 glycine residues without loss of the native dimer structure. The Rop variant with the longest insertion—Gly10—melted cooperatively at 50 °C,48 suggesting that it might tolerate an embedded thiazole or thiazoline heterocycle (Figure 5A). To test this hypothesis, we expressed and purified three Rop variants containing a single CAYD sequence embedded near the N-terminus (RopN), the C-terminus (RopC), or centrally (RopM) within a 10-residue glycine-rich loop (Figure 5A, Supplementary Table 3, Supplementary Figure 14, and Supplementary Figure 15). All three Rop variants exhibited high α-helical content at 20 μM as judged by wavelength-dependent CD measurements (Figure 5B). RopC and RopM migrated as discrete dimers at 50 μM as judged by size-exclusion chromatography (SEC) and melted cooperatively and reversibly with TM values of 28 and 32 °C (Supplementary Figure 16). RopN, by contrast, migrated as a heterogeneous mixture upon SEC and melted noncooperatively, albeit at a slightly higher apparent TM (43 °C) perhaps because of disulfide formation (Supplementary Figure 16).49
Figure 5.

MicD-F and ArtGox act in tandem to install thiazoline and thiazole backbones within globular proteins. (A) Cartoons illustrating the sequences of RopC, RopM, and RopN and the conditions used for MicD-F-catalyzed cyclodehydration (left arrow) or tandem cyclodehydration/dehydrogenation catalyzed by MicD-F and ArtGox (right arrow). (B) Wavelength-dependent circular dichroism spectra of RopC, RopM, and RopN at [monomer] = 20 μM in 10 mM phosphate, 100 mM NaCl, 150 μM TCEP, pH 7.0 and 25 °C. (C) LC/MS analysis of the reaction of RopC, RopN, and RopM with MicD-F under the conditions shown in panel (A) above. The characteristic loss of 18 mass units upon cyclodehydration is evident for both RopC and RopM; the reaction of RopM was incomplete under these conditions. (D) Treatment of RopC with MicD-F and ArtGox under the conditions shown in panel (A) above led to clean conversion into the corresponding thiazole (RopC-Z). (E) The wavelength-dependent CD spectra of RopC-U and RopC-Z compared to RopC; these data are not corrected for contributions due to the thiazoline or thiazole linkage. (F) The melting temperatures of RopC, RopC-U, and RopC-Z (after refolding) are almost identical.
Although RopC, RopN, and RopM all contained the same CAYD recognition sequence, only one—RopC—underwent clean conversion into the corresponding thiazoline upon treatment with 50 mol % MicD-F (pH 9.0, 37 °C, 16 h). RopM reacted partially under these conditions, and RopN was unreactive (Figure 5C). Reaction of RopC to generate thiazoline RopC-U proceeded more slowly at 25 °C (Supplementary Figure 17). RopC could be converted directly into the thiazole RopC-Z upon treatment with 50 mol % MicD-F and 80 mol % ArtGox (Figure 5D, Supplementary Figure 18). No reaction was observed when the Cys residue within the RopC reaction site was replaced with Ala or when the C-terminal AYD sequence was replaced by GGG (Supplementary Figure 19).
The products of the reaction of RopC with MicD-F (RopC-U) and with MicD-F and ArtGox (RopC-Z) were purified, confirmed via protease digest (Supplementary Figure 20), and analyzed by size-exclusion chromatography and wavelength- and temperature-dependent CD. Thiazoline-containing RopC-U was a homogeneous dimer as judged by SEC (Supplementary Figure 16) and retained a significant level of α-helical structure (Figure 5E). It also melted cooperatively and reversibly with a TM value of 27 °C, a value almost identical to that of RopC itself (28 °C) (Figure 5F). Thiazole-containing RopC-Z displayed more complex behavior; it was less homogeneous as judged by SEC and melted cooperatively (TM = 24 °C) but only after a refolding step (Supplementary Figure 21). These results indicate that the MicD-F can post-translationally install a thiazoline within a backbone of a helical bundle protein, and that ArtGox can oxidize this substrate to install a fully aromatic thiazole unit.
Computational Analysis of the Effects of Thiazoline/Thiazole Formation on Local Backbone Flexibility
To explore the effects of thiazoline/thiazole formation on local backbone flexibility, we examined the conformational space of the tetrapeptide Ac-AACA-NH2. The use of this simplified substrate allowed the inherent peptide backbone energetics to be evaluated without the complications of side chain fluctuations. Molecular mechanics methods (Macromodel, OPLS4 force field, implemented in Schrödinger Maestro software) were first used to generate and minimize large populations of conformers for cysteine-, thiazoline-, and thiazole-containing analogues (Figure 6A). For each species, 10 000 starting structures were sampled using the mixed torsional/low-mode method. All conformers within 4 kcal/mol of each global minimum were then subjected to geometry optimization using DFT (Jaguar: B3LYP-D3/6-31G**). An SM8 method was used to determine the relative energies in aqueous media.50 All nonredundant conformers were then ranked based on these energies and compared.
Figure 6.
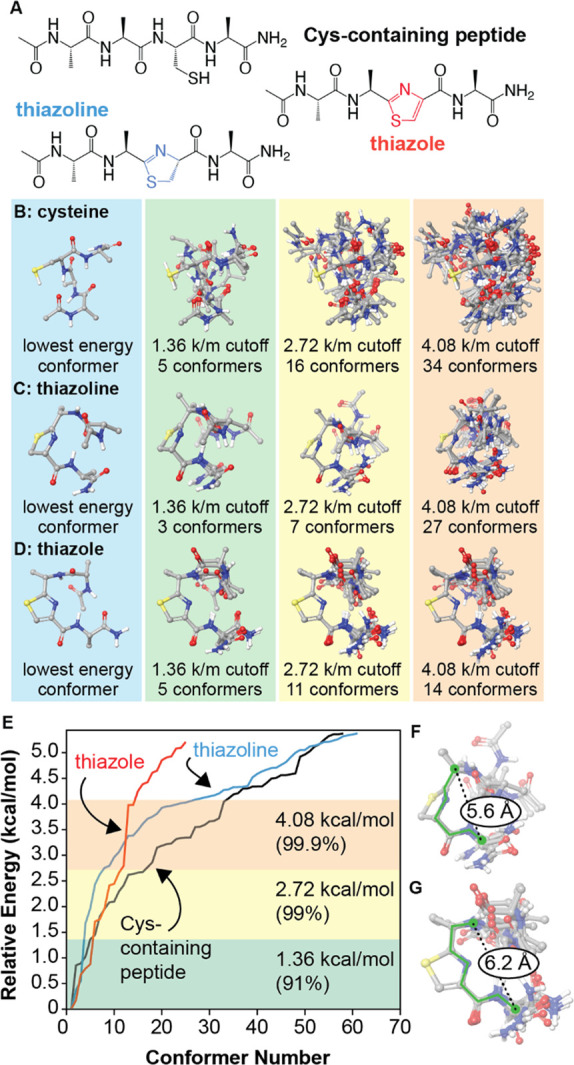
Conformational effects of thiazoline and thiazole formation. (A) The open chain and cyclized analogues Ac-AACA-NH2 were examined. Initial conformational searches were conducted using MacroModel (OPLS4 force field). All species within 4 kcal/mol of the global minimum were geometry optimized using DFT (B3LYP/6-31G**, SM8 solvent model) and reranked. (B–E) Lowest energy conformers are superimposed for different energy cutoff values. (F) A rigid six-bond motif (green) describes all identified conformers within 2.72 kcal/mol of the global minimum for the thiazoline. (G) A similar seven-bond motif describes the thiazole conformers. k/m = kcal/mol. For images of all structures within 4.08 kcal/mol of each global minimum, see Supporting Information.
The results of the conformational analysis appear in Figure 6B–E, sorted by progressive energy cutoffs relative to each global minimum. The noncyclized, cysteine-containing peptide exhibits the greatest flexibility, with 5 conformers being identified within 1.36 kcal/mol of the global minimum (91% of the population) and 16 within 2.72 kcal/mol (99%). Moreover, the identified conformers are largely non-superimposable, indicating that a high degree of conformational space is accessible within these energy ranges. In contrast, the thiazoline exhibits the most significant reduction in flexibility, with only three conformers identified at the 1.36 kcal/mol cutoff level and only seven identified at a cutoff of 2.72 kcal/mol. Superposition of the thiazoline rings of these conformers reveals a rigid six-bond motif that is preserved in all cases (Figure 6F). The thiazole analogue exhibits similarly reduced flexibility, with 11 conformers being identified within 2.72 kcal/mol of the global minimum. In this case, a rigid seven-bond motif can be identified (Figure 6G). These evaluations provide the basis of models that could be used to predict the conformational effects of backbone cyclization on larger sequences and could be used to predict sequence locations in which cyclizations are more likely to be successful. In current experiments, we are combining molecular dynamics studies with experimental data to examine the longer-range effects that result from introducing thiazoline and thiazole groups in longer peptides and intact proteins. Such information could be used to apply this chemistry more generally to improve the physiochemical properties of therapeutic proteins.
Conclusions
One can imagine two mutually synergistic strategies to introduce non-natural monomers into polypeptide and protein oligomers.51 One “bottom-up” approach relies on extant or engineered ribosomes to accept and process tRNAs carrying diverse noncanonical α-amino or non-α-amino acids.52 Hundreds of noncanonical α-amino acids (as well as α-hydroxy acids53−56) have been introduced into proteins in cells and animals using genetic code expansion,57,58 which usually relies on novel orthogonal aminoacyl tRNA synthetases to generate the requisite acylated tRNAs. Select noncanonical α-amino acids59 and one β-amino acid27 have also been incorporated into proteins in vivo using endogenous α-aminoacyl tRNA synthetases. Alternatively, many noncanonical α-amino acids, as well as certain non-α-amino acids, including β-amino acids29,60 and certain polyketide precursors,30 can be introduced into short peptides in vitro and on a small scale using genetic code reprogramming, in which a stoichiometric RNA coreagent (Flexizyme61) generates the requisite acylated tRNA.
The second “top-down” approach is reminiscent of late-stage functionalization reactions used to manipulate complex small molecule natural products62,63 and the natural biosynthetic strategy used to assemble ribosomally synthesized and post-translationally modified peptides (RiPPs).6 In this approach, enzymes, chemical reagents, or chemical catalysts are employed to post-translationally modify a peptide5 or protein51 to install a new or modified monomer. Examples of this approach include reactions of natural or noncanonical protein side chains or modification of the N- or C-terminus.64−67 The only backbone-focused nonenzymatic reaction of which we are aware is the O-mesitylenesulfonylhydroxylamine-promoted oxidative elimination of Cys residues to generate a dehydroalanine backbone68 that is subsequently modified. We note that the top-down and bottom-up strategies are complementary, and both have the potential to operate in vivo where very high protein titers are possible.69
Here, we show that a constitutively active form of MicD and ArtGox, two enzymes used in the biosynthesis of cyanobactin natural products,70 are sufficiently promiscuous to process substrates containing diverse backbone-modified monomers within substrate polypeptides, even at positions immediately preceding or following the site of cyclization/dehydrogenation. The backbone-modified monomers compatible with MicD-F and ArtGox include many accepted by extant ribosomes in small-scale in vitro reactions, including aminobenzoic acid derivatives and β2- and β3-amino acids. The products of these reactions are sequence-defined chemical polymers with multiple, diverse, non-α-amino acid monomers. We show further that cyclodehydration and dehydrogenation can install thiazoline or thiazole backbones within protein loops and linkers without disrupting the native tertiary fold. Calculations reported here reveal the extent to which these heterocycles restrict conformational space; they also eliminate a peptide bond—both features could improve the stability or add function to linker sequences now commonplace in emerging biotherapeutics. Moreover, as thiazoles and thiazoline heterocycles are replete in natural products,71−73 small molecule drugs,42,43 and peptide-mimetic therapeutics,74 their installation in protein-based biotherapeutics could improve or augment performance, activity, stability, and/or selectivity. This work represents a general strategy to expand the chemical diversity of the proteome without need for genetic manipulations.
Acknowledgments
This work was supported by the NSF Center for Genetically Encoded Materials (C-GEM), CHE 2002182. We are grateful to Professor James Naismith (Oxford University) for graciously sharing plasmids and advice, and to Professor Susan Marqusee and her laboratory for generously sharing CD instrumentation and advice.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c01577.
The authors declare the following competing financial interest(s): J.W. and A.S. have filed a patent related to this work.
Supplementary Material
References
- McGivern J. G. Ziconotide: A Review of Its Pharmacology and Use in the Treatment of Pain. Neuropsychiatric Disease and Treatment 2007, 3 (1), 69–85. 10.2147/nedt.2007.3.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A.; Lötsch J.; Freynhagen R.; Geisslinger G. Ziconotide for Treatment of Severe Chronic Pain. Lancet 2010, 375 (9725), 1569–1577. 10.1016/S0140-6736(10)60354-6. [DOI] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30 (1), 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M. A.; van der Donk W. A. New Insights into the Biosynthetic Logic of Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products. Cell Chemical Biology 2016, 23 (1), 31–44. 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Suga H. Engineering of RiPP Pathways for the Production of Artificial Peptides Bearing Various Non-Proteinogenic Structures. Curr. Opin. Chem. Biol. 2018, 46, 82–90. 10.1016/j.cbpa.2018.06.014. [DOI] [PubMed] [Google Scholar]
- Montalbán-López M.; Scott T. A.; Ramesh S.; Rahman I. R.; van Heel A. J.; Viel J. H.; Bandarian V.; Dittmann E.; Genilloud O.; Goto Y.; et al. New Developments in RiPP Discovery, Enzymology and Engineering. Nat. Prod. Rep. 2021, 38 (1), 130–239. 10.1039/D0NP00027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J.; Bent A. F.; Houssen W. E.; Mann G.; Jaspars M.; Naismith J. H. The Structural Biology of Patellamide Biosynthesis. Curr. Opin. Struct. Biol. 2014, 29, 112–121. 10.1016/j.sbi.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E. W.; Nelson J. T.; Rasko D. A.; Sudek S.; Eisen J. A.; Haygood M. G.; Ravel J. Patellamide A and C Biosynthesis by a Microcin-like Pathway in Prochloron Didemni, the Cyanobacterial Symbiont of Lissoclinum Patella. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (20), 7315–7320. 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia M. S.; Ravel J.; Schmidt E. W. A Global Assembly Line for Cyanobactins. Nat. Chem. Biol. 2008, 4 (6), 341–343. 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tianero Ma. D. B.; Donia M. S.; Young T. S.; Schultz P. G.; Schmidt E. W. Ribosomal Route to Small-Molecule Diversity. J. Am. Chem. Soc. 2012, 134 (1), 418–425. 10.1021/ja208278k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J.; Morawitz F.; Bent A. F.; Houssen W. E.; Shirran S. L.; Fuszard M. A.; Smellie I. A.; Botting C. H.; Smith M. C. M.; Jaspars M.; et al. An Enzymatic Route to Selenazolines. ChemBioChem. 2013, 14 (5), 564–567. 10.1002/cbic.201300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Suga H. In Vitro Biosynthesis of Peptides Containing Exotic Azoline Analogues. ChemBioChem. 2020, 21 (1–2), 84–87. 10.1002/cbic.201900521. [DOI] [PubMed] [Google Scholar]
- Kato Y.; Kuroda T.; Huang Y.; Ohta R.; Goto Y.; Suga H. Chemoenzymatic Posttranslational Modification Reactions for the Synthesis of Ψ[CH 2 NH]-Containing Peptides. Angew. Chem., Int. Ed. 2020, 59 (2), 684–688. 10.1002/anie.201910894. [DOI] [PubMed] [Google Scholar]
- Hudson G. A.; Zhang Z.; Tietz J. I.; Mitchell D. A.; van der Donk W. A. In Vitro Biosynthesis of the Core Scaffold of the Thiopeptide Thiomuracin. J. Am. Chem. Soc. 2015, 137 (51), 16012–16015. 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming S. R.; Bartges T. E.; Vinogradov A. A.; Kirkpatrick C. L.; Goto Y.; Suga H.; Hicks L. M.; Bowers A. A. Flexizyme-Enabled Benchtop Biosynthesis of Thiopeptides. J. Am. Chem. Soc. 2019, 141 (2), 758–762. 10.1021/jacs.8b11521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S.; Ozaki T.; Asamizu S.; Ikeda H.; O̅mura S.; Oku N.; Igarashi Y.; Tomoda H.; Onaka H. Genome Mining Reveals a Minimum Gene Set for the Biosynthesis of 32-Membered Macrocyclic Thiopeptides Lactazoles. Chem. & Biol. 2014, 21 (5), 679–688. 10.1016/j.chembiol.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Vinogradov A. A.; Shimomura M.; Goto Y.; Ozaki T.; Asamizu S.; Sugai Y.; Suga H.; Onaka H. Minimal Lactazole Scaffold for in Vitro Thiopeptide Bioengineering. Nat. Commun. 2020, 11 (1), 2272. 10.1038/s41467-020-16145-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Ito Y.; Kato Y.; Tsunoda S.; Suga H. One-Pot Synthesis of Azoline-Containing Peptides in a Cell-Free Translation System Integrated with a Posttranslational Cyclodehydratase. Chemistry & Biology 2014, 21 (6), 766–774. 10.1016/j.chembiol.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Koehnke J.; Mann G.; Bent A. F.; Ludewig H.; Shirran S.; Botting C.; Lebl T.; Houssen W. E.; Jaspars M.; Naismith J. H. Structural Analysis of Leader Peptide Binding Enables Leader-Free Cyanobactin Processing. Nat. Chem. Biol. 2015, 11 (8), 558–563. 10.1038/nchembio.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oman T. J.; Knerr P. J.; Bindman N. A.; Velásquez J. E.; van der Donk W. A. An Engineered Lantibiotic Synthetase That Does Not Require a Leader Peptide on Its Substrate. J. Am. Chem. Soc. 2012, 134 (16), 6952–6955. 10.1021/ja3017297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y.; Czekster C. M.; Miller O. K.; Botting C. H.; Schwarz-Linek U.; Naismith J. H. Insights into the Mechanism of the Cyanobactin Heterocyclase Enzyme. Biochemistry 2019, 58 (16), 2125–2132. 10.1021/acs.biochem.9b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueis E.; Nardone B.; Jaspars M.; Westwood N. J.; Naismith J. H. Synthesis of Hybrid Cyclopeptides through Enzymatic Macrocyclization. ChemistryOpen 2017, 6 (1), 11–14. 10.1002/open.201600134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueis E.; Adamson C.; Mann G.; Ludewig H.; Redpath P.; Migaud M.; Westwood N. J.; Naismith J. H. Derivatisable Cyanobactin Analogues: A Semisynthetic Approach. ChemBioChem. 2015, 16 (18), 2646–2650. 10.1002/cbic.201500494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S.; Ge Y.; Bent A. F.; Schwarz-Linek U.; Naismith J. H. Oxidation of the Cyanobactin Precursor Peptide Is Independent of the Leader Peptide and Operates in a Defined Order. Biochemistry 2018, 57 (41), 5996–6002. 10.1021/acs.biochem.8b00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart B. J.; Kakkar N.; Hudson G. A.; van der Donk W. A.; Mitchell D. A. Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent. Sci. 2017, 3 (6), 629–638. 10.1021/acscentsci.7b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz L.; Koehnke J. Leader Peptide Exchange to Produce Hybrid, New-to-Nature Ribosomal Natural Products. Chem. Commun. 2021, 57 (52), 6372–6375. 10.1039/D0CC06889F. [DOI] [PubMed] [Google Scholar]
- Melo Czekster C.; Robertson W. E.; Walker A. S.; Söll D.; Schepartz A. In Vivo Biosynthesis of a β-Amino Acid-Containing Protein. J. Am. Chem. Soc. 2016, 138 (16), 5194–5197. 10.1021/jacs.6b01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini R.; Chowdhury S. R.; Dedkova L. M.; Roy B.; Daskalova S. M.; Paul R.; Chen S.; Hecht S. M. Protein Synthesis with Ribosomes Selected for the Incorporation of β-Amino Acids. Biochemistry 2015, 54 (23), 3694–3706. 10.1021/acs.biochem.5b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T.; Suga H. Ribosomal Incorporation of Consecutive β-Amino Acids. J. Am. Chem. Soc. 2018, 140 (38), 12159–12167. 10.1021/jacs.8b07247. [DOI] [PubMed] [Google Scholar]
- Ad O.; Hoffman K. S.; Cairns A. G.; Featherston A. L.; Miller S. J.; Söll D.; Schepartz A. Translation of Diverse Aramid- and 1,3-Dicarbonyl-Peptides by Wild Type Ribosomes in Vitro. ACS Cent. Sci. 2019, 5 (7), 1289–1294. 10.1021/acscentsci.9b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Schwieter K. E.; Watkins A. M.; Kim D. S.; Yu H.; Schwarz K. J.; Lim J.; Coronado J.; Byrom M.; Anslyn E. V.; et al. Expanding the Limits of the Second Genetic Code with Ribozymes. Nat. Commun. 2019, 10 (1), 5097. 10.1038/s41467-019-12916-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T.; Suga H. Ribosomal Elongation of Aminobenzoic Acid Derivatives. J. Am. Chem. Soc. 2020, 142, 16518–16522. 10.1021/jacs.0c05765. [DOI] [PubMed] [Google Scholar]
- Chen S.; Ji X.; Gao M.; Dedkova L. M.; Hecht S. M. In Cellulo Synthesis of Proteins Containing a Fluorescent Oxazole Amino Acid. J. Am. Chem. Soc. 2019, 141 (14), 5597–5601. 10.1021/jacs.8b12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemert N.; Ishida K.; Quillardet P.; Bouchier C.; Hertweck C.; de Marsac N. T.; Dittmann E. Microcyclamide Biosynthesis in Two Strains of Microcystis Aeruginosa: From Structure to Genes and Vice Versa. AEM 2008, 74 (6), 1791–1797. 10.1128/AEM.02392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia M. S.; Schmidt E. W. Linking Chemistry and Genetics in the Growing Cyanobactin Natural Products Family. Chemistry & Biology 2011, 18 (4), 508–519. 10.1016/j.chembiol.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W.; Dong S.-H.; Sarkar S.; Nair S. K.; Schmidt E. W.. The Biochemistry and Structural Biology of Cyanobactin Pathways: Enabling Combinatorial Biosynthesis. In Methods in Enzymology; Elsevier, 2018; Vol. 604, pp 113–163. 10.1016/bs.mie.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp J. M.; Ad O.; Amikura K.; Ward F. R.; Garcia E. M.; Cate J. H. D.; Schepartz A.; Söll D. Initiation of Protein Synthesis with Non-Canonical Amino Acids In Vivo. Angew. Chem., Int. Ed. 2020, 59 (8), 3122–3126. 10.1002/anie.201914671. [DOI] [PubMed] [Google Scholar]
- Tharp J. M.; Vargas-Rodriguez O.; Schepartz A.; Söll D. Genetic Encoding of Three Distinct Noncanonical Amino Acids Using Reprogrammed Initiator and Nonsense Codons. ACS Chem. Biol. 2021, 16 (4), 766–774. 10.1021/acschembio.1c00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Do T.; Link A. J. Mechanisms of Action of Ribosomally Synthesized and Posttranslationally Modified Peptides (RiPPs). Journal of Industrial Microbiology and Biotechnology 2021, 48 (3–4), kuab005. 10.1093/jimb/kuab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. C. K.; Burrows L. L. Thiopeptides: Antibiotics with Unique Chemical Structures and Diverse Biological Activities. J. Antibiot. 2021, 74 (3), 161–175. 10.1038/s41429-020-00387-x. [DOI] [PubMed] [Google Scholar]
- Süssmuth R. D.; Mainz A. Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chem., Int. Ed. 2017, 56 (14), 3770–3821. 10.1002/anie.201609079. [DOI] [PubMed] [Google Scholar]
- Taylor R. D.; MacCoss M.; Lawson A. D. G. Rings in Drugs: Miniperspective. J. Med. Chem. 2014, 57 (14), 5845–5859. 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals: Miniperspective. J. Med. Chem. 2014, 57 (24), 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Staś M.; Broda M. A.; Siodłak D. Thiazole–Amino Acids: Influence of Thiazole Ring on Conformational Properties of Amino Acid Residues. Amino Acids 2021, 53 (5), 673–686. 10.1007/s00726-021-02974-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner N. C.; Campbell R. E.; Steinbach P. A.; Giepmans B. N. G.; Palmer A. E.; Tsien R. Y. Improved Monomeric Red, Orange and Yellow Fluorescent Proteins Derived from Discosoma Sp. Red Fluorescent Protein. Nat. Biotechnol. 2004, 22 (12), 1567–1572. 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Rana M. S.; Wang X.; Banerjee A. An Improved Strategy for Fluorescent Tagging of Membrane Proteins for Overexpression and Purification in Mammalian Cells. Biochemistry 2018, 57 (49), 6741–6751. 10.1021/acs.biochem.8b01070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner D. W.; Kokkinidis M.; Tsernoglou D. Structure of the ColE1 Rop Protein at 1.7 Å Resolution. J. Mol. Biol. 1987, 196 (3), 657–675. 10.1016/0022-2836(87)90039-8. [DOI] [PubMed] [Google Scholar]
- Nagi A. D.; Regan L. An Inverse Correlation between Loop Length and Stability in a Four-Helix-Bundle Protein. Folding and Design 1997, 2 (1), 67–75. 10.1016/S1359-0278(97)00007-2. [DOI] [PubMed] [Google Scholar]
- Amprazi M.; Kotsifaki D.; Providaki M.; Kapetaniou E. G.; Fellas G.; Kyriazidis I.; Perez J.; Kokkinidis M. Structural Plasticity of 4- -Helical Bundles Exemplified by the Puzzle-like Molecular Assembly of the Rop Protein. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (30), 11049–11054. 10.1073/pnas.1322065111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113 (18), 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Hoyt E. A.; Cal P. M. S. D.; Oliveira B. L.; Bernardes G. J. L. Contemporary Approaches to Site-Selective Protein Modification. Nature Reviews Chemistry 2019, 3 (3), 147–171. 10.1038/s41570-019-0079-1. [DOI] [Google Scholar]
- Noren C.; Anthony-Cahill S.; Griffith M.; Schultz P. A General Method for Site-Specific Incorporation of Unnatural Amino Acids into Proteins. Science 1989, 244 (4901), 182–188. 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- England P. M.; Zhang Y.; Dougherty D. A.; Lester H. A. Backbone Mutations in Transmembrane Domains of a Ligand-Gated Ion Channel: Implications for the Mechanism of Gating. Cell 1999, 96 (1), 89–98. 10.1016/S0092-8674(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Guo J.; Wang J.; Anderson J. C.; Schultz P. G. Addition of an α-Hydroxy Acid to the Genetic Code of Bacteria. Angew. Chem., Int. Ed. 2008, 47 (4), 722–725. 10.1002/anie.200704074. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Yanagisawa T.; Sakamoto K.; Yokoyama S. Recognition of Non-α-Amino Substrates by Pyrrolysyl-TRNA Synthetase. J. Mol. Biol. 2009, 385 (5), 1352–1360. 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- Li Y.-M.; Yang M.-Y.; Huang Y.-C.; Li Y.-T.; Chen P. R.; Liu L. Ligation of Expressed Protein α-Hydrazides via Genetic Incorporation of an α-Hydroxy Acid. ACS Chem. Biol. 2012, 7 (6), 1015–1022. 10.1021/cb300020s. [DOI] [PubMed] [Google Scholar]
- Chin J. W. Expanding and Reprogramming the Genetic Code. Nature 2017, 550 (7674), 53–60. 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- Young D. D.; Schultz P. G. Playing with the Molecules of Life. ACS Chem. Biol. 2018, 13 (4), 854–870. 10.1021/acschembio.7b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.; Lu Y.; Van Deventer J.; Tirrell D. Residue-Specific Incorporation of Non-Canonical Amino Acids into Proteins: Recent Developments and Applications. Current Opinion in Chemical Biology 2010, 14 (6), 774–780. 10.1016/j.cbpa.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adaligil E.; Song A.; Hallenbeck K. K.; Cunningham C. N.; Fairbrother W. J. Ribosomal Synthesis of Macrocyclic Peptides with B2- and B2,3-Homo-Amino Acids for the Development of Natural Product-Like Combinatorial Libraries. ACS Chem. Biol. 2021, 16 (6), 1011–1018. 10.1021/acschembio.1c00062. [DOI] [PubMed] [Google Scholar]
- Morimoto J.; Hayashi Y.; Iwasaki K.; Suga H. Flexizymes: Their Evolutionary History and the Origin of Catalytic Function. Acc. Chem. Res. 2011, 44 (12), 1359–1368. 10.1021/ar2000953. [DOI] [PubMed] [Google Scholar]
- Blakemore D.; Castro L.; Churcher I.; Rees D.; Thomas A.; Wilson D.; Wood A. Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nature Chemistry 2018, 10 (4), 383–394. 10.1038/s41557-018-0021-z. [DOI] [PubMed] [Google Scholar]
- Bostrom J.; Brown D.; Young R.; Keseru G. Expanding the Medicinal Chemistry Synthetic Toolbox. Nature Reviews Drug Discovery 2018, 17 (10), 709–727. 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- Bloom S.; Liu C.; Kölmel D. K.; Qiao J. X.; Zhang Y.; Poss M. A.; Ewing W. R.; MacMillan D. W. C. Decarboxylative Alkylation for Site-Selective Bioconjugation of Native Proteins via Oxidation Potentials. Nat. Chem. 2018, 10 (2), 205–211. 10.1038/nchem.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobba M. J.; Fellmann C.; Marmelstein A. M.; Maza J. C.; Kissman E. N.; Robinson S. A.; Staahl B. T.; Urnes C.; Lew R. J.; Mogilevsky C. S.; et al. Site-Specific Bioconjugation through Enzyme-Catalyzed Tyrosine–Cysteine Bond Formation. ACS Cent. Sci. 2020, 6 (9), 1564–1571. 10.1021/acscentsci.0c00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen C. B.; Francis M. B. Targeting the N Terminus for Site-Selective Protein Modification. Nat. Chem. Biol. 2017, 13 (7), 697–705. 10.1038/nchembio.2416. [DOI] [PubMed] [Google Scholar]
- Marmelstein A. M.; Lobba M. J.; Mogilevsky C. S.; Maza J. C.; Brauer D. D.; Francis M. B. Tyrosinase-Mediated Oxidative Coupling of Tyrosine Tags on Peptides and Proteins. J. Am. Chem. Soc. 2020, 142 (11), 5078–5086. 10.1021/jacs.9b12002. [DOI] [PubMed] [Google Scholar]
- Bernardes G.; Chalker J.; Errey J.; Davis B. Facile Conversion of Cysteine and Alkyl Cysteines to Dehydroalanine on Protein Surfaces: Versatile and Switchable Access to Functionalized Proteins. Journal of the American Chemical Society 2008, 130 (15), 5052–5053. 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- Gonzalez S. S.; Ad O.; Shah B.; Zhang Z.; Zhang X.; Chatterjee A.; Schepartz A. Genetic Code Expansion in the Engineered Organism Vmax X2: High Yield and Exceptional Fidelity. ACS Cent. Sci. 2021, 7 (9), 1500–1507. 10.1021/acscentsci.1c00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czekster C.; Ge Y.; Naismith J. Mechanisms of Cyanobactin Biosynthesis. Current Opinion in Chemical Biology 2016, 35, 80–88. 10.1016/j.cbpa.2016.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S.; Herrmann J.; Raju R.; Steinmetz H.; Mohr K. I.; Hüttel S.; Harmrolfs K.; Stadler M.; Müller R. Cystobactamids: Myxobacterial Topoisomerase Inhibitors Exhibiting Potent Antibacterial Activity. Angew. Chem., Int. Ed. 2014, 53 (52), 14605–14609. 10.1002/anie.201409964. [DOI] [PubMed] [Google Scholar]
- Groß S.; Schnell B.; Haack P. A.; Auerbach D.; Müller R. In Vivo and in Vitro Reconstitution of Unique Key Steps in Cystobactamid Antibiotic Biosynthesis. Nat. Commun. 2021, 12 (1), 1696. 10.1038/s41467-021-21848-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planke T.; Cirnski K.; Herrmann J.; Müller R.; Kirschning A. Synthetic and Biological Studies on New Urea and Triazole Containing Cystobactamid Derivatives. Chem.—Eur. J. 2020, 26 (19), 4289–4296. 10.1002/chem.201904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrele C.; Martinek T. A.; Reiser O.; Berlicki Ł. Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J. Med. Chem. 2014, 57 (23), 9718–9739. 10.1021/jm5010896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.